
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic properties of high entropy oxides
Abhishek
Sarkar
 *ab,
Robert
Kruk
*b and
Horst
Hahn
*abc
*ab,
Robert
Kruk
*b and
Horst
Hahn
*abc
aJoint Research Laboratory Nanomaterials – Technische Universität Darmstadt & Karlsruhe Institute of Technology, Otto-Berndt-Str. 3, 64206 Darmstadt, Germany. E-mail: abhishek.sarkar@kit.edu; horst.hahn@kit.edu
bInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: robert.kruk@kit.edu
cDepartment of Materials Science and Engineering, University of California Irvine, 92697 Irvine, USA
First published on 13th January 2021
Abstract
High entropy oxides (HEOs) are single phase solid solutions consisting of five or more elements in equiatomic or near-equiatomic proportions incorporated into the cationic sub-lattice(s). The uniqueness of the HEOs lies in their extreme chemical complexity enveloped in a single crystallographic structure, which in many cases results in novel functionalities. From the local structure perspective, HEOs consist of an unusually large number of different metal–oxygen–metal couples. Consequently, magnetic correlations in HEOs that inherently depend on the coordination geometry, valence, spin state and type of the metal cations that are hybridized with the bridging oxygen, are naturally affected by an extreme diversity of neighboring ionic configurations. In these conditions, a complex magneto-electronic free-energy landscape in HEOs can be expected, potentially leading to stabilization of unconventional spin-electronic states. This Frontier article provides an overview of the unique magnetic features stemming from the extreme chemical disorder in HEOs along with the possible opportunities for further research and exploration of potential functionalities.
Introduction
Utilization of chemical disorder in the quest for novel functional oxide materials started with the discovery of high entropy oxides (HEOs) in 2015.1 The presence of multiple elements, often five or more, in equiatomic or near-equiatomic proportions, on the cationic sub-lattice(s) makes the HEOs unique in contrast to the conventional oxide systems.1–5 The incorporation of multiple elements in a phase pure system on one hand results in the enhancement of the configurational entropy (Sconfig) that can be calculated using Boltzmann's entropy formula, while on the other hand facilitate tailoring of the functional properties.2,4,6,7 Especially, the tolerance towards high chemical complexity and large defect concentrations, such as oxygen non-stoichiometry, can be considered as the key assets of the HEOs. The growing interest in the field of HEOs is illustrated by the numerous studies focusing on their different structural and functional aspects that have already been reported within the five years since their discovery. Currently, there are eight major classes of HEOs that can be categorized based on the crystallographic structures: rocksalt,1,3,8,9 fluorite,10–15 bixbyite,10,12,16 perovskite (cubic, orthorhombic, rhombohedral),5,17–19 spinel,20,21 pyrochlore,7,22–24 layered (Ruddlesden–Popper and delafossite)25–27 and magnetoplumbite.28,29 Likewise, a broad range of properties, such as electrochemical,2,30–32 optical,11,33–35 magnetic,21,29,34,36–42,43 electronic and ionic transport,44–46 mechanical,47,48 thermal14,47,49 and catalytic50,51 are also reported for different classes of HEOs. In a few cases, the HEOs exhibit substantially improved functionalities compared to the conventional binary oxides or their doped variants,14,32,37,44,47 showcasing the advantages of the high configurational disorder and motivating further research on these chemically complex oxide systems.The key aspects of HEOs, such as the high entropy based nomenclature, the role of entropy in phase stabilization and related thermodynamic parameters, synthesis routes, structural and some of the functional features, have already been highlighted in the few review articles available on HEOs.2,4,6,7,52–55 In contrast, the focus of this brief Frontier article is the magnetic properties of HEOs that has recently started to receive an increasing attention. The fundamentals of magnetism in metal oxides that are of relevance for the understanding of magnetism in HEOs are briefly discussed to be followed by specific case studies on three important magnetic subclasses of HEOs. A short summary of magnetism in hitherto lesser investigated HEO subclasses is also provided. Finally, we surmise our perspective on the future prospects of research on magnetism in HEOs.
Magnetic interactions in metal oxides
Unlike in metallic systems, the direct magnetic interactions between the metal cations in oxides are rare, such as in CrO2 and SrRuO3. Indirect exchange interactions between cations that take place via the coordinating oxygen (anion) are common.56,57 Exchange interactions depend on the overlap of exponentially decaying orbital wave functions, in 3d-transition metals (TM) the 3d orbitals, of the nearest-neighbor (NN) cations that interact through the filled 2p-orbitals of the neighboring O2−.56 The exchange interaction between the participating ions in simplified form can be described by the Heisenberg Hamiltonian, (as shown in eqn (1)).
(as shown in eqn (1)). | (1) |
The  is the exchange parameter coupling the two cations a and b, whose spins are Sa and Sb, respectively. The sign of the exchange parameter determines the nature of the magnetic interactions where
is the exchange parameter coupling the two cations a and b, whose spins are Sa and Sb, respectively. The sign of the exchange parameter determines the nature of the magnetic interactions where  results in ferromagnetic (FM) interaction and
results in ferromagnetic (FM) interaction and 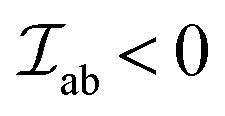 results in antiferromagnetic (AFM) interaction. The principle exchange interactions in oxides are summarized below:
results in antiferromagnetic (AFM) interaction. The principle exchange interactions in oxides are summarized below:
Super exchange interactions
This type of interactions is common for insulating metal oxides and can either be AFM or FM. The interaction involves two-step virtual electron transfers, where the first electron is excited from the O2− 2p6 shell to an adjacent cation, leading to a virtual 3dn+1 state in case of 3d TM cations.56 Then the uncompensated 2p oxygen spin becomes engaged in the direct interaction with the neighboring cation. Hence, the spins of two cations coordinated via O2− are coupled. The predominant superexchange interaction is AFM in nature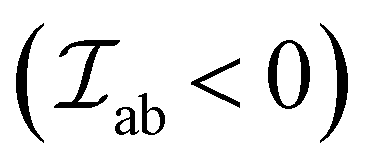 , as the antiparallel arrangement of the cations spins allows the electron in the O2− 2p orbital to spread out into unoccupied orbitals of the metal cations.58 Depending upon the metal–oxygen–metal (M–O–M) bond angle or the occupancy and orbital degeneracy of the metal cation
, as the antiparallel arrangement of the cations spins allows the electron in the O2− 2p orbital to spread out into unoccupied orbitals of the metal cations.58 Depending upon the metal–oxygen–metal (M–O–M) bond angle or the occupancy and orbital degeneracy of the metal cation  can also be greater than 0 leading to FM interactions. Following is a semi-empirical set of rules proposed by Goodenough and Kanamori (G–K rules), reformulated by Anderson, that largely dictate the type and strength of superexchange interactions:56
can also be greater than 0 leading to FM interactions. Following is a semi-empirical set of rules proposed by Goodenough and Kanamori (G–K rules), reformulated by Anderson, that largely dictate the type and strength of superexchange interactions:56
i. If the coupled cations have singly occupied (say 3d) orbital lobes that point toward each other, resulting in large overlap, the exchange is strong and AFM in nature  . This is typical for M–O–M with bond angles between 120°–180°.
. This is typical for M–O–M with bond angles between 120°–180°.
ii. If the coupled cations have a direct overlap integral between singly occupied (3d) and O2− 2p orbitals which is zero by symmetry, the effective indirect exchange becomes FM 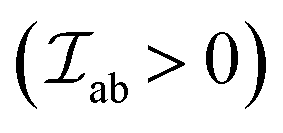 and is relatively weak. This is, for instance, the case for M–O–M with bond angles close to 90°.
and is relatively weak. This is, for instance, the case for M–O–M with bond angles close to 90°.
iii. If the coupled cations have an indirect overlap, via O2− bridge of 120°–180°, between singly occupied (3d) orbitals with an empty or doubly occupied orbitals (3d) of the same type, the exchange is also weakly FM.
