
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi, Pd, and Pt complexes of a tetradentate dianionic thiosemicarbazone-based O^N^N^S ligand†
Alexander
Haseloer
 a,
Luca Mareen
Denkler
a,
Rose
Jordan
a,
Max
Reimer
b,
Selina
Olthof
b,
Ines
Schmidt
b,
Klaus
Meerholz
b,
Gerald
Hörner
*c and
Axel
Klein
*a
a,
Luca Mareen
Denkler
a,
Rose
Jordan
a,
Max
Reimer
b,
Selina
Olthof
b,
Ines
Schmidt
b,
Klaus
Meerholz
b,
Gerald
Hörner
*c and
Axel
Klein
*a
aUniversität zu Köln, Department für Chemie, Institut für Anorganische Chemie, Greinstraße 6, D-50939 Köln, Germany. E-mail: axel.klein@uni-koeln.de; Tel: +49-221-470-4006
bUniversität zu Köln, Department für Chemie, Institut für Physikalische Chemie, Greinstrasse 4-6, D-50939 Köln, Germany
cInstitut für Chemie, Anorganische Chemie IV, Universität Bayreuth, Universitätsstraße 30, D-95440 Bayreuth, Germany. E-mail: gerald.hoerner@uni-bayreuth.de
First published on 3rd March 2021
Abstract
New tetradentate phenolate O^N^N^S thiosemicarbazone (TSC) ligands and their Ni(II), Pd(II) and Pt(II) complexes were studied. The diamagnetic and square planar configured orange or red complexes show reversible reductive electrochemistry and in part reversible oxidative electrochemistry at very moderate potentials. DFT calculations show essentially pyridyl-imine centred lowest unoccupied molecular orbitals (LUMO) while the highest occupied molecular orbitals (HOMO) receive contributions from the phenolate moiety, the metal d orbitals and the TSC thiolate atom in keeping with UV-vis spectroelectrochemistry. DFT calculations in conjunction with IR spectra showed details of the molecular structures, the UV-vis absorptions were modelled through TD-DFT calculation with very high accuracy. UPS is fully consistent with UV-vis absorption and TD-DFT calculated data and shows decreasing HOMO–LUMO gaps along the series Pd > Pt > Ni.
Introduction
Thiosemicarbazones (TSC) are very versatile ligands in coordination chemistry with applications in catalysis and medical chemistry.1–6 A lot of research on TSC is devoted to their chemical modification. Thus, the basic N^S chelate binding of TSC can be enlarged to tri-,7–12 tetra-,13–16 and polydentate binding chelators to further expand the coordination chemist's toolbox.17–22 Some of these higher coordination numbers are achieved by bis-thiosemicarbazone ligands. Good examples for the tetradentate coordination of a bis-TSC ligand are the Cu-ATSM (Scheme 1A)23–27 and the bis-TSC Ni(II) complex (B) reported recently as an electrocatalyst for H2 evolution.28 In contrast to these symmetric S^N^N^S-coordinating bis-TSCs, unsymmetric N^N^N^S coordination of Pt(II) was very recently accomplished with a phenanthroline-TSC ligand (C).29 An early example of a tetradentate mono-TSC complex with an O^N^N^S coordination was reported by Mak in 1997.30 Here, Ru(II) is coordinated to a tetradentate TSC ligand derived from 1,5-diacetylpyridine (Scheme 1D). From the 1980s on the tetradentate 1-(2-pyridyl-methylideneamino)-3-(salicyl-ideneamino)thiourea (PST) ligand (E) was used for the spectrometric determination of divalent metal ions like Pd(II), Fe(II), Cd(II), and Hg(II).31,32 This ligand and the related resorcinol derivative (PPRT)33 owe their use to the phenolate function in addition to the TSC thiolate function compensating for the charge of the divalent metal. Five years ago Ni(II) complexes containing salicylidene and aminophenol modified TSC ligands making up a O^N^N^S dianionic coordination (F) were reported.34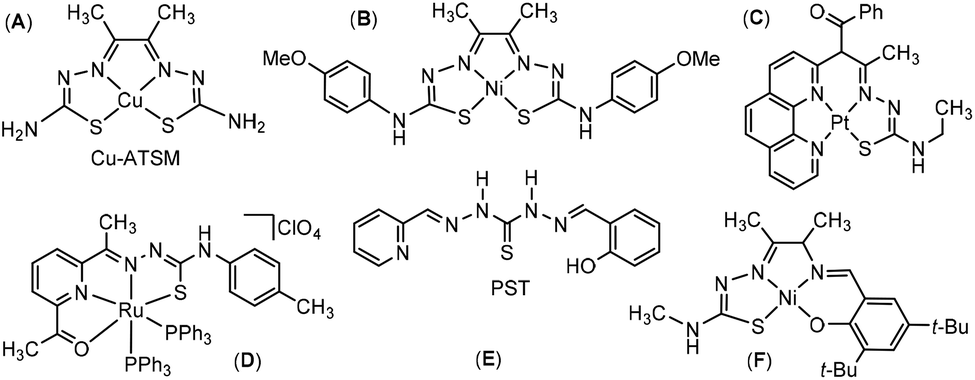 | ||
| Scheme 1 Structure of tetradentate TSC ligands and complexes. Cu-ATSM (A),23–27 a very similar Ni(II) complex (B)28 neutral Pt(II) complex consisting of a phenanthroline backbone (C)29 the [Ru(ONNS)(PPh3)2]+ complex (D),30 the ligand 1-(2-pyridylmethylideneamino)-3-(salicyl-idene-amino)thiourea (PST) (E),31,32 and a Ni(II) complex with an aminophenol modified TSC ligand (F).34 | ||
To our surprise no TSC ligands with a 2-(pyridin-2-yl)phenol side arm allowing for tetradentate O^N^N^S coordination (Scheme 2) were ever reported. Such tetradentate dianionic ligands are well suited to coordinate divalent metals with a planar coordination preference.
 | ||
| Scheme 2 Synthesis of the metal complexes [M(L)] and [M(tBuL)] with the 6-(2-hydroxyphenyl)pyridyl-2-carbaldehyde-TSC ligands. | ||
Herein, we report the synthesis of the first examples of such tetradentate O^N^N^S ligands based on TSC containing the chiral (S)-N4-(α-methylbenzyl) moiety (Scheme 2), H2L and H2tBuL. The chirality of this residue is not of importance for the present study but was relevant for a recent biochemical study using related TSC ligands.35 We also studied the coordination of the group 10 divalent metals Ni(II), Pd(II), and Pt(II) and report their complexes [M(L)] and [M(tBuL)] with structural, spectroscopic and electrochemical characterisation and detailed density functional theory (DFT) calculations.
Results and discussion
Ligand synthesis
2,6-Dibromopyridine was reacted with n-BuLi and DMF giving 6-bromo-2-pyridinecarbaldehyde in 24% yield (Scheme 3, details in the Experimental section). | ||
| Scheme 3 Synthesis of 6-bromo-2-pyridinecarbaldehyde. | ||
Subsequent Suzuki coupling using 2-methoxyphenyl boronic acid gave 6-(2-methoxyphenyl)picolinaldehyde in 88% or quantitative yield (Scheme 4, a), for R = H or t-Bu, respectively. In the same way, we tried 6-(2-hydroxyphenyl)picolinaldehyde but obtained only 5% yield (b). Both were converted into the thiosemicarbazones (TSC) through reaction with (S)-N4-α-methylbenzyl thiosemicarbazide in 84 or 67% yield, respectively (c). The protecting methoxy group was successfully converted to the hydroxy function using BBr3 (d). Alternatively, we also tested a route in which the 2-formyl-6-bromo-pyridine was reacted with the thiosemicarbazide (Scheme 4, bottom, c). However, the final Suzuki reactions (a and b) did not work with this substrate and we received only traces of the hydroxy and methoxy products. The overall yield of the “methoxy route” (upper reaction sequence) was 18 times higher than the direct route (middle reaction sequence) which gave only 2.5% overall yield for the protoligand (ligand prior to deprotonation) H2L.
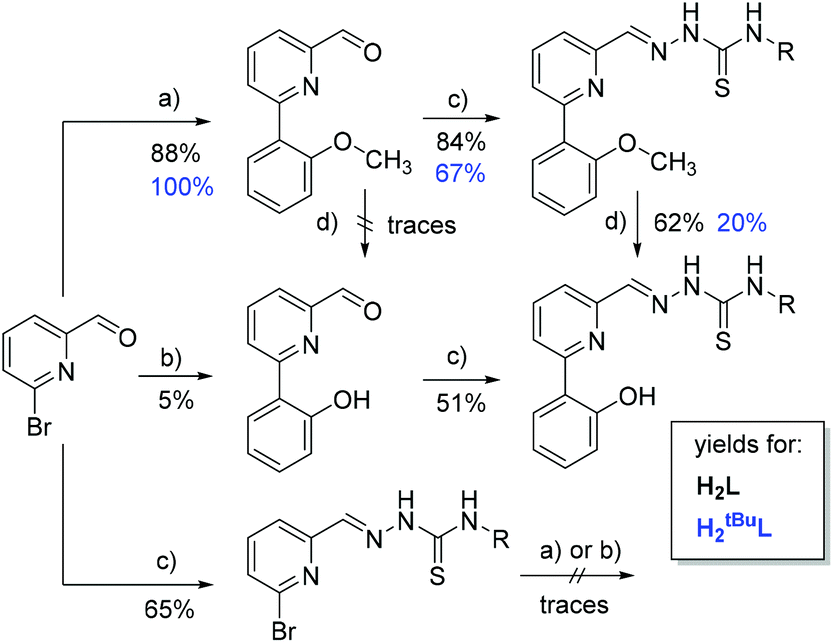 | ||
| Scheme 4 Synthesis routes and yields. Conditions: (a) 2-methoxyphenyl or 2-methoxy(3,5-di-tert-butyl-phenyl) boronic acid, (b) 2-hydroxyphenyl or 2-hydroxy(3,5-di-tert-butyl-phenyl) boronic acid. All with [Pd(PPh3)4], K2CO3 in boiling toluene/EtOH for 20 h; (c) N4-methylbenzyl thiosemicarbazide in MeOH, glacial acetic acid at 60 °C in ultrasonic bath for 2 h; (d) BBr3 in CH2Cl2 at 298 K for 4 h. | ||
For the 3,5-di-tert-butylphenol derivative H2tBuL we followed exclusively the “methoxy route” with an overall yield of 13%. Despite the better yield in the Suzuki coupling (Scheme 4, a), the conversion of OMe to OH (d) turned out to be less favourable for sterically hindered substituents.
