
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective protonation of the one-electron reduced cofactor in [FeFe]-hydrogenase†
Konstantin
Laun
ab,
Iuliia
Baranova
ac,
Jifu
Duan
 d,
Leonie
Kertess
d,
Florian
Wittkamp
e,
Ulf-Peter
Apfel
ef,
Thomas
Happe
d,
Moritz
Senger
*ag and
Sven T.
Stripp
*a
d,
Leonie
Kertess
d,
Florian
Wittkamp
e,
Ulf-Peter
Apfel
ef,
Thomas
Happe
d,
Moritz
Senger
*ag and
Sven T.
Stripp
*a
aDepartment of Physics, Freie Universität Berlin, 14195 Berlin, Germany. E-mail: sven.stripp@fu-berlin
bDepartment of Chemistry, Technische Universität Berlin, 10623 Berlin, Germany
cFaculty of Physics, St. Petersburg State University, 198504 St. Petersburg, Russian Federation
dFaculty of Biology and Biotechnology, Ruhr-Universität Bochum, 44801 Bochum, Germany
eFaculty of Chemistry and Biochemistry, Ruhr-Universität Bochum, 44801 Bochum, Germany
fFraunhofer UMSICHT, 46047 Oberhausen, Germany
gDepartment of Chemistry, Uppsala University, 75120 Uppsala, Sweden. E-mail: moritz.senger@kemi.uu.se
First published on 22nd February 2021
Abstract
Hydrogenases are bidirectional redox enzymes that catalyze hydrogen turnover in archaea, bacteria, and algae. While all types of hydrogenase show H2 oxidation activity, [FeFe]-hydrogenases are excellent H2 evolution catalysts as well. Their active site cofactor comprises a [4Fe–4S] cluster covalently linked to a diiron site equipped with carbon monoxide and cyanide ligands. The active site niche is connected with the solvent by two distinct proton transfer pathways. To analyze the catalytic mechanism of [FeFe]-hydrogenase, we employ operando infrared spectroscopy and infrared spectro-electrochemistry. Titrating the pH under H2 oxidation or H2 evolution conditions reveals the influence of site-selective protonation on the equilibrium of reduced cofactor states. Governed by pKa differences across the active site niche and proton transfer pathways, we find that individual electrons are stabilized either at the [4Fe–4S] cluster (alkaline pH values) or at the diiron site (acidic pH values). This observation is discussed in the context of the complex interdependence of hydrogen turnover and bulk pH.
[FeFe]-Hydrogenases are gas-processing metalloenzymes that have been found in bacteria and green algae.1–4 They serve various roles in the hydrogen metabolism of prokaryotes, including oxidation of H2 as an energy carrier and proton reduction (H2 evolution) to maintain the cellular redox equilibrium.5 In the chloroplast of green algae, they are part of the photosynthetic electron transport chain, coupling H2O oxidation and H2 evolution at the reducing end of photosystem I.6 The first crystal structures of [FeFe]-hydrogenase helped identifying accessory and catalytic iron-sulfur clusters as well as gas channels and potential proton transfer (PT) pathways.7–13 Additionally, various biophysical techniques were employed to characterize the electronic structure of the active site cofactor, the so-called ‘H-cluster’ (Fig. 1).1 This iron-sulfur compound is formed by a [4Fe–4S] cluster connected to a bimetallic iron site via a bridging cysteine residue. The diiron site has been shown to bind carbon monoxide (CO) and cyanide ligands (CN−) that are a unique feature of hydrogenase.14–16 The aminodithiolate ligand (ADT) that bridges the metal ions of the diiron site was suggested to act as an inner-sphere hydrogen-bonding donor to a number of apical ligands at the distal iron ion (Fed).17 Moreover, the secondary amine of the ADT ligand serves as proton relay between the diiron site and the amino acid residues of the catalytic proton transfer pathway (Fig. 1).18–23 The latter includes arginine and glutamic acid residues connected by a cysteine, a serine, and a small water cluster.24
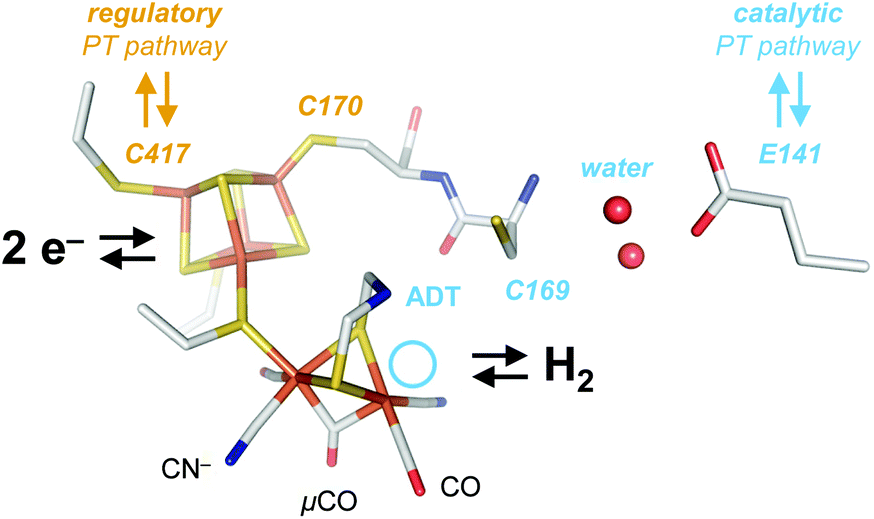 | ||
| Fig. 1 Cofactor and active site niche. The H-cluster comprises a [4Fe–4S] cluster linked to the catalytic diiron site. In the crystallized Hox state, the H-cluster carries two terminal CO and CN− ligands and a single μCO ligand (pdb ID 4XDC). The ADT ligand serves as a proton relay between the distal iron ion (Fed) and the catalytic PT pathway (light blue) including C169, a water cluster, E141, and other residues. The light blue circle marks the open coordination site at Fed in the oxidized state, Hox. At the [4Fe–4S] cluster, C417 may receive a proton directly from the solvent via the regulatory PT pathway (brown). | ||
The diiron site and the [4Fe–4S] cluster exist in an oxidized and reduced form each, resulting in a total of four different H-cluster species: the oxidized state, Hox, the 1e−-reduced states Hred′ (reduced [4Fe–4S] cluster) and Hred (reduced diiron site) as well as the 2e−-reduced state, Hsred.25–32 An additional state, Hhyd, is comprised of a reduced [4Fe–4S] cluster and a formally over-oxidized diiron site with a terminal hydride ligand (H−).33–35 Additional states include the CO-inhibited states Hox-CO and Hred′-CO and the oxidized protonated state, HoxH.29,32 While most authors agree on the importance of Hred′ and Hhyd in hydrogen turnover (Fig. 2), the protonation state, cofactor geometry, and involvement in catalysis of Hred and Hsred are under discussion.36
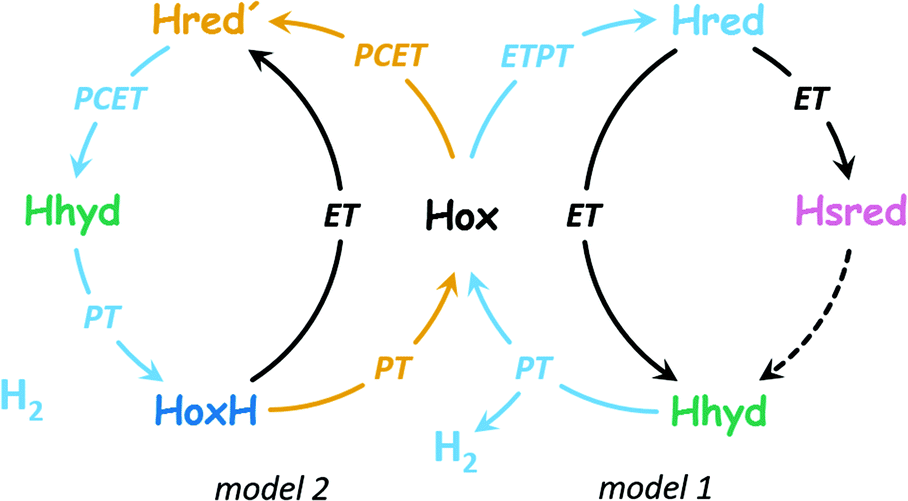 | ||
