
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination chemistry of the bench-stable tris-2-pyridyl pnictogen ligands [E(6-Me-2-py)3] (E = As, As![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O, Sb)†
O, Sb)†
Alex J.
Plajer
*a,
Daniel
Crusius
 a,
Rajesh B.
Jethwa
a,
Álvaro
García-Romero
b,
Andrew D.
Bond
a,
Raúl
García-Rodríguez
*b and
Dominic S.
Wright
*a
a,
Rajesh B.
Jethwa
a,
Álvaro
García-Romero
b,
Andrew D.
Bond
a,
Raúl
García-Rodríguez
*b and
Dominic S.
Wright
*a
aChemistry Department, Cambridge University, Lensfield Road, Cambridge, CB2 1EW, UK
bGIR MIOMeT-IU Cinquima-Química Inorgánica, Facultad de Ciencias, Campus Miguel, Delibes, Universidad de Valladolid, 47011 Valladolid, Spain. E-mail: raul.garcia.rodriguez@uva.es
First published on 27th November 2020
Abstract
Tripodal ligands with main group bridghead units are well established in coordination chemistry and single-site organometallic catalysis. Although a large number of tris(2-pyridyl) main group ligands [E(2-py)3] (E = main group element, 2-py = 2-pyridyl) spanning across the whole p-block are now synthetically acessible, only limited work has been done on the coordination chemistry on the tris(2-pyridyl) group 15 ligands for the heavier elements (As, Sb). In the current study we investigate the coordination chemistry of the ligand family E(6-Me-2-py)3 (E = As, Sb) and of the As(V) ligand O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) As(6-Me-2-py)3. The air- and mositure-stability of all of these main group ligands makes them especially attractive in future catalytic applications.
As(6-Me-2-py)3. The air- and mositure-stability of all of these main group ligands makes them especially attractive in future catalytic applications.
1. Introduction
Tripodal ligands are very common as facially coordinating 6-electron donor ligands in single-site homogeneous transition metal catalysis and biomimetic chemistry.1,2 One of the most prominent types of tripodal ligands employed are tris-pyrazolyl borates (Fig. 1a), as a result of the ease by which the steric and electronic properties of these ligands can be tuned by substituents on the pyrazolyl rings.3–5 Although not as well developed as a ligand class, tris-pyridyl ligands [E(2-py′)3] (Fig. 1b), containing a range of main group element bridgehead atoms (E), have also emerged.6,7 Changing the bridgehead atoms in these ligands not only allows the incorporation of a further donor centre, but also provides an additional tool for altering ligand bite and donor strength. Earlier work in this area had concentrated on ligands containing non-metal bridgehead atoms (e.g., CR, COR, CH, N, P, PO).8
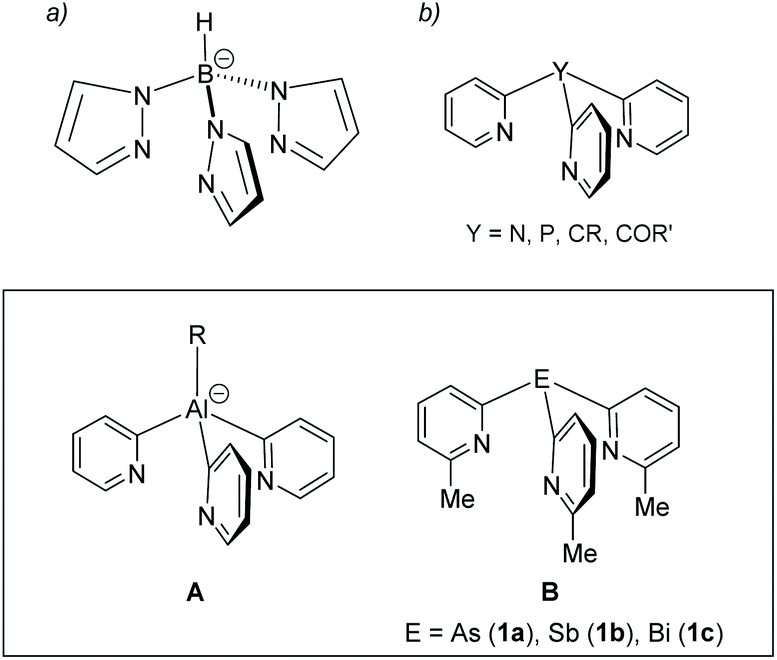 | ||
| Fig. 1 (a) The parent tris-pyrazolyl-borate anion and (b) the family of neutral unsubstituted tris (2-pyridyl) ligand containing non-metal bridgeheads. The inset shows two examples of metal-based analogues. | ||
Our focus has been on the most metallic p-block elements (Fig. 1, lower panel).9–13 Coordination of metal-based ligands of this type to a range of main group and transition metal atoms provides a simple approach to the assembly of heterometallic complexes. In addition, introducing electropositive metal atoms also has a large effect on the charge distribution within the ligand frameworks and their reactivity. For example, aluminates of type A (Fig. 1, lower panel) are useful 2-pyridyl transfer reagents which are stable at room temperature.14,15 Their acid–base reactivity with alcohols makes them useful as reagents in the quantitative determination of enantiomeric excess of mixtures of chiral alcohols.16 We also showed recently that the increase in Lewis acidity moving down group 15 in the pnictogen ligands B (E = As, Sb, Bi) (Fig. 1, lower panel) can drastically alter the 6-methyl-2-pyridyl ligand coordination mode and can be used to change their net donor character systematically.17 However, this previous work only explored Cu(I) complexes of these heavier group 15 ligands.