Double exchange interactions
These interactions are also mediated by oxygen but they result in metallic conductivity accompanied by FM coupling. In broad terms, this interaction originates from correlated transfer of the cation (say 3d) electron to O2− 2p orbital with the simultaneous move of the other electron from the O2− 2p to the neighboring cation. Furthermore, it was observed by Zener that the free electron hopping resulting in metallicity is energetically favorable when the conducting electrons carry their own spin unchanged and they move in an environment of spins pointing in the same direction, thereby resulting in FM.59,60 If the spins of the coupled cations are antiparallel, then a free hopping of electron is energetically costly, thereby restricting double exchange interactions. For double exchange interactions, it is important that the coupled cations have both localized and delocalized electrons, which is typically the case in a mixed-valence configuration of the cations, e.g., Mn3+–O–Mn4+ or Fe2+–O–Fe3+.56Antisymmetric exchange
This interaction, stemming from the relativity effects, was proposed by Dzyaloshinsky and Moriya and it results in a weak intrinsic FM in otherwise AFM systems.56,61,62 In many AFM ordered systems, especially in crystal systems with lower symmetry, a slight tilting/canting of the spins occurs. Hence, the system instead of being perfectly AFM exhibits a weak net FM moment in certain directions. The Dzyaloshinsky–Moriya interaction is dictated by eqn (2), which is similar to eqn (1) but here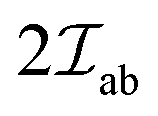 is replaced by D:
is replaced by D: | (2) |
D is the vector that lies along the axis of symmetry with its magnitude in the range of 1% of 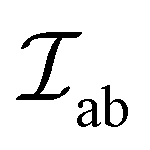 , making this coupling relatively weak.56
, making this coupling relatively weak.56
It can be seen that several parameters, in addition to the crystallographic structure, such as oxidation state of the cations, the metal oxygen bond length and bond angle are of importance for determining the stable magnetism in oxides. Furthermore, the effect of the crystal field, i.e., the O2− ligand field interactions should also be considered, especially for the unshielded 3d TM cations where the crystal field interactions are much stronger than the spin–orbit interactions. Thus, depending on the relative strength of the crystal field interactions versus electron–spin exchange correlations, high/low spin transitions of the cations triggered by the local Jahn–Teller (J–T) distortion can also be readily found.
Apart from the generic interactions discussed above, orbital and charge order are also observed in many oxides.56,57 In most of the HEOs reported till date, irrespective of the crystallographic structure, superexchange coupling accompanied by Dzyaloshinsky–Moriya interactions are found to be the most common ones. Besides, the possibilities of conducting double exchange interactions can also be anticipated in future via judicious choice of cation compositions in HEOs.
Magnetism in rocksalt-HEOs (R-HEOs)
(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O is the primary member of the R-HEOs family.1 Lab scale and synchrotron X-rays diffraction (XRD) coupled with neutron powder diffraction (NPD) indicate that (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O crystallizes in a phase pure rocksalt (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, 225) structure.1,9,32,38,39 X-ray absorption and photoelectron spectroscopies indicate the presence of all the cations in their respective divalent oxidation state, among which Co2+ (d7), Cu2+ (d9) and Ni2+ (d8) are the magnetic ions.3,8,38,39,63 The presence of the J–T active Cu2+ brings about slight deviation from the ideal rocksalt structure locally, observed in terms of relative intensity mismatch between the (111) and (200) reflections in the XRD pattern.3,38 Evidence of local structural distortion around the Cu2+ is also validated from extended X-ray absorption fine structure (EXAFS) and theoretical studies based on density functional theory (DFT) simulations.64–66 Nevertheless, NPD confirms absence of structural transition in (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O ruling out the presence of cooperative J–T distortion.38,39 EXAFS and atom probe tomography (APT) studies have confirmed the random and homogeneous distribution of the cations in the system.64,67 Interestingly, despite the high chemical disorder along with the presence of 40% of non-magnetic ions statistically distributed over the cationic sub-lattice, (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O still exhibits a long range AFM ordering akin to many of the constituent binary rocksalt oxides like CoO or NiO.38,39 The experimentally evaluated Néel temperature (TN) from magnetometry is found to be between 113–120 K, as highlighted in Fig. 1a. Crystallographic non-commensurate reflections in the NPD arising from the long range AFM ordering with the propagation wave vector k = [½ ½ ½] appear below 120 K.38,39 The magnetic transition in the (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O system is rather gradual as no sharp anomaly in the specific heat can be observed around the TN. Accordingly, a nominal deviation from the ideal linear Curie–Weiss fit of reverse susceptibility versus temperature is evident even above the TN.38 This indicates presence of short range magnetic correlations above the TN and likely so as many of the constituent magnetic ions are known to interact at temperatures well above 120 K.38,39 However, above 150 K absence of long range magnetic ordering is affirmed (Fig. 1b) as the neutron diffractogram can be adequately fitted solely using the Fmm crystallographic structure.38,39 The reason for this is believed to be a direct consequence of the chemical disorder that suppresses the long range ordering of magnetic correlations in the R-HEO lattice.38,39
