
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise collapse of a giant pore metal–organic framework†
Adam F.
Sapnik
 a,
Duncan N.
Johnstone
a,
Sean M.
Collins
ab,
Giorgio
Divitini
a,
Alice M.
Bumstead
a,
Christopher W.
Ashling
a,
Philip A.
Chater
c,
Dean S.
Keeble
c,
Timothy
Johnson
d,
David A.
Keen
e and
Thomas D.
Bennett
*a
a,
Duncan N.
Johnstone
a,
Sean M.
Collins
ab,
Giorgio
Divitini
a,
Alice M.
Bumstead
a,
Christopher W.
Ashling
a,
Philip A.
Chater
c,
Dean S.
Keeble
c,
Timothy
Johnson
d,
David A.
Keen
e and
Thomas D.
Bennett
*a
aDepartment of Materials Science and Metallurgy, University of Cambridge, Cambridge, CB3 0FS, UK. E-mail: tdb35@cam.ac.uk
bSchool of Chemical and Process Engineering & School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
cDiamond Light Source Ltd, Diamond House, Harwell Campus, Didcot, Oxfordshire OX11 0DE, UK
dJohnson Matthey Technology Centre, Blount's Court, Sonning Common, RG4 9NH, UK
eISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire OX11 0QX, UK
First published on 26th March 2021
Abstract
Defect engineering is a powerful tool that can be used to tailor the properties of metal–organic frameworks (MOFs). Here, we incorporate defects through ball milling to systematically vary the porosity of the giant pore MOF, MIL-100 (Fe). We show that milling leads to the breaking of metal–linker bonds, generating additional coordinatively unsaturated metal sites, and ultimately causes amorphisation. Pair distribution function analysis shows the hierarchical local structure is partially retained, even in the amorphised material. We find that solvents can be used to stabilise the MIL-100 (Fe) framework against collapse, which leads to a substantial retention of porosity over the non-stabilised material.
Introduction
Metal–organic frameworks (MOFs) are hybrid materials comprised of metal nodes connected via organic linkers. Systematic substitution of metal nodes and linkers, a practice known as crystal engineering, has afforded a diverse family of materials that have found potential applications in catalysis, gas sorption and drug delivery.1,2 Crystal engineering strategies have been employed to tune porosity in particular.3 This has led to the discovery of materials with specific surface areas in excess of 5000 m2 g−1.4 However, these strategies often require complex linkers with challenging syntheses and suffer the potential risk of structural interpenetration.3It has been known for many years that defects in inorganic materials often give rise to functional properties, for example the modification of electrical conductivity in semiconductors.5 More recently, defects such as linker vacancies, dislocations and surface heterogeneity have been investigated in MOFs to access materials with improved gas storage, separation or catalytic properties—referred to as defect engineering.6–11 Specific examples include using defects to enhance H2 and CO2 uptake,12,13 boost performance in catalytic oxidative addition reactions,14 modulate crystal morphology,15 increase hydrophilicity,16 and even induce long-range ferromagnetic ordering.17 Defects can be introduced through de novo syntheses. For example, modulated synthesis of UiO-66 is well documented to lead to the formation of correlated defect nanoregions.18,19 Defects can also be introduced post-synthetically to a crystalline MOF, for example via post-synthetic linker exchange, which has been used to improve CO2 adsorption capacity in UiO-66.20 Defects are often, inadvertently, incorporated into crystalline MOF powders when they are pressed into pellets to obtain the bulk materials that are commonly used in practical industrial applications.21 This is typically deemed to be an undesirable side effect.
Ball milling involves mechanical impact at high frequencies to apply shear stress to a material and has proven to be an easy way to incorporate defects through breaking metal–linker bonds.22 Prolonged ball milling has also been used to induce amorphisation, that is the loss of long-range order, in several MOFs.23–25 Few reports exist, however, which connect the introduction of framework defects to structural collapse.22,26,27
MIL-100 (Fe) has received attention due to its hierarchically structured, giant pore framework with large internal surface area, in addition to its high chemical and hydrothermal stability.28–30 MIL-100 (Fe) is formed from an oxo-centred trimer motif, where three FeO6 octahedra cluster around a single, shared, oxo anion [Fig. 1a]. Four of these trimer units assemble via the organic linker, 1,3,5-benezenetricarboxylate, to form a supertetrahedron [Fig. 1b]. These supertetrahedra form the nodes upon which the crystalline mtn network is constructed [Fig. 1c]. MIL-100 (Fe) possesses a cubic space group (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with a lattice parameter of approximately 73.3 Å, giving it a large unit cell volume in excess of 390
m) with a lattice parameter of approximately 73.3 Å, giving it a large unit cell volume in excess of 390![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å3.28 The structure includes two sizes of mesopore with internal diameters of 25 and 29 Å, accessible through respective microporous windows with diameters of 5.5 and 8.6 Å. MIL-100 (Fe) has permanent porosity between Type I and IV behaviour, indicating the presence of both micro- and mesopores, and a Brunauer–Emmett–Teller (BET) surface area in excess of 2000 m2 g−1.28 The nitrogen adsorption isotherm of MIL-100 (Fe) displays a slight secondary uptake due to the dual pore structure, with adsorption first occurring in the smaller pore, before filling the larger pore. The mesoporous cages can act as a water vapour adsorbent,31 whilst activation of the framework leads to coordinatively unsaturated metal sites.32,33
000 Å3.28 The structure includes two sizes of mesopore with internal diameters of 25 and 29 Å, accessible through respective microporous windows with diameters of 5.5 and 8.6 Å. MIL-100 (Fe) has permanent porosity between Type I and IV behaviour, indicating the presence of both micro- and mesopores, and a Brunauer–Emmett–Teller (BET) surface area in excess of 2000 m2 g−1.28 The nitrogen adsorption isotherm of MIL-100 (Fe) displays a slight secondary uptake due to the dual pore structure, with adsorption first occurring in the smaller pore, before filling the larger pore. The mesoporous cages can act as a water vapour adsorbent,31 whilst activation of the framework leads to coordinatively unsaturated metal sites.32,33
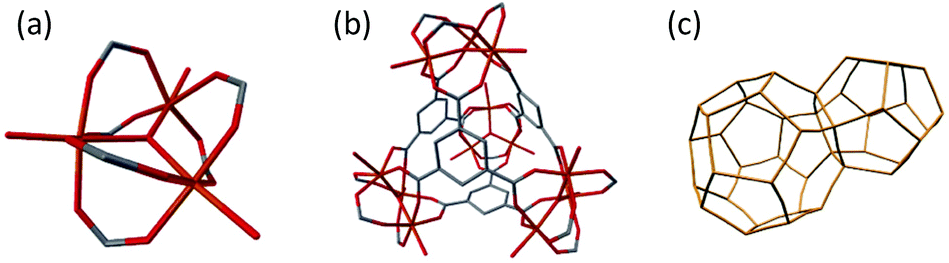 | ||
| Fig. 1 Hierarchical structure of MIL-100 (Fe). (a) Oxo-centred trimer of FeO6 octahedra, (b) supertetrahedron and (c) underlying mtn topology net of MIL-100 (Fe), supertetrahedra form the nodes of the connected network. Iron (orange), oxygen (red), carbon (grey), hydrogen atoms omitted for clarity. | ||
Furthermore, MIL-100 (Fe) has demonstrated promising separation of light hydrocarbons,34 and good activity as a Friedel–Crafts reaction catalyst.28,32,35 More recently, defects have been introduced into MIL-100 (Fe) using a mixed-linker synthetic approach which led to the formation of larger mesopores in addition to macropores, resulting in excellent performance for toluene adsorption.36 Coupled with its low acute toxicity and readily available reagents, the framework is an ideal candidate to investigate tuneable porosity through the incorporation of defects.37
Here, we characterise the defects introduced into MIL-100 (Fe) by ball milling and show that the controlled introduction of such defects can result in materials of varying porosity, which retain the chemical composition, thermal stability and local structure of the crystalline material. We demonstrate that increasing the proportion of framework defects ultimately leads to structural collapse and amorphisation. We also explore how solvents, acting as framework stabilising media, can provide access to an even wider range of porosity. Finally, we highlight the importance of defect engineering via ball milling as a powerful tool to readily augment the properties of crystalline MOFs in the search for new functional materials.
