
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bonding to metals as a probe for an inverted ligand field†
Alberto
Pérez-Bitrián
 ,
Miguel
Baya
,
José M.
Casas
,
Antonio
Martín
and
Babil
Menjón
*
,
Miguel
Baya
,
José M.
Casas
,
Antonio
Martín
and
Babil
Menjón
*
Instituto de Síntesis Química y Catálisis Homogénea (iSQCH), CSIC–Universidad de Zaragoza, Zaragoza, Spain. E-mail: b.menjon@csic.es
First published on 17th March 2021
Abstract
Electron-rich, late transition metals are known to act as hydrogen-bonding (HBd) acceptors. In this regard, Pt(II) centres in square-planar environments are particularly efficient. It is however puzzling that no convincing experimental evidence is currently available for the isoelectronic neighbour Au(III) being involved in HBd interactions. We report now on the synthesis and characterisation of two series of isoleptic and isoelectronic (d8) compounds [(CF3)3Pt(L)]− and (CF3)3Au(L), where the L ligands are based on the quinoline frame and have been selected to favour HBd with the metal centre. Strong HBd interactions were actually found in the Pt(II) compounds, based on structural and spectroscopic evidence, and they were further confirmed by theoretical calculations. In contrast, no evidence was obtained in the Au(III) case. In order to find the reason underlying this general disparity, we undertook a detailed theoretical analysis of the model systems [(CF3)3Pt(py)]− and (CF3)3Au(py). This study revealed that the filled dz2 orbital is the HOMO in the case of Pt(II), but is buried in the lower energy levels in the case of Au(III). The sharply different electronic configurations involve ligand-field inversion on going from Pt to the next element Au. This is not a gradual but an abrupt change, which invalidates Au(III) as a HBd-acceptor wherever ligand-field inversion occurs.
Introduction
Hydrogen bonding (HBd) is the secondary bonding interaction with the highest impact in chemistry, biochemistry, materials and biological sciences, as well as in geology and every aspect of life on Earth.1 The number and variety of HBd-acceptors have steadily widened over the years2 to include now a handful of metal centres.3,4 These non-conventional M⋯HE interactions (E being an electronegative main-group element) take place with electron-rich metal centres (mainly late transition metals, late-TM) featuring a suitable Lewis-base character. Square-planar d8 metal centres are particularly prone to act as HBd-acceptors.4
Results and discussion
Synthetic, structural and spectroscopic results
Compound [NBu4][(CF3)3Pt(NCMe)] was prepared in solution as a convenient synthon of the anionic fragment (CF3)3Pt⊖ (see the Experimental section in the ESI†). In this complex, the labile ligand MeCN was easily replaced by 8-hydroxyquinoline (hq) or 8-hydroxyquinaldine (hq′) to afford the organoplatinum(II) derivatives [NBu4][(CF3)3Pt(hq)] (1) and [NBu4][(CF3)3Pt(hq′)] (2), respectively (Scheme 1). Compounds 1 and 2 were isolated in good yields as fairly stable white solids. They were found to exhibit strong Pt⋯HO intramolecular interactions as confirmed from a number of structural and spectroscopic studies, which will be presented next. | ||
| Scheme 1 Synthesis of compounds 1–4. Where appropriate, the cation is [NBu4]+. See the ESI† for details. | ||
The crystal structure of 2 was determined by X-ray diffraction (XRD) methods. Reliable data obtained by low-temperature diffraction on good-quality single crystals enabled us to locate the OH atom directly from the difference density maps and the position was refined freely using no constraints. In the final position, the O–H vector is directed toward the Pt atom with a short Pt⋯H distance of 205(9) pm and a Pt⋯H–O angle of 158(8)° (Fig. 1a). The Pt⋯HO distance (Table 1) is comparable with that found in the related perfluorophenyl complex [NBu4][(C6F5)3Pt(hq′)] (219 pm)7 even though the CF3 group is more electronegative than C6F5.8 The non-bonding Pt⋯O distances are indistinguishable in both cases: ca. 300 pm. Additional structural parameters in the crystal structure of 2 are unexceptional and in agreement with other complexes of the (CF3)3Pt moiety.9
 | ||
| Fig. 1 (a) Displacement-ellipsoid diagram (50% probability) of the [(CF3)3Pt(hq′)]− anion as found in single crystals of 2; relevant structural parameters are given in Table 1. (b) 1H NMR signal of the OH nucleus (300.130 MHz, CD2Cl2, 253 K) with spectral parameters indicated; the whole spectrum is shown in Fig. S4.† | ||
| Compound | M–Cb/pm | av. M–C2c/pm | M–N/pm | M⋯E/pm | E–H/pm | M⋯H/pm | ∑∠d/° |
|---|---|---|---|---|---|---|---|
