
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-palindromic (C^C^D) gold(III) pincer complexes are not accessible by intramolecular oxidative addition of biphenylenes – an experimental and quantum chemical study†
Wolfram
Feuerstein
 and
Frank
Breher
*
and
Frank
Breher
*
Karlsruhe Institute of Technology (KIT), Institute of Inorganic Chemistry, Division Molecular Chemistry, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: breher@kit.edu
First published on 9th June 2021
Abstract
We herein report on the synthesis of biphenylenes substituted with a pyridine (N), a phosphine (P) and a carbene (C′) donor as well as a carbene donor with additional pyridine in the lateral position. We describe the synthesis and structures of derived gold(I) complexes, which we tried to use for the synthesis of non-palindromic [(C^C^D)AuIII] pincer complexes by means of an intramolecular oxidative addition of the strained biphenylene ring. However, the anticipated formation of gold(III) complexes failed due to kinetic and thermodynamic reasons, which we extensively investigated by quantum chemical calculations. Furthermore, we shed light on the oxidative addition of biphenylene to two different gold(I) systems reported in the literature. Our comprehensive quantum-chemical analysis is complemented by NMR experiments.
Introduction
Phosphorescent organic light-emitting diodes (OLEDs) based on transition metal complexes mainly incorporate the heavy metals iridium(III),1–4 ruthenium(II)5–7 or platinum(II).8–13 However, in the last 15 years, structures based on gold(III) have gained increasing interest. Most of these systems incorporate the 2,6-diphenylpyridine based (C^N^C) pincer motif,14–23 the rigid nature of which inhibits thermal relaxation.24,25 In addition, the (C^N^C) pincer ensures a high ligand field splitting, which, upon photoexcitation, avoids the population of metal-centred d-states regularly seen to be responsible for non-radiative relaxation processes.26,27Nevertheless, the (C^N^C) motif is by far not the only ligand suitable for the preparation of luminescent gold(III) structures. von Arx et al. described cyclometallated (C^C′) (C′ = N-heterocyclic carbene, NHC) gold(III) complexes with one or two phenyl alkynyl or pentafluorophenyl ligands.28 There are also reports on biphenyldiyl (C^C) complexes of gold(III), the first by Usón et al. in 1980,29 who prepared dibenzo stannoles and used the latter to transmetallate the biphenyl moiety to gold(III) salts. This approach was used by different groups for the preparation of luminescent gold(III) complexes.30–32
In 2015, Nevado and co-workers33 described the non-palindromic34 (C^C^N) analogue of the (C^N^C) pincer and proved that the corresponding gold(III) complexes outperform most palindromic congeners with regard to emission quantum yields.35 In subsequent reports it was shown that the [(C^C^N)Au(III)] complexes differentiate from their palindromic counterparts with respect to chemical properties as well.35–37 The (C^C^N) motif may be seen as a pincer variant of the bidentate biphenyl (C^C) ligand. Thus, only recently, we developed a synthetic access to pyridine and NHC substituted 2,2′-dihalobiphenyls, which are suitable pre-ligands for the preparation of highly luminescent (C^C^N) and (C^C^C′) gold(III) complexes by transmetallation of the respective stannoles.38 In addition, we could show that 2,2′-dihalobiphenyls may be applied to palladium using a double oxidative addition – comproportionation sequence.39
However, in the course of searching for new variants for introducing (C^C^D)-based pincer ligands, we followed a different idea as well: In 2015, Dean Toste and co-workers showed a gold(I) NHC complex to be able to undergo an oxidative addition into the strained ring of biphenylene, thereby forming the corresponding cyclometalated [(C^C)AuIII] complex 3 (Chart 1A),40 a reaction which was already shown to occur for group 10 metals.41–45 Only shortly thereafter, the group of Didier Bourissou succeeded in a similar reaction employing a bidentate 1,2-diphosphano-1,2-dicarba-closo-dodecaborane ligand (Chart 1B).46 Guy Bertrand and co-workers developed cyclic alkylamino carbenes (CAAC) substituted with an additional lateral donor, which enabled the oxidative addition of biphenylene to a gold(I) centre as well (Chart 1C).47 A lateral amine-donor was also employed by Bourissou and co-workers for the oxidative addition of aryl iodide and biphenylene to a gold(I) phosphine complex.48
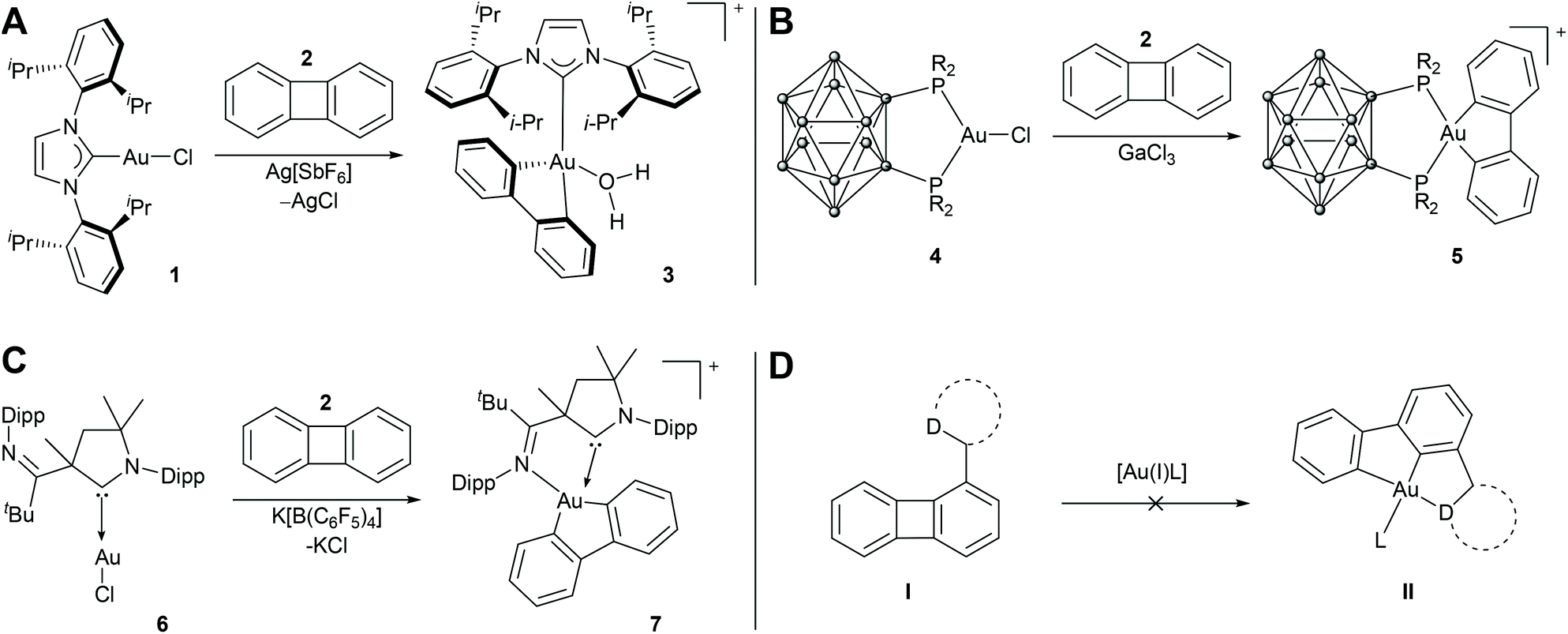 | ||
| Chart 1 Oxidative addition of biphenylene (2) to gold(I) complexes reported in the literature (A–C) and the unsuccessful approach to prepare [(C^C^D)AuIII] complexes II by intramolecular oxidative addition of 2-substituted biphenylenes I investigated in this study (D). Anions are omitted for clarity. R = Ph, iPr; iPr = isopropyl; Dipp = 2,6-diisopropylphenyl; tBu = tert-butyl; D = 2-pyridine, CH2PPh2, NHC; L = arbitrary neutral or anionic ligand or solvent molecule. | ||
Thus, to broaden the opportunities to introduce biphenyl-based (C^C^D) pincers to gold(III), we focused on the synthesis of donor substituted biphenylenes, which should serve to prepare the anticipated complexes by means of an intramolecular oxidative addition of the biphenylene motif (Chart 1D). This approach was shown to succeed for [(C^C^N)IrIII] complexes by Matsubara and co-workers.49
In this study, we describe the synthesis of such biphenylenes and discuss some of their gold(I) complexes. We show by comprehensive quantum chemical calculations that the intramolecular oxidative addition is impossible due to kinetic hindrance or the thermodynamic instability of reaction intermediates. Furthermore, we investigate the oxidative additions reported by Bertrand and Toste and discuss the conditions responsible for successful oxidative additions of biphenylenes to gold(I). NMR experiments complete our report.
