
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of diol isomer/water mixtures on the stability of Zn-MOF-74†
Carmen
González-Galán
a,
Salvador R. G.
Balestra
 *ab,
Azahara
Luna-Triguero
ac,
Rafael Maria
Madero-Castro
a,
Ana Paula
Zaderenko
a and
Sofia
Calero
*ad
*ab,
Azahara
Luna-Triguero
ac,
Rafael Maria
Madero-Castro
a,
Ana Paula
Zaderenko
a and
Sofia
Calero
*ad
aDepartment of Physical, Chemical, and Natural Systems, Universidad Pablo de Olavide, Ctra. Utrera km 1, ES-41013 Seville, Spain
bInstituto de Ciencia de Materiales de Madrid (ICMM), Consejo Superior de Investigaciones Científicas (CSIC), Sor Juana Inés de la Cruz 3, 28039 Madrid, Spain. E-mail: s.rodriguez.gomez@csic.es
cEnergy Technology, Department of Mechanical Engineering, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
dMaterials Simulation and Modelling, Department of Applied Physics, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: S.Calero@tue.nl
First published on 22nd December 2020
Abstract
The stability of metal–organic frameworks is a key factor in many applications in some fields that require working under harsh conditions. It is known that a large number of MOFs are vulnerable to humid air. It means that when they are exposed to water, a structural collapse of the crystal happens. In this work, Molecular Dynamics simulations using a reactive force field have been performed to study the stability of MOF-74 against the adsorption of catechol, resorcinol and hydroquinone in the presence of water. We reproduced the water instability of Zn-MOF-74 and we studied the resistance of the structure. Our simulations showed that the three isomers generate a volume change in the framework but the structural collapse does not happen. In contrast, for water–isomer mixtures, there is structural collapse. Not only do catechol, resorcinol and hydroquinone not behave as stabilizing agents but they do enhance the hydration effect on the structure.
Introduction
Metal–organic frameworks (MOFs) are a class of porous materials formed by metallic centres (or clusters) and organic ligands that are connected by a tridimensional network. The crystalline nature, high porosity and surface area and pore surface properties, which are subject to modification, make them promising materials for several applications in fields such as catalysis,1 separation and purification,2,3 luminescence,4 molecular sensing,5 targeted drug delivery6 or gas storage,7 among others. In particular, MOF-74/CPO-27 materials [M2(dhbdc), M = Zn2+, Mg2+, Ni2+, Co2+, Fe2+, Cu2+, Mn2+, Ca2+, Sr2+, and dhbdc = 2,5-dihydrohybenzenedicarboxylic acid] are a member of the best studied family of MOFs characterized by having unsaturated metal centers.8 The presence of unsaturated metal centres provides the structure with some special characteristics. For instance, a consequence of these open metal sites is the enhanced affinity to small molecules such as CO2, CH4 or H2.9–11 However, the application in this field needs working under harsh conditions requiring high thermal stability and humidity resistance. Therefore, in many cases this causes some problems.The water stability of certain MOFs, including the MOF-74 family,12 has been well-documented for being a restricting factor for applications; for example, flue gas is always saturated with H2O.13,14 While in some separation processes there may be strategies to dehumidify the mixtures, in other cases it remains a challenge. MOF-74 is just one of the many families of MOFs with members that are unstable in water. Other examples are HKUST-1 and members of the IRMOF family such as IRMOF-1 (MOF-5) or IRMOF-10.15–18 Nevertheless, water instability is not a limitation for all MOFs. MIL-53, MIL-100, MIL-101 (including in the MIL series), UiO-66, UiO-67, and UiO-68 (including in the UiO series) and MOFs containing soft base azolate ligands and acid metal ions (ZIFs) are known to be stable upon water adsorption.19 While any of these materials is an excellent choice for separation processes in wet environments and ensures the stability of the material, understanding the degradation mechanism and the strategies that try to avoid it using an atomistic view is an important insight in the designing and preparation of water robust MOFs.
There are two possible mechanisms to explain water instability: (a) hydrolysis: ML + H2O → M(OH) + LH and (b) ligand displacement: ML + H2O → M(OH2) + L, where M and L correspond to the metal and the ligand, respectively.8 Some members of the MOF-74 family are known to catalyse the dissociation of water molecules into H+ and OH− groups at low temperature and pressure (H2O → H+ + OH−). These catalytic reaction products are responsible for the structural collapse of the framework. The mechanism is based on two steps: (i) the –OH group remains bonded to the metal centre and (ii) the H atom binds with an O atom of the linker. The dissociation and breakage depend on the reactivity of the metal centre (Zn > Mg > Ni > Co),20 and as the size decreases, the H atoms (one of the products of the dissociation of water) get closer to the O atoms of the organic ligand causing an increase in the interaction between them and facilitating the structural collapse.12 For these reasons, within the MOF-74 family, Zn-MOF-74 exhibits the highest catalytic activity towards the water dissociation and thus it is the most affected by water adsorption.
Zn-MOF-74 was the first framework reported in this family in 2005 and there have been many studies since then.21 It consists of a trigonal geometry, with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group which is constructed by infinite helical secondary building units (SBUs) and dhbdc linkers where each Zn atom is linked via five O atoms: three carboxyl and two hydroxyl groups. It has mono-dimensional channels and open-metal sites that are primary adsorption sites (Fig. S1†). In this framework, water dissociation is the key step in the final structural collapse which affects many applications like CO2 adsorption (which is reduced in the presence of water).22,23
space group which is constructed by infinite helical secondary building units (SBUs) and dhbdc linkers where each Zn atom is linked via five O atoms: three carboxyl and two hydroxyl groups. It has mono-dimensional channels and open-metal sites that are primary adsorption sites (Fig. S1†). In this framework, water dissociation is the key step in the final structural collapse which affects many applications like CO2 adsorption (which is reduced in the presence of water).22,23
There are different strategies to improve the stability against water: (a) changing the metal centre (for example, using zirconium instead of zinc), (b) installing protective groups to prevent the access of water molecules to the metal centre, or (c) using a molecule that reacts with water (for example, performing the adsorption of CO in the presence of water to form formic acid inside the framework or controlling the water dissociation mechanism by the addition of the noble gas He).8,24,25 Many studies related to the stability of MOFs against water using molecular simulation have been reported.17,26–28 Huang et al.26 carried out Molecular Dynamics simulation in order to study the water stability and the influence of the content of water on CO2 adsorption in a set of 25 MOFs. Greathouse et al.17 used a flexible force field to analyse the stability of MOF-5 against water and observed structural collapse when the content of water was higher than 4%. Bellarosa et al.27 studied the mechanism involved in the disruption of IRMOF-1 with 6% water, obtaining a strong relation between the coordination sphere of Zn and the basicity of water. De Toni et al.28 used ab initio simulations to study the hydration process in a model IRMOF material.
