
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and complexes of a constrained-cavity Schiff-base dipyrrin macrocycle†
Karlotta
van Rees
and
Jason B.
Love
 *
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, The King's Buildings, Edinburgh, EH9 3FJ, UK. E-mail: Jason.Love@ed.ac.uk
First published on 22nd January 2021
Abstract
A new constrained-cavity [1 + 1] Schiff-base dipyrrin macrocycle comprising an N4 donor-pocket has been synthesised by spontaneous oxidation and in situ crystallisation. Access to Fe(II) and Zn(II) complexes is achieved by salt elimination reactions of the lithium salt. All compounds have been characterised by NMR and UV-vis spectroscopy, X-ray crystallography, and DFT analysis.
Dipyrrins, also known as dipyrromethenes, represent a fascinating class of organic chromophores and are readily synthesised via oxidation from their dipyrromethane analogues. The π-conjugation in these bis-pyrrole compounds facilitates effective absorption of visible light through π–π* transitions, and their boron difluoride adducts (BODIPY) belong to a family of fluorescent dyes with useful bioapplications.1–5 Dipyrrins incorporate both a mono-anionic and bidentate ligand environment creating rich and diverse coordination and supramolecular chemistry, forming stable, highly crystalline complexes with a variety of metal ions, as highlighted by Baudron and co-workers.6–13 In addition, Betley and co-workers have demonstrated that transition metal complexes of dipyrrins act as catalysts for C–H amination reactions.14–18
Due to their planar N4 donor-pockets, Schiff-base dipyrrins are similar to porphyrins and their expanded porphyrin cousins and display a rich coordination chemistry along with unique redox-activity and photo-physical properties. Previously, we reported the donor-expanded acyclic dipyrrin 1 and its uranyl(VI) complex and showed that this ligand acts as an electron conduit for inner- and outer-sphere redox reactions (Fig. 1).19–21 Sircoglou, Aukauloo and co-workers investigated the photo-physical properties of an acyclic zinc(II) and nickel(II) dipyrrin–pyridine complex.22 In contrast to acyclic ligands, non-porphyrinic, macrocyclic dipyrrins are rare. Naruta and co-workers reported the phenanthroline-dipyrrin macrocycle 2(Fig. 1) and described the photo-physical properties of its magnesium(II) complex.23,24 Sessler, Katayev, and co-workers studied competitive binding of palladium(II) with the expanded Schiff-base dipyrrin-diamide macrocycle 3 (Fig. 1).25 Accordion-like dimanganese(II) Schiff-base dipyrrin macrocycles have been investigated by Bowman-James and co-workers as a functional model for catalases.26,27 Smaller cavity macrocycles such as the benziporphyrins have also been prepared by Latos-Grażyński and co-workers for which metal-assisted C–H bond activation chemistry has been revealed.28–31
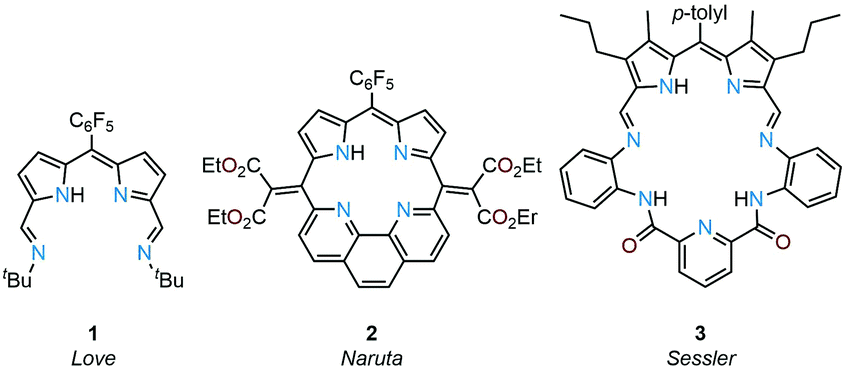 | ||
| Fig. 1 Structures of reported dipyrrin compounds with N4-donor pockets. | ||
The chemical and photochemical properties of the dipyrrin framework combined with the unique control of coordination and organometallic chemistry afforded by a macrocyclic environment led us to investigate the formation of macrocyclic analogues of 1. We report here the straightforward formation and isolation of a unique constrained-cavity [1 + 1] Schiff-base dipyrrin macrocycle HL that comprises an N4 dipyrrin-imine donor set and describe the formation and properties of its lithium, iron, and zinc complexes.