m, 225) structure.1,9,32,38,39 X-ray absorption and photoelectron spectroscopies indicate the presence of all the cations in their respective divalent oxidation state, among which Co2+ (d7), Cu2+ (d9) and Ni2+ (d8) are the magnetic ions.3,8,38,39,63 The presence of the J–T active Cu2+ brings about slight deviation from the ideal rocksalt structure locally, observed in terms of relative intensity mismatch between the (111) and (200) reflections in the XRD pattern.3,38 Evidence of local structural distortion around the Cu2+ is also validated from extended X-ray absorption fine structure (EXAFS) and theoretical studies based on density functional theory (DFT) simulations.64–66 Nevertheless, NPD confirms absence of structural transition in (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O ruling out the presence of cooperative J–T distortion.38,39 EXAFS and atom probe tomography (APT) studies have confirmed the random and homogeneous distribution of the cations in the system.64,67 Interestingly, despite the high chemical disorder along with the presence of 40% of non-magnetic ions statistically distributed over the cationic sub-lattice, (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O still exhibits a long range AFM ordering akin to many of the constituent binary rocksalt oxides like CoO or NiO.38,39 The experimentally evaluated Néel temperature (TN) from magnetometry is found to be between 113–120 K, as highlighted in Fig. 1a. Crystallographic non-commensurate reflections in the NPD arising from the long range AFM ordering with the propagation wave vector k = [½ ½ ½] appear below 120 K.38,39 The magnetic transition in the (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O system is rather gradual as no sharp anomaly in the specific heat can be observed around the TN. Accordingly, a nominal deviation from the ideal linear Curie–Weiss fit of reverse susceptibility versus temperature is evident even above the TN.38 This indicates presence of short range magnetic correlations above the TN and likely so as many of the constituent magnetic ions are known to interact at temperatures well above 120 K.38,39 However, above 150 K absence of long range magnetic ordering is affirmed (Fig. 1b) as the neutron diffractogram can be adequately fitted solely using the Fmm crystallographic structure.38,39 The reason for this is believed to be a direct consequence of the chemical disorder that suppresses the long range ordering of magnetic correlations in the R-HEO lattice.38,39
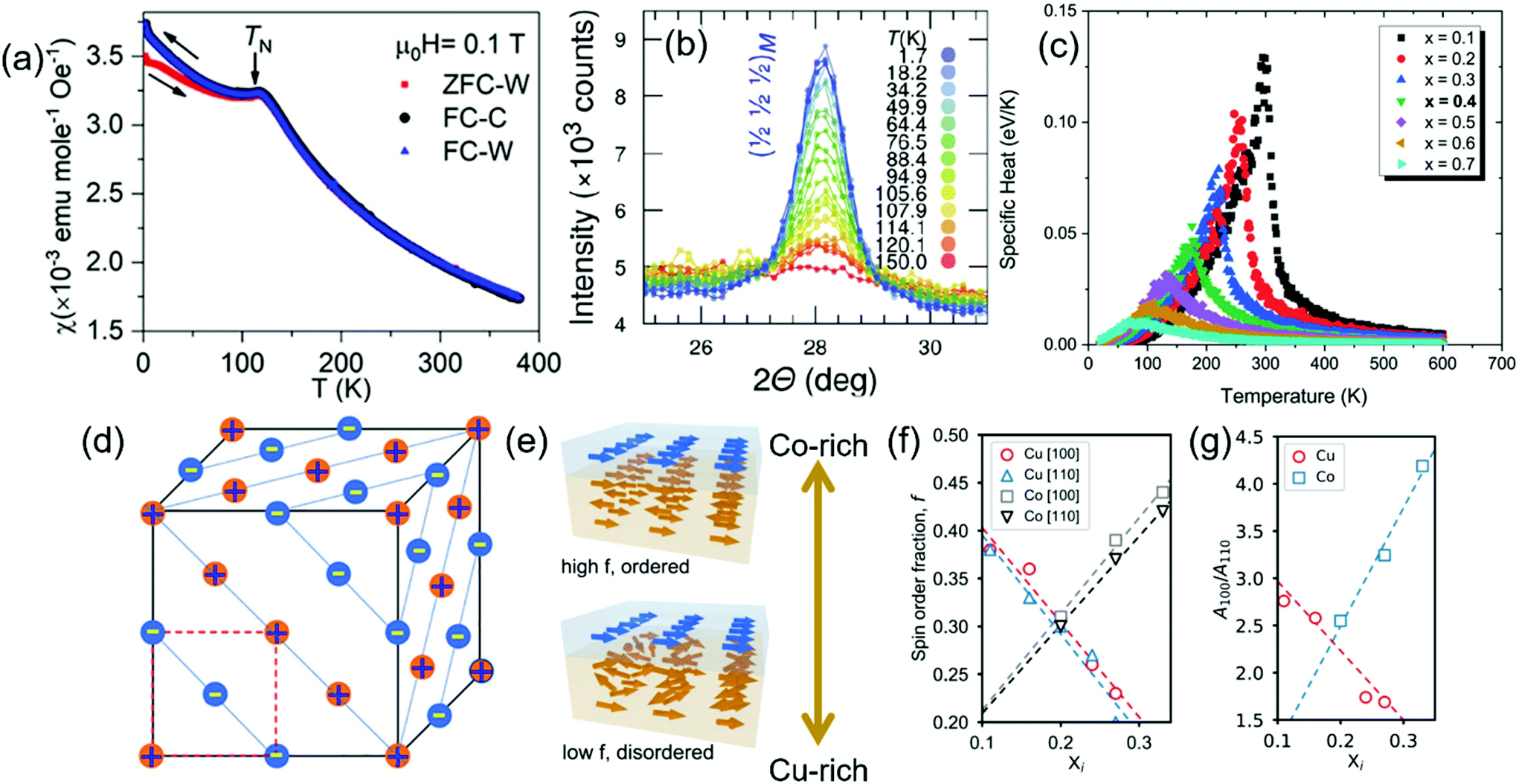 | ||
| Fig. 1 (a) Temperature (T) dependent magnetization (χ = magnetic susceptibility) measurement of (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O, indicating sharp AFM transition below 113 K.38 (b) The evolution of the magnetic reflection [½ ½ ½]M in (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O, stemming from the AFM ordering, as a function of temperature is shown.39 (c) The specific heat as a function of temperature, where the cusp indicates the magnetic ordering temperature, calculated for R-HEO with various degree of non-magnetic dilution.68 (d) Schematic of magnetic structure of R-HEO, the ‘+’ indicates spin up and the ‘−’ indicates spin down.38 Tailoring of the strength of the AFM with varying Co and Cu in R-HEO are shown in (e–g)43 where (e) schematically shows the increase of the glassiness with Cu enrichment, likewise (f) shows decrease/increase of the spin order with increase in Cu/Co and (g) indicates the change in the anisotropy energies for the [100] and [110] directions. | ||
Results from a rigorous theoretical study, where the lattice constants are estimated using DFT and the magnetic interactions are determined from first-principles methods in combination with Monte-Carlo simulations, are in good agreement with the experimentally observed long range AFM ordering.68 In order to better elucidate the magnetic ordering, the TN is estimated using three different fitting approaches: (a) using the exchange parameters of the binary and ternary oxides indicating the TN ≈ 170 K (Fig. 1c), (b) fitting the specific heat in the paramagnetic region with Curie–Weiss type behavior resulting in the TN ≈ 118 K and (c) calculated directly from the HEO system using the singularity in the heat capacity yielding a TN ≈ 135 K.68 The value identified from approach (b) is closest to the experimental value. Nevertheless, the other two approaches are also in close agreement with the experimental observations, especially if the short range correlations are considered. Thus, the theoretical approach on a whole indicates that magnetic interactions present in the simple binary oxides can be readily transferred to the HEO phase despite the chemical disorder and extreme non-magnetic dilution, as shown in Fig. 1c. In binary rocksalt oxides (like NiO or CoO), the first nearest-neighbor (FNN) TM-O-TM bond is 90°, which can ideally result in FM superexchange interactions as per the G–K rules discussed in the earlier section, while the dominant second nearest-neighbor (SNN) TM-O-TM bond is close to 180° that will ideally lead to AFM superexchange interactions.38,68 Akin to the constituent binary rocksalt oxides, it is predicted that the SNN AFM superexchange is the dominant magnetic interaction in R-HEO where the FM moments are arranged in (111) planes perpendicular to the [111] directions with the spins in adjacent planes oriented antiferromagnetically (Fig. 1d).68 Furthermore, the dominant AFM superexchange interaction is also coherent with the insulating nature of the R-HEO, which is known to exhibit a colossal dielectric constant.3
Exploiting the compositional flexibility, other magnetic (Fe3+, d5) and non-magnetic ions (Li+, Ga3+) are also added/substituted to the R-HEO lattice.39 Not only subtle difference in the magnetic transition temperature are observed but also alteration of the magnetic ground state (either AFM or spin-glass states) is evident, see Table 1. Irrespective of the compositions, all the R-HEOs exhibit distinct split in the field cooling (FC) and zero field cooling (ZFC) magnetization below the TN, which can be a result of the spin-canting similar to the Dzyaloshinsky and Moriya interactions. Apart from (Co0.19Cu0.19Mg0.19Ni0.19Zn0.19Li0.05)O, in all the other R-HEOs, either Ga3+ or Fe3+ is co-doped with Li+ that maintain the charge of the effective dopant as 2+. Thus, this kind of doping is not expected to alter the oxidation state of the constituent TM cations. Consequently, this kind of studies are meant to unravel the dependency of the magnetic features with respect to the structural parameters and concentration of non-magnetic ions. The experimentally observed trend of the lowering of the transition temperature with increasing amount of the non-magnetic ions (Table 1) is in good agreement with the Monte-Carlo simulations.39,68 In case of the systems with similar amount of non-magnetic ions the differences in the magnetic features can be plausibly related to the changes in the structural parameters. In case of doping with magnetic ions like Fe3+ in R-HEO, a complete elucidation of the magnetic ground state becomes challenging due to additional magnetic interaction of the Co2+, Cu2+ and Ni2+ with Fe3+. Nevertheless, such aliovalent doping of magnetic ions in R-HEOs can open up novel magneto-electronic functionalities similar Fe3+ doped NiO where it is proposed that conducting double exchange pathways through the doped Fe3+ and resulting cation vacancy that are created.69