We have focused on the strategy consisting of installing protective groups by using three isomers: catechol (1,2-dihydroxybenzene), resorcinol (1,3-dihydroxybenzene) and hydroquinone (1,4-dihydroxybenzene). In the same way as water, these molecules interact with the structure through a polar interaction. However, their large size prevents them from getting close enough to the linker and breaking the framework. Besides, the interaction with the framework could be different depending on the isomer because the position of the OH group changes in each molecule (ortho-, meta- or para-). The presence of diol molecules in the micropore will preserve the hydrophilic function of the structure unlike other strategies,25 but will still attract water molecules. The knowledge of these phenomena is important to understand the crystalline structure stabilisation/destabilisation processes (the presence of protective groups) in the presence of a second component in the second coordination sphere (water).
Simulation details
We used a reactive force field to model the interatomic and intramolecular interactions in our calculations. A reactive force field is characterized by calculating the bond order and the electrostatic point charge values as well in every Molecular Dynamics (MD) iteration.29 The use of reactive fields for structural stability studies is fully supported by the literature, and the reliability of its predictions has been increasing in recent years as a versatile method developed to simulate bond breaking/formation in large systems.30–35 Here, we have used the ReaxFF force field as implemented in the LAMMPS code with the reax/c package. This calculation is not based on a connectivity input for the chemical bonds, but it only requires the instantaneous interatomic positions. In this force field, each element is related to one atom type. For every pair of atoms, to explain the changing connectivity, non-bonded interactions are computed. To take into account the creation/deletion of bonds, electric charges are recalculated in every step of the MD simulations by using the charge equilibration Qeq method as described by Rappé and Goddard and as implemented in the LAMMPS code.36 We have added to the ESI† the input file for the LAMMPS code to perform energy minimization using the ReaxFF package (code S2).The ReaxFF parameters used in this work were taken from Han et al.37,38 and report the water dissociation and breakage of the Zn–ligand bond in this MOF, in agreement with the experiments. The LAMMPS-formatted set of parameters can be found in the original work.38 This set of parameters also includes all the essential information needed to simulate the host–host, guest–guest and host–guest interactions in this work: the Zn-MOF-74 structure, the pure diols (catechol, resorcinol and hydroquinone), the molecules of water and the mixtures of water and diols. We also selected the three isomeric benzenediols because they can function as stabilizing agents against the structural collapse. To validate this force field, we calculated the bulk density of the three diols at room conditions using MD simulations in the isothermal–isobaric (NPT) ensemble. The results reasonably agree with the experimental values—i.e. catechol: 1.20 g cm−3 (exp. 1.34 g cm−3), resorcinol: 1.18 g cm−3 (exp. 1.28 g cm−3) and hydroquinone: 1.17 g cm−3 (exp. 1.30 g cm−3). The calculated value of the density of water is 0.9634 g cm−3 (exp. 0.9982 g cm−3).
To study the guest-induced structural changes of Zn-MOF-74, NPT MD simulations were carried out. We calculated the volume change of the cell as a function of the content of molecules (pure diols, water or mixtures) at 300 K and 1 bar. To maintain the temperature and pressure, a Berendsen thermostat and barostat were used, respectively. These simulations were performed using the LAMMPS code39 during 5000 ps with a time step of 0.001 ps. This simulation time is sufficient to study the structural stability of Zn-MOF-74 because the collapse of MOFs induced by hydrolysis reactions is observed within 200–500 ps. The lattice parameters (a, b, and c) were changed independently while the lattice angles (α, β, and γ) were fixed.
The initial configurations, formed by the structure and the molecules inside, were generated using Monte Carlo (MC) simulations in the Canonical ensemble (NVT) with the RASPA code.40 MC simulations in the Grand-Canonical ensemble (μVT) were used to obtain the saturation loading. In this ensemble, the number of molecules can fluctuate until equilibrium. Universal Force Field (UFF)41 for metals and Dreiding42 for organic ligands were used. At this point, we modelled Zn-MOF-74 as a rigid structure using a simulation box with a 1 × 1 × 4 supercell. The interaction between the molecules and the framework was measured by coulombic and Lennard–Jones potentials. We used Transferable Potentials for Phase Equilibria Force Field (TraPPE)43 to model our molecules and the SPC Ewald flexible model for the molecule of water.44,45 For molecules, we have used flexible molecules. Note that in the case of diols, the aromatic ring is considered as rigid while the –OH groups are flexible.
We also obtained the heat of adsorption at infinite dilution for each isomer by using the Widom Test Particle insertion method.46 To insert the molecules in the framework, we used the same method explained above.
We estimated the average number of hydrogen bonds per molecule (nHB) to study the interaction between diols. For this purpose, we used a geometrical criterion described by J. Martí et al.47 and J. A. Padró et al.48 that is based on three cutoffs: (i) the distance between the two atoms of oxygen, (ii) the distance between the atoms of hydrogen and oxygen that are in different molecules and (iii) the angle between the atoms of hydrogen and oxygen of the first molecule and the oxygen of the second molecule involved should be less than 30°. Fig. S3† shows the (i) and (ii) cutoffs obtained from the first minimum of the Radial Distribution Function (RDF) of the MD simulations of pure diols in the bulk (dOO = 3.74 Å and dOH = 2.66 Å).