| Fig. 2 Two proposals of the catalytic cycle. In H2 evolution direction, both cycles start from Hox. Model 1 (right side, based on ref. 37) is characterized by ETPT to the diiron site in the 1st step (formation of Hred, protonation of the diiron site). This includes transient electron transfer (ET) via the [4Fe–4S] cluster and proton transfer (PT) in the formation of Hred. A subsequent reduction step (ET) may form Hhyd either directly or viaHsred by an unknown mechanism (dashed arrow). With a 2nd proton, H2 is released and Hox is restored. Model 2 (left side, based on ref. 36) is characterized by PCET to the [4Fe–4S] cluster in the 1st step (formation of Hred′, binding of a regulatory proton at the [4Fe–4S] cluster). Subsequently, PCET to the diiron site promotes formation of Hhyd, and with a 3rd proton, H2 is released and HoxH is formed. The latter may lose the regulatory proton to restore Hox or accept an electron to form Hred′ directly. | ||
Fig. 2 highlights that the 1e−-reduced H-cluster states Hred′ and Hred are enriched upon coupled or sequential electron- and proton transfer (PCET or ETPT). Due to the transient nature of electron tunnelling via the [4Fe–4S] cluster reduction, we will only use the PCET nomenclature in the following. In previous work, we suggested that a cysteine residue coordinating the [4Fe–4S] cluster may bind a proton in Hred′ (Fig. 1);31,38 however, there is no consensus regarding the nature of protonation and cofactor geometry in Hred. Sommer et al. presumed protonation of the ADT ligand (+NH2) and a shift of the μCO ligand into a ‘semi-bridging’ position at Fed as seen in hydrogenase crystals grown under 10% H2 and pressurized with 6 bar H2.39,40 Ratzloff et al. proposed a +NH2 geometry with a conserved μCO ligand in Hred,41 which was supported in recent infrared studies by Birrell et al. and Lorent et al. that identified a μCO ligand for Hred and Hsred at cryogenic temperatures.37,42 In contrast, our infrared evaluation of Hred and Hsred at ambient temperature implied the formation of a bridging hydride species (μH) and an apical CO ligand at Fed.30 A μH geometry was calculated to be rather unreactive.43–45 Therefore, such changes would exclude Hred and Hsred from the catalytic cycle (in contrast to Model 1 in Fig. 2) and favor a catalytic mechanism without the reduction of the diiron site (i.e., Model 2 in Fig. 2). Sanchez et al. demonstrated the kinetic competence of both Hred′ and Hred at ambient temperature46 but the spectroscopic marker bands that were used to follow Hred in their study are nearly identical for the μCO and the μH geometry. This impedes a kinetic discrimination of these isomers.37,42 We presume that the cryogenic states may represent kinetically trapped intermediates.24,47
The influence of bulk pH on Hox, Hred, and Hsred in the native [FeFe]-hydrogenase from Chlamydomonas reinhardtii (CrHydA1)32,39 and Hred′ in cofactor variant CrHydA1PDT was analysed before.31,48 To understand the equilibrium of Hred′ and Hred in native CrHydA1, we now investigate the pH-dependent accumulation of both 1e−-reduced H-cluster states under turnover conditions. Making use of operando attenuated total reflection Fourier-transform infrared (ATR FTIR) spectroscopy and spectro-electrochemistry under H2 oxidation or H2 evolution conditions, we found consistent trends for an accumulation of Hred′ towards alkaline pH values whereas the accumulation of Hred increases towards acidic pH values. This observation is explained by site-selective PCET to either the diiron site or the [4Fe–4S] cluster, guided by differences in proton affinity. Our findings are employed to distinguish catalytic from regulatory H-cluster states and inspire a molecular understanding of the pH-dependent hydrogen turnover of [FeFe]-hydrogenase.
Experimental
Protein purification and activation
All experiments involving CrHydA1 were performed under strictly anaerobic conditions. CrHydA1 apo-protein (wild-type and amino acid variants)49–51 and the synthetic mimics of the diiron site (ADT and PDT)19,20 were prepared as described previously. Activated CrHydA1 was eluted in 10 mM Tris/HCl (pH 8). Sodium dithionite was avoided to prevent accumulation of HoxH and Hhyd at low pH values.32,34 Each sample was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (∼0.5 mM CrHydA1) with mixed buffer containing 50 mM Tris, MES, and PIPPS to adjust the desired pH value.
:1 (∼0.5 mM CrHydA1) with mixed buffer containing 50 mM Tris, MES, and PIPPS to adjust the desired pH value.
ATR FTIR spectroscopy
The FTIR spectrometer (Tensor27, Bruker) was equipped with a triple-reflection ZnSe/Si crystal ATR cell (Smith Detection) and placed in an anaerobic chamber. Infrared spectra were recorded with 80 kHz scanning velocity at a spectral resolution of 2 cm−1 (MCT detection). Under these conditions, the time-resolution of data acquisition is in the range of seconds (i.e., five interferometer scans in forward/backward direction). ATR FTIR measurements were performed at 25 °C and on hydrogenase films derived by controlled dehydration and rehydration of 1 μl protein sample as reported earlier.29The anaerobically purified and activated [FeFe]-hydrogenase typically contained the H-cluster in various states. Hox was enriched in the film under a constant stream of N2 for 30–60 minutes. A constant gas stream (1.5 L min−1) was adjusted with digital mass flow controllers (SmartTrak, Sierra). Then, H2 was added to the N2 stream via separate flow controllers and passed through a wash bottle containing 150 mL mixed buffer (0.1–100% at ambient pressure). The resulting aerosol was fed into a gas-tight PCTFE compartment, attached on top of the ATR crystal plate and equipped with six optional gas inlets, a manometer for pressure control, and a glass window for UV/vis irradiaton.29 For each H2 concentration step, the film was equilibrated for 2.5 min to ensure a sufficiently stable composition of H-cluster states (Fig. S1†).