| Compositions | T mag | GS | Ref. |
|---|---|---|---|
#![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The exact magnetic ground state remains unknown; (5A0.2)(5B0.2)O3 is (Gd0.2La0.2Nd0.2Sm0.2Y0.2)(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3. The exact magnetic ground state remains unknown; (5A0.2)(5B0.2)O3 is (Gd0.2La0.2Nd0.2Sm0.2Y0.2)(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3. |
|||
Rocksalt HEOs (
Fm
![[3 with combining macron]](https://www.rsc.org/images/entities/b_char_0033_0304.gif) m
)
m
)
|
|||
| (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O | 120 | AFM | 38 and 39 |
| (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O (nano) | 106 | AFM | 41 |
| (Co0.19Cu0.19Mg0.19Ni0.19Zn0.19Li0.05)O | 95 | AFM | 39 |
| (Co0.2Cu0.2Ni0.2Zn0.2Li0.1Ga0.1)O | 125 | AFM | 39 |
| (Co0.2Cu0.2Mg0.2Ni0.2Li0.1Ga0.1)O | 155 | AFM | 39 |
| (Cu0.2Mg0.2Ni0.2Zn0.2Li0.1Ga0.1)O | 10 | SG | 39 |
| (Co0.2Mg0.2Ni0.2Zn0.2Li0.1Ga0.1)O | 60 | SG | 39 |
| (Co0.2Cu0.2Mg0.2Zn0.2Li0.1Ga0.1)O | 20 | SG | 39 |
| (Co0.16Cu0.16Mg0.16Ni0.16Zn0.16Li0.1Fe0.1)O | 100 | # | 39 |
| Orthorhombic perovskite HEOs ( Pbnm ) | |||
| La(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 | 185 | AFM | 37 |
| Gd(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 | 120 | AFM | 37 |
| Nd(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 | 145 | AFM | 37 |
| Sm(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 | 130 | AFM | 37 |
| Y(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 | 118 | AFM | 37 |
| (5A0.2)(5B0.2)O3# | 135 | AFM | 37 |
| (Gd.2La0.2Nd0.2Sm0.2Y0.2)CoO3 | — | PM | 42 and 46 |
| (Gd0.2La0.2Nd0.2Sm0.2Y0.2)CrO3 | 198 | AFM | 42 |
| (Gd0.2La0.2Nd0.2Sm0.2Y0.2)FeO3 | 675 | AFM | 42 |
|
Spinel HEOs (
Fd
m
)
|
|||
| (Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)3O4 | 425 | FM | 21 and 40 |
| (Co0.2Cu0.2Mg0.2Mn0.2Ni0.2)Fe2O4 | 735 | FM | 21 |
| (Co0.2Cu0.2Fe0.2Mn0.2Ni0.2)Fe2O4 | 715 | FM | 21 |
| (Co0.2Cu0.2Fe0.2Mg0.2Ni0.2)Fe2O4 | 774 | FM | 21 |
| (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)Fe2O4 | 648 | FM | 21 |
| (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)Cr2O4 | 36 | AFM | 21 |
| (Co0.2Cu0.2Fe0.2Mg0.2Ni0.2)Cr2O4 | 23 | AFM | 21 |
| (Co0.2Cu0.2Mg0.2Mn0.2Ni0.2)Cr2O4 | 30 | AFM | 21 |
|
Bixbyite HEOs (
Ia
)
|
|||
| (Gd0.2Tb0.2Dy0.2Ho0.2Er0.2)2O3 | — | PM | 16 |
|
Pyrochlore HEOs (
Fd
m)
|
|||
| (Yb0.2Tb0.2Gd0.2Dy0.2Er0.2)0.2Ti2O7 | <3 | SG | 24 |
| Magnetoplumbite HEOs ( P 6 3 / mmc ) | |||
| Ba(Fe6.0Ga1.26In1.17Ti1.20Cr1.22Co1.15)O19 | >350 | FM | 29 |
To explore the stability limits of the AFM phase, epitaxial thin films of R-HEOs with varying composition were deposited on MgO substrates with a heterogeneous top layer of FM permalloy.36,43 The magnetic heterogeneous interface so formed by the AFM R-HEOs and FM permalloy allows for strong exchange bias (EB) effects, which can be evaluated by estimating the shift of the hysteresis loop of the FM along the magnetic field axis due to interfacial exchange coupling with the adjacent AFM layer.70 The strength of the EB effect depends on several factors, among which one of the important feature is the robustness of the AFM phase. Tailoring the extent of the EB effect can be achieved via composition modulation of the deposited R-HEOs. Change in the concentration of Cu2+, apart from having a direct consequence on the magnetic exchange interactions purely due to composition, also results in significant change in the crystal structure due to its pronounced J–T effect and even changes the valence of Co in the resulting R-HEOs. These effects synergistically lead to alteration of the magnetic interactions, with increase in the amount of the Cu in R-HEO the glassiness of the AFM increases, likewise the spin order fraction at the interface decreases (Fig. 1e and f).43 Also the magnetic isotropy, evaluated by analyzing the ratio of the anisotropy energies for the [100] and [110] directions, increases with amount of J–T active Cu2+ in the R-HEO, as shown in Fig. 1g.43 Interestingly, increasing the amount of Co in the R-HEOs results in an inverse effect, the magnetic anisotropy becomes stronger and [110] becomes the preferred easy axis (Fig. 1f and g). Additionally, the robustness of the AFM is enhanced as the spin order fractions increase with the amount of Co in the R-HEO (Fig. 1e).36,43
Overall, the current studies already highlight an abundance of unique magnetic features exhibited by the R-HEOs: (a) experimentally and theoretically proven long AFM range order despite the extreme ionic disorder and high degree of dilution with non-magnetic ions, (b) gradual magnetic transitions spaced over wide range of temperature, (c) short range magnetic correlation present well over the TN and (d) controllable structural (like local J–T distortion) features retaining phase purity to alter the glassiness of AFM ordering. Additionally, R-HEOs show a clear entropy driven phase stabilization effect, which are widely discussed in the literature,1,2,52,71,72 unlike many other HEOs. This makes R-HEOs an ideal materials class for discerning the real effect entropy on the magnetic features of materials.
Magnetism in perovskite-HEOs (P-HEOs)
The rare-earth (RE)–transition metal (TM) perovskites (ABO3) constitute a vast and functionally very diverse class of oxides owing to the overwhelming richness of their magneto-electronic properties; such as colossal magnetoresistance, magneto-electronic phase separation, Mott transitions, spin–electron correlations, magneto-electric effect, etc. The interlink between their magneto-electronic properties and crystal structure has been extremely important as many types of structural features, e.g., lattice distortion or tilting of the TMO6 (BO6) octahedra, have often shown a decisive impact on the magnetic interactions. A well-known formulation that typically projects the degree of structural distortion (or deviation from ideal cubic structure) in perovskites is the Goldschmidt's tolerance factor t (calculated from eqn (3)). In eqn (3), rA and rB are the ionic radii of the cations at A-site and B-site, respectively, and rO is the radius of the oxygen ion (anion). | (3) |
The most common magnetic interaction present in RE–TM perovskites is the AFM superexchange interaction, which is the prevalent interaction among the cations in both inter- and intra-sites. However, the inter-site coupling between the RE and TM cations are rather weak compared to the intra-site coupling between the TM cations. Hence, the magnetic features in RE–TM perovskites strongly depend on the geometrical characteristics of the TM-O-TM (B–O–B) bond and the electronic configuration of the coupled TM ions.
