
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePentamethyl- and 1,2,4-tri(tert-butyl)cyclopentadienyl containing p-block complexes – differences and similarities†
Yi
Ding
ab,
Paul Niklas
Ruth
b,
Regine
Herbst-Irmer
b,
Dietmar
Stalke
 *b,
Zhi
Yang
*a and
Herbert W.
Roesky
*b
*b,
Zhi
Yang
*a and
Herbert W.
Roesky
*b
aSchool of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: zhiyang@bit.edu.cn
bInstitute of Inorganic Chemistry, Georg-August-Universität Göttingen, Göttingen Tammannstrasse 4, D-37077, Germany. E-mail: dstalke@chemie.uni-goettingen.de; hroesky@gwdg.de
First published on 18th January 2021
Abstract
The sterically encumbered cyclopentadienyl ligand 1,2,4-(Me3C)3C5H2 (Cp′′′) was used to stabilize efficiently the main group metals of Al, Ga, In, Ge and Sn, respectively. The σ-bonded gallium compounds [η1-Cp′′′Ga(μ-X)X]2 (X = Cl, 2; X = I, 3) and indium compound [η1-Cp′′′In(μ-Br)nBu]2 (7) exhibit dimers through halogen bridges. Reduction of 2 with 2 equivalents of KC8 leads almost to the same amount of η1-Cp′′′Ga(THF)Cl2 (4) and η5-Cp′′′Ga (5), respectively. The exception is compound 5, which is obtained by reducing 2 or 3 with 4 equivalents of KC8. Compound 5 as Lewis base reacts with GaI3 readily forming the Lewis acid–base adduct product η5-Cp′′′Ga → GaI3 (6). Moreover, compounds with the Cp′′′ ligand stabilize heavier low-valent group 14 elements for example [η5-Cp′′′EII]+[EIICl3]− (E = Ge 8, Sn 9), which are π-bonded ionic compounds that possess a low-valent cation and an anion. In the cation of [η5-Cp′′′EII]+, the Cp′′′ ligand adopts an η5-coordination mode with germanium and tin, respectively, which present half-sandwich complexes. While the EII fragment interacts with five π electrons from the Cp′′′ unit to generate an electron-octet arrangement at the respective element. All new reported structures are comparing well with the corresponding compounds containing the pentamethylcyclopentadienyl (Cp*) ligand.
Introduction
In 1951 the seminal discovery of ferrocene was reported,1 with the η5,η5 sandwich structure containing two cyclopentadienyl ligands. This class of compounds plays more and more an important role in the development of organometallic chemistry.2 The modification of the five-membered ring structure has been a topic of important research during the last 65 years. Especially the substitution of the hydrogen atoms by introducing more bulky substituents in the cyclopentadienyl ring resulted in mono-, di-, tri-, tetra-, and penta substituted cyclopentadienyl ring compounds C5H5−xRx (x = 1–5; R = Me, Et, iPr, tBu, benzyl), which can bind to a metal with different hapticity (η1–η5).3The first room-temperature stable Al(I) compound [Cp*Al]4 (Cp* = C5Me5) was reported by Schnöckel et al. in 1991.4 Two years later they reported the congener Cp*Ga.5 The subsequent research found that it is a monomer in the gas phase and in solution but a hexamer in the solid state.6 In 1993, Roesky et al.7 reported an easy route to [Cp*Al]4 in good yield that allowed to study the chemistry of this molecule on a broad scale.8 Furthermore [Cp*Al]4 and Cp*Ga were increasingly popular and extensively used by chemists.9 In 2004, Jutzi et al.10 isolated the first silylene cation [Cp*Si:]+[B(C6F5)4]−, which is thermally stable but extremely air- and moisture sensitive. Subsequently, the [Cp*Si:]+ ion became an important catalyst.11 Very recently, Heitkemper et al. reported a neutral Silicon(II) half-sandwich compound. It is interesting that in this compound the five-membered ring of the ligand consists of four carbon and one boron atoms and the distance between Si and the five-membered ring is closer than that in Cp*.12 Because of the excellent steric bulk, solubility, stability, and electron donor properties of Cp* ligand, it has been demonstrated versatility of the σ- (η3/2/1) or π-bonding (η5) in main group element chemistry.3,4,9–11,13
However, the 1,2,4-(Me3C)3C5H2 (Cp′′′) ligand with bulky substituents is rarely used for main group metals. Recently, Braunschweig et al. reported Cp′′′AlBr2, which was reduced by [Cp*Al]4 and then treated with cAAC to isolate the liquid low-valent monomer aluminum [η5-Cp′′′Al],14 which was studied for a number of interesting reactions under mild conditions.15 The main group metals supported by Cp′′′ ligand are depicted in Fig. 1.14–16 Encouraged by [η5-Cp′′′Al], we have initiated a research program based on other main group metals with the bulky Cp′′′ ligand.
