
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic antioxidant nanocomposites based on sequential adsorption of redox active metal complexes and polyelectrolytes on nanoclay particles†
Zoltán
Somosi
a,
Nóra V.
May
b,
Dániel
Sebők
c,
István
Pálinkó
 d and
István
Szilágyi
*a
d and
István
Szilágyi
*a
aMTA-SZTE Lendület Biocolloids Research Group, Interdisciplinary Research Center, Department of Physical Chemistry and Materials Science, University of Szeged, H-6720 Szeged, Hungary. E-mail: szistvan@chem.u-szeged.hu
bCentre for Structural Science, Research Centre for Natural Sciences, Hungarian Academy of Sciences, H-1117 Budapest, Hungary
cDepartment of Applied and Environmental Chemistry, University of Szeged, H-6720 Szeged, Hungary
dDepartment of Organic Chemistry, University of Szeged, H-6720 Szeged, Hungary
First published on 21st January 2021
Abstract
An antioxidant nanocomposite was prepared by successive adsorption of redox active metal complexes (copper(II)–bipyridyl and iron(III)–citrate) and polyelectrolytes (poly(styrene sulfonate) and poly(diallyldimethyl ammonium)) on layered double hydroxide nanoclay. The experimental conditions were optimized in each preparation step and thus, the final composite formed highly stable colloids, i.e., excellent resistance against salt-induced aggregation was achieved. Due to the synergistic effect of the metal complexes, the developed composite showed remarkable activity in the dismutation of superoxide radicals, close to the one determined for the native superoxide dismutase enzyme. The obtained composite is highly selective for superoxide radical dismutation, while its activity in other antioxidant tests was close to negligible. Structural characterization of the composite revealed that the excellent superoxide radical scavenging ability originated from the advantageous coordination geometry around the copper(II) center formed upon immobilization. The structure formed around the metal centers led to optimal redox features and consequently, to an improved superoxide dismutase-like activity. The catalytic antioxidant composite is a promising candidate to reduce oxidative stress in industrial manufacturing processes, where natural enzymes quickly lose their activity due to the harsh environmental conditions.
1. Introduction
Combating oxidative stress via decomposition of reactive oxygen species (ROS) attracts widespread contemporary interest due to its importance in both health-related and industrial processes.1 Accordingly, an elevated ROS level is responsible for the development of numerous diseases including cancer,2,3 while their presence in manufacturing processes leads to reduced-quality commercial products in the food, textile and cosmetics industries.4–6 The decomposition of ROS is naturally carried out by enzymatic (e.g., superoxide dismutase, catalase and various peroxidases) and molecular (e.g., flavonoids, vitamins and tannins) antioxidants.7 The most efficient ones are the enzymes, but their high sensitivity to temperature, pH, ionic strength and other factors makes them complicated to combat oxidative stress under harsh environmental conditions. This fact especially prevents their industrial application.Natural enzymes may be replaced with artificial ones with improved resistance against the above-mentioned effects. Two major directions exist to develop enzyme mimicking compounds. First, nanomaterials with antioxidant activity attract widespread interest in the scientific and technological communities.8–11 These nanomaterials, the so-called nanozymes, usually contain redox active metals such as iron,12 cerium13 or manganese,14 which are responsible for the reaction with ROS. Second, transition metal complexes with structures similar to the active centers of antioxidant enzymes can be synthesized and used as antioxidant agents.15–17 Literature data shed light on the fact that appropriate structural mimicking usually gave rise to considerable functional mimicking too, i.e., once the active center was modelled adequately, the complexes possessed high ROS scavenging ability.18–22
Although promising antioxidant nanomaterials and metal complexes have been reported in the past, various drawbacks (e.g., tendency for aggregation or considerable toxicity for nanoparticles and difficult separation or metal ion leakage for complexes) indicated that these research directions should be combined to develop efficient radical scavenging systems through immobilization of redox active coordination compounds on suitable nanoparticulate supports. Accordingly, considerable catalase mimicking activity was achieved by a manganese(II)-poly-L-histidine complex anchored to carboxylate modified multi-walled carbon nanotubes.23 Due to their biocompatibility and advantageous surface properties, silica particles were proven to be suitable solid supports for enzyme mimicking complexes; therefore, various copper(II) complexes were attached to their surfaces.24–29 Polymer-based latex particles were decorated with amino acids via covalent bonds and copper(II) complexes of considerable superoxide dismutase-like activity were prepared by adding metal salts to latex–amino acid hybrids.30 Lamellar cationic clays were also used to immobilize metal complexes through electrostatic interactions due to their considerable cation exchange capacity.31,32 However, the intercalation of the complexes between the lamellae hindered the diffusion of ROS to the catalytic centers and the elimination of the products, especially upon gas formation. Although a considerable amount of metal complexes could be immobilized with the above methods, their application in liquid environments was limited due to the tendency of the antioxidant composite particles for aggregation and subsequent sedimentation, which leads to significant loss of the enzyme-like activity.
One of the ways to immobilize metal complexes and to maintain colloidal stability of the resulting material is the use of polyelectrolytes for surface functionalization of the enzyme mimicking composites. Polyelectrolytes are applied as efficient stabilizing agents for particle dispersions through repulsive electrostatic and intermolecular steric interactions arising upon their adsorption on oppositely charged surfaces.33–38 For instance, in a recent contribution,39 a poly(vinylpyridine-b-methacrylic acid) block co-polyelectrolyte was used to functionalize layered double hydroxide (LDH) nanoclay particles. The poly(methacrylic acid) part was responsible for the strong attachment to the particle surface, while copper(II)–histamine complexes were anchored via coordination bonds between the metal ions and the nitrogen donor atoms of the poly(vinylpyridine) part. In this way, the polyelectrolyte part provided not only remarkable colloidal stability but also excellent structural (no metal ion leakage) and functional (the composite was able to scavenge superoxide radicals even at high temperatures, where the natural antioxidant enzymes quickly lost the activity) stabilities. These literature data prompted the idea to use polyelectrolytes for immobilization of metal complexes on nanoparticle supports via surface functionalization. Besides, to widen the spectrum of the antioxidant activity, attachment of complexes of various metal ions to the same particle support is desired.
Therefore, the aim of the present study was to develop nanocomposites by sequential adsorption of polyelectrolytes and metal complexes onto the surface of LDH particles. Such self-assembled multilayers of polyelectrolytes have been widely used to immobilize various ROS scavenging or other bioactive molecules, but mostly on planar surfaces.40–43 Only a few studies reported immobilization of enzymatic44,45 or molecular46 antioxidants on nanoparticles with this technique and no ROS scavenging complexes have been embedded into polyelectrolyte layers to date. It was demonstrated earlier in our group that building oppositely charged polyelectrolyte layers on LDH particles is feasible, once the appropriate dose of the polyelectrolytes is used in the steps of the sequential adsorption procedure.47 This strategy was accompanied by the optimization of colloidal stability to obtain homogeneously distributed composite particles. Here, LDH particles were also used as the substrate due to their many positive aspects such as variable composition, biocompatibility, tuneability of size down to the nanoscale with low polydispersity, and relatively easy and cheap synthesis.34,48–50 Two types of polyelectrolytes (poly(styrene sulfonate) (PSS) and poly(diallyldimethyl ammonium) (PDADMAC)) and metal complexes (copper(II)–bipyridyl (Cu(Bpy)2) and iron(III)–citrate (Fe(Cit)2)) were applied in the sequential adsorption process to build the hybrid catalytic material (denoted as COMP hereafter; see Scheme 1). The structural features were investigated by X-ray diffraction (XRD), scanning electron microscopy (SEM), and electron paramagnetic resonance (EPR) and infrared (IR) spectroscopy; the colloidal stability was probed by light scattering methods, while the antioxidant activity was assessed by colorimetric assays based on UV-Visible (UV-Vis) spectroscopy measurements. To the best of our knowledge, this is the first study reporting co-immobilization of catalytic metal complexes by the sequential adsorption method on LDH particles to combat oxidative stress.
