
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and biological activity of bis[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes†
Caroline Marie
Gallati‡
a,
Sina Katharina
Goetzfried‡
 a,
Anna
Ortmeier
abc,
Jessica
Sagasser
a,
Klaus
Wurst
d,
Martin
Hermann
e,
Daniel
Baecker
ab,
Brigitte
Kircher
bc and
Ronald
Gust
*a
a,
Anna
Ortmeier
abc,
Jessica
Sagasser
a,
Klaus
Wurst
d,
Martin
Hermann
e,
Daniel
Baecker
ab,
Brigitte
Kircher
bc and
Ronald
Gust
*a
aDepartment of Pharmaceutical Chemistry, Institute of Pharmacy, Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria. E-mail: ronald.gust@uibk.ac.at
bTyrolean Cancer Research Institute, Innrain 66, 6020 Innsbruck, Austria
cImmunobiology and Stem Cell Laboratory, Department of Internal Medicine V (Hematology and Oncology), Medical University Innsbruck, Anichstraße 35, 6020 Innsbruck, Austria
dInstitute for General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria
eDepartment of Anesthesiology and Critical Care Medicine, Medical University Innsbruck, Anichstraße 35, 6020 Innsbruck, Austria
First published on 8th February 2021
Abstract
A series of bis[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes (2a–f) containing methyl, fluoro or methoxy substituents at various positions in the 4-aryl ring was synthesized and evaluated for their anti-cancer properties in A2780 (wild-type and Cisplatin-resistant) ovarian carcinoma as well as LAMA 84 (imatinib-sensitive and -resistant) and HL-60 leukemia cell lines. The bis-NHC gold(I) complexes were more active compared to their related mono-NHC gold(I) analogues and reduced proliferation and metabolic activity in a low micromolar range. With the exception of 2d (3-F), the compounds displayed higher potency than the established drugs Auranofin and Cisplatin. The lack of effects against non-cancerous lung fibroblast SV-80 cells indicated a high selectivity towards tumor cells. All tested complexes generated reactive oxygen species in A2780cis cells; however, the induction of apoptosis was very low. Furthermore, thioredoxin reductase is not the main target of these complexes, because its inhibition pattern did not correlate with their biological activity.
Introduction
The successful application of Cisplatin in tumor therapy increased the interest in metal–organic compounds.1–5 Gold complexes came into the focus for drug design, as Auranofin, originally developed for the treatment of chronic polyarthritis, displayed strong anti-proliferative and cytotoxic effects in vitro and in vivo.6,7 These positive results led to the investigation of gold complexes as anti-tumor agents. Examples are presented in ref. 8–10. Analogous to Auranofin, many phosphines were chosen as ligands, due to their strong and stable bonding to the gold centre. The respective phosphine gold(I) complexes exhibited in part outstanding anti-cancer properties, but they also induced severe side effects, affecting the heart, liver and lung.11–13Considerable progress in the research on gold-based drugs has been made since the isolation of N-heterocyclic carbenes (NHCs) by Arduengo and colleagues.14 Bulky substituents at the N1 and N3 of the used 1,3-dihydro-2H-imdazol-2-ylidene were exploited to prevent dimerization.15 This discovery offered the possibility of synthesizing novel NHCs, which are suitable as ligands for coordination to metals.
The NHC–metal bond is regarded as more stable than those formed by phosphines.16–18 The high stability is achieved by three contributions, including the σ → d and π → d donation of the NHC to the metal as well as the d → π* metal to NHC back-donation.
NHC gold(I) complexes showed potential anti-tumor activity, whereby in most cases bis-NHC derivatives ([(NHC)2AuI]+) were more effective than their related mono-NHC analogues (e.g., (NHC)AuIX; X = Cl, Br, I, thiols).19–32 Many bis-NHC gold(I) complexes displayed a remarkable cytotoxicity in low micromolar or even nanomolar ranges (see e.g., ref. 33–36).
Thioredoxin reductase (TrxR) is discussed as a possible target for (NHC)AuIX complexes. Gold(I) complexes generally possess high affinity to Se- and S-containing proteins.31 Therefore, besides mono-NHC gold(I) complexes, their [(NHC)2AuI]+ derivatives act as strong TrxR inhibitors, too.18,37–42 However, also other modes of action are proposed for bis-NHC gold(I) species, e.g., interference with the mitochondrial permeability transition or the mitochondrial apoptotic pathway. After accumulation of [(NHC)2AuI]+ within the mitochondria, a selective induction of apoptosis occurs, mediated by a two-stage ligand exchange reaction with cysteine/selenocysteine.41,42 Furthermore, multimodal mechanisms of action such as alterations in the nucleolus on telomeres and actin stress fibers have been described.43,44
In a previous study, we reported on the synthesis and biological activity of bromido[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes. The compounds inhibited the growth and proliferation of A2780/A2780cis and HL-60 cells in the micromolar range.45 However, in aqueous solutions a ligand scrambling reaction to the related [(NHC)2AuI]+ species was noticed.46 Because these degradation products might participate in the biological activity of (NHC)AuIBr complexes, we synthesized a series of bis[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) derivatives bearing methyl, fluoro or methoxy substituents at position 4 of the 4-aryl ring (Scheme 1) and evaluated their anti-cancer effects using ovarian carcinoma and leukemia cell lines. The influence of the substitution pattern of the 4-aryl residue was exemplarily studied on 4/3-fluoro and 4/2-methoxy derivatives, which were easily available using conventional synthesis approaches.
 | ||
| Scheme 1 Synthesis pathway for complexes 2a–f. Method A: (1) compounds 1a–c, e, f (2.0 eq.), Ag2O (0.7 eq.), rt, dark, o/n, anhydrous DCM/MeOH (1/1). (2) LiBr (1.6 eq.), Me2SAuCl (1.0 eq.), rt, dark, 8 h. Method B: 3d (1.0 eq.), 1d (1.2 eq.), K2CO3 (2.0 eq.), rt, dark, 24 h, anhydrous acetone. | ||
Results and discussion
Chemistry
The formation of the ligands 1a–f followed the same multistep procedure as that published previously.45Two synthesis routes were applied to obtain the bis-NHC gold(I) complexes 2a–f (Scheme 1). The first one (Method A) followed the synthesis of (NHC)AuIX complexes, but using a 2-fold excess of the ligand.27 The coordination of the ligands 1a–c, e, f to gold(I) was achieved in a consecutive reaction with Ag2O and subsequent transmetalation with Me2SAuCl. The excess of ligand guaranteed the nearly quantitative formation of the bis-NHC gold(I) species 2a–c, e, f. To obtain the 3-F derivative 2d in satisfying yield, a second procedure (Method B) was performed, in which the (NHC)AuIBr complex 3d was reacted with the ligand 1d in the presence of K2CO3. The final complexes were purified by column chromatography (DCM/EtOAc (1/1)).
For the characterization of 2a–f, 1H NMR, 13C NMR and UV-Vis spectroscopy as well as electrospray ionisation mass spectrometry (ESI-MS) were employed.
Unfortunately, the chemical shift of the signals in the 1H NMR spectra of the bis-NHC gold(I) complexes (Fig. S1–S6, ESI†) did not differ from those of the mono-NHC gold(I) complexes.45 However, in the 13C NMR spectra, the signal of the C2 atom changed upon coordination of a second NHC from about 176 ppm ((NHC)AuIBr) to approximately 183 ppm (Fig. S7–S12, ESI†), indicating the formation of bis-NHC gold(I) compounds.
The UV-Vis spectra of the ligands altered as a consequence of coordination to gold(I), too (Fig. S13, ESI†). The free ligand 1e, for instance, showed in its spectrum (Fig. S13A, ESI†) an absorption at 233 nm, which was shifted in 3e to 226 nm. Strong absorptions at 254 nm (3e, Fig. S13B, ESI†) and 275 nm (2e, Fig. S13C, ESI†) characterized the (NHC)AuIBr and [(NHC)2AuI]+ complexes, respectively. The maxima at 226 nm and 233 nm can be assigned to the π→π* transition of the NHC ligand, while that at 254 nm and 276 nm correspond to the metal to ligand charge transfer.47
ESI-MS (Fig. S14–S19, ESI†) further confirmed the identity of the complexes.