The condensation of equimolar quantities of the mono-meso-substituted dipyrromethane dialdehyde 4 and bis(m-aminoisopropyl)benzene32 in acetonitrile at room temperature causes the macrocyclic product HL to crystallise directly from the reaction mixture as greenish-red crystals in 80% yield (Scheme 1 and ESI†). The 1H NMR spectrum of HL shows an imine proton resonance at 8.47 ppm and additional resonances indicative of C2-symmetry in solution. In addition, the disappearance of the meso-proton resonance from 4 reveals that spontaneous oxidation of the dipyrromethane to the dipyrrin has occurred.
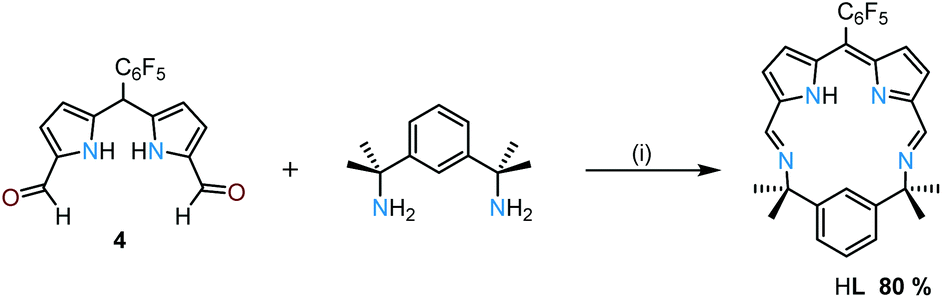 | ||
| Scheme 1 The synthesis of the dipyrrin macrocycle HL. (i) MeCN, rt, 16 h. | ||
The MALDI-TOF mass spectrum of HL shows the expected molecular ion at m/z 524.5 ([M + H]+) with no higher-order cyclisation products observed. Attempts to form HL using the dipyrrin analogue of 4 (4ox, ESI†) in a condensation with the diamine under similar conditions results in a mixture of the starting materials and a variety of imine-containing products. Significantly, the formation of 4ox requires oxidation with DDQ with photolysis under an inert atmosphere, which suggests that the simple aerobic oxidation that occurs during the synthesis of HL is driven by the formation of the macrocycle.33–35
The X-ray crystal structure of HL (Fig. 2) reveals a fully planer conformation of the N4 donor set with the meso-C6F5 substituent approximately orthogonal (82°) and the para-proton (H1) of the bridging phenyl group pointing into the macrocyclic cavity. The size of the cavity is defined by the pyrrole (N2⋯N3) and imine (N1⋯N4) separations of 2.672(2) Å and 4.391(2) Å, respectively, so resulting in a small and rigid trapezoid-shaped binding pocket. This is significantly different to porphyrins such as tetraphenylporphyrin (TPP) in which an approximately square cavity of 2.921 Å by 2.914 Å is seen.36
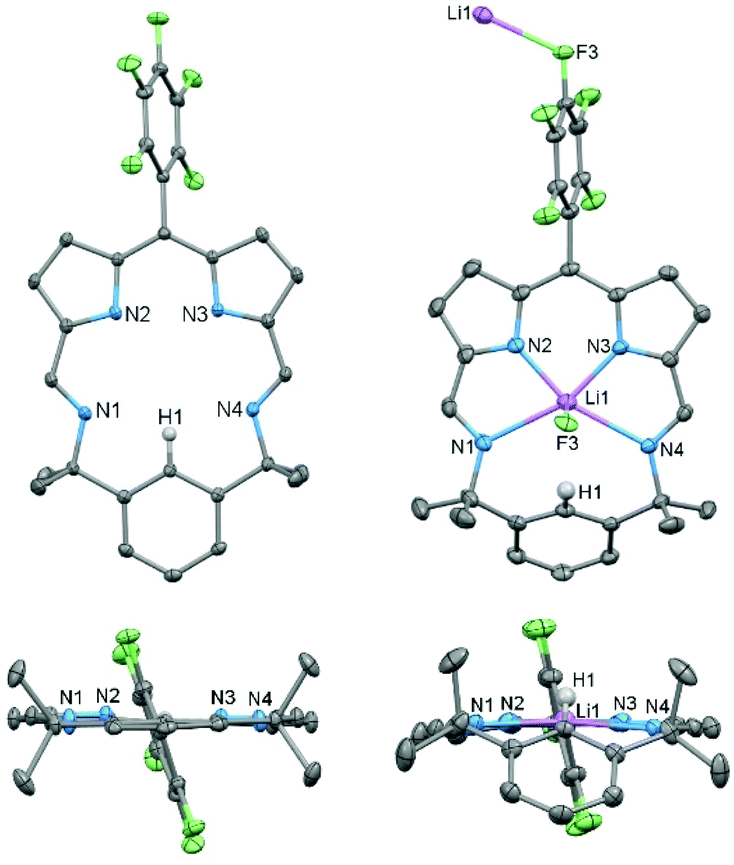 | ||