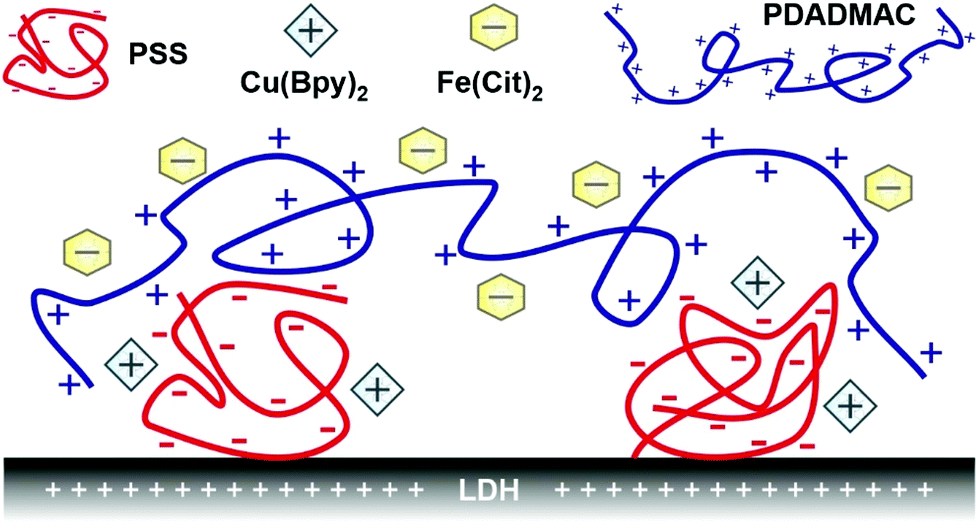 | ||
| Scheme 1 Illustration of the schematic structure of the COMP hybrid material composed of PSS, Cu(Bpy)2, Fe(Cit)2 and PDADMAC immobilized on LDH nanoparticles. | ||
2. Experimental
2.1. Materials
Magnesium(II) chloride hexahydrate, iron(III) chloride hexahydrate, copper(II) chloride dihydrate, 4 M sodium hydroxide (NaOH) stock solution, sodium chloride (NaCl), trisodium citrate dihydrate, ammonium acetate, methanol and the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical were all purchased from VWR International (Radnor, USA) with analytical purity and were used as received. Aluminum(III) chloride hexahydrate and xanthine were acquired in analytical grade from Alfa Aesar (Karlsruhe, Germany). 2,2′-Bipyridyl (99%) and nitro blue tetrazolium (90%, NBT) chloride monohydrate were purchased from Acros Organics (Geel, Belgium). Xanthine oxidase from bovine milk was purchased from Sigma-Aldrich. The sodium salt of poly(styrene sulfonate) (PSS, molecular mass of 10 kg mol−1) and 20 weight% aqueous poly(diallyldimethyl ammonium) chloride (PDADMAC, average molecular mass of 275 kg mol−1) solution were purchased from Sigma-Aldrich (Budapest, Hungary). Ultrapure water was produced by reverse osmosis and UV irradiation using a Puranity TU 3 + UV/UF system (VWR International).2.2. Synthesis of LDH particles
The LDH particles were composed of magnesium(II) and aluminum(III) ions at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio and chloride ions were the interlayer anions. The synthetic method was flash co-precipitation followed by hydrothermal post treatment to optimize the size distribution.51 Accordingly, 15 mL of a mixed salt solution of magnesium(II) (3.0 mmol) and aluminum(III) (1.5 mmol) chloride was poured into 60 mL of vigorously stirred NaOH solution (0.15 M), which was stirred for an additional 30 min under a N2 atmosphere to avoid carbonation. The freshly formed LDH precipitate was centrifuged to separate from the supernatant and washed five times with water. The obtained slurry was diluted with 50 mL water and was sonicated for 10 min in an ultrasonic bath, and then transferred to a stainless steel autoclave with a Teflon lining (Col-Int Tech, Irmo, USA), which was sealed and placed in a preheated oven at 120 °C for 24 h for hydrothermal post treatment. After the procedure was terminated, the autoclave was allowed to cool to room temperature, and the obtained material was centrifuged and washed with water several times. The final LDH material was dried and redispersed into a stock dispersion of 10 g L−1 particle concentration. The successful formation of LDH was confirmed by XRD (Fig. S1a†) and IR spectroscopy (Fig. S1b†). As shown in the ESI,† the diffractogram and the spectrum showed the characteristic peaks reported for LDH materials earlier.34,48,49,51,52
:1 molar ratio and chloride ions were the interlayer anions. The synthetic method was flash co-precipitation followed by hydrothermal post treatment to optimize the size distribution.51 Accordingly, 15 mL of a mixed salt solution of magnesium(II) (3.0 mmol) and aluminum(III) (1.5 mmol) chloride was poured into 60 mL of vigorously stirred NaOH solution (0.15 M), which was stirred for an additional 30 min under a N2 atmosphere to avoid carbonation. The freshly formed LDH precipitate was centrifuged to separate from the supernatant and washed five times with water. The obtained slurry was diluted with 50 mL water and was sonicated for 10 min in an ultrasonic bath, and then transferred to a stainless steel autoclave with a Teflon lining (Col-Int Tech, Irmo, USA), which was sealed and placed in a preheated oven at 120 °C for 24 h for hydrothermal post treatment. After the procedure was terminated, the autoclave was allowed to cool to room temperature, and the obtained material was centrifuged and washed with water several times. The final LDH material was dried and redispersed into a stock dispersion of 10 g L−1 particle concentration. The successful formation of LDH was confirmed by XRD (Fig. S1a†) and IR spectroscopy (Fig. S1b†). As shown in the ESI,† the diffractogram and the spectrum showed the characteristic peaks reported for LDH materials earlier.34,48,49,51,52
2.3. XRD
The XRD measurements were carried out using a PW 1830 diffractometer (Philips, Amsterdam, The Netherlands), which applies Cu–K radiation and operates in Bragg–Brentano geometry with a Ni filter at a voltage of 40 kV and a current of 30 mA. The diffractograms were recorded in the 4–80°2-θ range with a step size of 0.02°. The powder samples were placed on a glass zero background holder for the acquisition of the XRD patterns.2.4. Spectroscopy
The IR spectra were measured on a Bio-Rad Digilab Division FTS-65A/896 apparatus, equipped with a diffuse reflectance spectroscopy accessory. In a typical measurement, 256 scans were collected with 4 cm−1 resolution in the 4000–600 cm−1 wavenumber range.The antioxidant assays were carried out by recording the UV-Vis spectra with a Genesys 10S spectrophotometer (Thermo Scientific, Waltham, USA). The wavelength range used was 190–1100 nm with 0.1 nm scaling.
EPR spectra were recorded with a Bruker EleXsys E500 spectrometer (microwave frequency 9.54 GHz, microwave power 13 mW, modulation amplitude 5 G, modulation frequency 100 kHz). The details of the sample preparation used in the EPR measurements are presented in Table S1 (in the ESI†). To record frozen solution spectra, 0.20 mL samples were mixed with 0.05 mL of methanol (to avoid crystallization of water) and transferred into quartz EPR tubes, and the spectra were recorded in a Dewar flask containing liquid nitrogen (77 K). The spectra were simulated using the “epr” program.53 The spectra of Fe(Cit)2 were measured separately and for the iron-containing samples, this broad singlet background signal was eliminated in order to be able to simulate the copper(II) spectrum components. The anisotropic EPR spectra were analyzed considering axial or rhombic g-tensors (gx, gy, gz) and copper hyperfine tensors (Ax, Ay, Az). The nitrogen superhyperfine structure was determined taking into account a rhombic hyperfine tensor (aNx, aNy, aNz) in which the x, y, z directions referred to the g-tensor orientations. Orientation-dependent linewidth parameters (α, β, and γ) were used to fit the linewidths through the equation σMI = α + βMI + γM2I, where MI denotes the magnetic quantum number of the copper(II) ion. Since natural copper(II) chloride was used for the measurements, the spectra were calculated by the summation of the 63Cu and 65Cu spectra weighed by their natural abundances. The hyperfine and superhyperfine coupling constants and the relaxation parameters were obtained in field units (Gauss = 10−4 T).
