
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeteroleptic, polynuclear dysprosium(III)-carbamato complexes through in situ carbon dioxide capture†
Sören
Schlittenhardt
 a,
Eufemio
Moreno-Pineda
*ab and
Mario
Ruben
*acd
a,
Eufemio
Moreno-Pineda
*ab and
Mario
Ruben
*acd
aInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: mario.ruben@kit.edu; eufemio.moreno@up.ac.pa
bDepto. de Química-Física, Escuela de Química, Facultad de Ciencias Naturales, Exactas y Tecnología, Universidad de Panamá, Panamá
cInstitut de Science et d'Ingénierie Supramoléculaires (ISIS, UMR 7006), CNRS-Université de Strasbourg, 8 allée Gaspard Monge BP 70028 67083 Strasbourg Cedex France, France
dInstitute of Quantum Materials and Technologies (IQMT), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 15th March 2021
Abstract
Amine groups are among the most effective systems for carbon dioxide capture. Reminiscent of the activation of nature's most abundant enzyme RuBisCO, the treatment of amines with CO2 in the presence of oxophilic metal ions, e.g. Mg2+, results in the formation of carbamates. Here we report the synthesis, structure and magnetic properties of three new dysprosium-carbamato complexes. The reaction of gaseous CO2 with N,N-diisopropylamine and DyCl3(DME)2 (DME = Dimethoxyethane) in toluene leads to the formation of the tetrametallic complex [Dy4(O2CNiPr2)10(O-C2H4–OMe)2]. The addition of 2-hydroxy-3-methoxybenzaldehyde-N-methylimine yields the hexametallic compound [Dy6(O2CNiPr2)8(O-C2H4–OMe)2(CO3)2(C9O2NH10)4] in which the metal sites form a chair-like configuration; The same hexanuclear motif is obtained using N,N-dibenzylamine. We show that by employing CO2 as a feedstock, we are able to capture up to 2.5 molecules of CO2 per Dy ion. Magnetic measurements show a decreasing χMT at low temperatures. Combining the experimental magnetic data with ab initio calculations reveils tilting of the easy axes and implies the presence of antiferromagnetic interactions between the Dy(III) metal ions.
Introduction
The most common greenhouse gas in the earth's atmosphere is CO2. While atmospheric CO2-concentrations between 180 and 300 ppm were shown to be normal throughout earth's history by examination of Antarctic ice,1 present values are exceeding 400 ppm as a consequence of industrialisation, and are still increasing.2 It is well known that the high concentration of CO2 and other greenhouse gases are the main reason for the observed climate change of the past years and decades.3 In order to stop the enormous increase of CO2 in the atmosphere and by that help preserving our climate finding new ways of fixing CO2 or even better converting it to added value materials should be an important goal of modern research.In nature plants make use of CO2 using it as a C1-building block to form glucose in the Calvin-cycle of photosynthesis. The enzyme allowing for this process is earth's most abundant enzyme Ribulose-1,5-bisphosphate carboxylase-oxygenase better known as RuBisCO, in which the active site is based on an Mg2+ ion. The enzyme is activated when a single molecule of CO2 is inserted between the Mg2+-centre and the ε-N of a complexing lysine amino acid to form a carbamate (R2NCO2−), which then acts as a catalyst to convert CO2 to biomass. A variety of substances has been shown to fix CO2 directly as a coordinating ligand4 as well as by converting it into carbonates5 or – reminiscent of RuBisCO – into carbamates.6,7 While many of the reported materials in this regard are based on Mg2+ similar to RuBisCO or transition metals, also lanthanide ions have been shown to form carbamato complexes with high ratios of CO2 per metal ion due to their oxophilic nature.8–10
Not only do these materials serve the fixation of CO2 mentioned, lanthanides are also promising candidates for application in quantum technology as they have shown interesting physical properties besides their immense structural diversity. Examples of the fascinating properties observed in lanthanide systems are blocking of the magnetisation at high temperature,11,12 the ability to manipulate and read out nuclear spins13 or the so called ‘spin-ice’ effect.14 Based on their electronic structure ([Xe] 4fn) in combination with relatively strong spin–orbit-coupling lanthanide compounds are one of, if not, the most promising candidates for the synthesis of Single Molecule Magnets (SMMs) which have marked a growing research field in chemistry in the past years. SMM behaviour is expressed as an energy barrier to spin relaxation within a doublet ground state below a system specific blocking temperature. Due to these traits, lanthanide-based systems could be used to build quantum data storage devices. Several systems have been reported to be promising candidates for quantum spintronic devices like spin valves15,16 and transistors13 as well as for qubits for quantum computing.17,18 The later no longer being only a proposal as Godfrin et al. were able to implement Grover's search algorithm on a TbPc2-SMM.19,20
Herein we explore the range of lanthanide-based compounds using CO2 as a renewable feedstock. We report the synthesis and structure of three new dysprosium(III)-carbamato complexes [Dy4(O2CNiPr2)10(O-C2H4–OMe)2] 1, [Dy6(O2CNiPr2)8(O-C2H4–OMe)2(CO3)2(C9O2NH10)4] 2 and [Dy6(O2CNBz2)8(O-C2H4–OMe)2(CO3)2(C9O2NH10)4] 3 obtained by the reaction of gaseous CO2 with N,N-diisopropylamine or N,N-dibenzylamine in toluene in the presence of DyCl3(DME)2, where DME![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Dimethoxyethane. The magnetic properties of these systems are investigated by means of SQUID measurements as well as via ab initio calculations. We explore the effect of the introduced co-ligands on both the structure and the properties compared to known molecules obtained in comparable fashion.