 | ||
| Fig. 1 The main group metals supported by Cp′′′ ligand. | ||
Herein, we report on σ-bonded gallium and indium, π-bonded gallium, germanium, and tin metals stabilized by Cp′′′ ligand. The σ-bonded gallium and indium are μ2-bridging halogen dimers. The π-bonded Cp′′′Ga as Lewis base to afford Lewis acid–base adduct of composition (GaI → GaIII). While the π-bonded germanium and tin have both the cationic [η5-Cp′′′EII]+ (E = Ge, Sn) as well as the anionic [MIICl3]− (E = Ge, Sn) species of their respective low-valent group 14 elements. To the best of our knowledge, these are the first X-ray structures of ionic compounds with low-valent group 14 elements of Cp′′′, when compared with those containing the ionic feature of Cp species. We also discuss the differences and similarities between the Cp′′′ and Cp* metal compounds.
Results and discussion
Compound 1 is easily synthesized (Scheme 1) and characterized by NMR. In the 1H NMR spectrum, there is a resonance at δ 6.60 ppm for the two aromatic protons and two resonances at δ 1.39 and 1.20 ppm in a ratio of 18 to 9 protons of the tBu groups. The 27Al NMR spectrum exhibits a sharp singlet at δ = −27.09 ppm, which is considerably downfield shifted compared to that of [Cp*AlCl2]2 (δ = −51 ppm)17 and Cp′′′AlBr2 (δ = −42 ppm).14 The spectroscopic method strongly confirms compound 1. Regrettably, we were not able to obtain suitable single crystals for measurement. Reduction of 1 with 2.2 equivalents of KC8 was successful. However, according to the complicated 1H NMR spectrum of Cp′′′Al, it cannot be traced. This is exactly the same result as that reported by Braunschweig and co-workers.14 | ||
| Scheme 1 Synthesis of aluminum compound 1. | ||
Halogen bridged compound [η1-Cp′′′Ga(μ-X)X]2 (X = Cl, 2; X = I, 3) was synthesized according to Scheme 2. The reaction proceeded quickly within 3 h at ambient temperature, due to the high solubility of the magnesium precursor in hexane. The 1H NMR spectrum at δ 6.54 ppm exhibits a resonance for the aromatic protons, while for the tBu groups in [η1-Cp′′′Ga(μ-Cl)Cl]2 (2) two resonances are observed at δ 1.34 and 1.20 ppm. However, in [η1-Cp′′′Ga(μ-I)I]2 (3) the less electronegative iodine caused upfield shifts to δ 6.32, 1.30, and 1.17 ppm in the 1H NMR spectrum. Single crystals suitable for X-ray diffraction analysis are obtained by cooling a saturated n-hexane solution at −32 °C for 12 h. Molecular structures of 2 and 3 are presented in Fig. 2 and 3.
 | ||
| Scheme 2 Synthesis of gallium compounds 2–5. | ||
 | ||
| Fig. 2 Molecular structure of 2. Anisotropic displacement parameters are depicted at 50% probability level. The hydrogen atoms as well as the second molecule within the asymmetric unit are omitted for clarity. | ||
 | ||
| Fig. 3 Molecular structure of 3. Anisotropic displacement parameters are depicted at 50% probability level. The hydrogen atoms are omitted for clarity. | ||
The structures of 2 and 3 show isostructural dimeric arrangements with two bridging halogen atoms, which connect two gallium to constitute a planar four-membered Ga2X2 ring. While the asymmetric unit of 2 contains two halves of the molecule, with the other half generated by an inversion center, the asymmetric unit of 3 consists of a single complete molecule. Two Cp′′′ ligands and terminal halogen atoms residing alternately at both sides of the symmetric Ga2X2 ring. These are similar to the corresponding compounds supported by Cp* ligand. The gallium centers have distorted tetrahedral environments. The angles between the planes of the Cp′′′ ring and the Ga2X2 ring are 33.23(6)° and 33.29(6)° in 2 and 29.30(7)° and 28.61(7)° in 3. The angles between the Ga–C bond and the plane of the Cp′′′ rings reach 99.1° and 98.0° in 2 and 101.0° and 101.2° in 3, while the angles to Ga2X2 reach the values of 131.3° and 130.6° for the two molecules in the asymmetric unit in compound 2, and 128.0° and 130.1° in compound 3. For a similar series of dimeric halogen bridged molecules, regular changes in bond distances and bond angles as well as in distances between similar but nonbonded atoms in the four-membered rings are shown in Table 1. In the structure of 2 the Ga–Cl bridging bond distances [2.3497(10) to 2.3817(9) Å] are longer than the Ga–Cl terminal bond distance [2.1520(8) and 2.1518(8) Å]. In [Cp*Ga(μ-Cl)Cl]2,18 [Cp*Ga(μ-I)I]2,19 and 3, the arrangements are also the same when compared with 2. It is obvious that for both chlorine and iodine bridging bonds are longer than the terminal Ga–X bonds where the sterically more demanding ligand is involved. Similarly, Cp* and Cp′′′ ligands with the same structure center, reveal the bond lengths in the compound with more sterically hindered Cp′′′ ligand to be slightly longer.