2.5. Light scattering
Electrophoretic mobility values were measured by the phase analysis-based electrophoretic light scattering technique using a LiteSizer 500 (Anton Paar, Graz, Austria) device. Omega-shaped plastic capillary cuvettes (Anton Paar) were used for the measurements. The sample preparation process involved the mixing of a calculated amount of water, the polyelectrolyte and NaCl stock solutions to adjust the polyelectrolyte concentration and the ionic strength. The total volume of these mixtures was always 1.8 mL. The process was completed by adding 0.2 mL of 100 mg L−1 LDH stock dispersion. The samples were then allowed to rest for 2 h at room temperature. The capillary cuvettes were flushed with the majority of the dispersions and the last 0.35 mL was used for the actual experiment. The reported electrophoretic mobility values were the average of five individual measurements and their mean error was about 5%.The time-resolved dynamic light scattering (DLS) technique was used to study the aggregation processes under different experimental conditions. The measurements were carried out with a Litesizer 500 instrument (Anton Paar) equipped with a 40 mW semiconductor laser operating at 658 nm wavelength in the backscattering mode at a scattering angle of 175°. The hydrodynamic radii were calculated with the Stokes–Einstein equation from the diffusion coefficients, which were determined by applying the cumulant method to fit the correlation function.54 In an aggregating dispersion, the slopes obtained by the linear fits on the hydrodynamic radius versus time data were used to calculate the apparent aggregation rates.55 The colloidal stability of the particles was expressed in terms of the stability ratio,56 which is 1 by definition for unstable and quickly aggregating dispersions. Higher stability ratios refer to slower aggregation and more stable systems. The sample preparation procedures in the time-resolved DLS study were the same as in the electrophoretic mobility measurements, with the exception that the experiments were commenced immediately after the addition of the LDH stock dispersion followed by quick vortexing.
2.6. Microscopy
The morphology of the solids was characterized by using a Hitachi S-4700 scanning electron microscope. The surface of the samples was covered with gold via physical vapor deposition before the measurements which were conducted with 10 kV accelerating voltage. The samples were pre-emptively sonicated for 10 min prior to the experiments to eliminate particle aggregates. The dispersions were dried in the instrument.2.7. Dismutation of superoxide radicals
For determining the superoxide radical scavenging activity of the adsorbed complexes, the Fridovich method was used.57 The test systems contained xanthine and xanthine oxidase to produce superoxide radicals and NBT as a scavenger indicator. To carry out a probe reaction, 0.1 mL of 3 mM xanthine, 0.1 mL of 3 mM NBT and 0.3 mL of 3 mg mL−1 xanthine oxidase solutions were mixed together followed by the addition of 0.1–2.5 mL of the composite stock dispersion. The above solutions were all prepared in phosphate buffer at pH 7.5 applying a total phosphate concentration of 1 mM. Upon the reduction of the NBT by superoxide radicals, its color changed from yellow to blue and the appearance of an absorption peak at 565 nm was followed with a spectrophotometer. Blank (without added catalyst) measurements were carried out and changes in the absorbance in these reference experiments were used to calculate the inhibition of the NBT–superoxide radical reaction at several concentrations of the antioxidant material.2.8. Scavenging of DPPH radicals
The DPPH scavenging activity of the catalytic composites was evaluated by a standard assay.58 Accordingly, in a typical test, 60 μM of DPPH was dissolved in 3500 μL of methanol and 100 μL of the catalytic material was added into the system followed by quick homogenization. During the reaction, the change in the concentration of violet-colored DPPH was followed by measuring the absorbances at 517 nm wavelength with a spectrophotometer, i.e., decreases in the absorbances corresponded to the decrease in the radical concentration. The remaining DPPH percentage was then calculated to quantify the activity of the composite material.3. Results and discussion
3.1. Assessment of charging and aggregation features during sequential adsorption
For the preparation of COMP, the sequential adsorption process, which included PSS, Cu(Byp)2, PDADMAC and Fe(Cit)2 immobilization on the LDH surface, was applied. To optimize the experimental conditions in each step, electrophoretic mobilities and stability ratios were measured at different concentrations of the adsorbing substance (Fig. 1). The results were crucial to determine the dose of the polyelectrolytes and the concentration of the metal complexes to obtain stable particle dispersions in the individual steps.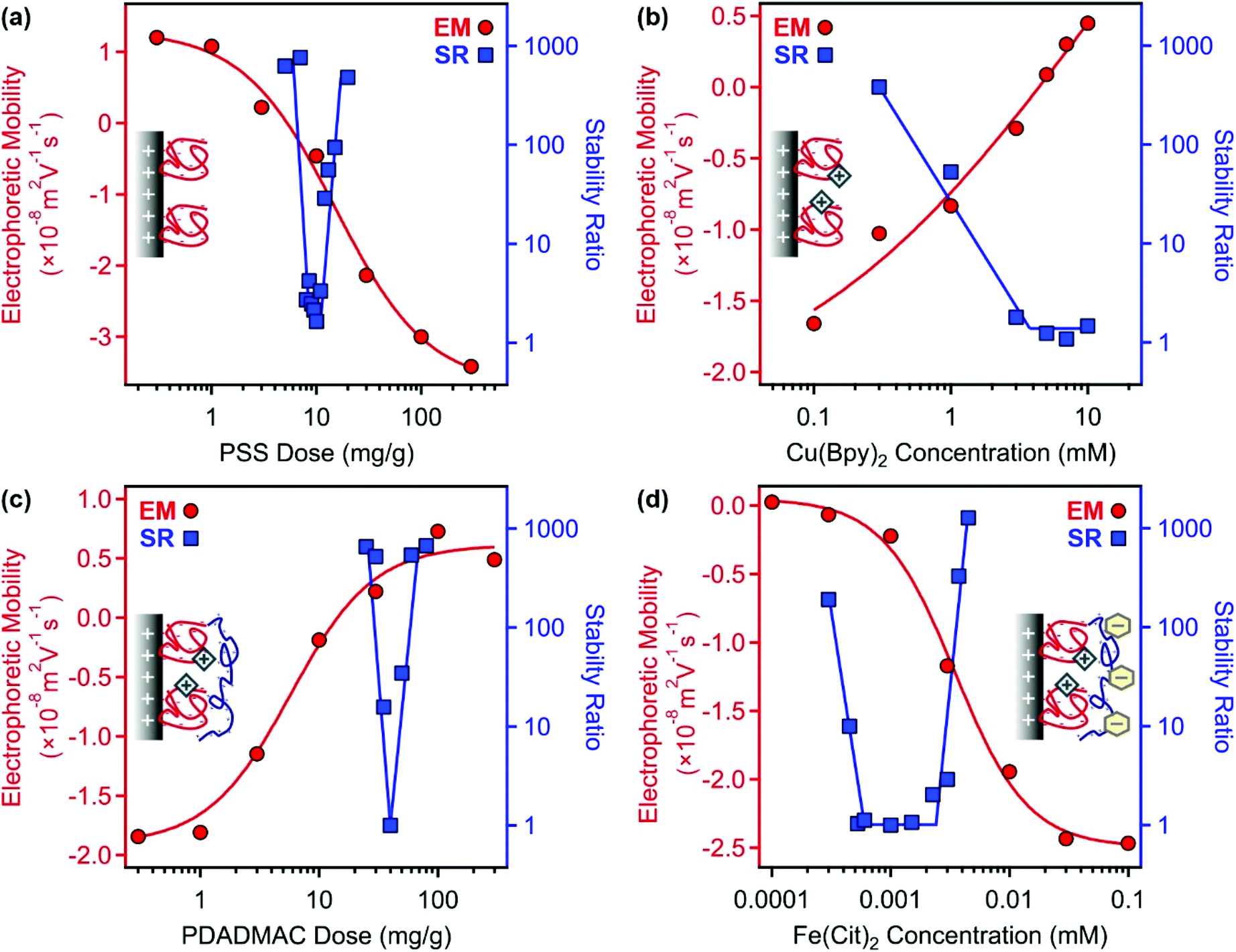 | ||
| Fig. 1 Electrophoretic mobilities (EM, circles) and stability ratios (SR, squares) determined in the LDH–PSS (a), LDH–PSS–Cu(Bpy)2 (b), LDH–PSS–Cu(Bpy)2–PDADMAC (c) and LDH–PSS–Cu(Bpy)2–PDADMAC–Fe(Cit)2 (COMP) (d) systems. The particle concentration was always 10 mg L−1 and the solid lines are just eye guides. The mg g−1 unit on the x-axis of (a) and (c) indicates mg polyelectrolyte per 1 gram of particle. | ||
First, PSS was adsorbed on the LDH support; the corresponding electrophoretic mobility and stability ratio data are shown in Fig. 1a. The LDH particles possess positive structural charge,34 which is reflected in positive electrophoretic mobilities at low PSS doses. On increasing the polyelectrolyte loading, the mobility data showed a decrease due to the adsorption of PSS on the oppositely charged surface. The surface charge was neutralized around 10 mg g−1 and further increase in the PSS dose gave rise to charge reversal. The adsorption process continued until surface saturation, which occurred after 100 mg g−1. Beyond this dose, there was no significant change in electrophoretic mobility values indicating that the further added PSS remained dissolved in the solution.
