DOI:
10.1039/D1DT00658D
(Paper)
Dalton Trans., 2021,
50, 6857-6866
Graphitic nitrogen in carbon catalysts is important for the reduction of nitrite as revealed by naturally abundant 15N NMR spectroscopy†
Received
26th February 2021
, Accepted 12th April 2021
First published on 12th April 2021
Abstract
Metal-free nitrogen-doped carbon is considered as a green functional material, but the structural determination of the atomic positions of nitrogen remains challenging. We recently demonstrated that directly-excited solid state 15N NMR (ssNMR) spectroscopy is a powerful tool for the determination of such positions in N-doped carbon at natural 15N isotope abundance. Here we report a green chemistry approach for the synthesis of N-doped carbon using cellulose as a precursor, and a study of the catalytic properties and atomic structures of the related catalyst. N-doped carbon (NH3) was obtained by the oxidation of cellulose with HNO3 followed by ammonolysis at 800 °C. It had a N content of 6.5 wt% and a surface area of 557 m2 g−1, and 15N ssNMR spectroscopy provided evidence for graphitic nitrogen besides regular pyrrolic and pyridinic nitrogen. This structural determination allowed probing the role of graphitic nitrogen in electrocatalytic reactions, such as the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), and nitrite reduction reaction. The N-doped carbon catalyst (NH3) showed higher electrocatalytic activities in the OER and HER under alkaline conditions and higher activity for nitrite reduction, as compared with a catalyst prepared by the carbonization of HNO3-treated cellulose in N2. The electrocatalytic selectivity for nitrite reduction of the N-doped carbon catalyst (NH3) was directly related to the graphitic nitrogen functions. Complementary structural analyses by means of 13C and 1H ssNMR, scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, and low-temperature N2 adsorption were performed and provided support to the findings. The results show that directly-excited 15N ssNMR spectroscopy at natural 15N abundance is generally capable of providing information on N-doped carbon materials if relaxation properties are favorable. It is expected that this approach can be applied to a wide range of solids with an intermediate concentration of N atoms.
Introduction
When fabricating carbon materials with tailored functionalities, the structure–property relationship at the atomic level should be considered.1–8 These considerations are important for the synthesis of metal-free and carbon-based catalysts, where the activity stems from the enhanced exposure of the structural features being catalytically most active.9–17 The absence of metal-containing entities has intensified the consideration of metal-free carbon catalysts as “green” catalysts.18,19 The physical and chemical properties of carbon catalysts can be altered by modifying their surface states with carbon–oxygen bonds,20,21 which in turn modifies their catalytic activity. Another approach to modify the catalytic activity of carbon is to incorporate dopants into the bulk carbon structure, such as doping with nitrogen.22–28 Although it has been postulated that any element applied as a dopant would improve the catalytic activity,29–37 a control of the dopant distribution at the atomic level is key to bridging the gap between the atomically precise understanding of homogeneous catalysts and the materials chemistry of heterogeneous catalysts.38,39 Numerous studies of N-doped carbon atoms for catalysis have been performed; for example, Zhao et al. demonstrated that several synthetic approaches for the fabrication of “green” carbon catalysts could be used.40
Nitrogen-doped carbon catalysts have indeed emerged as promising heterogeneous catalysts, such as for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). Despite the progress in developing N-doped carbon catalysts, the structural determination of nitrogen positions at the atomic level has remained highly challenging. The lack of analytical methods for this structural determination has rendered the interpretation of catalytic activity elusive.41–43 As nitrogen and carbon are neighbors in the periodic table, they are difficult to distinguish by scanning transmission electron microscopy with energy dispersive X-ray spectroscopy (STEM EDX) mapping,43,44 and in this spectroscopy there is also a problem with choosing the representative spots of the sample.
The local structures of the nitrogen atoms in N-doped carbons can be studied by 15N ssNMR, and by typically enriching the carbons in 15N.45–47 The obvious disadvantages of 15N labeling are the higher costs accompanied by more complexity of synthesis, particularly for methods using NH3. Only a few 15N ssNMR studies using N-doped carbon materials at the natural abundance of 15N have been reported in the literature including the study by Fang et al., and our very recent study.48,49 In the latter, we reported the synthesis of N-doped mesoporous carbon by using porous silica as a template and sugar as a carbon precursor.49 Upon treatment with HNO3 and carbonization under a flow of NH3, followed by the dissolution of the silica template in hydrofluoric acid (HF), an N-doped carbon catalyst with 3.7 wt% of N was produced. 15N ssNMR spectroscopy at natural 15N isotope abundance revealed that N was present in pyrrolic and pyridinic moieties.
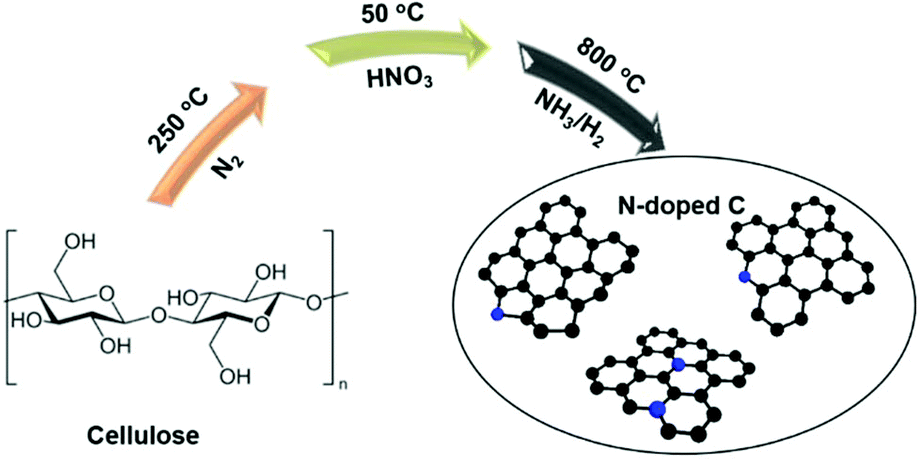
Since HF is toxic, we here explore a comparably sustainable route to the synthesis of N-doped carbons and circumvent the applications of HF, and we also involve a renewable cellulose substrate as the carbon precursor.50–56 The synthetic pathway is illustrated in Scheme 1. It does not cover all principles of Green Chemistry,57 as NH3 is toxic, however, NH3 is a common base chemical and allows the introduction of high amounts of N. We report an unexpected incorporation of graphitic N in the N-doped carbon and its detection by using 15N ssNMR spectroscopy. Among the two studied catalysts, the N-doped carbon derived via ammonolysis shows the highest catalytic activities in electrochemical reactions, such as the HER, OER and nitrite reduction, and the role of graphitic N is assessed.