Synthesis of the complexes
The complexes were obtained through mixing of equimolar amounts of 6-(2-hydroxyphenyl)-2-formyl-pyridine N4-methyl-benzyl TSC (H2L) dissolved in MeCN with aqueous solutions of NiCl2, K2PdCl4, or K2PtCl4 in the presence of NEt3 as base (Scheme 2). Upon mixing, a rapid colour change to orange (Pd) or red (Ni, Pt) occurred, indicating a successful coordination of the metal ions. All complexes were purified by column chromatography on silica with ethyl acetate as eluent. In keeping with a conserved complex polarity, all products eluted with very similar Rf values. In the same manner, Ni, Pd and Pt complexes of the sterically more demanding ligand 6-(3,5-di-tert-butyl-2-hydroxyphenyl)-2-formyl-pyridine N4-methyl-benzyl TSC (H2tBuL) were obtained as red or orange solids. All attempts failed to receive single crystals of the products of sufficiently high quality for X-ray structure elucidation. However, elemental analyses and MS are in keeping with the overall composition of neutral complexes with fully deprotonated ligands. IR spectra show clearly the C–S stretching frequency in line with a single bond (see more below). Sharp resonances in the 1H NMR spectra in the range of 1.0 < δ < 9.0 ppm, clearly support the diamagnetic d8 electron configuration with square planar coordination. All resonances could be unambiguously assigned to the proton sites of the complexes. (NMR spectra in the electronic ESI, Fig. S4–S6,† data in the Experimental section.)Molecular structures of the complexes
Lacking suitable single crystals for an XRD structure determination, we embarked on modelling the geometric and electronic structures with Density Functional Theory (DFT) methods (Fig. 1, data in Tables S2 and S3, ESI†). | ||
| Fig. 1 Optimised structures and selected bond lengths of the complexes [Ni(tBuL)] (a), [Pd(tBuL)] (b), [Ni(tL)] (c), and [Pt(tBuL)] (d) (further figures in Fig. S22, ESI.† | ||
In order to substantiate and validate the quality of the structure prediction, we included to the study related systems; that is, we re-visited compound F (Scheme 1), which has been reported by Thomas et al.34 and the bis-TSC Ni(II) complex reported by Artero and Orio et al. (B, metrical data in Table S1, ESI†).28
We found excellent agreement of predicted and experimental metrics across the entire reference set (error in bond lengths <2 pm). Taken together with the general close compliance between model and experiment of planar d8 configured complexes,36–39 the predicted metrics of [M(L)] and [M(tBuL)] can be interpreted on reliable grounds. This notion is corroborated by the close match of experimental and computed IR spectra, as is exemplarily shown for [Ni(L)] (Fig. 2; spectra of all [M(L)] and [M(tBuL)] are provided in Fig. S20 and S21, ESI†).
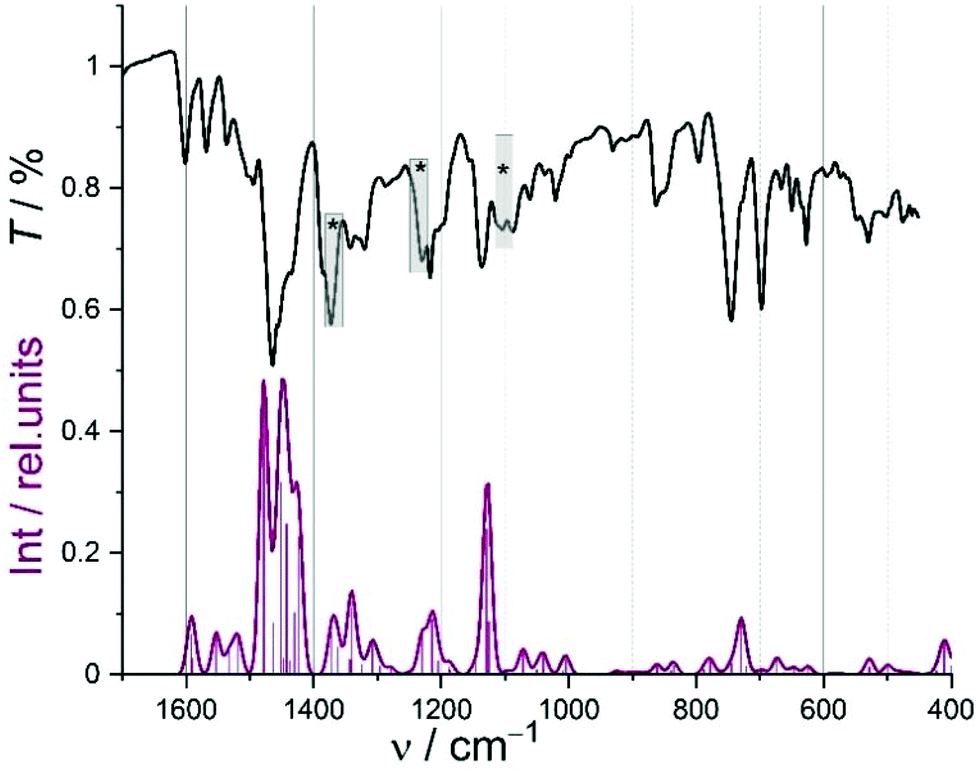 | ||
Fig. 2 Experimental (ATR; grey) and DFT-calculated (red; line: broadened convolution; sticks: modes) IR spectrum of [Ni(L)]; asterisks denote modes due to contamination with ethyl acetate (diagnostic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O mode at 1730 cm−1, not shown). O mode at 1730 cm−1, not shown). | ||
As could be concluded from strictly diamagnetic behaviour of the complexes in the 1H NMR spectra, all complexes exhibit close-to-ideal square planar coordination of the S^N^N^O donor set. The bond length of the coordinated C–S moiety cluster at 1.78 ± 0.01 Å, indicating dominant ene-thiolate character. It is noted that the structural changes along the series do not follow the formal order 3d < 4d < 5d. Instead we consistently find the metrics of the Pt complex rather close to those of the Ni case, suggesting an ordering 3d ≪ 5d < 4d. Similar observations have been previously made for the d8 triade in planar coordination.36,40–42
UV-vis absorption spectroscopy
The electronic structure of parent protoligands and of the metal complexes has been addressed through UV-vis absorption spectroscopy with dilute solutions in CH3CN at ambient temperature. Spectra recorded for the series deriving from H2L are shown in Fig. 3 (for the tBuL series, see Fig. S24, ESI†). The spectra of both protoligands H2L and H2tBuL are dominated by intense absorptions in the near UV regime, peaking around 330 nm (Fig. 3). These transitions are safely assigned to π–π* transitions carrying some CT character. When coordinating the metals, these broad bands are replaced by sharper bands of similar peak intensity. In case of Pd, the band is split into two bands of lower intensity. | ||
| Fig. 3 UV-vis absorption spectra of H2L black, [Ni(L)] dark red, [Pd(L)] orange and [Pt(L)] red, in MeCN. | ||
The intense UV bands peaking at 220 nm and 250 nm show the typical coordination-induced red-shift of ligand-borne π–π* transitions (Table 1). The relative intensity of these two bands clearly responds to metal; i.e., the transitions at ca. 270 nm is dominant for Ni but only features as a low-energy shoulder for Pd and Pt. More diagnostic is the metal-dependent response in the visible region >400 nm which defines the orange or red colour of the complexes. Energies, intensities and the partial recovery of fine-structure point to conserved phenolate-to-(pyridine-imine) charge transfer (ILCT) character. Nevertheless, the maxima and overall structure of these bands are highly depending on the metal. Clearly the energy and the intensity (1200 > ε [M−1 cm−1] > 600) of the leading visible transitions decrease along the series Pt (405 nm) > Ni (413 nm) > Pd (441 nm) (Table 1). This sequence is counter-intuitive on first view as it places the values for Pd not between Ni and Pt. However, this seemingly deviating ordering has been observed before along the d8 triade and is usually explained in terms of energy matching of the metal-borne binding orbitals in the case of Ni and Pt.36,43–46
| λ 1 | λ 2 | λ 3 | λ 4 | |
|---|---|---|---|---|
| a Measured in MeCN. Full data in Table S4, ESI.† | ||||
| H2L | 249 | 274 | 327 | |
| [Ni(L)] | 261 | 337 | 413 | 560 |
| [Pd(L)] | 229 | 316/360 | 441 | 497 |
| [Pt(L)] | 236 | 333 | 405 | 547 |
| H2tBuL | 248 | 279 | 331 | |
| [Ni(tBuL)] | 267 | 339 | 387/420 | 577 |
| [Pd(tBuL)] | 231 | 316/366 | 455 | 518 |
| [Pt(tBuL)] | 239 | 335 | 408 | 567 |
The low-energy tailing of these bands reveals additional weaker transitions in the case of Ni and Pt which extend to λ = 650 nm. Based on the similarity to the recently studied Pt(II) complexes of the pyridinecarbaldehyde (S)-N4-(α-methyl-benzyl)thiosemicarbazone (HTSCmB) ligands,35 these broad bands are preliminarily assigned to metal-to-ligand charge transfer (MLCT) transitions. Results from TD-DFT studies support this assignment.