HPLC (Fig. S20–S25, ESI†) documented a purity of >99%. (NHC)AuIBr species possessed retention times of tret = 5.80–6.02 min, if an RP18 column and an acetonitrile/water (0.1% TFA) eluent with a gradient from 70 to 90% acetonitrile was used. The corresponding [(NHC)2AuI]+ complexes were well separated with tret = 6.63–8.52 min.
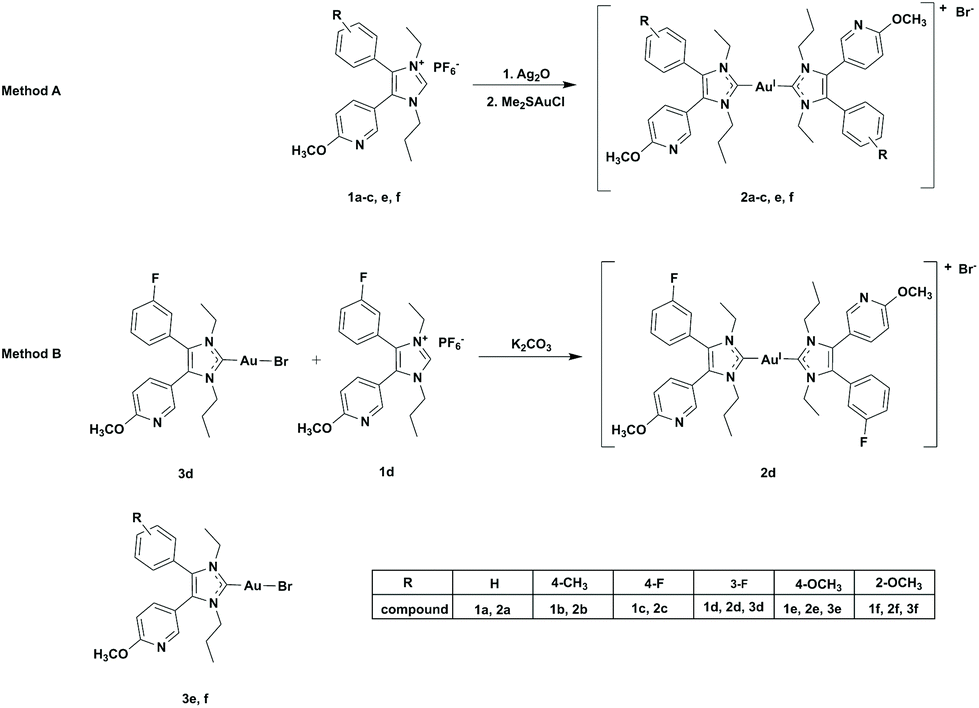
The structure of the bis-NHC gold(I) complexes was closely investigated using the example of 2a. It was possible to solve its X-ray crystal structure. Selected data are summarized in Table 1 and Fig. 1 shows the molecular (ORTEP) plot. 2a is a linear complex with conversely arranged ligands. The distance between the respective carbene carbons and the gold(I) centre amounts to 2.03 Å, very similar to that of the related mono-NHC gold(I) complexes (e.g., 3f: 2.00 Å).45
 | ||
| Fig. 1 Molecular (ORTEP) plot of 2a. | ||
| Crystal system | Monoclinic |
| Space group | C2/c (no. 15) |
| Unit cell dimensions | a = 15.3982(3) Å, α = 90° |
| b = 9.2903(3) Å, β = 94.218(2)° | |
| c = 29.2782(8) Å, γ = 90° | |
| Volume | 4177.02(19) Å3 |
| Z | 4 |
| Density (calculated) | 1.941 mg m−3 |
| Absorption coefficient | 5.780 mm−1 |
| F(000) | 2328 |
| Crystal size | 0.070 × 0.050 × 0.030 mm3 |
| Θ range for data collection | 2.562 to 24.996° |
| Index ranges | −18 ≤ h ≤ 18, −11 ≤ k ≤ 11, −34 ≤ l ≤ 34 |
| Reflections collected | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 106 106 |
| Independent reflections | 3624 [R(int) = 0.0640] |
| Absorption correction | None |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3624/0/238 |
| Goodness-of-fit on F2 | 1.021 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0358, wR2 = 0.0563 |
| R indices (all data) | R 1 = 0.0695, wR2 = 0.0624 |
| Largest diff. peak and hole | 0.689 and −0.569 e Å−3 |
Biological activity
The influence of the bis-NHC gold(I) complexes 2a–f on the proliferation and metabolic activity was evaluated using the Cisplatin-sensitive and -resistant ovarian carcinoma cell lines A2780 and A2780cis, as well as the leukemic cell lines LAMA-84 (STI-sensitive and -resistant) and HL-60. Moreover, the activation of caspases 3/7 as an indication of apoptotic processes was exemplarily analyzed in A2780cis cells. The complexes were further examined regarding their ability to induce the generation of reactive oxygen species (ROS) and their potential to inactivate TrxR.Determination of anti-proliferative effects
The impact of the compounds on the proliferation was investigated based on the incorporation of the radioactive-labelled nucleoside [3H]-thymidine into DNA during the S-phase of the cell cycle.As it has already been reported, the ligands displayed scarcely any anti-proliferative activity.45
Auranofin and/or Cisplatin were used in each experiment as references. Both compounds diminished the proliferation of A2780 cells at 1 μM to about 4%, but reduced the growth of A2780cis cells only to 50–60% (Fig. 2).
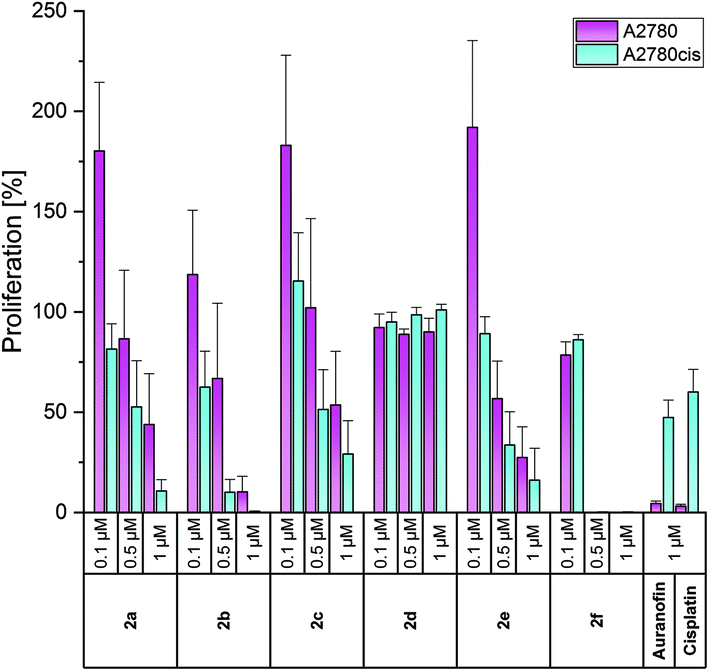 | ||
| Fig. 2 Anti-proliferative effects of 2a–f at concentrations of 0.1, 0.5 and 1 μM in A2780 (purple) and A2780cis (blue) cells, determined after incubation for 72 h. Auranofin and Cisplatin (1 μM) served as references. Proliferation in the absence of the compounds was set at 100%. The mean proliferation + standard error was calculated from three independent experiments. | ||
The complexes 2a–e were less active in Cisplatin-sensitive A2780 cells at 1 μM and even enhanced the proliferation at the lowest concentration tested (0.1 μM) in cases of 2a–c, e. In contrast, in A2780cis cells, all compounds, except for 2d, demonstrated high activity at 0.5 and 1 μM. These concentrations are about 10-fold lower than those of the respective (NHC)AuIBr species required to achieve the same effects. The complexes 2a and 2b almost blocked the proliferation of A2780cis cells at 1 μM. Even at 0.5 μM, 2b caused a reduction to <10%. The 4-OCH3-substituted complex 2e was marginally less active (1 μM: 16%; 0.5 μM: 33%). Weaker anti-proliferative effects were observed for the 4-F derivative 2c (1 μM: 29%; 0.5 μM: 51%).