The speed of aggregation was investigated at different PSS doses in time-resolved DLS experiments, in which the stability ratios were determined.55 Their values were close to unity in the case of fast (or diffusion controlled) aggregation, while high stability ratios refer to slower aggregation. In other words, the inverse of the stability ratio equals to the fraction of particle collisions, which results in dimer formation. The data shown in Fig. 1a indicate fast particle aggregation at doses close to the charge neutralization point, while the stability ratios increased away from this region, either at the low or high PSS dose sides. These results can be interpreted well within the DLVO (Derjaguin, Landau, Verwey and Overbeek) theory.33–35 Accordingly, the presence of appropriately high surface charges leads to particle stabilization through electrical double layer forces, while attractive van der Waals forces predominate once the surface charges and thus the electrical double layer vanish, like the conditions at the charge neutralization point in the present system. These surface charge and aggregation features are typical for systems containing particles and oppositely charged polyelectrolytes.33–35,38,39,47 For further experiments, a PSS dose of 100 mg g−1 was chosen (denoted as LDH–PSS thereafter), since these particles possess high negative charge and form stable dispersions under these experimental conditions.
Second, adsorption of Cu(Bpy)2 was followed on the LDH–PSS particles. To prepare the complex, the metal ions and the ligands were dissolved and mixed in a 1:2 molar ratio. Literature data59–61 suggest that in Cu(Bpy)2, the copper(II) ions are coordinated by four nitrogen atoms of the ligands in solutions. Besides, Cu(Bpy) complexes may also be present in a smaller extent. Therefore, the overall charge of the complexes is considered to be positive. Indeed, the electrophoretic mobility values increased with progressive addition of the complexes due to their adsorption on the oppositely charged LDH–PSS (Fig. 1b). Charge neutralization and slight charge reversal were observed at high complex concentrations, which were close to the solubility limit of the ligand. The stability ratios decreased by increasing the Cu(Bpy)2 concentration and became unity after a threshold value, which resembles the critical coagulation concentration (CCC) reported for charged particles dispersed in salt solutions.33,34,56 Since the goal of this study was to identify the conditions in which the Cu(Bpy)2-decorated LDH–PSS forms a stable dispersion, 0.1 mM complex concentration was applied in further experiments. The obtained composite was named LDH–PSS–Cu(Bpy)2. Note that this substance has a net negative charge.
Third, PDADMAC was adsorbed on the LDH–PSS–Cu(Bpy)2 particles. In general, very similar tendencies were observed to the ones in Fig. 1a measured for PSS adsorption. Electrophoretic mobility data clearly indicate that PDADMAC strongly adsorbs on the oppositely charged surface (Fig. 1c). Charge neutralization and reversal occurred at appropriate PDADMAC doses and the adsorption process continued until surface saturation, which was indicated by a plateau in the mobility values. The onset of this plateau was defined at 100 mg g−1 loading. Stability ratio measurements revealed that the dispersions were highly stable at this PDADMAC concentration, i.e., their values were extremely high at 100 mg g−1 dose. Therefore, this dose was chosen (LDH–PSS–Cu(Bpy)2–PDADMAC) for the final functionalization with the Fe(Cit)2 complexes.
Fourth, electrophoretic mobilities and stability ratios were determined for the LDH–PSS–Cu(Bpy)2–PDADMAC particles at different Fe(Cit)2 concentrations (Fig. 1d). Speciation in aqueous solutions containing iron(III) and citrate ions was frequently investigated in the past. The published results indicate that mixed hydroxy–citrate complexes may also form in the system; however, the species are all negatively charged.62,63 The mobility data underline this information, since they decreased with the complex concentration due to adsorption on the oppositely charged surface. Charge neutralization occurred already at very low Fe(Cit)2 loadings indicating its very strong affinity to the LDH–PSS–Cu(Bpy)2–PDADMAC particles leading to charge neutralization and very pronounced charge reversal at certain complex concentrations. Such a strong adsorption originates from electrostatic and hydrogen bonding interactions. The trend of the stability ratios resembles the ones measured upon polyelectrolyte adsorption for PSS and PDADMAC in the first and third steps of the sequential adsorption process. Accordingly, once the charges were low, i.e., close to the charge neutralization point, the dispersions were unstable and fast particle aggregation was observed indicated by stability ratios of about 1. On the other hand, stable dispersions formed at Fe(Cit)2 concentrations away from the charge neutralization point. Such a trend is qualitatively in line with the DLVO theory, as discussed above. To form the final COMP hybrid particle, 0.1 mM Fe(Cit)2 concentration was chosen, under which conditions the composite possesses significant negative charge (zeta potential is −31.6 mV) and forms a highly stable dispersion, i.e., stability ratios could not be determined in its aqueous dispersion at low ionic strength. Therefore, COMP was used in further structural and antioxidant investigations.
3.2. Structural and colloidal characterization of COMP
The SEM images were taken after drying the COMP dispersions. The morphology of the hybrid is similar to LDH materials prepared by the combined co-precipitation–hydrothermal treatment method earlier.48,51 Accordingly, platelets of rounded edges can be discovered in the images. The lateral size of COMP was in the 150–200 nm range (Fig. 2). | ||
| Fig. 2 SEM images of COMP. The measurements were performed after drying the dispersions. | ||
EPR spectroscopy was used to explore the coordination geometry around the iron(III) and copper(II) ions with the help of samples prepared with and without nanoparticulate support. The EPR spectra of Fe(Cit)2 (Fig. S2†) was a broad singlet line at 77 K due to fast relaxation; therefore, this background signal was eliminated from the iron(III) containing spectra and copper(II) spectra have only been simulated. The EPR spectra of Cu(Byp)2 was measured in aqueous solution at pH ∼ 7 and this spectrum could be simulated by the sum of 25% mono- and 75% bis-ligand complex spectra (Fig. 3a).