Methods
Materials
All chemicals were obtained from commercial suppliers and used as received. 1,3,5-Benzenetricarboxylic acid (95%), sodium hydroxide pellets (98%), iron(II) chloride tetrahydrate (99.99%), ethanol (99.8%) and ammonium fluoride (99.99%) were all purchased from Sigma Aldrich.Synthesis
MIL-100 (Fe), which has the chemical formula Fe3(OH)(H2O)2O[(C6H3)(CO2)3]2·nH2O, and is subsequently referred to as MIL-100, was synthesised following the procedure in ref. 38. Briefly, 1,3,5-benzenetricarboxylic acid (1.676 g, 7.976 mmol) was dissolved in an aqueous 1 M solution of sodium hydroxide (23.72 g), and iron(II) chloride tetrahydrate (2.26 g, 11.37 mmol) was separately dissolved in water (97.2 mL). The linker solution was then added dropwise to the metal solution. A green suspension formed immediately, which slowly turned brown over 24 hours whilst being stirred at room temperature. The suspension was centrifuged, and the resulting powder washed thoroughly with ethanol (3 × 20 mL). The powder was purified in accordance with ref. 30 to increase the surface area. The powder was dispersed and heated for 3 hours successively in water (700 mL at 70 °C), ethanol (700 mL at 65 °C) and aqueous 38 mM ammonium fluoride solution (700 mL at 70 °C) and was recovered between each stage by centrifugation. The resultant powder was then dried overnight at 60 °C.Ball milling
Prior to ball milling, MIL-100 was activated at 150 °C under dynamic vacuum overnight. For each duration of ball milling, 100 mg of activated MIL-100 was sealed inside a 10 mL stainless steel jar with one 7 mm diameter stainless steel ball bearing at room temperature. The jar was then placed in a Retsch MM400 grinder mill and ball milled at 25 Hz for 1, 2, 3, 4, 5, 10, 15 and 30 minute durations. After milling, the powder was recovered and characterised.Powder X-ray diffraction
Powder X-ray diffraction data were collected at room temperature on a Bruker D8 diffractometer using Cu Kα1 (λ = 1.5406 Å) radiation and a LynxEye position sensitive detector with Bragg–Brentano parafocusing geometry. Samples of powder were packed into circular (5 mm diameter) low background silicon sample holders, ensuring comparable amounts of sample were measured by the beam. They were then loaded onto the rotating stage of the diffractometer. Data were collected over the angular range 5° < 2θ < 45°. Pawley refinements were carried out using TOPAS Academic (V6) software.39 The unit cell parameters were compared with those previously reported for MIL-100.28 A modified Thompson-Cox-Hasting pseudo-Voigt peak shape and simple axial divergence correction were employed. Refinements were carried out over the angular range 5° < 2θ < 45°.Scanning electron microscopy
Scanning electron microscopy was carried out on a ZEISS Crossbeam 540 equipped with a Gemini 2 column. Finely ground samples were dispersed onto carbon tape on an aluminium specimen stub. A 1 kV accelerating voltage was used at a working distance of approximately 4.5 mm, and images were obtained under secondary electron imaging conditions.High-resolution scanning transmission electron microscopy
Finely ground powder samples were dispersed by drop-casting from a suspension in methanol onto lacey carbon grids. Aberration corrected high-resolution scanning transmission electron microscopy (HR-STEM) was performed using a JEOL ARM300CF microscope equipped with a cold field emission electron source and JEOL aberration correctors in both the probe-forming and image-forming optics, located in the electron Physical Sciences Imaging Centre (ePSIC) at the Diamond Light Source (UK). The microscope was operated at 200 kV with a convergence semi-angle of approximately 14 mrad. Micrographs were recorded using the annular bright-field (ABF) STEM signals obtained using a combination of an ABF aperture and bright-field detector, with a camera length selected such that the outer edge of the direct beam disk was aligned with the outer edge of the ABF aperture. The inner angle of the ABF aperture was approximately 50% of the outer angle, giving a collection window of approximately 7 to 14 mrad. Annular dark-field (ADF) STEM images were acquired simultaneously with an inner collection angle >40 mrad.Helium pycnometry
Sample densities were measured using a Micromeritics Accupyc 1340 helium pycnometer with a cylindrical 1 cm3 insert. Typically, 100 to 200 mg of sample was used and apparent pycnometric densities were reported as an average of 10 measurements. Prior to measurement, samples were degassed at 150 °C under dynamic vacuum overnight.Thermogravimetric analysis
Thermogravimetric analysis was performed on a TA Instruments TGA-Q400, under an air atmosphere, using a heating rate of 10 °C min−1 from room temperature up to 850 °C. For each measurement around 10 mg of sample was used. Prior to measurement, samples were degassed at 150 °C under dynamic vacuum overnight.Fourier-transform infrared spectroscopy
A Bruker Tensor 27 Infrared Spectrometer, equipped with a diamond attenuated total reflection module, was used to collect Fourier-transform infrared spectroscopy data. Data were collected between 590 and 4000 cm−1. A background was collected and subtracted from all spectra.Elemental analysis
Around 1.8 mg of sample was weighed into tin capsules and sealed. Analysis was conducted using an Exeter Analytical CE-440 Elemental Analyser with gas mixtures analysed using thermal conductivity detectors.X-ray total scattering and pair distribution function