Results and discussion
Synthesis of biphenylene ligands
Starting from commercially available 1,3-dibromobenzene (8), we prepared 1-bromobiphenylene (11) according to a modified literature procedure (Scheme 1).50–53 | ||
| Scheme 1 Synthesis of 2-bromobiphenylene (11) starting from 1,3-dibromobenzene (8). Conditions: (a) (1) diisopropylamine, nBuLi, THF, −78 °C, 2 h (2) I2, −78 °C → rt; (b) (1) tBuLi, THF, −100 °C, 1 h (2) 1,2-dibromobenzene, −78 °C → rt, 16 h; (c) (1) nBuLi, THF, −78 °C, 2 h (2) ZnBr2, −50 °C (3) CuCl2, −78 °C → rt, 16 h. Bottom right: Molecular structure of 11 in the solid state. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Only one molecule of the asymmetric unit is shown. Selected bond lengths (pm) and angles (°): C6–C7 149–6(3), C6–C5 142–0(3), C7–C12 142–0(3), C12–C5 151–4(4), C5–C6–C7 90–9(2), C6–C7–C12 89–9(2), and C7–C12–C5 90–1(2)–C12–C5–C6 89–1(2). | ||
Biphenylene 11 forms by the CuII-mediated ring closure of a zinkacyclopentadiene.53 The latter is accessible in situ by the twofold lithiation of 10 and successive transmetallation with ZnBr2. This kind of reaction is highly temperature-sensitive leading to a significantly reduced yield when upscaled (10 mmol ∼ 50%).50 However, we could overcome this problem by very slow addition of all reagents and by the use of a cooled (−75 °C) dropping funnel. Thus, we could perform this synthesis with excellent yields of 91% on scales >15 mmol.
1-Bromobiphenylene (11) served as the starting point to synthesize 2-(biphenylen-1-yl)pyridine (12) according to Koga et al.49 and to obtain diphenylphosphine 15 in a three-step procedure (Scheme 2).
 | ||
| Scheme 2 Synthesis of phosphine 15. Conditions: (a) (1) tBuLi, Et2O, −78 °C, 1 h (2) paraformaldehyde, −78 °C → rt, 16 h; (b) thionylchloride, pyridine, Et2O, reflux, 2 h; (c) (1) HPPh2, nBuLi, THF, −78 °C → rt, 2 h (2) 14, THF, −78 °C → rt, 16 h. | ||
Furthermore, imidazole 16 was obtained in excellent yields by Pd-catalysed C–N-coupling using a Cu-based catalyst developed by Buchwald and co-workers (Scheme 3).54
 | ||
| Scheme 3 Synthesis of imidazole 16. Conditions: imidazole, Cu2O (5 mol%), 4,7-dimethoxy-1,10-phenanthroline (15 mol%), Cs2CO3, PEG 400, butyronitrile, 110 °C, 24 h. | ||
Quaternization of 16 was accomplished using [Et3O][BF4] or 2-bromomethylpyridine to obtain the imidazolium salts 17 and 18 (Scheme 4).
 | ||
Scheme 4 Quaternization of imidazole 16. Conditions: (a) (1) (2-bromomethyl)pyridine hydrobromide, NaHCO3, H2O, Et2O, 0 °C, 10 min (2) 16, MeOH, rt, 16 h; (b) [Et3O][BF4], CH2Cl2, rt, 16 h; (c) Dowex® (chloride form), MeOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), rt, 16 h. :1), rt, 16 h. | ||
We anticipated pyridine substituted imidazolium salt 18 to serve as a tetradentate (C^C^C′^N) ligand able to stabilize cationic AuIII centres after oxidative addition into the biphenylene.55
Intramolecular oxidative addition
Phosphine 15 readily displaces tetrahydrothiophene of [(tht)AuCl] to form complex 19 in excellent yields (Scheme 5). 19 crystallizes from CH2Cl2/n-hexane in the form of colourless prisms suitable for X-ray diffraction analysis (Fig. 1). The Au–P bond length is comparable to other Au(I) phosphine complexes.56 The molecular structure does not indicate any interaction of the gold atom with the biphenylene unit. We also prepared NHC gold(I) complex 20 using imidazolium salt 17b (Scheme 6). Crystals suitable for X-ray diffraction analysis were obtained from CH2Cl2/n-hexane (Fig. 2). In contrast to phosphine complex 19, the gold atom of 20 is tilted in the same direction as the biphenylene unit and the C′–Au–Cl bond axis is slightly bent (177°). However, this is not indicative of a significant gold–biphenylene interaction.
 | ||
| Scheme 5 Preparation of phosphine complex 19. Conditions: [(THT)AuCl], CH2Cl2, rt, 24 h. | ||
 | ||
| Fig. 1 Molecular structure of 19 in the solid state. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (pm) and angles (°): P–Au 222.5(1) and Au–Cl 227.8(1). | ||
 | ||
| Scheme 6 Preparation of NHC gold(I) complex 20. The silver NHC complex 17c was prepared in situ and not isolated. Conditions: (a) Ag2O, CH2Cl2, rt, 16 h; (b) [(THT)AuCl], 3 h. | ||
 | ||
| Fig. 2 Molecular structure of 20 in the solid state. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (pm) and angles (°): C13–Au 197.9(4), Au–Cl 228.6(1), and C6–C1–N1–C13 50.0(6). | ||
Since neutral AuI pyridine complexes are rare57 due to the incompatibility of the hard pyridine donor and the soft gold(I) atom, we prepared cationic AuI complex 21 with pyridine ligand 12 (Scheme 7).
 | ||
| Scheme 7 Synthesis of cationic Au(I) complex 21. Conditions: Ag[SbF6], CH2Cl2, rt, 24 h. The [SbF6]-anion is omitted for clarity. | ||
 | ||
| Scheme 8 Attempts to prepare [(C^C^P)Au(III)] complex 22. L: Arbitrary ligand or solvent molecule. Conditions (all reactions at rt): (i) Ag[BF4], CH2Cl2; (ii) Ag[BF4], MeCN; (iii) Ag[SbF6], CH2Cl2; (iv) K[B(C6F5)4], CH2Cl2; (v) Ag[Al(OC(CF3)3)4], CH2Cl2; (vi) Ag[SbF6], MeCN; (vii) K[B(C6F5)4], MeCN; (viii) Ag[Al(OC(CF3)3)4], MeCN; (ix) Ag[SbF6], DMF; (x) Ag[SbF6] or Ag[NTf2], CH2Cl2/MeCN (1:1); (xi) Ag[SbF6] or AgNTF2, DMSO; (xii) Ag[SbF6] or AgNTF2, MeCN/H2O (1:1); (xiii) Ag[SbF6], pyridine, CH2Cl2; (xiv) benzalaniline, Ag[SbF6], CH2Cl2; (xv) Ag[SbF6], THF. The reactions failed in the presence of water as well. | ||
:2 NHC complex 23 could be isolated in low yields (Fig. 3). This may be indicative of a slightly longer lifetime of the intermediate cationic gold(I) species after chloride abstraction. As already reported for the previous experiments, the addition of CsF, AgF or water did not promote the desired oxidative addition, neither the presence of oxygen nor the absence of light. Even at −90 °C, only decomposition was observed when treating 20 with a chloride abstraction reagent.
 | ||
| Fig. 3 Molecular structure of 23 in the solid state. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms and the triflate anion are omitted for clarity. Selected bond lengths (pm) and angles (°): C–Au 201.5(6), 201.6(7), and C–Au–C 173.6(3). | ||
We tried to use imidazolium salt 18 with a lateral pyridine donor for the preparation of the corresponding AuI and AuIII complexes as well. Unfortunately, even the isolation of the respective NHC AuI complex failed due to decomposition over the course of two days after transmetallation of the Ag NHC complex of 18. The addition of reagents for chloride abstraction after the in situ preparation of this NHC Au(I) complex resulted in the instantaneous decomposition to elemental gold as well.