| [Cp*GaCl2]2 | 2 | [Cp*GaI2]2 | 3 | 4 | |
|---|---|---|---|---|---|
| a Xt = terminal halogen atom. b Xb = bridging halogen atom. c First molecule in asymmetric unit. d Second molecule in asymmetric unit. | |||||
| Ga⋯Ga | 3.382 | 3.4338(10)c | 3.745 | 3.8672(7) | — |
| 3.4215(10)d | |||||
| Ga–C | 1.97(1) | 2.005(2)c | 2.008(6) | 2.034(3) | 2.021(3)c |
| 2.003(2)d | 2.030(3) | 2.021(3)d | |||
Ga–Xt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
2.124(4) | 2.1520(8)c | 2.4895(11) | 2.5197(6) | 2.1869(9)c |
| 2.1518(8)d | 2.5186(6) | 2.1886(9)c | |||
| 2.1863(9)d | |||||
| 2.1891(9)d | |||||
| Ga–Xbb |
2.352(3); 2.373(3) | 2.3497(10)c | 2.7150(11); 2.7131(10) | 2.7039(6) | |
| 2.3817(9)c | 2.7103(6) | ||||
| 2.3486(9)d | 2.7610(5) | ||||
| 2.3753(10)d | 2.7736(5) | ||||
| Xb⋯Xb | 3.300 | 3.2552(13)c | 3.931 | 3.8749(6) | |
| 3.2572(13)d | |||||
| Xb–Ga–Xb | 88.6(1) | 86.94(3)c | 92.78(2) | 90.042(17) | |
| 87.18(3)d | 90.176(16) | ||||
| Ga–Xb–Ga | 91.4(1) | 93.06(3)c | 87.23(2) | 90.081(17) | |
| 92.82(3)d | 89.684(16) | ||||
Inspired by [η1-Cp*2GaCl]2,18 we tried to react with equivalent amounts of GaCl3 with Cp′′′2Mg to get the dimeric compound of composition [η1-Cp′′′2GaCl]2. Unfortunately, the crystals we obtained were still compound 2. Presumably, the bulk of Cp′′′ was the reason we failed to get [η1-Cp′′′2GaCl]2.
With σ-bonded compound [η1-Cp′′′Ga(μ-Cl)Cl]2 (2) in hand, we next tried to synthesize [η1-Cp′′′Ga(μ-Cl)]2 and Cp′′′Ga (5), respectively, which are accessible via direct reductive dehalogenation with KC8. First of all, 2 reacts with two equivalents of KC8 in THF, after the reaction the solvent THF was removed in a vacuum, and the product was extracted with n-hexane. We obtained colorless crystals of composition of two η1-Cp′′′Ga(THF)Cl2 (4), with a THF molecule strongly coordinated to the gallium per asymmetric unit. However, after isolating the crystals from the mother liquor, the liquid residue is treated in a vacuum to get a yellow-green liquid. The 1H NMR spectrum of the yellow-green liquid exhibits an aromatic proton signal at δ 5.93 ppm and two tBu groups resonate at δ 1.37 and 1.20 ppm. This pattern is very similar to monomeric Cp′′′Al reported by Braunschweig et al.14 The reduction of 2 with two equivalents of KC8 produces η1-Cp′′′Ga(THF)Cl2 (4) as well as η5-Cp′′′Ga (5). In the structure of 4 (Fig. 4), the Ga–Cl bond distances [2.1869(9), 2.1886(9), 2.1891(9), and 2.1863(9) Å] are considerably longer than Ga–Cl terminal distance [2.1520(8) Å] in compound 2, however, shorter than Ga–Cl bridging distances [2.3497(10) to 2.3817(9) Å].