The stability of the series of group 15 ligands [E(6-Me-2-py)3] (B) under ambient conditions, unlike the majority of metal-based ligands of this type, makes them particularly attractive, for example, as far as their development in the area of catalysis is concerned. Although the coordination chemistry of the parent, unsubstituted [E(2-py)3] ligands has been investigated extensively for the lighter elements (E = N and P), there have been almost no structural studies of tris-2-pyridyl ligands containing the heavier group 15 elements and the only such studies of the Sb and Bi ligands were reported by us recently (and, as previously noted, were limited to coordination of CuI).8,17 In this paper we explore the coordination chemistry of the group 15 tris-2-pyridyl ligands [E(6-Me-2-py)3] (E = As, Sb) and, in the case of the AsIII ligand, the effects of oxidising the bridgehead atom in the series of AsV ligands [X = As(6-Me-2-py)3] (X = O, S, Se). This work provides a basis for understanding the principal coordination modes for the ligands containing the heavier elements and how these compare to the N- and P-analogues.
2. Results and discussion
As we noted in our initial paper in this area,17 the incorporation of the 6-Me group into the 2-pyridyl units of the heavier group 15 ligands [E(6-Me-2-py)3] [E = As (1a), Sb (1b)] leads to a significant increase in their stability compared to unsubstituted 2-py groups. The parent AsIII-bridged tris-pyridyl ligand [As(2-py)3] has been prepared previously,18,19 and ligands 1a and 1b are readily obtained in good yields (60–64%) using the reactions of lithiate 6-Me-2Li-py with ECl3 in THF (Scheme 1).17 Contrary to our expectations, during the initial stages of the current work we found that, due to the stabilising effect the 6-Me substituents in the 2-pyridyl framework, both ligands are indefinitely stable in air under ambient conditions. | ||
| Scheme 1 Synthesis of the heavier group 15 ligand 1a and 1b. | ||
Since we had shown previously that changes in the Lewis acidity of the group 15 element had a major effect on the donor strength and coordination mode of the ligands, we were also interested to assess the potential effects of oxidation of the AsIII atom of 1a on charge distribution and coordination preference. The arsenic oxide ligand [OAs(6-Me-2-py)3] (1a-O) is obtained in 72% yield by the oxidation of 1a with H2O2 in THF, after removal of the solvent under vacuum. This new ligand was characterised using 1H, 13C NMR, IR, UV-Vis and elemental analysis, prior to obtaining its single-crystal X-ray structure. Comparison of the IR spectrum with that of 1a allowed the assignment of the AsO stretching frequency in 1a-O at 902 cm−1 (Ph3AsO 880 cm−1).20 Oxidation of the As bridgehead in 1a has a large effect on the solubility of the ligands. While 1a and 1a-O are indefinitely stable under ambient conditions in air, 1a-O is also soluble and stable in water as a solvent (as shown by 1H NMR spectroscopy). The solid-state structure of 1a-O shows that incorporation of the O-atom at the bridgehead position, not unexpectedly, pushes the 6-Me substituents away due to repulsion with the AsO bond, consistent with a simple VSEPR model (Fig. 2, left). The orientation of the 6-Me-2-py groups in 1a-O is different from that in the parent ligand 1a (reported previously17) in which the 6-Me-groups are all orientated (upwards) towards the AsIII atom (Fig. 2, right). Oxidation also has the effect of reducing the bridgehead As–Cα bond lengths by approximately 0.04 Å and increasing the bridgehead C–As–C angles compared to the parent ligand 1a (from 92.3(2) in 1a to 102.4(1)–106.3(1)° in 1a-O). The latter can be seen as a direct consequence of the more sterically congested orientation of the 6-Me groups in 1a-O, but is also in-line with VSEPR arguments concerning the effect of the increase in the electronegativity of the As centre on C–As/C–As bonding-pair repulsion.