Quantum chemical analysis
None of the presented attempts to prepare non-palindromic [(C^C^D)AuIII] complexes by intramolecular oxidative addition of biphenylene substituted ligands according to Chart 1D was successful. Thus, we were questioning ourselves whether kinetic or thermodynamic reasons were responsible for the failure of this approach and addressed this question by quantum chemical calculations.By means of DFT calculations, Bourissou and co-workers investigated the oxidative addition of benzocyclobutenone to the gold atom of the cationic [(DPCb)AuI]+ complex, which forms by abstraction of chloride from [(DPCb)AuICl] (4, Chart 1B).46 The flexibility of the bidentate DPCb diphosphine ligand is crucial for the success of this reaction: the bite angle may adjust according to the coordination tendencies of AuI (linear) and AuIII (square-planar), thereby enabling the change of the coordination environment upon oxidative addition of biphenylene or benzocyclobutenone. Thus, they found a P–Au–P bite angle of 112° for [(DPCb)AuI]+ and 87° to 90° (isomer dependent) after oxidative addition and the formation of the respective AuIII complex. The ligands investigated in this study do not show this kind of flexibility; however, neither do the ligands used by Toste (Chart 1A)40 or Bertrand (Chart 1C).47
We calculated the intramolecular oxidative addition of different biphenylene ligands according to Chart 1D at the RI60-TPSSh61,62 D3(BJ)/def2-TZVPP63–65 level of theory. The meta-GGA functional TPSS62 was shown to yield reliable results for the structures of transition metal complexes in general66,67 and in combination with dispersion correction (D3)68 and Becke–Johnson damping (BJ)69 for gold heteroatom bonds in particular.70 Since we were interested in transition state barriers, we have chosen the hybrid variant TPSSh because activation barriers are often overestimated by pure functionals and the inclusion of exact exchange (10% for TPSSh) may give more reasonable results.71 We considered solvent effects of CH2Cl2 by use of the SMD model (single point calculations using gas-phase optimized structures).72
We focused on pyridine, PPh2 and NHC (methyl instead of ethyl in the calculated model) ligand as experimentally prepared and a hypothetical PMe2 ligand. The Gibbs free energy of the transition state barrier ΔGTS and the Gibbs free energy of the oxidative addition product ΔGII for the different systems are listed in Table 1.
|
|
||||
|---|---|---|---|---|
| Entry | System | L | ΔGTS (kJ mol−1) | ΔGII (kJ mol−1) |
| a No meaningful structure; the AuI atom is rather coordinated in a η2 manner by the biphenylene than by the pyridine's nitrogen atom. Thus, relative energies are of no meaning. b The calculation was done with a NHC complex with the methyl group instead of the ethyl group. c Coordination via oxygen atom. | ||||
| 1 | Pyridinea | No | 61 (60) | −160 (−153) |
| 2 | NHCb | MeCN | 204 (191) | −53 (−49) |
| 3 | NHC | No | 95 (86) | −101 (−98) |
| 4 | PPh2 | Cl− | 179 (199) | −56 (−66) |
| 5 | PPh2 | No | 95 (92) | −61 (−59) |
| 6 | PMe2 | Cl− | 182 (192) | −60 (−71) |
| 7 | PMe2 | Pyridine | 177 (176) | −63 (−49) |
| 8 | PMe2 | OTf−c | 171 (173) | −64 (−90) |
| 9 | PMe2 | MeCN | 168 (169) | −60 (−48) |
| 10 | PMe2 | No | 93 (89) | −68 (−57) |

All calculated oxidative additions are thermodynamically favoured (ΔGII < 0), thus, the failure of the synthetic idea discussed in this study is not because of thermodynamic hindrance. The very low Gibbs free energy of the pyridine system (entry 1) is due to the instability of the hypothetical cationic AuI complex of pyridine ligand 12, which underlines a main problem of this synthetic approach: the biphenylene backbone coordinates via a donor – N, P or C′ – to the soft AuI centre; however, this donor must be suitable to stabilize a hard AuIII centre after oxidative addition as well.
Neutral chloride complexes (entries 4 and 6) show barriers about twice as high compared to the hypothetical monocoordinated AuI complexes without ligand or solvent molecule L (entries 5 and 10). However, even if the barriers are affected by the ligand L and decrease in the order Cl− > pyridine > OTf− > MeCN (entries 6–9), neither of the barriers is sufficiently low to kinetically allow the intramolecular oxidative addition. The biphenylene unit is not able to effectively bend the linear gold coordination towards a square-planar coordination. In the absence of any ligating molecule, the barrier is much lower (entries 3, 5 and 10), but the respective cationic AuI complexes are purely hypothetical. Mono-coordinated gold(I) complexes instantaneously undergo reactions with surrounding molecules, mostly with reduction to elemental gold as it was found during all our experiments. Obviously, the biphenylene unit cannot effectively approach the AuI centre to pre-empt decomposition pathways.
In summary, the intramolecular oxidative addition is thermodynamically favoured but fails due to the instability of mono coordinated AuI complexes or the kinetic barrier accompanied by linearly coordinated AuI centres.
The Bertrand-system
The oxidative addition of biphenylene to [(CAAC)AuI] complexes described by Bertrand and co-workers (Chart 1C) is only possible in the case of CAAC ligands with an additional lateral donor.47 After chloride abstraction, intermediary formed cationic AuI complexes may be stabilized by coordination of π bonds as it is often seen in AuI catalysis.73 Thus, the linear coordination of biphenylene to AuI is certainly the initial step of a C–C activation at the strained four-membered ring as it was already shown for other metals.74,75 We optimized the structure of cationic [(CAAC)AuI] biphenylene complex 6* at the RI-TPSSh-D3(BJ)/def2-TZVPP level of theory and identified no interaction of the AuI atom with the CAAC's lateral imine donor, thus the η2 coordination of biphenylene is sufficient to form an adequately stabilized AuI intermediate preventing decomposition to elemental gold (Scheme 9). The transition state of the oxidative addition 6TS is characterized by an imaginary stretching vibration of the four-membered biphenylene ring towards a five membered aura-cycle. We did not identify any interaction of the imine donor with gold at the transition state. Quite the contrary is true; the Au–Nimine distance is found to be longer at the transition state (dAu–N = 343 pm) than in reactant complex 6* (dAu–N = 338 pm). Only for product 7, the imine donor comes into play by stabilizing the AuIII centre (dAu–N = 226 pm) and ensuring a square-planar coordination environment, thus, thermodynamically stabilizing the product of oxidative addition. Since Bertrand and co-workers report that no C–C bond activation occurs without a lateral donor at the CAAC ligand, the oxidative addition itself is maybe not thermodynamically favoured.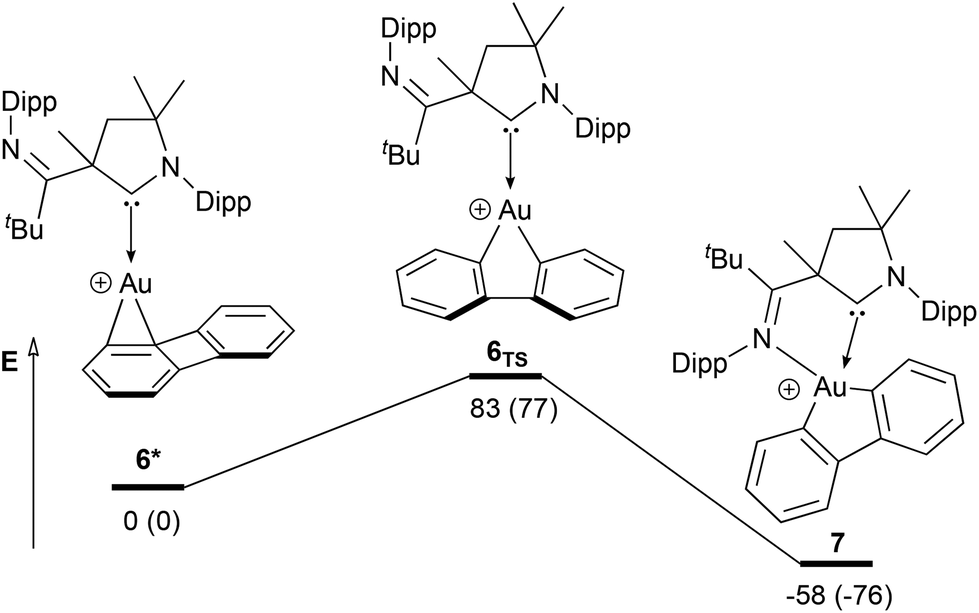 | ||
| Scheme 9 Oxidative addition of biphenylene to [(CAAC)Au(I)] complex 6* calculated at the RI-TPSSh-D3(BJ)/def2-TZVPP level of theory. Gibbs free energies (electronic energies in parentheses) are given in kJ mol−1 relative to reactant complex 6*. Solvent effects of CH2Cl2 were considered by means of the SMD model (single point calculations only). | ||
The Toste system
At first sight, the former analysis contrasts with the oxidative addition of biphenylene to [(IPr)AuI]+ (Chart 1A). The IPr ligand employed by Toste and co-workers does not have any lateral donor like the CAACs employed in the Bertrand group, which is a requirement for the oxidative addition of biphenylene. However, Toste and co-workers report on water molecules coordinating to AuIII after C–C bond activation thereby stabilizing the AuIII oxidation state in a square-planar coordination environment. The authors used commercially available Ag[SbF6] for chloride abstraction at [(IPr)AuCl] (1), which regularly contains significant amounts of water as we noticed during our own studies. Thus, we assumed the oxidative addition of biphenylene to [(IPr)AuI]+ may be only possible due to water impurities. To prove this assumption, we investigated this reaction by DFT calculations as well. However, since in the Toste case inter-molecular processes, i.e. water coordination or anion stabilizing effects may be of significant importance, for these calculations we have chosen to optimize all structures with the inclusion of solvent effects by the C-PCM model.76 Furthermore, we did not only focus on the cationic Au(I)-species but included the [SbF6]− anion as well. A comparison with calculations done with gas phase optimized structures is given in the ESI.†After chloride abstraction, the cationic AuI centre may be stabilized by the [SbF6]− anion via coordination of one fluorine atom, by η2 coordination of biphenylene or by a water molecule. Compared to the biphenylene coordination, the [SbF6]− coordination is disfavoured by a Gibbs free energy of 33 kJ mol−1 (electronic energy: 98 kJ mol−1). Water coordination is disfavoured as well by 26 kJ mol−1 (electronic energy: 36 kJ mol−1), i.e., water does not hinder oxidative addition. We emphasize at this point that we performed all our own oxidative addition experiments described in the foregoing section without success in the presence of water.