Magnetic orthorhombic (Pbnm) P-HEOs, whose structural features can be controlled effectively via composition, consists of a large number of RE and TM cations on the A- and the B-sites, respectively.17,37,42 The tilting of the BO6 (TMO6) octahedral and the degree of orthorhombic distortion in P-HEOs scales with their Goldschmidt's tolerance factor t. Furthermore, a single phase P-HEO can be stabilized even with 10 different cations, 5 RE on the A-site and 5 TM on the B-site. APT studies confirm the homogeneous distribution of the 10 different cations in P-HEOs, again highlighting the tolerance of HEOs towards extreme chemical complexity.67
The high chemical disorder, especially on the TM based B-site, is of immediate interest as a wide number of competing exchange interactions are possible leading to the creation of exotic magnetic states. The P-HEOs belonging to the group of five TM cations (5B0.2 = Co0.2Cr0.2Fe0.2Mn0.2Ni0.2) on B-site with a single RE cation, A(5B0.2)O3, were initially investigated.37 For comparison, the 10-cationic P-HEO, (Gd0.2La0.2Nd0.2Sm0.2Y0.2)(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3, is also studied. The magnetic transition temperatures and the ground states are summarized in Table 1. In accordance to G–K rules, a predominant superexchange AFM behavior (at lower temperature) was observed in the P-HEOs. It was found that the magnetic properties of P-HEO can be mostly understood by only considering the interactions within the TM cation sub-lattice. Several of the RE cations, like La3+ or Y3+ were non-magnetic, thus did not influence the magnetic response to the external field and temperature, whereas contributions from magnetic RE cations like, Gd3+, Nd3+ or Sm3+ were only visible at very low temperatures.37 It is worth noting the special case for Sm(5B0.2)O3, where a significant decrease of the magnetization below 25 K can be observed leading almost to a complete magnetization reversal. The exact reason for this behavior still remains unsolved, however, it can plausibly be attributed to long-range ordering of Sm3+ spins, which couple AFM to the (canted) magnetic moment of the TM cations. Apart from these lower temperature features, the effect of A-cations is clearly accounted for in a clear dependency between the TN and the tolerance factor (t), where the size of the A-cations determines the angle the TM-O-TM bond.37 It should be noted that similar to the R-HEOs, a slight deviation from the ideal Curie–Weiss type behavior could also be observed in P-HEOs above the TN, which can result from short range magnetic correlations. Additionally, distinct FM contributions leading to strong vertical exchange bias in all the B-site disordered P-HEOs are observed. As discussed in the earlier section, this kind of behavior is typically observed in heterogeneous structures, like FM–AFM multilayers or core–shell structures. Thus, the presence of such interactions in phase-pure P-HEOs is revealing. The possible origin of an intrinsic exchange bias effect in P-HEOs supported by the results of Mössbauer spectroscopy is schematically explained in Fig. 2a. The FM contributions in P-HEOs can be attributed to the presence of weakly interacting FM clusters with a higher and sharper transition temperature compared to the AFM matrix in which they are embedded. It is highly probable that the FM clusters are a consequence of the disordered B-site with multiple competing FM and AFM exchange interactions; for instance, couples like Fe3+–O–Cr3+, are known to interact ferromagnetically.37
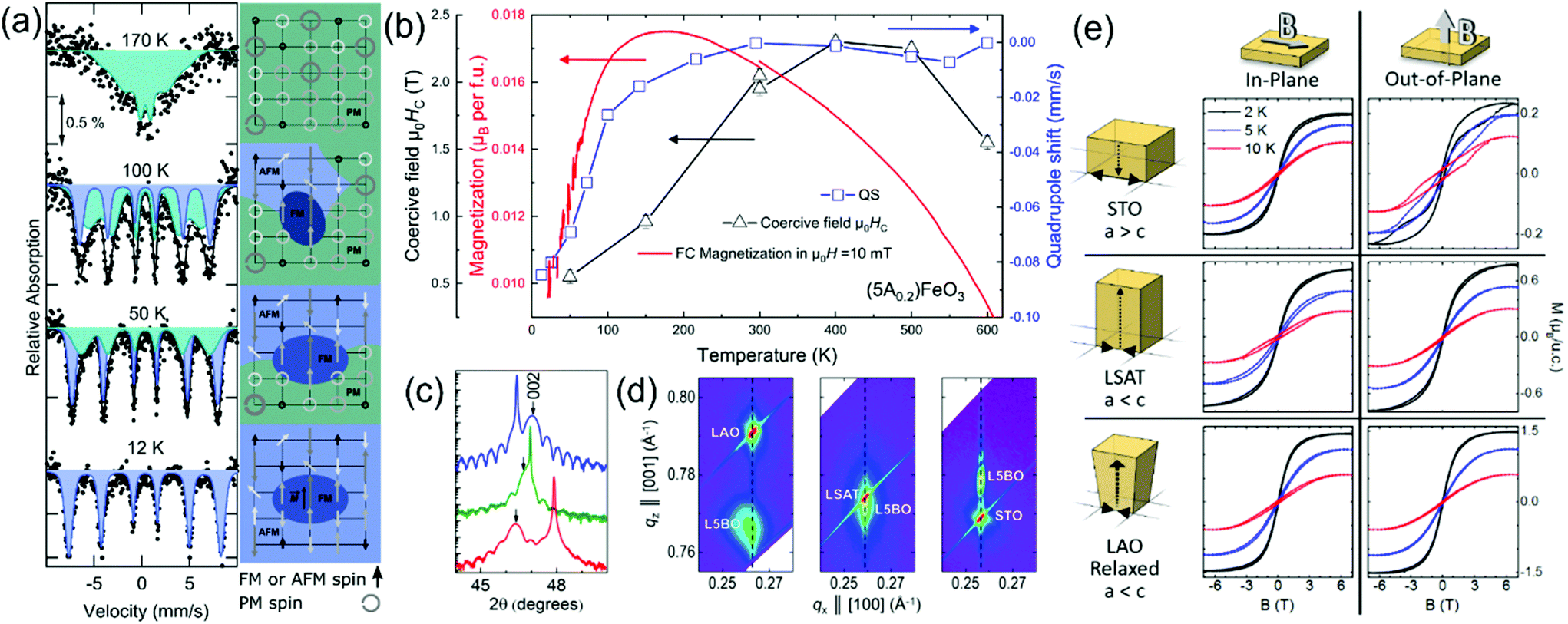 | ||
| Fig. 2 (a) The temperature dependent 57Fe Mössbauer spectroscopy of La(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 is presented, where the left column is the Mössbauer spectra represented with two sextets: one broad spectrum (green) representing dynamic fluctuating spins (on the characteristic timescale of the measurement) and one well defined subspectrum (blue) from static magnetic order. The right column presents a sketch of the evolution of the proposed magnetic structure resembling magnetic phase separation.37 (b) Highlight of the temperature dependency of the coercive field μ0HC, magnetization and the quadrupole coupling constant of the magnetic sextet determined from 57Fe Mössbauer spectra in (Gd0.2La0.2Nd0.2Sm0.2Y0.2)FeO3.42 (c) (002) XRD reflection of epitaxially grown La(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 on SrTiO3 (STO-blue curve), (LaAlO3)0.3(SrAl0.5Ta0.5O3)0.7 (LSAT-green curve) and LaAlO3 (LAO-red curve).19 (d) Reciprocal space mapping around the (103) reflections of La(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 films on different substrates indicating different kinds of epitaxial strains, while (e) indicates the changes in the magnetization as a function of magnetic field on the differently strained films highlighting a strong influence of strain on the magnetic easy axis direction and absolute magnetization.19 | ||
The magnetic properties of the P-HEOs, with a single TM cation and a disordered A-site with multiple RE cations, of (5A0.2)BO3 where 5A0.2 = Gd0.2La0.2Nd0.2Sm0.2Y0.2, have been investigated in a subsequent study.42 In general, (5A0.2)BO3 exhibit similarities to the magnetic properties of the parent perovskite oxides, namely, the ortho-cobaltites, -chromites, and -ferrites, despite the extreme chemical disorder on the RE site (see Table 1). This observation is intuitive to a certain extent as the magnetic properties of RE–TM perovskites are mostly dictated by the interactions in the TM sites. However, distinct differences and interesting magnetic properties are also evidenced. Most of the parent RE-cobaltites show transition of the Co3+ from low to high spin states or intermediate states, while in case of the (5A0.2)CoO3 an instability of the spin state is suppressed, resulting in a stable low spin state of Co3+ in the temperature range from 4.5 to 320 K.42 The (5A0.2)FeO3 exhibit one of the highest TN of all the investigated HEO materials.42 A synopsis of the results obtained from magnetometry and Mössbauer spectroscopy for (5A0.2)FeO3 is presented in Fig. 2b. It shows signatures of a spin-reorientation transition that is supported by Mössbauer spectroscopy, but more significant is the large increase of the coercive field of the canted ferromagnetic moment with increasing temperatures reaching a value of about 2 T at ambient temperature.
In addition to internal inherent strains present in P-HEOs stemming from composition and variation in tolerance factor, a recent study focused on tailoring the magnetic anisotropy in P-HEOs via external strains effects.19 Single-crystal thin films of La(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 were deposited on different substrates with different lattice parameters resulting in varying epitaxial strains in the P-HEO thin film, as shown in Fig. 2c and d.19 In congruence with the bulk counterpart, the La(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 thin films also exhibit magnetic phase separation, i.e., the presence of weak FM signatures in a predominant AFM matrix. In this work the FM order is actively manipulated via strain engineering, taking advantage of the internal fragility of the FM ground state. It is observed that depending upon the nature of the epitaxial strain, be it compressive or tensile, and the direction of the applied magnetic field, the magnetic characteristics of the FM phase can be strongly affected. Likewise, the magnetic anisotropy of the FM phase in the P-HEO can be controlled via epitaxial strain as shown in Fig. 2e. This initial study reveals the promises of magneto-electronic tunability in P-HEOs via conventional and effective strain engineering techniques.
In summary, similar to R-HEOs, several distinctive magnetic features are identified in P-HEOs. Long range magnetic ordering despite chemical disorder, gradual magnetic transitions and short range correlations well above the magnetic transition temperature appear to be universal characteristics of HEOs. Besides, competing magnetic interactions in P-HEOs with disordered B-site sublattice result in very local magnetic phase separations giving rise to a substantial degree of intrinsic vertical exchange bias. This encapsulation of different magnetic and spin electronic states in a single crystallographic structure of P-HEOs certainly motivates further investigation.
