
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic structure study of divanadium complexes with rigid covalent coordination: potential molecular qubits with slow spin relaxation†
Stephen
Sproules

WestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: stephen.sproules@glasgow.ac.uk
First published on 22nd March 2021
Abstract
The electronic structures of homovalent [V2(μ-S2)2(R2dtc)4] (R = Et, iBu) and mixed-valent [V2(μ-S2)2(R2dtc)4]+ are reported here. The soft-donor, eight-coordinate ligand shell combined with the fully delocalised ground state provides a highly rigid and covalent environment that will nurture long spin relaxation times in vanadyl-based molecular qubits.
Advances in computer processing speed and data storage capacity require miniaturising components down to the atomic level.1 At the nanoscale, classical physics gives way to quantum phenomena, and for matter-based hardware, involves assembling and aligning electron spins.2 Computer processing at the quantum level utilises a quantum bit, or qubit, which in its most elementary form is an atom with an unpaired electron. In contrast to a classical bit which is either 0 or 1, a qubit can be any value in between, a property called superposition, and refers to the alignment of the unpaired spin. The efficacy of a matter-spin qubit rests in the lifetime of the aligned spin, which is evaluated by measuring the spin–lattice (T1) and decoherence (T2) relaxation times. Over the last decade studies of paramagnetic coordination complexes has produced a detailed inventory of the design principles that promote slow relaxation. Initial work was invested in improving T2, where optimising the ligand field, steric crowding the paramagnetic centre, and eliminating deleterious nuclear spins has seen molecular qubits reaching millisecond decoherence times.3
Integrating these molecules into a prototypical device requires arranging qubits into multi-dimensional arrays,4 and this has brought spin–lattice relaxation into focus. Long spin–lattice relaxation times are necessary as T1 limits T2.5 Recent examination of spin–lattice relaxation dynamics in molecular qubits has revealed the structural and compositional factors required for use at room temperature. These include: (i) using a rigid or multiply-bonded ligand as it greatly perturbs relaxation pathways;6 (ii) increasing metal–ligand covalency that stabilises the ground state;7 (iii) choosing a ligand field that orientates the singly-occupied orbital in the plane of molecule;8 and (iv) avoiding heavy atoms which expedite relaxation via spin–orbit coupling.9,10 Here, the electronic structures of homovalent V(IV,IV) dimer [V2(μ-S2)2(R2dtc)4] (dtc = dithiocarbamate; R = ethyl, 1-Et; i-butyl, 1-Bu;) and the oxidised mixed-valent V(V,IV) species, 2-Et and 2-Bu, are investigated using a combined spectroscopic and computational approach. This system, with its high-coordination number and rigid nuclear-spin deficient ligands,11 possesses the requisite features to deliver enviable spin–lattice and decoherence relaxation times.