Dimethoxyethane. The magnetic properties of these systems are investigated by means of SQUID measurements as well as via ab initio calculations. We explore the effect of the introduced co-ligands on both the structure and the properties compared to known molecules obtained in comparable fashion.
Experimental section
Synthetic procedure
Crystallography
Single crystal X-ray diffraction measurements were performed on a STOE StadiVadi 25 diffractometer using a GeniX 3D HF micro focus with MoKα-radiation (λ = 0.71073 Å) and a CCD image plate detector. Crystals were mounted using crystallographic oil and placed in a cold nitrogen stream. All data were corrected for absorption using CrysAlisPro.23 The structures were solved by direct methods and refined against F2 using the SHELXL-97 package24 in Olex2.25 All non-hydrogen atoms were refined anisotropically and hydrogens were placed based on a riding model approach. Full crystallographic details can be found in CIF format: see the Cambridge Crystallographic Data Centre database 2049538–2049540 for 1–3.†Magnetic measurements
Magnetic susceptibility data were collected at temperatures between 2–300 K using a Quantum Design MPMS-XL SQUID magnetometer. Magnetisation data was collected with a Quantum Design MPMS3 SQUID magnetometer equipped with a 7 T magnet. The samples were grounded and fixed in a gelatine capsule using small amounts of eicosane to avoid any movement of the sample. The data obtained were corrected for diamagnetic contributions of the eicosane, the gelatine capsule and the sample holder. Diamagnetic corrections for the sample were estimated using Pascal's constants.Ab initio calculations
[9,7]-CASSCF calculations were performed on Dy1-fragments of the obtained molecules using MOLCAS 8.2.26 The basis sets employed were of ANO-RCC27 quality with a size of VTZP for Dy, VDZP for O and N and VDZ for C and H. The orbital optimisation was carried out including 21 sextets, 224 quartets and 490 doublets of which 21, 128 and 130 were mixed by spin–orbit-coupling respectively. All input geometries were taken from crystal structure refinement without further optimisation.Results and discussion
Synthesis
The reactions occurring between secondary amines and carbon dioxide have been studied thoroughly.28,29 In a first step a derivate of carbamic acid is formed, that reacts with a second amine yielding dialkylammonium dialkylcarbamate (eqn (1) and (2)).| R2NH + CO2 ⇌ R2NCOOH | (1) |
| R2NCOOH + R2NH ⇌ [R2NH2] [R2NCO2] | (2) |
Protonation of the carbamate ion favours the left-hand side of the equilibrium, resulting in a high sensitivity of carbamate compounds towards water and other protic solvents. Also, in the presence of water, direct hydrolysis of the carbamate to bicarbonate can occur (eqn (3)).