Magnetism in spinel-HEOs (S-HEOs)
Similar to perovskites, spinels (AB2O4) also consist of two different cation sites. Spinels can also be represented as M3O4 or MM2O4. Ideally in spinels, the 2+ cations occupy the tetrahedral A-sites while the 3+ cations occupy the octahedral B-sites. However, unlike in perovskites, a low energy barrier of forming anti-site defects or in other words an inversion where the 2+ cations occupy the octahedral sites and vice versa is rather common. Likewise, the inter-site magnetic interactions are also of utmost importance. Typically, the spins of the cations on the respective sites (intra-sites) are in parallel, while antiparallel interactions exist between the A- and B-site cations. Thus, the net magnetization (Mnet) is the difference moments of the A- and B-site (Mnet = |A − B|).(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)3O4 was the first S-HEO system (Fdm) to be reported.20 (Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)3O4 exhibits ferrimagnetism at room temperature with the magnetic transition temperature close to 420 K for its bulk variant.21 Several other S-HEO compositions are now known, as shown in Table 1.21,40 The compositional variation and the resulting impact on the site occupations result in drastic changes in the magnetic properties. Most of the Cr-rich system, where Cr is expected to solely occupy the B-site, showed predominant AFM ordering at low temperatures. For instance, (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)Cr2O4 is AFM below 36 K. In contrast, the Fe-rich system, showed high magnetic transition temperatures close to or above 650 K. The nature of the magnetic interactions in chemically complex S-HEOs is largely unraveled, as the exact occupation and oxidation states of the cations on the different sites needs to be precisely known, which is inherently challenging. Although X-ray absorption spectroscopy and X-ray magnetic linear dichroism employed in few of these S-HEO systems ((Mg0.2Mn0.2Co0.2Ni0.2Cu0.2)Fe2O4, (Mg0.2Fe0.2Co0.2Ni0.2Cu0.2)Cr2O4) shed light on the occupation and oxidation state of the cations, this information remains largely inaccessible in a vast majority of magnetic S-HEOs.21 Thus, future research relying on combining spectroscopic methods, focusing on detailed charge and spin characteristics of S-HEOs is still needed to open the ways for further tuning of their magnetic properties.
Magnetism in other HEOs
Apart from R-HEOs, P-HEOs and S-HEOs, magnetic properties of pyrochlore (polycrystalline and also bulk single crystal)24, RE based bixbyite16 and magnetoplumbite-hexaferrite29 type HEOs are also explored, as summarized in Table 1. Identical to the constituent binary pyrochlore or bixbyite oxides, the pyrochlore (Fdm) and bixbyite (Ia) HEOs also exhibit predominant paramagnetic (PM) behavior down to 3 and 5 K, respectively. Interestingly, lower temperature alternating current susceptibility measurements, carried at 0.3–1.7 K, indicate spin-glass magnetic ground state of the pyrochlore HEO system.24 The hexaferrite HEO (P63/mmc) exhibits a ferrimagnetic ordering at room temperature similar to its parent BaFe12O19, however, magnetic phase separation as observed in P-HEOs is also evident in the hexaferrite HEO.29
Conclusions and outlook
The last two years of active research in the field of magnetism of HEOs has provided a sufficiently large database from which certain general trends can be discerned. So far, it can be stated that the dominant features of magnetism in the HEOs largely resemble the isostructural conventional oxides, such as AFM in transition metal based rocksalt or rare earth-transition metal based orthorhombic perovskites, PM in rare earth based bixbyite or pyrochlore and ferrimagnetism in transition metal based spinels or hexaferrites. Although from the viewpoint of the single crystallographic structure and composition these results are rather intuitive and too certain extent predictive, the presence of a stable long range magnetic ordering despite the extremely high chemical disorder is certainly intriguing. Furthermore, the HEOs in most studied cases show definite magnetic features, overlying the magnetic states inherited from parent oxides, that are distinct from the conventional binary or the conventionally doped oxides. Building upon these noticeable differences, we present our viewpoints on future research directions.Composition based tailoring of magnetism
Altering the chemical composition by proper selection of metal cations is the most common way of tailoring the magnetic properties in any metal oxide and in that sense the observed differences in the transition temperature or magnetic ground state via chemical substitution in HEOs might look rather natural. However, the strength and uniqueness of HEOs lies in the extensive compositional flexibility while retaining the phase purity.39,73 The highly disordered cation sub-lattice(s) can effectively result in creation of numerous, often competing, local constellations of exchange interactions. For example, the strong vertical exchange bias, unusual in one-phase material, observed in phase pure disordered B-site P-HEOs can be a result of such locally competing interactions.37,42 Essentially, the complex exchange-energy landscape in HEOs can have many local minima with hitherto unknown physical properties, which are not present in the parent systems. Based on these premises the internal coupling between the local magnetic phases can be induced and further strengthened via conventional (compositional) doping. For instance, judicious choice of aliovalent (holes/electrons) doping even with non-magnetic ions can result change in oxidation state of constituent cations or oxygen stoichiometry. Especially in HEOs containing several TMs that can afford multiple valence states, the charge compensation can be expected to be in many more varieties than in parent oxides thus resulting in unconventional spin-electronic states. Likewise, for precise evaluation of the spin-electronic states in an element specific way in multicomponent HEOs, magnetic characterization technique such as X-Ray magnetic circular/linear dichroism can be of immense importance as it can complement spatially averaging conventional magnetometry techniques.Strain based tailoring magnetism
HEO feature pronounced local structural distortions with fluctuating degree of strains due to the presence of multiple cations of different ionic sizes. A result of which can be observed in the magnetic characteristics of R-HEO, where the transition temperature and the extent of exchange coupling with a FM interface can be tailored via controlled local J–T distortion.34,43 Likewise, the tilting of the TMO6 octahedra in P-HEOs and the geometry of TM-O-TM bonds can result in distinct changes in the magnetic feature, one such e.g., is the gradual magnetic transition, spreading coherently over a broad temperature range.37,39,42 The compositional variation, relying on the difference of the ionic sizes of cations, is one way to influence these inherent local structural features. Another way is the application of external strains (like epitaxial strain engineering), which is also found to be a viable approach to alter the local structure and thereby the magnetic features of HEOs. Simultaneously, for better elucidation the magnetic properties in HEOs, a strong support from a right combination of high-resolution structural characterization techniques are extremely crucial for unravelling the intricate details of local features of the cation-oxygen-cation bonds.Magnetoionics and electrochemical tuning of magnetism in HEOs
Many of the HEO crystal classes are known to afford stable Li-storage possibilities, either via, conversion, intercalation or insertion.26,30–32,74,75 Systems like the spinels, are known to possess two electrochemical windows, one for intercalation and another for conversion. Utilizing the intercalation windows, in some of earlier studies magneto-ionic or electrochemically driven reversible tuning of magnetism in conventional spinels has been shown.76–78 Likewise, magneto-ionic effects in Li-based conversion kind of electrodes are also reported.79 R-HEOs and S-HEOs, are known to be tolerant to reversible lithiation and delithiation, while layered HEOs are can reversibly accommodate both Li and Na.26,27,31,32,75 Thus, stable reversible control of magnetism via the electrochemically-driven insertion/extraction of ions might be feasible. The chemical disorder can be of advantage to possibly making it easier to reach, conventionally inaccessible, magneto-electric free-energy minima.The aforementioned approaches will be useful to map the structural-magnetic phase diagrams of HEOs as a function composition or strain along with the possibility to reversibly control the magnetic features in HEOs. Furthermore, the magnetic properties of the oxides are intimately correlated to their electronic and optical features. Hence, the quest for tuning the magnetic features in HEOs in combination with their vast composition space can be a stepping-stone for the discovery of a plethora of unknown physical phenomena.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AS and HH acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) project HA 1344/43-1. The financial support by Helmholtz Association is gratefully acknowledged. Part of this work was made possible by the collaboration with the UC Irvine MRSEC, Center for Complex and Active Materials, under National Science Foundation award DMR-2011967. The authors acknowledge Ralf Witte, Richard Brand, Ben Breitung, Qingsong Wang, Subramshu S. Bhattacharya, Leonardo Velasco, Miriam Botros, and Mohammad Reda Chellali for fruitful discussions.References
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J.-P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef.