X-ray total scattering data were collected at the I15-1 beamline at the Diamond Light Source, UK (λ = 0.161669 Å, 76.7 keV). A small amount of finely ground sample was loaded into a borosilicate capillary (inner diameter of 1.17 mm) to a height of 3.5 cm. Capillaries were sealed and mounted onto the instrument. Data were collected at room temperature for each sample, an empty capillary and the blank instrument over the region ∼0.4 < Q < ∼26 Å−1. The raw total scattering data were corrected for background, multiple scattering, container scattering and Compton scattering, along with absorption corrections, between 0.6 < Q < 24 Å−1 using GudrunX following standard procedures.40 The Fourier transform was then taken to produce the real-space pair distribution function, G(r). We also make use of the D(r) formulation of the pair distribution function to accentuate correlations at high-r.41 Partial pair distribution functions were calculated using the RMCProfile software package, using the previously reported structure of MIL-100.28,42 Partial PDFs were combined, using Q-independent weighting terms based on the number of electrons and proportion of each atom type in the material, to produce the total PDF of MIL-100. Longer-ranged oscillations in the PDF due to density fluctuations in the MIL-100 structure, which were not measured experimentally, were accounted for by subtracting the Fourier transform of the calculated low-Q scattering from the calculated PDF before comparison with the experimental PDF data.Nitrogen sorption porosimetry
Nitrogen adsorption measurements were carried out at 77 K using a Micromeritics TriStar II instrument. Approximately 40 mg of each sample was degassed at 120 °C for 12 hours prior to measurement. The specific surface area of the material was calculated using the multipoint BET method applied to the adsorption branch, while taking into account the Rouquerol consistency criteria.43–45Non-local density functional theory (NL-DFT) can be used to extract pore size distributions from nitrogen adsorption isotherms.46,47 Classical fluid density functional theory is used to construct adsorption isotherms in ideal pore geometries and then solve the adsorption integral equation.48 Several inherent limitations persist however, including the absence of a specific kernel available for MOFs.49 In lieu of this, activated carbon kernels are often used, given the vast majority of accessible surface within MOFs is organic in nature.50 Furthermore, whilst the use of NL-DFT for amorphous systems has previously demonstrated value, it cannot be scrutinised with the same level of detail as for crystalline systems.49 Micropore size distributions were estimated using an NL-DFT approach, employing a carbon—N2, 2D heterogeneous surface kernel, within the SAIEUS software.49,51
Results and discussion
Powder X-ray diffraction
MIL-100 was synthesised following the procedure in ref. 38 [see Methods]. Pawley refinement of the powder X-ray diffraction data was then used to confirm the phase purity and crystalline nature of MIL-100 [Fig. S1 and Table S1†]. Following this, samples of evacuated MIL-100 were subject to ball milling at 25 Hz for 1, 2, 3, 4, 5, 10, 15 and 30 minutes. Powder X-ray diffraction data from these samples revealed a relatively fast decrease in Bragg peak intensity as the duration of ball milling increased [Fig. 2a & S2†]. The relative integrated area of the (333) Bragg peak, one of the stronger isolated Bragg peaks, was used to quantify the effect of ball milling [Fig. 2b and Table S2†]. This simple measure may be interpreted in terms of “crystallinity”, but, of course, the peak profile itself results from a convolution of factors, for example crystallite size and static disorder such as the presence of defects and microstrain. The relative intensity displayed a 65% reduction after 5 minutes of ball milling. After 10 minutes most Bragg peaks showed almost negligible intensity, with over a 95% reduction in intensity. The rate of decreasing intensity slowed as milling progressed, with approximately a 5% decrease between 10 and 30 minutes. After 30 minutes, only weak diffuse scattering was observed and we denote this material as ‘amorphous’, labelling it amMIL-100 in accordance with prior literature [Fig. 2a].24 This gradual collapse is consistent with the incorporation of defects into the structure, which in turn causes a transition from the ordered, crystalline system, to the disordered, amorphous state. | ||
| Fig. 2 (a) Power X-ray diffraction data for MIL-100 as-synthesised (blue) and amMIL-100, after 30 minutes of ball milling (red). (b) “Crystallinity” as a function of milling time based on the intensity of the (333) Bragg peak (labelled in (a)). Grey line is a guide. | ||
Scanning electron microscopy
Whilst ball milling was used here to introduce structural defects, it is also a common technique to prepare homogeneous mixtures of materials, and also to reduce the particle size of materials from the sub-micrometre scale down to nanometre dimensions.52,53 The delineation between particle size reduction and the introduction of static disorder (e.g. defects and microstrain) by powder X-ray diffraction is notoriously difficult. Several examples of nanocrystalline MOF systems have been reported in the literature.54–56 Their powder X-ray diffraction patterns give rise to regions of broad scattering centred on the positions of Bragg scattering in the crystalline material, with the peak width inversely proportional to the crystallite size. This weak scattering is often misinterpreted as diffuse scattering from an amorphous system.25We therefore used scanning electron microscopy (SEM) to differentiate between amorphisation upon ball milling and decreasing particle size. Images of crystalline MIL-100, pre-ball milling, revealed sub 20 μm aggregates of particles around 0.5 to 1 μm in diameter, consistent with the reported synthesis [Fig. 3a].38 These particles displayed some degree of faceting, however the morphology itself remained ill-defined. For amMIL-100, i.e. the sample after 30 minutes ball milling, slightly fewer of the larger agglomerates were observed, yet the constituent particles themselves appear unchanged [Fig. 3b]. The qualitatively consistent size between samples pre- and post-ball milling suggests the weak Bragg scattering observed upon milling is linked to structural disordering, and not particle size reduction [see Fig. S3† for additional images].