The π-interaction between the molecules of the diol was also computed by using a homemade code. For that, we considered all the possible aromatic stacking arrangements (parallel face-centred, parallel offset, perpendicular t-shaped and perpendicular y-shaped).49 For the parallel arrangement, we used a geometrical criterion based on two cutoffs: (i) the distance between the two aromatic rings and (ii) the angle between the aromatic rings that should be between 0 and 30°. For the perpendicular arrangement, the geometrical criterion is also based on two cutoffs: (i) the distance between the two aromatic rings and (ii) the angle between the aromatic rings which should be between 60 and 120°.50 The procedure followed to obtain the mentioned distances is the same as that for the hydrogen bonds (first minimum of the RDF).
We also studied the electrostatic field lines existing in the framework using a line integral. We obtained the electrostatic field at a certain point and we displaced this point a differential distance (dl) in the direction of the field. This method provides us with a curve where the electrostatic is always tangential, meaning a field line. The calculation is based on the Coulomb interaction and the principle of superposition of the electric field generated by all the particles with partial charges in the structure.
Results and discussion
To study the stability of Zn-MOF-74 against pure diols, we inserted the molecules within the framework to get the initial point of the MD simulations. We studied the adsorption loading at 300 K and 109 Pa and we obtained the saturation loading for each isomer (51 for catechol and 54 for both resorcinol and hydroquinone). The difference in the number of molecules at saturation could be related to a different stacking between molecules within the framework.Taking into account the saturation loading, we choose different numbers of molecules to study the behaviour of the framework against diols. In this way, the number of molecules selected is 1, 2, 5, 10, 16, 18, 24, 36 and 48. We repeated the entire process three times in all cases; that is, for each number of molecules selected there are three configurations. These configurations are generated to explore the compatible and as diverse as possible microstates. Table 1 shows these configurations of the molecules within the pore that we have selected to see if the position of the molecules within the framework and the number of molecules are important factors to consider in structural stability. When the number of molecules is high, the interaction between molecules is stronger than the interaction with the structure, so the initial configuration is not so important.
| N | Molecule (catechol, resorcinol or hydroquinone) | ||
|---|---|---|---|
| Config. 1 | Config. 2 | Config. 3 | |
| 2 | 1-1-0 | 2-0-0 | 2-0-0 |
| 5 | 2-2-1 | 3-2-0 | 5-0-0 |
| 10 | 4-4-2 | 5-4-1 | 10-0-0 |
| 16 | 5-5-6 | 8-8-0 | 16-0-0 |
As shown Fig. 1, in the case that one molecule of the diol is located inside the framework, the position of the molecule depends on the type of isomer. Catechol, which is the closest molecule to the metal, interacts with the unsaturated metals with one hydroxyl group (O). Resorcinol and hydroquinone, due to the position of hydroxyl groups in the molecule, interact with the framework using the two hydroxyl groups (O1 and O2). For this reason, the distances between the hydroxyl groups and Zn metallic centres are larger for resorcinol and hydroquinone than for catechol (dO–Zn,catechol = 2.45 Å, dO1–Zn,resorcinol = 2.48 Å and dO2–Zn,resorcinol = 2.50 Å, dO1–Zn,hydroquinone = 2.45 Å and dO2–Zn,hydroquinone = 2.47 Å).
 | ||
| Fig. 1 Schematic representation of the most stable configuration of catechol, resorcinol and hydroquinone (from left to right) inside Zn-MOF-74. | ||
The most stable configuration of one molecule of diols inside Zn-MOF-74 was also confirmed by the study of the electrostatic field lines using a homemade code. There is a strong interaction between the hydroxyl group of the molecule of catechol and the metallic centre. Resorcinol and hydroquinone have the same interaction but with the two hydroxyl groups due to the position of these groups in each isomer (Fig. 2).
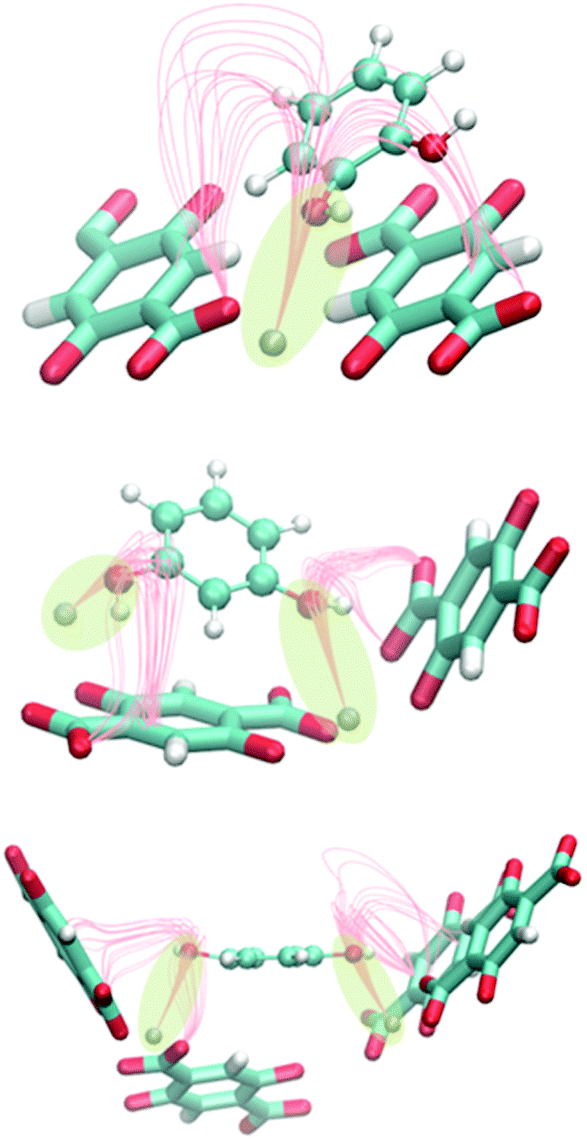 | ||
| Fig. 2 Schematic representation of the interaction of catechol, resorcinol and hydroquinone (from top to bottom) with the metallic centres of Zn-MOF-74 based on the electrostatic field lines. | ||
The dipolar moment that the C–O–H group of each molecule contains is aligned with the electrostatic field generated by the atoms of the structure. This creates a minimum in the electrostatic potential energy generating a stable configuration. The geometry of each isomer plays an important role in adsorption. Resorcinol and hydroquinone interact with the framework through their two –OH groups, the interaction being greater than that with catechol. This observation is reflected in the heat of adsorption. The value of the enthalpy of adsorption is similar for the meta- and para-isomers (−100.3 and −103.1 kJ mol−1, respectively) and higher than for the ortho-isomer (−78.9 kJ mol−1).