The homovalent divanadium complexes 1-Et and 1-Bu result from an esoteric induced internal redox reaction between tetrathiovanadate (V) and dithiuram disulfide.12 Cleavage of the S–S bond in the latter releases the dithiocarbamate ligands making the reaction applicable to a multitude of functional groups.13 Both 1-Et and 1-Bu have been structurally characterised. The V(IV) centres reside on the two-fold axis (Fig. 1), bridged by two disulfide ligands giving a separation of ca. 2.9 Å.12,14 Each V(IV) ion is eight-coordinate with two chelating dtc ligands. The disposition of the dtc ligands produced ΛΛ and ΔΔ enantiomers (racemic) in 1-Bu whereas the ΛΔ diastereomer (meso) is adopted in 1-Et, which are distinguished by their D2 and C2h point symmetry. The geometry about each vanadium centre is identical, resembling an endohedral vanadium ion housed in an S8 cluster. Continuous shape measurement (CShM) define the 12-vertex polyhedron as triangular dodecahedral (Table S1†).15 This is the exact same topology for monovanadium species with an S8 coordination sphere.16 However, specific to the dimeric nature of 1-Et and 1-Bu, the description as bicapped trigonal prismatic is preferred.17,18 Here the two-fold axis is along the V⋯V vector, orthogonal to the uncapped face of the trigonal prism, and defined as the x-axis for the electronic structure assignment (Fig. 1). Oxidation to the heterovalent V(V/IV) species occurs at the mild potential of 80 mV (vs. Fc+/0),19 underscoring the facile nature of vanadium-based redox processes.20 One variant has been structurally characterised, that being 2-Bu, with only s slight increase in the V⋯V separation by 0.084 Å. Unlike 1-Bu, the cation in 2-Bu crystallises as the meso isomer. The dimension of each VS8 unit remains unchanged, supporting their description as Class III mixed-valent species.21
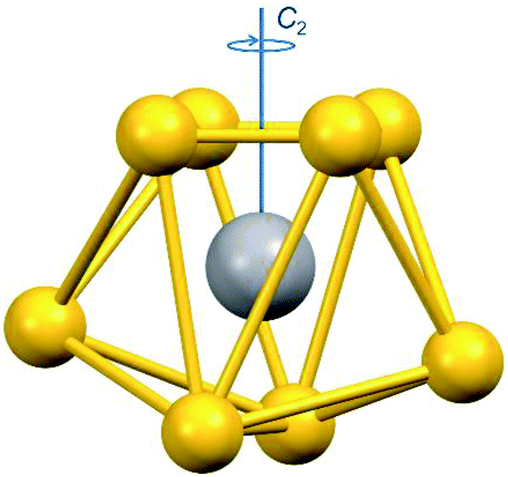 | ||
| Fig. 1 Depiction of the VS8 coordination sphere defined as the C2h symmetric bicapped trigonal prism where x is defined as the unique axis. | ||
The electronic spectra of 1-Et and 2-Et are overlaid in Fig. 2. The analogous spectra for 1-Bu and 2-Bu displayed in Fig. S1† are identical, confirming the electronic structure is independent of the dtc substituents. Dark brown 1-Et exhibits two weak features at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 and 18400 cm−1 (ε = 200 and 530 M−1 cm−1) that lie at the foot of the dominant peak at 21050 cm−1 (4300 M−1 cm−1). The intensity of the lowest energy band is commensurate with a ligand field transition. This assignment is consistent with mononuclear VS8 compounds, though their first LF transition appears in the range 12000–13500 cm−1 depending on the composition of the monoanionic S,S-chelate.22,23 The intense band at 21050 cm−1 is indicative of ligand-to-metal charge transfer (LMCT) from filled ligand π MOs to vanadium-based MOs (i.e. d orbitals) of matching symmetry. This corresponding transition is observed at 20400 cm−1 in [V2(μ-S2)2(CS3)4]4− and 20850 cm−1 in [V2(μ-S2)2(S2CSMe)4].24 The assignment is corroborated by time-dependent density functional theoretical (TD-DFT) calculations (vide infra).
300 and 18400 cm−1 (ε = 200 and 530 M−1 cm−1) that lie at the foot of the dominant peak at 21050 cm−1 (4300 M−1 cm−1). The intensity of the lowest energy band is commensurate with a ligand field transition. This assignment is consistent with mononuclear VS8 compounds, though their first LF transition appears in the range 12000–13500 cm−1 depending on the composition of the monoanionic S,S-chelate.22,23 The intense band at 21050 cm−1 is indicative of ligand-to-metal charge transfer (LMCT) from filled ligand π MOs to vanadium-based MOs (i.e. d orbitals) of matching symmetry. This corresponding transition is observed at 20400 cm−1 in [V2(μ-S2)2(CS3)4]4− and 20850 cm−1 in [V2(μ-S2)2(S2CSMe)4].24 The assignment is corroborated by time-dependent density functional theoretical (TD-DFT) calculations (vide infra).