The optical spectra of [M(L)] and [M(tBuL)] were modelled with TD-DFT methods using the TPSSh hybrid functional. These settings gave excellent agreement with experiment in several previous studies on square-planar d8-based systems.37–39 In keeping with this, the experimental transition energy and intensity could be satisfyingly matched for all complexes in this study (cf., Fig. 4 for Ni; for more spectra see Fig. S25–S29, ESI†). Accordingly, the leading vis bands in the experimental spectrum [Ni(L)] at λ = 413 and 560 nm can be attributed to intense LLCT transitions calculated at λ = 420 and 540 nm (an additional weak band toward the NIR is predicted for all complexes). Difference densities of these transitions indicate substantial CT character with the diimine unit acting as the acceptor. Ni(II) generally features prominently as an electron source in all transitions in the visible range, giving mixed ILCT/MLCT character.
 | ||
| Fig. 4 TD-DFT calculated optical spectrum of [Ni(L)]; line: convoluted spectrum; verticals: transitions. Inset: difference densities of selected transitions. | ||
By contrast, the broad near UV transition envelope from 280–360 nm is identified as a convolute of numerous individual transitions of varying character; nevertheless, the dominating transitions possess sharply decreased metal character and utilise donor orbitals located rather on the TSC moiety, rendering the transition(s) ILCT like. Similar conclusions and assignments hold for all complexes. However, the metal contribution to the leading transitions decreases along the series Pt > Ni > Pd. The complete spectral material is assembled in the ESI (Fig. S25–S29; Table S5, ESI†).
Ultraviolet photo electron spectroscopy (UPS)
UPS measurements of thin films of three complexes [M(L)] with M = Pt, Pd or Ni (Fig. 5) were used to determine the work function Wf and the ionisation energy IE. Alluding to Koopmans theorem the latter represents the binding energy of electrons in the highest occupied molecular orbital (HOMO). | ||
| Fig. 5 Ultraviolet photoelectron spectra (UPS) of [M(L)] with M = Pt, Pd, Ni showing the high binding energy cut-off (HBEC, left) and the HOMO region (right). | ||
The work function Wf was calculated from the difference of the energy of the excitation source hν and the difference of the high binding energy cut-off (HBEC) and the Fermi level EF (eqn (1)).
| Wf = hν − (HBEC − EF) | (1) |
The ionisation energy is calculated by adding the Wf and the difference of the HOMO onset and the Fermi level EF which is positioned at 0 eV.
| IE = Wf + (HOMO onset − EF) | (2) |
The values for the HBEC and the HOMO onset are determined by using a linear fit of the cut off and the background and of the HOMO onset and the background, respectively.
The ionisation energy IE decreased in the series Pd > Ni > Pt (Table 2). This notion is fully corroborated by DFT-based analysis of the electronic structure. The optical HOMO–LUMO gaps derived from the above absorption spectra (Fig. 3) are calculated as 2.494 (Pd), 2.266 (Pt), and 2.214 (Ni) eV, respectively.
| [Ni(L)] | [Pd(L)] | [Pt(L)] | |
|---|---|---|---|
| Work function Wf/eV | 3.99 | 3.84 | 3.94 |
| HOMO onset vs. EF/eV | 1.67 | 1.92 | 1.67 |
| Ionisation energy IE/eV | 5.66 | 5.76 | 5.61 |
Oxidation and reduction – DFT computation and cyclic voltammetry
The occupied frontier orbitals (blue bars in Fig. 6 for [M(L)] are qualitatively quite similar, irrespective of the metal centre. HOMO and HOMO−1 both are largely ligand-like, whereas non-bonding d-like metal orbitals feature ca. 1 eV below the HOMO in all cases (HOMO-2 for Ni and HOMO-3 for the others).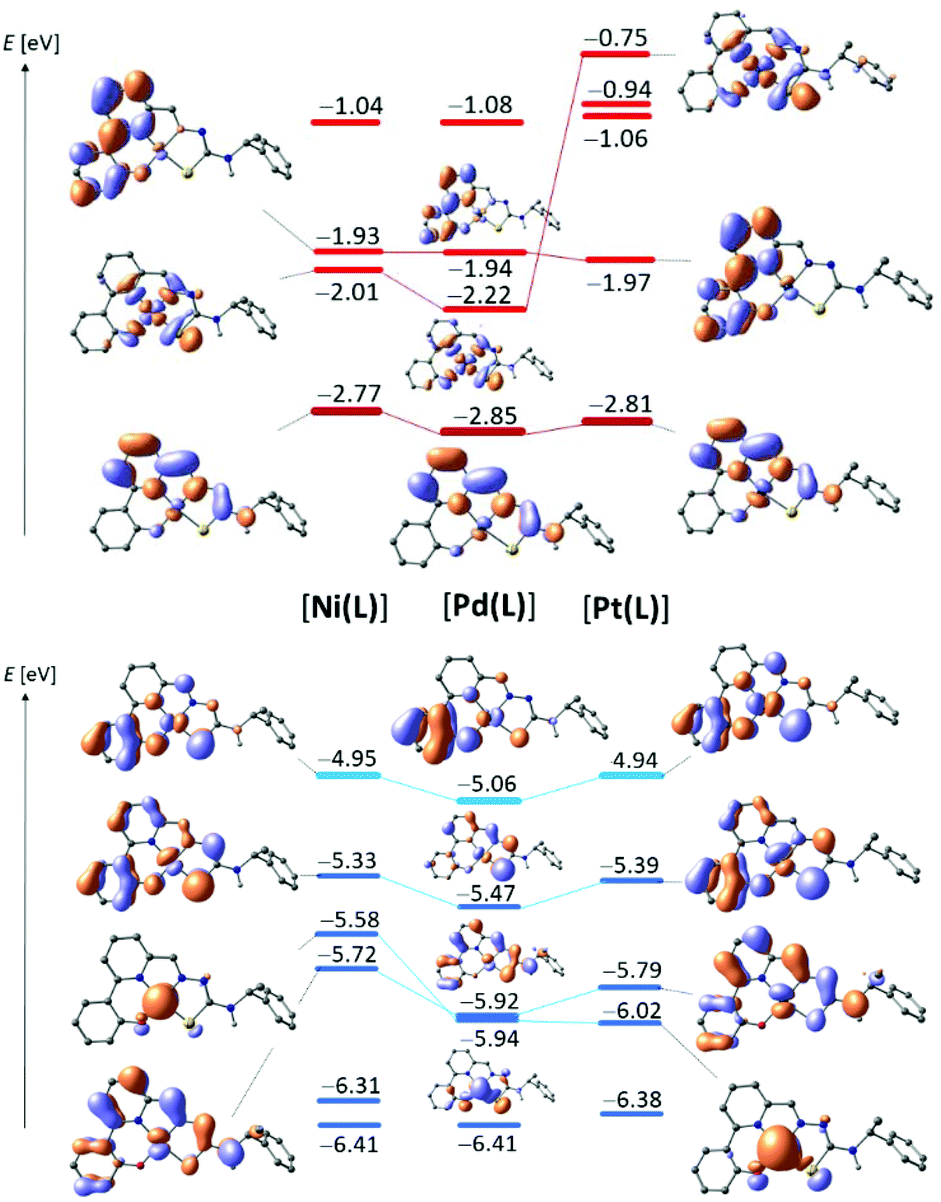 | ||
| Fig. 6 DFT calculated energies of virtual frontier MOs (top) and occupied orbitals (bottom) for [Ni(L)] (left), [Pd(L)] (centre) and [Pt(L)] (right). | ||
The HOMO is largely localised on the phenolate moiety in all cases, but subtle differences point out the influence of the metal on the energy and the nature of the HOMO. Firstly, in the Pt and Ni complexes significant contributions to the HOMO arise from the S atom of the TSC moiety and from the metal itself; all of these metal-borne components are π-antibonding in nature. Whereas such contributions persist in the Pd compounds, they are substantially smaller. In keeping with the diminished metal-orbital contribution of Pd, the HOMO of [Pd(L)] is stabilised by ca. 100 mV with respect to both, [Pt(L)] and [Ni(L)].