We simulated the volume change of Zn-MOF-74 as a function of diol content (for all the cases studied in the CMC simulations) using MD simulations at 300 K. The diol content was obtained by taking into account the number of molecules in each simulation (1, 2, 5, 10, 16, 18, 24, 36, and 48) and the number of molecules at saturation loading (51 for catechol and 54 for both resorcinol and hydroquinone). The simulations were performed for up to 5000 ps with a time step of 0.001 ps. Fig. 3 shows the volume change (Boltzmann average) for Zn-MOF-74 as a function of the content of diols. We have used the Boltzmann average to consider, with a high weight in the calculation, the most stable configuration in every case. This means that for each value of N, there are three repetitions and the highest contribution to the volume change is related to the most stable configuration. We observe that the framework is not destroyed in any case, unlike what happens with the molecules of water.38 We found that the volume change is higher at low loading. This is due to the fact that molecules of the diol interact more strongly with the structure through the metal. At this loading, catechol has the largest effect in the volume change for being the molecule that is closest to the metal (Fig. S4† shows the most distorted structure calculated, circa 30% volume change). When the number of molecules of diol in the structure increases, the interaction with the metallic centres decreases. This is because the molecules are interacting between them by forming hydrogen bonds or via π-stacking and so the variations in volume are lower.
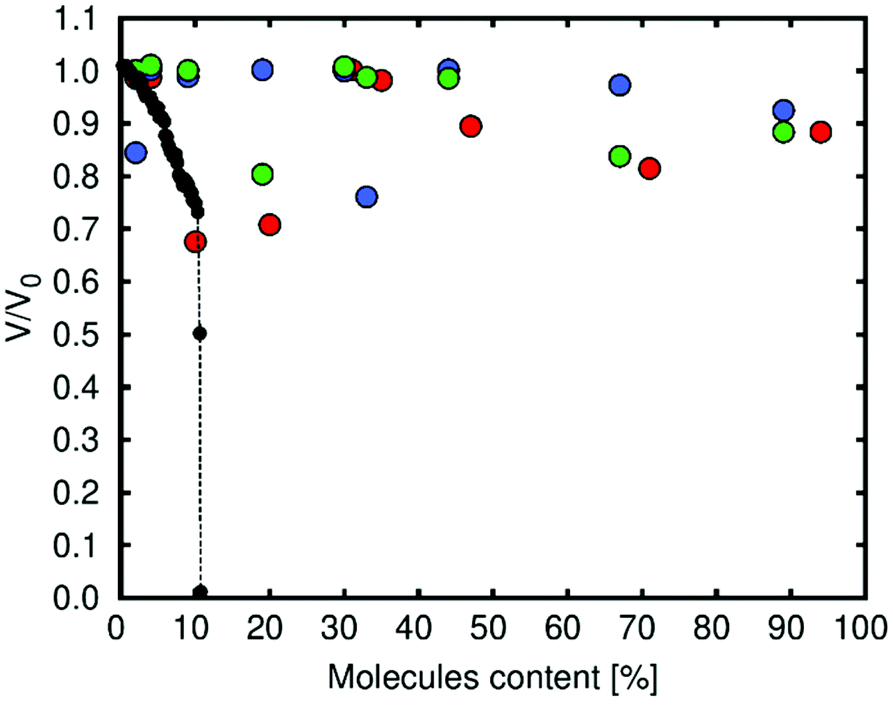 | ||
| Fig. 3 Simulated volume change as a function of diol content (red: catechol, blue: resorcinol and green: hydroquinone). Black points, which show the behaviour of the framework in the presence of water, are taken from Han et al.38 | ||
We calculated the host–guest energy using MD simulations for the same conditions. The host–guest energy (Boltzmann average) is related to the heat of adsorption at low loading, while this energy is related to the vaporization enthalpy of diols when the loading is high. The energy mainly depends on two factors: the energy of deformation of the framework (due to the interaction between the framework and the molecules) and the guest–guest energy (related to the interaction of the molecules between them). As shown in Fig. 4, at low loading the contribution related to the deformation of the structure is higher because the molecules of the diol go to the metallic centres and interact with them. This observation is also related to the volume change at the same loading. However, the guest–guest energy is the most important contribution at high loading. At this point, the molecules of the diol interact between them by forming hydrogen bonds or via π-stacking and the deformation of the framework is not the most important contribution.