| a The closely related compound [NBu4][(C6F5)3Pt(hq′)] (see Ref. 7) is also included for comparison. b The M–C distance trans to the neutral N-donor ligand is indicated here. c Average of the two independent M–C bond lengths in a trans arrangement. d Summation of all adjacent E–M–E′ angles as a measure of planarity. e No estimated experimental error is indicated here because the H position was refined under constraints. f The O–H unit interacts with a R2O molecule (R = Et, H), where the R2O⋯H–O angles amount to 178(6)° and 176(4)°, respectively, and the O⋯O′ separation is 263.6(4) and 265.0(3) pm in each case. g The closest M⋯H distance is indicated here. | |||||||
| [NBu4][(CF3)3Pt(hq′)] (2) | 202.0(6) | 206.6(7) | 213.2(5) | 297.2(5) | 97(9) | 205(9) | 360.1(3) |
| [NBu4][(C6F5)3Pt(hq′)]a | 200.8(5) | 206.1(5) | 212.6(4) | 297.5(4) | 82e | 219e | 360.0(2) |
| (CF3)3Au(hq·OEt2) (3·OEt2) | 202.3(4) | 207.6(4) | 210.9(3) | 268.8(3) | 77(5) | —f | 360.02(16) |
| (CF3)3Au(hq·OH2) (3·OH2) | 202.3(3) | 208.4(3) | 210.4(2) | 265.8(2) | 81.9(5) | —f | 360.01(13) |
| [NBu4][(CF3)3Pt(mq)] (5) | 199.9(8) | 205.7(8) | 212.7(7) | 309(1) | 98e | 273e,g | 360.3(3) |
| (CF3)3Au(mq) (6) | 202.9(3) | 208.9(3) | 211.2(3) | 305.1(3) | 98e | 267e,g | 360.04(13) |
The Pt⋯HO interaction found in the crystal structure of 2 is not merely due to crystal packing effects, since it is maintained in solution. In the 1H NMR spectrum of 2 (Fig. 1b), the OH signal (δH = 11.10 ppm) appears downfield shifted (ΔδH = 2.8 ppm) with respect to the free ligand (δH = 8.3 ppm).10 The sole signal shift might not be sufficient to prove the existence of HBd to the metal.11 However, the observed coupling between the involved atoms with a large coupling constant, J(195Pt,H) = 100 Hz, clearly shows that the Pt⋯HO interaction is preserved along the measuring process (NMR time scale, which is a fairly long one). The 1H NMR spectrum of 1 is qualitatively similar (Fig. S2†). Thus, the OH signal is downfield shifted to roughly the same region (δH = 10.82 ppm) and it also shows large coupling to platinum: J(195Pt,H) = 82.7 Hz, which can be taken as experimental evidence of mutual penetration of the two atoms involved. The ortho-H (Ho) signal appearing at δH = 9.29 ppm shows 195Pt-satellites as well. It must be noted that the splitting of this signal, 3J(195Pt,H) ≈ 25 Hz, is much smaller than that observed in the OH signal, even though the latter is located 5 bonds apart from the metal centre vs. 3 in the case of Ho. The extra contribution can be assigned to the direct Pt⋯H–O interaction.
The isoleptic and isoelectronic (d8) organogold(III) compounds (CF3)3Au(hq) (3) and (CF3)3Au(hq′) (4) were prepared from the neutral solvate (CF3)3Au·OEt2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 by using similar procedures to those mentioned above (Scheme 1) and were isolated in good yields as stable white solids. Our many attempts to obtain single crystals for XRD purposes under various conditions resulted in the formation of the solvates 3·OEt2 and 3·OH2. Their molecular structures (Fig. 2) show that both the Et2O and H2O molecules act as HBd-acceptors with canonical, nearly linear R2O⋯H–O arrangements (>176°; Table 1). As a result, the O–H vectors point away from the gold centre in an unusual s-trans conformation. Apart from this crucial difference, the remaining geometric features around the metal centre are very similar to those found in the Pt compound 2 described above. Compound 3·OH2 lends experimental support to the monohydrate hq·OH2 in the less common s-trans conformation, which has been recently studied by calculation.13 The experimental separation between the oxygen atoms found in 3·OH2 (O⋯O′ 265.0(3) pm) is substantially shorter than that calculated for hq·OH2 in the same conformation (O⋯O′ 280.5 pm), which implies a stronger HBd interaction in the organogold(III) complex.
12 by using similar procedures to those mentioned above (Scheme 1) and were isolated in good yields as stable white solids. Our many attempts to obtain single crystals for XRD purposes under various conditions resulted in the formation of the solvates 3·OEt2 and 3·OH2. Their molecular structures (Fig. 2) show that both the Et2O and H2O molecules act as HBd-acceptors with canonical, nearly linear R2O⋯H–O arrangements (>176°; Table 1). As a result, the O–H vectors point away from the gold centre in an unusual s-trans conformation. Apart from this crucial difference, the remaining geometric features around the metal centre are very similar to those found in the Pt compound 2 described above. Compound 3·OH2 lends experimental support to the monohydrate hq·OH2 in the less common s-trans conformation, which has been recently studied by calculation.13 The experimental separation between the oxygen atoms found in 3·OH2 (O⋯O′ 265.0(3) pm) is substantially shorter than that calculated for hq·OH2 in the same conformation (O⋯O′ 280.5 pm), which implies a stronger HBd interaction in the organogold(III) complex.
