DOI:
10.1039/D1DT00121C
(Paper)
Dalton Trans., 2021,
50, 4890-4903
Quasilinear 3d-metal(I) complexes [KM(N(Dipp)SiR3)2] (M = Cr–Co) – structural diversity, solution state behaviour and reactivity†
Received
13th January 2021
, Accepted 2nd March 2021
First published on 22nd March 2021
Abstract
The synthesis and characterization of neutral quasilinear 3d-metal(I) complexes of chromium to cobalt of the type [KM(N(Dipp)SiMe3)2] (Dipp = 2,6-di-iso-propylphenyl) are reported. In solid state these metal(I) complexes either occur as isolated molecules (Co) or are part of a potassium ion linked 1D-coordination polymer (Cr–Fe). In solution the potassium cation is either ligated within the ligand sphere of the metal silylamide or is separated from the complex depending on the solvent. For iron, we showcase that it is possible to use sodium or lithium metal for the reduction of the metal(II) precursor. However, in these cases the resulting iron(I) complexes can only be isolated upon cation separation using an appropriate crown-ether. Further, the neutral metal(I) complexes are used to introduce NBu4+ as an organic cation in the case of cobalt and iron. The impact of the intramolecular cation complexation was further demonstrated upon reaction with diphenyl acetylene which leads to bond formation processes and redox disproportionation instead of η2-alkyne complex formation.
Introduction
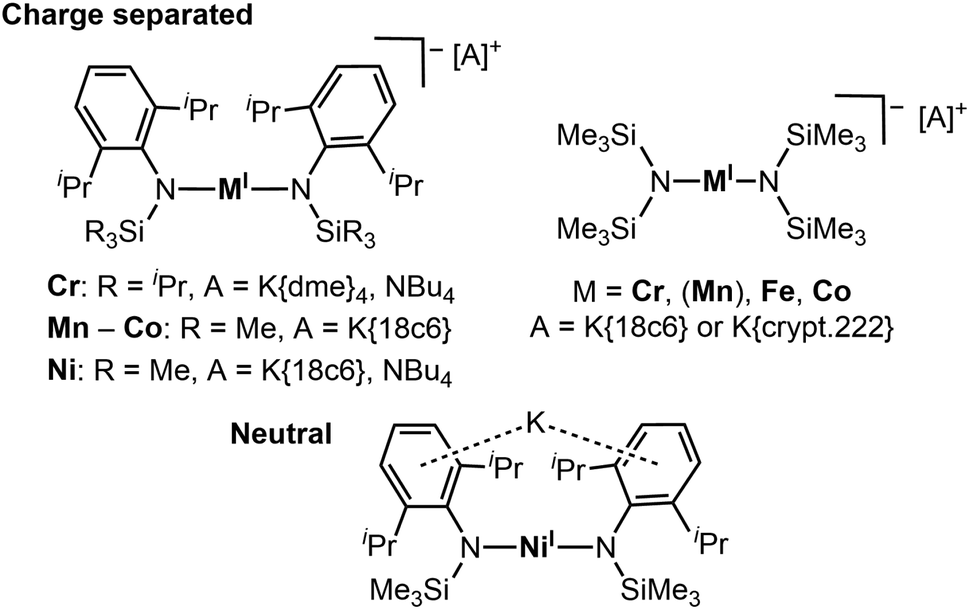
Complexes with two-coordinate, open-shell 3d-metal(I) ions, known for chromium to nickel, are a young and rare class of compounds in coordination chemistry.1,2–11 They combine an uncommon coordination motif with an unusual oxidation state. The isolation of such compounds relies mostly on the use of sterically encumbering and/or electronically stabilizing ligands, such as bulky amides or N-heterocyclic carbenes. Thereby intramolecular dispersion forces are generally thought to be crucial for their kinetic stabilisation of two-coordinate metal ions.11,12 Given the labile nature of two-coordinate metal(I) complexes, the physical properties, as well as their reactivity concerning small molecules and various substrates is only partially explored. Reports concerning their respective behaviour indicate a high potential, e.g. remarkable single molecule magnetic properties.2,4 These stem from the fact that these compounds can exhibit magnetic moments higher than the expected spin-only values, due to unquenched orbital momentum, as well as a strong magnetic anisotropy, attributed to their (near) linear ligand arrangement.4,13–15 Further, linear metal(I) complexes are shown to mediate intriguing bond activation processes like cleavage of H2, P–aryl, and C–F bonds or trimerisation of alkynes.7,9,10,16 The number of homoleptic, linear metal(I) complexes bearing anionic ligands is very limited, and restricted to silylmethanides and silylamides. For silylamides this gave so far few examples of complex salts of the type [K{18c6}][M(N(Dipp)SiR3)2] (Cr – Cu; 18c6 = 18-crown-6; Dipp = 2,6-di-iso-propylphenyl) or [K{m}][M(N(SiMe3)2)2] (Cr, Fe, Co; m = 18c6 or crypt.222).2,3,6,8,11 Their synthesis is achieved by reacting a two-coordinate metal(II) precursor with potassium graphite in the presence of a cryptand or crown-ether. The latter is generally thought to be needed to sequestrate the cation, and by that to prevent decomposition of the homoleptic complex anion, which was demonstrated in case of [Fe(N(SiMe3)2)2]−.2 A notable exception was reported by Tilley and co-workers in the case of the nickel(I) complex [KNi(N(Dipp)SiMe3)2],5 for which the monovalent state is comparably stable. Here the potassium ion is ligated within the ligand sphere (Fig. 1), leading to an overall neutral compound.5,6
 |
| | Fig. 1 Known anionic or neutral quasilinear open-shell 3d-metal(I) silylamides. | |
Given the fundamental interest of expanding the coordination chemistry of two-coordinate 3d-metal ions we wanted to elucidate the synthesis of monovalent compounds of the type [KM(L)2] of the earlier 3d-transition metals. The presence of an unmasked alkali metal cation in the vicinity of a 3d-metal is prospective of distinct reactivity due to synergistic effects, as shown for example for low-coordinate alkali metal ferrates or manganates,17 may labilise the 3d-metal–amide bond or can be used for introduction of further functionalities or other cations.
Herein we report the isolation of quasilinear complexes of the type [KM(L)2] (L1 = N(Dipp)SiiPr3 (Cr); L2 = N(Dipp)SiMe3 (Mn – Co)), with intra- or intermolecular complexation of the potassium cation, leading to unusual coordination polymers bearing open-shell, two-coordinate metal(I) ions. For iron we showcase the first use of lithium or sodium metal as reductants, whereas respective compounds are unstable and have to be stabilized by masking of the alkali metal cation. In due course a large structural variety of the ligand arrangement within the quasilinear silyl amide complexes can be observed that depends on interligand as well as cation⋯anion interactions. We provide insights into the solution state dependence of the [KM(L)2] complexes, also revealing how the alkali metal ion is extracted in donor solvents. First studies on the reactivity of [KM(L)2] complexes towards diphenyl acetylene hint to the consequences of the lack of persistent cation separation.
Results and discussion
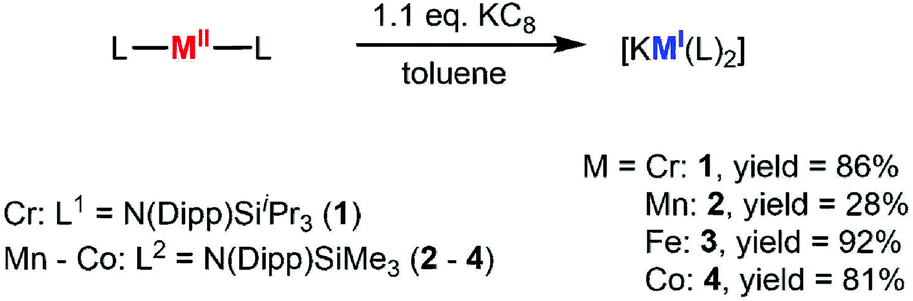
Synthesis and structure of [KM(L)2] complexes
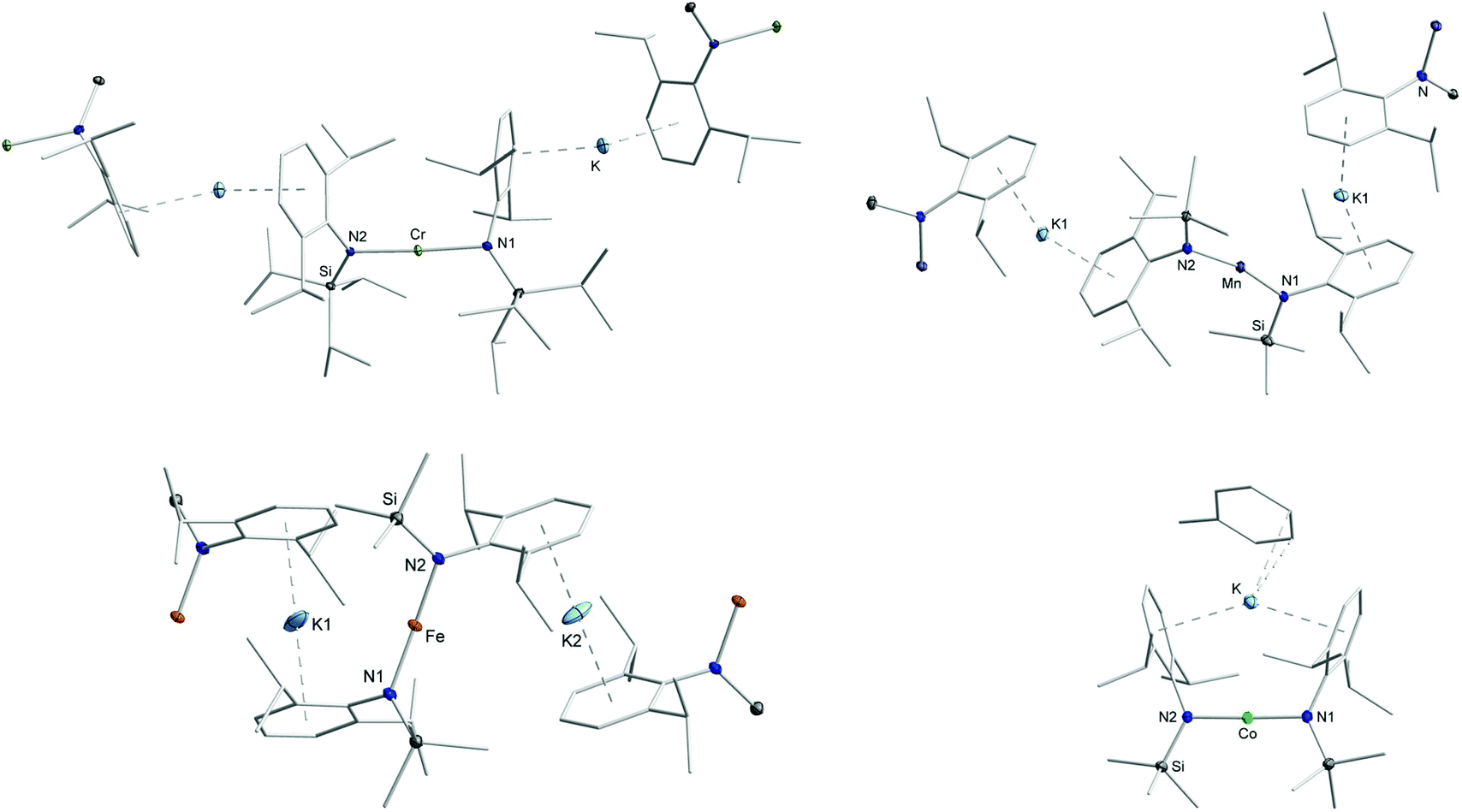
The quasilinear metal(II) complexes [M(L)2] (L1 = N(Dipp)SiiPr3 (Cr, due to reported instability of the SiMe3 derivative);5 L2 = N(Dipp)SiMe3 (Mn – Co)) were reacted with 1.1 equivalent KC8 in toluene at room temperature (Scheme 1) leading to an instantaneous change of colour in each case (Cr: dark red → red; Mn: colourless → dark violet; Fe: red → greenish brown; Co: red violet → green). Crystalline material was obtained from diffusion of n-pentane into a toluene solution of each compound at −40 °C giving green [KCr(L1)2], 1, violet [KMn(L2)2], 2, red brown [KFe(L2)2], 3, and green [K(toluene)Co(L2)2], 4. X-Ray diffraction analysis of compounds revealed for compounds 1–3 a polymeric structure (Fig. 2), where formally [M(L)2]− anions are linked via potassium cations, which are situated between the aryl rings of two neighbouring molecules. The potassium ion are located at the outer (1, Cr), inner and outer (2, Mn) or inner (3, Fe) side of the respective aryl ring. In case of compound 3 (Fe), the potassium ion is situated directly between two aryl rings with no apparent Fe⋯K interaction (Fe–K 4.3564(3) Å), whereas in 2 (Mn) it is clearly oriented towards the transition metal (Mn–K 3.8423(5) Å). These structural features lead to a zig-zag 1D-polymer chain for 2 (Mn) and 3 (Fe), and a more linear chain for 1 (Cr). This is also reflected by the intermolecular M–M′ distances, which gives for 1 (Cr) a Cr–Cr′ distance of 12.4099(9) Å within and of 9.9095(8) Å between the chains (Table 1). For 2 (Mn) and 3 (Fe), the situation is inverse with shorter intra- (2: 9.8350(5) Å; 3: 8.7128(5) Å) than interchain M–M′ distances (2: 11.0784(5) Å; 3: 11.6773(6) Å). The chain-like arrangement in solid state for compounds 1–3 is a so far unknown feature of complexes with open-shell, two-coordinate transition metal ions. It contrasts the situation of the related [KNi(L)2],5 where the potassium ion is ligated in an intramolecular fashion. Such a situation is observed for the cobalt derivative 4 (Co), where a toluene molecule is completing the coordination sphere of the potassium ion (Co–K distance of 3.5652(6) Å). The N–M–N bond angles within the polymeric compounds are almost linear in the case of chromium (177.44(10)°, 1) and slightly bent in manganese (165.56(6)°, 2) and iron (170.59(5)°, 3). For the monomeric cobalt compound 4, the N–Co–N bond angle amounts to 178.43(8)°. The M–N bond lengths shorten along the series from ca. 2.07 Å (1, Cr) to 1.96 Å (2, Mn) and down to 1.88 Å (4, Co).