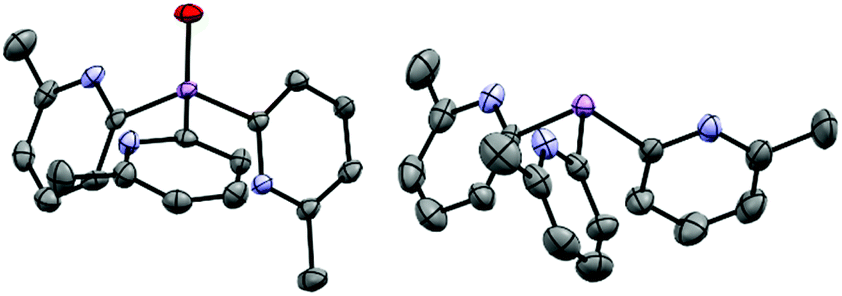 | ||
| Fig. 2 (Left) molecular structure of the ligand 1a-O, showing displacement ellipsoids at 50% probability, with H-atoms omitted. Selected bond lengths (Å) and angles (°): AsO 1.641(2), As–Cpyridyl range 1.939(3)–1.943(3), Cpyridyl–As–Cpyridyl range 105.9(1)–106.3(1), As–Cpyridyl–N range 113.8(2)–114.4(2). Colour key, As (pink), nitrogen (blue). (right) X-ray structure of the ligand 1a. All 6-Me groups and N-atoms of the pyridyl ring units are orientated (“upwards”) towards the arsenic bridgehead. X-ray data taken from reference.17 | ||
Coordination studies of 1a, 1a-O and 1b were undertaken with a range of transition metal salts. The results are summarized in Fig. 3. The new complexes [(1a-O) Cu(CH3CN)]PF6 (2), [Ag@Agx(1a)4](O2CCF3)1+x (3), [(1a-O)Ag(O2CCF3)] (4), [(1a)CoBr2] (5), [(1a-O)2CoBr](CoBr3NCCH3) (6), [(1a)NiBr2] (7), [(1a-O)NiBr2] (8), [(1a)(Cymene)RuCl2] (9a) and [(1b)(Cymene)RuCl2] (9b), and [(1a)2PtCl2] (10a) and [(1b)2PtCl2] (10b) have been synthesized and characterised by 1H and 13C{1H} NMR (where applicable), IR, UV-Vis and elemental analysis. Their single-crystal X-ray structures were also obtained (see Fig. 3 for their structural formulae).