Starting from complex 1* (Scheme 10), the barrier to the transition state 1TS of 88 kJ mol−1 (82 kJ mol−1) is slightly higher compared to the Bertrand system shown in Scheme 9. Interestingly, the Gibbs free energy of the oxidative addition to form 3* is found to be 10 kJ mol−1 (−2 kJ mol−1); hence, the oxidative addition itself is not thermodynamically favoured. Confirming our assumption, the Gibbs free energy for displacing [SbF6]− and coordination of one molecule water to the AuIII centre of 3* is −37 kJ mol−1 making the whole process of oxidative addition thermodynamically favourable (electronic energy: −79 kJ mol−1; see Computational Details for a discussion on the discrepancy between electronic and Gibbs free energy).
 | ||
| Scheme 10 Oxidative addition of biphenylene (2) to [(IPr)Au(I)]+ (1*) calculated at the RIJCOSX-TPSSh-D3(BJ)/def2-TZVPP level of theory. Gibbs free energies (electronic energies in parentheses) are given in kJ mol−1 relative to the reactant complex 1*. Solvent effects of CH2Cl2 were considered by means of the C-PCM model. | ||
Thus, the calculations support our hypothesis that without water impurities the oxidative addition of biphenylene to [(IPr)Au]+ (1*) would probably not be possible.
Nevertheless, we were not satisfied by these pure quantum chemical findings. Therefore, we performed the oxidative addition of biphenylene to [(IPr)AuI]+ by ourselves in dry and wet CD2Cl2 according to the procedure of Toste and co-workers, using meticulously dried Ag[SbF6]. We monitored the reaction via1H NMR spectroscopy (Fig. 4).
 | ||
| Fig. 4 1H NMR monitoring (500 MHz, CD2Cl2, 298.15 K) of the equimolar reaction of biphenylene (2) with [(IPr)Au(I)]+ in the absence (left) and presence (right) of water (ca. 1.5 equivalents) (cf.Chart 1A and Scheme 10). The bottom spectra show the signals of biphenylene and [(IPr)AuCl] (1) before the addition of Ag[SbF6]. After the addition of Ag[SbF6], the reaction mixture was instantaneously frozen and thawed just before the first NMR measurement (0 min). | ||
After chloride abstraction in dry CD2Cl2, no change of the biphenylene's 1H signals (2-position: δ = 6.65 ppm, 1-position: δ = 6.75 ppm) is observed after 120 min nor after control measurements at 24 h and 48 h. However, after 48 h, the majority of [(IPr)AuI]+'s signals had disappeared due to decomposition and the formation of elemental gold.
In wet CD2Cl2, the biphenylene's signals are notably affected: the signal of the protons at the 1-position broadens during the reaction indicating some dynamic process. Interestingly, in the high field region, there is no difference of the isopropyl group's signals (δ = 2.46, 1.30, 1.25 ppm) between dry and wet conditions. Thus, based on our NMR analysis, there are hints to dynamic processes enabled in the presence of water. However, we could not without doubt assign the NMR signature in wet CD2Cl2 to the oxidative addition of biphenylene and the clean formation of oxidative addition product 3* and/or 3. Therefore, a comprehensive clarification of the underlying mechanisms of this oxidative addition remains lacking.
Conclusions
The synthetic access to different substituted biphenylenes covering common donor motifs – phosphine, pyridine, and NHC – will probably be useful in the realm of transition metal complexes. However, our idea to use these compounds for the preparation of non-palindromic [(C^C^D)AuIII] complexes failed. The reasons for that are rooted in the kinetic hindrance of the intramolecular oxidative addition of biphenylene due to the linear coordination of AuI that usually takes place. This avoids an effective bending of the biphenylene unit to the AuI centre. By quantum chemical calculations, the oxidative addition is found to be only possible for monocoordinated AuI complexes, which are purely hypothetical species. These results stand in line with other findings about the oxidative addition of AuI to carbon–halogen bonds.56The oxidative additions of biphenylene to AuI complexes reported in the literature are enabled by additional factors and do not occur only due to the strain of the biphenylenes four-membered ring. In one case, a lateral imine donor (Bertrand) or the bite angle's flexibility of a diphosphine ligand (Bourissou) stabilizes the AuIII centre after oxidative addition. In the other case, water, which is a donor too hard to coordinate to the soft AuI atom, enables dynamic processes and probably stabilizes a hard AuIII centre after the oxidative addition of biphenylene, as it is in accordance with our DFT calculations. However, our NMR experiments do not conclusively prove this assumption.
Thus, our study besides many others shows the versatility of the chemistry of gold; however, even if there are reports on catalytic transformations involving oxidative additions to AuI, this kind of reaction still remains a phenomenon enabled by suitable conditions or specifically tailored ligands rather than being a common process.77
Experimental
General details
Manipulations were performed under a dry and oxygen-free argon or nitrogen atmosphere using standard Schlenk techniques.78 Solvents were dried and degassed. All reagents were purchased from commercial sources and used as received except for Ag[SbF6] which was recrystallized from dry toluene and dried in vacuo at 120 °C for three hours. 1-Pyridylbiphenylene (12) was prepared according to a literature procedure.49 Solution NMR spectra were recorded at 298.15 K using Bruker Avance Instruments operating at 1H Larmor frequencies of 300, 400 or 500 MHz; chemical shifts are given in parts per million (ppm) relative to TMS for 13C and 1H, to H3PO4 for 31P. NMR samples were prepared in oven-dried 5 mm NMR tubes. Air or moisture sensitive samples were prepared inside a glovebox using screw cap or Young valve NMR tubes or by vacuum transfer of the solvent to the samples and melting off the NMR tube.IR spectra were recorded on a Bruker Alpha FT-IR spectrometer using the ATR technique (attenuated total reflection) on the bulk material, and the data are quoted in wavenumbers (cm−1).
Mass spectra were recorded using a Varian MAT 3830 (70 eV) by electron ionization (EI).
Elemental analyses were done by the institutional technical laboratories of the Karlsruhe Institute of Technology (KIT).
Melting points were measured with a Thermo Fisher Scientific digital melting point apparatus of the IA9300 series and are not corrected.