 |
| | Scheme 1 Synthesis of solvent and cryptand free [KM(L)2] (M = Cr–Co) (1–4) (Dipp = 2,6-di-iso-propylphenyl). | |
 |
| | Fig. 2 Sections of the crystal structure of 1 (top left), 2 (top right), 3 (bottom left) and 4 (bottom right). H atoms are omitted for clarity. For compounds 1–3 the amide unit of neighbouring complex molecules is shown. | |
Table 1 Selected structural metrics of compounds 1–9 (L2 = –N(Dipp)SiMe3)
| Metal |
Compound |
M–N1/Å |
M–N2/Å |
M–M′/Å |
N1–M–N2/° |
Torsion angle/° |
Shortest, interligand C–C distance |
|
M–M′ distances along the 1D chain.
Generated via a crystallographic inversion centre on the metal atom.
|
| Cr |
1
|
2.076(2) |
2.070(2) |
9.9095(8)a |
177.44(10) |
59.97(23) |
3.719(4) (CH3(SiiPr3)⋯CHMe2(SiiPr3)) |
| 12.4099(9) |
3.796(3) (Ph⋯CH3(Dipp)) |
| 3.680(4) (CH3(Dipp)⋯CHMe2(SiiPr3)) |
|
5
|
2.0576(14) |
2.0528(14) |
11.1564(9) |
174.12(5) |
42.41(12) |
3.954(2) (CH3(SiiPr3)⋯CH3(SiiPr3)) |
| 3.536(1) (Ph⋯CH3(Dipp)) |
| Mn |
2
|
1.9653(16) |
1.9681(15) |
9.8350(5) |
165.56(6) |
161.68(14) |
3.979(2) (Ph⋯CH3(SiMe3)) |
| 11.0784(5)a |
| [K{18c6}][Mn(L2)2]8 |
1.961(3) |
1.954(3) |
10.8687(11) |
167.12(14) |
49.1(2) |
3.867(5) (Ph⋯CH3(SiMe3)) |
| 4.661(7) (CH3(SiMe3)⋯CH3(SiMe3)) |
| Fe |
3
|
1.9005(15) |
1.9014(13) |
8.7128(5) |
170.59(5) |
123.65(15) |
4.139(3) (CH3(Dipp)⋯CH3(SiMe3)) |
| 11.6773(6)a |
|
3·2DMAP |
1.9121(3) |
1.9121(3)b |
10.7640(19) |
180b |
16.8(3) |
4.469(7) (CH3(SiMe3)⋯CH3(SiMe3)) |
|
6
|
1.911(3) |
1.915(3) |
10.5043(9) |
175.34(13) |
148.9(3) |
4.825(5) (Ph⋯CH3(SiMe3)) |
| 4.200(10) (CH3(Dipp)⋯CH3(SiMe3)) |
|
7
|
1.894(4) |
1.903(4) |
11.518(2) |
170.82(14) |
7.0(4) |
3.804(9) (CH3(Dipp)⋯CH3(Dipp)) |
| 4.136(9) (CH3(SiMe3)⋯CH3(SiMe3)) |
|
8
|
1.915(2) |
1.918(2) |
10.8837(18) |
175.27(7) |
111.35(17) |
3.604(2) (Bu4N⋯Ph) |
| 4.089(4) (CH3(Dipp)⋯CH3(SiMe3)) |
[K{18c6}][Fe(L2)2]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 3 |
1.9135(14) |
1.9147(14) |
10.625(17) |
172.65(6) |
95.05(15) |
3.936(2) (CH3(Dipp)⋯SiCH3) |
| Co |
4
|
1.8787(23) |
1.8782(24) |
7.7857(8) |
178.43(8) |
1.22(23) |
3.416(4) (CH3(Dipp)⋯CH3(Dipp)) |
| 4.391(5) (CH3(SiMe3)⋯CH3(SiMe3)) |
|
9
|
1.878(2) |
1.884(2) |
11.1161(7) |
176.28(10) |
3.93(25) |
3.500(3) (Bu4N⋯Ph) |
| 3.497(4) (CH3(Dipp)⋯CH3(Dipp)) |
| 4.121(4) (CH3(SiMe3)⋯CH3(SiMe3)) |
| [K{18c6}][Co(L2)2]3 |
1.8835(10) |
1.8835(10)b |
10.5574(6) |
180b |
180b |
3.951(2) (CH3(Dipp)⋯CH3(SiMe3)) |
A large variety was observed for the Caryl–N–N′–C′aryl torsion angles (Table 1). While in 1 the opposing aryl rings twist with a torsion angle of 60(2)°, the rings are standing more or less trans to each other in 2 (161.68(14)°) and 3 (123.65(15)°). These large differences originate likely from respective aryl–K interaction as well as inter- or intramolecular dispersion forces between an iso-propyl group and an aryl ring of a second ligand. In the cobalt complex 4, the aryl rings of both ligands are facing each other (torsion angle of 1.22(23)°) which is enforced by the potassium ion. The intramolecular K⋯arene distances in 4 are with 3.1419(7) Å longer than in polymeric 1–3 (approx. 2.8 Å), probably due to the enforced proximity of opposing iso-propyl groups. Together with literature known [M(L2)2]− complexes, as well as further examples presented below, it reveals a rotational flexibility of the amide ligands around the N–M–N axis. This structural diversity contrasts the situation of the divalent precursors which exhibit a (symmetry generated) linear N–M–N axis with trans-oriented aryl rings only.18 As such, the amide ligand orientation is dictated by K-arene and not by interligand dispersion interactions via CH3⋯arene or CH3⋯CH3 units.
Solution state behaviour of [KM(L)2] complexes
1H NMR spectroscopy.
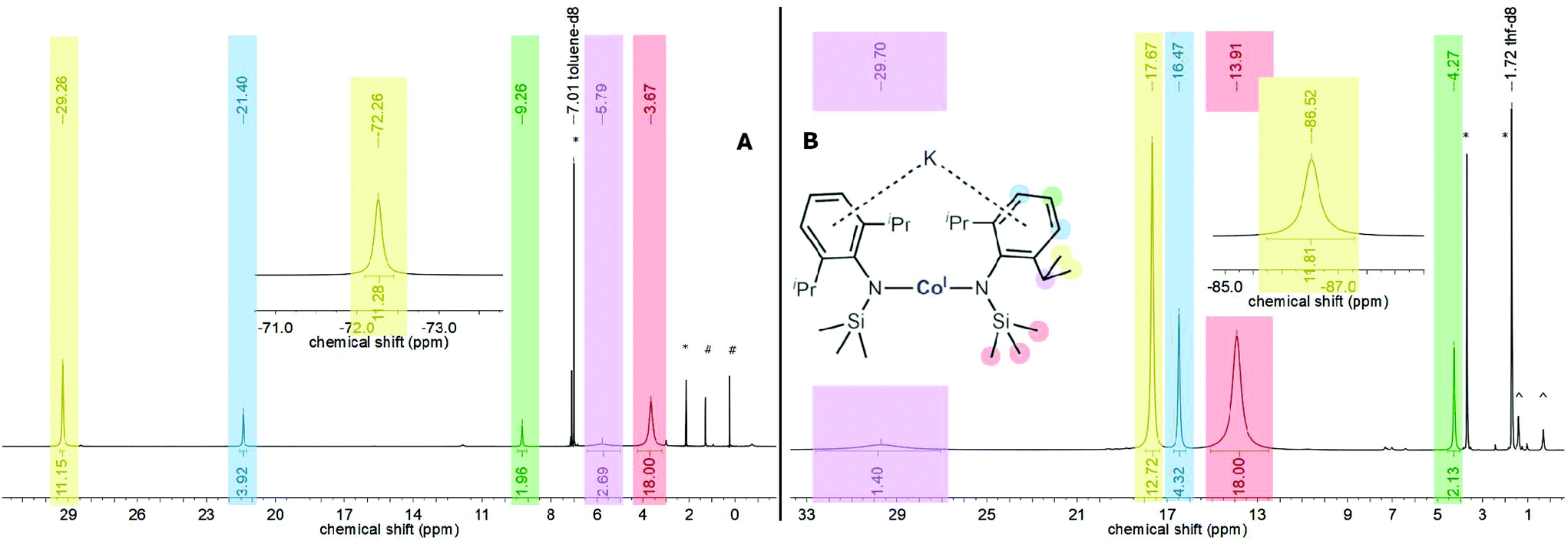
Given the observed structural variability of compounds 1–4 in solid state and their envisioned use for substrate activation, we sought further insights into their behaviour in solution with respect to the potassium cation speciation. Thereby it is important to note that 1–4 are highly soluble in toluene or Et2O, contrasting the congeners with 18-crown-6 masked potassium cations (only slightly soluble in Et2O). All complexes were first examined by 1H NMR spectroscopy in non-coordinating C6D6 revealing their paramagnetic nature by strongly shifted (in comparison with their expected diamagnetic signal position) and broadened proton signals. The paramagnetic shift comes primarily from interactions of the respective proton with the metal(I) ion via through-bond (contact shift) and through-space (pseudo-contact shift) interactions.19 In some cases, signal assignments were thus hampered (Table 2), e.g. for chromium (1) and manganese (2) only very broad signatures were detected. The proton spectra of 3 (iron, Fig. S5†) and especially 4 (cobalt, Fig. 3, left) are better behaved allowing for signal assignment via signal positions and integral intensities (Table 2). In 4, which shows rather sharp signatures, the signal belonging to the SiMe3 fragment in 4 is found at 3.67 ppm whereas the iso-propyl groups give rise to two signals at −72.3 and 29.3 ppm. Looking at the solid state structure the latter two signals likely represents the inward and outward positioned methyl functions of each iso-propyl group, suggesting suppression of the free rotation of the iso-propyl groups in solution. The huge paramagnetic shift difference between these two signals is remarkable and likely comes from pseudo-contact interactions, given the identical bond connection but different spacial distances to the metal ion (average d(Co–Cin) 4.3 Å vs. d(Co–Cout) 5.1 Å). Together with differences in line-broadening it suggests that the stronger paramagnetically influenced signal at −72.3 ppm belongs to the inward oriented methyl group and that the pseudo-contact and contact shifts have opposing signs. The meta-positioned protons of the aromatic systems are shifted downfield to 21.4 ppm, whereas the impact of the paramagnetic centre to the para-positioned ones (9.26 ppm) are weaker. The signal of the methine protons (5.79 ppm) are again strongly broadened due to the proximity of the cobalt(I) ion. Given the well-behaved 1H NMR spectrum of 4 (Co), we conducted further experiments to get qualitative insights into the speciation in solution, especially with respect to the location of the potassium ion (within the complex or as separated cation). The switch to the coordinating solvent THF-d8 impacted significantly the signal positions in comparison to the non-coordinating solvent toluene-d8 (Fig. 3, right). The highest impact of changing the solvent was detected for the methine protons, which are now found at 29.7 ppm (Δδ = 26 ppm). While the signal of the trimethylsilyl protons is shifted downfield by approx. 10 ppm to 13.9 ppm, signals of the methyl groups of the Dipp-units are shifted by around 12 ppm to higher field and are found at −86.5 ppm and 17.7 ppm, respectively (toluene-d8: −72.3, 29.3 ppm). The spectrum of 4 (Co) in THF-d8 is thereby analogous to the one obtained for the potassium ion separated complex [K{18c6}][Co(L2)2] (THF-d8, Fig. S19†),7 speaking to the complete solvation of the potassium ion by THF. Thereby, the process of the extraction of the potassium ion by THF requires a large excess of THF as addition of up to 20 equivalents of THF to a toluene-d8 solution of 4 (Co) led just to a slight shift of its 1H proton signals. In contrast, upon Et2O addition no shifting signals were observed, even using pure Et2O. This reflects the weak ability of THF and inability of Et2O to fully solvate the potassium cation. At low temperatures, the spectra of 4 in THF-d8 and toluene-d8 did not coincide (Fig. S20 and S22†), which indicated a persistent diverging potassium cation speciation between these solvents. All paramagnetic signals of 4 (Co) follow the Curie–Weiss law (δ(T) ∼ 1/T) in THF-d8 (Fig. S23†) as well as in toluene-d8 (Fig. S21†). Together with the absence of signal splitting,2,8 this showed that, like the cation/anion interaction, the electronic situation and complex geometry remained unchanged over the examined temperature range.