| Fig. 2 Solid-state structure of HL and Li(L) viewed from side and top. For clarity, all hydrogen atoms (except H1) and solvent of crystallisation are omitted (displacement ellipsoids are drawn at 50% probability). Li(L) top view is depicted including close-contact atoms of adjacent molecule. Crystals of Li(L) were grown by slow evaporation of a concentrated n-hexane solution. | ||
Reaction between HL and one equivalent of LiN(SiMe3)2 in toluene cleanly generates the lithium complex Li(L) which was isolated as a dark purple solid in 91% yield. Its formation is apparent by the disappearance of the pyrrole proton in the 1H NMR spectrum and the appearance of a broad singlet at 1.57 ppm in the 7Li NMR spectrum. The X-ray crystal structure of Li(L) shows that the lithium coordinates to all four N-donor atoms within the macrocyclic cavity with an axial Li⋯F3′ interaction from an adjacent molecule (Li⋯F3′ 2.354(3) Å), forming an infinite chain structural motif (Fig. 2 and ESI†). The presence of the Li cation causes the bridging phenyl ring to tilt significantly, from 176.56° in HL to 112.79° in Li(L), resulting in a C1⋯F3′ distance of 3.570(2) Å. Attempts to doubly deprotonate HL (e.g., at C1–H1) using two equivalents of LiN(SiMe3)2 were unsuccessful. The twist of the bridging aryl group such that the unique H-atom H1 avoids interaction with the Li cation within the N4-doner set shows that, unlike the porphyrin analogues, the macrocyclic cavity is size-compromised.
The reactions between Li(L) and either ZnBr2 or FeBr2 cleanly generate the metal bromide complexes ZnBr(L) and FeBr(L), respectively (Scheme 2); the isolated yield for FeBr(L) is low due to its poor solubility. HRMS of ZnBr(L) and FeBr(L) show molecular ion peaks at m/z 667.025720 ([M + H]+) and 577.110887 ([M − Br]+), respectively. The 1H NMR spectrum of ZnBr(L) shows mirror symmetry with the geminal methyl groups appearing as two singlets at 1.80 ppm and 1.34 ppm, indicative of top/bottom asymmetry; this asymmetry is also seen in the 19F NMR spectrum which displays five fluorine resonances.