The shift of the 4-F and 4-OCH3 substituents led to contrary results. The 3-F derivative 2d was entirely inactive at all concentrations used, whereas 2f (2-OCH3 derivative) represented the most active complex and inhibited the proliferation of A2780cis cells already at 0.5 μM to 0.29% (Fig. 2).
To test if the complexes were able to overcome also other drug resistances, their anti-proliferative effects were analyzed against chronic myeloid leukemia LAMA-84 cells, sensitive and resistant to the tyrosine kinase inhibitor imatinib mesylate (STI), a drug, which revolutionized the treatment of this disease.48
Auranofin was inactive on the LAMA-84 STI-resistant cell line and reduced the proliferation of STI-sensitive cells at 1 μM only to approximately 50% (Fig. 3).
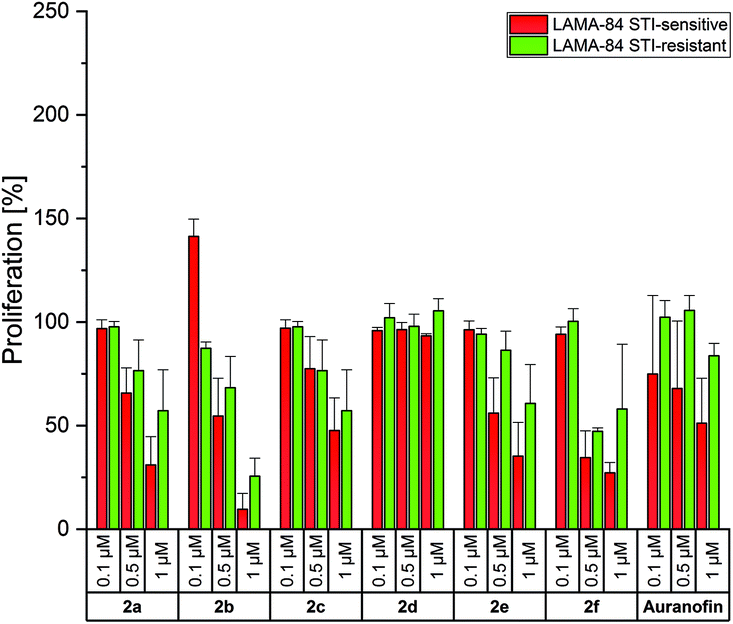 | ||
| Fig. 3 Anti-proliferative effects of 2a–f at concentrations of 0.1, 0.5 and 1 μM in LAMA-84 STI-sensitive (red) and LAMA-84 STI-resistant (green) cells, determined after incubation for 72 h. Auranofin served as a reference. Proliferation in the absence of the compounds was set at 100%. The mean proliferation + standard error was calculated from three independent experiments. | ||
In general, the bis-NHC gold(I) complexes were less effective in LAMA-84 cells in relation to A2780 cells. A strong inhibition of proliferation was caused only by 2b (4-CH3) at 1 μM (LAMA-84 STI-sensitive: 10%; LAMA-84 STI-resistant: 25%). The positive results of 2f, as discussed above, were not confirmed on these cell lines (proliferation at 1 μM, LAMA-84 STI-sensitive: 27%; LAMA-84 STI-resistant: 58%). Therefore, it can be assumed that the circumvention of resistance in A2780 cells with 2a–f followed a specific mode of action, which has to be elucidated further.
The activity of the bis-NHC gold(I) complexes was also analyzed in HL-60 cells (Fig. 4). Auranofin did not influence these cells at concentrations ≤1 μM. In contrast, 2a–c reduced the proliferation at 0.1 μM to about 52–66%. The 2-OCH3-substituted complex 2f was again the most active one at 0.5 and 1 μM (inhibition to 8% and 2%, respectively). Its 4-OCH3 derivative 2e interfered with the proliferation only at 1 μM (proliferation: 34%).
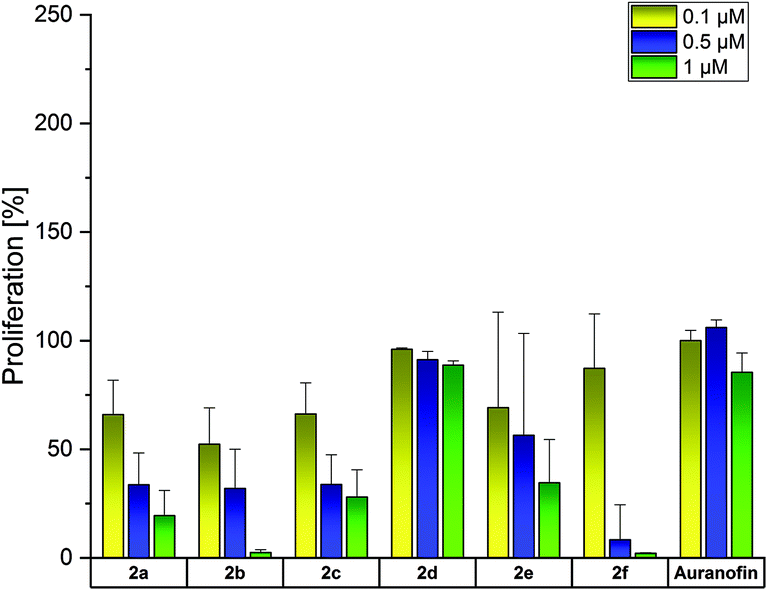 | ||
| Fig. 4 Anti-proliferative effects of 2a–f at concentrations of 0.1, 0.5 and 1 μM in HL-60 cells, determined after incubation for 72 h. Auranofin served as a reference. Proliferation in the absence of the compounds was set at 100%. The mean proliferation + standard error was calculated from three independent experiments. | ||
For a better evaluation of the anti-proliferative effects, IC50 values were calculated on the basis of the data presented in Fig. 2–4. From Table 2 it can be deduced that especially the Cisplatin-resistant A2780cis cell line and HL-60 cells were highly sensitive to the complexes. Based on the above-mentioned results, the following descending order of effectiveness can be obtained for the 4-substituted derivatives: 2b (4-CH3) > 2a (H) > 2e (4-OCH3) > 2c (4-F). The complexes were active with IC50 values <1 μM. Interestingly, the shift of the 4-OCH3 substituent (2e) to position 2 (2f) further increased the activity, while the change of the fluorine substituent (4-F → 3-F) strongly suppressed the anti-proliferative capacity. The 3-F-substituted complex 2d reduced the proliferation only at concentrations >1 μM. This finding contradicts that of the related (NHC)AuIBr complexes. The mono-NHC gold(I) complexes were only effective at concentrations >5 μM and the substituents enhanced the efficacy in the order 4-OCH3 > 4-F > 4-CH3 > H. The shift of the substituents in the 3/2 position of the 4-aryl ring led in each case to an increase of potency.45
| Compound | A2780 | A2780cis | LAMA-84 STI-sensitive | LAMA-84 STI-resistant | HL-60 |
|---|---|---|---|---|---|
| 2a | 0.93 | 0.53 | 0.67 | >1 | 0.21 |
| 2b | 0.65 | 0.16 | 0.55 | 0.73 | 0.12 |
| 2c | >1 | 0.51 | 0.98 | >1 | 0.21 |
| 2d | >1 | >1 | >1 | >1 | >1 |
| 2e | 0.53 | 0.26 | 0.63 | >1 | 0.58 |
| 2f | 0.19 | 0.21 | 0.44 | 0.98 | 0.23 |
In a recently published study, the stability of the 4-OCH3-substituted (NHC)AuIBr complex 3e was investigated. The complex partially degraded in aqueous acetonitrile/water mixtures within 72 h to the [(NHC)2AuI]+ complex 2e and its gold(III) derivative [(NHC)2AuIIIBr2]+.46 It was postulated that this reaction also occurs under cell-culture conditions and that 2e is involved in the anti-proliferative capacity. Indeed, 2e was >10-fold more effective than 3e and might contribute to the effects of 3e. In contrast, 2a was one of the most potent bis-NHC gold(I) complexes in this study, while its mono-NHC derivative was completely inactive up to a concentration of 10 μM. These discrepancies require a more detailed assessment of the stability of (NHC)AuIX (X = Cl, Br, I) complexes under physiological conditions. Furthermore, it is necessary to know more about the effectiveness of [(NHC)2AuI]+ complexes in the presence of biomolecules. These studies are currently ongoing and will be part of forthcoming papers.