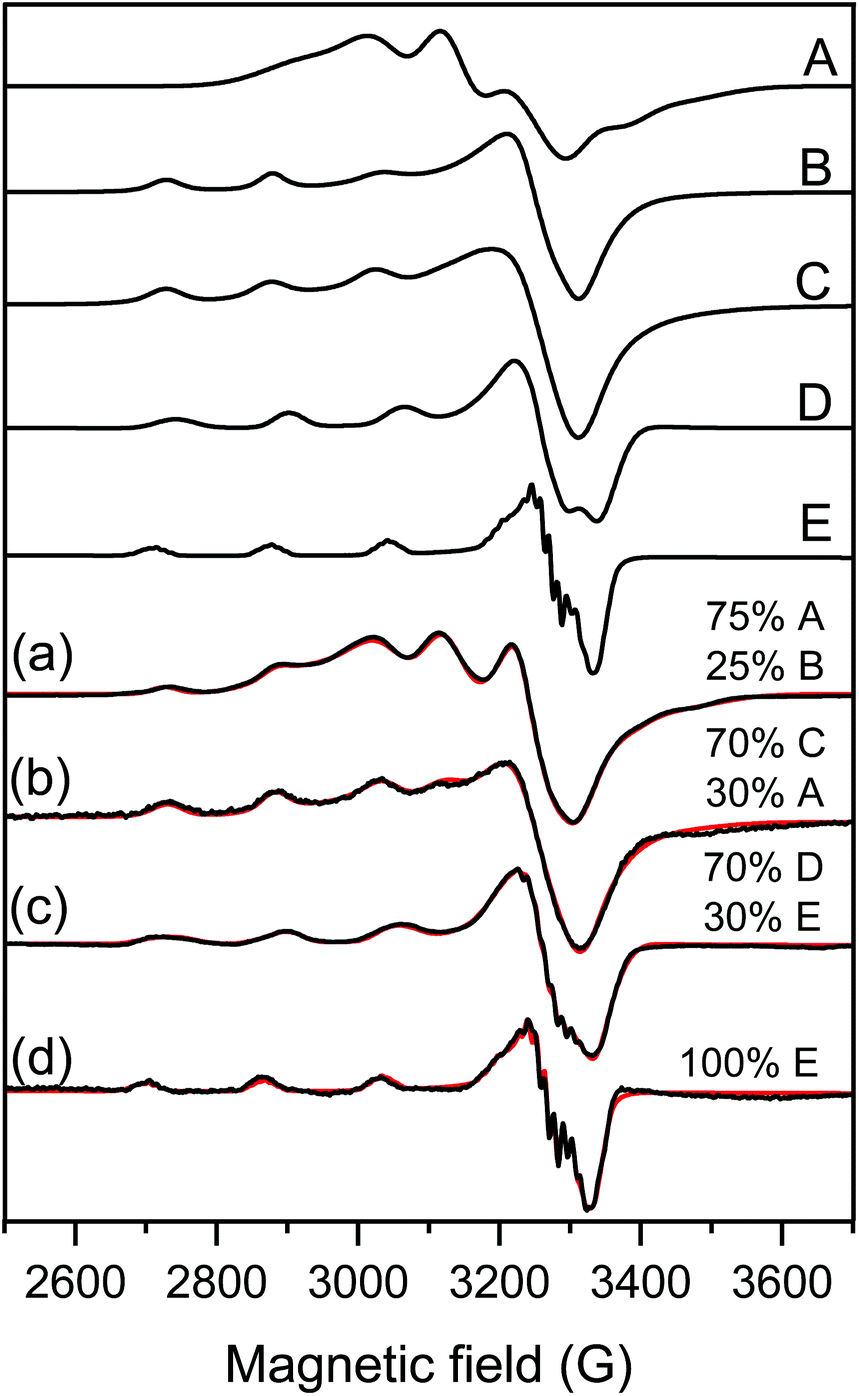 | ||
| Fig. 3 Measured (black) and simulated (red) EPR spectra of samples containing (a) Cu(Bpy)2 solution, (b) 100 ppm LDH–PSS–Cu(Bpy)2, (c) [Cu(Bpy)2] and [Fe(Cit)2] in aqueous solution and (d) 100 ppm COMP. For the details of sample preparation, see Table S1.† The spectra were simulated with the help of component spectra A: Cu(Bpy)2, B: Cu(Bpy), C: LDH–PSS–Cu(Bpy)2, D: Cu(Bpy)2(Cit) and E: Cu(Bpy)(Cit) described with the EPR parameters listed in Table 1. | ||
The spectra of the copper(II)–bipyridyl solution have already been measured by Garribba et al.,64 and it was found that the mono-ligand Cu(Bpy) complex possesses the usual g-tensor values (gx ∼ gy < gz) showing an elongated octahedral geometry, while the bis-ligand Cu(Bpy)2 complex spectra show an opposite g-tensor trend (gx ∼ gy > gz), which suggests trigonal bipyramidal geometry for this complex (Table 1).
| Coordination compound | g x | g y | g z | A x (G) | A y (G) | A z (G) | a N1 x (G) | a N2 x (G) |
|---|---|---|---|---|---|---|---|---|
| a N1 y (G) | a N2 y (G) | |||||||
| a N1 z (G) | a N2 z (G) | |||||||
| a The experimental errors were ±0.001 for g and ±1 G for A and aN. | ||||||||
| Cu(Bpy)2 | 2.177 | 2.177 | 2.017 | 83.7 | 83.7 | 75.8 | ||
| Cu(Bpy) | 2.066 | 2.066 | 2.274 | 12.6 | 12.6 | 154.7 | ||
| LDH–PSS–Cu(Bpy)2 | 2.062 | 2.062 | 2.280 | 14.0 | 14.0 | 147.8 | ||
| Cu(Bpy)2(Cit) | 2.063 | 2.063 | 2.259 | 23.5 | 23.5 | 157.0 | ||
| Cu(Bpy)(Cit) | 2.065 | 2.051 | 2.283 | 4.8 | 14.6 | 163.8 | 11.4 | 12.7 |
| 12.6 | 7.7 | |||||||
| 10.6 | 13.8 | |||||||
This latter structure was found in the solid state where two Bpy coordinated in the equatorial-axial position and one water molecule occupied the third equatorial position in the single crystal structure of Cu(Bpy)2(H2O)·2PF6.65 For LDH–PSS–Cu(Bpy)2 (Fig. 3b), the obtained spectrum could be simulated with the sum of 30% Cu(Byp)2 and 70% of a new component with parameters close to the mono-complex (Table 1). Therefore, this new component is most likely the mono-complex Cu(Bpy). The EPR spectra of Cu(Bpy)2 were recorded after adding Fe(Cit)2 in solution without the nanoparticles (Fig. 3c) and it was found that the spectra were significantly different. These spectral changes can be due to the coordination of the Cit ligand to the copper(II)–bipyridyl complexes resulting in the mixed ligand complexes Cu(Byp)(Cit) and Cu(Byp)2(Cit), both with elongated octahedral geometries. For COMP (Fig. 3d), the EPR spectra could be fitted with the Cu(Bpy)(Cit) component spectra suggesting that adsorption on COMP does not affect the EPR parameters significantly. However, it makes this mixed ligand complex exclusive in this complex system. The nitrogen superhyperfine structure clearly confirms the binding of two nitrogen donor atoms in the equatorial plane. The EPR spectra were also measured at 400 ppm COMP concentration (Fig. S2†) and dimerization or aggregation of paramagnetic centers on the COMP surface can be ruled out as the line broadening effect or the doublet or half field signal was not detected.
The resistance against salt-induced aggregation is an important parameter for catalytic materials applied in liquid environments, which is the case for most of the antioxidant compounds used in both medical treatments and industrial manufacturing processes. Therefore, this issue was addressed by determining the electrophoretic mobilities (Fig. 4a) and stability ratios (Fig. 4b) for the bare LDH and COMP at different ionic strengths adjusted with NaCl.