| R2NCO2− + H2O ⇌ R2NH + HCO3− | (3) |
The immediate formation of bicarbonate and carbonate has to be considered too, as the high concentration of amine pushes the equilibrium strongly towards the right-hand side of eqn (4). Typically, this process is very slow, however, in nature it is sped up drastically by carbonic anhydrases.30
| H2O + CO2 ⇌ H2CO3 ⇌ H+ + HCO3− ⇌ 2H+ + CO32− | (4) |
Introducing lanthanide chloride salts into the carbamate system can result in the formation of lanthanide-carbamato-complexes by exchanging the chloride with the respective carbamato anion. The resulting Dialkylammonium-chloride is barely soluble in toluene and can be removed by simple filtration. Due to the formation of carbonates and the general reactivity of carbamates towards water, homoleptic lanthanide-carbamato complexes of the general formula Ln4(O2CNR2)12 are prepared under strictly anhydrous conditions. LnCl3(DME)2 has been reported as an easily accessible precursor to anhydrous lanthanide chlorides employed in this type of synthesis.21 In order to obtain the Ln4(O2CNR2)12 compounds, DME can be removed from the precursor by drying at 120 °C under vacuum for 2 h before CO2 capture,8 or LnCl3(DME)2 can be introduced after treatment of the amine system with CO2.29 In this work we introduced the DME containing precursor prior to the CO2 uptake, without the initial drying step. DyCl3(DME)2 was suspended in toluene and treated with iPr2NH followed by the uptake of gaseous carbon dioxide. The presence of DME during the treatment with CO2 led to the cleavage of DME into methoxy-ethoxide, which is found as a bridging ligand in compounds 1, 2 and 3. Multiple other lanthanide complexes, both homoleptic in the form of a Dy10 wheel and heteroleptic, have been reported with methoxy-ethoxide as a ligand.31–33 However, in all cases the alkoxide is obtained from deprotonation of methoxy-ethanol, not from DME. To our knowledge, the cleavage of DME to methoy-ethoxide has previously been only observed in Li-containing systems.34,35 Very likely Dy3+, which can be seen as a very hard Lewis-acid similar to Li+, promotes the cleavage by coordination to the methoxy-O. With the introduction of the methoxy-ethoxide anion into the iPr2NH/CO2 system we obtained the tetranuclear complex 1 where the four Dy(III) ions form a rectangle due to the μ3-bridging ethoxide, vide infra. Alongside DME, we have introduced 2-hydroxy-3-methoxybenzaldehyde-N-methylimine (Fig. S1†) as a second co-ligand to the synthetic route. These types of o-vanilin derived ligands are known to function as ligands for several lanthanide complexes.36,37 Deprotonation of the hydroxyl group gives an anionic ligand bridging two Dy3+. Together with the ligand, small amounts of water have been introduced into our system. As the amount of water is low in respect to the high concentration of amine in our reaction, the equilibrium of eqn (4) is heavily shifted to the carbonate side upon addition of CO2. The resulting combination of carbonates, carbamates, methoxy-ethoxide and deprotonated 2-hydroxy-3-methoxybenzaldehyde-N-methylimine yields the hexanuclear Dy complex 2. The same hexanuclear motif was observed in the benzyl-analogue 3 employing dibenzylamine instead of diisopropylamine.
Infrared spectroscopic measurements (Fig. S5†) unveil very strong absorption of the iso-propyl groups between 2800 and 3000 cm−1 for compounds 1 and 2. More specifically the absorption in this region reveals three distinct peaks at 2875 cm−1, 2930 cm−1 and 2965 cm−1. The weakest one at 2875 cm−1 corresponds to the C–H stretching at the tertiary carbon, while the other two can be attributed to the symmetric and antisymmetric C–H stretching modes of the CH3-groups, respectively. For 3 the strong absorption of iPr cannot be observed, instead the overtone of the CO modes around 1500 cm−1 becomes visible at 3000 cm−1. All three compounds show very strong absorption in the range of 1300–1700 cm−1 which is the typical region for the CO modes of carbamates. At 1650 cm−1 a single sharp absorption is observed for compounds 2 and 3 that we attribute to the CO32− anion, as it is not observed in 1. The multitude of different ligands and coordination modes cause a very high number of absorption peaks in the low energy fingerprint region of the spectrum, which cannot be clearly attributed to single groups or modes.