 | ||
| Fig. 2 Displacement-ellipsoid diagram (50% probability) of the solvates 3·OEt2 (a) and 3·OH2 (b). Relevant structural parameters are given in Table 1. | ||
In the absence of structural evidence, any spectroscopic information acquires particular relevance in order to decide on the existence of a possible Au⋯HO interaction. The IR spectra of 3 and 4 show sharp absorptions at 3581 and 3575 cm−1, respectively, with 19 and 14 cm−1 half-width values, which are due to ν(O–H) vibrations (Fig. S18 and S19†). These high-frequency, sharp absorptions are characteristic of OH groups with little (if any) association.14 They appear well above the ν(O–H) absorption observed for the free hq ligand both in dilute solutions in non-polar solvents (3423–3400 cm−1)13 and in the gas phase (3462 cm−1),15 where intermolecular associations are minimized. It is worth noting that no absorption above 3000 cm−1 was, in turn, observed in the homologous Pt(II) complexes 1 and 2 (Fig. S16 and S17†). In the 1H NMR spectra of compounds 3 and 4 (Fig. S6 and S9†), the resonances corresponding to the hydroxyl groups appear as broad signals at δH = 7.03 and 6.66 ppm, respectively, giving therefore no indication of the existence of any Au⋯HO interaction. However, the addition of Et2O to the samples under measurement produces dramatic downfield shifts of these signals to δH > 10 ppm in both cases (Fig. S8 and S11†). The marked downfield shifts observed in the OH signals upon Et2O addition are in agreement with the Et2O⋯HO interaction detected in the solid state (Fig. 2a). It is clear that the OH groups in compounds 3 and 4 favour unsupported intermolecular HBd interactions with peripheral R2O molecules (R = H, Et) over the expected intramolecular M⋯HO interactions in analogy to the Pt homologues 1 and 2. This different behaviour will be studied by theoretical methods later on. It is worth noting that Au⋯HE HBd interactions involving auride Au(–I) centres16 or linear d10 Au(I) complexes have been recently reported or predicted.17 The nature of related Au⋯HE interactions in gold clusters is still much debated.11a,18
The 8-methylquinoline (mq) ligand has the same skeleton as hq, but the C(sp3)–H bond of the CH3 group is less polarized and therefore less prone to establish HBd interactions. Thus, a couple of compounds [NBu4][(CF3)3Pt(mq)] (5) and (CF3)3Au(mq) (6) was prepared as blank references following similar procedures to those mentioned above (see the Experimental section†). They were characterised by XRD methods (Fig. 3).
 | ||
| Fig. 3 Displacement-ellipsoid diagram (50% probability) of the [(CF3)3Pt(mq)]− (a) and (CF3)3Au(mq) (b) entities as found in the single crystals of 5 and 6, respectively. Only one set of rotationally disordered CF3 groups is shown in the Pt complex. Relevant structural parameters are given in Table 1. | ||
In the solid state, the axial methyl groups take on a staggered conformation with respect to the metal and thus no H atom points directly to it. The associated M⋯HC(sp3) distances are 272.6/275.1 pm (M = Pt) and 266.5/276.0 pm (M = Au), respectively. In our opinion, they are too long to evidence the existence of an intramolecular M⋯HC(sp3) interaction, considering that the sum of the corresponding van der Waals radii (H + M) amounts to 292 (M = Pt) and 286 (M = Au) pm.19 The remaining structural parameters around the metal including the non-bonding M⋯C(sp3) separation are indistinguishable within the experimental error (Table 1). In solution, there is no hint of hindered rotation of the methyl groups about the C(sp3)–C(sp2) single bond. Thus, the CH3 signals in the 1H and 13C NMR spectra of compounds 5 (δH/δC = 3.83/22.7 ppm) and 6 (δH/δC = 3.44/20.5 ppm) appear as singlets and suffer only slight downfield shifts with respect to the free mq ligand (δH/δC = 2.83/18.15 ppm).20 These moderate shifts may well be due to just anisotropic deshielding caused by the near metal centre.11 In fact, the couple 5 and 6 shows neither structural nor spectroscopic evidence for the existence of any substantial M⋯HC(sp3) interaction.
If we compare all the structural information presented in this work (Table 1), it becomes clear that the geometric arrangement of the ligands in the coordination plane shows little variation within the whole series under study, with the major differences being observed along the axial direction. With the aim to decide on the nature of the M⋯HO interaction in compounds 1–4 and to estimate the corresponding associated energy, we have carried out a number of theoretical studies, which will be presented next. Thus, we can benefit from the following advantageous features of our system: (1) the Pt/Au pairs are isoleptic and isoelectronic; (2) the CF3 ligands are fairly small and can be suitably modelled; and (3) the molecular core of the quinoline ligands is quite rigid, which greatly reduces the number of variables.