- D. Bérardan, S. Franger, D. Dragoe, A. K. Meena and N. Dragoe, Phys. Status Solidi RRL, 2016, 10, 328–333 CrossRef.
- B. L. Musicó, D. Gilbert, T. Z. Ward, K. Page, E. George, J. Yan, D. Mandrus and V. Keppens, APL Mater., 2020, 8, 040912 CrossRef.
- S. Jiang, T. Hu, J. Gild, N. Zhou, J. Nie, M. Qin, T. Harrington, K. Vecchio and J. Luo, Scr. Mater., 2018, 142, 116–120 CrossRef CAS.
- C. Oses, C. Toher and S. Curtarolo, Nat. Rev. Mater., 2020, 5, 295–309 CrossRef CAS.
- A. J. Wright and J. Luo, J. Mater. Sci., 2020, 55, 9812–9827 CrossRef CAS.
- D. Bérardan, A. K. Meena, S. Franger, C. Herrero and N. Dragoe, J. Alloys Compd., 2017, 704, 693–700 CrossRef.
- A. Sarkar, R. Djenadic, N. J. Usharani, K. P. Sanghvi, V. S. K. Chakravadhanula, A. S. Gandhi, H. Hahn and S. S. Bhattacharya, J. Eur. Ceram. Soc., 2017, 37, 747–754 CrossRef CAS.
- R. Djenadic, A. Sarkar, O. Clemens, C. Loho, M. Botros, V. S. K. Chakravadhanula, C. Kübel, S. S. Bhattacharya, A. S. Gandhi and H. Hahn, Mater. Res. Lett., 2017, 5, 102–109 CrossRef CAS.
- A. Sarkar, C. Loho, L. Velasco, T. Thomas, S. S. Bhattacharya, H. Hahn and R. Djenadic, Dalton Trans., 2017, 46, 12167–12176 RSC.
- L. Spiridigliozzi, C. Ferone, R. Cioffi, G. Accardo, D. Frattini and G. Dell'Agli, Materials, 2020, 13, 558 CrossRef CAS.
- K. Chen, X. Pei, L. Tang, H. Cheng, Z. Li, C. Li, X. Zhang and L. An, J. Eur. Ceram. Soc., 2018, 38, 4161–4164 CrossRef CAS.
- J. Gild, M. Samiee, J. L. Braun, T. Harrington, H. Vega, P. E. Hopkins, K. Vecchio and J. Luo, J. Eur. Ceram. Soc., 2018, 38, 3578–3584 CrossRef CAS.
- M. Anandkumar, S. Bhattacharya and A. S. Deshpande, RSC Adv., 2019, 9, 26825–26830 RSC.
- K. Tseng, Q. Yang, S. J. McCormack and W. M. Kriven, J. Am. Ceram. Soc., 2020, 103, 569–576 CrossRef CAS.
- A. Sarkar, R. Djenadic, D. Wang, C. Hein, R. Kautenburger, O. Clemens and H. Hahn, J. Eur. Ceram. Soc., 2018, 38, 2318–2327 CrossRef CAS.
- Y. Sharma, B. L. Musico, X. Gao, C. Hua, A. F. May, A. Herklotz, A. Rastogi, D. Mandrus, J. Yan, H. N. Lee, M. F. Chisholm, V. Keppens and T. Z. Ward, Phys. Rev. Mater., 2018, 2, 060404 CrossRef CAS.
- Y. Sharma, Q. Zheng, A. R. Mazza, E. Skoropata, T. Heitmann, Z. Gai, B. Musico, P. F. Miceli, B. C. Sales, V. Keppens, M. Brahlek and T. Z. Ward, Phys. Rev. Mater., 2020, 4, 014404 CrossRef CAS.
- J. Dąbrowa, M. Stygar, A. Mikuła, A. Knapik, K. Mroczka, W. Tejchman, M. Danielewski and M. Martin, Mater. Lett., 2018, 216, 32–36 CrossRef.
- B. Musicó, Q. Wright, T. Z. Ward, A. Grutter, E. Arenholz, D. Gilbert, D. Mandrus and V. Keppens, Phys. Rev. Mater., 2019, 3, 104416 CrossRef.
- Z. Teng, L. Zhu, Y. Tan, S. Zeng, Y. Xia, Y. Wang and H. Zhang, J. Eur. Ceram. Soc., 2020, 40, 1639–1643 CrossRef CAS.
- K. Zhang, W. Li, J. Zeng, T. Deng, B. Luo, H. Zhang and X. Huang, J. Alloys Compd., 2020, 817, 153328 CrossRef CAS.
- C. Kinsler-Fedon, Q. Zheng, Q. Huang, E. S. Choi, J. Yan, H. Zhou, D. Mandrus and V. Keppens, Phys. Rev. Mater., 2020, 4, 104411 CrossRef CAS.
- W. Zhang, A. R. Mazza, E. Skoropata, D. Mukherjee, B. Musico, J. Zhang, V. M. Keppens, L. Zhang, K. Kisslinger, E. Stavitski, M. Brahlek, J. W. Freeland, P. Lu and T. Z. Ward, ACS Nano, 2020, 14, 13030–13037 CrossRef.
- J. Wang, Y. Cui, Q. Wang, K. Wang, X. Huang, D. Stenzel, A. Sarkar, R. Azmi, T. Bergfeldt, S. S. Bhattacharya, R. Kruk, H. Hahn, S. Schweidler, T. Brezesinski and B. Breitung, Sci. Rep., 2020, 10, 18430 CrossRef CAS.
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y. Hu, Angew. Chem., 2020, 59, 264–269 CrossRef CAS.
- D. A. Vinnik, E. A. Trofimov, V. E. Zhivulin, O. V. Zaitseva, S. A. Gudkova, A. Y. Starikov, D. A. Zherebtsov, A. A. Kirsanova, M. Häßner and R. Niewa, Ceram. Int., 2019, 45, 12942–12948 CrossRef CAS.
- D. A. Vinnik, A. V. Trukhanov, F. V. Podgornov, E. A. Trofimov, V. E. Zhivulin, A. Y. Starikov, O. V. Zaitseva, S. A. Gudkova, A. A. Kirsanova, S. V. Taskaev, D. A. Uchaev, S. V. Trukhanov, M. A. Almessiere, Y. Slimani and A. Baykal, J. Eur. Ceram. Soc., 2020, 40, 4022–4028 CrossRef CAS.
- N. Qiu, H. Chen, Z. Yang, S. Sun, Y. Wang and Y. Cui, J. Alloys Compd., 2019, 777, 767–774 CrossRef CAS.
- D. Wang, S. Jiang, C. Duan, J. Mao, Y. Dong, K. Dong, Z. Wang, S. Luo, Y. Liu and X. Qi, J. Alloys Compd., 2020, 844, 156158 CrossRef CAS.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef.
- A. Sarkar, B. Eggert, L. Velasco, X. Mu, J. Lill, K. Ollefs, S. S. Bhattacharya, H. Wende, R. Kruk, R. A. Brand and H. Hahn, APL Mater., 2020, 8, 051111 CrossRef CAS.
- G. N. Kotsonis, P. B. Meisenheimer, L. Miao, J. Roth, B. Wang, P. Shafer, R. Engel-Herbert, N. Alem, J. T. Heron, C. M. Rost and J.-P. Maria, Phys. Rev. Mater., 2020, 4, 100401 CrossRef CAS.
- B. Cheng, H. Lou, A. Sarkar, Z. Zeng, F. Zhang, X. Chen, L. Tan, V. Prakapenka, E. Greenberg, J. Wen, R. Djenadic, H. Hahn and Q. Zeng, et al. , Commun. Chem., 2019, 2, 114 CrossRef.
- P. B. Meisenheimer, T. J. Kratofil and J. T. Heron, Sci. Rep., 2017, 7, 3–8 CrossRef.
- R. Witte, A. Sarkar, R. Kruk, B. Eggert, R. A. Brand, H. Wende and H. Hahn, Phys. Rev. Mater., 2019, 3, 034406 CrossRef CAS.