X-ray crystallography
Compound 1 (Fig. 1a and b, S2†) crystallises in the monoclinic space group C2/c with half the molecule in the asymmetric unit (Table 1). In total the molecule consists of four Dy(III)-ions, ten N,N-diisopropylcarbamates and two methoxy-ethoxides. The Dy(III)-ions form a rhombic plane with Dy(1)–Dy(2) and Dy(1)–Dy(2)′ distances along the edges of 3.7134(6) Å and 3.8759(5) Å, respectively. The distances across the plane are 3.8560(6) Å and 6.5387(6) Å for Dy(1)–Dy(1)′ and Dy(2)–Dy(2)′. On opposing sites of the plane the alkoxy-group of the two methoxy-ethoxide ligands are μ3-bridging the Dy(1)–Dy(1)′–Dy(2) and Dy(1)–Dy(1)′–Dy(2)′ triangle with Dy–O distances between 2.370(2) Å and 2.441(2) Å. The methoxy-O is coordinating towards Dy(2) with a distance of 2.516(2) Å. Along the edges the metal centres are bridged by two carbamato groups per edge with one of the carbamates coordinating as 2.11 and the other one as 2.21 following Harris’ notation38 with Dy–O distances between 2.233(2) Å and 2.558(2) Å. The two remaining carbamate ligands cap the Dy(2)-corner in a 1.11 fashion. The coordination geometries for Dy(1) and Dy(2) are best described as triangular dodecahedrons with CShM values of 3.228 and 1.791, respectively (Table S1†). Similar to the homoleptic compound Dy4(O2CNiPr2)12 we observe a tetranuclear compound. However, in Dy4(O2CNiPr2)12 the four Dy(III) ions form a distorted tetrahedron rather than a plane. | ||
| Fig. 1 (a) and (b) Side and Top view of the crystal structure of 1; (c) and (d) Side and Top view of the crystal structure of 2. Colour code: Dy: green; O: red; N: blue; C: black (H and iPr are omitted for clarity). | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| Chemical formula | Dy4N10O24C76H154·C7H8 | Dy6N12O34C100H166·4C7H8 | Dy6N12O34C164H166·2.5C7H8 |
| Fw | 2334.22 | 3423.97 | 4053.76 |
| Temperature [K] | 180 | 180 | 180 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c | P21/c |
| a [Å] | 25.3429(3) | 27.6199(2) | 15.05782(16) |
| b [Å] | 16.74118(16) | 28.7305(3) | 20.9377(2) |
| c [Å] | 25.2476(3) | 20.34422(16) | 29.0794(3) |
| α [°] | 90 | 90 | 90 |
| β [°] | 102.0169(10) | 92.4511(7) | 97.1774(9) |
| γ [°] | 90 | 90 | 90 |
| V [Å3] | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 477.09(18) 477.09(18) |
16129.1(2) |
9096.21(16) |
| Z | 4 | 4 | 2 |
| P cacld [g cm−3] | 1.480 | 1.372 | 1.430 |
| μ [mm−1] | 2.888 | 2.811 | 2.505 |
| R 1 | 0.0254 | 0.0292 | 0.0400 |
| wR2 | 0.0566 | 0.0711 | 0.1164 |
Swapping out two of the carbamato ligands with two methoxy-ethoxides is enough to completely change the geometry, as the alkoxide is capable of μ3-bridging Dy3 triangles.
Compound 2 (Fig. 1c and d, S3†) crystallises in the monoclinic space group C2/c with half the molecule in the asymmetric unit (Table 1). The molecule is composed of six Dy(III)-ions, eight N,N-diisopropylcarbamates, four deprotonated 2-hydroxy-3-methoxybenzaldehyde-N-methylimines, two deprotected DMEs and two carbonates. A rectangle is formed between four of the six metal ions with the remaining Dy(2) and Dy(2)′ being located above and below the long edges reminiscent of the chair-conformation in cyclohexane. The Dy(1)–Dy(2) and Dy(2)–Dy(3) distances along the long edges are 3.7228(8) Å and 3.8417(7) Å, while the short edge Dy(1)–Dy(3)′ distance equals 4.4852(7) Å. The two carbonate ions are located within the Dy6-chair connecting Dy(1)–Dy(2)–Dy(2)′–Dy(3)′ and Dy(1)′–Dy(2)–Dy(2)′–Dy(3). The alkoxy-group of 2-hydroxy-3-methoxybenzaldehyde-N-methylimine bridges Dy(1)–Dy(2) and Dy(2)–Dy(3) with the -OMe group coordinating Dy(1)/Dy(2) and the = NMe group coordinating Dy(2)/Dy(3). Dy(1)–Dy(2) is additionally bridged by the alkoxy-group of DME with the –OMe group joining the coordination of Dy(1). Four of the eight carbamates bridge Dy(2)–Dy(3) and Dy(1)–Dy(3)′ via 2.11 coordination, while the other four coordinate Dy(1) and Dy(3) as 1.11. The Dy–O distances towards bridging O's are between 2.243(3) Å and 2.433(3) Å. Towards non-bridging atoms the distances are between 2.359(3) Å and 2.418(3) Å for 1.11-carbamates and between 2.532(3) Å and 2.619(3) Å for methoxy and imine groups. The resulting coordination geometries are best described as biaugmented trigonal prisms for Dy(1) and Dy(3) with CShM values of 2.847 and 2.620 and a square antiprism for Dy(2) with a CShM value of 0.994 (Table S2†).