 | ||
| Fig. 4 Molecular structure of 4. Anisotropic displacement parameters are depicted at 50% probability level. The hydrogen atoms are omitted for clarity. | ||
Therefore, a reduction of 2 or 3 with four equivalents of KC8 has tried again in toluene, and pure compound 5 can be obtained in high yield as a yellow-green liquid. This is in contrast to the reduction of Cp′′′AlBr2 with KC8 that does not yield Cp′′′Al. The 71Ga NMR spectrum of 5 exhibits a resonance at δ −676.06 ppm, this value is observed between the CpGa20 (−714 ppm) and Cp*Ga5 (−653 ppm) reported by Schnöckel et al. The slight shift indicates that 5 also exists in the monomeric [η5-Cp′′′Ga] form in solution.21
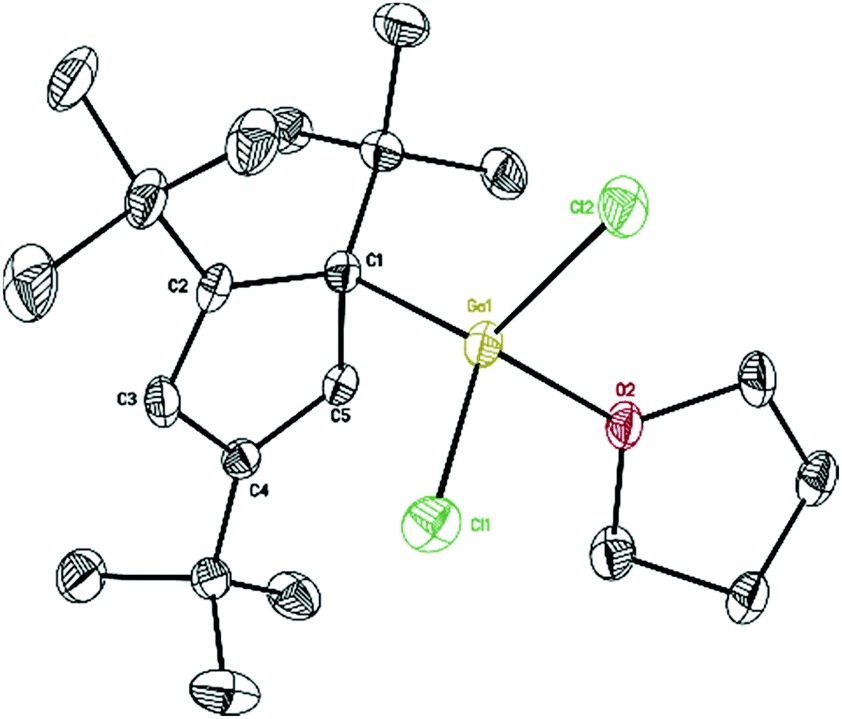
The Lewis acid GaI3 was reacted smoothly with 5 to afford the Lewis acid–base adduct product 6 (Scheme 3). However, the Lewis base PPh3 did not react with 5 according to the 1H NMR spectroscopy. This indicated that compound 5, like Cp′′′Al and other monovalent Ga(I) species, is a Lewis base. The 1H NMR spectrum of 6 shows an aromatic proton signal at δ 6.12 ppm, two tBu group resonances at δ 1.19 and 1.09 ppm. Compared with 5, the aromatic proton shifted downfield, but the tBu group shifted upfield. The 71Ga NMR spectrum of 6 exhibits a resonance at δ −279.55 ppm. Surprisingly, the 71Ga NMR signal of 6 (at ca. −240 to −320 ppm) is broader than that of 5 (at ca. −650 to −700 ppm) and strongly downfield shifted. This 71Ga resonance is for the tetracoordinate Ga(III) fragment while the signal of the Ga(I) center was not detected. This observation is in good agreement to that of 27Al NMR spectrum, reported for its Al analogue.14 The Ga(I)–Ga(III) distance (Fig. 5) in the structure of 6 is 2.4563(9) Å, which is very similar with that of [Li(THF)4][GaI3-GaI(PiPr2CH2)2BPh2] (2.4521(11) Å)22 and corresponds to the Ga–Ga coordination bond. Remarkably, these distances fall into the range of covalent Ga–Ga bond distances in complex (dpp-Bian)Ga–Ga(dpp-Bian) (2.3598(3) Å) (dpp-Bian = 1,2-bis[(2,6-diisopropylphenyl)imino]-acenaphthene),23 [RGaCl]2 (2.445(9) Å) (R = [(Me3Si)2C(Ph)C(Me3Si)N]),24 and the following compounds: [(tBuN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2]IGa–GaI[(tBuNCH)2] (2.4232(7) Å),25 (dpp-Bian)IGa–GaI(dpp-Bian) (2.4655(5) Å),26 [(ArNCH)2]IGa–GaI[(ArNCH)2] (2.5755(16) Å),27 [(Me3Si)3SiGaCl]4 (2.509(12) Å),28 [((Me3Si)2CH)2Ga]2 (2.541(1) Å),29 [(2,4,6-i-Pr3C6H2)2 Ga]2 (2.513(3) Å),30 [2,4,6-t-Bu3C6H2GaCl]2 (2.438(6) Å),31 and [(2,4,6-(CF3)3C6H2)2Ga]2 (2.479(1) Å).32 After the success with Cp′′′Ga we became interested in the corresponding Cp′′′In compound. InBr3 was treated with 0.5 equivalents of Cp′′′2Mg in hexane at ambient temperature for 6 h. However, we were not able to isolate any pure product. Next, we tried a one-pot reaction with Cp′′′2Mg, InBr3 and nBuLi. First, Cp′′′2Mg was treated with 2.2 equivalents of InBr3 in hexane at ambient temperature for 6 h. Then 2.2 equivalents of nBuLi were added to react at −78 °C. Finally, the mixture was warmed to room temperature and stirring was continued for 6 h. The filtered solution was stored at −32 °C in a freezer for 3 days (Scheme 4).