Thus, DFT nicely captures the discontinuous order which became evident also experimentally from the UV-vis absorption spectra and the UPS ionisation energies. Interestingly, analysis of the series [M(tBuL)] echoes this ordering but indicates a much closer range of HOMO energies. In keeping with the clustered HOMO energies of [M(tBuL)], electrochemical oxidation reveals only minor differences in the redox potentials (Table 3). Due to irreversible electrochemical oxidation of [M(L)] the computed HOMO energies cannot be correlated to the CV pattern in quantitative terms. It is nevertheless noted that the anodic peak potentials indeed follow the trends suggested by DFT.
| E 1/2-Red2 | E 1/2-Red1 | E 1/2-Ox1 | E pa-Ox2 | |
|---|---|---|---|---|
| a From cyclic voltammetry, electrochemical potentials in V (uncertainties ∼1–3 mV), half-wave potentials E1/2 for reversible and partially reversible redox waves, anodic (Epa) or cathodic (Epc) peak potentials for irreversible (irr) waves; measured in 0.1 M n-Bu4NPF6/MeCN at 298 K, scan rate 100 mV s−1. Full data in Table S6, ESI.† | ||||
| H2L | −2.36 irr | −1.94 irr | 0.94 irr | — |
| [Ni(L)] | −2.37 | −1.70 | 0.66 irr | — |
| [Pd(L)] | −2.34 | −1.66 | 0.72 irr | — |
| [Pt(L)] | −2.36 | −1.65 | 0.69 irr | — |
| H2tBuL | −2.40 irr | −2.02 irr | 0.59 | 1.14 irr |
| [Ni(tBuL)] | −2.42 | −1.75 | 0.56 | 0.99 irr |
| [Pd(tBuL)] | −2.35 | −1.70 | 0.55 | >1.3 |
| [Pt(tBuL)] | −2.36 | −1.69 | 0.55 | 0.96 irr |
In their cathodic voltammetric scans, the six complexes show two one-electron reduction waves and one or two one-electron oxidation waves in the range +1.3 to −2.7 V (vs. ferrocene/ferrocenium) (Table 3 and Fig. 7, further material, Fig. S31–S33 and Table S6, ESI.† The introduction of the tert-butyl substituents resulted in slightly more negative reduction potentials in keeping with the electron-donating character of these groups.47,48 It is noted that the Ni(II) compounds proved to be hardest to reduce, in agreement with the computed LUMO energies. With respect to the protoligands, reduction potentials of the complexes are shifted anodically by 0.4 to 0.6 V pointing to a substantial stabilisation of the LUMOs upon coordination. In fact, the cyclic voltammograms of the protoligands H2L and H2tBuL showed two reduction waves in the range from −1.9 to −2.4 V that were irreversible (Fig. 7). For the complexes, the first reduction wave gains markedly in reversibility and both were shifted anodically by about 0.25 V. From the high similarity of the reduction behaviour of the protoligands and their complexes we conclude, that all reduction processes occurred essentially at the pyridine-imine moiety of the TSC. This conclusion is in line with the behaviour of the recently studied pyridinecarbaldehyde (S)-N4-(α-methylbenzyl)-thiosemicarbazones (HTSCmB) and their Pt(II) complexes.35 It is corroborated also by DFT computation. On the TPSSh/TZVP level of theory, the series [M(L)] gives indeed similar virtual orbital pattern (red in Fig. 6). In particular, the LUMO energies vary by only 80 mV. The shape of the LUMO which is essentially localised on the diimine moiety in all cases and the conserved LUMO energy is in line with the similarity that prevailed in the UV-vis absorption spectra. Thus, it also matches the close similarity in the electrochemical reduction potentials.
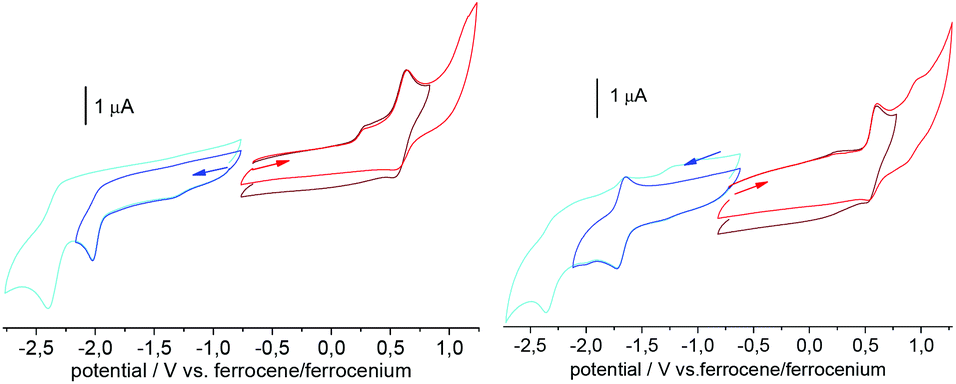 | ||
| Fig. 7 Cyclic voltammogramms of H2tBuL and [Pt(tBuL)] in 0.1 M n-Bu4NPF6 MeCN solution. | ||
For H2L one irreversible oxidation was observed at 0.94 V. For H2tBuL this wave became reversible, shifted to 0.59 V, and a second oxidation wave appeared. This is clearly due to the chemical stabilisation of the oxidised ligand through introduction of the tert-butyl groups at the 3,5-phenolate positions.47,48
The oxidation potentials recorded in the complex series [M(L)] are shifted cathodically by about 0.3 V when compared with the protoligand. For the series [M(tBuL)] this shift is very small (∼0.03 V). For the Ni and Pt complexes [M(tBuL)] the second oxidation is slightly shifted to lower potentials when compared with the protoligand H2tBuL. By contrast, Pd does not give a second oxidation wave in the covered potential window and an oxidation potential of >1.15 V must be assumed. With a view to the HOMOs depicted in Fig. 6, we associate the first oxidation mainly with a phenolate/phenoxy (PhO−/PhO˙) redox couple. For M = Pd, the oxidation site is particularly well localised on the phenolate with only little contributions from the metal and other donor sites. Inspection of the HOMO−1 suggests the second oxidation in the series derived from H2tBuL to be another phenolate-based event with significant metal contributions. Evidence for a metal contribution comes from the observation that the second oxidation wave shifted markedly in potential for the Pt and Ni complexes compared with the ligand H2tBuL, but nor for Pd. As these second oxidation events are irreversible for all compounds, however, we refrain from deep-going interpretation.
Spectroelectrochemical UV-vis absorption spectroscopy
The spectroscopic signature of reduced and oxidised states of the parent complexes has been studied through UV-vis absorption spectroscopy during reductive and oxidative electrolysis in dilute MeCN solution (spectroelectrochemistry). As is exemplarily shown for [Pt(tBuL)] (Fig. 8), the intense near UV pattern at 320 nm is completely bleached upon oxidation. The oxidised species, presumably the cation [Pt(tBuL)]+, gives no new diagnostic bands in the vis region but rather echoes the pattern of the parent neutral complex, though slightly shifted and with tailing to the NIR. Bands ranging between 300 and 500 nm, akin to those in Fig. 8 (and Fig. S34–S41, ESI†) were previously assigned to the oxidation of the phenolate group forming a coordinated phenoxy radical.34,47–52 In keeping with this, the spin density computed for doublet [Pt(tBuL)]+ (inset in Fig. 8) is largely localised on the phenolate/phenoxy moiety. A similar pattern prevails for the other complexes [M(tBuL)]+. While the assignment of the oxidised species as planar mono-cation is probably justified for Pd and Pt, reconstruction of the coordination sphere upon oxidation must be expected for Ni. Expansion of the coordination sphere of the oxidised complex through axial ligation has been previously found to switch the nature from ligand-radical like to formal Ni(III). Indeed, computation of [Ni(tBuL)(CH3CN)2]+ indicates localised metal oxidation. The second, irreversible oxidation of H2tBuL and its complexes leads to a general bleaching of the entire visible and near UV spectral range.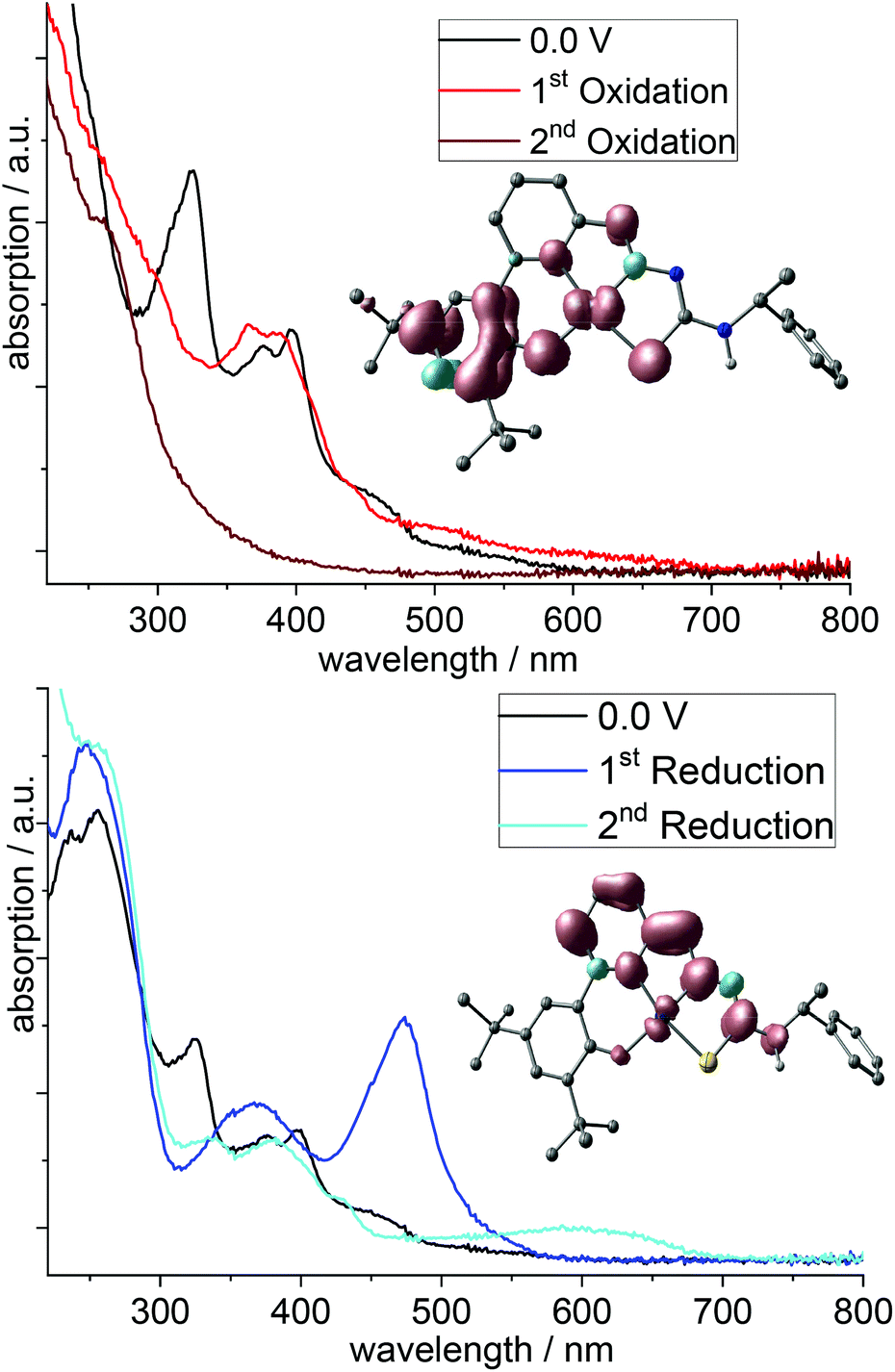 | ||
| Fig. 8 UV-vis absorption spectra recorded during electrolysis of [Pt(tBuL)] in 0.1 M n-Bu4NPF6 MeCN solution; top: oxidation; bottom: reductions. | ||
The first reduction of the protoligands and the complexes leads to pronounced absorption bands around 500 nm. They are largely insensitive to the presence of a metal and were thus assigned to π*–π* transitions in the TSC pyridine-imine moiety. In much the same way, we observed them in the complexes [Pt(R-TSCmB)Cl].35 Computed spin densities of mono-anionic [Pt(tBuL)]− (inset in Fig. 8, bottom) fully support this interpretation.