The complexes 2a, 2b, 2d and 2f were selected for investigations on the SV-80 lung fibroblast cell line to analyze the influence on the growth of non-cancerous cells (Fig. 5).
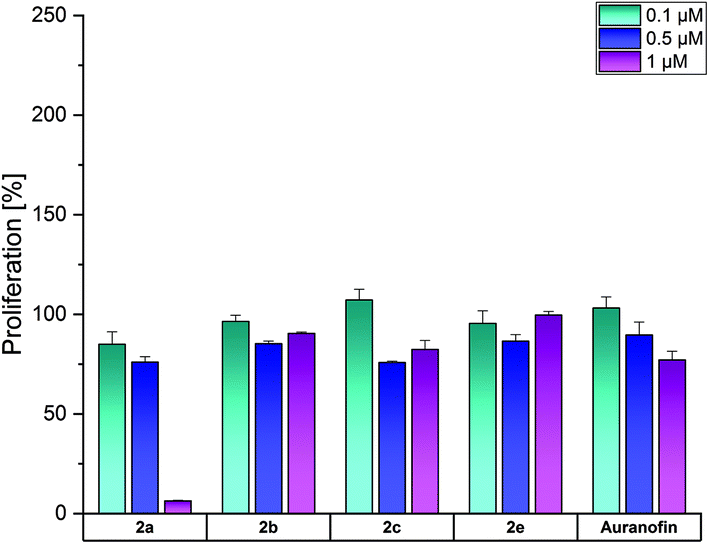 | ||
| Fig. 5 Anti-proliferative effects of 2a, 2b, 2d and 2f at concentrations of 0.1, 0.5 and 1 μM in SV-80 cells, determined after incubation for 72 h. Auranofin served as a reference. Proliferation in the absence of the compounds was set at 100%. The mean proliferation + standard error was calculated from three independent experiments. | ||
Among the complexes, only 2a and Auranofin reduced at 1 μM the proliferation of SV-80 cells to 6% and 77%, respectively. The other complexes did not affect the cell growth, indicating high tumor selectivity, as demonstrated in the examples of ovarian cancer and leukemic cell lines (especially HL-60).
Effect on metabolic activity
To evaluate if the anti-proliferative activity was also accompanied by cell death, the metabolic activity of the cells exposed to the complexes was measured after incubation for 72 h employing a modified MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay as previously adapted for the corresponding mono-NHC gold(I) complexes.45 In living cells, a bright yellow tetrazolium salt is transformed by cellular oxidoreductase enzymes to an insoluble orange formazan derivative, which is quantified after dissolution by photometric analysis and correlated with the metabolic activity and as a consequence with the viability of the cells.The effects on the metabolic activity were slightly weaker than those on the proliferation (Fig. 6 and S26–S27, ESI†), but show the same trend in all cell lines. Therefore, only the effects on A2780 and A2780cis cells are discussed below.
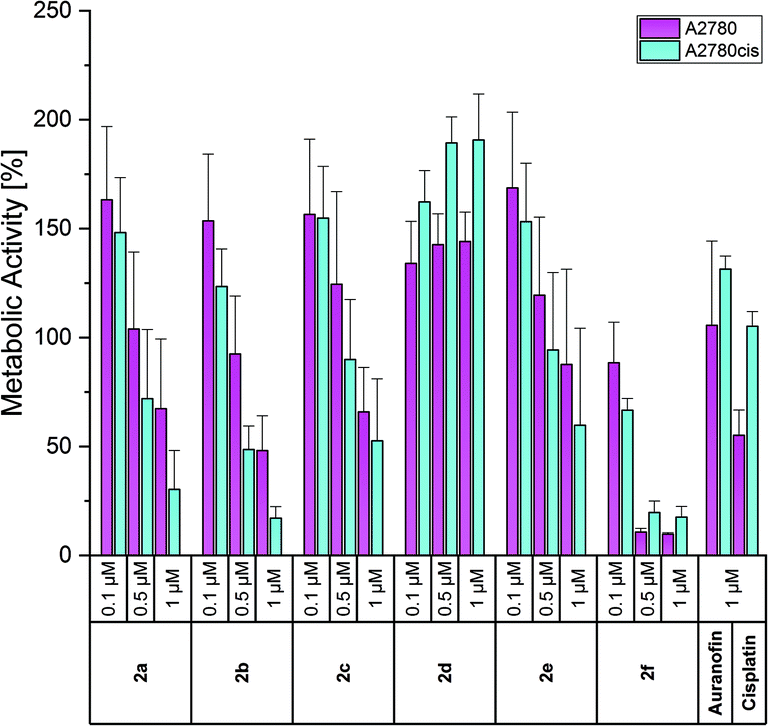 | ||
| Fig. 6 Anti-metabolic effects of 2a–f at concentrations of 0.1, 0.5 and 1 μM in A2780 (purple) and A2780cis (blue) cells, determined after incubation for 72 h. Auranofin and Cisplatin served as references. Metabolic activity in the absence of the compounds was set at 100%. The mean metabolic activity + standard error was calculated from three independent experiments. | ||
Auranofin was nearly inactive at a concentration of 1 μM, while Cisplatin inhibited the metabolic activity of A2780 cells to 55% (Fig. 6). In general, and in agreement with the proliferation data, all compounds stimulated at 0.1 μM the metabolic activity. As the most potent complex, 2f reduced at 0.5 and 1 μM the viability of wild-type and resistant cells to about 10% and 20%, respectively. Compounds 2a–c were slightly less effective, but indicated at 1 μM a better response of resistant cells (metabolic activity of A2780cis cells: <20%; A2780 cells: 25–50%). The 4-OCH3 derivative 2e caused lower effects than 2b (4-CH3) and 2c (4-F). Besides the proliferation, the 3-F isomer 2d also stimulated the metabolic activity of the cells (e.g., about 140% (A2780) and 190% (A2780cis) at 0.5 and 1 μM).
In this assay, too, the bis-NHC gold(I) complexes were more effective than their (NHC)AuIBr analogues. The latter influenced the metabolic activity only at concentrations >7.5 μM. The exception was the 4-OCH3-substituted complex 3e, which reduced the viability of A2780cis cells at 5 μM to 15%.45
It is worth mentioning that the metabolic activity of the non-cancerous lung fibroblast cell line SV-80 was not diminished by the compounds (Fig. S28, ESI†), which again confirmed their selectivity towards tumor cells.
Analysis of caspases 3/7 activity
Reduced metabolic activity may be an indicator of apoptosis induction. Therefore, A2780cis cells were exposed for 24 h to various concentrations (0.1, 0.5 and 1 μM) of compounds 2a–c, e and apoptosis induction was estimated by the activity of the effector caspases 3/749 in a luminescence assay. Only the unsubstituted 2a induced a slightly enhanced activity of caspases 3/7, however, solely at a concentration of 1 μM. These low effects and the high standard deviation suggest that induction of apoptosis is not a preferred mode of action exerted by this bis-NHC gold(I) series (Fig. 7).