ATR FTIR spectro-electrochemistry
The pH-dependent reduction of CrHydA1 in the absence of H2 was analyzed by ATR FTIR spectro-electrochemistry.30,31 For this, 1 μL protein sample (diluted with 50 mM mixed buffer pH 9–5) was injected into a 9 μm thin gold mesh on top of an ATR silicon crystal. The mesh was covered with an 8 kDa dialysis membrane to protect the film from dilution. A custom-made PCTFE electrochemical cell was attached to the ATR crystal plate and filled with 3 mL electrolyte buffer (50 mM mixed buffer pH 9–5 including 500 mM KCl as electrolyte) that was purged with N2 throughout the whole experiment. After 60–90 minutes, the film was fully hydrated and stable. The gold mesh was connected with the working electrode, a platinum wire was used as counter electrode, and an Ag/AgCl electrode served as reference (+230 mV vs. SHE, as determined with 1 mM methyl viologen at pH 7).31 After complete oxidation at −100 mV vs. SHE, the potential was lowered incrementally from −150 mV to −850 mV in steps of 50 mV with a fixed duration of 20 minutes for each step (Fig. S2†) until no further spectral changes were observed. Midpoint potentials were estimated from bi-sigmoidal fits. At strongly reducing potentials, smaller changes in current hinted at imperfect equilibria (Fig. S2†).Data treatment
All absorbance spectra were derived from single channel spectra of ‘reference’ (ZnSe/Si) and ‘sample’ (ZnSe/Si + protein) in OPUS software. Then, data was exported to a home-written routine as described previously.32 In the frequency regime of the H-cluster (2150–1750 cm−1), absorbance spectra were subtracted with a polynomial function simulating the low-frequency combination band of liquid water underneath the ‘sharp’ CO/CN− bands of the H-cluster. This gave rise to background-corrected spectra as shown in Fig. S1–S3.† Reference spectra of pure redox states (Fig. 3 and Fig. S3†) allowed determining fit parameters for all observed redox states (frequency, intensity, bandwidth, and peak ratio, see Table S1†), as described earlier.32 The sum of band area (2 CN− + 3 CO) for a given redox state was obtained by simulation of spectral data with a fixed set of parameters that represent the population in relation to the other redox states. This value (%) and was plotted against time illustrating how the system converges into new redox equilibria upon disturbance (i.e., changes in H2 concentration in Fig. S1† or electrochemical potential in Fig. S2†). In the final step, the population of redox states was plotted as a function of H2 concentration or electrochemical potential. | ||
| Fig. 3 Characterization of key H-cluster states. (A) Background-corrected spectra of Hox (grey) and Hred′ (brown) recorded under alkaline conditions[:] (pH 9) at −300 mV vs. SHE and −600 mV vs. SHE, respectively. Additionally, the figure shows background-corrected spectra recorded either upon illumination in the presence of eosin Y and EDTA (Hred, light blue) or under acidic conditions (pH 5) at −800 mV vs. SHE (Hsred, rose). (B) Proposed H-cluster geometries. The rectangle represents the diiron site including the ADT ligand (NH), the Fe–Fe bridging ligand (μCO, μH), and the apical binding site of Fed (vacant or occupied with CO). Observed charges are formally +4 to +2. The cube represents the [4Fe–4S] cluster (charges +2 or +1). Colors hint at differences relative to Hox (color code: Hox (black), HoxH (dark blue), Hred′ (brown), Hred (light blue), Hsred (rose). | ||
Results
All experiments were performed with the [FeFe]-hydrogenase CrHydA1 under ambient conditions. In the first step, ATR FTIR spectroscopy and spectro-electrochemistry29–31 were employed to extract the IR signatures of all relevant redox states (Table 1 and Fig. S3†). Fig. 3A shows spectra of Hox and Hred′ recorded under alkaline conditions (pH 9) at −300 mV vs. SHE and −600 mV vs. SHE, respectively. A pure spectrum of Hred′ in native CrHydA1 has not been reported before. Additionally, Fig. 3A shows spectra recorded either upon illumination in the presence of eosin Y and EDTA (Hred, see ref. 52 for the protocol) or under acidic conditions (pH 5) at −800 mV vs. SHE (Hsred). The overall downshift of the cofactor bands from Hox → Hred′ and Hred → Hsred has been attributed to a reduction of the [4Fe–4S] cluster (depicted as a cubane in Fig. 3B).27,28 Fig. S4† emphasized that the Hred → Hsred difference spectrum shows no signal around 1800 cm−1. This highlights the lack of a μCO ligand at the reduced diiron site (depicted as a rectangle in Fig. 3B) and confirms the assignment of bands at 1961 cm−1 and 1953 cm−1 to Hred and Hsred, respectively. A detailed discussion of the IR spectrum of Hred can be found in ref. 47. Based on previous work,30Hred and Hsred are depicted with a terminal CO ligand and a μH ligand in Fig. 3B.| CN−/cm−1 | CO/cm−1 | [4Fe] | [2Fe] | ||||
|---|---|---|---|---|---|---|---|
| Hox | 2088 | 2070 | 1964 | 1940 | 1802 | +2 | +3 |
| Hox-CO | 2092 | 2082 | 1968 | 1962 | 1812 | +2 | +3 |
| Hred′ | 2084 | 2066 | 1962 | 1933 | 1792 | +1 | +3 |
| Hhyd | 2082 | 2068 | 1978 | 1960 | 1860 | +1 | +4 |
| Hred | 2072 | 2034 | 1961 | 1915 | 1891 | +2 | +2 |
| Hsred | 2068 | 2026 | 1953 | 1918 | 1882 | +1 | +2 |
To address the pH-dependent population of H-cluster states under H2 oxidation conditions, we investigated CrHydA1 by ATR FTIR spectroscopy at different H2 concentration (without external potential control). Direct proof for the hydrogenase-catalysed cleavage of H2 came from D2 oxidation experiment in an aqueous environment.34 Moreover, H2 oxidation induces an accumulation of reduced H-cluster states so that we were able to follow the increase and decrease of redox state populations as a function of atmospheric H2 and time. As an example, Fig. 4A depicts a series of background-corrected ATR FTIR absorbance spectra of the H-cluster recorded after 2.5 min under 0–100% H2 (compare Fig. S1†). We note that residual, unidentified H-cluster states may be present in the spectra. As these contributions did not to interfere with the global fit analysis (i.e., χ2 < 10−4) we did consider them any further. Broader features in the corrected spectra stem from the ‘combination band’ of liquid water. For a spectroscopically unique identification of H-cluster states, see Fig. 3A and ESI.†Fig. 4B illustrates how increasing the H2 concentration to 0.1% resulted in an accumulation of 1e−-reduced states Hred′ and Hred, which remained fairly stable between 0.1–3% H2. At higher concentrations of H2, the 2e−-reduced Hsred state dominated the spectrum. Only minor traces of the 2e−-reduced Hhyd state were observed, most likely due to the lack of sodium dithionite in the sample (see Experimental section).34
 | ||
| Fig. 4 Composition of H-cluster states under H2 oxidation conditions (pH 7). All data recorded on a hydrated film of CrHydA1 at ambient temperature and 0–100% H2 in the gas phase. (A) Series of baseline-corrected FTIR absorbance spectra of the H-cluster obtained after 2.5 min at each step. (B) State populations as a function of [H2]. Between 0.1–3% H2, the population of Hred′ and Hred was relatively stable. Hsred dominated for [H2] > 10%. | ||
Analogous to the experiment shown in Fig. 4, six individual CrHydA1 protein films between pH 10–5 were analyzed (Fig. 5). Largely independent of pH, the oxidized state Hox was the most prominent species in the absence of H2 while Hsred dominated the spectrum for [H2] > 10% (Fig. S5†). The steady-state population of Hred′ and Hred is plotted as a function of [H2] and at different pH values in Fig. 5AB. Here, we observed diverging trends for the accumulation of the 1e−-reduced states: Hred dominated at acidic conditions whereas Hred′ was promoted under alkaline pH values. Fig. 5C depicts the accumulation of Hred′ and Hred at 3% H2 as a function of pH, which clearly illustrates this trend. At low pH and [H2] > 10%, an increasing accumulation of Hhyd was observed, which may explain the mild suppression of Hsred that was otherwise expected to follow the same pH dependence as Hred (Fig. S5†). Upon removal of H2 from the gas stream, the 2e−-reduced states Hhyd and Hsred converted transiently into the 1e−-reduced states Hred′ and Hred, indicating intermolecular electron transfer in the dense films,32 before the equilibrium shifted back towards Hox upon auto-oxidation.17 The diverging pH dependence of Hred′ and Hred was found to be well conserved in this transient increase, emphasizing the robustness of all observed trends (Fig. S6†).