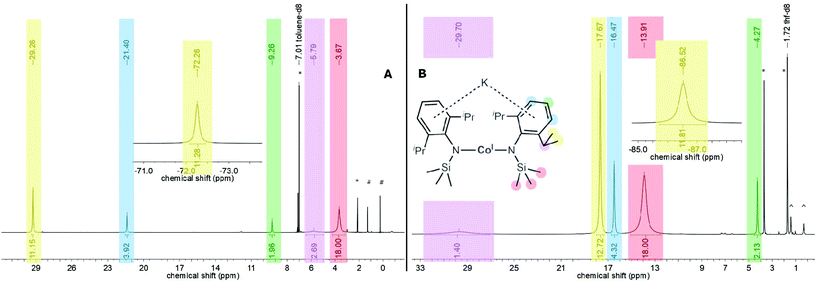 |
| | Fig. 3
1H NMR spectrum (500.1 MHz, 300 K) of 4 in toluene-d8 (left (A), * solvent, #n-pentane) and THF-d8 (right (B), ^ impurities). | |
Table 2
1H NMR signals of the complex anions of 1–9 with R = iPr (Cr), Me (Mn–Co) in ppm. The signal(s) belonging to the K{18c6} or NBu4 cations are found around their diamagnetic positions
| Metal |
Compound |
solvent |
SiR3 |
CH3 |
CHMe2 |
m-Ph |
p-Ph |
| Cr |
1
|
C6D6 |
6.81 |
No signal attribution possible |
| THF-d8 |
No signal attribution possible |
|
5
|
THF-d8 |
No signal attribution possible |
| Mn |
2
|
C6D6/THF-d8 |
No signal attribution possible |
| Fe |
3
|
C6D6 |
−1.65 |
−78.0/39.7 |
59.3 |
23.4 |
4.33 |
| THF-d8 |
−0.09 |
−103.1/— |
— |
— |
— |
|
3·2DMAP |
C6D6 |
−3.63 |
−78.6/39.4 |
— |
26.6 |
19.4 |
|
6
|
THF-d8 |
−1.03 |
−99.8/— |
— |
— |
— |
|
7
|
THF-d8 |
−0.49 |
−102.2/— |
— |
— |
— |
|
8
|
THF-d8 |
— |
−102.3/— |
— |
— |
— |
| Co |
4
|
Toluene-d8 |
3.67 |
−72.3/29.3 |
5.79 |
21.4 |
9.26 |
| THF-d8 |
13.9 |
−86.5/17.7 |
29.7 |
16.5 |
4.27 |
|
9
|
THF-d8 |
14.0 |
−88.0/17.4 |
30.1 |
16.0 |
7.31 |
With these results for 4 (Co) in mind, the solvent dependencies of the 1H NMR signals were also examined for complexes 1 (Cr), 2 (Mn) and 3 (Fe). For all three compounds similar the spectra in toluene-d8 or C6D6 (also in Et2O for 3) differed significantly from those in THF-d8. The latter are thereby identical to the 1H NMR spectra of the [K{18c6}][M(L)2] complexes (Table 2), whereas [K{18c6}][Cr(L1)2], 5, had to be synthesized first.3,6,8 Overall, 1H NMR spectroscopy showed for the [KML2] compounds that in non/weakly-coordinating solvents the K+ cation is likely connected to the complex anion, presumably residing between the aryl rings as in the solid state structure of 4, whereas in THF it is present as a solvent-separated counter-ion.
UV/Vis spectroscopy.
Given the observed 1H NMR spectroscopic features of compounds 1–4, a variable-temperature UV/Vis spectroscopic analysis was conducted. In case of the chromium compound 1, a UV/Vis spectrum recorded in THF showed three absorption bands at 286 nm (ε > 7930 L mol−1 cm−1), 343 nm (ε = 4440 L mol−1 cm−1) and 426 nm (ε = 3320 L mol−1 cm−1). These bands can be ascribed to ligand to metal charge transfer (LMCT) transitions (Table 3),3,12 and mimic those of 5 (Cr). When switching to toluene or Et2O only one absorption maximum was observed for 1 (Cr) (toluene: 431 nm, ε ≈ 5210 L mol−1 cm−1; Et2O: 435 nm, ε ≈ 6940 L mol−1 cm−1), speaking for a similar electronic situation for 1 in these two solvents. Cooling the respective solutions to −110 °C had no significant effect. For 2 (Mn) examinations were restricted to diethyl ether, due to decomposition in toluene and THF under these dilute conditions. Two absorption maxima at 448 nm (ε ≈ 1610 L mol−1 cm−1) and 565 nm (ε ≈ 2410 L mol−1 cm−1) were observed that superpose those of [K{18c6}][MnL22].8 For complex 3 (Fe) maxima at 421 nm (ε = 2120 L mol−1 cm−1), 610 nm (ε = 160 L mol−1 cm−1) and 773 nm (ε = 120 L mol−1 cm−1) were observed in THF (Fig. 4), whose positions are akin to the ones found for [K{18c6}][Fe(L2)2].3 The latter two maxima are tentatively assigned to d–d transitions.3,12 When switching to toluene as solvent, both LMCT bands at 368 nm (ε ≈ 3380 L mol−1 cm−1) and 432 nm (ε ≈ 3490 L mol−1 cm−1) appear as two distinct absorption maxima. Cooling these solutions down to −110 °C had no considerable effect on the position of the transitions. The UV/Vis spectrum of 3 (Fe) in Et2O resembled the one in THF with a LMCT band at 442 nm (ε ≈ 1540 L mol−1 cm−1) and a d–d transition at 601 nm (ε ≈ 100 L mol−1 cm−1). Upon cooling this solution to −110 °C the reversible disappearance of the LMCT band could be observed, whose origin is yet unclear.
 |
| | Fig. 4 UV/Vis spectrum (290–700 nm) of 3 in different solvents at 22 °C. | |
Table 3 UV/Vis absorption maxima of 1–9 and [K{18c6}][M(L2)2] (M = Fe, Co, L2 = –N(Dipp)SiMe3); 280–900 nm, (RT)
| Metal |
Compounds |
Solvent |
λ(ε)/nm(L mol−1 cm−1) |
|
UV/Vis spectrum was recorded but no absorption coefficients were given in the original report.
|
| Cr |
1
|
Toluene |
431 (5210) |
| THF |
286 (>7930), 343 (4440), 426 (3320) |
| Et2O |
435 (6940) |
|
5
|
THF |
288 (7710), 338 (3880), 421 (2160) |
| Mn |
2
|
Et2O |
448 (1610), 565 (2410), 849 (2650) |
| Fe |
3
|
Toluene |
368 (3380), 432 (3490), 602 (140) |
| THF |
421 (2120), 610 (160), 773 (120) |
| Et2O |
442 (1540), 601 (100) |
|
3·2DMAP |
Toluene |
360 (3230), 434 (3190), 607 (130) |
|
6
|
THF |
422 (1990), 771 (170) |
|
7
|
THF |
420 (5020), 613 (320), 771 (260) |
|
8
|
THF |
364 (1660), 420 (1900) |
| [K{18c6}][FeL22]3 |
THF |
428 (4000), 626 (100) |
| Co |
4
|
Toluene |
385 (3080) |
| THF |
336 (2800), 385 (2300) |
| Et2O |
393 (3560) |
|
9
|
THF |
340 (4770), 390 (4120) |
| [K{18c6}][CoL22]a3 |
THF |
337 (2380), 391 (2030), 629 (100) |
The UV/Vis spectrum of 4 (Co) in toluene exhibited one LMCT band at 385 nm (ε ≈ 3080 L mol−1 cm−1) which splits at −110 °C into two bands at 387 nm (ε ≈ 3910 L mol−1 cm−1) and 410 nm (ε ≈ 3850 L mol−1 cm−1). Measurements in THF showed two absorption bands at 336 nm (ε ≈ 2800 L mol−1 cm−1) and 385 nm (ε ≈ 2300 L mol−1 cm−1) at room temperature, which is comparable to the UV/Vis spectrum of [K{18c6}][Co(L2)2].3 A UV/Vis spectrum of 4 (Co) in diethyl ether showed a broad band at 393 nm with additional low-intensity d–d transitions (ε < 100 L mol−1 cm−1) above 600 nm. Cooling a solution of 4 (Co) in THF or Et2O had no considerable effect onto its UV/Vis spectroscopic properties. Overall the UV/Vis spectroscopic examinations showed markedly different spectra for complexes 1 (Cr), 3 (Fe) and 4 (Co) for either THF or toluene solutions. Thereby the spectra in THF coincided with those of cation separated complexes mimicking the 1H NMR spectroscopic results. In Et2O the UV/Vis spectroscopic situation is ambiguous. Whereas for 1 and 4 the respective spectra resembled those in toluene, implicating that the potassium ion is also not separated from the complex anion, for the iron complex 3 the spectroscopic features in Et2O are similar to those in THF. As this is not in line with the NMR spectroscopic observations, it speaks for an additional solvent effect, such as solvent coordination.