- J. Zhang, J. Yan, S. Calder, Q. Zheng, M. A. McGuire, D. L. Abernathy, Y. Ren, S. H. Lapidus, K. Page, H. Zheng, J. W. Freeland, J. D. Budai and R. P. Hermann, Chem. Mater., 2019, 31, 3705–3711 CrossRef CAS.
- M. P. Jimenez-Segura, T. Takayama, D. Bérardan, A. Hoser, M. Reehuis, H. Takagi and N. Dragoe, Appl. Phys. Lett., 2019, 114, 122401 CrossRef.
- A. Mao, H.-Z. Xiang, Z.-G. Zhang, K. Kuramoto, H. Zhang and Y. Jia, J. Magn. Magn. Mater., 2020, 497, 165884 CrossRef CAS.
- A. Mao, H.-Z. Xiang, Z.-G. Zhang, K. Kuramoto, H. Yu and S. Ran, J. Magn. Magn. Mater., 2019, 484, 245–252 CrossRef CAS.
- R. Witte, A. Sarkar, L. Velasco, R. Kruk, R. A. Brand, B. Eggert, K. Ollefs, E. Weschke, H. Wende and H. Hahn, J. Appl. Phys., 2020, 127, 185109 CrossRef CAS.
- P. B. Meisenheimer, L. D. Williams, S. H. Sung, J. Gim, P. Shafer, G. N. Kotsonis, J. P. Maria, M. Trassin, R. Hovden, E. Kioupakis and J. T. Heron, Phys. Rev. Mater., 2019, 3, 1–9 Search PubMed.
- D. Bérardan, S. Franger, A. K. Meena and N. Dragoe, J. Mater. Chem. A, 2016, 4, 9536–9541 RSC.
- N. Osenciat, D. Bérardan, D. Dragoe, B. Léridon, S. Holé, A. K. Meena, S. Franger and N. Dragoe, J. Am. Ceram. Soc., 2019, 102, 6156–6162 CrossRef CAS.
- P. A. Krawczyk, M. Jurczyszyn, J. Pawlak, W. Salamon, P. Baran, A. Kmita, Ł. Gondek, M. Sikora, C. Kapusta, T. Strączek, J. Wyrwa and A. Żywczak, ACS Appl. Electron. Mater., 2020, 2, 3211–3220 CrossRef CAS.
- J. L. Braun, C. M. Rost, M. Lim, A. Giri, D. H. Olson, G. N. Kotsonis, G. Stan, D. W. Brenner, J.-P. Maria and P. E. Hopkins, Adv. Mater., 2018, 30, 1805004 CrossRef.
- W. Hong, F. Chen, Q. Shen, Y. Han, W. G. Fahrenholtz and L. Zhang, J. Am. Ceram. Soc., 2019, 102, 2228–2237 CAS.
- R. Banerjee, S. Chatterjee, M. Ranjan, T. Bhattacharya, S. Mukherjee, S. S. Jana, A. Dwivedi and T. Maiti, ACS Sustainable Chem. Eng., 2020, 8, 17022–17032 CrossRef CAS.
- H. Chen, J. Fu, P. Zhang, H. Peng, C. W. Abney, K. Jie, X. Liu, M. Chi and S. Dai, J. Mater. Chem. A, 2018, 6, 11129–11133 RSC.
- H. Chen, W. Lin, Z. Zhang, K. Jie, D. R. Mullins, X. Sang, S.-Z. Yang, C. J. Jafta, C. A. Bridges, X. Hu, R. R. Unocic, J. Fu, P. Zhang and S. Dai, ACS Mater. Lett., 2019, 1, 83–88 CrossRef CAS.
- S. J. McCormack and A. Navrotsky, Acta Mater., 2021, 202, 1–21 CrossRef CAS.
- P. B. Meisenheimer and J. T. Heron, MRS Adv., 2020, 1–18 Search PubMed.
- A. Sarkar, B. Breitung and H. Hahn, Scr. Mater., 2020, 187, 43–48 CrossRef CAS.
- N. Dragoe and D. Bérardan, Science, 2019, 366, 573–574 CrossRef CAS.
- J. M. D. Coey, M. Venkatesan and H. Xu, in Functional Metal Oxides: New Science and Novel Applications, ed. S. B. Ogale, T. V. Venkatesan and M. G. Blamire, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013, pp. 221–244 Search PubMed.
- J. M. D. Coey, Magnetism and magnetic materials, 2010 Search PubMed.
- P. W. Anderson, Phys. Rev., 1950, 79, 350–356 CrossRef.
- P. W. Anderson and H. Hasegawa, Phys. Rev., 1955, 100, 675–681 CrossRef CAS.
- C. Zener, Phys. Rev., 1954, 96, 1335–1337 CrossRef CAS.
- T. Moriya, Phys. Rev., 1960, 120, 91–98 CrossRef CAS.
- I. Dzyaloshinsky, J. Phys. Chem. Solids, 1958, 4, 241–255 CrossRef CAS.
- M. Fracchia, P. Ghigna, T. Pozzi, U. Anselmi-Tamburini, V. Colombo, L. Braglia and P. Torelli, J. Phys. Chem. Lett., 2020, 11, 3589–3593 CrossRef CAS.
- C. M. Rost, Z. Rak, D. W. Brenner and J.-P. Maria, J. Am. Ceram. Soc., 2017, 100, 2732–2738 CrossRef CAS.
- Z. Rak, C. M. Rost, M. Lim, P. Sarker, C. Toher, S. Curtarolo, J. P. Maria and D. W. Brenner, J. Appl. Phys., 2016, 120, 1–3 CrossRef.
- Z. Rák, J.-P. Maria and D. W. Brenner, Mater. Lett., 2018, 217, 300–303 CrossRef.
- M. R. Chellali, A. Sarkar, S. H. Nandam, S. S. Bhattacharya, B. Breitung, H. Hahn and L. Velasco, Scr. Mater., 2019, 166, 58–63 CrossRef CAS.
- Z. Rák and D. W. Brenner, J. Appl. Phys., 2020, 127, 185108 CrossRef.
- Y.-H. Lin, J. Wang, J. Cai, M. Ying, R. Zhao, M. Li and C.-W. Nan, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 193308 CrossRef.
- J. Nogués and I. K. Schuller, J. Magn. Magn. Mater., 1999, 192, 203–232 CrossRef.
- A. V. Saghir, S. M. Beidokhti, J. V. Khaki and A. Salimi, One-step synthesis of single-phase (Co, Mg, Ni, Cu, Zn) O High entropy oxide nanoparticles through SCS procedure: Thermodynamics and experimental evaluation, J. Eur. Ceram. Soc., 2021, 41, 563–579 CrossRef CAS.
- A. D. Dupuy, X. Wang and J. M. Schoenung, Mater. Res. Lett., 2019, 7, 60–67 CrossRef CAS.
- N. J. Usharani, R. Shringi, H. Sanghavi, S. Subramanian and S. S. Bhattacharya, Dalton Trans., 2020, 49, 7123–7132 RSC.
- Q. Wang, A. Sarkar, D. Wang, L. Velasco, R. Azmi, S. S. Bhattacharya, T. Bergfeldt, A. Düvel, P. Heitjans, T. Brezesinski, H. Hahn and B. Breitung, Energy Environ. Sci., 2019, 12, 2433–2442 RSC.
- Z. Lun, B. Ouyang, D. H. Kwon, Y. Ha, E. E. Foley, T. Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clément, H. Ji and G. Ceder, Nat. Mater. DOI:10.1038/s41563-020-00816-0.
- S. Dasgupta, B. Das, M. Knapp, R. A. Brand, H. Ehrenberg, R. Kruk and H. Hahn, Adv. Mater., 2014, 26, 4639–4644 CrossRef CAS.
- S. Dasgupta, B. Das, Q. Li, D. Wang, T. T. Baby, S. Indris, M. Knapp, H. Ehrenberg, K. Fink, R. Kruk and H. Hahn, Adv. Funct. Mater., 2016, 26, 7507–7515 CrossRef CAS.
- A. Molinari, H. Hahn and R. Kruk, Adv. Mater., 2019, 31, 1806662 CrossRef.
- R. Singh, R. Witte, X. Mu, T. Brezesinski, H. Hahn, R. Kruk and B. Breitung, J. Mater. Chem. A, 2019, 7, 24005–24011 RSC.
| This journal is © The Royal Society of Chemistry 2021 |