Synthesis
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 = 3272 (vw), 3180 (vw), 3059 (vw), 3027 (vw), 2891 (vw), 2839 (vw), 1908 (vw), 1881 (vw), 1835 (vw), 1786 (vw), 1590 (vw), 1499 (vw), 1456 (vw), 1444 (w), 1409 (w), 1386 (vw), 1346 (w), 1277 (w), 1203 (vw), 1149 (w), 1111 (vw), 1089 (vw), 1062 (w), 1044 (m), 1010 (w), 968 (w), 916 (vw), 885 (vw), 873 (vw), 795 (vw), 753 (m), 733 (vs), 699 (m), 652 (w), 622 (w), 578 (w), 530 (vw), 477 (w), 401 (vw), 383 (vw). MS (EI, 70 eV), m/z (%): 182.1 (100%) [M]+, 152.1 (70%) [M − CH3O]+. Elemental analysis calculated (%) for C13H10O: C 85.69, H 5.53, found: C 85.91, H 5.245.
/cm−1 = 3064 (vw), 3029 (vw), 2967 (vw), 2860 (vw), 1919 (vw), 1882 (vw), 1853 (vw), 1836 (vw), 1785 (vw), 1726 (vw), 1659 (vw), 1616 (vw), 1582 (vw), 1534 (vw), 1457 (vw), 1442 (w), 1410 (m), 1378 (w), 1343 (vw), 1323 (vw), 1274 (w), 1257 (m), 1246 (m), 1190 (vw), 1159 (vw), 1149 (m), 1111 (w), 1087 (w), 1055 (vw), 1018 (w), 969 (m), 916 (m), 893 (w), 865 (w), 805 (vw), 765 (m), 734 (vs), 713 (vs), 706 (vs), 679 (vs), 595 (s), 552 (m), 481 (vw), 399 (vw), 384 (w). MS (EI, 70 eV), m/z (%): 200.0 (100%) [M]+, 165.0 (100%) [M − Cl]+. Elemental analysis calculated (%) for C13H9Cl: C 77.81, H 4.52, found: C 77.93, H 4.468.
/cm−1 = 3070 (vw), 3051 (vw), 2931 (vw), 2896 (vw), 2186 (vw), 2171 (vw), 2012 (vw), 1970 (vw), 1663 (vw), 1601 (vw), 1584 (vw), 1479 (vw), 1459 (vw), 1434 (w), 1414 (w), 1379 (vw), 1327 (vw), 1304 (vw), 1266 (vw), 1198 (vw), 1148 (vw), 1098 (vw), 1067 (vw), 1044 (vw), 1026 (vw), 999 (vw), 969 (w), 908 (vw), 889 (vw), 865 (vw), 836 (vw), 802 (vw), 766 (m), 735 (vs), 721 (m), 708 (w), 691 (vs), 612 (vw), 597 (vw), 580 (vw), 554 (vw), 506 (m), 497 (w), 472 (w), 429 (w), 396 (vw), 382 (vw). MS (EI, 70 eV), m/z (%): 350.1 (42%) [M]+, 165.1 (100%) [M − PPh2]+. Elemental analysis calculated (%) for C25H19P: C 85.69, H 5.47, found: C 85.70, H 5.480.
:1) to obtain 207 mg (95%) of a yellowish solid. Rf = 0.43. M.p. = 96 °C. 1H NMR (300 MHz, MeOH-d4): δ = 8.07 (dd, J = 1.4, 1.0 Hz, 1 H), 7.49 (t, J = 1.4 Hz, 1 H), 7.19 (dd, J = 1.5, 1.0 Hz, 1 H), 7.03–6.90 (m, 2 H), 6.87–6.83 (m, 2 H), 6.82–6.73 (m, 2 H), 6.66 (dd, J = 6.2, 1.2 Hz, 1H). 13C NMR (75 MHz, MeOH-d4): δ = 153.4 (Cq), 151.2 (Cq), 149.5 (Cq), 140.8 (Cq), 136.4 (CH), 132.1 (CH), 130.4 (CH), 130.3 (CH), 130.0 (CH), 128.1 (Cq), 121.6 (CH), 119.0 (2 CH), 118.9 (CH), 117.1 (CH). IR (ATR): /cm−1 = 3157 (vw), 3131 (vw), 3050 (vw), 2233 (vw), 2196 (vw), 2183 (vw), 2152 (vw), 2135 (vw), 2050 (vw), 2022 (vw), 2003 (vw), 1988 (vw), 1943 (vw), 1907 (vw), 1664 (w), 1603 (vw), 1588 (w), 1497 (s), 1445 (w), 1432 (w), 1386 (w), 1317 (m), 1297 (w), 1287 (w), 1261 (vw), 1244 (m), 1202 (vw), 1157 (w), 1099 (w), 1047 (m), 1021 (w), 973 (vw), 957 (w), 934 (vw), 903 (w), 879 (vw), 853 (vw), 816 (m), 795 (w), 753 (vs), 739 (vs), 726 (s), 698 (m), 653 (s), 623 (w), 614 (w), 590 (w), 522 (w), 484 (vw), 462 (vw), 426 (vw), 416 (vw), 403 (vw), 394 (vw). MS (ESI), m/z (%): 219.1 (100%) [M + H]+. Elemental analysis calculated (%) for C15H10N2: C 82.55, H 4.62, N 12.84 found: C 82.10, H 4.448, N 12.67.
/cm−1 = 3272 (vw), 3180 (vw), 3059 (vw), 3027 (vw), 2891 (vw), 2839 (vw), 1908 (vw), 1881 (vw), 1835 (vw), 1786 (vw), 1590 (vw), 1499 (vw), 1456 (vw), 1444 (w), 1409 (w), 1386 (vw), 1346 (w), 1277 (w), 1203 (vw), 1149 (w), 1111 (vw), 1089 (vw), 1062 (w), 1044 (m), 1010 (w), 968 (w), 916 (vw), 885 (vw), 873 (vw), 795 (vw), 753 (m), 733 (vs), 699 (m), 652 (w), 622 (w), 578 (w), 530 (vw), 477 (w), 401 (vw), 383 (vw). MS (EI, 70 eV), m/z (%): 182.1 (100%) [M]+, 152.1 (70%) [M − CH3O]+. Elemental analysis calculated (%) for C13H10O: C 85.69, H 5.53, found: C 85.91, H 5.245.
/cm−1 = 3064 (vw), 3029 (vw), 2967 (vw), 2860 (vw), 1919 (vw), 1882 (vw), 1853 (vw), 1836 (vw), 1785 (vw), 1726 (vw), 1659 (vw), 1616 (vw), 1582 (vw), 1534 (vw), 1457 (vw), 1442 (w), 1410 (m), 1378 (w), 1343 (vw), 1323 (vw), 1274 (w), 1257 (m), 1246 (m), 1190 (vw), 1159 (vw), 1149 (m), 1111 (w), 1087 (w), 1055 (vw), 1018 (w), 969 (m), 916 (m), 893 (w), 865 (w), 805 (vw), 765 (m), 734 (vs), 713 (vs), 706 (vs), 679 (vs), 595 (s), 552 (m), 481 (vw), 399 (vw), 384 (w). MS (EI, 70 eV), m/z (%): 200.0 (100%) [M]+, 165.0 (100%) [M − Cl]+. Elemental analysis calculated (%) for C13H9Cl: C 77.81, H 4.52, found: C 77.93, H 4.468.
/cm−1 = 3070 (vw), 3051 (vw), 2931 (vw), 2896 (vw), 2186 (vw), 2171 (vw), 2012 (vw), 1970 (vw), 1663 (vw), 1601 (vw), 1584 (vw), 1479 (vw), 1459 (vw), 1434 (w), 1414 (w), 1379 (vw), 1327 (vw), 1304 (vw), 1266 (vw), 1198 (vw), 1148 (vw), 1098 (vw), 1067 (vw), 1044 (vw), 1026 (vw), 999 (vw), 969 (w), 908 (vw), 889 (vw), 865 (vw), 836 (vw), 802 (vw), 766 (m), 735 (vs), 721 (m), 708 (w), 691 (vs), 612 (vw), 597 (vw), 580 (vw), 554 (vw), 506 (m), 497 (w), 472 (w), 429 (w), 396 (vw), 382 (vw). MS (EI, 70 eV), m/z (%): 350.1 (42%) [M]+, 165.1 (100%) [M − PPh2]+. Elemental analysis calculated (%) for C25H19P: C 85.69, H 5.47, found: C 85.70, H 5.480.