 | ||
| Fig. 2 Overlay of the electronic spectra of 1-Et and 2-Et recorded in dichloromethane solution at ambient temperature. | ||
Oxidation to mixed-valent 2-Et produces a suite of low-energy CT transitions (Fig. 2). The lowest energy feature appears as two overlapping transitions at 8300 and 9150 cm−1 with the latter slightly less intense (ε = 2350 and 2240 M−1 cm−1). This near-IR band is characteristic of an intervalence charge transfer (IVCT) transition that underpins the description of these monocationic complexes as mixed-valent V(V,IV) dimers.25 The appearance of this band as two overlapping peaks separated by 850 cm−1 is a consequence of a mixture of the meso and racemic isomers in solution.26 The higher energy IVCT band is likely that of the meso isomer,27 and based on the relative intensity, the racemic/meso isomeric mixture is estimated at 55:45. The next peak in the absorption spectrum also consists of overlapping bands at 13400 and 14250 cm−1, and given the same 850 cm−1 separation, likely stems from individual isomers.
Verification that oxidation of 1-Et is metal-centred is provided by X-ray absorption spectroscopy. In this instance, the lower energy sulfur K-edge is preferred as it has a higher resolution than the vanadium K-edge.28 The sulfur K-edge results from electric dipole-allowed 1s → 4p transitions and a pre-edge spectrum that also results from allowed transitions to the 3p level. This is used to diagnose valence holes in ligand-based orbitals through covalent bonding with metals or oxidation of π-type ligands. While dithiocarbamates are redox inert,29 the redox-activity of disulfide is well documented,30 and would be observed in the pre-edge. The spectra of 1-Et and 2-Et are overlaid in Fig. 3, and are identical to the 1-Bu and 2-Bu complexes (Fig. S3†). As expected the pre-edge region is nondescript because of the overlapping transitions to π orbitals of four dtc and two μ-S22− ligands, which confirms the latter are dianionic in both 1-Et and 2-Et. The salient observation is a shift of the first peak by 0.7 eV to lower energy for 2-Et and 1.0 eV for 2-Bu. The edge shift is in response to oxidation of the divanadium core where the increase in nuclear charge stabilises the frontier orbitals relative to the 1s core orbital of sulfur.28
 | ||
| Fig. 3 Overlay of the normalised sulfur K-edge spectra of 1-Et and 2-Et. | ||
The presence of a paramagnetic V(IV) d1 ion in 2-Et was established by EPR spectroscopy. The room temperature spectrum is dominated by hyperfine coupling to equivalent 51V nuclei (I = 7/2, ∼100% abundant) generating a 15-line signal (Fig. S4†). The Class III behaviour of this compound is evident in the symmetrical profile and equal splitting of the hyperfine lines. Simulation yielded giso = 2.0066 and a hyperfine coupling constant Aiso = 29.6 × 10−4 cm−1, identical to spectra for other analogues.19,31 It is important to note that the hyperfine coupling constant is exactly half that for [V(Et2dtc)4], signifying the unpaired electron is evenly distributed over the vanadium ions (Table S2†). The frozen solution spectrum of 2-Et recorded in a CH2Cl2/toluene glass is typical of an axial V(IV) species (Fig. 4). The 20 K signal is identical to that measured at 77 K;31 the temperature independence is the hallmark of Class III behaviour.21 The aforementioned axiality is borne out in the principal g-values where g|| (=gz) > g⊥ (=gx, gy) > ge, where the latter is the free electron ge = 2.0023 (Table 1). The small disparity between gx and gy attest to a slight orthorhombicity that stems from the ligand field with dithiocarbamates on one side and disulfides on the other. This pattern is unique among V(IV) species and derives from the dimeric composition of 2-Et (Table S2†). The paramagnetic V(IV) ions in square pyramidal [VO(Et2dtc)2] and dodecahedral [V(Et2dtc)4] have g⊥ > g||, with both less than ge; the former is a benchmark oxovanadium(IV) electron spin qubit that functions at room temperature.6 The trigonal vanadyl ion in tris(dithiolene)vanadium complexes also give g|| > g⊥, and like the other monomeric species, both are less than ge.3,32,33 The hyperfine splitting is also axial with A|| > A⊥, and these are the parameters that describe the dx2−y2 ground state akin that of [VO(Et2dtc)2] (Table S3†). With trigonal symmetry, the singly-occupied orbital (SOMO) is dz2, a configuration with efficient relaxation pathways that comprises performance at elevated temperatures.6 Overall the principal A-values in 2-Et are almost one-third smaller than those in [VO(Et2dtc)2], which attenuates decoherence pathways utilising the hyperfine interaction of the spin host.