The second reduction leads to a complete bleach of the intense visible feature, which is replaced by a blue-shifted much weaker band (Fig. 8). We assign also these bands to π*–π* transitions in the TSC pyridine-imine moiety. The long-wavelength feature for the complexes compared with the protoligands is probably due to the planarisation through coordination.
Conclusions
We herein present the synthesis of square planar Ni(II), Pd(II) and Pt(II) complexes [M(L)] and [M(tBuL)] of dianionic tetradentate O^N^N^O coordinating 6-(2-phenolato)picolinaldehyde thiosemicarbazone ligands. The ligands differ in their substitution pattern of the 3,5-R-phenolate group with R = H or t-Bu. The complexes are all air stable and can be purified by column chromatography. Lacking of single crystals we were able to study the molecular structures by a combination of DFT calculations and IR and NMR spectroscopy. With the aid of TD-DFT the weak long-wavelength absorptions between 500 and 600 nm in the UV-vis spectra were assigned to metal-to-ligand(pyridine-imine) charge transfer (MLCT) transition, while the strong absorptions in the 300–500 nm range, which are responsible for the orange (Pd) and red colour (Ni, Pt) of the materials, are traced to metal-perturbed intraligand(phenolate)-to-ligand(pyridine-imine) charge transfer (ILCT) transition. For both ILCT and MLCT band energies the Ni and the Pt complexes are far more similar and differ largely from the Pd complexes. The series of the MLCT energies is clearly Pd ≫ Pt > Ni. Akin to previous findings, we ascribe this to better orbital overlap for Ni and Pt. The work function Wf from UV photoelectron spectroscopy (UPS) sets the series Pd ≪ Pt < Ni in complete agreement with this idea. From the three possible redox centres of the complexes, the divalent metal M(II), the phenolate group and the pyridine-imine only the latter two were addressed in the oxidation and reduction processes observed in the range from +1.3 to −2.7 V. The introduction of the tert-butyl substituents in [M(tBuL)] led to several effects. As expected, the first oxidation gets reversible and the potentials are lowered, thus allowing also the observation of a second oxidation in the same potential range. In turn, tert-butyl substitution of the ligand led also to completely metal indifferent oxidation potentials for these complexes, while the unsubstituted complexes [M(L)] show a metal-dependence of the oxidation potentials Ni < Pt < Pd in keeping with the sequence from the MLCT energies and the UPS. Thus, the tert-butyl substitution lead to a shift of the character of the highest occupied molecular orbitals from having a small (L) to almost no (tBuL) metal character. The assignment of the loci for oxidation and reduction was further substantiated by UV-vis spectroelectrochemistry and the calculation of the frontier orbitals of the assumed radical cations [M(L)]+ or [M(tBuL)]+ and anions [M(L)]− or [M(tBuL)]−.In future work we will explore potential applications of these complexes. As a first idea we can state that the electrochemical data of the three [M(tBuL)] complexes looks promising in view of Negishi type of C–C cross coupling catalysis.53 The potentials are quite similar and the variation of the metals Ni–Pd–Pt is very interesting concerning differences in their specific reactivity towards C–C coupling.
Experimental section
Materials
All solvents and substances were purchased from commercial sources and used without further purification if not otherwise stated. N4-methylbenzyl thiosemicarbazide was prepared as previously reported.35Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 5:1). The product was isolated as a pale yellow solid. Yield: 0.94 g (186.01 g mol−1, 5.05 mmol, 24%). Rf: 0.56 (c-hex:EtOAc, v:v = 5:1). 1H NMR (300 MHz, CDCl3) δ = 10.01 (s, 1H), 7.93 (dt, 1H, J = 6.3, 1.8 Hz), 7.80–7.70 (m, 2H) ppm.
:v = 5:1). The product was isolated as a pale yellow solid. Yield: 0.94 g (186.01 g mol−1, 5.05 mmol, 24%). Rf: 0.56 (c-hex:EtOAc, v:v = 5:1). 1H NMR (300 MHz, CDCl3) δ = 10.01 (s, 1H), 7.93 (dt, 1H, J = 6.3, 1.8 Hz), 7.80–7.70 (m, 2H) ppm.
Syntheses of 6-phenyl substituted picolinaldehydes – general description
1 eq. 6-bromo-picolinaldehyde, 2 eq. K2CO3, 0.02 eq. [Pd(PPh3)4] and 1.1 eq. of the corresponding boronic acid were dissolved in 15 mL abs. toluene and 10 mL EtOH and heated to 90 °C for 20 h. The mixture was cooled to ambient temperature before 30 mL of H2O were added and the mixture was stirred for additional 30 min. The aqueous layer was extracted with CH2Cl2. The combined organic phases were dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (silica, EtOAc). The products were isolated as yellow oils.:EtOAc, v:v = 3:1). 1H NMR (300 MHz, CDCl3): δ = 10.19 (s, 1H), 8.03 (dd, 1H, J = 7.0, 2.0 Hz), 7.94–7.86 (m, 2H), 7.55 (d, 1H, J = 2.5 Hz), 7.44 (d, 1H, J = 2.5 Hz), 3.32 (s, 3H), 1.45 (s, 9H), 1.36 (s, 9H) ppm. 13C NMR (75 MHz, CDCl3): δ = 194.1, 159.1, 155.4, 153.1, 137.2, 132.3, 130.7, 129.3, 126.6, 125.6, 119.7, 61.7, 35.5, 31.7, 31.3 ppm.
:EtOAc, v:v = 2:1). 1H NMR (300 MHz, CDCl3): δ = 10.17 (s, 1H), 8.14–8.05 (m, 2H), 7.99–7.88 (m, 3H), 7.55–7.40 (m, 3H) ppm. 13C NMR (75 MHz, CDCl3): δ = 194.1, 158.1, 152.9, 138.3, 137.9, 129.8, 129.1, 127.1, 124.6, 119.9 ppm.
Synthesis of the thiosemicarbazones – general procedure
1 eq. of the corresponding aldehyde (about 2 mmol) were dissolved in 3 mL MeOH. To that solution 1 eq. of N4-methylbenzyl thiosemicarbazide and 0.1 mL HOAc were added. The mixture was sonicated at 60 °C for 2 h and then cooled to ambient temperature. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (silica; c-hex:EtOAc v:v = 2:1). The products were isolated as yellow solids.
:EtOAc, v:v = 2:1). 1H NMR (300 MHz, CDCl3): δ = 8.09–7.90 (m, 1H), 7.88–7.72 (m, 3H), 7.48–7.23 (m, 6H), 7.13–6.96 (m, 2H), 5.77 (dt, 1H, J = 8.5, 6.5 Hz), 4.12 (q, 1H, J = 7.1 Hz), 3.86 (d, 3H, J = 9.9 Hz), 1.66 (dd, 3H, J = 10.7, 7.0 Hz). 13C NMR (75 MHz, CDCl3): δ = 143.3, 137.1, 136.1, 133.0, 131.6, 131.1, 131.0, 130.3, 128.8, 128.6, 128.6, 127.5, 127.3, 126.4, 125.9, 125.5, 123.2, 121.6, 118.4, 60.4, 26.9. EI-MS [m/z] = 390 [M]+.
:EtOAc, v:v = 5:1). 1H NMR (499 MHz, CDCl3): δ = 9.41 (s, 1H), 7.99 (s, 1H), 7.79 (d, 1H, J = 8.6 Hz), 7.78–7.75 (m, 3H), 7.47–7.37 (m, 6H), 7.30 (t, 1H, J = 7.3 Hz), 5.76 (quint, 1H, J = 6.9 Hz), 3.31 (s, 3H), 1.69 (s, 3H), 1.43 (s, 9H), 1.34 (s, 9H) ppm.