:1) to obtain 207 mg (95%) of a yellowish solid. Rf = 0.43. M.p. = 96 °C. 1H NMR (300 MHz, MeOH-d4): δ = 8.07 (dd, J = 1.4, 1.0 Hz, 1 H), 7.49 (t, J = 1.4 Hz, 1 H), 7.19 (dd, J = 1.5, 1.0 Hz, 1 H), 7.03–6.90 (m, 2 H), 6.87–6.83 (m, 2 H), 6.82–6.73 (m, 2 H), 6.66 (dd, J = 6.2, 1.2 Hz, 1H). 13C NMR (75 MHz, MeOH-d4): δ = 153.4 (Cq), 151.2 (Cq), 149.5 (Cq), 140.8 (Cq), 136.4 (CH), 132.1 (CH), 130.4 (CH), 130.3 (CH), 130.0 (CH), 128.1 (Cq), 121.6 (CH), 119.0 (2 CH), 118.9 (CH), 117.1 (CH). IR (ATR): /cm−1 = 3157 (vw), 3131 (vw), 3050 (vw), 2233 (vw), 2196 (vw), 2183 (vw), 2152 (vw), 2135 (vw), 2050 (vw), 2022 (vw), 2003 (vw), 1988 (vw), 1943 (vw), 1907 (vw), 1664 (w), 1603 (vw), 1588 (w), 1497 (s), 1445 (w), 1432 (w), 1386 (w), 1317 (m), 1297 (w), 1287 (w), 1261 (vw), 1244 (m), 1202 (vw), 1157 (w), 1099 (w), 1047 (m), 1021 (w), 973 (vw), 957 (w), 934 (vw), 903 (w), 879 (vw), 853 (vw), 816 (m), 795 (w), 753 (vs), 739 (vs), 726 (s), 698 (m), 653 (s), 623 (w), 614 (w), 590 (w), 522 (w), 484 (vw), 462 (vw), 426 (vw), 416 (vw), 403 (vw), 394 (vw). MS (ESI), m/z (%): 219.1 (100%) [M + H]+. Elemental analysis calculated (%) for C15H10N2: C 82.55, H 4.62, N 12.84 found: C 82.10, H 4.448, N 12.67.
To exchange [BF4] for chloride, 17a (200 mg) was dissolved in MeOH (20 ml) and H2O (8 ml) and passed through a column (2 cm inner diameter) packed with about 3 g of Dowex® (chloride form). The column was extracted with MeOH/H2O (1:1) (3 × 20 ml) and the combined filtrates evaporated to dryness. The residue was taken up in CH2Cl2 (50 ml), dried over MgSO4 and finally all solvent was removed under reduced pressure. After drying in vacuo 169 mg (quant.) of a yellowish solid were isolated (17b). 17a: M.p. = 136 °C. 1H NMR (300 MHz, Aceton-d6): δ = 9.54 (t, J = 1.6 Hz, 1 H), 8.08 (s, 1 H), 8.08 (s, 1 H), 7.18 (dd, J = 8.8, 0.7 Hz, 1 H), 7.09 (dd, J = 8.8, 6.7 Hz, 1 H), 7.02–6.83 (m, 5 H), 4.57 (q, J = 7.3 Hz, 2 H, CH2Me), 1.67 (t, J = 7.3 Hz, 3 H, CH2CH3). 13C NMR (75 MHz, Acetone-d6): δ = 153.1 (Cq), 150.8 (Cq), 147.9 (Cq), 142.8 (Cq), 135.4 (Cq), 132.4 (CH), 131.0 (CH), 130.2 (CH), 125.8 (Cq), 124.5 (CH), 122.2 (CH), 122.0 (CH), 120.1 (CH), 119.6 (CH), 119.1 (CH), 46.5 (CH2), 15.5 (CH3). IR (ATR): /cm−1 = 3156 (vw), 2575 (vw), 2516 (vw), 2501 (vw), 2451 (vw), 2386 (vw), 2325 (vw), 2282 (vw), 2265 (vw), 2232 (vw), 2221 (vw), 2207 (vw), 2184 (vw), 2176 (vw), 2148 (vw), 2115 (w), 2081 (vw), 2064 (w), 2019 (vw), 1990 (vw), 1965 (vw), 1956 (vw), 1941 (vw), 1926 (vw), 1902 (vw), 1889 (vw), 1861 (vw), 1665 (vw), 1603 (vw), 1576 (vw), 1548 (vw), 1464 (vw), 1445 (vw), 1418 (vw), 1320 (vw), 1277 (vw), 1192 (vw), 1159 (vw), 1047 (vs), 1033 (vs), 967 (w), 881 (vw), 863 (vw), 837 (w), 793 (vw), 746 (vs), 707 (vw), 696 (w), 670 (vw), 633 (w), 616 (w), 596 (vw), 585 (vw), 565 (w), 543 (w), 520 (m), 501 (w), 492 (w), 477 (w), 463 (vw), 452 (w), 443 (w), 432 (w), 422 (w), 409 (w), 400 (w), 385 (m). MS (ESI), m/z (%): 247.1 (100%) [M]+. Elemental analysis calculated (%) for C17H15N2BF4: C 61.11, H 4.53, N 8.38 found: C 60.09, H 4.329, N 8.37. 17b: M.p. = 177 °C. 1H NMR (300 MHz, CD2Cl2): δ = 9.19 (t, J = 1.7 Hz, 1 H), 7.55 (d, J = 1.6 Hz, 2H), 7.03 (s, 1 H), 7.02 (d, J = 2.9 Hz, 1 H), 6.99–6.82 (m, 2 H), 6.82–6.76 (m, 3 H), 4.45 (q, J = 7.2 Hz, 2 H, CH2Me), 1.63 (t, J = 7.3 Hz, 3 H, CH2CH3). MS (ESI), m/z (%): 247.1 (100%) [M]+. Elemental analysis calculated (%) for C17H15N2Cl: C 72.21, H 5.35, N 9.91 found: C 72.19, H 5.151, N 9.73.
/cm−1 = 3035 (vw), 2287 (vw), 2186 (vw), 2166 (vw), 2129 (vw), 2098 (vw), 2024 (vw), 1998 (vw), 1984 (vw), 1935 (vw), 1610 (vw), 1590 (vw), 1568 (vw), 1545 (w), 1468 (vw), 1437 (w), 1419 (w), 1367 (vw), 1331 (vw), 1267 (vw), 1229 (vw), 1189 (w), 1155 (vw), 1100 (vw), 1069 (vw), 1052 (vw), 1020 (vw), 996 (vw), 972 (vw), 875 (vw), 817 (vw), 796 (w), 741 (vs), 697 (m), 653 (m), 638 (m), 619 (m), 595 (w), 582 (w), 520 (w), 476 (w), 464 (w), 449 (w), 431 (w), 422 (w), 402 (w), 393 (w), 385 (w). MS (ESI), m/z (%): 310.1 (100%) [M]+. Elemental analysis calculated (%) for C21H16N3Br: C 64.63, H 4.13, N 10.77 found: C 64.88, H 4.041, N 10.99.
/cm−1 = 3070 (vw), 3051 (vw), 2931 (vw), 2896 (vw), 2186 (vw), 2171 (vw), 2012 (vw), 1970 (vw), 1663 (vw), 1601 (vw), 1584 (vw), 1479 (vw), 1459 (vw), 1434 (w), 1414 (w), 1379 (vw), 1327 (vw), 1304 (vw), 1266 (vw), 1198 (vw), 1148 (vw), 1098 (vw), 1067 (vw), 1044 (vw), 1026 (vw), 999 (vw), 969 (w), 908 (vw), 889 (vw), 865 (vw), 836 (vw), 802 (vw), 766 (m), 735 (vs), 721 (m), 708 (w), 691 (vs), 612 (vw), 597 (vw), 580 (vw), 554 (vw), 506 (m), 497 (w), 472 (w), 429 (w), 396 (vw), 382 (vw). MS (EI. 70 eV), m/z (%): 582.0 (16%) [M]+, 349.1 (94%) [M − AuCl]+, 165.1 (100%) [M − AuCl − PPh2]+. Elemental analysis calculated (%) for C25H19P1AuCl: C 51.52, H 3.29 found: C 51.77, H 3.134.