 | ||
| Fig. 4 X-band EPR spectrum of 2-Et recorded in CH2Cl2/toluene solution at 20 K. The simulation is shown in red with the experimental spectrum in back (experimental conditions: frequency, 9.4355 GHz; power, 0.063 mW; modulation, 0.8 mT). | ||
| Experimental | Calculated | |
|---|---|---|
| a ZORA/TPSSh/aug-cc-pVTZ-J+CPCM(CH2Cl2) DFT calculations. b A-Values in ×10−4 cm−1; the sign is negative owing to the dominant Fermi-contact contribution. | ||
| g x | 2.0021 | 1.9949 |
| g y | 2.0069 | 1.9955 |
| g z | 2.0152 | 2.0044 |
| 〈g〉 | 2.0081 | 1.9983 |
| A xx | −18.0 | −23.3 |
| A yy | −18.0 | −25.0 |
| A zz | −52.9 | −54.0 |
| 〈A〉 | −29.6 | −34.1 |
The efficacy of 2-Et and 2-Bu as molecular spin qubits is further evaluated by comparing the covalency with related V(IV) systems (Table S3†).7,10 The principle g- and A-values are used to calculate β2, which defines the overlap or mixing of the vanadium d and ligand p orbitals that quantifies π bonding and therein covalency of the system. The value for 2-Et is 0.66, which is the same as [V(S2C2(CN)2)3]2− at 0.65. These complexes are 10% more covalent than [V(Et2dtc)4], and a sizable 20% more than [VO(Et2dtc)2].22 This reveals a direct correlation between covalency and the number of thiolate (RS−) donors in the coordination sphere, with two in [VO(Et2dtc)2], four in [V(Et2dtc)4], and six in 2-Et and [V(S2C2(CN)2)3]2−.
The metal–sulfur bonds are accurately reproduced in the optimised structures of 1-Et and 1-Bu though the V⋯V separation is underestimated by 0.18 and 0.13 Å, respectively (Table S8†). The electronic structure was calculated on the optimised geometry using the spin-unrestricted broken symmetry (BS) with the TPSSh functional. The result gave the BS singlet diradical (MS = 0) lower in energy than the triplet state (MS = 1). The BS solution comes about via antiferromagnetic coupling of the unpaired electron on each V(IV) ion located in the dx2–y2 orbital (Fig. S9†). Importantly the BS solution is 7.4 kcal mol−1 more stable than the spin-restricted singlet generated by a V–V single bond. Although the solid state structure is C2h symmetric, the electronic configuration lowers the orbital symmetry to C2v producing inequivalent V(IV) centres. Therefore the ground state of 1-Et and 1-Bu comprises the overlap of the singly occupied dx2–y2 orbitals that transform as a1 in C2v symmetry (Fig. S4 and S5†). An orbital overlap integral of S = 0.58 is computed for both, and represents the extent of spatial overlap of the two SOMOs. The multiconfigurational composition of the ground state is reflected in the small contribution to each SOMO from the neighbouring metal ion, and gave a Mayer bond order of 0.16. This small direct overlap in conjunction with the superexchange pathway via the efficient μ-S22− ligands generates a robust intramolecular antiferromagnetic interaction; the exchange coupling constant is estimated at 2755 cm−1.