Structural effects of donor solvents on [KM(L)2] complexes
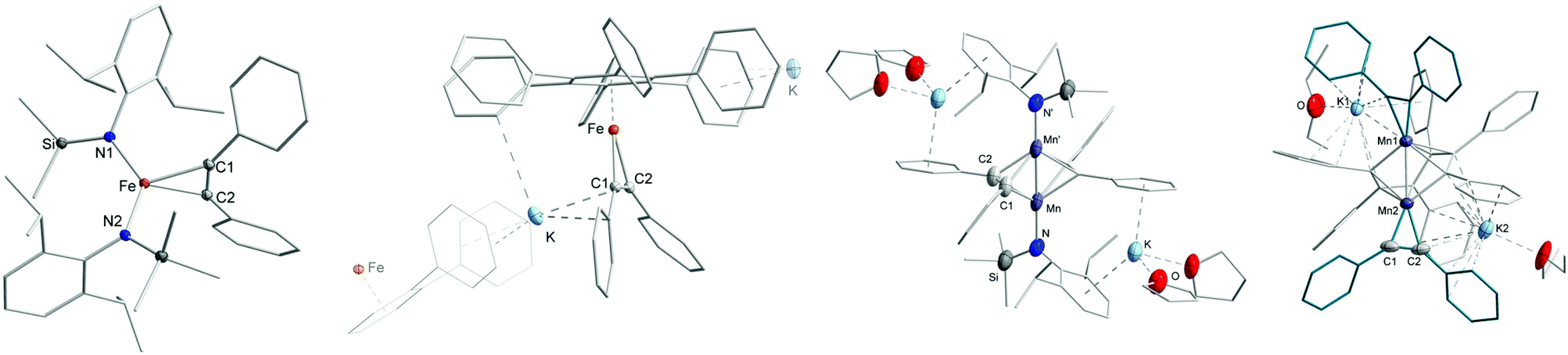
Having observed the 1H NMR and UV/Vis spectroscopic changes for complexes 1–4 when going from non-coordinating to coordinating solvents, we were interested if this could be somewhat retraced on a structural level. This turned out to be highly challenging as small amounts of donor solvents such as Et2O or THF lead to a very high solubility of the respective compounds 1–4 even in n-pentane, indicative of solvent coordination. Upon rigorous drying of respective solutions, the donor solvents could be removed as these complexes were again insoluble in n-pentane. Nonetheless, in due course we could identify for iron the highly soluble diethyl ether adduct 3·Et2O (Fe) using X-ray diffraction analysis (Fig. 5, left). As the crystal data suffers from some flaws, its general features are only shortly discussed. In 3·Et2O (Fe) the potassium cation is coordinated in an intramolecular fashion by the two aryl rings, as observed for 4 (Co) and [KNiL22].5 It thus shows how the polymeric form is transformed to a monomer in solution. A diethyl ether molecule completes the coordination sphere of the potassium ion. As a result of the intramolecular potassium cation fixation the N–Fe–N bond axis is nearly linear with a negligible CAr–N–N′–C′Ar torsion angle. Given the difficulties in the crystallization progresses, which we attributed to the volatility of the employed Et2O in conjunction with the high solubility of 3·Et2O (Fe), we used DMAP as an exemplary donor ligand. This gave the stable adduct 3·2DMAP (Fe) (Fig. 5, middle) as green crystals in yields up to 75%. Similar to 3·Et2O (Fe), the potassium cation is located between the two aryl rings in an intramolecular fashion and is further coordinated by two DMAP molecules. Looking at the UV/Vis spectrum of 3·2DMAP, two distinct LMCT bands at 360 nm (ε ≈ 3230 L mol−1 cm−1) and 434 nm (ε ≈ 3190 L mol−1 cm−1) are present. This mimics the behaviour of pure 3 in toluene, corroborating for the latter a persistent intramolecular potassium cation complexation. For chromium, the monomeric complex 1·3THF (Cr) was obtained as revealed by X-ray diffraction analysis. In 1·3THF (Cr) the potassium cation resides on the external side of one of the aryl rings of the silylamide ligand set with three tetrahydrofuran molecules completing its coordination sphere. Together with 3·Et2O (Fe) and 3·2DMAP (Fe), 1·3THF (Cr) thus gives insight on how the potassium cation is extracted out of the aryl pocket of the [KM(L)2] complexes upon addition of donor solvents. Remarkably, no coordination of donor solvent molecules to the 3d-metal ion takes place, which is observed for their divalent counterparts.20
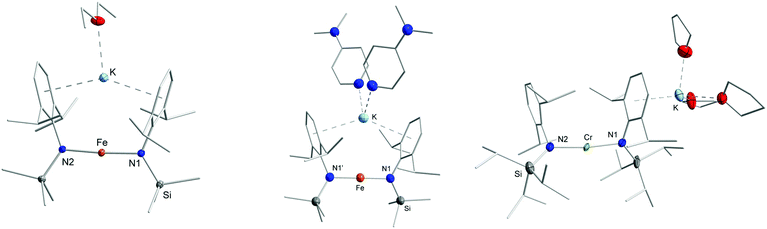 |
| | Fig. 5 Sections of the crystal structure of 3·Et2O (left), 3·2DMAP (middle) and 1·3THF (right). H atoms are omitted for clarity. In case of 1·3THF disorders found for one THF molecule as well as for two iso-propyl groups are not depicted. | |
Sodium, lithium and NBu4+ as counter ions
The synthesis of homoleptic quasilinear metal(I) silylamides was so far exclusively approached using potassium graphite as the reductant. In view of the different solid state structures of the neutral compounds we wanted to introduce lithium and sodium as reductants for such complexes, as these smaller ions might enforce a different solid or solution state behaviour. To probe this possibility we chose the iron derivative as a representative. Treatment of [Fe(L2)2] with lithium or sodium (5% Na dispersed in NaCl)21 led to a slow (Li), respectively rapid (Na) colour change from orange/red to brownish green (Scheme 2). Attempts to obtain the presumed neutral reaction products [AFe(L2)2] (A = Li, Na) led to their decomposition. This was evidenced by a colour change of the reaction solutions overnight at −40 °C or upon evaporation of the solvent and was additionally verified via the subsequent recrystallization of the metal(II) precursor. The initial formation of [AFe(L2)2] could be proven via1H NMR spectroscopy when performing the reduction in THF-d8. Adding either 12-crown-4 (for lithium) or 18-crown-6 (for sodium) to a solution of [AFe(L2)2] led, after work-up, to the isolation of the respective ion-separated products [Li{12c4}2][Fe(L2)2], 6, and [Na{18c6}(Et2O)][Fe(L2)2], 7, as shown in Fig. 6.
 |
| | Scheme 2 Reduction of [Fe(L2)2] (L2 = N(Dipp)SiMe3) with lithium (6) or sodium (7), respectively. | |
 |
| | Fig. 6 Sections of the crystal structure of 7 (left), 8 (middle) and 9 (right). H atoms and a disorder of the 18-crown-6 fragment of 7 are omitted for clarity. For 8 and 9 the shortest CH3–arene interactions are depicted (dashed line). | |
Structural analysis via X-ray diffraction on suitable crystals showed most notably different orientations of the amide ligands within the [Fe(L2)2]− complex anion. In 6 (Fe), the aryl functions are pointing in the opposite direction (torsion angle 148.9(3)°), whereas in 7 (Fe), they are in an eclipsed position (torsion angle 7.0(4)°). As expected, the UV/Vis and paramagnetic 1H NMR spectra in THF-d8 for both 6 and 7 resembled those of [K{18c6}][Fe(L2)2]. Similar to the extraction of the alkali metal ion by crown-ethers, we also wanted to use the [KM(L)2] complexes to introduce an organic cation (Scheme 3). This was already shown in case of the related nickel complex [KNi(L2)2] as well as in the case of chromium [K{dme}4][Cr(L1)2].5,6
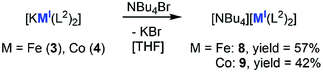 |
| | Scheme 3 Introduction of the organic cation NBu4+ in [NBu4][M(L2)2] (M = Fe (8), Co (9), L2 = N(Dipp)SiMe3). | |
The reaction of 2 (Mn) with NBu4Br or PPh4Br led to discolouring of the reaction mixture as well as to precipitation of small amounts of a black solid on the stirring bar. This indicated an insufficient stability of the cation in the presence of the highly reducing manganese(I) complex [MnL22]− (Ered ≈ −2.5 V vs. Fc/Fc+).8 The reaction of 3 (Fe) and 4 (Co) with NBu4Br led, after work-up, to the isolation of the corresponding compounds [NBu4][Fe(L2)2], 8, and [NBu4][Co(L2)2], 9 (Scheme 3). X-Ray diffraction analysis revealed that the exchange of the cation has no significant influence on the M–N bond lengths and the N–M–N angles in the anion (Table 1). However, larger differences in the torsion angles were notable. Whereas the ligand set of 9 (Co) remains in an eclipsed conformation with a torsion angle of 3.93(25)°, a rotation around the N–M–N axis took place in 8 (Fe), resulting in a staggered conformation (111.35(17)°). Both features can be attributed to interactions of the aryl rings with one of the methyl groups of the NBu4+ cation, whose distances are in part even shorter than intramolecular CH3⋯aryl/CH3⋯CH3 distances (Table 1). It implicates that intramolecular dispersion forces, thought as essential for the stability of two-coordinated complexes, are here of lesser importance. The impact of attracting intramolecular dispersion interactions is further discussed to influence M–N distances.12,22 However, the cobalt complexes 4 (KCo), 9 (NBu4Co) and [K{18c6}][Co(L2)2], for example, exhibit virtually identical Co–N distances, despite bearing drastically differing ligand orientations and interactions in solid state.
Magnetic properties in solution
Having understood the speciation of the complexes 1–4 in solution, we were briefly interested if the intramolecular potassium ion complexation of the [KML2] complexes has a discernible impact onto the solution state magnetic properties in comparison with to their cation separated counterparts. Using Evans method (Table 4) the effective magnetic moments of 1 (Cr) and 2 (Mn) in C6D6 were determined to be 5.17μB (μS.O.(S=5/2) = 5.92μB) and 4.68μB (μS.O.(S=2) = 4.82μB). These are lower than the expected spin-only values (CrI: μS.O.(S=5/2) = 5.92μB, MnI: μS.O.(S=2) = 4.92μB) but are in the range of other linear chromium(I) and manganese(I) complexes (e.g. [K{dme}4][Cr(L1)2]: 5.2μB, K{18c6}[Mn(L2)2]: 4.98).6,8 Higher than spin-only values were measured for complexes 3 (Fe) (μeff = 5.31μB, μS.O.(S=3/2) = 3.87μB), 3·2DMAP (Fe) (μeff = 4.89μB), and 4 (Co) (μeff = 4.50μB, μS.O.(S=1) = 2.83μB) which is expected for linear complexes with unquenched orbital contributions.13,14,23 When switching to THF-d8 a slight drop in the respective magnetic susceptibilities was observed for 1 (4.92μB), 3 (4.34μB) and 4 (3.93μB). The values in THF are comparable to those found for the respective cation separated complexes (5–9), stressing that the complex anion is more or less unaffected by a separated counter ion in solution. Within the limits of the Evans method this indicates a beneficial factor of intramolecular potassium ion complexation, either by blocking of the free amide rotation or fixation of a near-linear N–Fe–N axis which would overall enhance orbital contributions.
Table 4 Solution state magnetic susceptibilities of complexes 1–9 (L2 = N(Dipp)SiMe3) in THF-d8 and C6D6via the Evans method. Theoretical spin-only values are given for the high-spin case
| Metal |
Compound |
Cation |
μ
eff [μB] in THF-d8 |
μ
eff [μB] in C6D6 |
| Cr [μS.O. = 5.92μB] |
1
|
K |
4.92 |
5.17 |
|
5
|
K{18c6} |
4.21 |
Insoluble |
| Mn [μS.O. = 4.90μB] |
2
|
K |
Partial decomposition |
4.68 |
| [K{18c6}][Mn(L2)2] |
K{18c6} |
4.988 |
Insoluble |
| Fe [μS.O. = 3.87μB] |
3
|
K |
4.34 |
4.85 |
|
3·2DMAP |
K(DMAP)2 |
— |
4.89 |
|
6
|
Na{18c6} |
4.74 |
Insoluble |
|
7
|
Li{12c4}2 |
4.24 |
Insoluble |
|
8
|
NBu4 |
4.30 |
Insoluble |
| Co [μS.O. = 2.83μB] |
4
|
K |
3.93 |
4.18 |
|
9
|
NBu4 |
3.58 |
Insoluble |
Reactivity of [KM(L)2] complexes towards diphenylacetylene
Lastly, we conducted first experiments concerning the impact of the intramolecular potassium ion complexation on the reactivity of the metal(I) complexes for manganese to cobalt given the identical ligand set. For that we chose diphenyl acetylene (PhCCPh) as a probe, given our recent examination of anionic metal(I) silylamides [M(N(SiMe3)2)2]− towards alkynes, that yielded predominantly side-on alkyne complexes ([M(η2-RCCR)(N(SiMe3)2)2]−) but also some bond reduction and disproportionation processes in case of manganese.24 In this context the –N(Dipp)SiMe3 ligand set was already probed for manganese yielding relatively stable complexes of the type [Mn(η2-RCCR)(L2)2] (R = Et, Ph).24 As such we pursued first the isolation of the iron and cobalt derivatives. Reaction of [K{18c6}][Fe(L2)2] with PhCCPh led to the quantitative formation of the side-on alkyne complex [K{18c6}][Fe(L2)2(η2-PhCCPh)], 10 (Scheme 4). The same reaction with [K{18c6}][Co(L2)2] yielded in an initial color change to red brown, but only the employed cobalt(I) precursor was isolated upon crystallisation. This is not surprising as weak and reversible binding of alkynes to cobalt silylamides was already observed in case of sterically less demanding [Co(N(SiMe3)2)2]−.24 Treating 4 (KCo) with PhCCPh afforded also only the starting materials. In contrast, when 3 (Fe) was treated with PhCCPh in Et2O, crystallization yielded the mixed arene/alkyne iron complex [K(D)(Fe(C6Ph6)(PhCCPh)], 11·D, (D = none, Et2O) (Fig. 7, second left, ESI†) as well as inseparable free hexaphenylbenzene (HPB). Following the reaction of 3 (Fe) in C6D6 by 1H NMR spectroscopy showed the rapid consumption of PhCCPh and formation of minor amounts of free HPB. 11·D corresponds to the known iron compound [K{18c6}][Fe(HPB)(η2-PhCCPh)], obtained by reaction of the iron(–I) synthon [K{18c6}][Fe(C10H8)2] with PhCCPh,25 as well as the manganese analogue [K{18c6}][Mn(HPB)(η2-PhCCPh)].24 The latter was observed during the reaction of the manganese(I) complex [Mn(N(SiMe3)2)2]− with PhCCPh under redox disproportionation and ligand redistribution. For 3 (Fe) a similar mechanism is plausible, whereas 3 acts as reductant as well as source of an iron atom under formal release of KL2 and [Fe(L2)2].