CH)2]IGa–GaI[(tBuNCH)2] (2.4232(7) Å),25 (dpp-Bian)IGa–GaI(dpp-Bian) (2.4655(5) Å),26 [(ArNCH)2]IGa–GaI[(ArNCH)2] (2.5755(16) Å),27 [(Me3Si)3SiGaCl]4 (2.509(12) Å),28 [((Me3Si)2CH)2Ga]2 (2.541(1) Å),29 [(2,4,6-i-Pr3C6H2)2 Ga]2 (2.513(3) Å),30 [2,4,6-t-Bu3C6H2GaCl]2 (2.438(6) Å),31 and [(2,4,6-(CF3)3C6H2)2Ga]2 (2.479(1) Å).32 After the success with Cp′′′Ga we became interested in the corresponding Cp′′′In compound. InBr3 was treated with 0.5 equivalents of Cp′′′2Mg in hexane at ambient temperature for 6 h. However, we were not able to isolate any pure product. Next, we tried a one-pot reaction with Cp′′′2Mg, InBr3 and nBuLi. First, Cp′′′2Mg was treated with 2.2 equivalents of InBr3 in hexane at ambient temperature for 6 h. Then 2.2 equivalents of nBuLi were added to react at −78 °C. Finally, the mixture was warmed to room temperature and stirring was continued for 6 h. The filtered solution was stored at −32 °C in a freezer for 3 days (Scheme 4).
 | ||
| Fig. 5 Molecular structure of 6. Anisotropic displacement parameters are depicted at 50% probability level. The hydrogen atoms are omitted for clarity. | ||
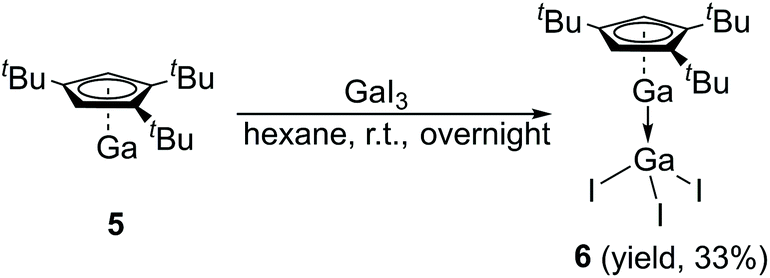 | ||
| Scheme 3 Synthesis of Lewis acid–base adduct gallium compound 6. | ||
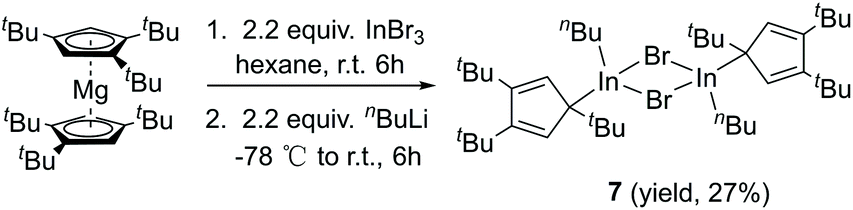 | ||
| Scheme 4 Synthesis of indium compound 7. | ||
The molecular structure of 7 is presented in Fig. 6, while selected bond distances can be found in the figure caption. We obtained colorless crystals of compound 7 (Fig. 6), reminiscent of the halogen bridged gallium compound [η1-Cp′′′Ga(μ-X)X]2. The dimeric molecule of 7 has two bridging bromine atoms, forming together with the indium atoms a planar four-membered In2Br2 ring with a center of symmetry. The two Cp′′′ ligands and the two n-butyl substituents reside alternately above and below the In2Br2 ring.