Energetics of the axial M⋯HO interaction
Geometry optimizations at the DFT/M06 level were performed under no symmetry constraints. The final atomic coordinates are given in the ESI.† The calculated structure of the anion [(CF3)3Pt(hq′)]− is in excellent agreement with that experimentally found in the crystal of 2 (Table S6†), which indicates that the observed Pt⋯HO interaction in 2 is not merely due to packing effects. The NMR data in solution clearly confirm this reasoning.Once the geometries were optimized, we performed calculations of the overall energy of the [(CF3)3Pt(hq)]− and (CF3)3Au(hq) entities as a function of the H–O–C–C torsion angle, τ, in the coordinated hq ligand. Thus, a stepwise variation of τ was introduced as the only geometric constraint. The resulting energy profiles are represented in Fig. 4. It is clearly observed that the Pt⋯HO interaction is attractive and maximized at an optimal value of τ = 0° (Fig. 4a), which is close to the experimental value found in the crystal of 2: τ = −7°. The energy difference between the minimum at τ = 0° and the rotamers at τ = ±180° was calculated to be ∼15.0 kcal mol−1. This value gives a reasonable estimate of the Pt⋯HO bond strength: it is in the turning point from the so-called strong (4–15 kcal mol−1) to very strong (15–40 kcal mol−1) HBd interactions.2a
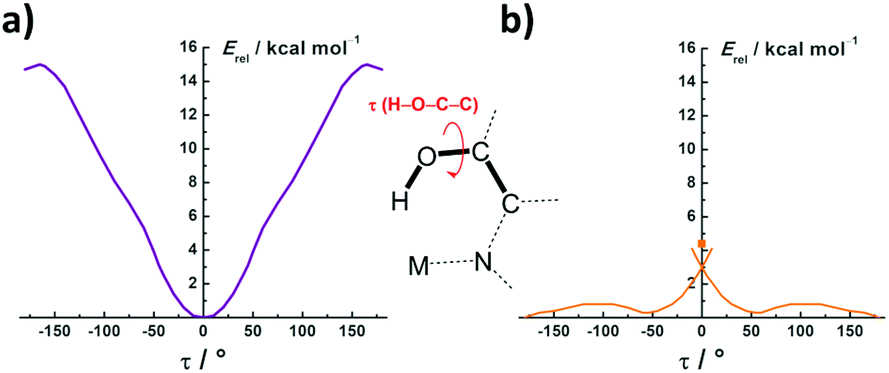 | ||
| Fig. 4 Energy profiles obtained for [(CF3)3Pt(hq)]− (a) and (CF3)3Au(hq) (b) as a function of the H–O–C–C torsion angle τ. | ||
A similar profile might be expected for the isoelectronic Au(III) centre, perhaps with a weaker interaction since the oxidation-state increase on going from Pt(II) to Au(III) and the overall charge variation should result in diminished Lewis basicity of the HBd-acceptor site.21 As a matter of fact, in a recent study on the hydration of various Pt(II) substrates, it was found that not only the anionic [Pt(CN)4]2− or neutral trans-Cl2Pt(NH3)(glycine) substrates, but also the dipositive cation [Pt(NH3)4]2+, all favour Pt⋯HOH over Pt⋯OH2 interactions with the surrounding H2O molecules regardless of the 4-unit change in the overall charge.22 Similar results were further obtained in the aquation of neutral and cationic platinum anticancer drugs.23 In our case, however, a totally unexpected profile was obtained on going from Pt(II) to Au(III). As shown in Fig. 4b, the Au⋯HO interaction is clearly repulsive. In fact, the rotamer with the OH group pointing directly to the Au centre (τ = 0°) is a transition state and appears located +4.5 kcal mol−1 above the non-interaction level (τ = ± 180°). The waving shape of the profile at intermediate τ values is due to some tilting of the O–H unit towards the nearest F atoms to establish weak O–H⋯F contacts. This effect is also present in the Pt case, but is less noticeable in the corresponding profile (Fig. 4a).
The optimized compounds were further studied with the aid of Bader's quantum theory of atoms in molecules (QTAIM).24 The topological analysis of the electron density function ρ(r) of the platinum compounds 1 and 2 (Fig. 5) shows bond paths (BPs) and bond critical points (BCPs) along the Pt⋯HO line, which are characteristic of a HBd interaction.25 The corresponding ρ(r) values (0.0401 and 0.0448; Table 2) are higher than those previously found in related systems.26 It is worth noting that the calculated Pt⋯H bond length in the analysed entity 2 (205.5 pm) exactly matches with that experimentally found in the crystal: 205(9) pm (Table S6†). The penetration of the BCP into the Pt shell is also in accordance with the sizable internuclear J(195Pt,H) coupling experimentally observed in solution (see above). In the optimized minima of the gold compounds 3 and 4, the OH group points away from the metal centre, an arrangement which is obviously not suited for intramolecular Au⋯HO interactions. The topological analysis reveals, however, that a BP is located instead on the Au⋯O line (Fig. 5). This means that the gold centre acts rather as a Lewis acid along the axial direction. On the other hand, the topological analysis of the 3·OEt2 adduct (Fig. S22†) confirmed the presence of a canonical intermolecular O⋯HO interaction, as experimentally observed in the solid state (Fig. 2a) and in solution (Fig. S8†). The positive values of ∇2ρ(r) in all cases indicate that the HBd interactions are mainly electrostatic. However, the slightly negative values of the Pt⋯H local energy density function H(r) in compounds 1 and 2 denote partial covalence, and the same applies to the Et2O⋯HO bond in the 3·OEt2 adduct.
 | ||
| Fig. 5 2D contour-line ∇2ρ(r) diagrams obtained from QTAIM analysis of the optimized compounds 1–4. Cross sections contain the Pt/Au, O and N atoms in each case. Bond critical points (orange spheres) and bond paths (black straight lines) are shown as obtained using weak-CP (0.03) and non-CP (0.02) thresholds. | ||
| Complex | 1 | 2 | 3 | 3·Et2O | 4 | |
|---|---|---|---|---|---|---|
| CP (A–B) | Pt–H | Pt–H | Au–Ohq | Au–Ohq | Et2O–H | Au–Ohq′ |
| ρ(r)/au | 0.0401 | 0.0448 | 0.0243 | 0.0296 | 0.0533 | 0.0268 |
| ∇2ρ(r) | 0.0753 | 0.0830 | 0.0975 | 0.1230 | 0.1660 | 0.1107 |
| Ellipticity | 0.021 | 0.022 | 0.111 | 0.112 | 0.029 | 0.123 |
| A–B/pm | 210 | 205 | 273 | 264 | 163 | 268 |
| BP length/pm | 214 | 208 | 273 | 264 | 166 | 268 |
| G(r)/au | 0.0251 | 0.0289 | 0.0230 | 0.0294 | 0.0429 | 0.0262 |
| V(r)/au | −0.0314 | −0.0369 | −0.0216 | −0.0281 | −0.0442 | −0.0247 |
| H(r)/au | −0.0063 | −0.0081 | 0.0014 | 0.0013 | −0.0013 | 0.0015 |
| G(r)/ρ(r) | 0.63 | 0.64 | 0.95 | 0.99 | 0.80 | 0.98 |
Up to this point, we have presented experimental and theoretical evidence demonstrating that the platinum compounds 1 and 2 exhibit strong M⋯HO interactions, whereas no evidence was found in the homologous gold compounds 3 and 4. We have also seen that compounds 1–4 show much structural resemblance in the coordination plane, but largely differ in the axial direction depending on the metal. All other factors being equal, the difference must rely on the metal itself.