 | ||
| Fig. 3 Transition metal complexes of ligand 1a, 1a-O and 1b obtained in the current work. The face-capping ligands have been omitted form the structure of 3 for clarity. | ||
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric reaction of [Cu(CH3CN)4]PF6 with 1a-O in CH3CN at room temperature yields the complex [(1a-O)Cu(CH3CN)]PF6 (2) in 31% isolated yield after crystallization from the concentrated reaction mixture at −14 °C. The 1H NMR spectrum at room temperature shows a single pyridyl environment, but with very broad resonances that sharpen upon cooling to −30 °C. This is in contrast to the previously reported CuPF6 complex of the un-oxidised ligand 1a, [(1a)Cu(CH3CN)]PF6, for which sharp resonances are observed even at room temperature and might suggest that a fluxional process is occurring in 2 in which the coordination of a pyridyl-N atom to CuI is exchanging with AsO coordination. N,O-coordination is observed later in this study in a number of other complexes of 1a-O. In the solid state, 2 adopts an ion-separated structure containing [{OAs(6-Me-2-py)3}CuCH3CN]+ cations, in which the CuI atom is chelated by all three of the pyridyl-N atoms, and PF6− anions (Fig. 4). This metal coordination mode is commonplace for tris-2-pyridyl ligands and is the same as that found in the previously reported CuPF6 complexes of 1a and 1b.17 The only noticeable differences between the cations [(1a) Cu(CH3CN)]+ and [(1a-O) Cu(CH3CN)]+ stem from the larger bridgehead bite angles in 1a-O, the bridgehead C–As–C angles for 1a-O being ca. 5° greater than in the [(1a-O)Cu(CH3CN)]+ cation, resulting in an associated large increase in the Npyridyl–Cu–Npyridyl angles (by ca. 15–23°) compared to the [(1a)Cu(CH3CN)]+ cation.
:1 stoichiometric reaction of [Cu(CH3CN)4]PF6 with 1a-O in CH3CN at room temperature yields the complex [(1a-O)Cu(CH3CN)]PF6 (2) in 31% isolated yield after crystallization from the concentrated reaction mixture at −14 °C. The 1H NMR spectrum at room temperature shows a single pyridyl environment, but with very broad resonances that sharpen upon cooling to −30 °C. This is in contrast to the previously reported CuPF6 complex of the un-oxidised ligand 1a, [(1a)Cu(CH3CN)]PF6, for which sharp resonances are observed even at room temperature and might suggest that a fluxional process is occurring in 2 in which the coordination of a pyridyl-N atom to CuI is exchanging with AsO coordination. N,O-coordination is observed later in this study in a number of other complexes of 1a-O. In the solid state, 2 adopts an ion-separated structure containing [{OAs(6-Me-2-py)3}CuCH3CN]+ cations, in which the CuI atom is chelated by all three of the pyridyl-N atoms, and PF6− anions (Fig. 4). This metal coordination mode is commonplace for tris-2-pyridyl ligands and is the same as that found in the previously reported CuPF6 complexes of 1a and 1b.17 The only noticeable differences between the cations [(1a) Cu(CH3CN)]+ and [(1a-O) Cu(CH3CN)]+ stem from the larger bridgehead bite angles in 1a-O, the bridgehead C–As–C angles for 1a-O being ca. 5° greater than in the [(1a-O)Cu(CH3CN)]+ cation, resulting in an associated large increase in the Npyridyl–Cu–Npyridyl angles (by ca. 15–23°) compared to the [(1a)Cu(CH3CN)]+ cation.
 | ||
| Fig. 4 Molecular structure of the cation of 2. H-atoms and the PF6− anion omitted for clarity, with displacement ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°): AsO 1.635(2), As–Cpyridyl range 1.927(3)–1.930(3), Cu–Npyridyl range 2.081(3)–2.136(2), Cu–NCMe 1.950(3), Cpyridyl–As–Cpyridyl range 105.6(1)–105.6(9), As–Cpyridyl–N range 116.8(2)–117.0(2), Npyridyl–Cu–Npyridyl range 98.69(7)–100.6(1). Colour key, Cu (orange), As (pink), N (blue), O (red). | ||
The UV-Vis absorption spectrum of 2 shows two distinct bands at 265 nm and 333 nm which can be assigned to a ligand π–π* transition and a CuI-to-ligand charge transfer transition (MLCT).21–23 These values are similar to those observed in the previously reported AsIII analogue [(1a)Cu(CH3CN)]PF6 (270 nm and 359 nm, respectively). While the π–π* transitions in [(1a)Cu(CH3CN)]PF6 and 2 are identical to that found in the ligands 1a (269 nm) and 1a-O (265 nm), respectively, the MLCT band shows a small blue shift comparing [(1a)Cu(CH3CN)]PF6 to 2. This can be attributed to an increase in ligand-field strength, similar to conclusions made by Kodera et al. for the family of [RC(6-Me-2-py)3]Cu(CH3CN)]PF6 complexes (R = H, Me, Et).24
The 1:1 stoichiometric reaction of AgO2CCF3 with 1a in CH3CN at room temperature yields the cluster [Ag@Agx(1a)4](O2CCF3)1+x (3) in an isolated crystalline yield of 21%, after layering the concentrated reaction mixture with Et2O. The arrangement of 3 was established via single-crystal X-ray analysis (Fig. 5). The cationic cluster in 3 [Ag@Agx(1a)4](1+x)+ has a very similar structural arrangement to the previously reported compound [Ag@Ag4(P(2-py)3)4]5+,25 which comprises an Ag-centred Ag4 tetrahedral core, with four AsIII ligands (1a) coordinating three Ag+ cations over each face of the tetrahedron. Unlike [Ag@Ag4(P(2-py)3)4]5+, however, in which the Ag+ cations are also coordinated by PF6− anions or solvent molecules, the presence of the sterically-blocking 6-Me groups in 1a prevents further coordination of the Ag centres, so that they remain three coordinate (trigonal planar).