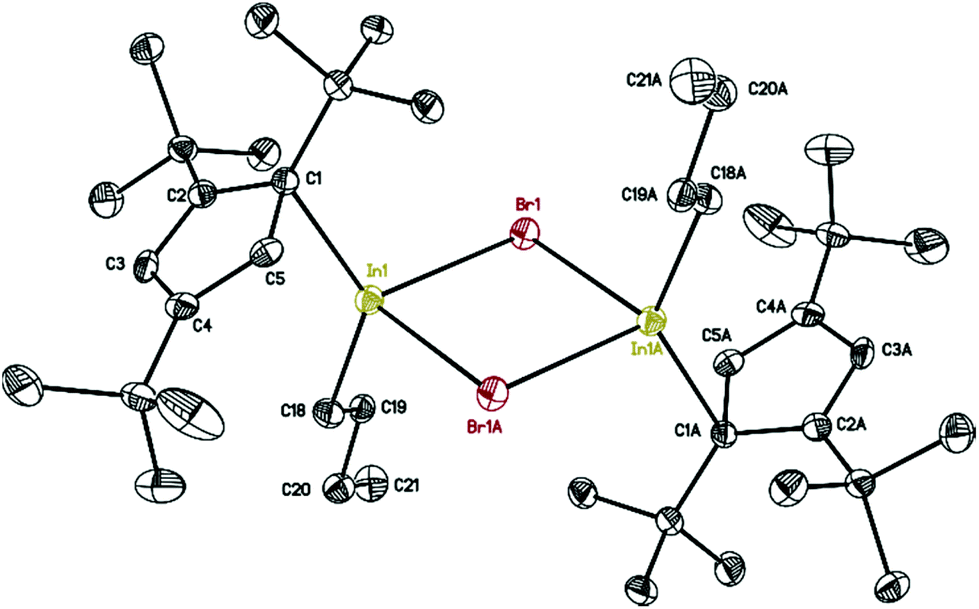 | ||
| Fig. 6 Molecular structure of 7. Anisotropic displacement parameters are depicted at 50% probability level. The hydrogen atoms are omitted for clarity. In–In 4.0476(14) Å; In–C18: 2.158(3) Å. | ||
After group 13 metals supported by Cp′′′ ligand, we extended the Cp′′′ chemistry to stabilize group 14 metals. Germanium [Cp*Ge]+[X]− (X = AlCl4,33 BF4,34 C5(CO2Me)5,35 GeCl3,36 SnCl3.37) and tin [Cp*Sn]+[X]− (X = AlCl4,38 B(C6F5)4,39 BF4,36,38,40 CF3SO3.38) ionic compounds supported by the Cp* ligand have been reported successively since 1980. Most of them were characterized by NMR and mass spectrometry. However, single-crystal X-ray structural analysis of ionic compounds of group 14 metals are rare.
The simple salt elimination reaction of Cp′′′Li with ECl2 (E = Ge, Sn) resulted in a poor conversion of the products. However, compounds 8 (E = Ge) and 9 (E = Sn) were isolated in good yields by reacting Cp′′′2Mg in Et2O solution (Scheme 5). The same reactions were also carried out with Cp′′′2Mg and SiCl4 as well with SiHCl3. However, with silicon chlorides no reaction was observed. The reactions were monitored by 1H NMR spectroscopy. The corresponding ionic compound of [η5-Cp′′′EII]+[EIICl3]− was obtained when Cp′′′2Mg was reacted with GeCl2·dioxane or SnCl2 in a ratio of 1:2 and 1:4, respectively.
 | ||
| Scheme 5 Synthesis of germanium and tin compounds 8 and 9. | ||
The 1H NMR the spectrum of compounds 8 and 9 exhibit resonances for the aromatic protons at δ 6.03 and 6.07 ppm and two resonances for the tBu groups at δ 1.14, 1.09 and 1.25, 1.17 ppm, respectively. Compounds 8 and 9 each possess a cationic charge. The electrophilicity of the metals is higher than those in neutral compounds. Due to this, the protons of Cp′′′ ligands of compounds 8 and 9 are shifted upfield in the 1H NMR, when compared with the corresponding resonances of compounds 2–4. However, this observation is similar to that of compound 5, which also indicates that 5 exhibits an η5-coordination mode.
Suitable single crystals of 8 were obtained from saturated toluene solution at −32 °C and those of 9 were formed in a mixture of toluene and Et2O at 4 °C. The structures of 8 (Fig. 7 left) and 9 (Fig. 7 right) contain each a half-sandwich cationic [η5-Cp′′′EII]+ (E = Ge, Sn) unit and their respective low-valent element of anionic [EIICl3]− species are located in the same crystal lattice. In the cationic [η5-Cp′′′EII]+ E(II) coordinates through η5-coordination to a Cp′′′ ligand. In the solid-state structure of compound 8, the GeII is coordinated by the three chlorine atoms of GeCl3−, which in turn points to the Cp′′′ moiety with a distance of 3.8706(13) Å (see Fig. S15†). In contrast, the solid-state structure of 9 does not exhibit this ordering in one-dimensional chains. The distance between the two metals in cation and anion is 3.8265(8) Å in compound 8 and 4.5920(6) Å in compound 9. These exceed the single bond length of Ge–Ge and Sn–Sn.41 The distances of the Ge and Sn atom to the center of the Cp′′′ ring are 1.9336(8) and 2.2056(10) Å; the Ge–C(Cp′′′) distances are 2.2529(16) to 2.3233(16) Å as well as Sn–C(Cp′′′) distances are 2.4759(14) to 2.5856(14) Å. The Cp′′′ rings are planar and exhibit η5-coordination to germanium and tin as shown by the Cring–Cring bond length patterns within each Cp′′′ ring. In the anion [EIICl3]−, three chlorine atoms (Cl1, Cl2, and Cl3) are attached to the metal ion EII, resulting in a trigonal pyramidal shape of the anion.