Inverted ligand field
In order to ascertain the underlying reason for the substantial difference found between the Pt and Au couples under study, we analysed the electronic structures of the model compounds [(CF3)3Pt(py)]− (7) and (CF3)3Au(py) (8), which have the same in-plane coordination skeleton as compounds 1–4, but no axial ligands. The molecular geometries of the model compounds 7 and 8 were optimized under imposed Cs symmetry (Fig. 6). Symmetrisation greatly simplifies the analysis of the electronic structures at a negligible energy cost (ΔE < 1 kcal mol−1). The obtained results are shown in Fig. 7. | ||
| Fig. 6 Structures of the model compounds [(CF3)3Pt(py)]− (7-Cs) and (CF3)3Au(py) (8-Cs) optimized at the DFT/M06 level under imposed Cs symmetry. | ||
 | ||
| Fig. 7 Electronic structures of the model compounds 7-Cs (left) and 8-Cs (right) obtained by calculation at the DFT/M06 level. Contributions to the most relevant MOs are indicated by the following colour code: Pt (purple), Au (golden), CF3 (green), and py (blue). See the ESI† for details. | ||
In the platinum compound 7-Cs (Fig. 7, left), the energy levels with major metal contribution (purple labelling) roughly follow the standard splitting of the d metal orbitals under a square-planar ligand field.27 Even in ideal MX4 entities with D4h symmetry, the precise d-ordering depends on the metal ion and the type of ligand involved.27–29 In any case, the dx2−y2 orbital, which is oriented towards the ligands, is always greatly destabilized and remains empty in most d8 metal complexes.27 This is the case of complex 7-Cs, where the MO with main dx2−y2 contribution is located well above the LUMO. Here, the dxy orbital appears twofold stabilized below the dxz, dyz orbitals.28a,29 Most importantly, dz2 is always located in between and generally filled in d8 metal complexes. This is also the case of complex 7-Cs, where the MO with main dz2 contribution is identified as the HOMO (Fig. 8a).
 | ||
| Fig. 8 HOMO of the model compounds 7-Cs (a) and 8-Cs (b) as obtained at the DFT/M06 level of calculation. | ||
In d8 metal complexes with a square-planar geometry, [M], the dz2 orbital has suitable symmetry to interact along the axial direction with Lewis acids, A, wherever the energy balance is favourable. This dative [M]→A interaction was theoretically studied by Aullón and Álvarez, who scrutinized a wide range of cases and derived important consequences therefrom.30 A simple example is given with the NO+ cation, which affords a number of [M]→NO metal nitrosyls.31 Cationic metal fragments, QLnq+, or simple metal cations (n = 0) with an obvious Lewis-acidic character are also known to enable manifold [M]→A interactions.32 Similar [M]→X2 interactions with halogens are key intermediates in SN2 oxidative–addition processes eventually affording X–[M]–X derivatives.33 The range of Lewis acids has been recently expanded to incorporate EX3 neutral molecules of Group 13 elements (E = B, Al, Ga; X = H, F, Cl, Br).34 In the same way, the HOMO of 7-Cs with main 5dz2 contribution is most suited to establish an axial Pt⋯HO interaction,35 as experimentally observed in compounds 1 and 2. This Pt⋯HO interaction can be considered as an incipient stage in proton transfer from oxygen to the metal.
When we turn to the gold compound 8-Cs, a quite different electronic structure is found (Fig. 7, right). The observed difference is not just quantitative but essentially qualitative and evidences ligand-field inversion on going from Pt to Au. The ‘inverted ligand field’ concept (ILF) has been only recently introduced.36 It is the last stage in the evolution of the ligand field theory along nearly a century. This theory, which had its origin in a purely ionic model (crystal field theory),37 later incorporated the tools of the MO theory, thereby enabling some degree of covalence in the chemical bonding between the central metal ion and the surrounding ligands.38 Now, the term ILF is coined36 to convey that the MOs with major metal d contribution are not at the frontier, as is usually the case (e.g.7-Cs), but submerged in the low energy levels of bonding MOs. This phenomenon was first identified in the organocopper(III) complex [Cu(CF3)4]− and is also found in the heavier-metal homologues [Ag(CF3)4]− and [Au(CF3)4]−,39 as well as in other high-valent late-TM derivatives with a high degree of covalency.36 The electronic structure variations found in simple halide complexes, such as [PtBr4]2−vs. [AuBr4]−,40 might also be taken as an indication of the ILF.