Compound 3 (Fig. S3†) crystallises in the monoclinic space group P21/c with half the molecule in the asymmetric unit (Table 1). The overall structural motif is similar to 2 with eight N,N-dibenzylcarbamato groups instead of the diiopropyl-analogue. The Dy(1)–Dy(2) and Dy(2)–Dy(3) distances along the long edges of the rectangle are on average shorter than in 2 with distances of 3.8224(6) Å and 3.7088(8) Å, respectively. The Dy(1)–Dy(3)′ distance along the short edge is longer with 4.4977(6) Å. The Dy–O distances range between 2.231(1) Å and 2.444(8) Å for non-bridging oxygen donors. For the terminal 1.11-carbamates, the distances lie in a range of 2.357(2) Å to 2.441(5) Å and towards methoxy- and imine-groups, the distances are between 2.511(5) Å and 2.642(7) Å. With the small changes to the geometry compared to 2 the coordination of Dy(1) and Dy(3) in 3 is best described as a triangular dodecahedron with CShM values of 2.493 and 2.276. Dy(2) is, same as for 2, best described as a square antiprism with a CShM value of 0.997 (Table S3†). The most remarkable feature are the two central carbonate ions which bridge four Dy(III) ions. Multiple other lanthanide carbonato compounds have been reported, however, coordination towards four ions is unusual. In those compounds the carbonate forms a regular triangle of three metal centres39,40 or even connects two lanthanide ions to a transition metal ion.41
Magnetic data
We explored the magnetic properties of compounds 1–3 by measuring of the magnetic susceptibility upon cooling the sample from 300 to 2 K in a 1 kOe DC field (0.1 T). The data for all three compounds are shown as χMT vs. T plots in Fig. 2. At room temperature the χMT values observed are 53.25 cm3 K mol−1, 74.11 cm3 K mol−1 and 79.60 cm3 K mol−1, respectively. For all compounds 1–3 those are about 10% below the expected values derived from Curie's law of 56.7 cm3 K mol−1 and 85.0 cm3 K mol−1 for four and six non-interacting Dy(III)-ions, respectively. Below 100 K the χMT values start dropping most likely due to depopulation of Stark levels and possibly antiferromagnetic interactions. The magnetisation versus field has been measured at 2, 3, 4 and 5 K; the data plots are shown in Fig. 2. The magnetisation approaches saturation for all three compounds, however, none of the samples reaches saturation at 7 T, due to the interplay with excited states. Similar to the susceptibility the magnetisation values are in good agreement with what is expected for four and six Dy(III)-ions of 5μB per Dy3+. | ||
| Fig. 2 (a): χMT vs. T for compounds 1–3; (b): M vs. H for 1; (c): M vs. H for 2; (d): M vs. H for 3 (solid lines represent CASSCF simulations). | ||
Single molecule magnet behaviour can be tested for by means of AC magnetic susceptibility measurements as they show whether a sample exhibits slow magnetic relaxation. We employed AC measurements at 2 K with and without applied DC fields to all compounds, however neither complex shows out-of-phase signals.
Theoretical description
Ab initio CASSCF calculations give us the opportunity to explore the electronic nature of the lanthanide ions as well as to simulate physical and magnetic properties. We calculated the χMT(T) and M(H) behaviour to compare it with the performed measurements. The simulations shown in Fig. 2 (solid lines) correspond to the magnetic properties of uncoupled systems. It can be seen that above 100 K the theoretical susceptibility fits the experimental data really well. The simulations are multiplied by a scaling factor of 0.957 for 1, 0.889 for 2 and 0.954 for 3 to fit the experimental data. Note that these scaling factors are not unusual and do not imply a wrong model.42 Below 100 K the measured susceptibility of all three compounds drops faster than the theoretical susceptibility. A possible explanation for the fast drop is the existence of antiferromagnetic interactions between the different Dy ions of the complex, which are neglected in the single-ion-fragment calculations. Employing crystal field parameters, it is possible to account for the susceptibility and magnetisation resulting from coupled ions.Unfortunately, due to the large Hilbert space we are not able to perform fitting of experimental data employing four and six highly anisotropic Dy ions. However, by employing the g-tensors and the magnetic frames obtained for each individual ion, it is possible to calculate the purely dipolar interactions between the Dy–Dy pairs using eqn (5).
| J1,2dip = μB2/R3 (g1g2 − 3 ((g1R)(Rg2)/|R|2)) | (5) |
Fig. 3 shows which interaction corresponds to which pair of ions. For 1 we obtained J1 = –0.858 cm−1, J2 = –0.681 cm−1 and J3 = –0.103 cm−1 considering a −2J formalism. For 2 and 3 the interactions found (Table S5†) are also all antiferromagnetic and in the magnitude of 10−1 cm−1. The interactions are significant enough that we can assume them as the reason for the fast decay of susceptibility at low temperatures. For SMM behaviour a well separated pure mJ = 15/2 (for Dy III) ground state is required, characterised by the magnetic g-tensors gx ≈ gy ≈ 0 and gz ≈ 20.