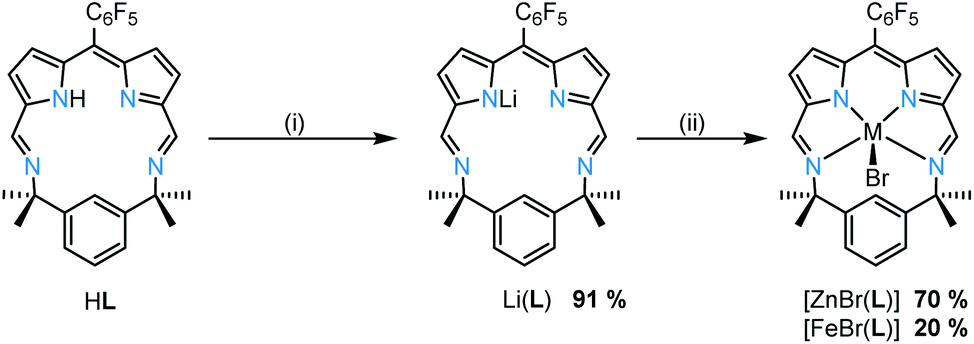 | ||
| Scheme 2 Metalation of HL. (i) 1 eq. LiN(SiMe3)2, toluene, RT, 16 h. (ii) ZnBr2, toluene, RT, 16 h or FeBr2, THF, RT, 16 h. | ||
The X-ray crystal structures of ZnBr(L) and FeBr(L) were determined (Fig. 3) and both exhibit square pyramidal geometries with axially coordinated bromide ligands. There is a noticeable difference in distance between the metal and the plane of the N4 donor set; for zinc this is 0.593 Å above the plane while for iron it is 0.304 Å. Similarities can be found in previously reported structures of iron chloride octaethylpophyrin37 and zinc chloride m-benziporphodimethene28,38 in which the Fe and Zn are ca. 0.47 Å and 0.51 Å, respectively, out of plane. In contrast, for iron chloride and zinc chloride TPP complexes the metal ion is coordinated on the macrocyclic N4 donor plane.39,40 This difference could be explained by the additional steric effect of the para-proton (H1) of the bridging phenyl group that points towards the cavity.
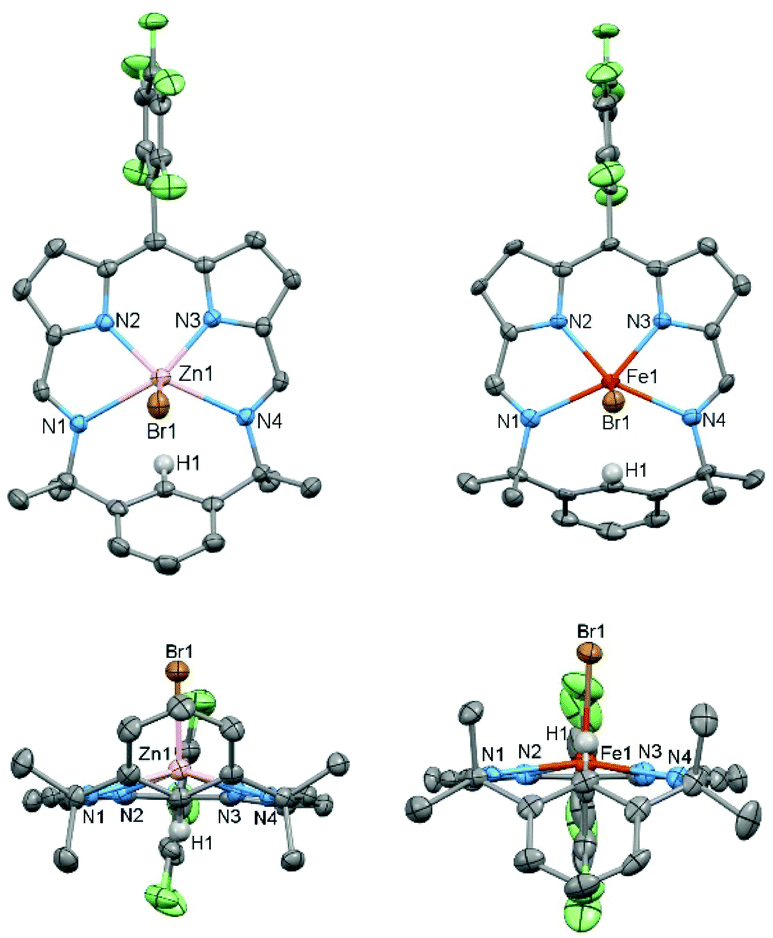 | ||
| Fig. 3 Solid-state structures of ZnBr(L) and FeBr(L), respectively, viewed from side and top. For clarity, all hydrogen atoms (except H1) and solvent of crystallisation are omitted (displacement ellipsoids drawn at 50% probability). Crystals were grown by slow evaporation of dilute, saturated n-hexane solutions. | ||
The UV-vis spectrum of HL shows two absorptions at 277 and 512 nm (Fig. 4) that are similar to those seen for other expanded dipyrrins and substituted BODIPY compounds.20,41,42 Upon deprotonation to form Li(L) and subsequent transmetallation to form ZnBr(L) and FeBr(L), significant red shifts occur in the UV-vis spectra, with the lower wavelength adsorption splitting into two. The experimental UV-Vis spectra (with the exception of FeBr(L)) were reproduced by TD-DFT calculations using B3LYP functional and 6-311G(d,p) basis set (Fig. 4, 5 and ESI†). The energies of the transition states are over estimated, as seen with previously reported hetero-diarylmethene ligands and their complexes.43 The electronic density of both the