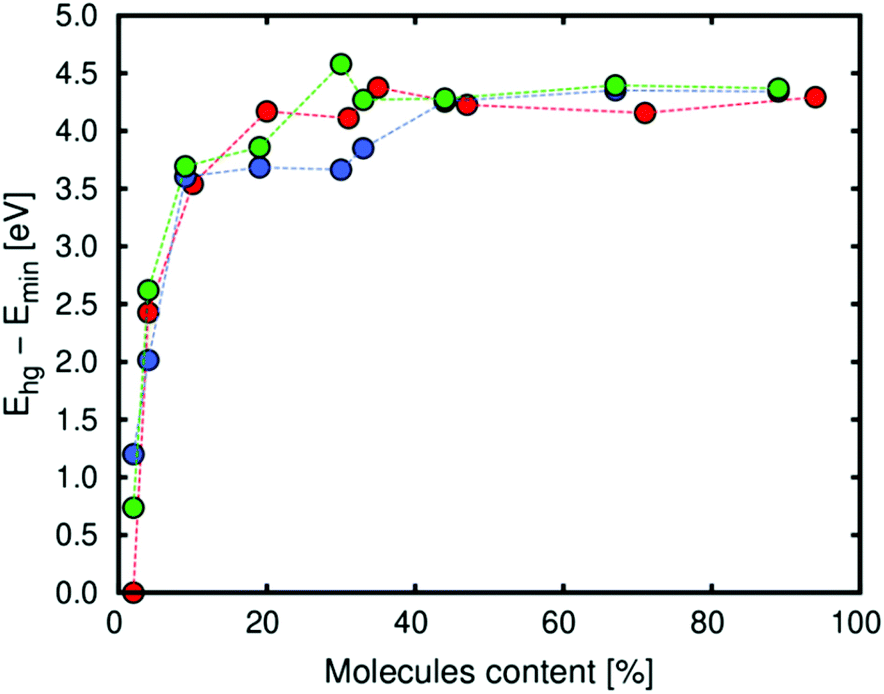 | ||
| Fig. 4 Boltzmann average of the host–guest energy (per molecule) between the molecules of diol and Zn-MOF-74 (red: catechol, blue: resorcinol and green: hydroquinone). | ||
Regarding the energy of deformation of Zn-MOF-74 due to the interaction with the molecules of the diol, Fig. 5 shows that this energy depends on the loading and the type of isomer. At low loading, the value of the energy strongly depends on the isomer. Catechol, which is the only isomer that interacts with the framework with one hydroxyl group, has the highest value for being closer to the metal than resorcinol and hydroquinone. The two isomers that interact with the framework with the two hydroxyl groups have similar values of energy since the distances between the hydroxyl groups and the metal are very similar and the interaction is practically the same. At high loading, the three isomers have similar behaviour due to the small interaction with the framework (the interaction between the molecules of diols is preferred).
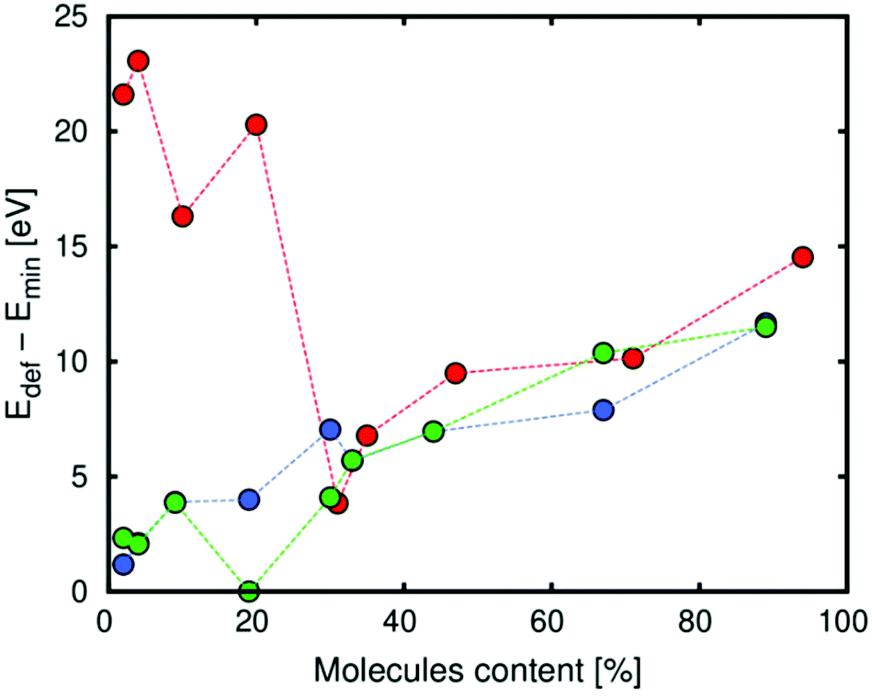 | ||
| Fig. 5 Energy of deformation of Zn-MOF-74 due to the interaction with catechol (red), resorcinol (green) and hydroquinone (blue). | ||
As we observe, there is a step in the case of catechol and resorcinol for 10% (N = 5) and 20% (N = 10), respectively. In both cases, this step corresponds with a configuration where the pore condensation degree has the highest value. It means that all the molecules are distributed in the same pore. We think that this step corresponds with a special configuration of the framework to compensate the deformation produced for the molecules and avoid structural collapse (Fig. S4†).
As shown in Fig. 5, the interaction between the molecules of diol plays a key role in the deformation (volume change) of the framework. High interaction between the molecules leads to low deformation of the structure, so we studied the interaction between molecules from the number of hydrogen bonds per molecule using a geometrical criterion. The number of hydrogen bonds increases with the number of molecules and the pore condensation degree.