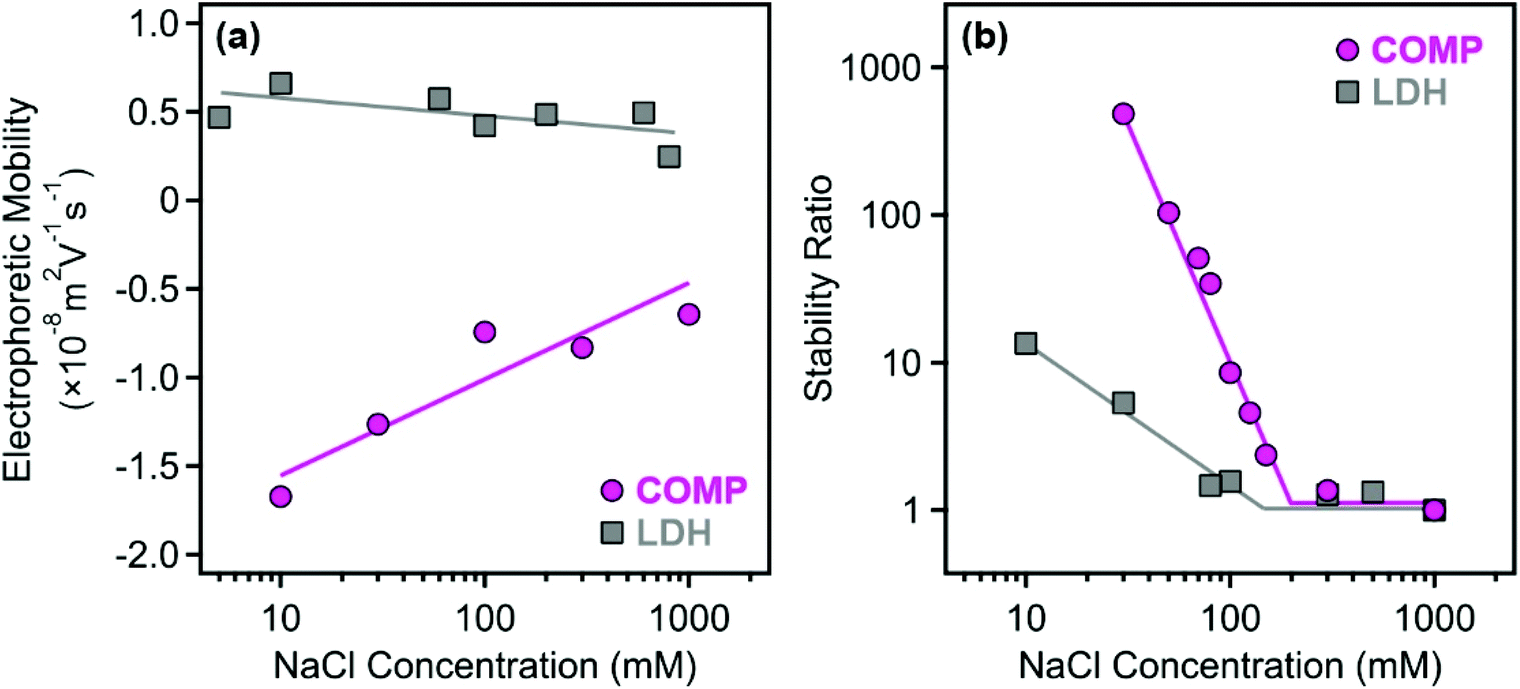 | ||
| Fig. 4 Electrophoretic mobility (a) and stability ratio (b) values determined for the bare LDH (squares) and for COMP (circles) at different NaCl concentrations at 10 mg L−1 particle concentration. The solid lines serve to guide the eyes and their breakpoint in (b) indicates the location of the CCC values. | ||
The magnitude of the mobilities followed the same tendency for both LDH and COMP. Accordingly, they decreased upon increasing the NaCl concentration, but no charge reversal was observed in the systems. This behavior is typical in dispersions containing charged particles and indifferent salts and can be explained by the screening effect of the salt constituents on the surface charges.34,39,56 Comparing the data obtained for both materials, one can observe that the magnitude of the mobilities at low NaCl concentrations is about three times higher for COMP indicating that surface functionalization by sequential adsorption increased the surface charge significantly. This is also reflected in the stability ratio data, since only limited stability, i.e., stability ratios were below 10 above 10 mM NaCl concentration, was observed for the bare LDH, while COMP formed highly stable dispersions at low ionic strengths. Besides, the shapes of the curves are somewhat similar and the stability ratios decreased by increasing the NaCl concentration until the CCC (the threshold salt concentration values, which separate fast and slow aggregation regimes) and remain unity thereafter. The onset of the fast aggregation is located at higher ionic strengths for COMP underlining the fact that it possesses significantly higher resistance against salt-induced aggregation.
Such a behavior can be described well by the DLVO theory, which states that higher charges lead to higher stability and elevated CCC values, since stronger electrical double layer forces can be screened at higher ionic strengths. An interesting discovery that deserves a short discussion is that the colloidal stability of COMP is somehow lower than the one reported earlier for the LDH–PSS–PDADMAC composite without immobilized metal complexes in the structure.47 In that system, the fast aggregation regime at a stability ratio of 1 could not be reached due to the presence of extensive steric repulsion owing to the loosely adsorbed polyelectrolyte chains on the surface. These forces are most likely not present in COMP since the stability ratios are unity within the experimental error beyond the CCC indicating the presence of DLVO-type interactions (electrical double layer and van der Waals) only. The most sensible explanation is that the adsorption of the metal complexes on the polyelectrolyte layers gave rise to a more compact structure of the mixed complex-polyelectrolyte layer on the surface of the particles and thus, the formation of polyelectrolyte tails and loops, which are responsible for steric effects, is not preferred.
3.3. Antioxidant activity
Antioxidant enzymes contain redox active transition metal ions coordinated by donor atoms from the side chains of peptide-building amino acids and hence their active center can be considered as a metal complex.16,17 Therefore, the Cu(Bpy)2 and Fe(Cit)2 complexes embedded in the COMP hybrid serve as the active centers in the present biocatalytic system. The polyelectrolyte environment may mimic the structure formation role of the peptide chains present in the native enzymes. Therefore, the superoxide dismutase-like activity was tested first (Fig. S3a†).The superoxide radicals were produced from the oxidation of xanthine to uric acid with xanthine oxidase.57 This colorimetric assay is based on the reduction of NBT to formazan by the forming superoxide radicals and consequently, the color change from yellow to blue. Once the radicals formed are captured by the antioxidant material COMP, the reduction of NBT is inhibited and hence, no color change occurs. Such an inhibition was calculated from the spectrophotometric data recorded during the development of the blue color in the assays.
Based on the results, one can notice that the developed COMP hybrid exhibited excellent activity in the assay. The inhibition increased sharply with the concentration and reached 100% at about 0.050 mg L−1. The usual measure for the superoxide dismutase-like activity is the so-called IC50 value, which gives the concentration of the catalyst necessary to dismutate 50% of the superoxide radicals forming in the assay. The IC50 of COMP was found to be 0.026 mg L−1 indicating remarkably high activity of the hybrid (Table 2).
| Compound | IC50 (mg L−1) | I max (%) | Ref. |
|---|---|---|---|
| Superoxide dismutase | 0.069 | 100 | 52 |
| COMP | 0.026 | 95 | Present study |
For instance, the measured IC50 is 63 times lower (meaning that the activity is 63 times higher) than the one reported earlier for Prussian blue antioxidant nanozymes.12 More importantly, COMP has comparable superoxide radical scavenging activity to the native copper(II)–zinc(II) superoxide dismutase enzyme, whose IC50 was determined to be 0.069 mg L−1 earlier, in the same biochemical assay.52 The maximum inhibitions (Imax) of the native enzyme and COMP were very similar (100% and 95%, respectively, see Table 2). However, one must note that the concentration of the redox active metal ions is much higher in COMP than in the native enzyme due to the extensive structure-forming peptide part in the latter case. Applying equivalent concentrations of the stand-alone complexes to the IC50 value of COMP, inhibition was observed neither for Cu(Bpy)2 nor for Fe(Cit)2. The same result was obtained when the mixture of the two complexes was applied in solution. These data indicate that a synergistic effect between the coordination compounds takes place upon co-immobilization giving rise to remarkable improvement in the superoxide radical scavenging activity. Considering the EPR results discussed earlier, this excellent activity originates from the Cu(Bpy)(Cit) complexes formed upon co-immobilization of the metal complexes and hence, this active center is mostly responsible for the increased activity.