/cm−1 = 3169 (vw). 3129 (vw). 3096 (vw). 2983 (vw). 2185 (vw). 1704 (vw). 1668 (vw). 1588 (vw). 1470 (w). 1450 (w). 1425 (w). 1407 (w). 1380 (vw). 1357 (vw). 1315 (vw). 1268 (w). 1258 (w). 1213 (w). 1156 (w). 1138 (vw). 1110 (vw). 1090 (vw). 1051 (vw). 979 (vw). 951 (vw). 921 (vw). 895 (vw). 874 (vw). 850 (vw). 799 (w). 765 (vw). 739 (vs). 703 (m). 688 (w). 674 (w). 644 (vw). 609 (w). 541 (vw). 486 (vw). 423 (vw). 385 (vw). MS (ESI), m/z (%): 448.3 (28%) [M − C2H6]+. Elemental analysis calculated (%) for C17H14N2AuCl: C 42.65, H 2.95, N 5.85 found: C 41.86, H 2.848, N 5.79.
/cm−1 = 2960 (vw). 2927 (vw). 2870 (vw). 1607 (vw). 1566 (vw). 1551 (vw). 1470 (w). 1423 (vw). 1387 (vw). 1366 (vw). 1330 (vw). 1287 (vw). 1256 (vw). 1215 (vw). 1182 (vw). 1154 (vw). 1120 (vw). 1060 (vw). 967 (vw). 949 (vw). 936 (vw). 807 (w). 783 (vw). 764 (m). 738 (m). 706 (w). 654 (vs). 550 (vw). 507 (vw). 451 (vw). 417 (vw). 394 (vw). MS (ESI), m/z (%): 814.3 (20%) [M]+, 653.3 [M − (i-Pr)2Ph]+. Elemental analysis calculated (%) for C44H47N3AuSbF6: C 50.30, H 4.51, N 4.00 found: C 49.11, H 3.931, N 3.75.
/cm−1 = 3123 (vw), 3095 (vw), 2979 (vw), 1668 (vw), 1607 (vw), 1588 (vw), 1568 (vw), 1470 (m), 1446 (w), 1417 (w), 1381 (w), 1357 (vw), 1315 (w), 1281 (m), 1261 (vs), 1222 (m), 1157 (m), 1140 (s), 1104 (w), 1088 (w), 1055 (w), 1031 (s), 976 (w), 964 (w), 928 (w), 898 (w), 822 (w), 800 (w), 774 (w), 744 (vs), 704 (m), 694 (m), 671 (w), 634 (vs), 604 (m), 572 (m), 548 (w), 536 (w), 517 (s), 496 (w), 474 (w), 448 (m), 436 (m), 423 (m), 411 (m), 395 (s), 386 (s). MS (ESI), m/z (%): 689.2 [M]+. Elemental analysis calculated (%) for C35H28N4AuSO3F3: C 50.13, H 3.37, N 6.68 found: C 47.61, H 3.211, N 6.69.
X-ray diffraction analysis
To avoid degradation, single crystals were mounted in perfluoropolyalkylether-oil on top of the edge of an open Mark tube and then brought in contact with the cold nitrogen stream of a low-temperature device (Oxford Cryosystems Cryostream unit) so that the oil solidified. Diffraction data were measured using a Stoe IPDS II diffractometer (graphite-monochromated MoKα (0.71073 Å) radiation). The structures were solved by dual-space direct methods with SHELXT,79 followed by full-matrix least-squares refinement using SHELXL-2014/7.80 All non-hydrogen atoms were refined anisotropically, with organic hydrogen atoms placed in calculated positions using a riding model. Absorption corrections were applied for all compounds (numerical). Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre.CCD-numbers of structures contained in this manuscript: 112018371, 192018369, S12018372, 202018370, 232018373.
Computational details
All quantum chemical calculations except for solvent effects (vide infra) were performed with the TURBOMOLE81,82 program package (version 7.4). Fine integration grids (m4) and SCF convergence criteria of 10−8 a.u., density convergence ≤10−7, and Cartesian gradient ≤10−4 were used. The TURBOMOLE module AOFORCE83 (analytically frequency calculations) was used. All optimized structures were checked for minima (no imaginary frequencies) or transition states (one imaginary frequency according to the bond-forming and -breaking processes connecting the respective minima). Thermal Gibbs free energy contributions with inclusion of zero point energies (ZPE) were calculated at 298 K and 1 bar with the TURBOMOLE module FREEH based on assumptions of statistical thermodynamics of an ideal gas and added to the total gas-phase electronic energies. Except for the Toste system, Gibbs free energies of solvation were calculated by means of single point calculations at the same level of theory as mentioned above with the gas phase optimized structures employing the SMD72 solvation model as implemented in ORCA 4.1.2.84,85 A detailed description about this kind of solvent effect inclusion can be found in the literature.86For the Toste system, all structure optimizations were done with the inclusion of solvent effects by the C-PCM solvation model using a Gaussian charge scheme with scaled van der Waals cavities as implemented in the ORCA 4.2.0 program package.87 The RIJCOSX-approximation was used for these calculations.88 We note that all structures were checked to be global minima or a transition state by inspection of the numerical hessian. The respective gas-phase optimized structures did not show any imaginary frequencies. However, upon inclusion of solvent effects, in few cases up to two small imaginary frequencies (<15 cm−1) corresponding to movements of the whole cationic or anionic fragment were identified. These frequencies were neglected for the calculation of thermodynamic properties. Thermodynamics in solution phase were calculated assuming concentrations of 1 mol L−1 at 298 K.
The discrepancy between electronic and Gibbs free energy upon water coordination to 3* (Scheme 10) may be understood by a common problem when dealing with association reactions and their thermochemistry: structure optimizations may be done on separate species.89 An association then results in a huge increase of entropy due to the loss of translational and rotational degrees of freedom. In solution this can lead to errors in the calculated entropy of up to 60 kJ mol−1.90 In solution, reactions only take place when reactants approach each other in the same solvent cage, thus, the majority of translational and rotational degrees of freedom are already suppressed due to interactions with solvent molecules. Therefore, the calculation of reactant complexes according to a (hypothetical) situation of not or only weakly interacting reactants is advisable.91 Unfortunately, for the association of complex 3* and water, we did not find any reasonable reactant complex. Even though the described entropy-related problem may to some extent be covered by the solvation model,86 −37 kJ mol−1 found for the association of water to complex 3* probably contains a notable error.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. F. thanks the Carl-Zeiss Stiftung for a PhD scholarship as well as the Studienstiftung des Deutschen Volkes for general support.References
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071104 CrossRef.
- H. J. Bolink, E. Coronado, S. Garcia Santamaria, M. Sessolo, N. Evans, C. Klein, E. Baranoff, K. Kalyanasundaram, M. Graetzel and M. K. Nazeeruddin, Chem. Commun., 2007, 3276–3278, 10.1039/b707879j.
- P. N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198–10207 CrossRef CAS PubMed.
- H. Rudmann, S. Shimada and M. F. Rubner, J. Am. Chem. Soc., 2002, 124, 4918–4921 CrossRef CAS PubMed.
- S. Welter, K. Brunner, J. W. Hofstraat and L. De Cola, Nature, 2003, 421, 54–57 CrossRef CAS PubMed.
- Y. Chi and P. T. Chou, Chem. Soc. Rev., 2007, 36, 1421–1431 RSC.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115–137 CrossRef CAS.
- S. C. Kui, F. F. Hung, S. L. Lai, M. Y. Yuen, C. C. Kwok, K. H. Low, S. S. Chui and C. M. Che, Chem. – Eur. J., 2012, 18, 96–109 CrossRef CAS PubMed.
- E. Turner, N. Bakken and J. Li, Inorg. Chem., 2013, 52, 7344–7351 CrossRef CAS PubMed.
- C. Cebrian and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef CAS PubMed.
- C. Lee, R. Zaen, K. M. Park, K. H. Lee, J. Y. Lee and Y. Kang, Organometallics, 2018, 37, 4639–4647 CrossRef CAS.
- K. M. Wong, X. Zhu, L. L. Hung, N. Zhu, V. W. Yam and H. S. Kwok, Chem. Commun., 2005, 2906–2908, 10.1039/b503315b.
- V. K. Au, K. M. Wong, D. P. Tsang, M. Y. Chan, N. Zhu and V. W. Yam, J. Am. Chem. Soc., 2010, 132, 14273–14278 CrossRef CAS PubMed.