HOMO and the LUMO of HL and ZnBr(L) comprise π orbitals that are centred on the dipyrrin unit and the imines, highlighting the extended conjugation in these molecules. The dominant absorption band in HL at 277 nm is assigned as a mixture of HOMO to LUMO+3 (41%) and HOMO to LUMO+1 (13%) transitions, with the HOMO–LUMO transition appearing at 512 nm. This latter transition is comparable with (Fig. 1) the acyclic dipyrrin ligand 1, which has the HOMO–LUMO transition at 540 nm.20 The red-shifted dominant absorption bands at 595 and 557 nm for Li(L) and ZnBr(L), respectively, correspond to the HOMO–LUMO transition with the HOMO–LUMO+3 transition shifting to 303 and 300 nm, respectively. The UV-vis spectrum of the ZnBr(L) resembles that of the deprotonated ligand Li(L), suggesting the absence of any metal-centred transitions.
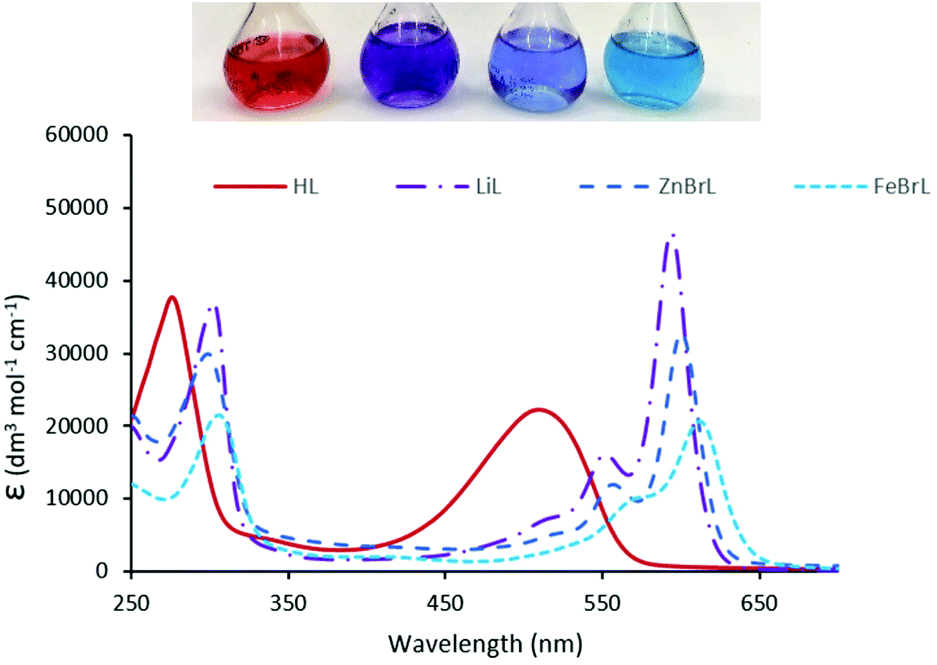 | ||
| Fig. 4 Electronic absorption spectrum for HL, Li(L), ZnBr(L), and FeBr(L). All spectra were recorded in dry THF. | ||
 | ||
| Fig. 5 Molecular orbital plots of HL and ZnBr(L) (B3LYP/6-311G(d,p)). ISO value of 0.02 a.u. Positive is blue; negative is red. (a) LUMO and (c) HOMO of HL, (b) LUMO and (d) HOMO of ZnBr(L). | ||
We have synthesised a new constrained-cavity [1 + 1] Schiff-base dipyrrin macrocycle that is spontaneously oxidised and crystallised during its preparation. The straightforward deprotonation of the macrocycle unveils a uninegative, N4-donor set that allows the formation of iron(II) and Zn(II) complexes; unlike porphyrinic analogues, these complexes retain the ancillary bromide ligands. The observation of ligand-centred electronic transitions suggests a potentially rich ligand-centred redox chemistry that, combined with the Lewis acidity of the metal, could lead to new chemical transformations. Our current investigations focus on the catalytic redox behaviour of this ligand and its complexes as well as exploring the possibility of using the NC(H)N pocket in the formation of dinuclear complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Edinburgh, the EPSRC (UK), and the EPSRC CRITICAT Centre for Doctoral Training (PhD studentship to K. v. R.; Grant EP/L016419/1) for financial support and the Mass Spectrometry Facility at the University of Edinburgh for carrying out air-sensitive HRMS analysis.Notes and references