 |
| | Scheme 4 Reactivity of [K{m}][M(L2)2] of manganese and iron (m = none or 18c6) towards PhCCPh (HPB = C6Ph6). | |
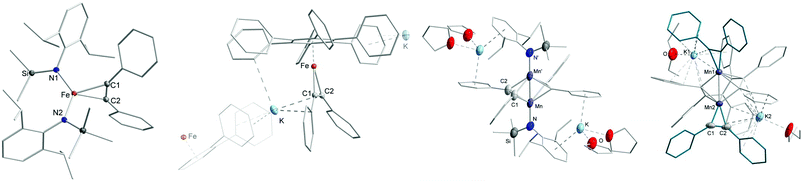 |
| | Fig. 7 Sections of the crystal structures of 10–13 (from left to right). H atoms and the K{18c6} unit of 10 are omitted for clarity. | |
Treatment of 2 (KMn) in toluene with PhCCPh resulted also in the trimerisation product HPB. Remarkably, small amounts of crystals of the dimeric complexes 12 and 13 (Fig. 7, right, ESI†) were obtained that give insights into this manganese mediated trimerisation process. In 12 two manganese ions are bridged by two PhCCPh units, which are oriented orthogonal towards the Mn–Mn axis. The distance between the manganese atoms are with 2.434(2) Å rather short and comparable to a ketimide linked MnII/MnII dimer.26 Similar to 11·D, the formation of 12 is probably the result of formal redox disproportionation of the employed 2 (Mn). Compound 12 is in close resemblance to a recently reported dinuclear iron complex ([(L2)Fe(μ-η2-PhCCPh)2Fe(L2)], which was observed upon reacting [Fe(IDipp)(L2)] (IDipp = 1,3-bis(2,6-di-iso-propylphenyl) imidazolin-2-ylidene) with PhCCPh.27 It exhibits significantly less distorted alkyne ligands (C–C 1.349(7) Å, C–C–Cphenyl bond angle of 133.3(4)°) which speaks to a stronger π-backbonding into the π*-orbitals of the alkyne within 12. In the other dimeric complex 13 two manganese atoms (Mn–Mn 2.5665(11) Å) are linked via a C6-chain, stemming from incomplete alkyne trimerisation. Judging from the bond lengths, the carbon fragment is best described as two allyl units linked via a C–C single bond giving it a formal dianionic charge.28–30 The coordination sphere of each manganese ion, that lacks any silylamide ligation, is completed by an alkyne ligand, which experiences only a moderate C–C bond elongation (1.267(7) Å and 1.287(8) Å). The presence of two potassium ions, each being located in a pocket composed of two aryl rings of the C6 fragment as well as one of an alkyne, leads to a formal oxidation state of Mn0 for both manganese ions. The formation of the trimerised C6 fragment in 13 is remarkable as it poses a snapshot in the final step of alkyne trimerisation. Metallacycloheptatriene-like intermediates in alkyne trimerisation were so far only observed for mononuclear complexes,28–31 whereas bimetallic derivatives were only postulated.27 Any attempts of isolating pure 12 or 13via adjustment of the reaction stoichiometry as well as reaction temperature failed so far, in part by the ubiquitous presence of the complete trimerisation product HPB. When the reaction mixture of 2 (KMn) with PhCCPh in Et2O was layered after several minutes with 18c6 in Et2O and stored at −35 °C, to promote crystallisation via cation separation, only the known η2-alkyne complex K{18c6}[Mn(L2)2(η2-PhCCPh)]24 can be isolated, showing that the formation of 12 and 13 is a slower process.
Conclusions
The syntheses and characterization of neutral quasilinear, homoleptic 3d-metal(I) silylamides of the type [KM(L)2] (L1 = N(Dipp)SiiPr3 (Cr); L2 = N(Dipp)SiMe3 (Mn – Co)) via the reduction of the respective quasilinear metal(II) silylamide with KC8 in non-coordinating solvents were presented. X-Ray diffraction analysis shows that the alkali metal ion is coordinating to the aryl rings in either an intramolecular (Co) or intermolecular intermolecular fashion (Cr, Mn, Fe). For the latter this results in the presence of unprecedented 1D-coordination polymers of linear, open-shell metal(I) complexes. Detailed 1H NMR spectroscopic examinations of the complexes in solution showed that, in non-coordinating solvents, the cation remains in the vicinity of the complex anion whereas, in good donor solvents like THF, these compounds exist as an ion pair. When using lithium or sodium as reductants the respective iron complexes [AFe(L2)2] (A = Li, Na) could be generated, which were isolated by sequestrating the alkali metal. Starting off the formally neutral metal(I) silylamides, the alkali metals could also be exchanged by an organic cation in case of iron and cobalt. Upon comparison of the structural metrics of all obtained and literature reported [M(L)2]− complexes, it is shown that the two amide ligands can virtually exhibit any orientation towards each other (cis, trans or orthogonal). This is dependent on the presence of not only intra- but also intermolecular interactions in solid states and has no considerable effect on the metal–amide bond length. It indicates that intramolecular dispersion forces are an important, but not an essential factor for the stabilization of homoleptic, two-coordinate metal(I) amides. First reactivity studies of the neutral compounds [KM(L2)2] (M = Mn –Co) towards diphenyl acetylene showed distinct differences with respect to their cation separated counterparts K{18c6}[M(L2)2]. Whereas the latter prefer the formation of η2-alkyne complexes, the neutral complexes serve as coordination sites as well as reductants. This leads ultimately to substrate trimerisation whereas unusual intermediates could be structurally characterized. It thus shows that the intramolecular potassium ion complexation labilises the metal amide bond and give rise to distinct reactivities The further use of neutral metal(I) silylamides as precatalysts as well as a detailed inspection of their magnetic properties is currently explored in our lab.
Experimental section
Materials and methods
All manipulations were carried out in a glovebox under a dry argon atmosphere, unless indicated otherwise. Used solvents were dried by continuous distillation over sodium metal for several days, degassed via three freeze–pump–thaw cycles and stored over molecular sieves 4 Å. Deuterated solvents were used as received, degassed via three freeze-pump cycles and stored over molecular sieves 4 Å. The 1H NMR spectra were recorded on a Bruker AV 500, a Bruker HD 500 or a Bruker HD 300 NMR spectrometer (Bruker Corporation, Billerica, USA). Chemical shifts are reported in ppm relative to the residual proton signals of the solvent (for 1H). w1/2 is the line width of a signal at half its maximum intensity. Integrals of the broad signals ligand set were obtained directly or by peak fitting (in case of overlapping signals) using the MestreNova software package (Mestrelab, Santiago de Compostela, Spain). IR measurements were conducted on a Bruker Alpha ATR-IR spectrometer (Bruker Corporation, Billerica, USA). The UV/VIS measurement were recorded on an AnalytikJena Specord S600 using WinASPECT software. Elemental analysis were performed by the “in-house” service of the Chemistry Department of the Philipps University Marburg, Germany using a CHN(S) analyser vario MICRO Cube (Elementar Analysensysteme GmbH, Langenselbold, Germany). Dispersed sodium (5% Na/NaCl),21 [Cr(N(Dipp)SiiPr3)2],6 [Mn(N(Dipp)SiMe3)2],8 [Fe(N(Dipp)SiMe3)2],14 and [Co(N(Dipp)SiMe3)2],12 were prepared according to literature procedures.
Synthesis and characterization
[KM(L)2] (L1 = N(Dipp)SiiPr3 (Cr); L2 = N(Dipp)SiMe3 (Mn – Co)).
One equivalent of [M(L)2] (M = Cr–Co) was dissolved in either 10 mL toluene or diethyl ether. After adding KC8 (1.1 equiv.) the reaction mixture was stirred for several minutes at room temperature, while a change in colour was observed (Cr: dark red → red; Mn: beige → dark violet; Fe: orange → red; Co: dark red → light green). The graphite was filtered off and all volatiles were removed under reduced pressure. After washing with n-pentane and drying in vacuo, [KM(L)2] (1–4) was obtained in yields of 28–92%.
[KCr(N(Dipp)SiiPr3)2] (1).
Using 500 mg of [Cr(N(Dipp)SiiPr3)2], 1 could be obtained as red solid. Yield: toluene: 391 mg (0.52 mmol, 74%), Et2O: 447 mg (0.59 mmol, 86%). Crystals, suitable for X-ray diffraction analysis, were obtained from a concentrated toluene solution of 1 at −40 °C. 1H NMR (500.1 MHz, C6D6, 300 K, ppm): δ = 22 (bs, w½ = 1900 Hz), 15 (bs, w½ = 1300 Hz), 12 (bs, w½ = 310 Hz), 7 (bs, 18 H, w½ = 2100 Hz, Si(CH(CH3)2)3), 4.0 (s, w½ = 42 Hz), 3.6 (s, w½ = 32 Hz), 2.4 (s, w½ = 20 Hz), 0.29 (s, w½ = 22 Hz), −16 (bs, w½ = 200 Hz) ppm. (300.2 MHz, THF-d8, 300 K, ppm): δ = 16 (bs, w½ = 920 Hz), 12 (bs, w½ = 810 Hz), 2.65 (s, w½ = 17.3 Hz). Elemental analysis: C42H76CrKN2Si2 (756.35 g mol−1): calcd: N 3.70, C 66.70, H 10.13; found: N 3.84, C 66.36, H 10.05%. IR (ATR, cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2941 (m), 2861 (m), 1576 (w), 1456 (m), 1407 (s), 1313 (m), 1244 (s), 1140 (w), 1101 (w), 992 (w), 925 (s), 878 (m), 804 (w), 767 (s), 719 (w), 640 (s), 554 (w), 525 (w), 487 (w), 418 (m). EVANS (500.1 MHz, 300 K, C6D6 + 1% TMS): μeff = 5.17μB; μS.O. = 5.92μB. (500.1 MHz, 300 K, C6D6 + 1% TMS): μeff = 4.92μB; μS.O. = 5.92μB.
= 2941 (m), 2861 (m), 1576 (w), 1456 (m), 1407 (s), 1313 (m), 1244 (s), 1140 (w), 1101 (w), 992 (w), 925 (s), 878 (m), 804 (w), 767 (s), 719 (w), 640 (s), 554 (w), 525 (w), 487 (w), 418 (m). EVANS (500.1 MHz, 300 K, C6D6 + 1% TMS): μeff = 5.17μB; μS.O. = 5.92μB. (500.1 MHz, 300 K, C6D6 + 1% TMS): μeff = 4.92μB; μS.O. = 5.92μB.
[KMn(N(Dipp)SiMe3)2] (2).