:EtOAc, v:v = 2:1). 1H NMR (300 MHz, CDCl3): δ = 13.59 (s, 1H), 9.25 (s, 1H), 7.78 (d, 1H, J = 1.0 Hz), 7.72–7.64 (m, 1H), 7.58 (t, 1H, J = 7.8 Hz), 7.52–7.46 (m, 1H), 7.45–7.35 (m, 4H), 7.34–7.27 (m, 1H), 5.83–5.64 (quint, 1H, J = 7.2 Hz), 1.68 (d, 3H, J = 6.9 Hz) ppm.
:EtOAc, v:v = 2:1). 1H NMR (300 MHz, CDCl3): δ = 14.88 (s, 1H), 9.05 (s, 1H), 8.01 (d, 2H, J = 8.1 Hz), 7.93 (d, 1H, J = 1.0 Hz), 7.83–7.68 (m, 4H), 7.56–7.31 (m, 7H), 5.77 (p, 1H, J = 7.6, 7.2, 6.8 Hz), 1.70 (d, 3H, J = 7.0 Hz) ppm.
:EtOAc, v:v = 2:1). 1H NMR (300 MHz, CDCl3): δ = 10.42 (s, 1H), 8.03 (d, 1H, J = 8.5 Hz), 7.90–7.76 (m, 4H), 7.56–7.48 (m, 2H), 7.42–7.21 (m, 5H), 7.03 (dd, 1H, J = 8.3, 1.3 Hz), 6.91 (ddd, 1H, J = 8.3, 7.2, 1.3 Hz), 5.83–5.61 (m, 1H), 1.73 (d, 3H, J = 6.9 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ = 176.64, 160.63, 157.96, 148.11, 142.67, 138.41, 137.99, 131.97, 128.73, 127.47, 126.54, 126.26, 120.63, 119.34, 118.88, 118.70, 118.17, 53.98, 22.28 ppm. EI-MS [m/z] = 376 [M]+.
:EtOAc, v:v = 1:1) yielding a yellow solid. Yield: 201 mg (C21H20N4OS, 376.48 g mol−1, 0.53 mmol, 62%). Rf = 0.29 (c-hex:EtOAc, v:v = 2:1). Anal. calcd (%): C, 67.00; H, 5.35; N, 14.88. Found: C, 67.04; H, 5.38; N, 14.91. 1H NMR (300 MHz, CDCl3): δ = 10.42 (s, 1H), 8.03 (d, 1H, J = 8.5 Hz), 7.90–7.76 (m, 4H), 7.56–7.48 (m, 2H), 7.42–7.21 (m, 5H), 7.03 (dd, 1H, J = 8.3, 1.3 Hz), 6.91 (ddd, 1H, J = 8.3, 7.2, 1.3 Hz), 5.83–5.61 (m, 1H), 1.73 (d, 3H, J = 6.9 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ = 176.64, 160.63, 157.96, 148.11, 142.67, 138.41, 137.99, 131.97, 128.73, 127.47, 126.54, 126.26, 120.63, 119.34, 118.88, 118.70, 118.17, 53.98, 22.28 ppm. EI-MS [m/z] = 376 [M]+.
:EtOAc v:v = 5:1). The ligand was isolated as a yellow solid. Yield: 40 mg (C29H36N4OS, 488.69 g mol−1, 0.082 mmol, 20%). Rf: 0.18 (c-hex:EtOAc, v:v = 5:1). Anal. calcd (%): C, 71.28; H, 7.43; N, 11.46. Found: C, 71.22; H, 7.41; N, 11.44. 1H NMR (499 MHz, CDCl3): δ = 14.69 (s, 1H), 9.59 (s, 1H), 7.88 (d, 1H, J = 8.4 Hz), 7.86 (m, 1H), 7.80–7.77 (m, 2H), 7.61 (d, 1H, J = 2.1 Hz), 7.39–7.35 (m, 4H), 7.29–7.26 (m, 2H), 7.20–7.17 (m, 1H), 5.69 (m, 1H), 1.65 (d, 3H, J = 6.9 Hz), 1.41 (s, 9H), 1.29 (s, 9H) ppm. 13C NMR (75 MHz, CDCl3): δ = 158.42, 156.83, 138.80, 138.16, 128.68, 127.41, 126.87, 126.33, 120.83, 120.37, 120.21, 53.73, 31.58, 29.61, 22.33 ppm. HR-ESI-MS [m/z] = 489.26828 ([M + H]+, calc. 489.2682590), 511.25050 ([M + Na]+, calc. 511.2502037).
Synthesis of the metal complexes – general procedure
5.0 mL of a stock solution of H2L or H2tBuL (c = 0.01 M) in MeCN was mixed first with Et3N (5 mmol, 1.0 eq.) before aqueous solutions of NiCl2, K2PdCl4 or K2PtCl4 were added. After stirring for 12 h at ambient temperatures, the solvent was removed, and the crude products were purified by column chromatography (silica, EtOAc for [M(L)], c-hex:EtOAc, v:v = 3:1 for [M(tBuL)]).
[Ni(L)]. From 2.0 mL of an aqueous NiCl2 (c = 0.01 M) solution. The product was isolated as red solid. Yield: 18.6 mg (C21H18N4ONiS, 433.16 g mol−1, 0.43 mmol, 86%). Rf = 0.8 (EtOAc). Anal. calcd (%): C, 58.23; H, 4.19; N, 12.93. Found: C, 58.31; H, 4.10; N, 12.95.1H NMR (300 MHz, CDCl3): δ = 8.07 (d, 1H, J = 8.5 Hz,). 7.81–7.92 (m, 2H), 7.31–7.40 (m, 4H), 7.17–7.31 (m, 3H), 7.15 (m, 1H), 7.02 (dd, 1H, J = 8.5, 1.0 Hz), 6.60–6.75 (m, 1H), 5.04 (m, 1H), 1.56 (d, 3H, J = 6.8 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ = 161.90, 151.51, 142.77, 137.13, 137.29, 131.50, 128.84, 127.63, 127.04, 126.25, 120.76, 118.90, 117.84, 115.62, 55.09, 29.84, 22.63 ppm. HR-ESI-MS(+) [m/z] = 433.06305 ([M + H]+, calc. 433.06276), 455.04496 ([M + Na]+, calc. 455.04470).
[Pd(L)]. From 2.0 mL of an aqueous K2PdCl4 (c = 0.01 M) solution. The product was isolated as orange solid. Yield: 21.6 mg (C21H18N4OPdS, 480.88 g mol−1, 0.45 mmol, 90%). Rf = 0.8 (EtOAc). Anal. calcd (%): C, 52.45; H, 3.77; N, 11.65. Found: C, 52.44; H, 3.81; N, 11.65. 1H NMR (300 MHz, CDCl3): δ = 8.05 (d, 1H, J = 8.6 Hz). 7.72–7.96 (m, 2H), 7.33–7.41 (m, 4H), 7.22–7.28 (m, 2H), 7.28–7.33 (m, 2H), 7.18 (dd, 1H, J = 8.6, 1.5, 1.0 Hz), 6.69 (ddd, 1H, J = 8.2, 6.5, 1.5 Hz), 5.07 (m, 1H), 1.56 (d, 3H, J = 6.8 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ = 163.25, 150.91, 143.12, 137.60, 137.80, 132.23, 128.90, 128.66, 127.74, 126.32, 124.30, 120.51, 119.13, 115.65, 53.93, 21.34 ppm. EI-MS(+) [m/z] = 480 [M]+, 465 [M − CH3]+.
[Pt(L)]. From 5.0 mL of an aqueous K2PtCl4 (c = 0.009 M) solution. The product was isolated as a red solid. Yield: 26.3 mg (C21H18N4OPtS 569.55 g mol−1, 0.46 mmol, 92%). Rf = 0.8 (EtOAc). Anal. calcd (%): C, 44.29; H, 3.19; N, 9.84. Found: C, 44.24; H, 3.20; N, 9.86. 1H NMR (300 MHz, CDCl3): δ = 8.16 (d, 1H, J = 8.6 Hz), 7.99 (d, 1H, J = 8.1 Hz), 7.91 (t, 1H, J = 8.0 Hz), 7.63 (s, 1H), 7.33–7.41 (m, 4H), 7.25–7.34 (m, 3H), 7.20 (d, 1H, J = 7.4 Hz), 5.19 (m, 1H), 6.67–6.85 (m, 1H), 1.58 (d, 3H, J = 6.9 Hz). ppm. 13C NMR (75 MHz, CDCl3): δ = 161.62, 147.25, 143.30, 136.32, 131.58, 128.88, 128.02, 127.70, 126.32, 124.49, 122.51, 121.76, 119.08, 116.37, 58.81, 22.82 ppm. EI-MS(+) [m/z] = 569 [M]+.