After the obtained excellent superoxide radical scavenging activity described above, the selectivity of the catalytic system was checked. The general radical scavenging activity of COMP was assessed in the standard DPPH test.58 The reaction was followed through the color change from violet (DPPH radical) to yellow (reaction product), which was monitored by measuring the absorbance at 517 nm with a spectrophotometer. As shown in Fig. S3b,† some activity was observed; however, COMP was only able to decompose 55% of the initial DPPH radicals within a reasonable timeframe, even at high catalyst concentrations. The effective concentration (EC50), which gives the COMP concentration needed to react with 50% of the DPPH radicals in the test, is 49 mg L−1. These activity data, especially the low maximum percentage in the decomposition of the radicals, indicate moderate DPPH scavenging activity compared to other particle-based antioxidant materials.66–68 Applying the equivalent concentration (calculated from the EC50 value) of the lone complex of Cu(Bpy)2 in solution led to only 5.9% scavenging activity, while for Fe(Cit)2, 9.7% was measured. In addition, the mixture of the two lone complexes resulted in 11.4% scavenged radicals. This result sheds light on the importance of the co-immobilization of the complexes; however, the DPPH decomposing activity of COMP is still moderate.
Moreover, the antioxidant abilities were tested in other probe reactions too, but the activities were detected neither in the guaiacol-based peroxidase assays69 nor in the CUPRAC assays.70 These assays were performed in purely aqueous solutions, where the solubility of the substrate is not a problem. The above results clearly indicate that COMP is exceptionally selective for superoxide radicals, while it fails in other antioxidant assays. Such a selectivity resembles the native enzyme, which is responsible solely for the decomposition of the superoxide radicals (and not other ROS) in living organisms.
4. Conclusions
An antioxidant nanocomposite was developed by the sequential adsorption technique involving 2 polyelectrolytes and 2 enzyme mimicking metal complexes immobilized successively on a LDH nanoclay support. The adsorption of the individual components occurred due to electrostatic interactions and their doses were optimized in order to obtain stable colloidal dispersions in the preparation steps. The obtained COMP hybrid possessed great resistance against salt-induced aggregation and the presence of all components was confirmed in the structure. The developed material showed remarkable activity in the dismutation of superoxide radicals; its mass normalized activity was close to the one reported for the native superoxide dismutase enzyme. Moreover, COMP represented negligible or zero activity in other antioxidant test reactions such as DPPH, peroxidase and CUPRAC assays indicating its selectivity towards superoxide radical dismutation. These facts make COMP a promising replacement of the superoxide dismutase enzyme, particularly in industrial processes, where native enzymes often denature and consequently, lose their antioxidant activity due to the harsh environmental conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Research, Development and Innovation Office (SNN131558) and by the Ministry of Human Capacities (20391-3/2018/FEKUSTRAT). The support from the University of Szeged Open Access Fund (5165) is gratefully acknowledged.References
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS.
- A. A. Alfadda and R. M. Sallam, J. Biomed. Biotechnol., 2012, 936486, DOI:10.1155/2012/936486.
- K. Brieger, S. Schiavone, F. J. Miller and K. H. Krause, Swiss Med. Wkly., 2012, 142, 13659 Search PubMed.
- K. M. Gupta, S. Das, P. S. Chow and C. Macbeath, ACS Appl. Nano Mater., 2020, 3, 5351–5361 CrossRef CAS.
- A. Bafana, S. Dutt, S. Kumar and P. S. Ahuja, Crit. Rev. Biotechnol., 2011, 31, 65–76 CrossRef CAS.
- J. W. Finley, A. N. Kong, K. J. Hintze, E. H. Jeffery, L. L. Ji and X. G. Lei, J. Agric. Food Chem., 2011, 59, 6837–6846 CrossRef CAS.
- B. N. Ames, M. K. Shigenaga and T. M. Hagen, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7915–7922 CrossRef CAS.
- L. Valgimigli, A. Baschieri and R. Amorati, J. Mater. Chem. B, 2018, 6, 2036–2051 RSC.
- S. I. Tsekhmistrenko, V. S. Bityutskyy, O. S. Tsekhmistrenko, V. M. Polishchuk, S. A. Polishchuk, N. V. Ponomarenko, Y. O. Melnychenko and M. Y. Spivak, Regul. Mech. Biosyst., 2018, 9, 469–476 CrossRef.
- S. Murath, N. B. Alsharif, S. Saringer, B. Katana, Z. Somosi and I. Szilagyi, Crystals, 2020, 10, 148 CrossRef CAS.
- K. Takahashi and L. Takahashi, Dalton Trans., 2016, 45, 3244–3246 RSC.
- N. B. Alsharif, G. F. Samu, S. Sáringer, S. Muráth and I. Szilagyi, J. Mol. Liq., 2020, 309, 113066 CrossRef CAS.
- A. Othman, L. Norton, A. S. Finny and S. Andreescu, Talanta, 2020, 208, 120473 CrossRef CAS.
- N. Singh, M. A. Savanur, S. Srivastava, P. D'Silva and G. Mugesh, Angew. Chem., Int. Ed., 2017, 56, 14267–14271 CrossRef CAS.
- F. Yan, Y. Mu, G. L. Yan, J. Q. Liu, J. C. Shen and G. M. Luo, Mini-Rev. Med. Chem., 2010, 10, 342–356 CrossRef CAS.
- A. J. Wu, J. E. Penner-Hahn and V. L. Pecoraro, Chem. Rev., 2004, 104, 903–938 CrossRef CAS.
- D. P. Riley, Chem. Rev., 1999, 99, 2573–2587 CrossRef CAS.
- A. Horn, G. L. Parrilha, K. V. Melo, C. Fernandes, M. Horner, L. D. Visentin, J. A. S. Santos, M. S. Santos, E. C. A. Eleutherio and M. D. Pereira, Inorg. Chem., 2010, 49, 1274–1276 CrossRef CAS.
- I. N. Jakab, O. Lorincz, A. Jancso, T. Gajda and B. Gyurcsik, Dalton Trans., 2008, 6987–6995, 10.1039/b811452h.
- G. Tabbi, W. L. Driessen, J. Reedijk, R. P. Bonomo, N. Veldman and A. L. Spek, Inorg. Chem., 1997, 36, 1168–1175 CrossRef CAS.
- I. Labádi, M. Benko, M. Kata and I. Szilagyi, React. Kinet. Catal. Lett., 2009, 96, 327–333 CrossRef.
- I. Szilagyi, I. Labadi, K. Hernadi, I. Palinko, N. V. Nagy, L. Korecz, A. Rockenbauer, Z. Kele and T. Kiss, J. Inorg. Biochem., 2005, 99, 1619–1629 CrossRef CAS.
- J. X. Zhou, Y. Chen, L. T. Lan, C. Zhang, M. X. Pan, Y. Y. Wang, B. K. Han, Z. H. Wang, J. Jiao and Q. Chen, Anal. Biochem., 2019, 567, 51–62 CrossRef CAS.