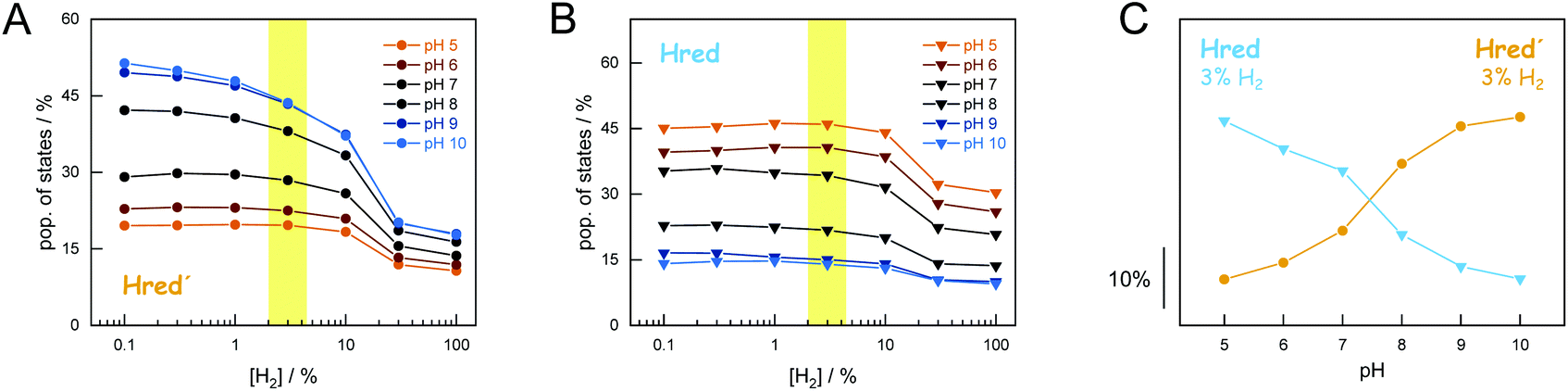 | ||
| Fig. 5 Accumulation of Hred′ and Hred as a function of H2 and pH. Population of Hred′ (panel A) and Hred (panel B) for six different pH values (pH 10–5) under 0.1–100% H2. Plotting the population of Hred′ and Hred at 3% H2 (yellow mark-up) against pH clearly illustrates the opposing pH dependency (panel C). For [H2] > 10%, Hsred and Hhyd are accumulated. | ||
The simultaneous presence of Hred′ and Hred complicates unique conclusions regarding the mechanism of H-cluster protonation. Therefore, additional experiments were performed. First, we probed the pH dependence of H2 oxidation with CrHydA1PDT. This cofactor variant lacks the secondary amine of the native ADT ligand (Fig. 1) and allows analysing the reduction of the [4Fe–4S] cluster (i.e., the Hox → Hred′ transition) independent from redox chemistry at the diiron site.28,31 Fig. S7† shows that the H2 oxidation activity of CrHydA1PDT increases between pH 10–8. While this cannot be explained by the stoichiometry of the catalysed reaction, our results support PCET chemistry at the [4Fe–4S] cluster.31 Earlier, the influence of cysteinyl ligand C417 on the catalytic properties of CrHydA1 has been addressed by site-directed mutagenesis.50,51 Now, we analyzed three cysteine variants to compare the composition of H-cluster states under H2 oxidation conditions (Fig. S8†). We made the following observations. (i) C417S behaved much like wild-type CrHydA1 but showed a reduced percentage of Hred′ under H2. (ii) Due to electron withdrawal from the [4Fe–4S] cluster by the imidazole ligand, C417H was reported with a less negative redox potential than wild-type CrHydA1.51 In agreement with earlier observations C417H adopted Hred′ as a resting state, reflecting the lack of H2 evolution activity of CrHydA1 C417H.51
In the presence of H2, the variant converted into Hsred (at pH 8) or Hhyd (pH 4) with no detectable traces of Hred. In the absence of H2, low pH conditions resulted in an accumulation of Hred′H. (iii) The spectral behavior of C417D was surprisingly similar to C417H, indicative of electron withdrawal. We speculate that this may be due to hydrogen-bonding between the aspartic acid side chain and the [4Fe–4S] cluster. In variance to the histidine variant, C417D slowly converted into Hox (pH 8) or HoxH (pH 4), reflecting the low but significant H2 evolution activity of CrHydA1 C417D.50
Overall, our data demonstrate how the equilibrium of redox states is affected by cluster ligation, proton concentration (pH), and the percentage of H2 in the gas phase. Under H2 oxidation conditions, virtually all H-cluster species were present at every step of the experiment, impeding an individual analysis of states. Thus, we investigated the pH-dependent population of redox states in CrHydA1 under H2 evolution conditions by injecting electrons into the systems in the absence of H2. We employed ATR FTIR spectro-electrochemistry to follow the evolution of state populations as a function of electrochemical potential. In contrast to conventional, Moss-type53 transmission cells, the ATR FTIR spectro-electrochemistry approach allowed H2 to be released from the protein-modified working electrode hence precluding product oxidation or product inhibition. Fig. 6 depicts how the oxidized states Hox and Hox-CO were lost at reductive potentials, followed by accumulation of the 1e−-reduced states Hred′ and Hred. Upon further reduction, accumulation of Hsred was observed; however, in contrast to the experiments performed under H2 oxidation conditions (Fig. 4), Hhyd was not observed.