A more rigorous correlated post-Hartree–Fock ab initio calculation was performed on 1-Et using a CAS(2,10) reference with two electrons in the ten d orbitals. The ground state consists of a 61% contribution from the symmetric σ MO and 39% from its antisymmetric counterpart, which translates as ∼20% input from the closed-shell singlet state (Fig. S13†). The diradical character of the ground state is estimated at 68% from the CAS(2,10) calculation, which nicely matches the 66% computed from DFT. The dominance of the singlet diradical to the ground state accommodates the lack of charge density between the vanadium ions that would constitute a single bond.18 Moreover, the C2v orbital symmetry, producing inequivalent vanadium centres, would account for the two signals in the 13C NMR spectrum of 1-Et and [V2(μ-S2)2(CS3)4]4−.24 It was suggested that interconversion of the meso and racemic isomers occurred in solution which are differentiated by NMR.34 Although both isomers have been identified in the solid state, there is no evidence of isomerisation for the homovalent species especially considering they do not react with alkynes in the same manner as Mo and W analogues, reactions that occur at the sulfur donor ligands.19 Hence, the mixed-valent species were sought as more likely candidates to give reaction of unsaturated organic molecules at the second coordination sphere.
The bond distances and angles in the optimised structure of 2-Bu are in excellent agreement with the experimental data (Table S8†). The V⋯V distance is underestimated by only 0.05 Å such that oxidation of 1-Bu sees an increase in the intermetal separation of 0.084 Å experimental and 0.154 Å computationally. These changes are small considering the long held viewpoint that a formal V–V single bond exists in 1-Bu and by extension, a two-centre one-electron bond in 2-Bu. The centrosymmetry of the monocations in 2-Et and 2-Bu is retained giving a delocalised ground state comprising equal contributions from the dx2–y2 orbital. Their overlap leads to a partial V–V bond with computed bond order of 0.36 (Fig. S6 and S7†). This is seen in the Mulliken spin density plot with +0.83 spins on each vanadium (Fig. 5). The additional +0.33 spins on each V ion stems from polarisation of the V–S bonds and is common in high-valent complexes.20,32 The SOMO is ag in C2h symmetry, and the lowest unoccupied orbital (LUMO) is the au antibonding counterpart with an energy gap of ca. 9000 cm−1 as defined by the IVCT band (Fig. 2).25 The veracity of this electronic structure is confirmed by calculation of electronic spectrum using a TD-DFT protocol. The calculated spectrum closely matches the experiment with the IVCT transition at 9157 cm−1. Only one is computed as the optimised structure is that of the meso isomer of 2-Et which corresponds to the experimental band at 9150 cm−1. The band at 8300 cm−1 is suggested to belong to the racemic isomer (vide supra). The correlated CAS(1,10) ab initio calculation also computed one IVCT band for the SOMO–LUMO excitation, though at 4950 cm−1, due a size limitation on the active space (Fig. S15†).