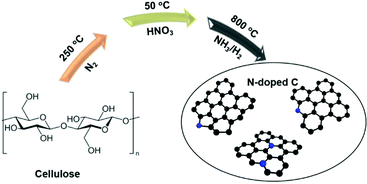 |
| | Scheme 1 Schematic illustration of the synthesis to create N-doped carbons. Before the introduction of nitrogen into the structure, the cellulose was dried at 250 °C under an inert atmosphere. | |
Experimental
Synthesis
Cellulose (microcrystalline, AquaBioSolve Stockholm AB) was used as a carbon source and the main raw material in the synthesis of two types of N-doped carbons. 3.0 g of cellulose was placed into a crucible and then dried in a tube furnace under a N2 flow (40 mL min−1) at 250 °C for 6 h. The dried product was oxidized by immersing it in a 30 mL of HNO3 solution (40 wt%) at 50 °C for 3 h. The resultant suspension was centrifuged and washed with deionized water until neutral pH was achieved, before being dried at 120 °C for 8 h under ambient conditions. For carbonization, 1 g of the obtained yellow product was placed into an alumina crucible in a tube furnace and then carbonized under a flow of NH3 (15 mL min−1) with H2 (5 mL min−1) at 800 °C for 2 h, at a heating and cooling rate of 10 °C min−1. The NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio has been chosen based on our experience with metal (oxy)nitride formation.58,59 The addition of H2 shifts the equilibrium more toward NH3 instead of N2 at such elevated temperatures. The resulting modified carbon was marked as N-doped C (NH3). Similarly, the complementary carbon obtained under the flow of N2 (40 mL min−1) was labelled as N-doped C (N2).
:H2 ratio has been chosen based on our experience with metal (oxy)nitride formation.58,59 The addition of H2 shifts the equilibrium more toward NH3 instead of N2 at such elevated temperatures. The resulting modified carbon was marked as N-doped C (NH3). Similarly, the complementary carbon obtained under the flow of N2 (40 mL min−1) was labelled as N-doped C (N2).
Characterization
N2 adsorption.
N2 adsorption analyses were conducted at −196 °C using an ASAP 2020 instrument (Micrometrics) for the samples previously degassed at 250 °C for 4 h under dynamic vacuum. The recorded data were analyzed using the following models: Brunauer–Emmett–Teller (surface area, SBET), Langmuir (surface area, SLangmuir), t-plot (micropore volume, Vmicro), and Barrett–Joyner–Halenda (mesopore volume, Vmeso). Moreover, the total pore volume (Vtotal) was determined by the single point method (p/p0 ∼ 0.99).
Powder X-ray diffraction (PXRD).
PXRD measurements were performed in transmission mode on an STOE STADI-P diffractometer (Cu Kα1 radiation) using a DECTRIS Mythen 1 K detector at a scan rate of 1° min−1 in a 2θ range from 10° to 60°.
Raman spectroscopy.
A LabRAM HR 800 Raman instrument equipped with a spectrograph having an 800 nm focal length was used for the Raman spectroscopic study. The detector was an air cooled (−70 °C) and back thinned CCD detector with 1024 × 256 pixels and a spectral range of 200–1050 nm. The laser used was an air cooled and double frequency Nd:YAG laser (532 nm/50 mW) whose intensity was adjusted by using a filter wheel with 6ND filters which was software controlled.
Electron microscopy (EM).
Scanning electron microscopy (SEM) micrographs were recorded using a Leo Supra 35VP SMT (Zeiss) thermal field emission scanning electron microscope. Transmission electron microscopy (TEM) images were recorded using a JEOL 2100F microscope.
Elemental analysis.
A Flash 2000 elemental analyzer containing a thermal conductivity detector and an autosampler was used to determine the content of C, N, and H. The quantitative analysis of the metal content in the carbon materials was carried out by inductively-coupled plasma optical emission spectrometry (ICP-OES).
X-ray photoelectron spectroscopy (XPS).
The surface composition of the carbonized samples was investigated by XPS using a system constructed by Prevac, equipped with a hemispherical analyzer (VG SCIENTA R3000) and a monochromatized aluminum source Al Kα (E = 1486.6 eV). No charge compensation was needed for the conductive samples. The Au 4f reference peak at 84.0 eV was used for the calibration of the binding energy scale. The collected spectra were analysed using the CasaXPS software.
ssNMR.
Magic-angle-spinning (MAS) NMR experiments were performed at a magnetic field of 14.1 T (Larmor frequencies of 600.12, 150.92, and 60.83 MHz for 1H, 13C, and 15N, respectively) using a Bruker Avance-III spectrometer with a wide-bore magnet. The 1H MAS NMR spectrum was acquired using a 1.3 mm probehead and a 60 kHz MAS rate. The acquisition of the 1H MAS NMR spectrum involved the use of a rotor-synchronized, double-adiabatic spin-echo sequence with a 90° excitation pulse of 1.25 μs followed by a pair of 50.0 μs tanh/tan short high-power adiabatic pulses (SHAPs) with a 5 MHz frequency sweep.49,60,61 Such a pulse sequence was applied to prevent proton background signals and to achieve a flat baseline in the acquired 1H MAS NMR spectrum. All pulses operated at a nutation frequency of 200 kHz, and 64 signal transients were acquired using a relaxation delay of 5 s. Directly-excited 13C and 15N MAS NMR spectra were recorded using a 7 mm probehead and a 7 kHz MAS rate. The 13C acquisition involved a single-pulse (Bloch-decay) protocol with a 90° excitation pulse of 4.0 μs, and 16384 signal transients were collected using a relaxation delay of 20 s. The 13C NMR chemical shifts were referenced with respect to TMS. The 15N acquisition involved a rotor-synchronized spin-echo sequence with 90° and 180° pulses of 6.5 and 13.0 μs, and 561800 signal transients were collected with a recycle delay of 1 s (∼7 days acquisition time). The 15N NMR chemical shifts are presented with respect to nitromethane. All NMR experiments were performed at natural isotope abundance.