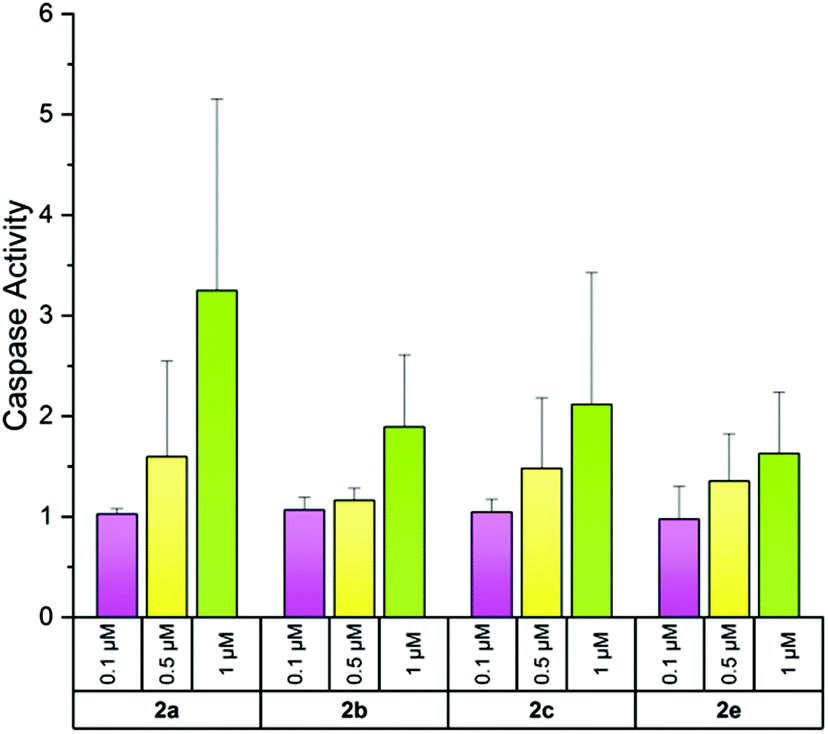 | ||
| Fig. 7 Induction of caspases 3/7 activity by the complexes 2a–c, e at 0.1, 0.5 and 1 μM after incubation for 24 h in A2780cis cells. Caspases 3/7 activity without compounds was set at 1. The mean activity + standard deviation was calculated from two independent experiments. | ||
Influence on the generation of ROS
It is well known that mitochondrial ROS (mROS) play an important role in cell-cycle progression. Low levels enhance cell proliferation and differentiation, while increased mROS, generated by external and internal factors, lead to cell death as a consequence of oxidative damage of lipids, proteins and DNA.50As gold(I) complexes can strongly affect the mROS content, the produced ROS in A2780cis cells were stained with MitoTrackerRed and analyzed with a confocal microscope according to a published procedure.51
All tested compounds (1 μM) induced the generation of mROS after 24 h (Fig. 8). Although quantification is not feasible, the obtained images indicate that 2c promoted the strongest oxidative stress. Only minimal production of mROS was observed upon incubation with 2b and Auranofin. Therefore, generation of ROS may be a general, not a specific, mode of action for these compounds.
 | ||
| Fig. 8 Formation of mROS in A2780cis cells after incubation for 24 h with the complexes 2a–c, e (1 μM). A negative control of untreated A2780cis cells and Auranofin served as references. | ||
Effects on the activity of thioredoxin reductase
The enzyme TrxR regulates the intracellular redox environment by reducing thioredoxin (Trx) and protects, e.g., proteins from oxidative aggregation and inactivation. It is well known that the binding tendency of Auranofin and gold complexes to the selenocysteine in the active site of the enzyme is high.52 Therefore, the complexes 2a–f (1 μM) were incubated with isolated TrxR and its activity was correlated with the ability to transform 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) to 5-thio-2-nitrobenzoic acid (TNB).Interestingly, the inhibition of TrxR by the complexes did not correspond to their anti-proliferative/anti-metabolic activity. The nearly inactive 2d (3-F) and the most cytotoxic 2f (2-OCH3) complexes revealed to be the most potent inhibitors of TrxR. Both compounds completely repressed the conversion of DTNB (Fig. 9). The complexes 2a (H) and 2e (4-OCH3) exhibited similar activity as Auranofin, while 2b (4-CH3) and 2c (4-F) showed the weakest inhibition of TrxR.
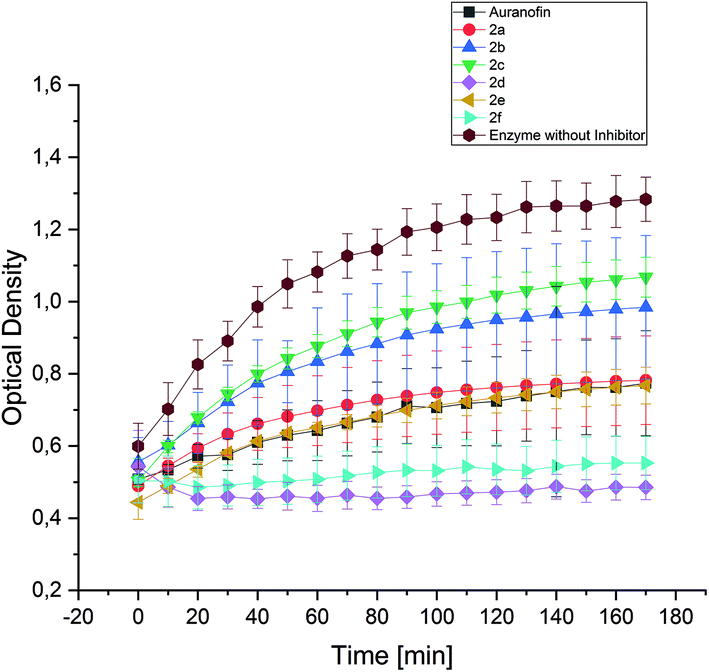 | ||
| Fig. 9 Inhibition of the TrxR-mediated conversion of 5,5′-dithiobis-(2-nitrobenzoic acid) to 5-thio-2-nitrobenzoic acid, quantified by UV measurement at 412 nm. The influence of 2a–f (1 μM) was observed over a period of 170 min. Auranofin served as a reference. The mean TrxR activity ± standard error was calculated from three independent experiments. | ||
The effects of 2c and 2e can be compared with that of their related (NHC)AuIBr analogues, both tested previously with the same assay set-up.452c and 2e showed a clearly lower potential to inhibit TrxR activity than related mono-NHC gold(I) complexes, which fully inactivated the enzyme. This finding can be attributed to the easier and faster substitution of the bromide ligand for selenium. This hypothesis, however, needs to be confirmed by reactivity studies, which are currently in progress in a further structure–activity relationship (SAR) study.
Based on these data, it appears that inhibiting TrxR is not the main mechanism of the anti-cancer action caused by the present bis-NHC gold(I) complexes.
Conclusion
A variety of bis[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes were synthesized, spectroscopically characterized and tested for their biological effects against ovarian cancer and leukemia cell lines. Compared to previously reported related (NHC)AuIBr complexes (active at concentrations >5 μM), representatives of this series inhibited the proliferation/metabolic activity of the cells already in a low micromolar range. They circumvented the Cisplatin resistance of A2780 ovarian cancer cells. For instance, complex 2b blocked at 1 μM the proliferation and reduced the metabolic activity of A2780cis cells to <10%. Apoptosis, however, may not be the cause of cell death, since only weak activity of caspases 3/7 was detected after 24 h.The highest effects of the complexes were observed on the HL-60 cell line (Table 2) as they diminished the proliferation to approximately the half already at a concentration of 0.1 μM. It should be highlighted that the complexes 2a–c, e did not influence non-cancerous lung fibroblast SV-80 cells at concentrations <1 μM, indicating selectivity towards tumor cells.
All compounds induced the generation of ROS, although to a varying degree, which did not correlate with the cell growth inhibitory effects. The impact on the activity of TrxR was investigated, too, as this enzyme is generally assumed to be the main target of anti-tumor active gold complexes. Indeed, 2d and 2f completely terminated the conversion mediated by the enzyme, while 2a and 2e displayed similar activity as Auranofin. 2b and 2c showed only marginal inhibition of TrxR. An accordance with their anti-proliferative/anti-metabolic potency is not given.