We would like to emphasize that covalency is a key factor determining ligand-field inversion. In the traditional ligand field theory, covalency has always been treated as a perturbative effect operating upon the main d-orbital splitting caused by the L ligands considered as ions or dipoles. The ILF phenomenon appears when the covalency of the M–L bonds becomes so marked that it can no longer be treated just as a perturbation. In those cases, the metal d orbitals make a significant and even predominant contribution to the low-lying bonding MOs, which are otherwise contributed mainly by the ligands. The M–L bond covalency is favoured with increasing electronegativity of the metal. In this regard, it cannot be overlooked that gold is the most electronegative metal41 and therefore more likely to form M–L bonds with a higher degree of covalency. Accordingly, the bonding MO with major 5dz2 contribution in the gold compound 8-Cs appears buried well below the HOMO, which is now centred on the mutually trans-standing CF3 groups (Fig. 8b). This considerable energy lowering makes the dz2 unsuited for axial interaction with any Lewis acid in general, and with a polarized OHδ+ unit in particular. This electronic arrangement seriously limits the role of Au(III) as a HBd-acceptor and provides a convincing explanation for our experimental observations.
Conclusions
In this work, the isoleptic and isoelectronic compounds [(CF3)3Pt(L)]− and (CF3)3Au(L) containing L ligands with the rigid quinoline skeleton have been prepared: 1–4 (Scheme 1). They are sensitive touchstones to assay the extent to which the isoelectronic d8 centres Pt(II) and Au(III) may act as HBd-acceptors. Using a combined experimental and theoretical approach, we demonstrate that the Pt⋯HO interactions detected in compounds 1 and 2 match every geometry and energy requirement stipulated for a genuine HBd.1 Conversely, none of them is fulfilled in the gold compounds 3 and 4. As a matter of fact, the corresponding Au⋯HO interaction is found to be repulsive in nature (Fig. 4b). The origin of this disparity was found in the sharply different electronic structures of the model compounds [(CF3)3Pt(py)]− and (CF3)3Au(py) (Fig. 7). The electronic structure of the Pt(II) complex follows the standard sequence expected for a d8 transition metal in a square-planar environment. In contrast, the Au(III) homologous compound exhibits ligand-field inversion. The energy drop of the MO with major 5dz2 contribution greatly diminishes the axial basicity of Au(III) and invalidates it as a HBd-acceptor. It further limits the reactivity of Au(III) towards electrophiles. In fact, the Au(III) centre should rather behave as a Lewis acid and react preferentially with nucleophiles along the axial direction.The drastic alteration in the electronic structure found here on going from Pt to Au was totally unexpected for contiguous transition metals and points to a deep break between elements of Groups 10 and 11. This fundamental disparity had been hitherto overlooked, probably because of the apparent structural similarity generally found between Pt(II) and Au(III) complexes, as in compounds 5 and 6 (Fig. 3). The chemistry of gold might therefore be determined not only by the well-known relativistic effect,42 but also by the inverted ligand field (ILF) phenomenon.36 The consequences of ILF on reactivity are just starting to emerge.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to Prof. Dr. Santiago Alvarez on the occasion of his 70th birthday in recognition of his enlightening contributions to the most diverse chemistry concepts. This work was supported by the Spanish MICIU/FEDER (Project PGC2018-094749-B-I00) and the Gobierno de Aragón (Grupo E17_20R). BIFI (Instituto de Biocomputación y Física de Sistemas Complejos) and CESGA (Centro de Supercomputación de Galicia) are acknowledged for allocation of computational resources. A. P.-B. also thanks the Spanish Ministerio de Educación, Cultura y Deporte for a grant (FPU15/03940).Notes and references
- (a) G. R. Desiraju, Angew. Chem., Int. Ed., 2011, 50, 52 CrossRef CAS PubMed; (b) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637 CAS; (c) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619 CAS.
- (a) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, Oxford University Press, 1999, sect. 3.5.1, pp. 271–277 Search PubMed; (b) I. Alkorta, I. Rozas and J. Elguero, Chem. Soc. Rev., 1998, 27, 163 RSC.
- (a) H. Schmidbaur, Angew. Chem., Int. Ed., 2019, 58, 5806 CrossRef CAS PubMed; (b) L. R. Falvello, Angew. Chem., Int. Ed., 2010, 49, 10045 CrossRef CAS PubMed; (c) M. J. Calhorda, in Hydrogen Bonding—New Insights, ed. S. J. Grabowski, Springer, 2006, ch. 6, pp. 245–262 Search PubMed; (d) L. Brammer, Dalton Trans., 2003, 3145 RSC; (e) A. Martín, J. Chem. Educ., 1999, 76, 578 CrossRef.
- (a) J. Kozelka, in Noncovalent Forces, ed. S. Scheiner, Springer, 2015, ch. 6, pp. 129–158 Search PubMed; (b) I. C. M. Wehman-Ooyevaar, D. M. Grove, H. Kooijman, P. van der Sluis, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 1992, 114, 9916 CrossRef CAS; (c) L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow, A. G. Orpen and T. F. Koetzle, J. Chem. Soc., Dalton Trans., 1991, 1789 RSC; (d) L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow, T. F. Koetzle and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1987, 443 RSC; (e) A. Albinati, C. G. Anklin and P. S. Pregosin, Inorg. Chim. Acta, 1984, 90, L37 CrossRef CAS.