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- Z. Shi, X. Han, W. Hu, H. Bai, B. Peng, L. Ji, Q. Fan, L. Li and W. Huang, Chem. Soc. Rev., 2020, 49, 7533–7567 RSC.
- R. S. Singh, R. P. Paitandi, R. K. Gupta and D. S. Pandey, Coord. Chem. Rev., 2020, 414, 213269 CrossRef CAS.
- H. Kim, A. Burghart, M. B. Welch, J. Reibenspies and K. Burgess, Chem. Commun., 1999, 1889–1890 RSC.
- S. A. Baudron, Dalton Trans., 2013, 42, 7498–7509 RSC.
- H. Ruffin, S. A. Baudron, D. Salazar-Mendoza and M. W. Hosseini, Chem. – Eur. J., 2014, 20, 2449–2453 CrossRef CAS PubMed.
- A. Poma, I. Grigioni, M. V. Dozzi, S. A. Baudron, L. Carlucci, M. W. Hosseini and E. Selli, New J. Chem., 2017, 41, 15021–15026 RSC.
- F. Zhang, S. A. Baudron and M. W. Hosseini, New J. Chem., 2018, 42, 6997–7004 RSC.
- F. Zhang, A. Fluck, S. A. Baudron and M. W. Hosseini, Dalton Trans., 2019, 48, 4105–4108 RSC.
- S. A. Baudron, Coord. Chem. Rev., 2019, 380, 318–329 CrossRef CAS.
- S. A. Baudron and H. Chevreau, CrystEngComm, 2019, 21, 1853–1856 RSC.
- S. A. Baudron, Dalton Trans., 2020, 49, 6161–6175 RSC.
- Y. Baek, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2019, 141, 16944–16953 CrossRef CAS PubMed.
- Y. Baek and T. A. Betley, J. Am. Chem. Soc., 2019, 141, 7797–7806 CrossRef CAS PubMed.
- K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S.-L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138 CrossRef CAS.
- Y. Dong, R. M. Clarke, G. J. Porter and T. A. Betley, J. Am. Chem. Soc., 2020, 142, 10996–11005 CrossRef CAS PubMed.
- D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599–15602 CrossRef CAS.
- J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108–116 RSC.
- J. R. Pankhurst, T. Cadenbach, D. Betz, C. Finn and J. B. Love, Dalton Trans., 2015, 44, 2066–2070 RSC.
- N. L. Bell, B. Shaw, P. L. Arnold and J. B. Love, J. Am. Chem. Soc., 2018, 140, 3378–3384 CrossRef CAS PubMed.