Electrochemical (EC) measurements
For the preparation of an electrode, 5 mg of N-doped C, 30 μL of Nafion (D-520 dispersion 5 wt%, Alfa Aesar), and 970 μL of ethanol were mixed in a glass bottle and dispersed by ultrasound for 30 min. 200 μL of the mixture was subsequently dropped on each side of the carbon fiber substrate (Toray carbon-paper TP-030, Quintech) in a thorough manner, multiple times. The electrode was subsequently obtained after drying the mixture at room temperature for 2 h. The EC experiments were performed with a potentiostat (Gamry instruments) using an electrochemical cell. This electrode, a platinum wire (or carbon paper), and a 1 M Ag/AgCl electrode were used as a working electrode, a counter electrode, and a reference electrode in a three-electrode system, respectively. All current values of the electrodes were recorded at E1M Ag/AgCl (V), which can be converted to ERHE (V) using the formula: ERHE (V) = 0.235 (V) + E1M Ag/AgCl (V) + [0.059 × pH] (V) at 25 °C. Milli-Q water (18.3 Ω cm) was used to prepare the electrolytes. Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were performed at a scan rate of 10 mV s−1, and chronoamperometry (CA) was performed at −0.84 V vs. RHE for the nitrite reduction reaction. NH3 was analyzed using the Nessler method and Merck Spectroquant 114423.62 The amount of nitrite was determined with the 114658 test kits (MColortest™). The analysis was performed using a UV-Vis spectrophotometer (20 Genesys™) (Fig. S1†). The NO2− conversion, selectivity, Faradaic efficiency and NH4+ yield of the product were calculated using the following equations:
| NO2− conversion = ΔCNO2−/C0 × 100% |
| Selectivity = (CNH4+/18)/(ΔCNO2−/46) × 100% |
| Faradaic efficiency = (6F × CNH4+ × V)/(18 × Q) × 100% |
| NH4+ yield = (CNH4+ × V)/(18 × t × m) |
where ΔCNO2− is the concentration difference of NO2− before and after electrolysis (ppm), C0 is the initial concentration of NO2− (50 ppm), CNH4+ is the final concentration of NH4+ (ppm), V is the electrolyte volume (50 mL), F is the Faradaic constant (96485 C mol−1), Q is the total charge used for the electrode (C), t is the electrolysis time (2 h), and m is the mass of the catalyst (1 mg).
Results and discussion
Catalysis
Doping a carbon-based catalyst with N atoms has been previously shown to enhance the catalytic activity for the OER and HER (vide supra),41,63 although the exact effect of N is still unclear due to the lack of structural information regarding the chemical form of N. It has been suggested that pyridinic N, pyrrolic N and graphitic N with the carbon atoms located at the armchair edge are the intrinsic active sites for the HER and OER.41,63–65 Here, we investigated two “green” carbon catalysts for the OER, HER and electrochemical nitrite reduction. LSV tests were conducted after extended CV measurements to obtain a stable CV curve (Fig. S2†). The LSV curves are summarized in Fig. 1, which also includes the response of the reference sample of the carbon cloth used as the substrate. N-doped C (NH3) had the highest amount of N (6.5 wt% based on elemental analysis) and showed higher catalytic activity than N-doped C (N2).
 |
| | Fig. 1 LSV curves of carbon fiber, N-doped C (N2) and N-doped C (NH3) for the OER (a) and HER (b) in a 0.05 M H2SO4 (pH 1) electrolyte and 0.1 M NaOH (pH 13) electrolyte; LSV curves (c) and CA at −0.84 V vs. RHE (d) of carbon fiber, N-doped C (N2) and N-doped C (NH3) for the nitrite reduction reaction in an aqueous electrolyte (0.1 M Na2SO4) with 50 ppm of NO2−. | |
For the OER, the catalytic activity of N-doped C (NH3) and N-doped C (N2) was investigated in 0.05 M H2SO4 (pH 1) and 0.1 M NaOH (pH 13) electrolytes (Fig. 1a). The anodic current density generated on the N-doped C (NH3) catalyst was significantly higher than those on both N-doped C (N2) and support (carbon fiber) at high potentials in the NaOH and H2SO4 electrolytes. The highest electrocatalytic activity was achieved for the N-doped C (NH3) sample at pH 13. Under these conditions, the achieved current density of this sample was higher than the sum of all other LSV curves. It should be noted that the current density increase below 1.23 V vs. RHE is related to pseudocapacitance. Since N-doped C (NH3) and N-doped C (N2) had similar textual properties, the significant enhancement in the activity of the N-doped C (NH3) catalyst in the OER was related to its much higher N-content, 6.5 wt% versus 0.6 wt%. The concentration of the transition metals was <10 ppm.
To further investigate the catalytic activity, we also assessed the HER in 0.05 M H2SO4 and 0.1 M NaOH electrolytes (Fig. 1b). A similar trend of superior electrocatalytic activity was observed during the HER for N-doped C (NH3) as compared with N-doped C (N2) and the support. However, the differences were not as large as that for the OER. The highest activity was observed under alkaline conditions (pH 13), which indicated that the pyridinic N, pyrrolic N, oxidized nitrogen, NH2 and graphitic N were activated in N-doped C (NH3) as in the OER.