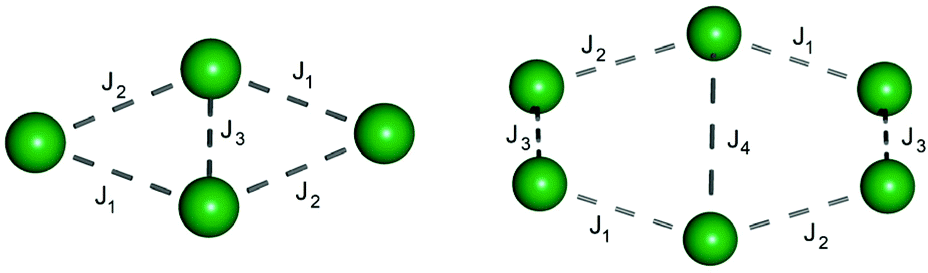 | ||
| Fig. 3 Exchange interaction scheme for complex 1 (left) and 2 and 3 (right). | ||
Through CASSCF calculations, we were able to get a deeper insight to the nature of the ground state. All g-tensors and separations towards the first excited doublet state are given in Table S4.† In 1 only Dy(2) has an essentially pure mJ = 15/2 ground state with gx, gy and gz being 0.07, 0.18 and 19.51, respectively. The separation to the first excited doublet is low (42.2 cm−1). As comparison the separation towards the first excited states in the reported carbamate based SMM Dy4(O2CNiPr2)128 are 102 cm−1 and 122 cm−1 for the two Dy(III)-centres. The differences are well understood if one considers the coordination geometry. In Dy4(O2CNiPr2)12 the coordination polyhedron is given as a capped trigonal prism, which is better suited to stabilise the oblate mJ states than the triangular dodecahedrons observed for 1.43 For 2 all three different sites Dy(1), Dy(2) and Dy(3) are close to pure mJ = 15/2 ground states with gz values of 19.72, 18.79 and 18.99 respectively. The separations to each first excited state are 91.9 cm−1, 58.0 cm−1 and 85.8 cm−1. Due to the close structural relationship to 2, Dy(1) and Dy(3) of compound 3 show very similar gz values at 19.67 and 19.22 and separations of 97.3 cm−1 and 93.0 cm−1 respectively. Surprisingly, Dy(2) which is the closest in geometry to its analogue in 2 (vide supra) shows the largest deviation with gz = 19.17 and ΔE = 95.56 cm−1. Comparing the Dy(2)'s in 2 and 3 reveals that the formed square antiprism is almost the same in both complexes. However, in 3 the central Dy ion is shifted towards the O3N square and away from the O4 square compared to 2. Being closer to the softer nitrogen donor than to the harder oxygen causes a more axial ligand field, which is beneficial for the oblate Dy(III) ion.
The separation values towards the first excited doublet imply that SMM behaviour could be possible in all three molecules. Thus, we can infer that the reason for the absence of blocking of magnetisation for all three compounds could be related to the Dy–Dy interactions, vide supra. In addition, Fig. S6–S8† show the directions of the anisotropy axes obtained from the CASSCF calculations. We observe a large tilting between the easy axes of the different Dy-sites in both the tetra- and hexanuclear structures. It is well known that co-linear alignment of the easy axes often leads to, while non co-linearity suppresses SMM behaviour.44 The transverse g values observed for the ground states of Dy(1) in 1 and Dy(2) in 2 and 3 allow quantum tunnelling of the magnetisation (QTM),45,46 and with the strong antiferromagnetic coupling towards the other Dy(III) ions, therefore, could provide an efficient way of relaxation for the whole molecule.