This SAR study clearly demonstrated that the biological effects of the bis-NHC gold(I) complexes depended on the substitution pattern of the 4-aryl ring. However, an evident correlation between the modality of substitution and the activity cannot be deduced. At this point, a clear statement about the role of the substituent cannot be given. However, it was found that the shift of the electron-donating OCH3 group from the 4- to the 2-position improved the efficiency of the compound. The ligand scrambling reaction has to be taken into account, as the bis-NHC gold(I) complexes displayed a different anti-cancer pattern from those of the respective mono-NHC gold(I) complexes. Therefore, additional intrinsic and extrinsic factors, e.g., substituent effects, incubation temperature and solvents, have to be examined to gain more insight into the efficacy of [(NHC)AuI]+ complexes.
Materials & methods
Chemical reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich, Fluka, Alfa Aesar and Acros) and used without further purification. Analytical thin layer chromatography on silica gel: Polygram® SIL G/UV254 (Macherey-Nagel) plastic backed plates (0.25 mm layer thickness) with a fluorescent indicator or Merck TLC silica gel 60 F254 aluminium backed plates. The spots were visualized using UV light (254 nm). Column chromatography: silica gel 60 (0.040–0.063 mm). Microwave assisted reactions: CEM Discover microwave (Software Synergy, CEM Corporation). NMR spectra: Bruker Avance 4 Neo operating at 400 MHz (1H NMR) and 100 MHz (13C NMR) (2 channels, rt BBFO probe) with a sample charger and Bruker Avance II+ (3 channels, liquid N2 cooled TCI Prodigy probe) systems. Deuterated solvents: Euriso-top®. Chemical shifts are given in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz). The centre of the solvent signal and the tetramethylsilane (TMS) signal served as the internal standard. Mass spectra: Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, USA) using direct infusion. HPLC experiments: Shimadzu prominence HPLC, equipped with a SIL-20A HT auto sampler, a CTO-10AS VP column oven, a DGU-20A degasser, a SPD-M20A detector and LC-20AD pumps a KNAUER 250 × 4 mm Eurospher 100–5 C18 column. The software for data processing was LabSolutions. Graphics were created with OriginPro 2018G (OriginLab Corporation, Northampton, MA, USA).Synthesis
1H NMR (400 MHz, CDCl3): δ = 0.90 (t, 6 H, J = 7.4 Hz), 1.39 (t, 6 H, J = 7.4 Hz), 1.82 (qt, 4 H, J = 7.4, 7.4 Hz), 3.92 (s, 6 H), 4.12–4.31 (m, 8 H), 6.77 (d, 2 H, J = 8.8 Hz), 7.23–7.29 (m, 4 H), 7.38–7.41 (m, 6 H), 7.52 (dd, 2 H, J = 8.8, 2.4 Hz), 8.02 (d, 2 H, J = 2.0 Hz). 13C NMR (100 MHz, CDCl3): δ = 11.3, 17.4, 25.3, 44.5, 50.8, 53.8, 111.5, 116.8, 127.2, 128.9, 129.1, 129.7, 130.7, 132.8, 140.7, 148.6, 164.6, 183.3. ESI-MS m/z: 839.3310 (M − Br)+, calculated: 839.3349.
1H NMR (400 MHz, CDCl3): δ = 0.89 (t, 6 H, J = 7.4 Hz), 1.38 (t, 6 H, J = 7.2 Hz), 1.81 (qt, 4 H, J = 7.4, 7.4 Hz), 3.81 (s, 3 H), 3.93 (s, 3 H), 4.10–4.28 (m, 4 H), 6.77 (dd, 1 H, J = 8.6, 0.6 Hz), 6.90 (d, 2 H, J = 8.8 Hz), 7.17 (d, 2 H, J = 9.0 Hz), 7.51 (dd, 1 H, J = 8.6, 2.4 Hz), 8.01 (dd, 1 H, J = 2.4, 0.6 Hz). 13C NMR (100 MHz, CDCl3): δ = 11.3, 17.4, 25.3, 44.3, 50.8, 53.8, 55.4, 111.5, 114.6, 116.9, 119.0, 128.8, 132.0, 132.7, 140.7, 148.6, 160.5, 164.5, 183.1. ESI-MS m/z: 867.3608 (M − Br)+, calculated: 867.3661.
1H NMR (400 MHz, CD3CN): δ = 0.89 (t, 6 H, J = 7.4 Hz), 1.38 (t, 6 H, J = 7.4 Hz), 1.81 (qt, 4 H, J = 7.4, 7.4 Hz), 3.93 (s, 6 H), 4.10–4.30 (m, 8 H), 6.78 (d, 2 H, J = 8.4 Hz), 7.10 (t, 4 H, J = 8.4 Hz), 7.24–7.31 (m, 4 H), 7.52 (dd, 2 H, J = 8.8, 2.6 Hz), 8.01 (d, 2 H, J = 2.0 Hz). 13C NMR (100 MHz, CD3CN): δ = 11.3, 17.3, 25.3, 44.4, 50.9, 53.8, 111.6, 116.5 (d, J = 21.8 Hz), 123.2 (d, J = 3.4 Hz), 129.2, 131.8, 132.7 (d, J = 8.7 Hz), 140.7, 148.6, 163.4 (d, J = 251.0 Hz), 164.6, 183.4. ESI-MS m/z: 875.3123 (M − Br)+, calculated: 875.3159.
1H NMR (400 MHz, CDCl3): δ = 0.91 (t, 6 H, J = 7.4 Hz), 1.43 (t, 6 H, J = 7.4 Hz), 1.79 (qt, 2 H, J = 7.4, 7.4 Hz), 3.93 (s, 6 H), 4.02–4.05 (m, 4 H), 4.15 (qt, 4 H, J = 7.4, 7.4 Hz), 6.79 (dd, 2 H, J = 8.8, 2.6 Hz), 7.10 (t, 1 H, J = 8.4 Hz), 7.12 (t, 1 H, J = 8.4 Hz), 7.39–7.43 (m, 4 H), 7.53 (dd, 2 H, J = 8.8, 2.6 Hz), 8.05 (d, 2 H, J = 2.0 Hz). 13C NMR (100 MHz, CDCl3): δ = 11.3, 17.4, 25.3, 44.6, 50.9, 53.8, 111.6, 117.5 (d, J = 68.3 Hz), 126.8 (d, J = 4.6 Hz), 129.2, 131.1 (d, J = 8.7 Hz), 131.4, 140.7, 148.6, 161.5, 163.9, 164.8, 183.7. ESI-MS m/z: 875.3123 (M − Br)+, calculated: 875.3159.
1H NMR (400 MHz, CDCl3): δ = 0.89 (t, 6 H, J = 7.4 Hz), 1.38 (t, 6 H, J = 7.2 Hz), 1.81 (qt, 4 H, J = 7.4, 7.4 Hz), 3.81 (s, 3 H), 3.93 (s, 3 H), 4.10–4.28 (m, 8 H), 6.77 (dd, 2 H, J = 8.6, 0.6 Hz), 6.90 (d, 4 H, J = 8.8 Hz), 7.17 (d, 4 H, J = 9.0 Hz), 7.51 (dd, 2 H, J = 8.6, 2.4 Hz), 8.01 (dd, 2 H, J = 2.4, 0.6 Hz). 13C NMR (100 MHz, CDCl3): δ = 11.3, 17.4, 25.3, 44.3, 50.8, 53.8, 55.4, 111.5, 114.6, 116.9, 119.0, 128.8, 132.0, 132.7, 140.7, 148.6, 160.5, 164.5, 183.1. ESI-MS m/z: 899.3539 (M − Br)+, calculated: 899.3559.