 | ||
| Fig. 3 Scanning electron microscopy images of (a) MIL-100 and (b) amMIL-100. | ||
High-resolution scanning transmission electron microscopy
To confirm the absence of structural periodicity in amMIL-100, high-resolution scanning transmission electron microscopy (HR-STEM) was used. HR-STEM allows for direct observation of lattice fringes, features that correspond to real-space structural periodicity within the sample. As expected, images of crystalline MIL-100 contained lattice fringes. The fringes exhibited a spacing of approximately 4.2 nm, which corresponds to the (111) crystallographic plane [Fig. 4a]. Numerous images were collected of amMIL-100, though no lattice fringes were observed [Fig. 4b]. Images of amMIL-100 generally exhibited a highly mottled contrast, suggesting variations in density or fragmentation of nanoscale particles at the surface. The absence of lattice fringes in numerous images of amMIL-100 acquired under conditions used to repeatedly record lattice fringes in MIL-100, though not definitive, was indicative of reduced periodic order in the samples after ball milling, consistent with the amorphous structure of amMIL-100 observed via powder X-ray diffraction measurements [see Fig. S4† for additional images].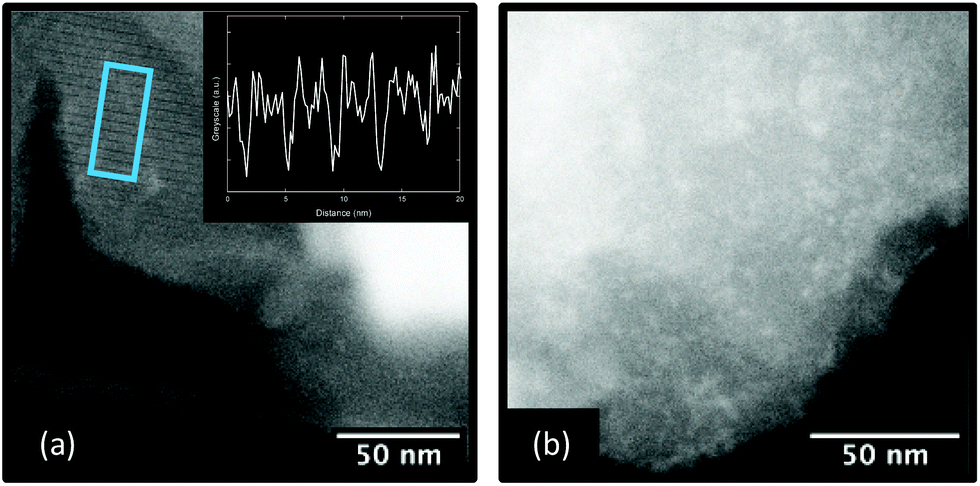 | ||
| Fig. 4 (a) High-angle annular dark-field (HAADF) HR-STEM image of MIL-100 exhibiting lattice fringes, highlighted by the blue box. The inset shows the intensity profile corresponding to the highlighted region, the distance between minima is 4.2 nm. (b) Example image of amMIL-100 where no lattice fringes were observed. | ||
Helium pycnometry
The amorphisation of MIL-100 was accompanied by a visible decrease in sample size. Helium pycnometry was used to quantify this densification by measuring the skeletal densities of MIL-100 and amMIL-100. Unexpectedly, we found the experimental density of MIL-100 to decrease slightly upon milling from 2.083(3) to 2.030(3) g cm−3 for amMIL-100. We ascribe this inconsistency to the highly porous nature of crystalline MIL-100. Helium is likely to penetrate the pores of MIL-100, overestimating the free space volume for the measurement. The measured slight decrease in density upon amorphisation, despite a visible volume decrease, suggests amMIL-100 becomes less penetrable by helium. In both materials, these densities are significantly higher than that of the crystal structure (0.69 g cm−3).28 This is likely due to the presence of unreacted linker and water molecules within the pores, which were subsequently confirmed in the thermogravimetric analysis and Fourier-transform infrared spectroscopy measurements below.Thermogravimetric analysis
Thermogravimetric analysis (TGA) was used to compare the effect of ball milling upon thermal stability, and in particular to examine the effect of defect incorporation. The TGA traces of crystalline MIL-100 and the ball-milled materials exhibited three characteristic mass losses, consistent with previous reports [Fig. 5a].28 Firstly, at around 100 °C water molecules within the pores are lost. At 200 °C, structural water molecules coordinated directly to the iron centres in the trimer unit are lost. Finally, at 365 °C the structure decomposes.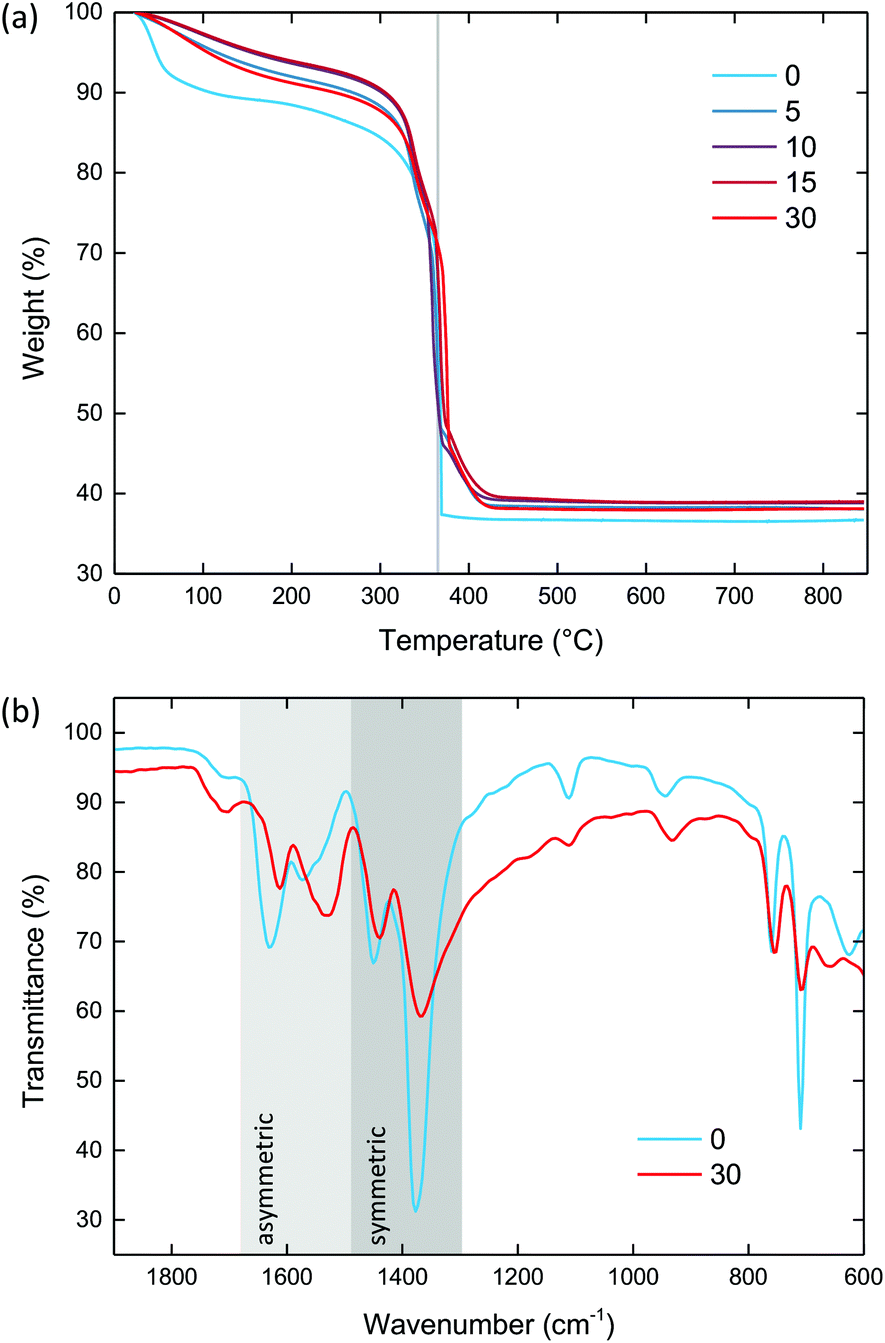 | ||
| Fig. 5 (a) Thermogravimetric data of the ball-milled MIL-100 materials, performed under air at 10 °C min−1. Vertical grey line highlights decomposition at 365 °C. (b) FT-IR spectra for MIL-100 (blue) and amMIL-100 (red). The asymmetric (light grey) and symmetric (dark grey) carboxylate regions are highlighted. Key in both figures shows the duration of ball milling in minutes. | ||
All samples were degassed prior to measurement. Despite this, crystalline MIL-100 still showed a significant amount of water within the pores (9.7%), consistent with uptake of water from the atmosphere within minutes after evacuation. For the ball-milled materials, there was a noticeable reduction in the amount of water within the pores to between 2.6 and 4.6%, which suggests some degree of collapsed porous interior.
As for the structural water, the ball-milled materials exhibited a higher mass loss (between 3.4 and 4.2%) than crystalline MIL-100 (1.9%). This is consistent with the breaking of metal–linker bonds upon milling, and hence the formation of extra unsaturated metal sites to which water can coordinate.
The temperature of decomposition was very similar for all materials, centred around 365 °C. The mass loss associated with linker decomposition was similar for all of the materials, within the 52 to 55% range, in agreement with previous reports.38 Crystalline MIL-100 showed a decomposition pathway that is consistent with the previously reported abrupt, single-step process. On the other hand, the ball-milled samples all decomposed via a two-step route that continued until 420 °C. This secondary step in the TGA has previously been observed for 1,3,5-benzentricarboxylic acid itself.38 Noting that MIL-100 does not display this feature, it may therefore be indicative of metal–linker bonds breaking generating undercoordinated linkers, as expected upon ball milling. Powder X-ray diffraction of the bright orange combustion product confirmed the decomposition of MIL-100 into the iron oxide hematite (Fe2O3) [Fig. S5 and Table S3†].