Conclusion
We have prepared three new dysprosium-carbamato-complexes from water-free DyCl3(DME)2, diisopropylamine or dibenzylamine and gaseous CO2. The introduction of DME as a second ligand alongside the carbamato anion led to the formation of a tetranuclear compound 1 in which the Dy4-structure is being pushed into a rhombic plane instead of the almost tetrahedral structure observed previously in Dy4(O2CNPri2)12.8–10 The oxophilic nature of the lanthanide is causing cleavage of DME turning it into the methoxy-ethoxide anion. This unusual cleavage of the DME has not been observed for other lanthanide systems so far. In 2 and 3 the addition of 2-hydroxy-3-methoxybenzaldehyde-N-methylimine led to the formation of hexanuclear clusters in which the DME is deprotected in a similar fashion. However, opposing to 1, DME is not μ3-bridging but connecting two Dy ions. On top of that, the molecular structures of 2 and 3 are built around two bridging carbonate ions, which are formed from CO2 and water introduced with the imine-ligand, which cannot be properly dried in vacuo without sublimation. Interestingly, in the basic surrounding the water reacts with CO2 to form CO32− instead of reacting with the present carbamates, which are normally very sensitive to water.The magnetic data of all three compounds show the common behaviour for χMT vs. T and M vs. H. However, neither can be considered an SMM. On the other hand, ab initio calculations show that the individual metal centres could very well show SMM behaviour. A possible reason to bring the magnetic data and calculated properties together could be the occurrence of antiferromagnetic interactions between the Dy-ions within the molecules and the tilting of their easy axes, which are known to induce fast relaxation in SMMs. The dipolar interactions between pairs of Dy(III) ions have been calculated and the interactions confirm our expectation of antiferromagnetic coupling.
Besides finding new ways of using carbon dioxide as a feedstock, our goal was to find new materials for application in quantum technologies. We successfully employed CO2 as a starting material to form molecules with CO2/Ln-ratios of 2.5 and 1.66. Despite none of the compounds reported showing SMM behaviour, the synthesis can be seen as a model step to the wide and promising variety of lanthanide-carbamato-compounds possible to obtain.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Karlsruhe Nano Micro Facility (KNMF) for access to instruments and laboratories. EMP thanks the Panamanian National Systems of Investigators (SNI, SENACYT) for support.References
- J. Petit, J. Jouzel, D. Raynaud, N. I. Barkov, J.-M. Barnola, I. Basile, M. Bender, J. Chappellaz, M. Davis, G. Delaygue, M. Delmotte, V. M. Kotlyakov, M. Legrand, V. Y. Lipenkov, C. Lorius, L. Pépin, C. Ritz, E. Saltzman and M. Stievenard, Nature, 1999, 399, 429–436 CrossRef CAS.
- P. M. Cox, R. A. Betts, C. D. Jones, S. A. Spall and I. J. Totterdell, Nature, 2000, 408, 184–187 CrossRef CAS PubMed.
- Climate Change 1995: The Science of Climate Change, ed. J. T. Houghton, et al., Cambridge Univ. Press, Cambridge, 1996 Search PubMed.
- M. N. Bochkarev, E. A. Fedorova, Y. F. Radkov, S. Y. Khorhev, G. S. Kalinina and G. A. Razuvaev, J. Organomet. Chem., 1983, 258, C29–C33 CrossRef CAS.
- X.-L. Tang, W.-H. Wang, W. Dou, J. Jiang, W.-S. Liu, W.-W. Qin, G.-L. Zhang, H.-R. Zhang, K.-B. Yu and L.-M. Zheng, Angew. Chem., Int. Ed., 2009, 48, 3499–3502 CrossRef CAS PubMed.
- M. Ruben, D. Walther, R. Knake, H. Görls and R. Beckert, Eur. J. Inog. Chem., 2000, 5, 1055–1060 CrossRef.
- D. B. Dell’ Amico, F. Calderazzo, B. Giovannitti and G. Pelizzi, J. Chem. Soc., Dalton Trans., 1984, 0, 647–652 RSC.
- E. Moreno Pineda, Y. Lan, O. Fuhr, W. Wernsdorfer and M. Ruben, Chem. Sci., 2017, 8, 1178–1185 RSC.
- D. B. Dell’ Amico, F. Calderazzo, F. Marchetti and G. Perego, J. Chem. Soc., Dalton Trans., 1983, 0, 483–487 RSC.
- U. Baisch, D. B. Dell’ Amico, F. Calderazzo, L. Labella, F. Marchetti and A. Mergio, Eur. J. Inorg. Chem., 2004, 6, 1219–1224 CrossRef.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- M. Gregson, N. F. Chilton, A.-M. Ariciu, F. Tuna, I. F. Crowe, W. Lewis, A. J. Blake, D. Collision, E. J. L. McInnes, R. E. P. Winpenny and S. T. Liddle, Chem. Sci., 2016, 7, 155–165 RSC.
- R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357–360 CrossRef CAS PubMed.
- A. P. Ramirez, A. Hayashi, R. J. Cava, R. Siddharthan and B. S. Shastry, Nature, 1999, 399, 333–335 CrossRef CAS.
- M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502–506 CrossRef CAS PubMed.