- V. K. Au, D. P. Tsang, K. M. Wong, M. Y. Chan, N. Zhu and V. W. Yam, Inorg. Chem., 2013, 52, 12713–12725 CrossRef CAS PubMed.
- M. C. Tang, D. P. Tsang, M. M. Chan, K. M. Wong and V. W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446–449 CrossRef CAS PubMed.
- M. C. Tang, D. P. Tsang, Y. C. Wong, M. Y. Chan, K. M. Wong and V. W. Yam, J. Am. Chem. Soc., 2014, 136, 17861–17868 CrossRef CAS PubMed.
- C. H. Lee, M. C. Tang, Y. C. Wong, M. Y. Chan and V. W. Yam, J. Am. Chem. Soc., 2017, 139, 10539–10550 CrossRef CAS PubMed.
- W. P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C. M. Che, Angew. Chem., Int. Ed., 2017, 56, 14036–14041 CrossRef CAS PubMed.
- C. H. Lee, M. C. Tang, W. L. Cheung, S. L. Lai, M. Y. Chan and V. W. Yam, Chem. Sci., 2018, 9, 6228–6232 RSC.
- L.-K. Li, M.-C. Tang, S.-L. Lai, M. Ng, W.-K. Kwok, M.-Y. Chan and V. W.-W. Yam, Nat. Photonics, 2019, 13, 185–191 CrossRef CAS.
- M. C. Tang, M. Y. Leung, S. L. Lai, M. Ng, M. Y. Chan and V. Wing-Wah Yam, J. Am. Chem. Soc., 2018, 140, 13115–13124 CrossRef CAS PubMed.
- C. J. Ballhausen, N. Bjerrum, R. Dingle, K. Eriks and C. R. Hare, Inorg. Chem., 1965, 4, 514–518 CrossRef CAS.
- L. J. Andrews, J. Phys. Chem., 1979, 83, 3203–3209 CrossRef CAS.
- C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC.
- E. S. Lam, W. H. Lam and V. W. Yam, Inorg. Chem., 2015, 54, 3624–3630 CrossRef CAS PubMed.
- T. von Arx, A. Szentkuti, T. N. Zehnder, O. Blacque and K. Venkatesan, J. Mater. Chem. C, 2017, 5, 3765–3769 RSC.
- R. Usón, J. Vicente, J. A. Cirac and M. T. Chicote, J. Organomet. Chem., 1980, 198, 105–112 CrossRef.
- K. T. Chan, G. S. M. Tong, Q. Wan, G. Cheng, C. Yang and C. M. Che, Chem. – Asian J., 2017, 12, 2104–2120 CrossRef CAS PubMed.
- L. Nilakantan, D. R. McMillin and P. R. Sharp, Organometallics, 2016, 35, 2339–2347 CrossRef CAS.
- B. David, U. Monkowius, J. Rust, C. W. Lehmann, L. Hyzak and F. Mohr, Dalton Trans., 2014, 43, 11059–11066 RSC.
- R. Kumar, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 14287–14290 CrossRef CAS PubMed.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- H. Beucher, S. Kumar, E. Merino, W.-H. Hu, G. Stemmler, S. Cuesta-Galisteo, J. A. González, J. Jagielski, C.-J. Shih and C. Nevado, Chem. Mater., 2020, 32, 1605–1611 CrossRef CAS.
- R. Kumar, J. P. Krieger, E. Gomez-Bengoa, T. Fox, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 12862–12865 CrossRef CAS.
- L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobarik and M. Bochmann, J. Am. Chem. Soc., 2018, 140, 8287–8302 CrossRef CAS PubMed.
- W. Feuerstein, C. Holzer, X. Gui, L. Neumeier, W. Klopper and F. Breher, Chem. – Eur. J., 2020, 26, 17156–17164 CrossRef CAS PubMed.
- W. Feuerstein and F. Breher, Chem. Commun., 2020, 56, 12589–12592 RSC.
- C. Y. Wu, T. Horibe, C. B. Jacobsen and F. D. Toste, Nature, 2015, 517, 449–454 CrossRef CAS PubMed.
- J. J. Eisch, A. M. Piotrowski, K. I. Han, C. Kruger and Y. H. Tsay, Organometallics, 1985, 4, 224–231 CrossRef CAS.
- B. L. Edelbach, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 1998, 120, 2843–2853 CrossRef CAS.
- B. L. Edelbach, D. A. Vicic, R. J. Lachicotte and W. D. Jones, Organometallics, 1998, 17, 4784–4794 CrossRef CAS.
- T. Schaub and U. Radius, Chem. – Eur. J., 2005, 11, 5024–5030 CrossRef CAS PubMed.
- T. Schaub, M. Backes and U. Radius, Organometallics, 2006, 25, 4196–4206 CrossRef CAS.
- M. Joost, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236–5240 CrossRef CAS PubMed.
- J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884–7887 CrossRef CAS PubMed.
- A. Zeineddine, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed.
- Y. Koga, M. Kamo, Y. Yamada, T. Matsumoto and K. Matsubara, Eur. J. Inorg. Chem., 2011, 2869–2878, DOI:10.1002/ejic.201100055.
- S. M. Kilyanek, X. D. Fang and R. F. Jordan, Organometallics, 2009, 28, 300–305 CrossRef CAS.
- F. Leroux, L. Bonnafoux and F. Colobert, Synlett, 2010, 2953–2955 CrossRef.
- L. Bonnafoux, F. R. Leroux and F. Colobert, Beilstein J. Org. Chem., 2011, 7, 1278–1287 CrossRef CAS PubMed.
- S. M. H. Kabir and M. Iyoda, Synthesis, 2000, 1839–1842, DOI:10.1055/s-2000-8239.
- R. A. Altman and S. L. Buchwald, Org. Lett., 2006, 8, 2779–2782 CrossRef CAS PubMed.
- S. Grundemann, A. Kovacevic, M. Albrecht, J. W. Faller and R. H. Crabtree, J. Am. Chem. Soc., 2002, 124, 10473–10481 CrossRef PubMed.
- M. Livendahl, C. Goehry, F. Maseras and A. M. Echavarren, Chem. Commun., 2014, 50, 1533–1536 RSC.
- R. Usón, A. Laguna, M. Laguna, B. R. Manzano, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1984, 285–292, 10.1039/dt9840000285.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS.
- T. A. Engesser, C. Friedmann, A. Martens, D. Kratzert, P. J. Malinowski and I. Krossing, Chem. – Eur. J., 2016, 22, 15085–15094 CrossRef CAS PubMed.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. M. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- Y. Minenkov, A. Singstad, G. Occhipinti and V. R. Jensen, Dalton Trans., 2012, 41, 5526–5541 RSC.
- P. Rydberg and L. Olsen, J. Phys. Chem. A, 2009, 113, 11949–11953 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Dohm, A. Hansen, M. Steinmetz, S. Grimme and M. P. Checinski, J. Chem. Theory Comput., 2018, 14, 2596–2608 CrossRef CAS PubMed.
- F. Jensen, Introduction to computational chemistry, John wiley & sons, 2017 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. S. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- G. Dong, Topics in Current Chemistry, Springer Berlin, 2014 Search PubMed.
- A. B. Chaplin, R. Tonner and A. S. Weller, Organometallics, 2010, 29, 2710–2714 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc., 2019, 141, 17414–17420 CrossRef CAS PubMed.
- D. F. Shriver and M. A. Drezdzon, The manipulation of air-sensitive compounds, Wiley, New York, 1986 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- TURBOMOLE V7.4 2019, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com, 2019.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- J. Ho and M. Z. Ertem, J. Phys. Chem. B, 2016, 120, 1319–1329 CrossRef CAS PubMed.
- A. W. Lange and J. M. Herbert, J. Chem. Phys., 2010, 133, 244111 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- J. Song, E. L. Klein, F. Neese and S. Ye, Inorg. Chem., 2014, 53, 7500–7507 CrossRef CAS.
- H. Fang, H. Jing, H. Ge, P. J. Brothers, X. Fu and S. Ye, J. Am. Chem. Soc., 2015, 137, 7122–7127 CrossRef CAS.
- B. Mondal, F. Neese and S. Ye, Inorg. Chem., 2015, 54, 7192–7198 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2018369–2018373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00953b |
| This journal is © The Royal Society of Chemistry 2021 |