[Ni(tBuL)]. From 2.0 mL of an aqueous NiCl2·6H2O (c = 0.01 M) solution. The product was isolated as dark red solid. Yield: 11.0 mg (C29H34N4ONiS, 545.37 g mol−1, 0.020 mmol, 100%). Rf = 0.86 (EtOAc). Anal. calcd (%): C, 63.87; H, 6.28; N, 10.27. Found: C, 63.84; H, 6.19; N, 10.24. 1H NMR (499 MHz, CDCl3): δ = 8.17 (d, 1H, J = 8.6 Hz), 7.89 (dd, 1H, J = 8.5, 7.5 Hz), 7.75 (d, 1H, J = 2.2 Hz), 7.43–7.32 (m, 5H), 7.31–7.26 (m, 1H), 7.19 (d, 1H, J = 7.2 Hz), 5.09 (m, 1H), 1.56 (d, 3H, J = 5.6 Hz), 1.34 (s, 9H), 1.30 (s, 9H). 1H NMR (300 MHz, DMSO-d6): δ = 8.52 (d, 1H, J = 7.3 Hz), 8.46 (d, 1H, J = 8.8 Hz), 8.14 (dd, 1H, J = 8.6, 7.4 Hz), 7.86 (d, 1H, J = 2.3 Hz), 7.75 (s, 1H), 7.41–7.29 (m, 5H), 7.26–7.25 (m, 1H), 7.23 (d, 1H, J = 2.4 Hz), 4.96 (m, 1H), 1.41 (d, 3H, J = 6.2 Hz), 1.30 (s, 9H), 1.21 (s, 9H) ppm. 13C NMR (75 MHz, CDCl3): δ = 152.51, 136.58, 136.55, 128.83, 127.61, 126.84, 126.20, 121.60, 120.80, 118.17, 117.11, 49.33, 31.68, 29.69, 22.50 ppm. EI-MS(+) [m/z] = 544 [M]+, 529 [M − CH3]+.
[Pd(tBuL)]. From 2.0 mL of an aqueous K2PdCl4 (c = 0.01 M) solution. The product was isolated as red solid. Yield: 10.0 mg (C29H34N4OPdS, 593.10 g mol−1, 0.017 mmol, 84%). Rf = 0.87 (EtOAc). Anal. calcd (%): C, 58.73; H, 5.78; N, 9.45. Found: C, 58.74; H, 5.77; N, 9.41. 1H NMR (499 MHz, CDCl3): δ = 8.13 (d, 1H, J = 8.7 Hz), 7.87 (dd, 1H, J = 8.7, 7.4 Hz), 7.71 (d, 1H, J = 2.5 Hz), 7.44 (d, 1H, J = 2.5 Hz), 7.39–7.31 (m, 4H), 7.30–7.27 (m, 1H), 7.25 (d, 1H, J = 7.5 Hz), 5.14 (m, 1H), 1.57 (d, 3H, J = 6.8 Hz), 1.40 (s, 9H), 1.35 (s, 9H) ppm. 1H NMR (300 MHz, DMSO-d6): δ = 8.62 (d, 1H, J = 8.0 Hz), 8.44 (d, 1H, J = 8.7 Hz), 8.17 (t, 1H, J = 8.0 Hz), 7.92 (s, 1H), 7.82 (d, 1H, J = 2.3 Hz), 7.69 (d, 1H, J = 7.5 Hz), 7.44–7.33 (m, 3H), 7.32 (d, 1H, J = 2.3 Hz), 7.25 (m, 1H), 5.03 (m, 1H), 1.45 (d, 3H, J = 6.3 Hz), 1.31 (s, 9H), 1.24 (s, 9H) ppm. 13C NMR (75 MHz, CDCl3): δ = 150.09, 136.98, 128.71, 127.40, 126.50, 122.50, 114.47, 47.03, 31.52, 29.69, 22.41 ppm. EI-MS(+) [m/z] = 592 [M]+, 577 [M − CH3]+.
[Pt(tBuL)]. From 5.0 mL of an aqueous K2PtCl4 (c = 0.009 M) solution. The product was isolated as a red solid. Yield: 6.7 mg (C29H34N4OPtS, 681.76 g mol−1, 0.0098 mmol, 49%). Rf = 0.92 (EtOAc). Anal. calcd (%): C, 51.09; H, 5.03; N, 8.22. Found: C, 51.11; H, 5.09; N, 8.22. 1H NMR (300 MHz, CDCl3): δ = 8.33 (d, 1H, J = 8.6 Hz, JPt,H = 21.0 Hz), 8.01 (td, 1H, J = 7.5, 1.0 Hz), 7.92 (d, 1H, J = 2.4 Hz), 7.84 (s, 1H), 7.52 (d, 1H, J = 2.4 Hz), 7.43–7.32 (m, 4H), 7–32–7.27 (m, 2H), 5.28 (m, 1H), 1.60 (d, 3H, J = 6.8 Hz), 1.44 (s, 9H), 1.37 (s, 9H) ppm. 13C NMR (75 MHz, CDCl3): δ = 157.03, 136.01, 128.98, 127.67, 126.03, 126.24, 119.81, 51.82, 31.71, 29.77, 18.22 ppm. EI-MS(+) [m/z] = 681 [M]+, 666 [M − CH3]+.
Instrumentation
NMR spectra were recorded on a Bruker Avance II 300 MHz spectrometer, using a triple resonance 1H, nBB inverse probe head or a Bruker Avance III spectrometer at 499 MHz with a TCI prodigy 5 mm probe head with z-gradient (1H/19F 13C 15N 2H). The unambiguous assignment of the 1H and 13C resonances was obtained from 1H NOESY, 1H COSY, gradient selected 1H, 13C HSQC and 1H, 195Pt HMBC experiments. All 2D NMR experiments were performed using standard pulse sequences from the Bruker pulse program library. Chemical shifts were relative to TMS (1H, 13C), H2PtCl6 (195Pt), and BF3-OEt2 (11B). UV-vis absorption spectra were measured using a Varian 50 Scan UV-visible photometer. Electrochemical experiments were carried out in 0.1 M n-Bu4NPF6 solutions using a three-electrode configuration (glassy carbon working electrode, Pt counter electrode, Ag/AgCl pseudo reference) and a BioLogic SP-150 or a Metrohm μStat400 potentiostat. Experiments were run at a scan rate of 100 mV s−1, at ambient temperature. The ferrocene/ferrocenium couple served as internal reference. The number of electrons per wave was gauged using weighted samples of ferrocene. UV-vis spectroelectrochemical measurements were performed with an optical transparent thin-layer electrochemical (OTTLE) cell.55,56 EI(+)-Mass spectra were recorded on a Thermo Quest Finnigan MAT 95 at 70 eV.For ultraviolet photoelectron spectroscopy (UPS) and inverse photoelectron spectroscopy (IPES) measurements thin films were prepared on ITO-coated glass substrates (15 Ω per sq sheat resistance), which were cleaned with CHCl3, acetone, a 2% Mucasol soap solution and deionised H2O before use. Thin films were prepared out of CH2Cl2 solutions via spin-coating at a rate of 4000 rpm for 35 s, to yield layers with thicknesses of about 10 nm. Solid state UV-vis absorption spectra were recorded using a Varian Cary 50 Bio spectrometer. Photoelectron spectroscopy measurements were performed in an ultra-high vacuum chamber using a Phoibos 100 hemispherical analyser by Specs. For ultraviolet photoelectron spectroscopy (UPS) a monochromatic helium discharge lamp (He I at 21.22 eV) with a probing depth of around 2 nm at a pass energy of 2 eV and a resolution of 110 meV was used.
Computational details (DFT)
Electronic structure calculations on the complexes have been performed through density-functional theory (DFT) methods using the ORCA program package.57 For all optimisations triple-ξ-valence TZVP58 basis sets were used with the generalised gradient approximated functional BP86.59 Harmonic frequencies were computed along the series [ML] (a computed IR spectrum of [NiL] is shown in Fig. 2); the absence of imaginary modes proved the optimised geometries to be stationary points. Pertinent metrical data is arranged in Tables S2 and S3, ESI.† Molecular orbitals and electronic properties were extracted from single-point calculations in the optimised positions with the global hybrid functional TPSSh60 and triple-ξ-valence TZVP basis sets. Grimme's third generation D3 correction of dispersion was used;61,62 medium effects were approximated in a dielectric continuum approach (COSMO), parameterised for THF.63 Coordinates of the computed structures are assembled in the ESI† file COORDINATES, frontier orbital landscapes are shown in Fig. 6 and Fig. S25–S29 (ESI).† For each complex the 80 lowest optical electronic transitions were assessed with ORCA implemented TD-DFT methods within the Tamm-Dancoff approximation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. Dr Mathias Schäfer and Michael Neihs, Department of Chemistry, University of Cologne are acknowledged for the HR-ESI-MS measurements. The Deutsche Forschungsgemeinschaft DFG KL1194/15-1 and KL1194/16-1 [DFG Priority Programme 2102 “Light-controlled Reactivity of Metal Complexes”] is acknowledged for funding of this project. G.H. thanks DFG for financial support within SFB 840 (TP A10 and A2). Prof. Dr Bernd Neumaier, Department of Chemistry, Institute for Nuclear Chemistry, Cologne University Clinics, Institute of Radiochemistry, and Research Centre Jülich for financial support and consulting. We also like to thank the Regional Computing Centre of the University of Cologne (RRZK) for providing computing time on the DFG-funded High Performance Computing (HPC) system CHEOPS as well as for the support.References
- S. Padhyé and G. B. Kauffman, Coord. Chem. Rev., 1985, 63, 127–160 CrossRef.
- A. G. Quiroga and C. N. Ranninger, Coord. Chem. Rev., 2004, 248, 119–133 CrossRef CAS.
- T. S. Lobana, R. Sharma, G. Bawa and S. Khanna, Coord. Chem. Rev., 2009, 253, 977–1055 CrossRef CAS.
- J. R. Dilworth and R. Hueting, Inorg. Chim. Acta, 2012, 389, 3–15 CrossRef CAS.
- J. S. Casas, M. S. García-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
- P. Heffeter, V. F. S. Pape, É. A. Enyedy, B. K. Keppler, G. Szakacs and C. R. Kowol, Antioxid. Redox Signal., 2019, 30, 1062–1082 CrossRef CAS PubMed.