Fourier-transform infrared spectroscopy and elemental analysis
Fourier-transform infrared (FT-IR) spectroscopy was used to identify the bonding mode of the linker, and hence confirm the breaking of metal–linker bonds [Fig. 5b & S6†]. The FT-IR spectrum of MIL-100 contained two symmetric and two asymmetric bands between 1445 to 1370 and 1630 to 1564 cm−1, respectively, characteristic of the linker carboxylate groups binding in a syn–syn fashion.57,58 These bands were present in amMIL-100, however there was a significant shift of the asymmetric bands to lower wavenumbers. This shift indicates the carboxylate group has partially transitioned from syn–syn to a monodentate coordination mode, with the band at 1550 cm−1 characteristic of the asymmetric stretch of a monodentate coordinated carboxylate.59 This behaviour has previously been observed for UiO-66 under compression,59 and confirms that metal–linker bonds have been broken upon ball milling. Furthermore, the presence of defects resulted in a slightly broader spectrum in amMIL-100 than MIL-100 which may arise from the disordered structure. Three small bands were present from minor impurities of unreacted linker (1111, 1713 and 2361 cm−1). CHN analysis revealed the chemical composition remained the same upon ball milling [Table S4†].X-ray total scattering and pair distribution function
X-ray total scattering data were collected using the I15-1 (XPDF) beamline at the Diamond Light Source (UK) to examine how the local structure of the framework is affected upon milling. The structure factor for MIL-100 reflected its crystalline nature, displaying clear Bragg peaks [Fig. S7†]. The peak intensities decreased as the milling duration increased. The dominant Bragg peak at 0.7 Å−1, likely to include the peaks centred on the strong (733) Bragg reflection, exhibited a particularly large decrease in intensity. The rate of decrease was comparable to the (333) peak in the powder diffraction data. There was a notable absence of Bragg scattering after 10 minutes [Fig. S8†]. This suggests a significant proportion of metal–linker bonds are broken after 10 minutes allowing the structure to collapse and disrupt the long-range order.Following appropriate normalisation, the raw total scattering data were converted by Fourier transform to the real-space pair distribution function (PDF), which describes the atom–atom correlations in a material. The presence of long-range correlations, i.e. those beyond 20 Å, in the PDF for MIL-100 are consistent with the crystalline nature of the framework [Fig. 6a]. Firstly, the experimental PDF for MIL-100 was compared to the expected PDF calculated from the MIL-100 structure. We found good agreement between the experimental and calculated data.
 | ||
| Fig. 6 (a) Experimental MIL-100 (blue), experimental amMIL-100 (red) and calculated MIL-100 (black) pair distribution functions. The inset shows the 8 to 14 Å region for MIL-100 and amMIL-100. (b) Simulated pair distribution function of the trimer unit. Inset shows a trimer unit. (c) Simulated pair distribution function of the supertetrahedron unit. Inset shows a supertetrahedral unit. Iron (orange), oxygen (red), carbon (grey), hydrogens omitted for clarity. | ||
The PDF for amMIL-100 displayed a notable absence of long-range correlations, as expected due to its amorphous nature [Fig. 6a]. However, the PDF for amMIL-100 was very similar to crystalline MIL-100 at low-r (<15 Å). This suggests there is significant retention of the local structural geometry in the amorphous material. Comparison between the experimental amMIL-100 PDF and those simulated from individual oxo-centred trimer and supertetrahedron models revealed the extent of local structure in amMIL-100 [Fig. 6b & c]. Specifically, we find that amMIL-100 contains all of the correlations associated with the trimer unit (below 7 Å), indicating these are largely retained upon amorphisation [Fig. 6a]. There are very slight indications of correlations from the supertetrahedra present in the region of 8 to 12 Å of the PDF of amMIL-100 [Fig. 6a inset]. The low intensity of these correlations suggests that a significant proportion of the supertetrahedra are broken during the milling process, consistent with the breaking of metal–linker bonds.
The structure of MIL-100 is particularly complex, with the unit cell containing over 10 000 atoms. In order to help further interpret the PDFs, the six partial PDFs (Fe–Fe, Fe–O, Fe–C, O–O, C–O and C–C) of MIL-100 were calculated and weighted by their Q-independent contributions to the total PDF [Fig. 7]. These partial PDFs highlight the pairwise interactions between specific sets of atom types. Those containing H were disregarded due to their minimal scattering contribution. We note that even at low-r there are multiple overlapping contributions to each peak, reflecting the complexity of the MIL-100 structure. Nonetheless, this facilitated the assignment of the first three main peaks in the PDF at 1.98, 3.34 and 4.68 Å (labelled a to c respectively), the major contributions to which are ascribed to (a) Fe–O, (b) Fe–O and Fe–Fe and (c) Fe–O, Fe–C and C–O correlations [Fig. 7 inset]. This is consistent with those previously reported by Weckhuysen et al.46 Furthermore, the use of synchrotron radiation in this study provided us with extremely high real-space resolution, and so additional peaks at 2.98, 4.38 and 5.32 Å were also able to be resolved. The first and second of these were assigned to Fe–C distances. The origin of the third remained unclear, even after comparison with the partial PDFs.
 | ||
| Fig. 7 Partial pair distribution functions calculated from the crystalline structure of MIL-100, weighted by their Q-independent contribution to the total pair distribution function. Partials are offset for clarity. The inset shows how the partial pair distribution functions contribute to the low-r region of the experimental G(r) for MIL-100 (black, offset from partials for clarity). | ||
Furthermore, obtaining high-resolution PDFs for the entire series of ball-milled materials allows for valuable insight into the structural evolution of MIL-100 upon milling [Fig. 8 & S9†]. Upon first inspection we note that the PDFs of the ball-milled materials, i.e. those intermediate to the crystalline and amorphous materials, are broadly similar and contain peaks in the same positions with similar peak widths. In particular, the same correlations are observed in the 1 to 7 Å region, indicating clear retention of the trimer unit in all of the ball-milled materials. We also note the presence of substantial peaks between 7 and 12 Å. These correlations reflect the retention of the supertetrahedral units. Notably, their intensity, relative to amMIL-100, suggests a greater proportion of these assemblies remain intact in the intermediate materials. In turn, the greater proportion of supertetrahedral units points towards a greater retention of porosity. The dominant change upon milling is associated with the peak intensities. As the duration of milling increases, the overall intensity of the PDF peaks decreases; that is, the number of atom–atom correlations at a given distance decreases as defects are introduced. This has a more significant effect at higher-r, where long-range order within the structure is required for these correlations to be preserved.
 | ||
| Fig. 8 Pair distribution functions for the ball-milled MIL-100 materials, from crystalline MIL-100 (blue) to amMIL-100 (red). The Fe–O (a), Fe–O and Fe–Fe (b) and Fe–O, Fe–C and C–O (c) correlations have been marked. Key shows duration of ball milling in minutes. | ||
Closer inspection revealed the relative variation in peak intensity was not the same for all peaks. In order to probe the rate at which the peaks varied, a simple peak analysis was carried out. Firstly, the baseline of each PDF was calculated using the peak minima and subtracted from the D(r) to obtain Dsub(r) [Fig. 9a]. From Dsub(r) the peak maxima were extracted, giving the position and maximum intensity of each peak. For each peak position, a relative peak intensity was calculated from the initial intensity. This was performed for all PDFs of the ball-milled series. The relative peak intensities, as a function of milling time, for each peak position between 0 and 30 Å were then plotted [Fig. 9b]. Peaks with an initial intensity below 0.15 in Dsub(r) were excluded due to the sensitivity of these peaks to systematic errors in the background subtraction.
 | ||
| Fig. 9 (a) Baseline subtracted pair distribution functions of the ball-milled materials. Key shows the duration of milling in minutes. (b) Relative intensity of the correlations extracted from (a) as a function of milling time. The two correlations of interest are shown in blue (9.62 Å) and red (11.30 Å) and have been highlighted in (a) as vertical lines. | ||
As noted previously, peak intensities decrease upon milling. The amount by which the relative peak intensity decreases upon milling generally increases with r. Peak intensities within a PDF will typically decrease as a function of r, due to thermal disorder in the sample and the finite Q-resolution of the measurement. These lower intensity peaks are more sensitive to changes in intensity. Hence, greater changes in relative intensity are observed as peak position increases.