Fig. 6 shows that the number of hydrogen bonds increases with the number of total molecules inside the framework and with the condensation degree. The pore condensation degree is a way to measure the number of molecules adsorbed in the pore:
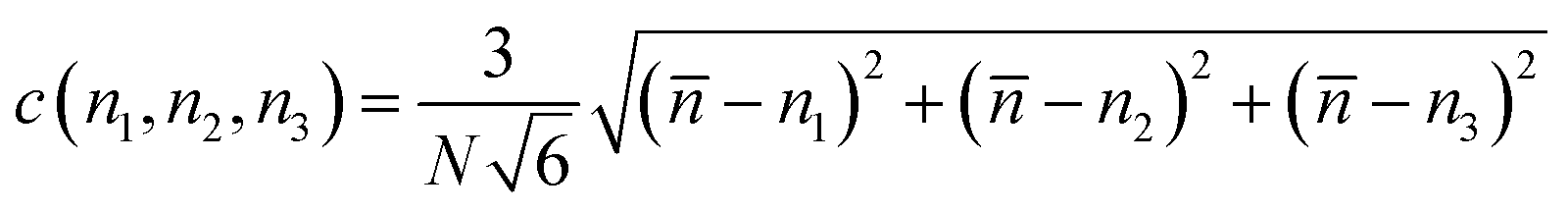 | (1) |
![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) the average number of molecules per pore and ni for i = 1, 2, 3, the number of molecules for each pore.
the average number of molecules per pore and ni for i = 1, 2, 3, the number of molecules for each pore.
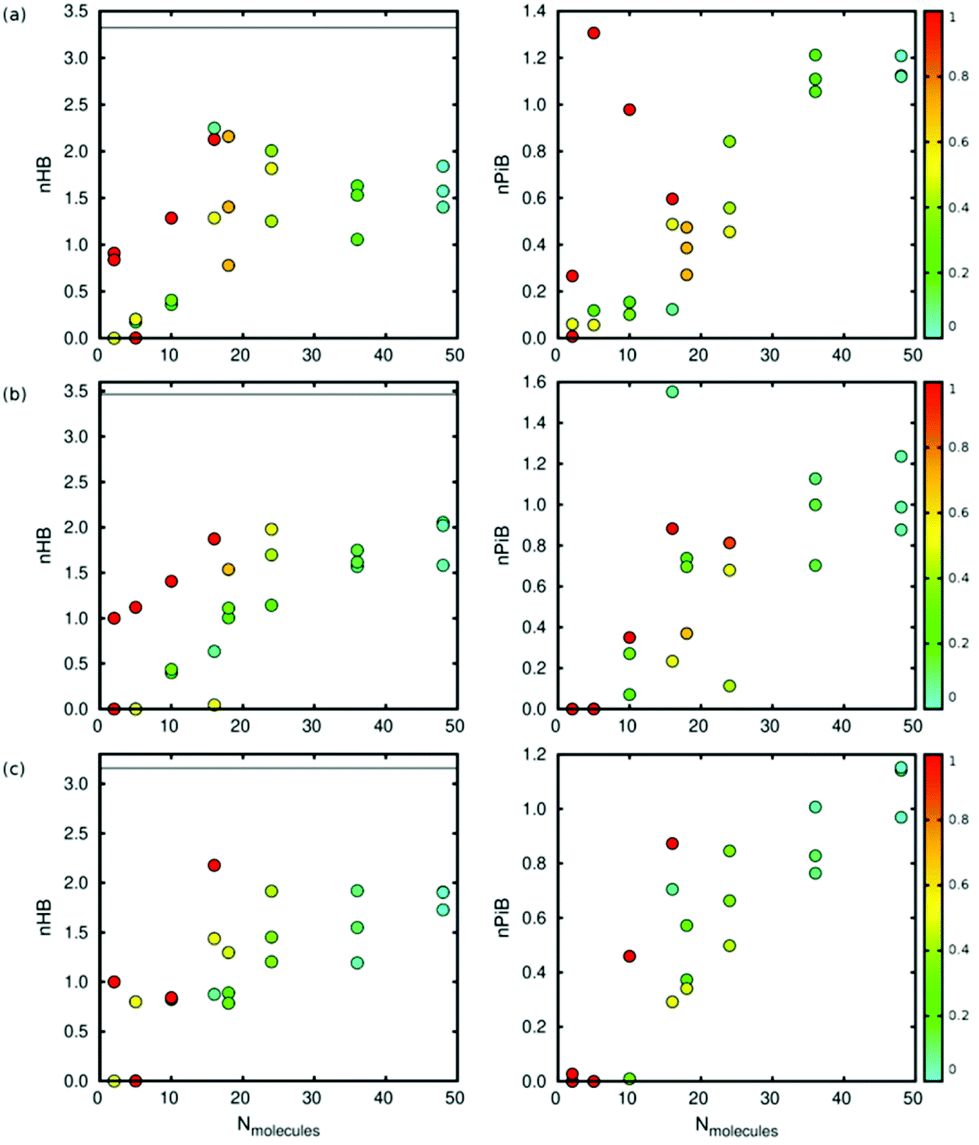 | ||
| Fig. 6 Average number of hydrogen bonds (left) and average number of π-bonds (right) per molecule of diol in Zn-MOF-74 at 300 K (from top to bottom: (a) catechol, (b) resorcinol, and (c) hydroquinone). The colour code depends on the condensation (1: the highest condensation (red) and 0: the lowest condensation (blue)). The black line indicates the average number of hydrogen bonds in the bulk. | ||
In the case of catechol, when the number of molecules inside the framework is 5 and the condensation degree has the highest value (all the molecules are in the same pore), there is no formation of hydrogen bonds. In this situation, π-stacking is preferred (Fig. S5†). We have also computed the number of hydrogen bonds per molecule in the bulk (black solid line in Fig. 6), taking into account the density of each isomer. We see that the value of nHB in the bulk is higher than that in a confined situation due to the presence of a higher number of molecules.
The π-interaction between the molecules is due to the presence of aromatic rings in all the adsorbates. As shown in Fig. 6, the number of this type of interaction increases with the number of molecules inside the pore and also with the pore condensation degree. Note that we are considering all the possible aromatic stacking arrangements. For five molecules of catechol in the same pore (the highest pore condensation degree), we observe that the π-interaction is preferred over the formation of hydrogen bonds.