To better understand the influence of the N-containing moieties on the selectivity during the electrocatalytic reactions, the electrochemical reduction of nitrite was studied. Our previous report on another type of N-doped carbon catalyst, containing only pyridinic N and pyrrolic N, showed no specific influence on the NH3 selectivity during the electrochemical reduction of nitrate.49 The reduction of nitrites is found to follow a similar reduction mechanism to that for nitrates, i.e. energetically-favored reduction from nitrate over N2 and/or NOx directly to NH3. We performed electrochemical denitrification in aqueous solutions with 50 ppm of nitrite and 0.1 M of Na2SO4. We did not choose higher concentrations, because it is unlikely that wastewater would contain higher concentrations of nitrite. The corresponding LSV curves, and CA experiments performed at −0.84 V vs. RHE, are presented in Fig. 1c and d. It should be kept in mind that the amount of generated NH3 has an influence on the potential vs. RHE, but a constant potential value is more realistic for potential commercial applications of nitrite reduction. After CA for 2 h, the N-doped C (NH3) catalyst showed the highest nitrite reduction efficiency of 58.4%, outperforming the N-doped C (N2) catalyst and carbon fiber substrate (Table 1).
Table 1 Electrochemical reduction of NO2− on carbon fiber, N-doped C (N2) and N-doped C (NH3) electrodes in 50 mL of 0.1 M Na2SO4 with a 50 ppm NO2− electrolyte
| Electrode |
CA conditions |
Initial concentration of NO2− (ppm) |
Final concentration of NO2− (ppm) |
Reduction efficiency of NO2− |
Final concentration of NH4+ (ppm) |
| Carbon fiber |
−0.84 V vs. RHE for 2 h (pH 7) |
50 |
38.3 |
23.6% |
1.0 |
| N-Doped C (N2) |
−0.84 V vs. RHE for 2 h (pH 7) |
50 |
30.1 |
39.8% |
1.9 |
| N-Doped C (NH3) |
−0.84 V vs. RHE for 2 h (pH 7) |
50 |
20.8 |
58.4% |
7.5 |
The selectivity toward NH3 was the highest for N-doped C (NH3) (65.6%) when compared to the N-doped C (N2) catalyst (24.4%) with a lower N content (Fig. S3†). The N-doped C (NH3) catalyst produced 7.5 ppm of NH3 from a nitrite concentration of 50 ppm, with the highest Faradaic efficiency of 58.3% and an NH4+ yield of 2.63 μmol h−1 mgcat.−1 (Fig. S3†), while the N-doped C (N2) catalyst produced only 1.9 ppm of NH3. Considering that pyridinic N and pyrrolic N have been shown to not influence the selectivity of the overall nitrite/nitrate reduction, this result indicates that the presence of graphitic N can on the other hand increase the nitrite-to-NH3 selectivity.
Spectroscopy
To assess the local atomic structure and rationalize the enhanced electrocatalytic activities of N-doped C (NH3) in the OER, HER, and reduction of nitrite, ssNMR experiments were performed according to our previously described methodology.49
For determining the nitrogen positions in the carbon matrix, ssNMR experiments were performed. The 1H MAS NMR spectrum (60 kHz MAS) of the N-doped C (NH3) sample shows three distinct signals (Fig. 2a). The broad resonance centered at 6.5 ppm originates from the protons terminating the carbon edges. The signal at 1.0 ppm with a shoulder at 1.8 ppm is attributed to the protons present in the –NH2 and hydroxyl groups. The resonance that shows a rather unusual, negative chemical shift of −2.8 ppm can be assigned to the nucleus-independent chemical shift (NICS) effect on the protons of H2O molecules confined to the internal cavities of the N-doped C particles.66–68 This is associated with ring currents that are induced perpendicularly to the plane of the aromatic system of delocalized π electrons in the external magnetic field. Such ring current effects may be responsible for abnormal proton chemical shifts in aromatic systems.69 The directly-excited 13C MAS NMR spectrum of N-doped C (NH3) is depicted in Fig. 2b. The single resonance from the sp2 hybridized carbon atoms shows an isotropic chemical shift of 122 ppm and spinning sideband manifold due to the chemical shift anisotropy, which collectively is in line with previous reports on graphite-based materials.49,70–73
 |
| | Fig. 2 NMR spectra of N-doped C (NH3): (a) 1H MAS NMR spectrum at a 60 kHz MAS rate and magnetic field strength of 14.1 T. Directly excited 13C (b) and 15N (c) MAS NMR spectra collected at 7 kHz MAS and 14.1 T, shown together with chemical shifts (in ppm) calculated for hypothetical structural motifs. Experimental chemical shifts are given in black, calculated 13C shifts are in red, and 15N shifts are in blue. The DFT-derived shifts are based on a previous work of the authors. | |
The directly-excited 15N MAS NMR spectrum of N-doped C (NH3) is presented in Fig. 2c. Although this experimental approach is very challenging due to the natural abundance of 15N being only 0.37%, it allows the detection of all types of N sites. We note that the 15N MAS NMR spectrum of Fig. 2c might not be quantitative due to the short relaxation interval employed, however, the choice of such compromise is supported by the relatively fast 13C T1 relaxation indicating the presence of paramagnetic centers (Fig. S4†). In our previous report on a related N-doped carbon catalyst,49 we could only detect pyridinic and pyrrolic groups with signal positions in excellent agreement with chemical shifts calculated on the respective models at the reliable DSD-PBEP86/pcS-2 level of theory.74,75 Serendipitously, the N-doped carbon (NH3) showed a significantly different 15N MAS NMR spectrum. It contains five distinct 15N resonances as can be observed in Fig. 2c, and the corresponding models with calculated chemical shifts (based on our previous work49) are shown above the spectrum. The signals at −74, −275, and −332 ppm could be unambiguously assigned to pyridinic N, pyrrolic N and –NH2, respectively. The resonance at −21 ppm originates probably from the oxidized nitrogen species, since its chemical shift is close to that of the nitromethane reference at 0 ppm. However, the unexpected broad signal between −160 and −180 ppm was here attributed to graphitic N. As indicated by the structural model, the chemical shifts of such N environments depend on the position of the N atom with respect to the carbon edge.76 The electronic environments might also be perturbed further by neighbouring structural motifs and result in a broad 15N NMR chemical shift distribution. Irrespectively, to the best of our knowledge, this observation of graphitic N is unprecedented at natural 15N isotope abundance. This shows, in turn, that 15N ssNMR spectroscopy can likely be applied on all types of N-doped carbons with sufficient N-content without expensive and tedious 15N isotope labelling, given that the nitrogen relaxation properties are favorable. Specific to N-doped carbon (NH3), the combination of 15N ssNMR spectroscopy and quantum chemical shift data showed a structure–property relationship when it came to the graphitic N and the electrocatalytic selectivity in nitrite reduction. The analysis and interpretation of the 15N ssNMR spectrum were fully corroborated by the complementary XPS analysis. However, we cannot exclude a degree of clustering of N atoms in the N-doped carbon (NH3), as the resonance for graphitic N was broad and a very weak signal at −110 ppm was observed, which could have stemmed from the clustering of pyridinic N with pyrrolic N.48
The XPS method gave additional insight into the distribution of N moieties formed on the surface of N-doped C (NH3). Consistent with the ssNMR results, it showed a comparatively large amount of graphitic N, which accounts for almost 35% of all N atoms. As would be expected, much of the N atoms remained incorporated in the pyridine and pyrrole-type moieties. There are features of four different types of N moieties in the XPS N 1s spectrum in Fig. 3: pyridinic (peak at 398.4 eV), pyrrolic (400.2 eV), graphitic (401.0 eV) and oxidized (403.2 eV).77 The relative contents of these individual N forms are given in Table 2. The complementary N-doped C (N2) XPS N 1s spectrum is characterized by a very low signal-to-noise ratio, and the potential nitrogen signals are lost in the baseline (Fig. 3). This shows that ammonolysis is required to obtain higher nitrogen contents in these materials.