Within this trend of decreasing relative peak intensity, the peaks cluster into three distinct regimes: correlations between 1.34 to 2.98 Å, 3.34 to 11.30 Å and 13.44 to 24.90 Å. The first set show an almost negligible change in intensity after 30 minutes. The second exhibit a decrease of approximately 50%, whilst the third have an almost 100% decrease.
The first regime includes the C–C and C–O of the linker, Fe–O (which comprises the shorter Fe–O(μ3) and Fe–O(carboxylate) distances) and the Fe–C correlation. Initially these peaks show an increase in intensity. However, this is not regarded to have any physical significance due to the lower statistics and susceptibility to systematic error in this low-r region of the PDF. Beyond this, these correlations appear to exhibit very minimal changes in relative intensity. These correlations predominantly originate from the trimer unit of MIL-100 [Fig. 6b]. This is consistent with the resistance to deformation and hence retention of the trimer unit upon milling.
The second regime of peak positions (3.34 to 11.30 Å) contains contributions from all six of the partial PDFs. Most peaks in this region have multiple overlapping contributions. The crucial observation is that peaks in this second regime are largely dominated by those originating from within individual supertetrahedra [Fig. 6c]. This is a clear indication of the greater susceptibility to collapse upon milling of the supertetrahedra with respect to the individual trimer units.
The final regime (13.44 to 24.90 Å) also involves numerous overlapping contributions to each peak. Importantly, we identify that these peaks arise from atomic interactions between supertetrahedra (i.e. those which depend on long-range order within the structure). It is these correlations that are impacted the most upon milling, demonstrating the disruption of periodic order between supertetrahedra.
There are two notable exceptions at 9.62 and 11.30 Å to the trends described above. These peaks arise in the supertetrahedral region of the PDF [see Fig. 6c], yet their relative decrease in intensity is more comparable to that of the extended structure. More specifically, both peaks are dominated by Fe–Fe interactions between trimers within a supertetrahedron, and also have considerable contributions from Fe–O and O–O interactions. The accelerated rate of decrease for these two peaks implies a particularly low mechanical stability of the supertetrahedra with respect to milling, more so than the second regime initially suggests.
This simple peak analysis has implications for our understanding of the relative structural stability of the hierarchical structure of MIL-100. The delineation in rates between the trimer, supertetrahedral and extended structure peaks is clear experimental evidence for the lower mechanical stability of the supertetrahedral unit. Given that the trimers within a supertetrahedron are assembled via the linker, and hence the presence of supertetrahedra is reliant upon the metal–linker coordination remaining intact, this is consistent with our evidence for metal–linker bond breakage. In other words, breaking metal–linker bonds through milling directly prevents the retention of supertetrahedral assemblies.
Finally, the peak analysis also revealed most peaks exhibit a very small (<0.2 Å) shift to higher-r upon milling. This suggests that minor structural distortions are occurring in addition to the structural collapse described above.
Nitrogen adsorption
Nitrogen adsorption isotherms were collected to probe the porosity of the materials, with a reduction expected due to the progressive collapse of the MIL-100 structure upon milling. The isotherms display a clear decrease in maximal nitrogen uptake upon ball milling [Fig. 10a]. The BET surface area (2240 m2 g−1) and total pore volume (0.991 cm3 g−1) for MIL-100 were higher than reported for this particular synthetic route, likely due to the purification procedure.38 On the other hand, amMIL–100 was essentially non-porous to nitrogen with a surface area of 2.02 m2 g−1 and a total pore volume of 0.049 cm3 g−1, consistent with the collapse of the porous interior. | ||
| Fig. 10 (a) Nitrogen adsorption (closed circles) and desorption (open circles) isotherms for the ball-milled MIL-100 materials. Key shows duration of ball milling in minutes. (b) Proportion of the BET surface area retained upon ball milling. | ||
At shorter milling times, where fewer defects were introduced, an appreciable retention of porosity was observed [Fig. 10b and Table S5†]. For example, milling for one minute resulted in a 30% retention in porosity, equivalent to a BET surface area of 673 m2 g−1, and a total pore volume of 0.357 cm3 g−1. After three minutes the BET surface area was 401 m2 g−1. After five minutes the surface area still remained above 100 m2 g−1.
The nitrogen adsorption isotherm of MIL-100 contains a visible signature of the dual pore structure at approximately 0.05 and 0.12 p/p0 [Fig. 10a]. These pore-dependent uptakes can be emphasised by plotting the amount adsorbed against log(p/p0) rather than p/p0 [Fig. S10†]. The secondary uptake at 0.12 p/p0 is more pronounced than the first, as expected from the greater nitrogen uptake possible from the larger pore cavity. From a simple visual inspection, the secondary uptake is difficult to resolve after ball milling for five minutes. This could suggest a weaker mechanical stability of the larger pore, which may collapse more readily upon defect incorporation.
Despite the limitations associated with NL-DFT and its suitability for amorphous MOFs [see Methods for discussion], we performed an analysis to yield a semi-quantitative insight into how pore size distribution varies upon defect incorporation in MIL-100. Consistent with prior literature on MIL-100, we used the Carbon – N2, 2D-NL-DFT, Heterogeneous Surface kernel to confirm the presence of the two pore system in crystalline MIL-100.50 Two pore sizes, with maxima at 10.9 and 17.7 Å, were observed [Fig. S11†]. These were smaller than the expected van der Waals pore dimensions, however NL-DFT has previously been reported to underestimate pore dimensions.60,61
Two pores were also evident within the samples ball milled for one to three minutes, consistent with the incorporation of localised defects. After five minutes, the distribution was significantly broader and the two pores were less prominent, suggesting significant structural disorder had been introduced disrupting the accessibility of the pore network [Fig. S12†]. This is consistent with the significant reduction in crystallinity observed via powder X-ray diffraction. No further analysis of the kinetics of pore collapse was performed given the uncertainties associated with NL-DFT outlined previously.
Solvent stabilisation
Given the short duration of ball milling required to elicit a substantial change in porosity, we sought to investigate whether it may be possible to access a wider range of surface areas by stabilising the MIL-100 structure with solvent molecules prior to milling. The sample milled for one minute was particularly attractive for this investigation, as it would enable us to obtain a material with maximum retained porosity. It has previously been shown that non-activated porous frameworks, those with solvent molecules in the pores, display greater resistance to structural collapse upon ball milling than evacuated materials.62 Such control would be beneficial for tuning the gas selectivity and separation abilities of MIL-100.Ethanol and toluene were selected as suitable solvents for the stabilisation of MIL-100. Their kinetic diameters, 0.45 and 0.61 nm respectively, mean that ethanol is able to enter both pores, whilst toluene may only enter the larger pore.63 The pore connectivity of the mtn topology ensures permeation of toluene throughout the larger pore network is possible.
Evacuated MIL-100 was soaked in either ethanol or toluene overnight. The samples, MIL-100-EtOH and MIL-100-Tol, respectively, were then filtered and air-dried overnight at room temperature. Analysis by TGA showed 25 to 30 wt% of solvent was retained in the pores [Fig. S13†]. Both solvent-stabilised samples were then ball milled for one minute. Following ball milling they were dried at 150 °C and evacuated prior to characterisation. MIL-100-EtOH and MIL-100-Tol were then compared to the MIL-100 sample that had been evacuated prior to one minute of ball milling (i.e. not stabilised by solvent).