To study the ability of the three isomers to prevent the structural collapse (it is reported to happen in the presence of water), we run MD simulations using a ReaxFF with several diol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixtures (including catechol, resorcinol and water). We used 10.4% water (that is the amount of water that destroys the framework) and different concentrations of diols (including the different number of molecules inside Zn-MOF-74). In all cases, for this content of water, the framework is destroyed regardless of the number of molecules of diol (see Fig. S6†). We also found structural collapse when the concentration of water is lower than 10.4% and concentrations of diols were the same as those we used in the previous case. In all cases, the dissociation of a molecule of water into OH− and H+ is observed and the hydrolytic stability of this MOF is affected by the presence of the diols. The mechanism of collapse is similar to that which occurs with pure water adsorption, i.e. dissociation of water, hydroxylation of Zn, weakening of the Zn–ligand bond and its subsequent breakage, but the strong interaction between the diols and water favours water being closer to the ZnO group, thus increasing the hydrogen bond between the O of the ZnO and the H of water, leading to more efficient breakage of the Zn–ligand. So, the diol acts as a catalyst, and water is more easily dissociated by a collective interaction.
:water mixtures (including catechol, resorcinol and water). We used 10.4% water (that is the amount of water that destroys the framework) and different concentrations of diols (including the different number of molecules inside Zn-MOF-74). In all cases, for this content of water, the framework is destroyed regardless of the number of molecules of diol (see Fig. S6†). We also found structural collapse when the concentration of water is lower than 10.4% and concentrations of diols were the same as those we used in the previous case. In all cases, the dissociation of a molecule of water into OH− and H+ is observed and the hydrolytic stability of this MOF is affected by the presence of the diols. The mechanism of collapse is similar to that which occurs with pure water adsorption, i.e. dissociation of water, hydroxylation of Zn, weakening of the Zn–ligand bond and its subsequent breakage, but the strong interaction between the diols and water favours water being closer to the ZnO group, thus increasing the hydrogen bond between the O of the ZnO and the H of water, leading to more efficient breakage of the Zn–ligand. So, the diol acts as a catalyst, and water is more easily dissociated by a collective interaction.
Liu et al.37 also explain the structural collapse of interpenetrated Zn-MOF-74 through the water dissociation reaction in confined spaces that is related to the interaction of the –OH group with the metallic centre (Zn) and the H atom with the oxygen of the linker. Moreover, the deprotonation of carboxylic acids in confined spaces has been reported here.51 We think that a similar reaction, due to the acidic character of diols, should happen with diols in the presence of water as the framework is destroyed with a low concentration of water in the presence of diols.
Conclusions
We studied the stability of Zn-MOF-74 with pure diols, water and diol:water mixtures. We reproduced the experimental instability of Zn-MOF-74 upon water adsorption and we tried to solve this limitation using catechol, resorcinol and hydroquinone as stabilizing agents. Molecular Dynamics simulations using ReaxFF were used for this purpose. We found that Zn-MOF-74 is stable against the molecules of diol selected as stabilizing agents. The interaction between molecules of diol and the structure depends on the type of isomer. The isomer with the closest –––OH groups, catechol, interacts with the framework with only one hydroxyl group, while resorcinol and hydroquinone interact with two hydroxyl groups. We also found that the deformation of the framework is less at high concentrations of the adsorbate. This is due to the formation of hydrogen bonds between the molecules. However, the simulations of mixtures of diol and water show that catechol, resorcinol and hydroquinone not only cease to act as stabilizing agents but also further increase the effect of water on the structure. The structural destabilization caused by a second guest component in the second sphere of coordination with the metal is a surprising and rarely reported result. Although the strategy followed does not bring a positive result to the stabilisation of MOF-74, our results can stimulate future work in this field. These findings can probably be extended to other similar structures and will undoubtedly help in the synthesis of MOFs with greater stability with water. This result also raises the question of whether the addition of other protective groups that do not change the hydrophilic nature of water (such as a molecule with a less acidic character) might be possible and encourages further research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministerio de Educación, Cultura y Deporte (MEFP) with a predoctoral fellowship (FPU16/04322) and Ministerio de Ciencia e Innovación (MICINN) under the PID2019-111189GB-I00 and CTQ2016-80206-P projects. SRGB also acknowledges support from MICINN for the Juan de la Cierva Formación fellowship under de FJC2018-035697-I project. We thank C3UPO for the HPC support.References
- H. Q. Zheng, C. Y. Liu, X. Y. Zeng, J. Chen, J. Lü, R. G. Lin, R. Cao, Z. J. Lin and J. W. Su, Inorg. Chem., 2018, 57, 9096–9104 CrossRef CAS
.