1H NMR: (400 MHz, CDCl3): δ = 0.89 (t, 6 H, J = 8.0 Hz), 1.35 (t, 6 H, J = 8.0 Hz), 1.81–1.89 (m, 4 H), 3.80 (s, 6 H), 3.91 (s, 6 H), 3.99–4.08 (m, 4 H), 4.13–4.19 (m, 4 H), 6.75 (d, 2 H, J = 8.4 Hz), 6.93–6.96 (m, 4 H), 7.10 (dd, 2 H, J = 7.7, 1.7 Hz), 7.39 (dt, 2 H, J = 8.4, 1.4 Hz), 7.51 (dd, 2 H, J = 8.5, 2.5 Hz), 7.99 (d, 2 H, J = 1.9 Hz). 13C NMR (100 MHz, CDCl3): δ = 11.3, 17.2, 25.4, 44.6, 50.8, 53.8, 55.6, 111.3 (twice), 115.8, 117.2, 121.1, 129.3, 132.0, 132.9, 140.7, 148.3, 158.2, 164.6, 183.2. ESI-MS m/z: 899.3539 (M − Br)+, calculated: 899.3559.
Crystallography
A Bruker D8 Quest Kappa diffractometer equipped with a Photon 100 detector was used to collect the single-crystal intensity data. Monochromatized MoKα radiation was generated using an Incoatec microfocus X-ray tube (50 kV/1 mA power settings) in combination with a multilayer optic. The crystallographic data of 2a were deposited as CCDC 1923123.†Cell lines
The acute myeloid leukemia HL-60 and the chronic myeloid leukemia LAMA-84 cell lines were purchased from DSMZ – German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany. The ovarian carcinoma cell lines A2780 and A2780cis (Cisplatin-resistant) were kindly provided by the Department of Gynecology, Medical University Innsbruck, and the non-cancerous lung fibroblast cell line SV-80 by the Department of Hematology, Medical University Innsbruck. All cell lines were grown in RPMI 1640 medium without phenol red (BioWhittaker, Lonza, Walkersville, MD, USA), supplemented with L-glutamine (2 mM), penicillin (100 U mL−1), streptomycin (100 μg mL−1) and fetal bovine serum (FBS; 10%; all from Invitrogen Corporation, Gibco, Paisley, Scotland) at 37 °C under a 5% CO2/95% air atmosphere and fed/passaged twice weekly. LAMA-84 cells have been made resistant to STI by treating with increasing concentrations of STI for several weeks. To maintain resistance, A2780cis and LAMA-84 STI-resistant cells were incubated every second week with Cisplatin (1 μM) and STI (1.5 μM), respectively.Analysis of metabolic activity and proliferation
Logarithmically growing cells were resuspended in cell-culture medium at a density of 1 × 106 cells per mL and plated in triplicate in U-bottomed or flat-bottomed microtiter plates (50 μL; Falcon, Becton Dickinson, Franklin Lakes, NJ, USA). Different concentrations of compounds were added in triplicate 2 h thereafter. After incubation for 72 h at 37 °C in a 5% CO2/95% air atmosphere, cultures were analyzed for metabolic activity using a modified MTT assay (EZ4U kit; Biomedica, Vienna, Austria) according to the manufacturer's instructions. For the analysis of cell proliferation each well was exposed to 2 μCi of [3H]-thymidine (2 Ci per mmol; Hartmann Analytic, Braunschweig, Germany) for the last 12–16 h of incubation. Then, the cells were harvested using a semi-automated device and [3H]-thymidine uptake into the cells expressed as counts per minute (cpm) was measured using a scintillation counter (MicroBeta TriLux, PerkinElmer, Waltham, MA, USA).The proliferation and the metabolic activity in the absence of the compounds were set at 100%.
Generation of mROS
A2780cis cells (2 × 104 cells per well) were grown on LabTek Chamber slides (Nalge Nunc International, Rochester, NY, USA) for 24 h at 37 °C under a 5% CO2/95% air atmosphere. Afterwards, complexes 2a–c, e and Auranofin, respectively, were added to achieve a concentration of 1 μM and the cells were incubated again for 24 h. Fifteen min prior to the visualization of mROS with a confocal microscope (Zeiss Axio Observer Z1), 5 μL of 1 M HEPES buffer (Biochrom, Berlin, Germany) and 2 μL of reduced MitoTrackerRed/H2XROS (200 nM; Introgen, Carlsbad, CA, USA) were added to each well.TrxR inhibition
All complexes were investigated for their potential to inhibit TrxR with a Thioredoxin Reductase Assay Kit (Sigma Aldrich Chemie GmbH, Steinheim, Germany). The assay was performed following a modified procedure as reported earlier.45 All compounds were evaluated at 1 μM on the isolated enzyme. The TrxR activity was determined by the reaction of DTNB in the presence of NADPH/H+ to two moles of TNB. After the addition of DTNB, the absorption of TNB was measured at 412 nm with a microplate reader (Enspire, PerkinElmer) every 5 min for 170 min.Caspases 3/7 assay
Exponentially growing cells were seeded in 96-well plates (2 × 104 cells per well, 50 μL per well) in triplicate. The compounds were added at appropriate concentrations after 2 h. Following cultivation for 24 h at 37 °C under a 5% CO2/95% air atmosphere, the supernatant was transferred into white 96-well plates and the induction of apoptosis was recorded by caspases 3/7 detection using the Caspase-Glo 3/7 kit (Promega, Madison, USA) according to the manufacturer's recommendations. Values were normalized to the luminescence values of the medium only. All values were calculated in relation to the caspases 3/7 activity of the untreated cells, which was set at 1.Abbreviations
| CDCl3 | Deuterated chloroform |
| CD3CN | Deuterated acetonitrile |
| CHCl3 | Chloroform |
| DCM | Dichloromethane |
| DTNB | 5,5′-Dithiobis-(2-nitrobenzoic acid) |
| eq. | Equivalents |
| EtOAc | Ethyl acetate |
| ESI-MS | Electrospray ionisation mass spectrometry |
| FBS | Fetal bovine serum |
| HPLC | High performance liquid chromatography |
| mROS | Mitochondrial ROS |
| MeOH | Methanol |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NHC | N-Heterocyclic carbene |
| NMR | Nuclear magnetic resonance |
| o/n | Overnight |
| ORTEP | Oak ridge thermal-ellipsoid plot |
| ROS | Reactive oxygen species |
| RPMI | Roswell park memorial institute |
| rt | Room temperature |
| SAR | Structure–activity relationship |
| STI | Imatinib mesylate |
| TFA | Trifluoroacetic acid |
| TNB | 5-Thio-2-nitrobenzoic acid |
| Trx | Thioredoxin |
| TrxR | Thioredoxin reductase |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Österreichische Forschungsförderungsgesellschaft FFG [West Austrian BioNMR 858017] is kindly acknowledged.References
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef PubMed.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef PubMed.
- M. Galanski, M. A. Jakupec and B. K. Keppler, Curr. Med. Chem., 2005, 12, 2075–2094 CrossRef PubMed.
- T. Boulikas and M. Vougiouka, Oncol. Rep., 2003, 10, 1663–1682 Search PubMed.
- A. Pasini and F. Zunino, Angew. Chem., Int. Ed. Engl., 1987, 26, 615–624 CrossRef.
- T. M. Simon, D. H. Kunishima, G. J. Vibert and A. Lorber, Cancer, 1979, 44, 1965–1975 CrossRef PubMed.
- C. K. Mirabelli, R. K. Johnson, C. M. Sung, L. Faucette, K. Muirhead and S. T. Crooke, Cancer Res., 1985, 45, 32–39 Search PubMed.
- M. Porchia, M. Pellei, M. Marinelli, F. Tisato, F. Del Bello and C. Santini, Eur. J. Med. Chem., 2018, 146, 709–746 CrossRef PubMed.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC.
- T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- G. D. Hoke, R. A. Macia, P. C. Meunier, P. J. Bugelski, C. K. Mirabelli, G. F. Rush and W. D. Matthews, Toxicol. Appl. Pharmacol., 1989, 100, 293–306 CrossRef PubMed.