Powder X-ray diffraction patterns of MIL-100-EtOH and MIL-100-Tol milled for one minute revealed both materials were crystalline in nature [Fig. 11a]. Additionally, SEM images of the two samples demonstrated little change in the particle morphology compared to the non-stabilised sample also milled for one minute. [Fig. S14†]. X-ray total scattering data were collected for the solvent-stabilised materials to compare the effect of different stabilising media on the framework structure [Fig. S15†]. PDF analysis revealed the trimer unit (peaks between 1 and 7 Å) and supertetrahedral assemblies (7 to 12 Å) were present in both materials.
 | ||
| Fig. 11 (a) Powder X-ray diffraction data for MIL-100-Tol (orange), MIL-100-EtOH (black) and non-stabilised MIL-100 (blue) after one minute of ball milling. (b) Nitrogen adsorption (closed) and desorption (open) isotherms for MIL-100-Tol (orange), MIL-100-EtOH (black) and non-stabilised MIL-100 (blue). | ||
We expected this stabilising effect of the solvent molecules to increase the porosity of the stabilised materials with respect to non-stabilised MIL-100. Indeed, the BET surface area for MIL-100-Tol displayed an increase of over 43%, compared with the non-stabilised MIL-100 that had also been milled for one minute, from 673 to 966 m2 g−1 [Fig. 11b, S16 and Table S6†]. For MIL-100-EtOH, a promising 63% increase in BET surface area was observed (1101 m2 g−1) over the non-stabilised sample. The maximal uptakes of MIL-100-Tol and MIL-100-EtOH were 362.8 and 372.8 cm3 g−1 respectively, both much higher than the non-stabilised sample's uptake of 231.2 cm3 g−1. Toluene was expected to stabilise the pore structure through π–π interaction between the aromatic system and the MIL-100 linker lining the surface of the pores.30,36,64 However, the smaller size of the ethanol molecule allowed it to penetrate throughout the pore network, providing a greater resistance to collapse than the toluene which had occluded only the larger pores.
NL-DFT calculations were performed on the nitrogen adsorption isotherms [Fig. S17†]. Both the solvent-stabilised and non-stabilised samples had similar pore size distributions, with maxima around 10.5 and 17.5 Å. The solvent-stabilised materials displayed a greater retention of both pore types compared to the non-stabilised sample. The peaks in the stabilised material appeared more well-defined than in the non-stabilised sample, again consistent with the reinforcement of the pore architecture by the solvent molecules.
Conclusions
Ball milling of MIL-100 was demonstrated to result in the progressive destruction of metal–linker bonds and hence the incorporation of increasing amounts of framework defects. This resulted in the eventual amorphisation of the framework. X-ray PDF analysis revealed the greater susceptibility of supertetrahedra towards collapse compared with the trimer unit, which were retained even in the amorphous material. Short durations of ball milling led to appreciably porous materials. The generation of additional coordinatively unsaturated metal sites, that are intrinsically linked to MIL-100's catalytic ability, in the defective materials is a particularly attractive benefit of these materials. Furthermore, the collapse of MIL-100 itself represents an appealing route for trapping potentially harmful guest species in the amorphous state where the local structure and thermal stability are retained. The use of solvents of different sizes, prior to ball milling, led to even greater degrees of porosity, with ethanol in particular resulting in an impressive 63% increase in BET surface area over the non-stabilised material that had been ball milled for the same time. This appears to be a particularly promising method to further control the porosity upon ball milling. As such the results here will form the basis of a future study exploring the power of solvent stabilisation in MOFs, in particular focusing on a variety of solvents and the effect of stabilisation at longer milling times. We have clearly demonstrated the ease with which defect engineering can be used to modify the properties of MIL-100, an approach that could easily be applied to other highly porous crystalline MOFs. Introducing defects into MOFs is an exciting avenue for further research; generation of coordinatively unsaturated metal sites through bond breaking may produce additional catalytically active sites, whilst modifying the surface chemistry of the pores will strongly affect the interactions with guest molecules, enhancing gas separation capabilities.Conflicts of interest
T. J. works for a company with interest in commercialisation of MOF materials.Acknowledgements
AFS acknowledges the EPSRC for a PhD studentship award under the industrial CASE scheme, along with Johnson Matthey PLC (JM11106). AMB acknowledges the Royal Society for funding (RGF\EA\180092) as well as the Cambridge Trust for a Vice Chancellor's Award (304253100). CWA would like to thank the Royal Society for a PhD studentship (RG160498) and the Commonwealth Scientific and Industrial Research Council for additional support (C2017/3108). TDB thanks the Royal Society for a University Research Fellowship (UF150021), the Leverhulme Trust for a Philip Leverhulme Prize, and the University of Canterbury Te Whare Wānanga o Waitaha, New Zealand, for a University of Cambridge Visiting Canterbury Fellowship. The authors would like to thank the JMTC analytical department and in particular Przemyslaw Magdziarz for CHN analysis. The ZEISS Crossbeam SEM is funded by the Henry Royce Institute Equipment Grant EP/P024947/1. We extend our gratitude to Diamond Light Source, Rutherford Appleton Laboratory, U.K., for access to beamline I15-1 (EE20038) and ePSIC (EM20198). We thank Dr Mohsen Danaie for his assistance at ePSIC and Dr Thomas Forrest for his help at I15-1. AFS would also like to thank Dr L. R. Owen (University of Cambridge) for useful discussions.Notes and references
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- U. Stoeck, S. Krause, V. Bon, I. Senkovska and S. Kaskel, Chem. Commun., 2012, 48, 10841–10843 RSC.
- H. J. Queisser and E. E. Haller, Science, 1998, 281, 945–950 CrossRef CAS PubMed.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS.
- M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- D. S. Sholl and R. P. Lively, J. Phys. Chem. Lett., 2015, 6, 3437–3444 CrossRef CAS.
- J. Ren, M. Ledwaba, N. M. Musyoka, H. W. Langmi, M. Mathe, S. Liao and W. Pang, Coord. Chem. Rev., 2017, 349, 169–197 CrossRef CAS.
- J. Canivet, M. Vandichel and D. Farrusseng, Dalton Trans., 2016, 45, 4090–4099 RSC.
- C. S. Tsao, M. S. Yu, C. Y. Wang, P. Y. Liao, H. L. Chen, U. S. Jeng, Y. R. Tzeng, T. Y. Chung and H. C. Wu, J. Am. Chem. Soc., 2009, 131, 1404–1406 CrossRef CAS.
- S. Gadipelli and Z. Guo, Chem. Mater., 2014, 26, 6333–6338 CrossRef CAS.
- G. Ye, D. Zhang, X. Li, K. Leng, W. Zhang, J. Ma, Y. Sun, W. Xu and S. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 34937–34943 CrossRef CAS PubMed.
- T. H. Park, A. J. Hickman, K. Koh, S. Martin, A. G. Wong-Foy, M. S. Sanford and A. J. Matzger, J. Am. Chem. Soc., 2011, 133, 20138–20141 CrossRef CAS PubMed.
- P. Ghosh, Y. J. Colón and R. Q. Snurr, Chem. Commun., 2014, 50, 11329–11331 RSC.
- L. Shen, S. W. Yang, S. Xiang, T. Liu, B. Zhao, M. F. Ng, J. Göettlicher, J. Yi, S. Li, L. Wang, J. Ding, B. Chen, S. H. Wei and Y. P. Feng, J. Am. Chem. Soc., 2012, 134, 17286–17290 CrossRef CAS PubMed.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F. X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- D. N. Johnstone, F. C. M. Firth, C. P. Grey, P. A. Midgley, M. J. Cliffe and S. M. Collins, J. Am. Chem. Soc., 2020, 142, 13081–13089 CrossRef CAS PubMed.