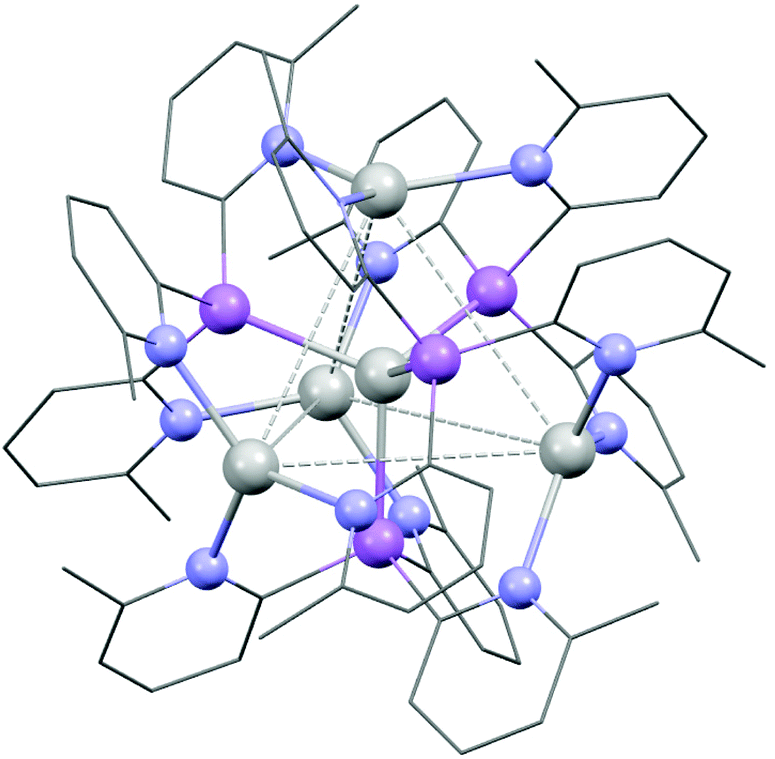 | ||
| Fig. 5 Structure of the cluster compound [{Ag@Agx{(6-Me-2-py)3}As}4(CF3COO)1+x] (3), with CF3COO− and H atoms omitted for clarity. Colour key, Ag (white), As (pink), N (blue). The X-ray crystal structure of 3 suggests partial site occupancy of the outer Ag+ atoms, and the exact nature of [Ag@Agx(1a)4](1+x)+ remains uncertain. | ||
The structure of 3 is probably viewed most appropriately as an [Ag(As(6-Me-2-py)3)4]+ cation, involving tetrahedral coordination of the central Ag+, with the outer Ag+ ions being captured by coordination to the pyridyl rings of the As(6-Me-2-py)3 ligands. The central [Ag(As(6-Me-2-py)3)4]+ core resembles the well-established tetrahedral ion [Ag(AsPh3)4]+. Supporting this, the Ag–As bond length in [Ag(AsPh3)4]+ span the range 2.645–2.668 Å (mean 2.653 Å), compared to the range 2.594(2)–2.691(2) Å seen in 3.26 The comparison to [Ag(AsPh3)4]+ indicates that the [Ag(As(6-Me-2-py)3)4]+ cation should exist independently from further coordination of the outer Ag+ ions. The ion therefore has the potential to span the range from +1 to +5, depending on the extent of Ag+ coordination in the pyridyl binding sites. This is consistent with the observation of partial site occupancy of the Ag+ ions within the tetrahedral unit of 3 (see ESI†). However, it is difficult to verify this hypothesis from the X-ray data, since the trifluoroacetate counterions required for charge balance are also poorly resolved. 1H and 13C NMR spectra at room temperature in MeCN solution show multiple, broad pyridyl environments. Since the analogous cluster [Ag@Ag4(P(2-py)3)4]5+ is reported to stay intact in MeCN,25 this may support the suggestion of variable coordination within an intact [Ag@Agx(1a)4](1+x)+ cluster, but it may also indicate that [Ag@Agx(1a)4](1+x)+ disassembles in solution. At the present time, the similarity to [Ag@Ag4(P(2-py)3)4]5+ is established, but the exact nature of [Ag@Agx(1a)4](1+x)+ remains uncertain.