- R. Shi, D. Lv, Y. Chen, H. Wu, B. Liu, Q. Xia and Z. Li, Sep. Purif. Technol., 2018, 207, 262–268 CrossRef CAS
- H. Jasuja, G. W. Peterson, J. B. Decoste, M. A. Browe and K. S. Walton, Chem. Eng. Sci., 2015, 124, 118–124 CrossRef CAS
- Y. T. Yan, J. Liu, G. P. Yang, F. Zhang, Y. K. Fan, W. Y. Zhang and Y. Y. Wang, CrystEngComm, 2018, 20, 477–486 RSC
- R. B. Lin, S. Y. Liu, J. W. Ye, X. Y. Li and J. P. Zhang, Adv. Sci., 2016, 3, 1–20 Search PubMed
- K. Jiang, L. Zhang, Q. Hu, D. Zhao, T. Xia, W. Lin, Y. Yang, Y. Cui, Y. Yang and G. Qian, J. Mater. Chem. B, 2016, 4, 6398–6401 RSC
- X. Yang and Q. Xu, Cryst. Growth Des., 2017, 17, 1450–1455 CrossRef CAS
- K. Tan, N. Nijem, Y. Gao, S. Zuluaga, J. Li, T. Thonhauser and Y. J. Chabal, CrystEngComm, 2015, 17, 247–260 RSC
- H. Wu, J. M. Simmons, G. Srinivas, W. Zhou and T. Yildirim, J. Phys. Chem. Lett., 2010, 1, 1946–1951 CrossRef CAS
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2009, 2, 4995–5000 CrossRef
- Y. Liu, H. Kabbour, C. M. Brown, D. A. Neumann and C. C. Ahn, Langmuir, 2008, 24, 4772–4777 CrossRef CAS
- S. Zuluaga, E. M. A. Fuentes-Fernandez, K. Tan, F. Xu, J. Li, Y. J. Chabal and T. Thonhauser, J. Mater. Chem. A, 2016, 4, 5176–5183 RSC
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS
- N. Al-Janabi, P. Hill, L. Torrente-Murciano, A. Garforth, P. Gorgojo, F. Siperstein and X. Fan, Chem. Eng. J., 2015, 281, 669–677 CrossRef CAS
- J. R. Álvarez, E. Sánchez-González, E. Pérez, E. Schneider-Revueltas, A. Martínez, A. Tejeda-Cruz, A. Islas-Jácome, E. González-Zamora and I. A. Ibarra, Dalton Trans., 2017, 46, 9192–9200 RSC
- J. M. Castillo, T. J. H. Vlugt and S. Calero, J. Phys. Chem. C, 2008, 112, 15934–15939 CrossRef CAS
- J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2006, 128, 10678–10679 CrossRef CAS
- L. Bellarosa, J. J. Gutiérrez-Sevillano, S. Calero and N. López, Phys. Chem. Chem. Phys., 2013, 15, 17696–17704 RSC
- M. Feng, P. Zhang, H. C. Zhou and V. K. Sharma, Chemosphere, 2018, 209, 783–800 CrossRef CAS
- K. Tan, S. Zuluaga, Q. Gong, P. Canepa, H. Wang, J. Li, Y. J. Chabal and T. Thonhauser, Chem. Mater., 2014, 26, 6886–6895 CrossRef CAS
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS
- J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. Levan, Langmuir, 2011, 27, 11451–11456 CrossRef CAS
- P. Li, J. Chen, J. Zhang and X. Wang, Sep. Purif. Rev., 2015, 44, 19–27 CrossRef CAS
- S. Zuluaga, E. M. A. Fuentes-Fernandez, K. Tan, C. A. Arter, J. Li, Y. J. Chabal and T. Thonhauser, J. Mater. Chem. A, 2016, 4, 13176–13182 RSC
- T. Wu, L. Shen, M. Luebbers, C. Hu, Q. Chen, Z. Ni and R. I. Masel, Chem. Commun., 2010, 46, 6120–6122 RSC
- H. Huang, W. Zhang, D. Liu and C. Zhong, Ind. Eng. Chem. Res., 2012, 51, 10031–10038 CrossRef CAS
- L. Bellarosa, J. M. Castillo, T. J. H. Vlugt, S. Calero and N. López, Chem. – Eur. J., 2012, 18, 12260–12266 CrossRef CAS
- M. De Toni, R. Jonchiere, P. Pullumbi, F. X. Coudert and A. H. Fuchs, ChemPhysChem, 2012, 13, 3497–3503 CrossRef CAS
- K. Chenoweth, A. C. T. van Duin, A. William and I. Goddard, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS
- L. Huang, K. L. Joshi, A. C. T. van Duin, T. J. Bandosz and K. E. Dubbins, Phys. Chem. Chem. Phys., 2012, 14, 11327–11332 RSC
- Y. Liu, J. Hu, H. Hou and B. Wang, Chem. Phys. Lett., 2020, 751, 137554 CrossRef CAS
- Y. Yang, Y. K. Shin, S. Li, T. D. Bennett, A. C. T. van Duin and J. C. Mauro, J. Phys. Chem. B, 2018, 122(41), 9616–9624 CrossRef CAS
- X. Q. Zang, E. Lype, S. V. Nedea, A. P. J. Jansen, B. M. Szyja, E. J. M. Hensen and R. A. van Santen, J. Phys. Chem. C, 2014, 118(13), 6882–6886 CrossRef
- K. Chenoweth, S. Cheung, A. C. T. van Duin, W. A. Goddard and E. M. Kober, J. Am. Chem. Soc., 2005, 127(19), 7192–7202 CrossRef CAS
- J. E. Mueller, A. C. T. van Duin and W. A. Goddard, J. Phys. Chem. C, 2010, 114(12), 5675–5685 CrossRef CAS
- A. K. Rappé and W. A. Goddard, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef
- X. Y. Liu, S. J. Pai and S. S. Han, J. Phys. Chem. C, 2017, 121, 7312–7318 CrossRef CAS
- S. S. Han, S. H. Choi and A. C. T. Van Duin, Chem. Commun., 2010, 46, 5713–5715 RSC
- H. M. Aktulga, J. C. Fogarty, S. A. Pandit and A. Y. Grama, Parallel Comput., 2012, 38, 245–259 CrossRef
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, Mol. Simul., 2016, 42, 81–101 CrossRef CAS
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114(25), 10024–10035 CrossRef
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS
- N. Rai and J. I. Siepmann, J. Phys. Chem. B, 2013, 117, 273–288 CrossRef CAS
- N. Schmid, A. P. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. E. Mark and W. F. Van Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 CrossRef CAS
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS
- B. Widom, J. Chem. Phys., 1963, 39, 2808–2812 CrossRef CAS
- J. Martí, J. A. Padró and E. Guàrdia, J. Chem. Phys., 1996, 105, 639–649 CrossRef
- J. A. Padró, L. Saiz and E. Guàrdia, J. Mol. Struct., 1997, 416, 243–248 CrossRef
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC
- I. Dance, New J. Chem., 2003, 27, 22–27 RSC
- H. L. Gao, H. Zhang, C. Y. Li and X. H. Xia, Electrochim. Acta, 2013, 110, 159–163 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0dt03787g |
| This journal is © The Royal Society of Chemistry 2021 |