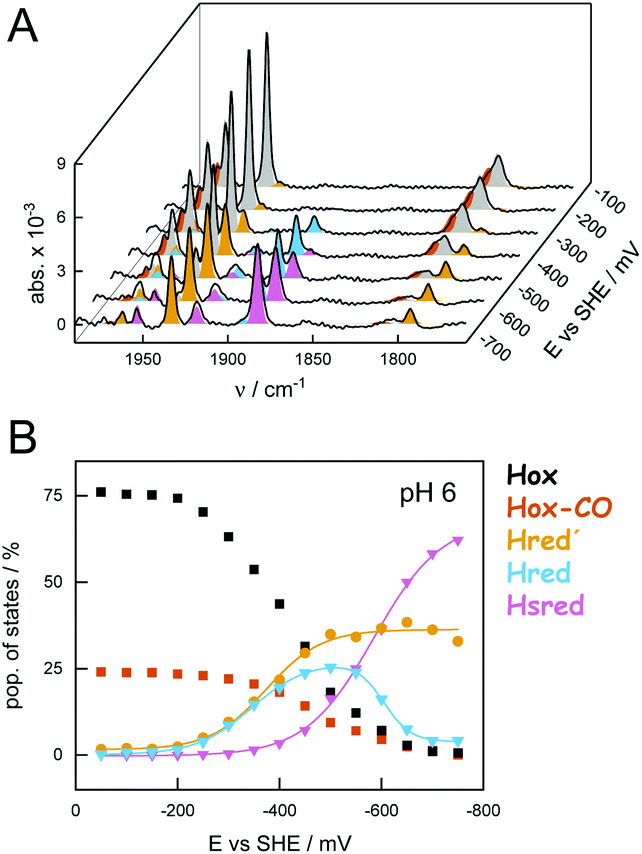 | ||
| Fig. 6 Composition of H-cluster states under H2 evolution conditions (pH 6). Exemplary data recorded on a hydrated film of CrHydA1 between −50 and −750 mV vs. SHE and constant N2 purging. (A) Series of baseline-corrected ATR FTIR absorbance spectra of the H-cluster obtained after 20 min at each increment of 50 mV (steady-state conditions). (B) State populations as a function of electrochemical potential. Sigmoidal fits allowed approximating the following midpoint potentials (vs. SHE): Hox → Hred′ −375 mV; Hox → Hred −345 mV; Hred → Hsred −585 mV (a second fit component at −605 mV may account for Hred′ → Hsred). | ||
Analogous to the experiment shown in Fig. 6, nine individual protein films between pH 9–5 were analyzed (Fig. 7). The maximal population of H-cluster states as a function of potential and pH value was used as the main observable. This approach allowed analyzing the 1e−- and 2e−-reduced states separately, which was not possible under H2 oxidation conditions (Fig. 5). The oxidized state Hox was the most prominent species at potentials more positive than −350 mV vs. SHE, largely independent of pH (i.e., in the absence of dithionite32). The accumulation of Hsred was found to be affected by pH more drastically, with a higher population at acidic pH values that reflects the lack of Hhyd in the experiment (Fig. S9†). Overall, we observed a mean midpoint potential around −650 mV vs. SHE for Hsred, which leaves a potential window of ∼300 mV to analyze the accumulation of the 1e−-reduced states.
 | ||
| Fig. 7 Accumulation of Hred′ and Hred as a function of electrochemical potential and pH. Population of Hred′ (panel A) and Hred (panel B) for nine different pH values (pH 9–5) between −50 and −750 mV vs. SHE. The opposite pH dependence is clearly illustrated plotting the population of Hred′ (–550 mV vs. SHE) and Hred (–350 mV vs. SHE) as function of pH in panel C (the yellow mark-up highlights the respective potentials). | ||
In Fig. 7AB the steady-state population of Hred′ and Hred is plotted as a function of potential and at different pH values. Like what has been observed under H2 oxidizing conditions, Hred dominated under acidic conditions whereas Hred′ was promoted at alkaline pH values. The population of states varies from 30–80% (−550 mV vs. SHE, Hred′) and 40–5% (−350 mV vs. SHE, Hred) for increasing pH values. Fig. 7C depicts the accumulation of Hred′ and Hred as a function of pH. This trend is strictly conserved in the aforementioned potential window (Fig. S9†) and facilitated an estimation of apparent proton affinities for the accumulation of Hred′ and Hred. Moreover, the Pourbaix diagram in Fig. S9† highlights the decrease in electrochemical driving force for Hox→Hred′ between pH 9–8 and Hox → Hred between pH 6–5. Interestingly, no pronounced pH dependency is observed around pH 7, which may reflect the concomitant formation of Hred′ and Hred.
Discussion
We analyzed the [FeFe]-hydrogenase HYDA1 from Chlamydomonas reinhardtii by ATR FTIR spectroscopy and spectro-electrochemistry under ambient conditions. We addressed the pH-dependent accumulation of various H-cluster states. Varying the sample pH under H2 oxidation and H2 evolution conditions established consistent trends for the accumulation of 1e−-reduced H-cluster states: we observed enrichment of Hred under acidic conditions whereas Hred′ prevailed at alkaline conditions. Our data on cofactor and amino acid variants highlight the importance of the [4Fe–4S] cluster for catalysis and the equilibrium of redox species.In earlier work, we identified the pH dependence of Hred′ formed upon reduction of the [4Fe–4S] cluster and protonation of a nearby cysteine, C417 in CrHydA1.31 Moreover, the Hred′ state is involved in the steady-state accumulation of HoxH,32 which proceeds under reducing conditions exclusively and represents the starting state for an enrichment of Hhyd in the presence of H2.34,54 While these data indicate PCET to the [4Fe–4S] cluster and emphasize the importance of redox chemistry adjacent to the diiron site, an understanding of the pH dependence of Hred′ and Hred with respect to hydrogen turnover is yet to be accomplished. Due to the simultaneous presence of various reduced H-cluster states under H2 (including Hhyd and Hsred), unraveling the PCET chemistry of Hred′ and Hred was found to be challenging.39 To this end, ATR FTIR spectro-electrochemistry facilitated analyzing the population of 1e−- and 2e−-reduced H-cluster states individually. The accumulation of Hred at acidic pH values and mildly reducing conditions (e.g., −350 mV vs. SHE) suggests a slightly acidic pKa, in agreement with the involvement of glutamic acid residues as ‘bottle neck’ in the catalytic PT pathway.21 In contrast, our data for the population of Hred′ at more reducing potentials (e.g., −550 mV vs. SHE) hints at a pKa in the alkaline, which reflects the greater ease of proton transfer to the [4Fe–4S] cluster via bulk solvent32,36 and is in excellent agreement with the formal pKa of 8.1 for a cysteine sidechain. This observation is evidence for the pH dependence of Hred′ in native [FeFe]-hydrogenase, in particular because the Hred state does not show dedicated pH dependence in this pH window. Surprisingly, Rodríguez-Macia et al. found the redox potential of the [4Fe–4S] cluster in CrHydA1PDT to be independent from bulk pH.48 The reasons for this discrepancy are unclear. In our hands, present and previous data31,38 support PCET in the formation of Hred′. We propose to distinguish a catalytic PT pathway to the diiron site from a regulatory pathway to the [4Fe–4S] cluster as a common feature of [FeFe]-hydrogenases.