Using 100 mg of [Mn(N(Dipp)SiMe3)2], 2 could be obtained as dark violet solid. Yield: toluene: 5 mg (0.009 mmol, 5%), Et2O: 30 mg (0.051 mmol, 28%). Crystals, suitable for X-ray diffraction analysis, were obtained from a n-pentane layered solution of 2 in toluene at −35 °C. 1H NMR (500.1 MHz, C6D6, 300 K, ppm): δ = 16 (bs, w½ = 1400 Hz), 12 (bs, w½ = 630 Hz), 2.11 (s, w½ = 8.09 Hz), 2.01 (s, w½ = 14.3 Hz), 1.4 (bs, w½ = 450 Hz), 1.21 (s, w½ = 8.17 Hz), 0.99 (s, w½ = 18.0 Hz), 0.41 (s, w½ = 15.4 Hz), 0.11 (s, w½ = 11.0 Hz), −7.3 (bs, w½ = 1400 Hz). (500.1 MHz, THF-d8, 300 K, ppm): δ = 15 (bs, w½ = 2100 Hz), 11.0 (bs, w½ = 470 Hz), 6.10 (s, w½ = 55.3 Hz), 3.9 (s, w½ = 56 Hz), 2.89 (s, w½ = 14.8 Hz), 2.29 (s, w½ = 6.31 Hz), 0.20 (bs, w½ = 220 Hz), −0.06 (s, w½ = 34 Hz), −13 (bs, w½ = 860 Hz). Elemental analysis C30H52MnKN2Si2 (590.96 g mol−1): calcd: N 4.74, C 60.97, H 8.87; found: N 5.00, C 61.30, H 8.65%. IR (ATR, cm−1): = 2953 (m), 2867 (w), 1577 (vw), 1459 (w), 1416 (s), 1380 (w), 1359 (w), 1313 (m), 1237 (vs), 1190 (s), 1105 (w), 1052 (w), 1039 (w), 958 (w), 916 (s), 879 (w), 824 (vs), 784 (vs), 741 (m), 666 (m), 619 (w), 571 (w), 534(m), 431 (m). EVANS (500 MHz, 300 K, C6D6 + 1% TMS): μeff = 4.68μB; μS.O. = 4.90μB.
[KFe(N(Dipp)SiMe3)2] (3).
Using 350 mg of [Fe(N(Dipp)SiMe3)2], 3 could be obtained as red solid. Yield: toluene: 339 mg (0.57 mmol, 90%), Et2O: 343 mg (0.58 mmol, 92%). Crystals, suitable for X-ray diffraction analysis, were obtained from a n-pentane layered solution of 3 in toluene at −35 °C. Crystals of 3·Et2O, suitable for X-ray diffraction analysis, were obtained from a n-pentane layered solution of 3 in minimal amounts of Et2O at −35 °C. The Et2O adduct is soluble in n-pentane whereas the one without is not. 1H NMR (500.1 MHz, C6D6, 300 K, ppm): δ = 60 (bs, 2 H, CH(CH3)2), 39.7 (bs, 12 H, w½ = 620 Hz, CH(CH3)2), 23.4 (bs, 4 H, w½ = 470 Hz, m-PhH), 4.3 (bs, 2 H, w½ = 290 Hz, p-PhH), 0.15 (s, w½ = 7.70 Hz), −1.7 (bs, 18 H, w½ = 1800 Hz, Si(CH3)3), −78 (bs, 12 H, w½ = 1200 Hz, CH(CH3)2). (300.2 MHz, THF-d8, 300 K, ppm): δ = 29 (bs, w½ = 560 Hz), 28 (bs, w½ = 360 Hz), 12.6 (bs, w½ = 240 Hz), 1.3 (bs, w½ = 33 Hz), 0.26 (bs, w½ = 20 Hz), −0.1 (bs, 18 H, w½ = 1300 Hz, Si(CH3)3), −103 (bs, w½ = 1500 Hz, CH(CH3)2). Elemental analysis C30H52FeKN2Si2 (591.87 g mol−1): calcd: N 4.73, C 60.88, H 8.86; found: N 4.91, C 60.97, H 8.63%. IR (ATR, cm−1): = 2947 (m), 2864 (w), 1582 (w), 1462 (w), 1417 (s), 1379 (w), 1357 (w), 1317 (m), 1238 (s), 1198 (m), 1141 (w), 1102 (m), 1051 (w), 1041 (w) 978 (m), 914 (s), 877 (m), 828 (vs), 787 (vs), 745 (m), 671 (m), 624 (w), 576 (w), 541 (w), 425 (m). EVANS (500 MHz, 300 K, C6D6 + 1% TMS): μeff = 4.85μB; μS.O. = 3.87μB. (500 MHz, 300 K, THF-d8 + 1% TMS): μeff = 4.34μB; μS.O. = 3.87μB.
[K(DMAP)2Fe(N(Dipp)SiMe3)2] (3·2DMAP).
Crystals, suitable for X-ray diffraction analysis, were obtained by adding two equivalents of DMAP (20.5 mg, 0.17 mmol, 2 eq.) to 50 mg of 3 (0.08 mmol, 1 equiv.) in Et2O, layering the solution with n-pentane and keeping it at −40 °C for several days. Yield: 49.7 mg (0.06 mmol, 75%). 1H NMR (500.1 MHz, C6D6, 300 K, ppm): δ = 61 (bs, w½ = 260 Hz), 39.4 (bs, 12 H, w½ = 600 Hz, CH(CH3)2), 26.2 (bs, 4 H, w½ = 480 Hz, m-PhH), 19.4 (bs, 2 H, w½ = 170 Hz, p-PhH), 1.8 (bs, w½ = 290 Hz), 1.2 (bs, w½ = 63 Hz), 0.98 (bs, w½ = 96 Hz), 0.23 (bs, w½ = 230 Hz), −1.75 (bs, w½ = 550 Hz), −3.6 (bs, 18 H, w½ = 1700 Hz, Si(CH3)3), −79 (bs, 12 H, w½ = 1300 Hz, CH(CH3)2). Elemental analysis C44H72FeKN6Si2 (591.87 g mol−1): calcd: N 10.05, C 63.20, H 8.68; found: N 10.38, C 62.83, H 8.50%. IR (ATR, cm−1): = 2949 (m), 2863 (m), 1603 (s), 1534 (m), 1461 (w), 1442 (w), 1417 (s), 1387 (m), 1357 (w), 1318 (m), 1230 (s), 1200 (m), 1154 (w), 1139 (w), 1106 (m), 1056 (w), 1042 (w), 992 (s), 951 (m), 928 (s), 881 (w), 834 (vs), 801 (vs), 781 (vs), 752 (m), 669 (m), 622 (m), 527 (m), 480 (w), 430 (m). EVANS (500 MHz, 300 K, C6D6 + 1% TMS): μeff = 4.89μB; μS.O. = 3.87μB.
[KCo(N(Dipp)SiMe3)2] (4).
Using 200 mg of [Co(N(Dipp)SiMe3)2], 4 could be obtained as light green solid. Yield: toluene: 159 mg (0.27 mmol, 74%), Et2O: 173 mg (0.29 mmol, 81%). Crystals, suitable for X-ray diffraction analysis, were obtained from a n-pentane layered solution of 4 in toluene at −25 °C. 1H NMR (500.1 MHz, toluene-d8, 300 K, ppm): δ = 29.3 (s, 12 H, w½ = 29.4 Hz, CH(CH3)2), 21.4 (s, 4 H, w½ = 26 Hz, m-PhH), 9.26 (s, 2 H, w½ = 22 Hz, p-PhH), 5.8 (bs, 4 H, w½ = 310 Hz, CH(CH3)2), 3.7 (s, 18 H, w½ = 78 Hz, Si(CH3)3), −72.3 (s, 12 H, w½ = 50 Hz, CH(CH3)2). (500.1 MHz, THF-d8, 300 K, ppm): δ = 30 (bs, 4 H, w½ = 800 Hz, CH(CH3)2), 17.7 (s, 12 H, w½ = 55 Hz, CH(CH3)2), 16.5 (s, 4 H, w½ = 43 Hz, m-PhH), 13.9 (s, 18 H, w½ = 210 Hz, Si(CH3)3), 4.27 (s, 2 H, w½ = 34 Hz, p-PhH), −86.5 (s, 12 H, w½ = 150 Hz, CH(CH3)2). Elemental analysis C30H52CoKN2Si2 (594.96 g mol−1): calcd: N 4.71, C 60.56, H 8.81; found: N 4.90, C 60.24, H 8.97%. IR (ATR, cm−1): = 2953 (m), 2865 (w), 1567 (w), 1453 (m), 1415 (vs), 1367 (m), 1345 (w), 1289 (w), 1232 (s), 1134 (w), 1105 (w), 1044 (w), 978 (s), 940 (m), 829 (vs), 806 (vs), 779 (s), 755 (s), 740 (m), 726 (m), 649 (m), 609 (w), 428 (w). EVANS (500.1 MHz, 300 K, C6D6 + 1% TMS): μeff = 4.18μB; μS.O. = 2.83μB. (500.1 MHz, 300 K, THF-d8 + 1% TMS): μeff = 3.93μB; μS.O. = 2.83μB.
[K{18c6}][Cr(N(Dipp)SiiPr3)2] (5).
50 mg (0.07 mmol, 1 equiv.) [Cr(N(Dipp)SiiPr3)2] were dissolved in 5 mL Et2O. After adding KC8 (0.08 mmol, 1.1 equiv.) it was stirred for several minutes at room temperature. The graphite was filtered off the red solution and the volatiles were reduced to a minimum. After layering with a solution of one equiv. 18-crown-6 in Et2O and storing at −40 °C for several days, [K{18c6}][Cr(N(Dipp)SiiPr3)2] (5) was obtained as orange crystals in a yield of 43%. Yield: 31 mg (0.03 mmol, 43%). 1H NMR (500.1 MHz, THF-d8, 300 K, ppm): δ = 23 (bs, w½ = 2000 Hz), 16 (bs, w½ = 1700 Hz), 3.6* (s, 24 H, 18c6), 0.4 (bs, w½ = 2400 Hz). * Signal is overlapping with the solvent peak, why no further information can be provided. Elemental analysis C54H100CrKN2O6Si2 (1020.67 g mol−1): calcd: N 2.74, C 63.55, H 9.88; found: N 2.72, C 63.10, H 9.66%. IR (ATR, cm−1): = 2953 (m), 2892 (m), 2849 (m), 1584 (w), 1469 (m), 1452 (w), 1413 (m), 1373 (w), 1350 (m), 1309 (w), 1283 (w), 1241 (s), 1196 (m), 1103 (vs), 1058 (w), 991 (m), 965 (m), 925 (m), 878 (m), 837 (m), 800 (w), 759 (s), 738 (m), 663 (m), 635 (s), 555 (m), 530 (w), 515 (w), 461 (w), 427 (m). EVANS (500.1 MHz, 300 K, THF-d8 + 1% TMS): μeff = 4.21μB; μS.O. = 5.92μB.
[Li{12c4}2][Fe(N(Dipp)SiMe3)2] (6).
100 mg (0.18 mmol, 1 equiv.) [Fe(N(Dipp)SiMe3)2] were dissolved in 5 mL THF. After adding a piece of lithium the reaction mixture was stirred for 2 hours at room temperature. Residual lithium was filtered off the dark red solution and the volatiles were reduced to a minimum. After layering with a solution of two equiv. 12-crown-4 in Et2O and storing at −40 °C for several days, [Li{12c4}2] [Fe(N(Dipp)SiMe3)2] (6) was obtained as green crystals in a yield of 57%. Yield: 94 mg (0.10 mmol, 57%). 1H NMR (300.2 MHz, THF-d8, 300 K, ppm): δ = 29.5 (bs, w½ = 320 Hz), 27.7 (bs, w½ = 480 Hz), 13 (bs, w½ = 200 Hz), 2.77 (s, 32 H, w½ = 19.4 Hz, 12c4), 0.12 (s, 16 H, w½ = 4.0 Hz), −1 (bs, 18 H, w½ = 1800 Hz, Si(CH3)3), −100 (bs, w½ = 1600 Hz, CH(CH3)2). Elemental analysis C46H84FeLiN2O8Si2 (912.14 g mol−1): calcd: N 3.07, C 60.57, H 9.28; found: N 3.38, C 60.19, H 9.09%. IR (ATR, cm−1): = 3035 (vw), 2950 (m), 2913 (m), 2862 (m), 1580 (w), 1482 (w), 1443 (m), 1420 (m), 1364 (m), 1351 (w), 1313 (m), 1287 (m), 1251 (s), 1194 (m), 1134 (s), 1094 (vs), 1051 (w), 1023 (s), 921 (vs), 881 (w), 827 (vs), 778 (s), 742 (m), 666 (m), 625 (w), 554 (m), 429 (m). EVANS (500.1 MHz, 300 K, THF-d8 + 1% TMS): μeff = 4.74μB; μS.O. = 3.87μB.
[Na{18c6}][Fe(N(Dipp)SiMe3)2] (7).