- (a) A. Behnia, M. A. Fard, P. D. Boyle and R. J. Puddephatt, Eur. J. Inorg. Chem., 2019, 2899 CrossRef CAS; (b) M. Baya, U. Belío and A. Martín, Inorg. Chem., 2014, 53, 189 CrossRef CAS PubMed; (c) S. Rizzato, J. Bergès, S. A. Mason, A. Albinati and J. Kozelka, Angew. Chem., Int. Ed., 2010, 49, 7440 CrossRef CAS PubMed.
- (a) Thematic Issue Chem. Rev., 2021, 121, still in preparation; ; (b) I. Chambrier, D. L. Hughes, R. J. Jeans, A. J. Welch, P. H. M. Budzelaar and M. Bochmann, Chem. – Eur. J., 2020, 26, 939 CrossRef CAS PubMed; (c) H. G. Raubenheimer and H. Schmidbaur, J. Chem. Educ., 2014, 91, 2024 CrossRef CAS; (d) H. Schmidbaur, H. G. Raubenheimer and L. Dobrzańska, Chem. Soc. Rev., 2014, 43, 345 RSC; (e) F. Kraus, H. Schmidbaur and S. S. Al-juaid, Inorg. Chem., 2013, 52, 9669 CrossRef CAS PubMed; (f) M. A. Cinellu, A. Zucca, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1995, 2865 RSC; (g) M. A. Cinellu, A. Zucca, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1996, 4217 RSC.
- J. M. Casas, L. R. Falvello, J. Forniés, A. Martín and A. J. Welch, Inorg. Chem., 1996, 35, 6009 CrossRef CAS.
- M. A. García-Monforte, S. Martínez-Salvador and B. Menjón, Eur. J. Inorg. Chem., 2012, 4945 CrossRef.
- S. Martínez-Salvador, J. Forniés, A. Martín, B. Menjón and I. Usón, Chem. – Eur. J., 2013, 19, 324 CrossRef PubMed.
- SDBS-1559, SDBSWeb: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?sdbsno=1559.
- (a) J. Vícha, C. Foroutan-Nejad and M. Straka, Nat. Commun., 2019, 10, 1643 CrossRef PubMed; (b) S. Scheiner, ChemPhysChem, 2016, 17, 2263 CrossRef CAS PubMed; (c) A. D. Buckingham and P. J. Stephens, J. Chem. Soc., 1964, 4583 RSC.
- A. Pérez-Bitrián, M. Baya, J. M. Casas, L. R. Falvello, A. Martín and B. Menjón, Chem. – Eur. J., 2017, 23, 14918 CrossRef PubMed.
- M. Amati, S. Belviso, P. L. Cristinziano, C. Minichino, F. Lelj, I. Aiello, M. la Deda and M. Ghedini, J. Phys. Chem. A, 2007, 111, 13403 CrossRef CAS PubMed.
- E. Pretsch, P. Bühlmann and M. Badertscher, Spektroskopische Daten zur Strukturaufklärung organischer Verbindungen, Springer, 6th edn, 2020, sect. 7.8.1, pp. 325–326 Search PubMed.
- G. N. Rodionova, Y. G. Tuchin and N. A. Partalla, Chem. Heterocycl. Compd., 1987, 23, 547 CrossRef.
- E. S. Kryachko, J. Mol. Struct., 2008, 880, 23 CrossRef CAS.
- (a) X. Lin and Y. Mo, Inorg. Chem., 2021, 60, 460 CrossRef CAS PubMed; (b) M. K. Pandey, H. S. Kunchur, D. Mondal, L. Radhakrishna, B. S. Kote and M. S. Balakrishna, Inorg. Chem., 2020, 59, 3642 CrossRef CAS PubMed; (c) M. Kumar and J. S. Francisco, J. Am. Chem. Soc., 2020, 142, 6001 CrossRef CAS PubMed; (d) S. K. Verma, S. N. Ansari, P. Kumari and S. M. Mobin, Organometallics, 2019, 38, 2591 CrossRef CAS; (e) M. Straka, E. Andris, J. Vícha, A. Růžička, J. Roithová and L. Rulíšek, Angew. Chem., Int. Ed., 2019, 58, 2011 CrossRef CAS PubMed; (f) M. Rigoulet, S. Massou, E. D. S. Carrizo, S. Mallet-Ladeira, A. Amgoune, K. Miqueu and D. Bourissou, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 46 CrossRef CAS PubMed; (g) R. J. F. Berger, J. Schoiber and U. Monkowius, Inorg. Chem., 2017, 56, 956 CrossRef CAS PubMed; (h) M. Teci, E. Brenner, D. Matt, C. Gourlaouen and L. Toupet, Chem. – Eur. J., 2015, 21, 10997 CrossRef CAS PubMed.
- (a) L. Estévez, Dalton Trans., 2020, 49, 4797 RSC; (b) M. A. Bakar, M. Sugiuchi, M. Iwasaki, Y. Shichibu and K. Konishi, Nat. Commun., 2017, 8, 576 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- SDBS-1834, SDBSWeb: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?sdbsno=1834.
- O. Kroutil, M. Předota and Z. Chval, Inorg. Chem., 2016, 55, 3252 CrossRef CAS PubMed.
- S. Aono, T. Mori and S. Sakaki, J. Chem. Theory Comput., 2016, 12, 1189 CrossRef CAS PubMed.
- T. Hirakawa, D. R. Bowler, T. Miyazaki, Y. Morikawa and L. A. Truflandier, J. Comput. Chem., 2020, 41, 1973 CrossRef CAS PubMed.