Both in vivo and in vitro, the pH dependence of H2 evolution of [FeFe]-hydrogenase shows a bell-shaped distribution with a maximal activity around neutral or mildly alkaline pH values.55–58 This has recently been confirmed by bulk electrochemistry.59 While the increase in H2 evolution activity between pH 9–7 can be attributed to rising proton concentration, our data on CrHydA1 now allows correlating the activity decrease between pH 7–5 to the formation of Hred, which clearly dominated over Hred′ at acidic pH values. Although our data does not report on enzymatic activity directly, this behavior is in agreement with Hred and Hsred as ‘H2-inhibited’ states60 that bind a bridging hydride at the diiron site30 and have been shown to play a key role in sensory [FeFe]-hydrogenases.61–63 The ligand flip required to form a reactive terminal hydride geometry disfavors fast catalysis.43–45 The present data support the theory that reduction and site-selective protonation at the [4Fe–4S] cluster adjusts the redox potential of the H-cluster to stabilizes a reactive geometry necessary for efficient hydrogen catalysis.36 We suggest that similar concepts may give rise to a novel generation of biomimetic hydrogen catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank all reviewers for the critical feedback. Konstantin was supported by the Einstein Foundation Berlin. The research stay of Iuliia was funded by G-RISC (Grant No. P-2016b-28). We thank the Studienstiftung des Deutschen Volkes for a Kekulé Mobility Fellowship (to Leonie) and a PhD fellowship (to Florian). Ulf acknowledges funding by the DFG (under Germanys’ Excellence Strategy – EXC 2033–390677874) and the Fraunhofer Internal Programs (Grant No. ATTRACT 097-602175). Thomas acknowledges financial support from the Volkswagen Stiftung (Grant No. Az 93412) and the DFG (under Germanys’ Excellence Strategy – EXC 2033–390677874 and GRK 2341 Microbial Substrate Conversion). The European Union's Horizon 2020 research and innovation program is gratefully acknowledged for funding to Moritz (Marie Skłodowska-Curie Grant No. 897555). Sven is funded by the German Research Foundation (DFG) within the framework of SPP 1927 priority program “Iron-Sulfur for Life” (Grant No. STR1554/5-1).References
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114(8), 4081–4148, DOI:10.1021/cr4005814.
- J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. Adams, [FeFe]- and [NiFe]-Hydrogenase Diversity, Mechanism, and Maturation, Biochim. Biophys. Acta, 2015, 1853(6), 1350–1369, DOI:10.1016/j.bbamcr.2014.11.021.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, Current State of [FeFe]-Hydrogenase Research - Biodiversity and Spectroscopic Investigations, ACS Catal., 2020, 10, 7069–7086, DOI:10.1021/acscatal.0c01614.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav and D. Siegmund, .; Apfel, U.-P. [FeFe]-Hydrogenases: Maturation and Reactivity of Enzymatic Systems and Overview of Biomimetic Models, Chem. Soc. Rev., 2021 10.1039/d0cs01089h , Advance article.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, Genomic and Metagenomic Surveys of Hydrogenase Distribution Indicate H 2 Is a Widely Utilised Energy Source for Microbial Growth and Survival, ISME J., 2016, 10(3), 761–777, DOI:10.1038/ismej.2015.153.
- A. Hemschemeier and T. Happe, Alternative Photosynthetic Electron Transport Pathways during Anaerobiosis in the Green Alga Chlamydomonas Reinhardtii, Biochim. Biophys. Acta, Bioenerg., 2011, 1807(8), 919–926, DOI:10.1016/j.bbabio.2011.02.010.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, X-Ray, Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium Pasteurianum to 1.8 Angstrom Resolution, Science, 1998, 282(5395), 1853–1858, DOI:10.1126/science.282.5395.1853.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Desulfovibrio Desulfuricans Iron Hydrogenase: The Structure Shows Unusual Coordination to an Active Site Fe Binuclear Center, Structure, 1999, 7(1), 13–23, DOI:10.1016/S0969-2126(99)80005-7.
- J. Cohen, K. Kim, P. W. King, M. Seibert and K. Schulten, Finding Gas Diffusion Pathways in Proteins: Application to O2 and H2 Transport in CpI [FeFe]-Hydrogenase and the Role of Packing Defects, Structure, 2005, 13(9), 1321–1329, DOI:10.1016/j.str.2005.05.013.
- P. Liebgott, F. Leroux, B. Burlat and S. Dementin, Relating Diffusion along the Substrate Tunnel and Oxygen Sensitivity in Hydrogenase, Nat. Chem. Biol., 2009, 6, 63–70, DOI:10.1038/nchembio.276.
- D. W. Mulder, E. M. Shepard, J. E. Meuser, N. Joshi, P. W. King, M. C. Posewitz, J. B. Broderick and J. W. Peters, Insights into [FeFe]-Hydrogenase Structure, Mechanism, and Maturation, Structure, 2011, 19(8), 1038–1052, DOI:10.1016/j.str.2011.06.008.
- T. Lautier, P. Ezanno, C. Baffert, V. Fourmond, L. Cournac, J. C. Fontecilla-Camps, P. Soucaille, P. Bertrand, I. Meynial-Salles and C. Léger, The Quest for a Functional Substrate Access Tunnel in FeFe Hydrogenase, Faraday Discuss., 2011, 148, 385, 10.1039/c004099c.
- M. Winkler, J. Esselborn and T. Happe, Molecular Basis of [FeFe]-Hydrogenase Function: An Insight into the Complex Interplay between Protein and Catalytic Cofactor, Biochim. Biophys. Acta, 2013, 1827(8–9), 974–985, DOI:10.1016/j.bbabio.2013.03.004.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, Importance of the Protein Framework for Catalytic Activity of [FeFe]-Hydrogenases, J. Biol. Chem., 2012, 287(2), 1489–1499, DOI:10.1074/jbc.M111.305797.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Hybrid [FeFe]-Hydrogenases with Modified Active Sites Show Remarkable Residual Enzymatic Activity, Biochemistry, 2015, 54(7), 1474–1483, DOI:10.1021/bi501391d.
- O. Lampret, A. Adamska-Venkatesh, H. Konegger, F. Wittkamp, U.-P. Apfel, E. J. Reijerse, W. Lubitz, O. Rüdiger, T. Happe and M. Winkler, Interplay between CN Ligands and the Secondary Coordination Sphere of the H-Cluster in [FeFe]-Hydrogenases, J. Am. Chem. Soc., 2017, 139(50), 18222–18230, DOI:10.1021/jacs.7b08735.