- G. D. Hoke, G. E. Bossard, J. V. McArdle, B. D. Jensen, C. K. Mirabelli and G. F. Rush, J. Biol. Chem., 1988, 263, 11203–11210 CrossRef.
- P. J. Smith, G. D. Hoke, D. W. Alberts, P. J. Bugelski, S. Lupe, C. K. Mirabelli and G. F. Rush, J. Pharmacol. Exp. Ther., 1989, 249, 944–950 Search PubMed.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef.
- P. Pyykkö, J. Li and N. Runeberg, Chem. Phys. Lett., 1994, 218, 133–138 CrossRef.
- A. T. Biju, M. N. Hopkinson and F. Glorius, in N–Heterocyclic Carbenes in Organocatalysis, ed. A. T. Biju, Wiley-VCH, Weinheim, 2019, ch. 1, pp. 1–36 Search PubMed.
- J. A. Mata, A. R. Chianese, J. R. Miecznikowski, E. P. M. Poyatos, J. W. Faller and R. H. Crabtree, Organometallics, 2004, 23, 1253–1263 CrossRef.
- S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778–787 CrossRef PubMed.
- A. M. Al-Majid, S. Yousuf, M. I. Choudhary, F. Nahra and S. P. Nolan, ChemistrySelect, 2016, 1, 76–80 CrossRef.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708–3715 RSC.
- D. Krishnamurthy, M. R. Karver, E. Fiorillo, V. Orrú, S. M. Stanford, N. Bottini and A. M. Barrios, J. Med. Chem., 2008, 51, 4790–4795 CrossRef PubMed.
- W. Liu, K. Bensdorf, M. Proetto, U. Abram, A. Hagenbach and R. Gust, J. Med. Chem., 2011, 54, 8605–8615 CrossRef PubMed.
- W. Liu, K. Bensdorf, M. Proetto, A. Hagenbach, U. Abram and R. Gust, J. Med. Chem., 2012, 55, 3712–3724 Search PubMed.
- C. Schmidt, B. Karge, R. Misgeld, A. Prokop, R. Franke, M. Brönstrup and I. Ott, Chem. – Eur. J., 2017, 23, 1869–1880 CrossRef PubMed.
- C. Schmidt, L. Albrecht, S. Balasupramaniam, R. Misgeld, B. Karge, M. Brönstrup, A. Prokop, K. Baumann, S. Reichl and I. Ott, Metallomics, 2019, 11, 533–545 CrossRef PubMed.
- J. Weaver, S. Gaillard, C. Toye, S. Macpherson, S. P. Nolan and A. Riches, Chem. – Eur. J., 2011, 17, 6620–6624 CrossRef PubMed.
- L. Kaps, B. Biersack, H. Müller-Bunz, K. Mahal, J. Münzner, M. Tacke, T. Mueller and R. Schobert, J. Inorg. Biochem., 2012, 106, 52–58 CrossRef PubMed.
- B. Bertrand, E. Bodio, P. Richard, M. Picquet, P. Le Gendre and A. Casini, J. Organomet. Chem., 2015, 775, 124–129 CrossRef.
- J. K. Muenzner, B. Biersack, H. Kalie, I. C. Andronache, L. Kaps, D. Schuppan, F. Sasse and R. Schobert, ChemMedChem, 2014, 9, 1195–1204 CrossRef PubMed.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef PubMed.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef PubMed.
- C. H. G. Jakob, B. Dominelli, E. M. Hahn, T. O. Berghausen, T. Pinheiro, F. Marques, R. M. Reich, J. D. G. Correia and F. E. Kühn, Chem. – Asian J., 2020, 15, 2754–2762 CrossRef PubMed.
- S. Sen, Y. Li, V. Lynch, K. Arumugam, J. L. Sessler and J. F. Arambula, Chem. Commun., 2019, 55, 10627–10630 RSC.
- F. Binacchi, F. Guarra, D. Cirri, T. Marzo, A. Pratesi, L. Messori, C. Gabbiani and T. Biver, Molecules, 2020, 25, 5446 CrossRef PubMed.
- D. Curran, H. Müller-Bunz, S. I. Bär, R. Schobert, X. Zhu and M. Tacke, Molecules, 2020, 25, 3474 CrossRef PubMed.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625–5635 CrossRef.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef PubMed.
- J. F. Arambula, R. McCall, K. J. Sidoran, D. Magda, N. A. Mitchell, C. W. Bielawski, V. M. Lynch, J. P. Sessler and K. Arumugam, Chem. Sci., 2016, 7, 1245–1256 RSC.
- R. McCall, M. Miles, P. Lascuna, B. Burney, Z. Patel, K. J. Sidoran, V. Sittaramane, J. Kocerha, D. A. Grossie, J. L. Sessler, K. Arumugam and J. F. Arambula, Chem. Sci., 2017, 8, 5918–5929 RSC.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 96, 1642–1647 CrossRef PubMed.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef PubMed.
- B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. Le Gendre, E. Warmerdam, M. H. De Jager and G. M. Groothuis, Inorg. Chem., 2014, 53, 2296–2303 CrossRef PubMed.
- S. Meier-Menches, B. Neuditschko, K. Zappe, M. Schaier, M. Gerner, K. Schmetterer, G. Del Favero, R. Bonsignore, M. Cichna-Makl, G. Koellensperger, A. Casini and C. Gerner, Chem. – Eur. J., 2020, 26, 15528–15537 CrossRef PubMed.
- C. M. Gallati, S. K. Goetzfried, M. Ausserer, J. Sagasser, M. Plangger, K. Wurst, M. Hermann, D. Baecker, B. Kircher and R. Gust, Dalton Trans., 2020, 49, 5471–5481 RSC.
- S. K. Goetzfried, C. M. Gallati, M. Cziferszky, R. A. Talmazan, K. Wurst, K. R. Liedl, M. Podewitz and R. Gust, Inorg. Chem., 2020, 59, 15312–15323 CrossRef PubMed.
- L. Messori, L. Marchetti, L. Massai, F. Scaletti, A. Guerri, I. Landini, S. Nobili, G. Perrone, E. Mini, P. Leoni, M. Pasquali and C. Gabbiani, Inorg. Chem., 2014, 53, 2396–2403 CrossRef PubMed.
- L. Kalmanti, S. Saussele, M. Lauseker, M. C. Müller, C. T. Dietz, L. Heinrich, B. Hanfstein, U. Proetel, A. Fabarius, S. W. Krause, S. Rinaldetti, J. Dengler, C. Falge, E. Oppliger-Leibundgut, A. Burchert, A. Neubauer, L. Kanz, F. Stegelmann, M. Pfreundschuh, K. Spiekermann, C. Scheid, M. Pfirrmann, A. Hochhaus, J. Hasford and R. Hehlmann, Leukemia, 2015, 29, 1123–1132 CrossRef PubMed.
- E. A. Slee, M. T. Harte, R. M. Kluck, B. B. Wolf, C. A. Casiano, D. D. Newmayer, H.-G. Wang, J. C. Reed, D. W. Nicholson, E. S. Alnemri, D. R. Green and S. J. Martin, J. Cell Biol., 1999, 144, 281–292 CrossRef PubMed.
- H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef PubMed.
- J. Sagasser, B. N. Ma, D. Baecker, S. Salcher, M. Hermann, J. Lamprecht, S. Angerer, P. Obexer, B. Kircher and R. Gust, J. Med. Chem., 2019, 62, 8053–8061 CrossRef PubMed.
- F. Saccocccia, F. Angelucci, G. Boumis, D. Carotti, G. Desiato, A. Miele and A. Bellelli, Curr. Protein Pept. Sci., 2014, 15, 621–646 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR and ESI-MS spectra of 2a–f, the HPLC chromatograms of 2a–f, and the anti-metabolic activity of 2a–f against LAMA-84 (STI-sensitive and -resistant) cells, HL-60 cells and non-cancerous SV-80 cells. CCDC 1923123. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03902k |
| ‡ These authors contributed equally to this publication. |
| This journal is © The Royal Society of Chemistry 2021 |