150 mg (0.27 mmol, 1 equiv.) [Fe(N(Dipp)SiMe3)2] were dissolved in 5 mL Et2O. After adding Na/NaCl (5% w/w) (0.30 mmol (Na), 1.1 equiv.) the reaction mixture was stirred for several minutes at room temperature. The residuals were filtered off and the resulting dark red solution was reduced in vacuo to approx. 1 ml. After layering with a solution of one equiv. 18-crown-6 in Et2O and storing at −40 °C for several days, [Na{18c6}][Fe(N(Dipp)SiMe3)2] (7) was obtained as red-brownish crystals in a yield of 79%. Yield: 180 mg (0.21 mmol, 79%). 1H NMR (300.2 MHz, THF-d8, 300 K, ppm): δ = 29.5 (bs, w½ = 390 Hz), 28.0 (s, w½ = 380 Hz), 12.8 (bs, w½ = 170 Hz), 3.45 (m, 4 H, Et2O), 1.92 (s, 24 H, 18c6), 1.17 (m, 6 H, Et2O), −0.5 (bs, w½ = 540 Hz, Si(CH3)3) −102 (bs, w½ = 540 Hz, CH(CH3)2). Elemental analysis C42H76FeN2NaO6Si2 (840.08 g mol−1): calcd: N 3.33, C 60.05, H 9.12; found: N 3.83, C 60.06, H 9.06%. IR (ATR, cm−1): = 3052 (vw), 3035 (vw), 2950 (m), 2879 (m), 2862 (m), 1581 (w), 1456 (m), 1419 (s), 1375 (w), 1353 (m), 1317 (m), 1294 (w), 1233 (s), 1194 (m), 1094 (vs), 1053 (m), 1039 (w), 948 (m), 920 (s), 882 (w), 832 (vs), 781 (s), 742 (m), 666 (m), 619 (w), 576 (w), 542 (w), 530 (w), 431 (m). EVANS (500.1 MHz, 300 K, THF-d8 + 1% TMS): μeff = 4.24μB; μS.O. = 3.87μB.
[NBu4][Fe(N(Dipp)SiMe3)2] (8).
70 mg (0.12 mmol, 1 equiv.) [KFe(N(Dipp)SiMe3)2] and 39 mg NBu4Br (0.12 mmol, 1 equiv.) were dissolved in 2 mL THF. After stirring over night at room temperature, the solvent was removed under reduced pressure. The green solid was resolved in Et2O and layered with n-pentane, before cooling to −40 °C for crystallization. After several days, the solution was decanted off. The remaining green crystals were dried in vacuo and crystalline [NBu4][Fe(N(Dipp)SiMe3)2] (8) was obtained in a yield of 57%. Yield: 54 mg (0.07 mmol, 57%). 1H NMR (300.2 MHz, THF-d8, 300 K, ppm): δ = 28 (bs, w½ = 550 Hz), 12.7 (s, w½ = 180 Hz), 2.49 (s, 8 H, w½ = 65 Hz, NBu4+), 1.0 (bs, 8 H, w½ = 66 Hz, NBu4+), 0.57 (s, 8 H, w½ = 81 Hz, NBu4+), 0.18 (m, 12 H, w½ = 48 Hz, NBu4+), −36.1 (s, w½ = 12.4 Hz), −56.1 (bs, w½ = 12.4 Hz), −102 (bs, w½ = 1100 Hz, CH(CH3)2). Elemental analysis C46H88FeN3Si2 (795.25 g mol−1): calcd: N 5.28, C 69.48, H 11.15; found: N 5.43, C 69.04, H 10.76%. IR (ATR, cm−1): = 2958 (s), 2863 (s), 1581 (w), 1482 (m), 1459 (m), 1419 (s), 1378 (w), 1356 (w), 1314 (m), 1239 (vs), 1195 (m), 1149 (w), 1102 (w), 1041 (w), 997 (w), 923 (s), 879 (m), 837 (s), 777 (s), 736 (m), 668 (s), 623 (m), 575 (w), 539 (w), 519 (w), 432 (s). EVANS (500.1 MHz, 300 K, THF-d8 + 1% TMS): μeff = 4.30μB; μS.O. = 3.87μB.
[NBu4][Co(N(Dipp)SiMe3)2] (9).
70 mg (0.12 mmol, 1 equiv.) [KCo(N(Dipp)SiMe3)2] and 39 mg NBu4Br (0.12 mmol, 1 equiv.) were dissolved in 2 mL THF. After stirring over night at room temperature, the solvent was removed under reduced pressure. The green solid was resolved in Et2O and layered with n-pentane, before cooling to −40 °C for crystallization. After several days, the solution was decanted off. The remaining green crystals were dried in vacuo and crystalline [NBu4][Co(N(Dipp)SiMe3)2] (9) was obtained in a yield of 42%. Yield: 40 mg (0.05 mmol, 42%). 1H NMR (300.2 MHz, THF-d8, 300 K, ppm): δ = 30 (bs, w½ = 800 Hz, CH(CH3)2), 17.4 (s, 12 H, w½ = 64 Hz, CH(CH3)2), 16.0 (s, w½ = 38 Hz, m-PhH), 14.0 (bs, 18 H, w½ = 210 Hz, Si(CH3)3), 7.31 (s, w½ = 9.48 Hz, p-PhH), 3.01 (bs, 8 H, w½ = 120 Hz, NBu4+), 0.53–1.50 (m, 28 H, NBu4+), −88.0 (bs, 12 H, w½ = 110 Hz, CH(CH3)2). Elemental analysis C46H88CoN3Si2 (798.33 g mol−1): calcd: N 5.26, C 69.21, H 11.11; found: N 5.65, C 68.77, H 10.63%. IR (ATR, cm−1): = 2950 (m), 2863 (m), 1581 (w), 1477 (w), 1460 (w), 1420 (m), 1375 (w), 1318 (m), 1245 (s), 1229 (s), 1199 (m), 1142 (w), 1103 (w), 1042 (w), 1025 (w), 939 (s), 838 (vs), 778 (s), 664 (m), 616 (w), 543 (w), 436 (m). EVANS (500.1 MHz, 300 K, THF-d8 + 1% TMS): μeff = 3.58μB; μS.O. = 2.83μB.
[K{18c6}][Fe(N(Dipp)SiMe3)2(η2-PhCCPh)] (10).
50 mg (0.06 mmol, 1 equiv.) [K{18c6}][Fe(N(Dipp)SiMe3)2] and 10 mg diphenyl acetylene (0.06 mmol, 1 equiv.) were dissolved in 2 mL THF. After stirring for several minutes at room temperature, the solution was layered with n-pentane, before cooling to −40 °C for crystallization. After several days, the solution was decanted off. The remaining dark red crystals were dried in vacuo and crystalline [K{18c6}] [Fe(N(Dipp)SiMe3)2(η2-PhCCPh)] (10) was obtained in a yield of 71%. Yield: 36 mg (0.04 mmol, 71%). 1H NMR (300.2 MHz, THF-d8, 300 K, ppm): δ = 27.0 (bs, w½ = 87 Hz), 4.67 (bs, w½ = 530 Hz), 3.50 (s, 24 H, w½ = 37 Hz, 18c6), 3.50 (bs, w½ = 44 Hz), 0.8 (bs, w½ = 99 Hz), −3.5 (bs, w½ = 330 Hz), −8.83 (bs, w½ = 42 Hz), −25.0 (bs, w½ = 57 Hz). Elemental analysis C42H76FeKN2O6Si2 (1034.43 g mol−1): calcd: N 2.71, C 65.02, H 8.38; found: N 3.19, C 65.09, H 8.35%. IR (ATR, cm−1): = 3054 (w), 3038 (w), 3002 (w), 2953 (m), 2911 (m), 2888 (m), 2862 (m), 1816 (w), 1587 (m), 1472 (w), 1461 (m), 1421 (s), 1376 (w), 1351 (m), 1312 (m), 1283 (w), 1235 (s), 1191 (m), 1154 (w), 1131 (w), 1104 (vs), 1054 (m), 1040 (m), 1024 (w), 997 (w), 962 (m), 928 (s), 912 (s), 879 (m), 831 (s), 777 (s), 766 (w), 760 (m), 738 (m), 695 (m), 666 (m), 640 (w), 618 (m), 591 (w), 572 (w), 554 (w), 530 (m), 507 (w), 490 (w), 438 (m). EVANS (500.1 MHz, 300 K, THF-d8 + 1% TMS): μeff = 3.88μB; μS.O. = 3.87μB.
Reaction of [KMn(L2)2] (2) with diphenyl acetylene.
53 mg (0.10 mmol, 1.0 equiv.) [Mn(N(Dipp)SiMe3)2] and 21 mg KC8 (0.15 mmol, 1.5 equiv.) were dissolved in 2 mL toluene in the presence or absence of small amounts of THF, respectively. After stirring for several minutes at room temperature, the dark violet solution was filtered in a solution of diphenyl acetylene (20 mg, 0.11 mmol, 1.1 equiv.) in the same solvent. It was layered with n-pentane and allowed to crystallize at room temperature. After several days, dark red crystals of both 12 and 13, besides pale yellow crystals of hexaphenylbenzene, could be obtained, which were suitable for X-ray diffraction analysis.
Reaction of [KFe(L2)2] (3) with diphenyl acetylene.
59 mg (0.10 mmol, 1.0 equiv.) [KFe(N(Dipp)SiMe3)2] (3) and 18 mg diphenyl acetylene (0.10 mmol, 1.0 equiv.) were dissolved in 2 mL diethyl ether. It was stirred for several minutes at room temperature. The brownish solution was filtered and layered with n-pentane, before crystallizing at room temperature. After several days, single crystals of [K(D)Fe(η6-HPB)(η2-PhCCPh)] (11·D) (D = none, Et2O) could be obtained, suitable for X-ray diffraction analysis.
[K{18c6}][Fe(N(Dipp)SiMe3)2]3.
100 mg (0.18 mmol, 1 equiv.) [Fe(N(Dipp)SiMe3)2] were dissolved in 5 mL Et2O. After adding KC8 (0.20 mmol, 1.1 equiv.) the reaction mixture was stirred for several minutes at room temperature, while the colour changed to red. The graphite was filtered off and it was layered with a solution of 18-crown-6 (1.1 equiv.) in Et2O. After storing at −40 °C for several days [K{18c6}][Fe(N(Dipp)SiMe3)2] was obtained as orange crystals in a yield of 64%. Yield: 99 mg (0.12 mmol, 64%). 1H NMR (500.1 MHz, THF-d8, ppm): δ = 29.0 (bs, w½ = 370 Hz), 27.5 (s, w½ = 420 Hz), 12.4 (bs, w½ = 157 Hz), 2.58 (bs, 24 H, w½ = 40 Hz, 18c6), 1.14 (s, w½ = 17.0 Hz), 0.11 (s, w½ = 11.1 Hz), −0.7 (bs, w½ = 1500 Hz, Si(CH3)3), −101 (bs, w½ = 1400 Hz, CH(CH3)2). The spectroscopic data coincide with 1H NMR measurements of [K{18c6}][Fe(N(Dipp)SiMe3)2] synthesized via literature procedure.3
[K{18c6}][Co(N(Dipp)SiMe3)2]3.