- I. G. Santos, U. Abram, R. Alberto, E. V. Lopez and A. Sanchez, Inorg. Chem., 2004, 43, 1834–1836 CrossRef CAS PubMed.
- P. Chellan, S. Nasser, L. Vivas, K. Chibale and G. S. Smith, J. Organomet. Chem., 2010, 695, 2225–2232 CrossRef CAS.
- S. F. F. dos Santos, A. A. Oliveira, G. R. Santos, G. Mahmoudi, F. A. Afkhami, P. S. Santiago, R. B. Viana, A. B. F. da Silva and R. H. A. Santos, J. Mol. Struct., 2019, 1197, 393–400 CrossRef CAS.
- N. Arefyeva, A. Sandleben, A. Krest, U. Baumann, M. Schäfer, M. Kempf and A. Klein, Inorganics, 2018, 6, 51 CrossRef , 1–18.
- L. Fuentes, A. G. Quiroga, J. A. Organero and A. I. Matesanz, J. Inorg. Biochem., 2020, 203, 110875 CrossRef CAS , 1–11.
- P. Paul, R. J. Butcher and S. Bhattacharya, Inorg. Chim. Acta, 2015, 425, 67–75 CrossRef CAS.
- Z. Moradi-Shoeili and M. Zare, Kinet. Catal., 2018, 59, 203–210 CrossRef CAS.
- J. C. Lima, R. D. Nascimento, L. M. Vilarinho, A. P. Borges, L. H. F. Silva, J. R. Souza, L. R. Dinelli, V. M. Deflon, A. E. da Hora Machado, A. L. Bogado and P. I. S. Maia, J. Mol. Struct., 2020, 1199, 126997 CrossRef CAS , 1–10.
- C. González-García, A. Mata, F. Zani, M. A. Mendiola and E. López-Torres, J. Inorg. Biochem., 2016, 163, 118–130 CrossRef.
- T. B. Y. Nguyen, C. T. Pham, T. N. Trieu, U. Abram and H. H. Nguyen, Polyhedron, 2015, 96, 66–70 CrossRef CAS.
- M.-X. Li, M. Yang, J.-Y. Niu, L.-Z. Zhang and S.-Q. Xie, Inorg. Chem., 2012, 51, 12521–12526 CrossRef CAS PubMed.
- N. C. Kasuga, K. Sekino, M. Ishikawa, A. Honda, M. Yokoyama, S. Nakano, N. Shimada, C. Koumo and K. Nomiya, J. Inorg. Biochem., 2003, 96, 298–310 CrossRef CAS.
- K. Nomiya, K. Sekino, M. Ishikawa, A. Honda, M. Yokoyama, N. C. Kasuga, H. Yokoyama, S. Nakano and K. Onodera, J. Inorg. Biochem., 2004, 98, 601–615 CrossRef CAS PubMed.
- M. R. Bermejo, R. Pedrido, M. I. Fernandez, A. M. González-Noya, M. Maneiro, M. J. Rodríguez, M. J. Romero and M. Vázquez, Inorg. Chem. Commun., 2004, 7, 4–8 CrossRef CAS.
- B. M. Paterson, J. M. White and P. S. Donnelly, Dalton Trans., 2010, 39, 2831–2837 RSC.
- V. V. Pavlishchuk, S. V. Kolotilov, A. W. Addison, R. J. Butcher and E. Sinn, J. Chem. Soc., Dalton Trans., 2000, 335–341 RSC.
- Y. Fujibayashi, H. Taniuchi, Y. Yonekura, H. Ohtani, J. Konishi and A. Yokoyam, J. Nucl. Med., 1997, 38, 1155–1160 CAS.
- A. L. Vāvere and J. S. Lewis, Dalton Trans., 2007, 2007, 4893–4902 RSC.
- J. S. Lewis, R. Laforest, F. Dehdashti, P. W. Grigsby, M. J. Welch and B. A. Siegel, J. Nucl. Med., 2008, 49, 1177–1182 CrossRef PubMed.
- C. P. W. Soon, P. S. Donnelly, B. J. Turner, L. W. Hung, P. J. Crouch, N. A. Sherratt, J.-L. Tan, N. K.-H. Lim, L. Lam, L. Bica, S. Lim, J. L. Hickey, J. Morizzi, A. Powell, D. I. Finkelstein, J. G. Culvenor, C. L. Masters, J. Duce, A. R. White, K. J. Barnham and Q.-X. Li, J. Biol. Chem., 2011, 286, 44035–44044 CrossRef CAS PubMed.
- R. Anjum, D. Palanimuthu, D. S. Kalinowski, W. Lewis, K. C. Park, Z. Kovacevic, I. U. Khan and D. R. Richardson, Inorg. Chem., 2019, 58, 13709–13723 CrossRef CAS PubMed.
- T. Straistari, J. Fize, S. Shova, M. Réglier, V. Artero and M. Orio, ChemCatChem, 2017, 9, 2262–2268 CrossRef CAS.
- A. C. R. Gonçalves, A. R. Rettondin, C. G. Oliveira, A. O. T. Patrocinio, A. E. H. Machado, V. M. Deflon, U. Abram and P. I. S. Maia, Dalton Trans., 2020, 49, 9564–9567 RSC.
- M. Maji, S. Ghosh, S. K. Chattopadhyay and T. C. W. Mak, Inorg. Chem., 1997, 36, 2938–2943 CrossRef CAS PubMed.
- D. Rosales, I. Millan and J. L. G. Ariza, Talanta, 1986, 33, 607–610 CrossRef CAS PubMed.
- D. Rosales, J. L. G. Ariza and A. G. Asuero, Analyst, 1986, 111, 449–453 RSC.
- R. B. Lucena, E. Morales and J. L. G. Ariza, Farmaco, 1994, 49, 291–295 CAS.
- A. Kochem, G. Gellon, O. Jarjayes, C. Philouze, A. du Moulinet d'Hardemare, M. van Gastel and F. Thomas, Dalton Trans., 2015, 44, 12743–12756 RSC.
- A. Haseloer, T. Lützenburg, J. P. Strache, J. Neudörfl, I. Neundorf and A. Klein, ChemBioChem, 2021, 22, 694–704 CrossRef CAS PubMed.
- N. M. Mews, M. Reimann, G. Hörner, M. Kaupp, H. Schubert and A. Berkefeld, Dalton Trans., 2020, 49, 9735–9742 RSC.
- A. Berkefeld, M. Fröhlich, M. Kordan, G. Hörner and H. Schubert, Chem. Commun., 2020, 56, 3987–3990 RSC.
- L. Kletsch, G. Hörner and A. Klein, Organometallics, 2020, 39, 2820–2829 CrossRef CAS.
- H. Kurz, K. Schötz, I. Papadopoulos, F. W. Heinemann, H. Maid, D. M. Guldi, G. Hörner and B. Weber, J. Am. Chem. Soc., 2021 DOI:10.1021/jacs.0c12568.
- K. Ray, T. Petrenko, K. Wieghardt and F. Neese, Dalton Trans., 2007, 2007, 1552–1566 RSC.
- K. Ray, T. Weyhermuller, F. Neese and K. Wieghardt, Inorg. Chem., 2005, 44, 5345–5360 CrossRef CAS PubMed.
- D. Herebian, E. Bothe, F. Neese, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 9116–9128 CrossRef CAS PubMed.
- S. Samiee and S. Taghvaeian, Appl. Organomet. Chem., 2019, 33, e4626 CrossRef , 1–12.
- Y. Shimazaki, T. D. P. Stack and T. Storr, Inorg. Chem., 2009, 48, 8383–8392 CrossRef CAS PubMed.
- A. Klein, Z. Anorg. Allg. Chem., 2001, 627, 645–650 CrossRef CAS.
- H. Masai, K. Sonogashira and N. Hagihara, Bull. Chem. Soc. Jpn., 1971, 44, 2226–2230 CrossRef CAS.
- F. Thomas, Dalton Trans., 2016, 45, 10866–10877 RSC.
- K. Butsch, Th. Günther, A. Klein, K. Stirnat, A. Berkessel and J. Neudörfl, Inorg. Chim. Acta, 2013, 394, 237–246 CrossRef CAS.
- K. Butsch, A. Klein, S. Nitsche, K. Stirnat, J. R. Hawkett, E. J. L. McInnes and M. Bauer, Dalton Trans., 2012, 41, 11464–11475 RSC.
- L. Benisvy, G. M. Zats, O. Fleker, D. S. Yufit, M. Orio, G. Romanenko and V. Ovcharenko, Dalton Trans., 2015, 4, 17924–17926 RSC.
- H. Oshita, T. Suzuki, K. Kawashima, H. Abe, F. Tani, S. Mori, T. Yajima and Y. Shimazaki, Dalton Trans., 2019, 48, 12060–12069 RSC.
- K. Kamaraj, E. Kim, B. Galliker, L. N. Zakharov, A. L. Rheingold, A. D. Zuberbühler and K. D. Karlin, J. Am. Chem. Soc., 2003, 125, 6028–6029 CrossRef CAS PubMed.
- Y. H. Budnikova, D. A. Vicic and A. Klein, Inorganics, 2018, 6, 18 CrossRef , 1–18.
- K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed.
- Spectroelectrochemistry, ed. W. Kaim and A. Klein, RSC Publishing, Cambridge, UK, 2008 Search PubMed.
- W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373–3382 RSC.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. Schäfer, H. Horn and R. J. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed , 1–19.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt00272d |
| This journal is © The Royal Society of Chemistry 2021 |