 | ||
| Fig. 7 Molecular structure of 8 (left) and 9 (right). Anisotropic displacement parameters are depicted at 50% probability level. The hydrogen atoms and the smaller disordered faction are omitted for clarity. | ||
Conclusions
In summary, we report halogen bridged σ-bonded dimers of gallium and indium, π-bonded aluminum, gallium, germanium, and tin compounds supported by the bulky 1,2,4-tri(tert-butyl)cyclopentadienyl ligand. Reducing the halogen bridged σ-bonded dimers of gallium with KC8 to afford the π-bonded Cp′′′Ga, which exhibits a Lewis base that forms an acid–base Ga(I)/Ga(III) adduct of Cp′′′Ga → GaI3. The ionic compounds contain half-sandwich cationic [η5-Cp′′′EII]+ as well as trigonal pyramidal anionic [EIICl3]− with the same elements. Due to the larger steric hindrance and poor solubility, Cp′′′ ligand is difficult to form metallocene-type molecules of main group metals. Comparing the Cp* substituted main group metal compounds with those containing Cp′′′ ligands, the latter are much more difficult to synthesize. For metallocenes, the Cp* ligand is a very good choice, however, for ionic compounds the Cp′′′ ligand is the preferable ligand.Experimental section
All manipulations were carried out using standard Schlenk and glove-box techniques under an atmosphere of high purity dinitrogen. THF, hexane, and toluene, respectively, were distilled over Na/K alloy (25:75), while diethyl ether was distilled over potassium mirror. Deuterated NMR solvent C6D6 was dried by stirring for 2 days over Na/K alloy followed by distillation in a vacuum and degassed. 1H, 13C{1H}, 27Al and 71Ga NMR spectra were recorded on Bruker Avance 400 or 500 MHz NMR spectrometer and were referenced to the resonances of the solvent used. Microanalyses were performed by the Analytisches Labor für Anorganische Chemie der Universität Göttingen. Melting points were determined in sealed glass capillaries under dinitrogen, and are uncorrected. The starting material Cp′′′2Mg was synthesized by following literature procedure,42 and Cp′′′Li was prepared by reaction of Cp′′′H with nBuLi in hexane at 85 °C overnight. All other reagents were used as received.
General synthesis of [η1-Cp′′′M(μ-X)X]2
A mixture of Cp′′′2Mg (490 mg, 1 mmol) and of two millimoles of MX3 was taken in a 100 mL round bottom flask and 40 mL of hexane were added at ambient temperature. The reaction mixture was stirred for 3 h to give corresponding compounds. After filtration of insoluble residue, the solvent was concentrated to 10 mL under vacuum. The solution was stored at −32 °C for 12 h in a freezer to get solid of 1 and X-ray quality crystals of 2, and 3, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. W. R. thanks the Deutsche Forschungsgemeinschaft for financial support RO224/70-1. Y. Ding thanks the China Scholarship Council (CSC) for the fellowship (201906030003). D. S. thanks the Danish National Research Foundation (DNRF93) funded Centre for Materials Crystallography (CMC) for partial support. P. N. R. is grateful for financial support by the RTG “BENCh”, which was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455. Dedicated to Professor Dr Thomas M. Klapötke.Notes and references
-
(a) T. Keally and P. Pauson, Nature, 1951, 168, 1039–1044 CrossRef
; (b) S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632–635 RSC
-
(a) H. Werner, Angew. Chem., Int. Ed., 2012, 51, 6052–6058 CrossRef CAS
- P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969–990 CrossRef CAS
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564–565 CrossRef
- D. Loos and H. Schnöckel, J. Organomet. Chem., 1993, 463, 37–40 CrossRef CAS
-
(a) D. Loos, E. Baum, A. Ecker, H. Schnöckel and A. J. Downs, Angew. Chem., Int. Ed. Engl., 1997, 36, 860–862 CrossRef CAS
- S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., Int. Ed. Engl., 1993, 32, 1729–1731 CrossRef
-
(a) S. Schulz, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, J. Organomet. Chem., 1995, 493, 69–75 CrossRef CAS