- J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, E. Hofmann, U.-P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, Geometry of the Catalytic Active Site in [FeFe]-Hydrogenase Is Determined by Hydrogen Bonding and Proton Transfer, ACS Catal., 2019, 9(10), 9140–9149, DOI:10.1021/acscatal.9b02203.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, 14N HYSCORE Investigation of the H-Cluster of [FeFe] Hydrogenase: Evidence for a Nitrogen in the Dithiol Bridge, Phys. Chem. Chem. Phys., 2009, 11(31), 6553–6554, 10.1039/b913085n.
- G. Berggren, A. Adamska-Venkatesh, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J.-M. Mouesca, E. J. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Biomimetic Assembly and Activation of [FeFe]-Hydrogenases, Nature, 2013, 499(7456), 66–69, DOI:10.1038/nature12239.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. F. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Spontaneous Activation of [FeFe]-Hydrogenases by an Inorganic [2Fe] Active Site Mimic, Nat. Chem. Biol., 2013, 9, 607–609, DOI:10.1038/nchembio.1311.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Crystallographic and Spectroscopic Assignment of the Proton Transfer Pathway in [FeFe]-Hydrogenases, Nat. Commun., 2018, 9, 4726, DOI:10.1038/s41467-018-07140-x.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, How [FeFe]-Hydrogenase Facilitates Bidirectional Proton Transfer, J. Am. Chem. Soc., 2019, 141(43), 17394–17403, DOI:10.1021/jacs.9b09225.
- O. Lampret, J. Duan, E. Hofmann, M. Winkler, F. A. Armstrong and T. Happe, The Roles of Long-Range Proton-Coupled Electron Transfer in the Directionality and Efficiency of [FeFe]-Hydrogenases, Proc. Natl. Acad. Sci. USA, 2020, 117(34), 20520–20529, DOI:10.1073/pnas.2007090117.
- H. Tai, S. Hirota and S. T. Stripp, Proton Transfer Mechanisms in Bimetallic Hydrogenases, Acc. Chem. Res., 2021, 54, 232–241, DOI:10.1021/acs.accounts.0c00651.
- A. L. De Lacey, C. Stadler, C. Cavazza, E. C. Hatchikian and V. M. Fernandez, FTIR Characterization of the Active Site of the Fe-Hydrogenase from Desulfovibrio Desulfuricans, J. Am. Chem. Soc., 2000, 122(45), 11232–11233, DOI:10.1021/ja002441o.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, The Active Site of the [FeFe]-Hydrogenase from Desulfovibrio Desulfuricans. II. Redox Properties, Light Sensitivity and CO-Ligand Exchange as Observed by Infrared Spectroscopy, J. Biol. Inorg. Chem., 2006, 11(1), 102–118, DOI:10.1007/s00775-005-0040-2.
- A. Adamska-Venkatesh, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Identification and Characterization of the “Super-Reduced” State of the H-Cluster in [FeFe] Hydrogenase: A New Building Block for the Catalytic Cycle?, Angew. Chem., Int. Ed., 2012, 51(46), 11458–11462, DOI:10.1002/anie.201204800.
- A. Adamska-Venkatesh, D. Krawietz, J. F. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, New Redox States Observed in [FeFe] Hydrogenases Reveal Redox Coupling within the H-Cluster, J. Am. Chem. Soc., 2014, 136(32), 11339–11346, DOI:10.1021/ja503390c.
- M. Senger, S. Mebs, J. Duan, F. Wittkamp, U.-P. Apfel, J. Heberle, M. Haumann and S. T. Stripp, Stepwise Isotope Editing of [FeFe]-Hydrogenases Exposes Cofactor Dynamics, Proc. Natl. Acad. Sci. USA, 2016, 113(30), 8454–8459, DOI:10.1073/pnas.1606178113.
- S. Mebs, M. Senger, J. Duan, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, S. T. Stripp and M. Haumann, Bridging Hydride at Reduced H-Cluster Species in [FeFe]-Hydrogenases Revealed by Infrared Spectroscopy, Isotope Editing, and Quantum Chemistry, J. Am. Chem. Soc., 2017, 139(35), 12157–12160, DOI:10.1021/jacs.7b07548.
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U.-P. Apfel and S. T. Stripp, Proton-Coupled Reduction of the Catalytic [4Fe-4S] Cluster in [FeFe]-Hydrogenases, Angew. Chem., Int. Ed., 2017, 56(52), 16503–16506, DOI:10.1002/anie.201709910.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Protonation/Reduction Dynamics at the [4Fe–4S] Cluster of the Hydrogen-Forming Cofactor in [FeFe]-Hydrogenases, Phys. Chem. Chem. Phys., 2018, 20(5), 3128–3140, 10.1039/C7CP04757F.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, Identification of a Catalytic Iron-Hydride at the H-Cluster of [FeFe]-Hydrogenase, J. Am. Chem. Soc., 2016, 139(1), 83–86, DOI:10.1021/jacs.6b11409.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Accumulating the Hydride State in the Catalytic Cycle of [FeFe]-Hydrogenases, Nat. Commun., 2017, 8(16115), 1–7, DOI:10.1038/ncomms16115.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, Direct Observation of an Iron-Bound Terminal Hydride in [FeFe]- Hydrogenase by Nuclear Resonance Vibrational Spectroscopy, J. Am. Chem. Soc., 2017, 139(12), 4306–4309, DOI:10.1021/jacs.7b00686.
- M. Haumann and S. T. Stripp, The Molecular Proceedings of Biological Hydrogen Turnover, Acc. Chem. Res., 2018, 51(8), 1755–1763, DOI:10.1021/acs.accounts.8b00109.
- C. Lorent, S. Katz, J. Duan, C. Julia Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, Shedding Light on Proton and Electron Dynamics in [FeFe] Hydrogenases, J. Am. Chem. Soc., 2020, 142(12), 5493–5497, DOI:10.1021/jacs.9b13075.
- K. Laun, S. Mebs, J. Duan, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Spectroscopical Investigations on the Redox Chemistry of [FeFe]-Hydrogenases in the Presence of Carbon Monoxide, Molecules, 2018, 23(7), 1669, DOI:10.3390/molecules23071669.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, Proton Coupled Electronic Rearrangement within the H-Cluster as an Essential Step in the Catalytic Cycle of [FeFe] Hydrogenases, J. Am. Chem. Soc., 2017, 139(4), 1440–1443, DOI:10.1021/jacs.6b12636.
- Y. Nicolet, A. L. De Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, Crystallographic and FTIR Spectroscopic Evidence of Changes in Fe Coordination upon Reduction of the Active Site of the Fe-Only Hydrogenase from Desulfovibrio Desulfuricans, J. Am. Chem. Soc., 2001, 123(8), 1596–1601, DOI:10.1021/ja0020963.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, CO-Bridged, H-Cluster Intermediates in the Catalytic Mechanism of [FeFe]-Hydrogenase CaI, J. Am. Chem. Soc., 2018, 140(24), 7623–7628, DOI:10.1021/jacs.8b03072.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. Debeer, Spectroscopic and Computational Evidence That [FeFe] Hydrogenases Operate Exclusively with CO-Bridged Intermediates, J. Am. Chem. Soc., 2020, 142(1), 222–232, DOI:10.1021/jacs.9b09745.