- G. C. Shearer, J. G. Vitillo, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 7190–7193 CrossRef CAS.
- J. Hou, A. F. Sapnik and T. D. Bennett, Chem. Sci., 2020, 11, 310–323 RSC.
- T. D. Bennett, T. K. Todorova, E. F. Baxter, D. G. Reid, C. Gervais, B. Bueken, B. Van De Voorde, D. De Vos, D. A. Keen and C. Mellot-Draznieks, Phys. Chem. Chem. Phys., 2016, 18, 2192–2201 RSC.
- T. D. Bennett, S. Cao, J. C. Tan, D. A. Keen, E. G. Bithell, P. J. Beldon, T. Friščić and A. K. Cheetham, J. Am. Chem. Soc., 2011, 133, 14546–14549 CrossRef CAS PubMed.
- S. Cao, T. D. Bennett, D. A. Keen, A. L. Goodwin and A. K. Cheetham, Chem. Commun., 2012, 48, 7805–7807 RSC.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555–1562 CrossRef CAS PubMed.
- T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F. X. Coudert, Nat. Chem., 2016, 9, 11–16 CrossRef PubMed.
- A. K. Cheetham, T. D. Bennett, F. X. Coudert and A. L. Goodwin, Dalton Trans., 2016, 45, 4113–4126 RSC.
- P. Horcajada, S. Surblé, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 100, 2820–2822 RSC.
- G. Zhong, D. Liu and J. Zhang, Cryst. Growth Des., 2018, 18, 7730–7744 CrossRef CAS.
- Y. K. Seo, J. W. Yoon, J. S. Lee, U. H. Lee, Y. K. Hwang, C. H. Jun, P. Horcajada, C. Serre and J. S. Chang, Microporous Mesoporous Mater., 2012, 157, 137–145 CrossRef CAS.
- F. Jeremias, A. Khutia, S. K. Henninger and C. Janiak, J. Mater. Chem., 2012, 22, 10148–10151 RSC.
- A. Dhakshinamoorthy, M. Alvaro, P. Horcajada, E. Gibson, M. Vishnuvarthan, A. Vimont, J. M. Grenèche, C. Serre, M. Daturi and H. Garcia, ACS Catal., 2012, 2, 2060–2065 CrossRef CAS.
- H. Leclerc, A. Vimont, J. C. Lavalley, M. Daturi, A. D. Wiersum, P. L. Llwellyn, P. Horcajada, G. Férey and C. Serre, Phys. Chem. Chem. Phys., 2011, 13, 11748–11756 RSC.
- B. Yuan, X. Wang, X. Zhou, J. Xiao and Z. Li, Chem. Eng. J., 2019, 355, 679–686 CrossRef CAS.
- J. N. Hall and P. Bollini, ACS Catal., 2020, 10, 3750–3763 CrossRef CAS.
- C. Duan, Y. Yu, P. Yang, X. Zhang, F. Li, L. Li and H. Xi, Ind. Eng. Chem. Res., 2020, 59, 774–782 CrossRef CAS.
- T. Baati, L. Njim, F. Neffati, A. Kerkeni, M. Bouttemi, R. Gref, M. F. Najjar, A. Zakhama, P. Couvreur, C. Serre and P. Horcajada, Chem. Sci., 2013, 4, 1597–1607 RSC.
- K. Guesh, C. A. D. Caiuby, Á. Mayoral, M. Díaz-García, I. Díaz and M. Sanchez-Sanchez, Cryst. Growth Des., 2017, 17, 1806–1813 CrossRef CAS.
- A. A. Coelho, TOPAS-Academic, version 6 (computer software), Tech. rep, Coelho Software, Brisbane Search PubMed.
- A. K. Soper, Rutherford Appleton Lab. Tech. Rep., 2011, RAL-TR-2011-013.
- D. A. Keen, J. Appl. Crystallogr., 2001, 34, 172–177 CrossRef CAS.
- S. M. G. Tucker, D. A. Keen, M. T. Dove, A. L. Goodwin and Q. Hui, J. Phys.: Condens. Matter, 2007, 19, 335218 CrossRef PubMed.
- F. Rouquerol, J. Rouquerol and K. S. W. Sing, Adsorption by Powders and Porous Solids, Academic Press, Oxford, 2014 Search PubMed.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef CAS.
- F. Ambroz, T. J. Macdonald, V. Martis and I. P. Parkin, Small Methods, 2018, 2, 1800173 CrossRef.
- M. Rivera-Torrente, M. Filez, R. Hardian, E. Reynolds, B. Seoane, M. V. Coulet, F. E. Oropeza Palacio, J. P. Hofmann, R. A. Fischer, A. L. Goodwin, P. L. Llewellyn and B. M. Weckhuysen, Chem. – Eur. J., 2018, 24, 7498–7506 CrossRef CAS PubMed.
- X. S. Wang, L. Li, J. Liang, Y. B. Huang and R. Cao, ChemCatChem, 2017, 9, 971–979 CrossRef CAS.
- J. Jagiełło, Langmuir, 1994, 10, 2778–2785 CrossRef.
- G. Kupgan, T. P. Liyana-Arachchi and C. M. Colina, Langmuir, 2017, 33, 11138–11145 CrossRef CAS.
- D. W. Bruce, D. O'Hare and R. I. Walton, Multi Length–Scale Characterisation, Wiley, 2014 Search PubMed.
- Micromeritics, SAIEUS, version 3 (computer software), GA Search PubMed.
- N. Kotake, M. Kuboki, S. Kiya and Y. Kanda, Adv. Powder Technol., 2011, 22, 86–92 CrossRef CAS.
- Q. Han, R. Setchi and S. L. Evans, Powder Technol., 2016, 297, 183–192 CrossRef CAS.
- B. S. Barros, O. J. de Lima Neto, A. C. de Oliveira Frós and J. Kulesza, ChemistrySelect, 2018, 3, 7459–7471 CrossRef CAS.
- S. Billinge, Physics, 2010, 3, 25 CrossRef.
- S. J. L. Billinge and I. Levin, Science, 2007, 316, 561–565 CrossRef CAS PubMed.
- F. Zhang, J. Shi, Y. Jin, Y. Fu, Y. Zhong and W. Zhu, Chem. Eng. J., 2015, 259, 183–190 CrossRef CAS.
- C. Li, X. Lou, M. Shen, X. Hu, Z. Guo, Y. Wang, B. Hu and Q. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 15352–15360 CrossRef CAS PubMed.
- Z. Su, Y. R. Miao, G. Zhang, J. T. Miller and K. S. Suslick, Chem. Sci., 2017, 8, 8004–8011 RSC.
- W. Li, Y. Zhang, C. Zhang, Q. Meng, Z. Xu, P. Su, Q. Li, C. Shen, Z. Fan, L. Qin and G. Zhang, Nat. Commun., 2016, 7, 1–9 RSC.
- M. L. Ojeda, J. M. Esparza, A. Campero, S. Cordero, I. Kornhauser and F. Rojas, Phys. Chem. Chem. Phys., 2003, 5, 1859–1866 RSC.
- T. D. Bennett, J. Sotelo, J. C. Tan and S. A. Moggach, CrystEngComm, 2015, 17, 286–289 RSC.
- S. Van Der Perre, T. Van Assche, B. Bozbiyik, J. Lannoeye, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Langmuir, 2014, 30, 8416–8424 CrossRef CAS PubMed.
- Y. Y. Fu, C. X. Yang and X. P. Yan, J. Chromatogr. A, 2013, 1274, 137–144 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt00881a |
| This journal is © The Royal Society of Chemistry 2021 |