The reaction of AgO2CCF3 with 1a-O in CH3CN at room temperature gives the crystalline complex [(1a-O)Ag(O2CCF3)] (4) in 21% isolated yield after crystallization from the concentrated reaction mixture at −14 °C. The local C3-symmetric (N,N,N) coordination of AgI is apparent from the 1H NMR spectrum for 4 which shows only one sharp 6-Me-2-py environment. The single-crystal X-ray structure is that of an ion-paired complex in which the O2CCF3− anion also coordinates the metal centre (Fig. 6). The structure of 4 resembles the previously reported AgI complex [{Ag{(2-py)3PO}(PPh3)}BF4].27 In both cases, the presence of the P/AsO substituent precludes the ligand bonding mode present in the clusters [Ag@Ag4(P(2-py)3)4] and 3.
 | ||
| Fig. 6 Molecular structure of 4, H-atoms and a second crystallographically independent molecule are omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°) (range over both independent molecules): AsO 1.640(3), As–Cpyridyl range 1.932(5)–1.942(5), Ag–Npyridyl range 2.313(4)–2.465(4), Ag–O 2.274(4)-2.32(3), Cpyridyl–As–Cpyridyl range 106.3(2)–109.0(2), As–Cpyridyl–N range 117.2(3)–118.8(3), Npyridyl–Ag–Npyridyl range 87.2(1)–94.6(1). Colour key, Ag (white), As (pink), N (blue), O (red), F (yellow). | ||
The 1:1 stoichiometric reactions of CoBr2 and NiBr2 with 1a and subsequent crystallization from the concentrated reaction mixtures at −14 °C give the corresponding 1:1 complexes [(1a)NiBr2] (5) (27% yield) and [(1a)CoBr2] (6) (22% yield), respectively. The solid-state structures of both show bidentate coordination of the metal centres using two of the three pyridyl-N atoms of 1a (Fig. 7). The complexes are isostructural with each other and with the recently described complexes [{PhSi(6-Me-2-py)3}CoCl2] and [{PhSi(6-Me-2-py)3}FeCl2] containing the PhSi(6-Me-2-py)3 ligand.28 Similarly to these complexes, the adoption of a bidentate coordination mode is probably a consequence of the steric congestion of the 6-Me substituents, which prevents the coordination of smaller ions by all three of the pyridyl-N atoms. Although meaningful 1H NMR studies of 5 proved impossible due to the paramagnetic nature of the complex, six distinct pyridyl resonances were observed for 6 at room temperature in MeCN, corresponding to the bonded and non-bonded 6-Me-2-py groups. The spectrum did not change with temperature, suggesting that the complex is intact in solution and that no fluxional interchange of 2-pyridyl ligand coordination is occurring. This is in contrast with the behaviour of the previously reported complexes [{PhSi(6-Me-2-py)3}CoCl2] and [{PhSi(6-Me-2-py)3}FeCl2] which are fluxional at room temperature.