- G. Filippi, F. Arrigoni, L. Bertini, L. De Gioia and G. Zampella, DFT Dissection of the Reduction Step in H2 Catalytic Production by [FeFe]-Hydrogenase-Inspired Models: Can the Bridging Hydride Become More Reactive Than the Terminal Isomer?, Inorg. Chem., 2015, 54(19), 9529–9542, DOI:10.1021/acs.inorgchem.5b01495.
- C. Greco, M. Bruschi, L. Gioia and U. De; Ryde, A QM/MM Investigation of the Activation and Catalytic Mechanism of Fe-Only Hydrogenases, Inorg. Chem., 2007, 46(15), 5911–5921, DOI:10.1021/ic062320a.
- M. Bruschi, P. Fantucci and L. De Gioia, Density Functional Theory Investigation of the Active Site of Fe-Hydrogenases. Systematic Study of the Effects of Redox State and Ligands Hardness on Structural and Electronic Properties of Complexes Related to the [2Fe](H) Subcluster, Inorg. Chem., 2004, 43(12), 3733–3741, DOI:10.1021/ic035326y.
- M. L. K. Sanchez, C. Sommer, E. Reijerse, J. A. Birrell, W. Lubitz and R. B. Dyer, Investigating the Kinetic Competency of CrHydA1 [FeFe] Hydrogenase Intermediate States via Time-Resolved Infrared Spectroscopy, J. Am. Chem. Soc., 2019, 141(40), 16064–16070, DOI:10.1021/jacs.9b08348.
- S. T. Stripp, S. Mebs and M. Haumann, Temperature Dependence of Structural Dynamics at the Catalytic Cofactor of [FeFe]-Hydrogenase, Inorg. Chem., 2020, 59(22), 16474–16488, DOI:10.1021/acs.inorgchem.0c02316.
- P. Rodríguez-Maciá, N. Breuer, S. DeBeer and J. A. Birrell, Insight into the Redox Behavior of the [4Fe-4S] Subcluster in [FeFe] Hydrogenases, ACS Catal., 2020, 10, 13084–13095, DOI:10.1021/acscatal.0c02771.
- J. M. Kuchenreuther, S. J. George, C. S. Grady-Smith, S. P. Cramer and J. R. Swartz, Cell-Free H-Cluster Synthesis and [FeFe] Hydrogenase Activation: All Five CO and CN− Ligands Derive from Tyrosine, PLoS One, 2011, 6(5), e20346, DOI:10.1371/journal.pone.0020346.
- L. Kertess, A. Adamska-Venkatesh, P. Rodríguez-Maciá, O. Rüdiger, W. Lubitz and T. Happe, Influence of the [4Fe-4S] Cluster Coordinating Cysteines on Active Site Maturation and Catalytic Properties of C. Reinhardtii [FeFe]-Hydrogenase, Chem. Sci., 2017, 8(12), 8127–8137, 10.1039/c7sc03444j.
- P. Rodríguez-Maciá, L. Kertess, J. Burnik, J. A. Birrell, E. Hofmann, W. Lubitz, T. Happe and O. Rüdiger, His-Ligation to the [4Fe-4S] Subcluster Tunes the Catalytic Bias of [FeFe] Hydrogenase, J. Am. Chem. Soc., 2019, 141(1), 472–481, DOI:10.1021/jacs.8b11149.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, How [FeFe]-Hydrogenase Facilitates Bidirectional Proton Transfer, J. Am. Chem. Soc., 2019, 141(43) DOI:10.1021/jacs.9b09225.
- D. Moss, E. Nabedryk, J. Breton and W. Mäntele, Redox-Linked Conformational Changes in Proteins Detected by a Combination of Infrared Spectroscopy and Protein Electrochemistry. Evaluation of the Technique with Cytochrome C, Eur. J. Biochem., 1990, 187(3), 565–572, DOI:10.1111/j.1432-1033.1990.tb15338.x.
- L. Mészáros, P. Ceccaldi, M. Lorenzi, H. J. Redman, E. Pfitzner, J. Heberle, M. Senger, S. T. Stripp and G. Berggren, Spectroscopic Investigations under Whole Cell Conditions Provide New Insight into the Metal Hydride Chemistry of [FeFe]-Hydrogenase, Chem. Sci., 2020, 11(18), 4608–4617, 10.1039/d0sc00512f.
- F. P. Healey, The Mechanism of Hydrogen Evolution by Chlamydomonas Moewusii, Plant Physiol., 1970, 45(2), 153–159, DOI:10.1104/pp.45.2.153.
- U. Klein and A. Betz, Induced Protein Synthesis during the Adaptation to H2 Production in Chlamydomonas Moewusii, Physiol. Plant., 1978, 42(1), 1–4, DOI:10.1111/j.1399-3054.1978.tb01529.x.
- P. Roessler and S. Lien, Anionic Modulation of the Catalytic Activity of Hydrogenase from Chlamydomonas Reinhardtii, Arch. Biochem. Biophys., 1982, 213(1), 37–44, DOI:10.1016/0003-9861(82)90436-2.
- T. Happe and J. D. Naber, Isolation, Characterization and N–terminal Amino Acid Sequence of Hydrogenase from the Green Alga Chlamydomonas Reinhardtii, Eur. J. Biochem., 1993, 214(2), 475–481, DOI:10.1111/j.1432-1033.1993.tb17944.x.
- J. C. Ruth, R. D. Milton, W. Gu and A. M. Spormann, Enhanced Electrosynthetic Hydrogen Evolution by Hydrogenases Embedded in a Redox–Active Hydrogel, Chem. – Eur. J., 2020, 26(32), 7323–7329, DOI:10.1002/chem.202000750.
- V. Fourmond, C. Baffert, K. Sybirna, S. Dementin, A. Abou-Hamdan, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, The Mechanism of Inhibition by H2 of H2-Evolution by Hydrogenases, Chem. Commun., 2013, 49(61), 6840–6842, 10.1039/c3cc43297a.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rüdiger, W. Lubitz and H. Ogata, Unique Spectroscopic Properties of the H-Cluster in a Putative Sensory [FeFe] Hydrogenase, J. Am. Chem. Soc., 2018, 140(3), 1057–1068, DOI:10.1021/jacs.7b11287.
- H. Land, P. Ceccaldi, L. S. Mészáros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Discovery of Novel [FeFe]-Hydrogenases for Biocatalytic H2-Production, Chem. Sci., 2019, 10(43), 9941–9948, 10.1039/c9sc03717a.
- N. Chongdar, K. Pawlak, O. Rüdiger, E. J. Reijerse, P. Rodríguez-Maciá, W. Lubitz, J. A. Birrell and H. Ogata, Spectroscopic and Biochemical Insight into an Electron-Bifurcating [FeFe] Hydrogenase, J. Biol. Inorg. Chem., 2020, 25, 135–149, DOI:10.1007/s00775-019-01747-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt00110h |
| This journal is © The Royal Society of Chemistry 2021 |