 |
| | Fig. 3 XPS N 1s spectra of studied N-doped C (NH3) and N-doped C (N2). | |
Table 2 Distribution of different types of N species and their relative contents for N-doped C (NH3) from XPS
| N species (at%) |
| Pyridinic |
Pyrrolic |
Graphitic |
Oxidized |
Total N |
| 2.82 |
1.02 |
2.34 |
0.56 |
6.74 |
In the Raman spectra (Fig. 4) of both N-doped C (NH3) and N-doped C (N2), D and G bands were observed at 1360 cm−1 and 1575 cm−1, respectively. The D band is associated with the disordered sp2 hybridized carbon, while the G band arises from the crystalline graphitic carbon.78 The intensity ratio of D and G bands (ID/IG) is used to assess the degree of graphitization of porous carbon materials.79 The ID/IG ratio of N-doped C (NH3) (0.948) was higher than that of N-doped C (N2) (0.740), indicating more defects and disordered structures, which have been suggested to be beneficial for applications in electrocatalysis.13,63,64 The PXRD patterns for N-doped C (NH3) and N-doped C (N2) were similar and showed two broad diffraction peaks at 24° and 44°, which were assigned to the graphite facets (002) and (101) of carbon (Fig. S5†).
 |
| | Fig. 4 Raman spectra of N-doped C (N2) and N-doped C (NH3). | |
Textural data and EM study
The N2 adsorption/desorption isotherms for N-doped C (N2) and N-doped C (NH3) are presented in Fig. 5. Both catalysts showed type I isotherms of microporous materials,80 and were apparently independent of whether N2 or NH3 had been used for the carbonization of cellulose. The specific surface area was slightly higher for N-doped C (NH3) than for N-doped C (N2) (SBET = 557 m2 g−1; SLangmuir = 669 m2 g−1). It is most likely that the treatment of cellulose with HNO3 followed by carbonization in flowing NH3/H2 led to the formation of wide pores (mesopores) and, as a result, an increase in the total pore volume. A similar effect has been observed previously for sucrose-derived carbon replicas.49 The textural data derived via N2 adsorption/desorption experiments under cryogenic conditions are displayed in Table 3.
 |
| | Fig. 5 N2 adsorption–desorption isotherms of N-doped C (N2) and N-doped C (NH3). | |
Table 3 Textural parameters and N content from the elemental analysis of N-doped C (N2) and N-doped C (NH3)
| Sample |
S
BET (m2 g−1) |
S
Langmuir (m2 g−1) |
V
total (cm3 g−1) |
V
meso (cm3 g−1) |
V
micro (cm3 g−1) |
N content (wt%) |
| N-Doped C (N2) |
452 |
534 |
0.19 |
0.006 |
0.17 |
0.6 |
| N-Doped C (NH3) |
557 |
669 |
0.25 |
0.022 |
0.20 |
6.5 |
The features observed in the SEM micrographs (Fig. 6a and b) confirm the irregularity in the shape of the particles of N-doped C (NH3) and N-doped C (N2). However, the size of the particles was smaller for N-doped C (NH3) as is visible from the SEM micrographs at lower magnification (Fig. S6†). The complementary morphological analysis of N-doped C (NH3) by means of TEM provides evidence of a high degree of porosity with pore sizes of app. 1–2 nm (Fig. 6c and d), which was consistent with the analysis of N2 adsorption/desorption data. Moreover, structural analysis by TEM and XPS indicates that the textural characteristics of the N-doped C (NH3) catalyst remain unchanged and its structural stability is maintained after the electrocatalytic tests (Fig. S7†). It should be noted that the XPS spectrum in Fig. S7† was collected on the complete composite electrode instead of the N-doped carbon powder. As such, the signal-to-noise ratio is smaller and only the graphitic and pyridinic nitrogen signals could be detected.
 |
| | Fig. 6 SEM micrographs of N-doped C (NH3) (a) and N-doped C (N2) (b); (c, d) TEM images of N-doped C (NH3). | |
Elemental analysis
The developed procedure of cellulose carbonization, including initial oxidation with nitric acid and then ammonolysis at 800 °C, allowed the incorporation of 6.5 wt% of N into N-doped C (NH3), while N-doped C (N2) had only 0.6 wt% of N. N-doped C had the metal content of Cu, Co, Cr, Mn, Mo, Ni, V, W, Pt and Pd of < 10 ppm.