 | ||
| Fig. 7 Molecular structure of the complex [(1a)CoBr2] (5); [(1a)NiBr2] (6) is isostructural in the solid state. Displacement ellipsoids shown at 50% probability, with H atoms omitted for clarity. Selected bond lengths (Å) and angles (°): 5, As–Cpyridyl range 1.959(3)–1.965(3), Co–Npyridyl range 2.051(2)–2.052(2), Co–Br range 2.3684(6)–2.4080 (6), Cpyridyl–As–Cpyridyl range 96.1(1)–104.2(1), As–Cpyridyl–N range 122.5(2)–123.8(2) (coordinating pyridyl groups), As–Cpyridyl–N 115.1(3) (non-coordinating pyridyl group), N–Co–N 109.1(1), Br–Co–Br 120.46(2). 6, As–Cpyridyl range 1.949(4)–1.975(5), Ni–Npyridyl range 2.018(3)–2.022(4), Ni–Br range 2.3484(8)–2.4029(8), Cpyridyl–As–Cpyridyl range 96.0(2)–103.1(2), As–Cpyridyl–N range 122.3(3)–124.1(3) (coordinating pyridyl groups), As–Cpyridyl–N 115.3(3) (non-coordinating pyridyl group), N–Ni–N 107.1(1), Br–Ni–Br 130.23(3). Colour key, As (pink), N (light blue), Co (dark blue), Br (orange). | ||
Oxidation of the bridgehead atom of 1a has a marked effect on the coordination preferences of ligand 1a-O. The 1:1 stoichiometric reaction of CoBr2 with 1a-O in CH3CN and subsequent crystallization from the concentrated reaction mixture at −14 °C yields blue crystals of [(1a-O)2CoBr](CoBr3NCCH3) (7) in 74% yield. Surprisingly, the solid-state structure reveals an ion-separated arrangement containing [(1a-O)2CoBr]+ cations (Fig. 8) and [CoBr3NCCH3]− anions. Thus, overall, the complex has a 1:1 ratio of the ligand 1a to CoBr2. Within the [(1a-O)2CoBr]+ cations, the CoII centre adopts a trigonal bipyramidal geometry in which two 2-pyridyl-N atoms of separate 1a-O ligands bond at the axial positions. The CoII cation is further coordinated in the equatorial plane by the bridgehead O-atoms of the ligands and by a Br− ion. The anion (which is not shown in Fig. 8) has a tetrahedral CoII centre that is bonded to three Br− anions and a MeCN molecule. The paramagnetic 1H NMR spectrum of 7 at room temperature in MeCN shows two sets of pyridyl resonances, indicating that the solid-state structure is maintained in solution.
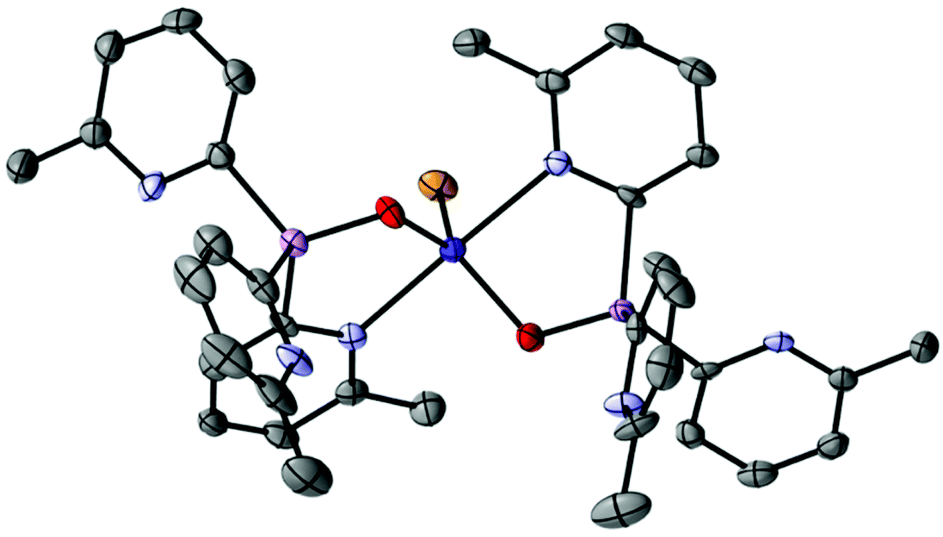 | ||
| Fig. 8 Molecular structure of the cation of [(1a-O)2CoBr](CoBr3NCCH3) (7), showing displacement ellipsoids at 50% probability, with H-atoms, lattice-bound CH3CN and the counter-anion omitted for clarity. Selected bond lengths (Å) and angles (°): As–Cpyridyl range 1.904(9)–1.940(9), AsO 1.672(6)–1.677(7), Co–Npyridyl range 2.240(8)–2.318(8), Co–Br 2.430(2), Co–O range 1.967(7)–1.971(7), Cpyridyl–As–Cpyridyl range 106.3(4)–112.5(4), As–Cpyridyl–N range 107.7(8)–113.3(7), N–Co–N 177.4(3), O–Co–O 117.3(3), O–Co–Br range 121.3(2)–121.4(2). Colour key, As (pink), N (light blue), O (red), Co (dark blue), Br (orange). | ||