- C. Ducloiset, P. Jouin, E. Paredes, R. Guillot, M. Sircoglou, M. Orio, W. Leibl and A. Aukauloo, Eur. J. Inorg. Chem., 2015, 5405–5410 CrossRef CAS.
- M. Ishida, Y. Naruta and F. Tani, Angew. Chem., Int. Ed., 2010, 49, 91–94 CrossRef CAS.
- M. Ishida, J. M. Lim, B. S. Lee, F. Tani, J. L. Sessler, D. Kim and Y. Naruta, Chem. – Eur. J., 2012, 18, 14329–14341 CrossRef CAS PubMed.
- E. A. Katayev, Y. A. Ustynyuk, V. M. Lynch and J. L. Sessler, Chem. Commun., 2006, 4682–4684 RSC.
- N. N. Gerasimchuk, A. Gerges, T. Clifford, A. Danby and K. Bowman-James, Inorg. Chem., 1999, 38, 5633–5636 CrossRef CAS PubMed.
- W. A. Reiter, A. Gerges, S. Lee, T. Deffo, T. Clifford, A. Danby and K. Bowman-James, Coord. Chem. Rev., 1998, 174, 343–359 CrossRef CAS.
- G.-F. Chang, C.-H. Wang, H.-C. Lu, L.-S. Kan, I. Chao, W. H. Chen, A. Kumar, L. Lo, M. A. C. de la Rosa and C.-H. Hung, Chem. – Eur. J., 2011, 17, 11332–11343 CrossRef CAS PubMed.
- M. Stępień, L. Latos-Grażyński, L. Szterenberg, J. Panek and Z. Latajka, J. Am. Chem. Soc., 2004, 126, 4566–4580 CrossRef.
- M. Stępień, L. Latos-Grażyński and L. Szterenberg, Inorg. Chem., 2004, 43, 6654–6662 CrossRef PubMed.
- M. Stępień and L. Latos-Grażyński, Chem. – Eur. J., 2001, 7, 5113–5117 CrossRef.
- L. Dahlenburg, H. Treffert and F. W. Heinemann, Inorg. Chim. Acta, 2008, 361, 1311–1318 CrossRef CAS.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Chem. – Eur. J., 2015, 21, 2855–2861 CrossRef CAS.
- S. M. Hubig, T. M. Bockman and J. K. Kochi, J. Am. Chem. Soc., 1997, 119, 2926–2935 CrossRef CAS.
- S. Guski, M. Albrecht, T. Willms, M. Albrecht, T. Nabeshima, F. Pan, R. Puttreddy and K. Rissanen, Chem. Commun., 2017, 53, 3213–3215 RSC.
- S. J. Silvers and A. Tulinsky, J. Am. Chem. Soc., 1967, 89, 3331–3337 CrossRef CAS.
- J. Ernst, J. Subramanian and J. H. Fuhrhop, Z. Naturforsch., 1977, 32a, 1129 CrossRef CAS.
- C.-H. Hung, G.-F. Chang, A. Kumar, G.-F. Lin, L.-Y. Luo, W.-M. Ching and E. W.-C. Diau, Chem. Commun., 2008, 978–980 RSC.
- W. R. Scheidt, J. U. Mondal, C. W. Eigenbrot, A. Adler, L. J. Radonovich and J. L. Hoard, Inorg. Chem., 1986, 25, 795–799 CrossRef CAS.
- G. J. Corban, S. K. Hadjikakou, A. C. Tsipis, M. Kubicki, T. Bakas and N. Hadjiliadis, New J. Chem., 2011, 35, 213–224 RSC.
- C. Bonnier, D. D. Machin, O. Abdi and B. D. Koivisto, Org. Biomol. Chem., 2013, 11, 3756–3760 RSC.
- R. M. Diaz-Rodriguez, K. N. Robertson and A. Thompson, Dalton Trans., 2019, 48, 7546–7550 RSC.
- M. Curcio, D. Henschel, M. Hüttenschmidt, S. Sproules and J. B. Love, Inorg. Chem., 2018, 57, 9592–9600 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2051272–2051276. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00175b |
| This journal is © The Royal Society of Chemistry 2021 |