The surfaces of the N-doped C (NH3) and N-doped C (N2) catalysts contained a significant amount of O atoms, and XPS allowed three different forms of such O atoms to be recognized. The photoemissions at 531.1, 532.4 and 533.7 eV in the O 1s spectra (Fig. 7) were assigned to O in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carbonyls), OC–O (esters, anhydrides and carboxylic acids) and C–O (phenols and ethers), respectively.81 N-doped C (N2) had more O atoms than N-doped C (NH3) (Table 4), with a total amount of 3.43 at% of surface O atoms, with predominantly carboxyl groups (66% of all O atoms). The reducing atmosphere (NH3/H2) during the preparation of N-doped C (NH3) seems to have suppressed the oxygenation of the surface.
O (carbonyls), OC–O (esters, anhydrides and carboxylic acids) and C–O (phenols and ethers), respectively.81 N-doped C (N2) had more O atoms than N-doped C (NH3) (Table 4), with a total amount of 3.43 at% of surface O atoms, with predominantly carboxyl groups (66% of all O atoms). The reducing atmosphere (NH3/H2) during the preparation of N-doped C (NH3) seems to have suppressed the oxygenation of the surface.
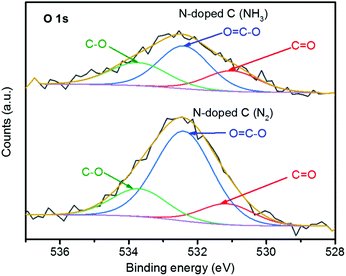 |
| | Fig. 7 XPS O 1s spectra of both studied N-doped C samples. | |
Table 4 Distribution of different types of O species on N-doped C (N2) and N-doped C (NH3)
| Sample |
O species (at%) |
| CO |
OC–O |
C–O |
Total O |
| N-Doped C (N2) |
0.49 |
2.26 |
0.68 |
3.43 |
| N-Doped C (NH3) |
0.50 |
0.98 |
0.61 |
2.09 |
Conclusions
The N-doped carbon catalyst prepared from cellulose with a combination of HNO3 treatment and ammonolysis had a high N content. It showed a higher electrocatalytic activity for the OER, HER, and nitrate reduction as compared with the reference material prepared by the carbonization of HNO3 treated cellulose in N2. The enhanced activity of N-doped C (NH3) was related to the high content of 6.5 wt% N, and the improved catalytic activity under alkaline conditions was tentatively related to the type of nitrogen function. Its high selectivity in the electrochemical reduction of nitrite was related to graphitic N, since pyridinic N and pyrrolic N had earlier been shown to not affect the electrochemical reduction of nitrite. The inclusion of graphitic N was ascribed to the comparably high temperature of ammonolysis and/or the use of cellulose as a carbon source. The presence of graphitic N in N-doped C (NH3) was determined by means of directly-excited 15N ssNMR spectroscopy.
Our study highlights the potential of using 15N ssNMR spectroscopy at natural 15N isotope abundance for a wide range of materials. Specifically, we identify the structure–property relationship for graphitic N and the enhanced selectivity in the electrochemical reduction of nitrite without costly and often tedious 15N labelling. The methodology provides such an opportunity to determine the local chemistry around N at the atomic level and to establish a structure–property relationship with respect to its catalytic activity in general. The serendipitous discovery of graphitic N in N-doped catalysts derived from cellulose has further potential for the synthesis of metal-free catalysts using renewable substrates. Further studies on the synthesis of carbon materials with solely graphitic N are currently being carried out in our laboratories.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Z. C. thanks the China Scholarship Council for a PhD scholarship. P. K. acknowledges the European Regional Development Fund in the framework of the Polish Innovation Operational Program (contract no. POIG.02.01.00-12-023/08) for financial support. We are indebted to Serhiy Budnyk for ICP analyses and Jedrzej Piatek for fruitful discussions. We thank the Swedish Foundation for Strategic Environmental Research (project: Mistra SafeChem, project number 2018/11) for financial support.
References
- H. Tan, Y. Zhao, W. Xia, J. Zhao, X. Xu, K. Wood, Y. Sugahara, Y. Yamauchi and J. Tang, Chem. Mater., 2020, 32, 4248–4256 CrossRef CAS.
- G. H. Wang, Z. Cao, D. Gu, N. Pfänder, A. C. Swertz, B. Spliethoff, H. J. Bongard, C. Weidenthaler, W. Schmidt, R. Rinaldi and F. Schüth, Angew. Chem., Int. Ed., 2016, 55, 8850–8855 CrossRef CAS PubMed.
- C. Lu, P. R. Jothi, T. Thersleff, T. M. Budnyak, A. Rokicinska, K. Yubuta, R. Dronskowski, P. Kuśtrowski, B. P. T. Fokwa and A. Slabon, Nanoscale, 2020, 12, 3121–3128 RSC.
- G. J. F. Cruz, L. Kuboňová, D. Y. Aguirre, L. Matějová, P. Peikertová, I. Troppová, E. Cegmed, A. Wach, P. Kustrowski, M. M. Gomez and L. Obalová, ACS Sustainable Chem. Eng., 2017, 5, 2368–2374 CrossRef CAS.
- G. Mestl, N. I. Maksimova, N. Keller, V. V. Roddatis and R. Schlögl, Angew. Chem., Int. Ed., 2001, 40, 2066–2068 CrossRef CAS PubMed.
- R. Pawlak, X. Liu, S. Ninova, P. D'Astolfo, C. Drechsel, S. Sangtarash, R. Haner, S. Decurtins, H. Sadeghi, C. J. Lambert, U. Aschauer, S.-X. Liu and E. Meyer, J. Am. Chem. Soc., 2020, 142, 12568–12573 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- K. Yuan, D. Lutzenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- H. W. Kim, H. Park, J. S. Roh, J. E. Shin, T. H. Lee, L. Zhang, Y. H. Cho, H. W. Yoon, V. J. Bukas, J. Guo, H. B. Park, T. H. Han and B. D. McCloskey, Chem. Mater., 2019, 31, 3967–3973 CrossRef CAS.
- H. Wang, Y. Shao, S. Mei, Y. Lu, M. Zhang, J.-K. Sun, K. Matyjaszewski, M. Antonietti and J. Yuan, Chem. Rev., 2020, 120, 9363–9419 CrossRef CAS PubMed.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li, C. Ma, T. Wu, J. Yuan, M. Antonietti and G. A. Ozin, Angew. Chem., Int. Ed., 2017, 56, 7847–7852 CrossRef CAS PubMed.