-
(a) P. Jutzi and G. Reumann, J. Chem. Soc., Dalton Trans., 2000, 2237–2244 RSC
- P. Jutzi, A. Mix, B. Rummel, W. W. Schoeller, B. Neumann and H.-G. Stammler, Science, 2004, 305, 849–851 CrossRef CAS
-
(a) P. Jutzi, Chem. – Eur. J., 2014, 20, 9192–9207 CrossRef CAS
- T. Heitkemper, J. Sarcevic and C. P. Sindlinger, J. Am. Chem. Soc., 2020, 142, 21304–21309 CrossRef CAS
- C. P. Sindlinger and P. N. Ruth, Angew. Chem., Int. Ed., 2019, 58, 15051–15056 CrossRef CAS
- A. Hofmann, T. Tröster, T. Kupfer and H. Braunschweig, Chem. Sci., 2019, 10, 3421–3428 RSC
-
(a) A. Hofmann, C. Pranckevicius, T. Tröster and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 3625–3629 CrossRef CAS
- H. Sitzmann, Y. Ehleiter, G. Wolmershäuser, A. Ecker, C. Üffing and H. Schnöckel, J. Organomet. Chem., 1997, 527, 209–213 CrossRef CAS
- C.-H. Wang, Y.-F. Lin, H.-C. Tseng, G.-S. Lee, S.-M. Peng and C.-W. Chiu, Eur. J. Inorg. Chem., 2018, 2232–2236 CrossRef CAS
- O. Beachley, R. Hallock, H. Zhang and J. Atwood, Organometallics, 1985, 4, 1675–1680 CrossRef CAS
- P. Jutzi, B. Neumann, G. Reumann and H.-G. Stammler, Organometallics, 1998, 17, 1305–1314 CrossRef CAS
- D. Loos, H. Schnöckel, J. Gauss and U. Schneider, Angew. Chem., Int. Ed. Engl., 1992, 31, 1362–1364 CrossRef
- C. Dohmeier, D. Loos and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1996, 35, 129–149 CrossRef CAS
- B. J. Malbrecht, J. W. Dube, M. J. Willans and P. J. Ragogna, Inorg. Chem., 2014, 53, 9644–9656 CrossRef CAS
- I. L. Fedushkin, A. N. Lukoyanov, S. Y. Ketkov, M. Hummert and H. Schumann, Chem. – Eur. J., 2007, 13, 7050–7056 CrossRef CAS
- K. S. Klimek, C. Cui, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2000, 19, 3085–3090 CrossRef CAS
- R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844–3850 RSC
- I. L. Fedushkin, A. A. Skatova, V. A. Dodonov, V. A. Chudakova, N. L. Bazyakina, A. V. Piskunov, S. V. Demeshko and G. K. Fukin, Inorg. Chem., 2014, 53, 5159–5170 CrossRef CAS
- R. J. Baker, R. D. Farley, C. Jones, D. P. Mills, M. Kloth and D. M. Murphy, Chem. – Eur. J., 2005, 11, 2972–2982 CrossRef CAS
- G. Linti and W. Köstler, Angew. Chem., Int. Ed. Engl., 1996, 35, 550–552 CrossRef CAS
- W. Uhl, M. Layh and T. Hildenbrand, J. Organomet. Chem., 1989, 364, 289–300 CrossRef CAS
- X. He, R. A. Bartlett, M. M. Olmstead, K. Ruhlandt-Senge, B. E. Sturgeon and P. P. Power, Angew. Chem., Int. Ed. Engl., 1993, 32, 717–719 CrossRef
- A. H. Cowley, A. Decken and C. A. Olaza, J. Organomet. Chem., 1996, 524, 271–273 CrossRef CAS
- R. D. Schluter, A. H. Cowley, D. A. Atwood, R. A. Jones, M. R. Bond and C. J. Carrano, J. Am. Chem. Soc., 1993, 115, 2070–2071 CrossRef CAS
-
(a) F. X. Kohl and P. Jutzi, J. Organomet. Chem., 1983, 243, 31–34 CrossRef CAS
-
(a)
P. Jutzi, Adv. Organomet. Chem., Elsevier, 1986, vol. 26, pp. 217–295 Search PubMed
- P. Jutzi, B. Hampel, M. B. Hursthouse and A. J. Howes, Organometallics, 1986, 5, 1944–1948 CrossRef CAS
- P. Jutzi, F. Kohl, P. Hofmann, C. Krüger and Y.-H. Tsay, Chem. Ber., 1980, 113, 757–769 CrossRef CAS
- J. Rouzaud, M. Joudat, A. Castel, F. Delpech, P. Riviere, H. Gornitzka, J. Manriquez and I. Chavez, J. Organomet. Chem., 2002, 651, 44–51 CrossRef CAS
- F. X. Kohl and P. Jutzi, Chem. Ber., 1981, 114, 488–494 CrossRef CAS
- J. N. Jones, J. A. Moore, A. H. Cowley and C. L. Macdonald, Dalton Trans., 2005, 3846–3851 RSC
- S. P. Constantine, G. M. De Lima, P. B. Hitchcock, J. M. Keates, G. A. Lawless and I. Marziano, Organometallics, 1997, 16, 793–795 CrossRef CAS
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC
- F. Weber, H. Sitzmann, M. Schultz, C. D. Sofield and R. A. Andersen, Organometallics, 2002, 21, 3139–3146 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of new compounds and complexes prepared during this study. CCDC 2049785–2049791. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt04412a |
| This journal is © The Royal Society of Chemistry 2021 |