- Y. C. Fang, Y. P. Chen, C. T. Chen, T. S. Lin and C. Y. Mou, J. Mater. Chem. B, 2013, 1, 6042–6052 RSC.
- I. Szilagyi, L. Horvath, I. Labadi, K. Hernadi, I. Palinko and T. Kiss, Cent. Eur. J. Chem., 2006, 4, 118–134 CAS.
- I. Szilagyi, I. Labadi, K. Hernadi, I. Palinko and T. Kiss, J. Mol. Struct., 2005, 744, 495–500 CrossRef.
- Z. Csendes, C. Dudas, G. Varga, E. G. Bajnoczi, S. E. Canton, P. Sipos and I. Palinko, J. Mol. Struct., 2013, 1044, 39–45 CrossRef CAS.
- Y. C. Fang, H. C. Lin, I. J. Hsu, T. S. Lin and C. Y. Mou, J. Phys. Chem. C, 2011, 115, 20639–20652 CrossRef CAS.
- M. Yang, W. Jiang, Z. Q. Pan and H. Zhou, J. Inorg. Organomet. Polym. Mater., 2015, 25, 1289–1297 CrossRef CAS.
- I. Szilagyi, O. Berkesi, M. Sipiczki, L. Korecz, A. Rockenbauer and I. Palinko, Catal. Lett., 2009, 127, 239–247 CrossRef CAS.
- I. Szilagyi, I. Labadi, K. Hernadi, T. Kiss and I. Palinko, in Molecular Sieves: From Basic Research to Industrial Applications, Pts a and B, ed. J. Cejka, N. Zilkova and P. Nachtigall, 2005, vol. 158, pp. 1011–1018 Search PubMed.
- I. Labadi, I. Szilagyi, N. I. Jakab, K. Hernadi and I. Palinko, Mater. Sci., 2003, 21, 235–244 CAS.
- S. Muráth, S. Sáringer, Z. Somosi and I. Szilagyi, Colloids Interfaces, 2018, 2, 32 CrossRef.
- M. Pavlovic, P. Rouster, T. Oncsik and I. Szilagyi, ChemPlusChem, 2017, 82, 121–131 CrossRef CAS.
- I. Szilagyi, G. Trefalt, A. Tiraferri, P. Maroni and M. Borkovec, Soft Matter, 2014, 10, 2479–2502 RSC.
- R. Meszaros, I. Varga and T. Gilanyi, Langmuir, 2004, 20, 5026–5029 CrossRef CAS.
- I. Safir, K. X. Ngo, J. N. Abraham, M. G. Afshar, E. Pavlova and C. Nardin, Polymer, 2015, 79, 29–36 CrossRef CAS.
- A. Tiraferri and M. Borkovec, Sci. Total Environ., 2015, 535, 131–140 CrossRef CAS.
- M. Pavlovic, M. Nafradi, P. Rouster, S. Murath and I. Szilagyi, J. Colloid Interface Sci., 2019, 543, 174–182 CrossRef CAS.
- O. S. Sakr and G. Borchard, Biomacromolecules, 2013, 14, 2117–2135 CrossRef CAS.
- T. Szabo, M. Szekeres, I. Dekany, C. Jackers, S. De Feyter, C. T. Johnston and R. A. Schoonheydt, J. Phys. Chem. C, 2007, 111, 12730–12740 CrossRef CAS.
- D. C. Kim, J. I. Sohn, D. J. Zhou, T. A. J. Duke and D. J. Kang, ACS Nano, 2010, 4, 1580–1586 CrossRef CAS.
- B. Wang, Y. J. Wang, X. F. Gao and Y. S. Li, J. Biomater. Sci., Polym. Ed., 2015, 26, 1312–1326 CrossRef CAS.
- F. Caruso and C. Schuler, Langmuir, 2000, 16, 9595–9603 CrossRef CAS.
- R. Saladino, M. Guazzaroni, C. Crestini and M. Crucianelli, ChemCatChem, 2013, 5, 1407–1415 CrossRef CAS.
- V. Vergaro, Y. M. Lvov and S. Leporatti, Macromol. Biosci., 2012, 12, 1265–1271 CrossRef CAS.
- Z. Somosi, M. Pavlovic, I. Palinko and I. Szilagyi, Nanomaterials, 2018, 8, 986 CrossRef.
- Z. B. Cao, B. Li, L. Y. Sun, L. Li, Z. P. Xu and Z. Gu, Small Methods, 2019, 1900343, DOI:10.1002/smtd.201900343.
- G. Arrabito, A. Bonasera, G. Prestopino, A. Orsini, A. Mattoccia, E. Martinelli, B. Pignataro and P. G. Medaglia, Crystals, 2019, 9, 361 CrossRef CAS.
- C. P. Chen, J. C. Buffet and D. O'Hare, Dalton Trans., 2020, 49, 8498–8503 RSC.
- Z. P. Xu, G. Stevenson, C. Q. Lu and G. Q. Lu, J. Phys. Chem. B, 2006, 110, 16923–16929 CrossRef CAS.
- M. Pavlovic, P. Rouster and I. Szilagyi, Nanoscale, 2017, 9, 369–379 RSC.
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS.
- P. A. Hassan, S. Rana and G. Verma, Langmuir, 2015, 31, 3–12 CrossRef CAS.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- M. Kobayashi, M. Skarba, P. Galletto, D. Cakara and M. Borkovec, J. Colloid Interface Sci., 2005, 292, 139–147 Search PubMed.
- C. Beaucham and I. Fridovich, Anal. Biochem., 1971, 44, 276–287 CrossRef.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT–Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- X. M. Zhang, M. L. Tong, M. L. Gong, H. K. Lee, L. Luo, K. F. Li, Y. X. Tong and X. M. Chen, Chem. – Eur. J., 2002, 8, 3187–3194 CrossRef CAS.
- T. Kohzuma, A. Odani, Y. Morita, M. Takani and O. Yamauchi, Inorg. Chem., 1988, 27, 3854–3858 CrossRef CAS.
- M. Juric, D. Pajic, D. Zilic, B. Rakvin, D. Milic and P. Planinic, Polyhedron, 2015, 98, 26–34 CrossRef CAS.
- R. B. Martin, J. Inorg. Biochem., 1986, 28, 181–187 CrossRef CAS.
- I. Shweky, A. Bino, D. P. Goldberg and S. J. Lippard, Inorg. Chem., 1994, 33, 5161–5162 CrossRef CAS.
- E. Garribba, G. Micera, D. Sanna and L. Strinna-Erre, Inorg. Chim. Acta, 2000, 299, 253–261 CrossRef CAS.
- Y. Shi, B. B. Toms, N. Dixit, N. Kumari, L. Mishra, J. Goodisman and J. C. Dabrowiak, Chem. Res. Toxicol., 2010, 23, 1417–1426 Search PubMed.
- S. Murath, S. Szerlauth, D. Sebok and I. Szilagyi, Antioxidants, 2020, 9, 153 Search PubMed.
- S. Saringer, R. A. Akula, A. Szerlauth and I. Szilagyi, J. Phys. Chem. B, 2019, 123, 9984–9991 CrossRef CAS.
- T. M. Chen, H. Zou, X. J. Wu, C. C. Liu, B. Situ, L. Zheng and G. W. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 12453–12462 CrossRef CAS.
- D. R. Doerge, R. L. Divi and M. I. Churchwell, Anal. Biochem., 1997, 250, 10–17 CrossRef CAS.
- M. Ozyurek, K. Guclu, E. Tutem, K. S. Baskan, E. Ercag, S. E. Celik, S. Baki, L. Yildiz, S. Karaman and R. Apak, Anal. Methods, 2011, 3, 2439–2453 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation method of samples for EPR measurements; XRD diffractogram; IR spectrum; measured and simulated EPR spectra. See DOI: 10.1039/d0dt04186f |
| This journal is © The Royal Society of Chemistry 2021 |