 | ||
| Fig. 5 Mulliken spin density plot for (a) 1-Et, and (b) 2-Et from spin unrestricted DFT calculations (red: α-spin, yellow: β-spin). | ||
EPR spin-Hamiltonian parameters were also calculated for 2-Et and gave an excellent match with the experiment including the degree of axiality in the g- and A-values (Table 1). The calculated orientation of the g-matrix sees the smallest value aligned parallel to C2 axis of the complex (Fig. 1). As this is designated gx, the molecular axes are rotated such that the x-axis is parallel to the V–V bond which is a non-standard alignment in C2h symmetry (Fig. S18†). This leaves the molecular z-axis and the largest g- and A-values aligned perpendicular to V–V but parallel to the S–S bonds, pointing towards the longer of the V–Sdtc bonds. This resultant orientation is identical to that for square pyramidal oxovanadium(IV) and square planar Cu(II) species.6,8,9 This analysis reveals that 2-Et and 2-Bu have the preferred ground state orbital that is completely shielded from the surrounding environment by the bicapped trigonal prismatic arrangement of the eight sulfur atoms. These soft donor, nuclear-spin deficient S,S-chelates provide a rigid and highly covalent enclosure for the paramagnetic V(V,IV) core, meeting the design specifications that will provide the long spin relaxation times required for room temperature quantum coherence. An investigation of the spin dynamics of these eight-coordinate vanadyl species is currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the University of Glasgow and the Scottish Funding Council for a Postgraduate and Early Career Researcher Exchange grant.Notes and references
- (a) T. D. Ladd, F. Jelezko, R. Laflamme, Y. Nakamura, C. Monroe and J. L. O'Brien, Nature, 2010, 464, 45 CrossRef CAS PubMed; (b) R. P. Feynman, Opt. News, 1985, 11, 11 CrossRef.
- S. Sproules, in Electron Paramagnetic Resonance, ed. V. Chechik and D. M. Murphy, The Royal Society of Chemistry, Cambridge, UK, 2017, vol. 25, p. 61 Search PubMed.
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, ACS Cent. Sci., 2015, 1, 488 CrossRef CAS PubMed.
- S. Cardonna-Serra and A. Gaita-Ariño, Dalton Trans., 2018, 47, 5533 RSC.
- K. Bader, D. Dengler, S. Lenz, B. Endeward, S.-D. Jiang, P. Neugebauer and J. van Slageren, Nat. Commun., 2014, 5, 5304 CrossRef CAS PubMed.
- (a) M. Atzori, E. Morra, L. Tesi, A. Albino, A. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 11234 CrossRef CAS PubMed; (b) M. Atzori, L. Tesi, S. Benci, A. Lunghi, R. Righini, A. Taschin, R. Torre, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2017, 139, 4338 CrossRef CAS PubMed.
- M. S. Fataftah, M. D. Krzyaniak, B. Vlaisavljevich, M. R. Wasieleski, J. M. Zadrozny and D. E. Freedman, Chem. Sci., 2019, 10, 6707 RSC.
- K. Bader, M. Winkler and J. van Slageren, Chem. Commun., 2016, 52, 3623 RSC.
- T. J. Pearson, D. W. Laorenza, M. Krzyaniak, M. R. Wasieleski and D. E. Freedman, Dalton Trans., 2018, 47, 11744 RSC.
- (a) J. McGuire, H. N. Miras, J. P. Donahue, E. Richards and S. Sproules, Chem. – Eur. J., 2018, 24, 17598 CrossRef CAS PubMed; (b) J. McGuire, H. N. Miras, E. Richards and S. Sproules, Chem. Sci., 2019, 10, 1483 RSC.
- The rigidity of the dithiocarbamate ligand relates to the formation of a four-membered metallodithiocarbamate ring with the exocyclic C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in the trigonal S2CN core of the ligand. This is in contrast to widely used 1,2-dithiolate ligands in V- and Cu-containing molecular qubits that provide the more conformationally flexible five-membered metallo-dithiolene ring. The overall rigidity of the complex is qualitatively measured by the higher coordination number about each vanadium ion with four chelating ligands compared with the five- and six-coordinate counterparts.
N bond in the trigonal S2CN core of the ligand. This is in contrast to widely used 1,2-dithiolate ligands in V- and Cu-containing molecular qubits that provide the more conformationally flexible five-membered metallo-dithiolene ring. The overall rigidity of the complex is qualitatively measured by the higher coordination number about each vanadium ion with four chelating ligands compared with the five- and six-coordinate counterparts. - T. R. Halbert, L. L. Hutchings, R. Rhodes and E. I. Stiefel, J. Am. Chem. Soc., 1986, 108, 6437 CrossRef CAS.