- The Quantum Theory of Atoms in Molecules, ed. C. F. Matta and R. J. Boyd, Wiley–VCH, 2007 Search PubMed.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835 CrossRef CAS.
- F. Groenewald, H. G. Raubenheimer, J. Dillena and C. Esterhuysen, Dalton Trans., 2017, 46, 4960 RSC see also ref. 4a and 18a.
- H. B. Gray, Transition Met. Chem., Ser. Adv., 1965, 1, 239 CAS.
- (a) J. J. Oppenheim, B. J. McNicholas, J. Miller and H. B. Gray, Inorg. Chem., 2019, 58, 15202 CrossRef CAS PubMed; (b) H. B. Gray and C. J. Ballhausen, J. Am. Chem. Soc., 1963, 85, 260 CrossRef CAS.
- J. Börgel, M. G. Campbell and T. Ritter, J. Chem. Educ., 2016, 93, 118 CrossRef.
- G. Aullón and S. Alvarez, Inorg. Chem., 1996, 35, 3137 CrossRef PubMed.
- (a) I. Ara, J. Forniés, M. A. García-Monforte, B. Menjón, R. M. Sanz-Carrillo, M. Tomás, A. C. Tsipis and C. A. Tsipis, Chem. – Eur. J., 2003, 9, 4094 CrossRef CAS PubMed; (b) J. Forniés, B. Menjón, R. M. Sanz-Carrillo and M. Tomás, Chem. Ber., 1994, 127, 651 CrossRef; (c) R. Usón, J. Forniés, M. Tomás, B. Menjón, R. Bau, K. Sünkel and E. Kuwabara, Organometallics, 1986, 5, 1576 CrossRef.
- (a) R. J. Puddephatt, J. Organomet. Chem., 2017, 849–850, 268 CrossRef CAS; (b) M. Baya, Ú. Belío, I. Fernández, S. Fuertes and A. Martín, Angew. Chem., Int. Ed., 2016, 55, 6978 CrossRef CAS PubMed; (c) J. Forniés and A. Martín, in Metal Clusters in Chemistry, Vol. 1: Molecular Metal Clusters, ed. P. Braunstein, L. A. Oro and P. R. Raithby, Wiley-VCH, 1999, ch. 1.22, pp. 417–443 Search PubMed; (d) R. Usón and J. Forniés, Inorg. Chim. Acta, 1992, 198–200, 165 CrossRef.
- (a) D. W. Shaffer, S. A. Ryken, R. A. Zarkesh and A. F. Heyduk, Inorg. Chem., 2012, 51, 12122 CrossRef CAS PubMed; (b) B. Menjón, S. Martínez-Salvador, M. A. Gómez-Saso, J. Forniés, L. R. Falvello, A. Martín and A. Tsipis, Chem. – Eur. J., 2009, 15, 6371 CrossRef PubMed; (c) L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735 CrossRef CAS PubMed.
- Z. Chval, O. Dvořáčková, D. Chvalová and J. V. Burda, Inorg. Chem., 2019, 58, 3616 CrossRef CAS PubMed.
- R. Sánchez-de-Armas and M. S. G. Ahlquist, Phys. Chem. Chem. Phys., 2015, 17, 812 RSC.
- R. Hoffmann, S. Alvarez, C. Mealli, A. Falceto, T. J. Cahill III, T. Zeng and G. Manca, Chem. Rev., 2016, 116, 8173 CrossRef CAS PubMed.
- H. Bethe, Ann. Phys., 1929, 395, 133 CrossRef.
- (a) B. N. Figgis and M. A. Hitchman, Ligand Field Theory and Its Applications, Wiley–VCH, 2000 Search PubMed; (b) C. J. Ballhausen, Introduction to Ligand Field Theory, McGraw-Hill, 1962 Search PubMed; (c) J. S. Griffith and L. E. Orgel, Q. Rev., Chem. Soc., 1957, 11, 381 RSC.
- M. Baya, D. Joven-Sancho, P. J. Alonso, J. Orduna and B. Menjón, Angew. Chem., Int. Ed., 2019, 58, 9954 CrossRef CAS PubMed.
- (a) W.-L. Li, Y. Li, C.-Q. Xu, X.-B. Wang, E. Vorpagel and J. Li, Inorg. Chem., 2015, 54, 11157 CrossRef CAS PubMed; (b) Y. M. Bosworth and R. J. H. Clark, J. Chem. Soc., Dalton Trans., 1975, 381 RSC; (c) W. R. Mason III and H. B. Gray, Inorg. Chem., 1968, 7, 55 CrossRef.
- P. Schwerdtfeger, O. R. Smits and P. Pyykkö, Nat. Rev. Chem., 2020, 4, 359 CrossRef CAS.
- (a) P. Pyykkö, Annu. Rev. Phys. Chem., 2012, 63, 45 CrossRef PubMed; (b) P. Schwerdtfeger and M. Lein, in Gold Chemistry: Applications and Future Directions in the Life Sciences, ed. F. Mohr, Wiley-VCH, 2009, ch. 4, pp. 183–247 Search PubMed; (c) P. Schwerdtfeger, Heteroat. Chem., 2002, 13, 578 CrossRef CAS; (d) N. Bartlett, Gold Bull., 1998, 31, 22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures (.pdf); atomic coordinates of the optimized structures (.xyz). CCDC 2040138–2040142. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00597a |
| This journal is © The Royal Society of Chemistry 2021 |