100 mg (0.18 mmol, 1 equiv.) [Co(N(Dipp)SiMe3)2] were dissolved in 5 mL. After adding KC8 (0.20 mmol, 1.1 equiv.) the reaction mixture was stirred for several minutes at room temperature, while the colour changed to green. The graphite was filtered off and it was layered with a solution of 18-crown-6 (1.1 equiv.) in Et2O. After storing at −40 °C for several days [K{18c6}][Co(N(Dipp)SiMe3)2] was obtained as light green crystals in a yield of 56%. Yield: 87 mg (0.10 mmol, 56%). 1H NMR (500.1 MHz, THF-d8, 300 K, ppm): δ = 29 (bs, 4 H, w½ = 900 Hz, CH(CH3)2), 17.3 (s, 12 H, w½ = 48 Hz, CH(CH3)2), 16.3 (s, 4 H, w½ = 36 Hz, m-PhH), 13.3 (s, 18 H, w½ = 210 Hz, Si(CH3)3), 4.04 (s, 2 H, w½ = 23 Hz, p-PhH), 2.85 (s, 24 H, w½ = 26 Hz, 18c6), −86.5 (s, 12 H, w½ = 140 Hz, CH(CH3)2). The spectroscopic data coincide with 1H NMR measurements of [K{18c6}][Co(N(Dipp)SiMe3)2] synthesized via literature procedure.3
X-Ray diffraction analysis
Data for compounds 1 (CCDC 2010661), 1·3THF (CCDC 2011333), 2 (CCDC 2011213), 3 (CCDC 2010659), 4 (CCDC 2010663), 5 (CCDC 2011200), 6 (CCDC 2011410), 10 (CCDC 2047637), 11 (CCDC 2047640), 11·Et2O (CCDC 2047641) and 13 (CCDC 2047638)† were collected at 100 K on a Bruker Quest D8 diffractometer (Bruker Corporation, Billerica, USA) using an Incoatec Microfocus Source Mo-Kα radiation and equipped with an Oxford Instrument Cooler Device (Oxford Instruments, Abingdon, UK) and Photon 100 detector. Data for compounds 3·2DMAP (CCDC 2010662), 7 (CCDC 2011409) and 12 (CCDC 2047639)† were collected at 100 K on a STOE IPDS2 diffractometer (STOE & Cie GmbH, Darmstadt, Germany) and data for compound 8 (CCDC 2010664), 9 (CCDC 2010665) and [K{18c6}][Fe(N(Dipp)SiMe3)2]·Et2O (CCDC 2010666)† were collected at 100 K on a STOE IPDS2T diffractometer using a graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) and equipped with an Oxford Instrument Cooler Device (Oxford Instruments, Abingdon, UK). Data for compound 3·Et2O (CCDC 2010651†) were collected at 100 K on a Bruker Kappa Apex2 using an Area graphite source Mo-Kα radiation equipped with an Oxford Instrument Cooler Device (Oxford Cryosystems open-flow nitrogen cryostat, Cosier & Glazer, 1986). The structures have been solved using either OLEX SHELXT V2014/132 and refined by means of least-squares procedures on an F2 (all complexes but 3·Et2O which was refined on F) with the aid of the program SHELXL-2016/633 included in the software package WinGX version 1.6334 or using CRYSTALS.35 The atomic scattering factors were taken from International Tables for X-ray crystallography.36 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were refined by using a riding model. Disorders were found for 1·3THF (a coordinating THF molecule and two iso-propyl groups), 5 (a coordinating THF molecule) and 7 (18-crown-6 unit) and were modelled accordingly. The structure of 9 was refined as an inversion twin. For 3·Et2O some data sets are probably incomplete due to strategy errors, however the structure is very good (R < 3%, 20 refl/parameter, no restrains, no disorder, low maximum/minimum residual density). Absorption corrections were introduced by using the MULTISCAN and X-Red programs.37 Drawings of molecules are performed with the program DIAMOND (Crystal Impact, Bonn, Germany) with 50% probability displacement ellipsoids for non-H atoms. Additional details are given in the ESI.†
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the DFG (grant WE 5627/1-1 and WE 5627/4-1 for C. G. W.), the Philipps-University Marburg, the CNRS and the ANR (programme blanc “IRONHYC” ANR-12 for C. G. W., S. S.-E. and S. B.) for funding.
Notes and references
-
(a) P. P. Power, Chem. Rev., 2012, 112, 3482–3507 CrossRef CAS PubMed;
(b) G. Ung, J. Rittle, M. Soleilhavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 8427–8431 CrossRef CAS PubMed;
(c) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed;
(d) Z. Mo, Z. Ouyang, L. Wang, K. L. Fillman, M. L. Neidig and L. Deng, Org. Chem. Front., 2014, 1, 1040–1044 RSC;
(e) P. P. Samuel, K. C. Mondal, N. Amin Sk, H. W. Roesky, E. Carl, R. Neufeld, D. Stalke, S. Demeshko, F. Meyer, L. Ungur, L. F. Chibotaru, J. Christian, V. Ramachandran, J. van Tol and N. S. Dalal, J. Am. Chem. Soc., 2014, 136, 11964–11971 CrossRef CAS PubMed;
(f) P. P. Samuel, R. Neufeld, K. Chandra Mondal, H. W. Roesky, R. Herbst-Irmer, D. Stalke, S. Demeshko, F. Meyer, V. C. Rojisha, S. De, P. Parameswaran, A. C. Stückl, W. Kaim, J. H. Christian, J. K. Bindra and N. S. Dalal, Chem. Sci., 2015, 6, 3148–3153 RSC;
(g) C.-Y. Lin, J. C. Fettinger, N. F. Chilton, A. Formanuik, F. Grandjean, G. J. Long and P. P. Power, Chem. Commun., 2015, 51, 13275–13278 RSC;
(h) C. A. Laskowski and G. L. Hillhouse, J. Am. Chem. Soc., 2008, 130, 13846–13847 CrossRef CAS PubMed;
(i) M. I. Lipschutz and T. D. Tilley, Angew. Chem., 2014, 126, 7418–7422 CrossRef;
(j) Y.-S. Meng, Z. Mo, B.-W. Wang, Y.-Q. Zhang, L. Deng and S. Gao, Chem. Sci., 2015, 12, 7156–7162 RSC.
- C. G. Werncke, P. C. Bunting, C. Duhayon, J. R. Long, S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2015, 54, 245–248 CrossRef CAS PubMed.
- C.-Y. Lin, J. C. Fettinger, F. Grandjean, G. J. Long and P. P. Power, Inorg. Chem., 2014, 53, 9400–9406 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577–581 CrossRef CAS PubMed.
- M. I. Lipschutz, X. Yang, R. Chatterjee and T. D. Tilley, J. Am. Chem. Soc., 2013, 135, 15298–15301 CrossRef CAS PubMed.
- I. C. Cai, M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2014, 50, 13062–13065 RSC.
- P. P. Samuel, K. C. Mondal, H. W. Roesky, M. Hermann, G. Frenking, S. Demeshko, F. Meyer, A. C. Stückl, J. H. Christian, N. S. Dalal, L. Ungur, L. F. Chibotaru, K. Pröpper, A. Meents and B. Dittrich, Angew. Chem., 2013, 125, 12033–12037 CrossRef.
- C. G. Werncke, E. Suturina, P. C. Bunting, L. Vendier, J. R. Long, M. Atanasov, F. Neese, S. Sabo-Etienne and S. Bontemps, Chem. – Eur. J., 2016, 22, 1668–1674 CrossRef CAS PubMed.
- C. A. Laskowski, D. J. Bungum, S. M. Baldwin, S. A. Del Ciello, V. M. Iluc and G. L. Hillhouse, J. Am. Chem. Soc., 2013, 135, 18272–18275 CrossRef CAS PubMed.
- M. I. Lipschutz, T. Chantarojsiri, Y. Dong and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 6366–6372 CrossRef CAS PubMed.
- C. L. Wagner, L. Tao, E. J. Thompson, T. A. Stich, J. Guo, J. C. Fettinger, L. A. Berben, R. D. Britt, S. Nagase and P. P. Power, Angew. Chem., Int. Ed., 2016, 55, 10444–10447 CrossRef CAS PubMed.
- C.-Y. Lin, J.-D. Guo, J. C. Fettinger, S. Nagase, F. Grandjean, G. J. Long, N. F. Chilton and P. P. Power, Inorg. Chem., 2013, 52, 13584–13593 CrossRef CAS PubMed.
- W. M. Reiff, A. M. LaPointe and E. H. Witten, J. Am. Chem. Soc., 2004, 126, 10206–10207 CrossRef CAS PubMed.
- M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, Chem. Sci., 2013, 4, 139–156 RSC.
-
(a) W. M. Reiff, C. E. Schulz, M.-H. Whangbo, J. I. Seo, Y. S. Lee, G. R. Potratz, C. W. Spicer and G. S. Girolami, J. Am. Chem. Soc., 2009, 131, 404–405 CrossRef CAS PubMed;
(b) M. Atanasov, D. Aravena, E. Suturina, E. Bill, D. Maganas and F. Neese, Coord. Chem. Rev., 2015, 289–290, 177–214 CrossRef CAS.
-
(a) C. G. Werncke, J. Pfeiffer, I. Müller, L. Vendier, S. Sabo-Etienne and S. Bontemps, Dalton Trans., 2019, 48, 1757–1765 RSC;
(b) I. Müller, C. Schneider, C. Pietzonka, F. Kraus and C. G. Werncke, Inorganics, 2019, 7, 117 CrossRef;
(c) C. G. Werncke and I. Müller, Chem. Commun., 2020, 56, 2268–2271 RSC.
-
(a) L. C. H. Maddock, T. Nixon, A. R. Kennedy, M. R. Probert, W. Clegg and E. Hevia, Angew. Chem., Int. Ed., 2018, 57, 187–191 CrossRef CAS PubMed;
(b) E. Nagaradja, F. Chevallier, T. Roisnel, V. Jouikov and F. Mongin, Tetrahedron, 2012, 68, 3063–3073 CrossRef CAS;
(c) V. L. Blair, W. Clegg, R. E. Mulvey and L. Russo, Inorg. Chem., 2009, 48, 8863–8870 CrossRef CAS PubMed;
(d) V. L. Blair, W. Clegg, B. Conway, E. Hevia, A. Kennedy, J. Klett, R. E. Mulvey and L. Russo, Chem. – Eur. J., 2008, 14, 65–72 CrossRef CAS PubMed.
- M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC.
-
(a)
NMR of paramagnetic molecules. Applications to metallobiomolecules and models, ed. I. Bertini, C. Luchinat, G. Parigi and E. Ravera, Elsevier, Amsterdam, Netherlands, 2017 Search PubMed;
(b) I. Bertini, C. Luchinat, G. Parigi and R. Pierattelli, ChemBioChem, 2005, 6, 1536–1549 CrossRef CAS PubMed;
(c) I. Bertini, C. Luchinat and G. Parigi, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 40, 249–273 CrossRef CAS;
(d) C. L. I. Bertini, Coord. Chem. Rev., 1996, 150, 1–28 CrossRef;
(e)
G. N. La Mar, W. D. Horrocks and R. H. Holm, NMR of Paramagnetic Molecules. Principles and Applications, Elsevier Science, Burlington, 1973 Search PubMed.
- C.-Y. Lin, J. C. Fettinger and P. P. Power, Inorg. Chem., 2017, 56, 9892–9902 CrossRef CAS PubMed.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810–4813 CrossRef CAS.
- C. L. Wagner, L. Tao, J. C. Fettinger, R. D. Britt and P. P. Power, Inorg. Chem., 2019, 58, 8793–8799 CrossRef CAS PubMed.
- W. Alexander Merrill, T. A. Stich, M. Brynda, G. J. Yeagle, J. C. Fettinger, R. D. Hont, W. M. Reiff, C. E. Schulz, R. D. Britt and P. P. Power, J. Am. Chem. Soc., 2009, 131, 12693–12702 CrossRef CAS PubMed.
- I. Müller, D. Munz and C. G. Werncke, Inorg. Chem., 2020, 59, 9521–9537 CrossRef PubMed.
- R. Wolf, N. Ghavtadze, K. Weber, E.-M. Schnöckelborg, B. de Bruin, A. W. Ehlers and K. Lammertsma, Dalton Trans., 2010, 39, 1453–1456 RSC.
- R. A. Lewis, S. Morochnik, A. Chapovetsky, G. Wu and T. W. Hayton, Angew. Chem., Int. Ed., 2012, 51, 12772–12775 CrossRef CAS PubMed.
- R. J. Witzke, D. Hait, K. Chakarawet, M. Head-Gordon and T. D. Tilley, ACS Catal., 2020, 10, 7800–7807 CrossRef CAS.
- W.-Y. Yeh, S.-M. Peng and G.-H. Lee, Organometallics, 2002, 21, 3058–3061 CrossRef CAS.
- F. Calderazzo, G. Pampaloni, P. Pallavicini, J. Straehle and K. Wurst, Organometallics, 1991, 10, 896–901 CrossRef CAS.
- K. Mach and S. I. Troyanov, J. Organomet. Chem., 1991, 414, C15–C18 CrossRef CAS.
- N. E. Schore, Chem. Rev., 1988, 88, 1081–1119 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
-
International Union of Crystallography, International tables for crystallography, Springer, Chester, England, New York, 1st edn, 2006 Search PubMed.
-
(a)
SADABS-2016/2, Bruker, 2016 Search PubMed;
(b)
X.-R. 1. X-Area, STOE, 2016 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Sylviane
Sabo-Etienne
b,
Sylviane
Sabo-Etienne