- G. Hogarth, Prog. Inorg. Chem., 2005, 53, 71 CrossRef CAS.
- E. R. T. Tiekink, X.-F. Yan and C. G. Young, Aust. J. Chem., 1992, 45, 897 CrossRef CAS.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575 CrossRef CAS PubMed.
- D. Attanasio, C. Bellitto, A. Flamini and G. Pennesi, Inorg. Chem., 1982, 21, 1461 CrossRef CAS.
- S. A. Duraj, M. T. Andras and P. A. Kibala, Inorg. Chem., 1990, 29, 1233 CrossRef.
- M. Costas, C. Bo and J. M. Poblet, Chem. Phys. Lett., 1992, 200, 8 CrossRef CAS.
- M. K. Taylor, D. J. Evans and C. G. Young, Chem. Commun., 2006, 4245 RSC.
- S. Sproules, Angew. Chem., Int. Ed., 2019, 58, 10043 CrossRef CAS PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247 CrossRef CAS.
- D. C. Bradley, I. F. Rendall and K. D. Sales, J. Chem. Soc., Dalton Trans., 1973, 2228 RSC.
- (a) O. Piovesana and G. Cappuccilli, Inorg. Chem., 1972, 11, 1543 CrossRef CAS; (b) R. D. Bereman and J. R. Dorfman, Polyhedron, 1983, 2, 1013 CrossRef CAS; (c) R. D. Bereman and D. Nalewajek, J. Inorg. Nucl. Chem., 1978, 40, 1309 CrossRef CAS.
- S. C. Sendlinger, J. R. Nicholson, E. B. Lobkovsky, J. C. Huffman, D. Rehder and G. Christou, Inorg. Chem., 1993, 32, 204 CrossRef CAS.
- The terms “IVCT transition” and “mixed-valence” are retained even though in this fully delocalised Class III the metal centres possess a partial oxidation state and the IVCT transition occurs within the molecular orbital manifolds of the systems which does not involve net charge transfer.
- F. R. Keene, Chem. Soc. Rev., 1998, 27, 185 RSC.
- D. M. D'Allessandro and F. R. Keene, Chem. – Eur. J., 2005, 11, 3679 CrossRef PubMed.
- S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2011, 255, 837 CrossRef CAS.
- C. Milsmann, S. Sproules, E. Bill, T. Weyhermüller, S. DeBeer George and K. Wieghardt, Chem. – Eur. J., 2010, 16, 3628 CrossRef CAS PubMed.
- C. A. P. Goodwin, B. L. L. Réant, G. F. Vettese, J. G. C. Kragskow, M. J. Giansiracusa, I. M. DiMucci, K. M. Lancaster, D. P. Mills and S. Sproules, Inorg. Chem., 2020, 59, 7571 CrossRef CAS PubMed.
- I. S. Fomenko, A. L. Gushchin, V. A. Nadolinny, N. N. Efimov, Y. A. Laricheva and M. Sokolov, Eur. J. Inorg. Chem., 2018, 2965 CrossRef CAS.
- S. Sproules, T. Weyhermüller, S. DeBeer and K. Wieghardt, Inorg. Chem., 2010, 49, 5241 CrossRef CAS PubMed.
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, J. Am. Chem. Soc., 2014, 136, 15841 CrossRef CAS PubMed.
- (a) M. Sokolov, A. Virovets, O. Oeckler, A. Simon and V. Fedorov, Inorg. Chim. Acta, 2002, 331, 25 CrossRef CAS; (b) M. N. Sokolov, A. L. Gushchin, S. V. Tkachev, D. Y. Naumov, P. Nuñez, P. Gili, J. G. Platas and V. P. Fedin, Inorg. Chim. Acta, 2005, 358, 2371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, continuous shape measurements, spectra and computational data. See DOI: 10.1039/d1dt00709b |
| This journal is © The Royal Society of Chemistry 2021 |