- M. Urdampilleta, S. Klyatskaya, M. Ruben and W. Wernsdorfer, ACS Nano, 2015, 9, 4458–4464 CrossRef CAS PubMed.
- F. Luis, A. Repollés, M. J. Martínez-Pérez, D. Aguilà, O. Roubeau, D. Zueco, P. J. Alonso, M. Evangelisti, A. Camón, J. Sesé, L. A. Barrios and G. Aromí, Phys. Rev. Lett., 2011, 107, 117203 CrossRef CAS PubMed.
- M. J. Martínez-Pérez, S. Cardona-Serra, C. Schlegel, F. Moro, P. J. Alonso, H. Prima-García, J. M. Clemente-Juan, M. Evangelisti, A. Gaita-Ariño, J. Sesé, J. van Slageren, E. Coronado and F. Luis, Phys. Rev. Lett., 2012, 108, 247213 CrossRef PubMed.
- C. Godfrin, A. Ferhat, R. Ballou, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Phys. Rev. Lett., 2017, 119, 187702 CrossRef CAS PubMed.
- A. Morello, Nat. Nanotechnol., 2018, 13, 9–10 CrossRef CAS PubMed.
- D. B. Dell’ Amico, F. Calderazzo, C. della Porta, A. Mergio, P. Biagini, G. Lugli and T. Wagner, Inorg. Chim. Acta, 1995, 240, 1–3 CrossRef.
- D. J. Hart, P. A. Cain and D. A. Evans, J. Am. Chem. Soc., 1978, 100, 1548–1557 CrossRef CAS.
- Agilent, CrysAlis PRO. Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fdez Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. Å. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotsky, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- B. O. Roos, R. Lindh, P. Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem., 2005, 109, 6575–6579 CrossRef CAS PubMed.
- D. B. Dell’ Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857–3897 CrossRef PubMed.
- L. Armelao, D. B. Dell’ Amico, P. Biagini, G. Bottaro, S. Chiaberge, P. Flavo, L. Labella, F. Marchetti and S. Samartani, Inorg. Chem., 2014, 53, 4861–4871 CrossRef CAS PubMed.
- W. Kaim and B. Schwederski, Bioanorganische Chemie, Teubner, Stuttgart, 1991 Search PubMed.
- L. Westin, M. Kritikos and A. Caneschi, Chem. Commun., 2003, 3, 1012–1013 RSC.
- S. Daniele and L. G. Hubert-Pfalzgraf, Polyhedron, 1993, 12, 2091–2096 CrossRef CAS.
- W. J. Evans, M. A. Greci and J. W. Ziller, Inorg. Chem., 1998, 37, 5221–5226 CrossRef CAS.
- D. Ballweg, Y. Liu, I. A. Guzei and R. West, Silicon Chem., 2002, 1, 57–60 CrossRef CAS.
- A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Angew. Chem., Int. Ed., 2012, 51, 12194–12198 CrossRef CAS PubMed.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed.
- R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 0, 2349–2356 RSC.
- X.-L. Tang, W.-H.- Wang, W. Dou, J. Jiang, W.-S. Liu, W.-W. Qin, G.-L. Zhang, H.-R. Zhang, K.-B. Yu and L.-M. Zheng, Angew. Chem., Int. Ed., 2009, 48, 3499–3502 CrossRef CAS PubMed.
- M. C. Majee, S. M. T. Abtab, D. Mobdal, M. Maity, M. Weselski, M. Witwicki, A. Bienko, M. Antkowiak, G. Kamieniarz and M. Chaudhury, Dalton Trans., 2018, 47, 3425–3439 RSC.
- A. Das, S. Goswami, R. Sen and A. Ghosh, Inorg. Chem., 2019, 58, 5787–5798 CrossRef CAS PubMed.
- R. Marx, F. Moro, M. Dörfel, L. Ungur, M. Waters, S. D. Jiang, M. Orlita, J. Taylor, W. Frey, L. F. Chibotaru and J. van Slageren, Chem Sci., 2014, 5, 3287–3293 RSC.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- E. Moreno-Pineda, N. F. Chilton, R. Marx, M. Dörfel, D. O. Sells, P. Neugebauer, S.-D. Jiang, D. Collison, J. van Slageren, E. J. L. McInnes and R. E. P. Winpenny, Nat. Commun., 2014, 5, 5243 CrossRef CAS PubMed.
- S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655 RSC.
- L. Ungur and L. F. Chibotaru, Phys. Chem. Chem. Phys., 2011, 13, 20086–20090 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2049538–2049540. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00063b |
| This journal is © The Royal Society of Chemistry 2021 |