- X. Hu, L. Zhong, C. Shu, Z. Fang, M. Yang, J. Li and D. Yu, J. Am. Chem. Soc., 2020, 142, 4621–4630 CrossRef CAS PubMed.
- K. Tian, J. Wang, L. Cao, W. Yang, W. Guo, S. Liu, W. Li, F. Wang, X. Li, Z. Xu, Z. Wang, H. Wang and Y. Hou, Nat. Commun., 2020, 11, 3884 CrossRef PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- X. Wang, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Adv. Mater., 2019, 31, 1803625 CrossRef PubMed.
- X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed.
- D. Liu, L. Dai, X. Lin, J.-F. Chen, J. Zhang, X. Feng, K. Müllen, X. Zhu and S. Dai, Adv. Mater., 2019, 31, 1804863 CrossRef PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- J. T. Grant, C. A. Goeltl, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Science, 2016, 354, 1570–1573 CrossRef CAS PubMed.
- Y. Subaşı, M. Somer, M. B. Yağcı, A. Slabon and S. Afyon, J. Solid State Electrochem., 2020, 24, 1085–1093 CrossRef.
- W. Hao, N. Keshavarzi, A. Branger, L. Bergström and N. Hedin, Carbon, 2014, 78, 521–531 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, G. Tian, J. Schmidt and A. Thomas, Chem. Mater., 2019, 31, 3274–3280 CrossRef CAS.
- C. Galeano, J. C. Meier, M. Soorholtz, H. Bongard, C. Baldizzone, K. J. J. Mayrhofer and F. Schüth, ACS Catal., 2014, 4, 3856–3868 CrossRef CAS.
- D. Deng, X. Pan, L. Yu, Y. Cui, Y. Jiang, J. Qi, W.-X. Li, Q. Fu, X. Ma, Q. Xue, G. Sun and X. Bao, Chem. Mater., 2011, 23, 1188–1193 CrossRef CAS.
- C. Maddi, F. Bourquard, V. Barnier, J. Avila, M.-C. Asensio, T. Tite, C. Donnet and F. Garrelie, Sci. Rep., 2018, 8, 3247 CrossRef PubMed.
- T. Schiros, D. Nordlund, L. Pálová, D. Prezzi, L. Zhao, K. S. Kim, U. Wurstbauer, C. Gutiérrez, D. Delongchamp, C. Jaye, D. Fischer, H. Ogasawara, L. G. M. Pettersson, D. R. Reichman, P. Kim, M. S. Hybertsen and A. N. Pasupathy, Nano Lett., 2012, 12, 4025–4031 CrossRef CAS PubMed.
- H. Luo, S. Dimitrov, M. Daboczi, J.-S. Kim, Q. Guo, Y. Fang, M.-A. Stoeckel, P. Samorì, O. Fenwick, A. B. J. Sobrido, X. Wang and M.-M. Titirici, ACS Appl. Nano Mater., 2020, 3, 3371–3381 CrossRef CAS.
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, J. Am. Chem. Soc., 2020, 142, 1417–1423 CrossRef CAS PubMed.
- L. Wang, Z. Sofer and M. Pumera, ACS Nano, 2020, 14, 21–25 CrossRef CAS PubMed.
- M. Borghei, J. Lehtonen, L. Liu and O. J. Rojas, Adv. Mater., 2018, 30, 1703691 CrossRef PubMed.
- H. Begum, M. S. Ahmed and Y.-B. Kim, Sci. Rep., 2020, 10, 12431 CrossRef CAS PubMed.
- L. Xu, Q. Yao, Y. Zhang and Y. Fu, ACS Sustainable Chem. Eng., 2017, 5, 2960–2969 CrossRef CAS.
- B. You, F. Kang, P. Yin and Q. Zhang, Carbon, 2016, 103, 9–15 CrossRef CAS.
- B. You, P. Yin, J. Zhang, D. He, G. Chen, F. Kang, H. Wang, Z. Deng and Y. Li, Sci. Rep., 2015, 5, 1–9 Search PubMed.
- B. You, P. Yin and L. An, Small, 2014, 10, 4352–4361 CAS.
- J. Zhang, G. Chen, Q. Zhang, F. Kang and B. You, ACS Appl. Mater. Interfaces, 2015, 7, 12760–12766 CrossRef CAS PubMed.
- B. You, X. Liu, G. Hu, S. Gul, J. Yano, D. E. Jiang and Y. Sun, J. Am. Chem. Soc., 2017, 139, 12283–12290 CrossRef CAS PubMed.
- M. Soorholtz, R. J. White, T. Zimmermann, M.-M. Titirici, M. Antonietti, R. Palkovits and F. Schüth, Chem. Commun., 2013, 49, 240–242 RSC.
- K. Qu, Y. Zheng, X. Zhang, K. Davey, S. Dai and S. Z. Qiao, ACS Nano, 2017, 11, 7293–7300 CrossRef CAS PubMed.
- L. Zhao, N. Baccile, S. Gross, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Carbon, 2010, 48, 3778–3787 CrossRef CAS.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- J. H. Warner, Y.-C. Lin, K. He, M. Koshino and K. Suenaga, ACS Nano, 2014, 8, 11806–11815 CrossRef CAS PubMed.
- C. Hofer, V. Skákalová, T. Görlich, M. Tripathi, A. Mittelberger, C. Mangler, M. R. A. Monazam, T. Susi, J. Kotakoski and J. C. Meyer, Nat. Commun., 2019, 10, 4570 CrossRef CAS PubMed.
- X. Li, I. V. Sergeyev, F. Aussenac, A. F. Masters, T. Maschmeyer and J. M. Hook, Angew. Chem., Int. Ed., 2018, 57, 6848–6852 CrossRef CAS PubMed.
- Y. Hu, Y. Shim, J. Oh, S. Park, S. Park and Y. Ishii, Chem. Mater., 2017, 29, 5080–5089 CrossRef CAS.
- S. Kuroki, Y. Hosaka and C. Yamauchi, Carbon, 2013, 55, 160–167 CrossRef CAS.
- X. Fang, J. Mao, E. M. Levin and K. Schmidt-Rohr, J. Am. Chem. Soc., 2009, 131, 1426–1435 CrossRef CAS PubMed.
- I. Szewczyk, A. Rokicińska, M. Michalik, J. Chen, A. Jaworski, R. Aleksis, A. J. Pell, N. Hedin, A. Slabon and P. Kuśtrowski, Chem. Mater., 2020, 32, 7263–7273 CrossRef CAS.
- W. Luo, B. Wang, C. G. Heron, M. J. Allen, J. Morre, C. S. Maier, W. F. Stickle and X. Ji, Nano Lett., 2014, 14, 2225–2229 CrossRef CAS PubMed.
- X. Wu, Z. Shi, R. Tjandra, A. J. Cousins, S. Sy, A. Yu, R. M. Berry and K. C. Tam, J. Mater. Chem. A, 2015, 3, 23768–23777 RSC.
- X. Du, Z. Zhang, W. Liu and Y. Deng, Nano Energy, 2017, 35, 299–320 CrossRef CAS.
- A. Wütscher, T. Eckhard, D. Hiltrop, K. Lotz, W. Schuhmann, C. Andronescu and M. Muhler, ChemElectroChem, 2019, 6, 514–521 CrossRef.
- B. P. Chhetri, D. Soni, A. B. RanguMagar, C. M. Parnell, H. Wayland, F. Watanabe, G. Kannarpady, A. S. Biris and A. Ghosh, J. Environ. Chem. Eng., 2017, 5, 2586–2596 CrossRef CAS.
- S. Zuo, W. Zhang, Y. Wang and H. Xia, Ind. Eng. Chem. Res., 2020, 59, 7527–7537 CrossRef CAS.
- R. Huang, C. Cao, J. Liu, D. Sun and W. Song, Chem. Commun., 2019, 55, 1935–1938 RSC.
- N. N. Sauer, J. Am. Chem. Soc., 2000, 122, 5419–5420 CrossRef.
- Z. Ma, A. Jaworski, J. George, A. Rokicinska, T. Thersleff, T. M. Budnyak, G. Hautier, A. J. Pell, R. Dronskowski, P. Kuśtrowski and A. Slabon, J. Phys. Chem. C, 2020, 124, 152–160 CrossRef CAS.
- Z. Ma, T. Thersleff, A. L. Görne, N. Cordes, Y. Liu, S. Jakobi, A. Rokicinska, Z. G. Schichtl, R. H. Coridan, P. Kustrowski, W. Schnick, R. Dronskowski and A. Slabon, ACS Appl. Mater. Interfaces, 2019, 11, 19077–19086 CrossRef CAS PubMed.
- T.-L. Hwang, P. C. M. van Zijl and M. Garwood, J. Magn. Reson., 1998, 133, 200–203 CrossRef CAS PubMed.
- G. Kervern, G. Pintacuda and L. Emsley, Chem. Phys. Lett., 2007, 435, 157–162 CrossRef CAS.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- Q. Lv, W. Si, J. He, L. Sun, C. Zhang, N. Wang, Z. Yang, X. Li, X. Wang, W. Deng, Y. Long, C. Huang and Y. Li, Nat. Commun., 2018, 9, 3376 CrossRef PubMed.
- J. J. Ternero-Hidalgo, J. M. Rosas, J. Palomo, M. J. Valero-Romero, J. Rodríguez-Mirasol and T. Cordero, Carbon, 2016, 101, 409–419 CrossRef CAS.
- A. R. MacIntosh, G. Jiang, P. Zamani, Z. Song, A. Riese, K. J. Harris, X. Fu, Z. Chen, X. Sun and G. R. Goward, J. Phys. Chem. C, 2018, 122, 6593–6601 CrossRef CAS.
- A. C. Forse, J. M. Griffin, V. Presser, Y. Gogotsi and C. P. Grey, J. Phys. Chem. C, 2014, 118, 7508–7514 CrossRef CAS.
- L. Cervini, O. D. Lynes, G. R. Akien, A. Kerridge, N. S. Barrow and J. M. Griffin, Energy Storage Mater., 2019, 21, 335–346 CrossRef.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- A. Lerf, H. He, T. Riedl, M. Forster and J. Klinowski, Solid State Ionics, 1997, 101, 857–862 CrossRef.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- Y. Li, H. Chen, L. Y. Voo, J. Ji, G. Zhang, G. Zhang, F. Zhang and X. Fan, J. Mater. Chem., 2012, 22, 15021–15024 RSC.
- Q. Zhang, K. Scrafford, M. Li, Z. Cao, Z. Xia, P. M. Ajayan and B. Wei, Nano Lett., 2014, 14, 1938–1943 CrossRef CAS PubMed.
- G. L. Stoychev, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2018, 14(9), 4756–4771 CrossRef CAS PubMed.
- P. Rzepka, Z. Bacsik, A. J. Pell, N. Hedin and A. Jaworski, J. Phys. Chem. C, 2019, 123, 21497–21503 CrossRef CAS.
- S. R. Kelemen, M. Afeworki, M. L. Gorbaty, P. J. Kwiatek, M. S. Solum, J. Z. Hu and R. J. Pugmire, Energy Fuels, 2002, 16, 1507–1515 CrossRef CAS.
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X.-D. Zhou, Angew. Chem., Int. Ed., 2015, 127, 13905–13909 CrossRef.
- A. C. Ferrari, Solid State Commun., 2007, 143, 45–47 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- S. Tian, P. Yan, F. Li, X. Zhang, D. Su and W. Qi, ChemCatChem, 2019, 11, 2073–2078 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calibration curves; CV curves; selectivity, Faradaic efficiency and NH4+ yield; T1 NMR relaxation curve for 13C; XRD; SEM micrographs; TEM images and XPS N 1s spectra. See DOI: 10.1039/d1dt00658d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Jianhong
Chen
b,
Tetyana M.
Budnyak
b,
Ireneusz
Szewczyk
c,
Anna
Rokicińska
b,
Jianhong
Chen
b,
Tetyana M.
Budnyak
b,
Ireneusz
Szewczyk
c,
Anna
Rokicińska