The same N,O-coordination mode as seen in 7 is also observed in the NiII complex [(1a-O)NiBr2] (8), obtained from the reaction of NiBr2 with 1a-O in CH3CN in low yield (2%). In the solid-state structure (Fig. 9), the NiII centre is chelated by the bridgehead-O and a pyridyl-N atom of the ligand 1a-O and further bonded to two Br− anions, giving a distorted tetrahedral geometry at the metal centre. The formation of 7 and 8 highlight that bridgehead oxidation can drastically change the ligand character of tris-pyridyl arsine ligands. This switch in coordination character from all-N to N,O (e.g., comparing the CoII complexes 5 and 7) is no doubt largely a consequence of the relative hardness or softness of the ligand modes with respect to the metal coordinated. However, there may also be a more subtle influence of the steric effect of the 6-Me-substution of the pyridyl groups in 1a-O on the preference for N-chelation versus N,O-chelation. It is noteworthy in this regard that the closely related, unsubstituted PV ligand [OP(2-py)3] (which has been studied extensively) has been observed to bond almost exclusively using its pyridyl-N atoms, the only example of N,O-chelation similar to that seen in 7 and 8 being seen in [MoBr(CO)2(CH2CHCH2){OP(2-py)3}].8,29
 | ||
| Fig. 9 Molecular structure of [(1a-O)NiBr2] (8). Displacement ellipsoids are shown at 50% probability. H-atoms and lattice bound CH2Cl2 are omitted. Selected bond lengths (Å) and angles (°): As–Cpyridyl range 1.908(7)–1.940(6), AsO 1.669(4), Ni–Npyridyl 2.021(6), Ni–Br range 2.342(1)–2.367(1), Ni–O 1.963(4), Cpyridyl–As–Cpyridyl range 106.2(3)–112.3(3), As–Cpyridyl–N 111.(5) (coordinating pyridyl groups), As–Cpyridyl–N range 111.0(5)–114.3(5) (non-coordinating pyridyl group), O–Ni–N 93.0(2), Br–Ni–Br 134.01(6). Colour key, As (pink), N (light blue), Ni (light green), Br (orange). | ||
Having probed the coordination preferences of the arsines 1a and 1a-O, we moved on to explore the Sb-bridged ligand 1b. Attempted oxidation of the Sb atom with H2O2 resulted in complicated mixtures of products. This was not pursued further since a localised SbO bonding arrangements (analogous to 1a-O) would in any case not be expected, on the basis of the low bond energy compared to Sb–O single bonds.30 Attempts to coordinate harder transition metal ions with 1b often lead to the formation of intractable mixtures, preventing further analysis. This appears to be due to the greater polarity of the bridgehead C–Sb bonds (which leads to 2-pyridyl transfer to the metal rather than coordination) and to greater sensitivity to hydrolysis by trace moisture present in the metal salts employed. Similar observations have been made by us recently in regard to the closely related Bi ligand [Bi(6-Me-2-py)3].31 For example, in the case of reaction of the Sb-ligand 1b with CoBr2 only the decomposition product [(6-Me-2-py)2CoBr2] could be isolated, as a result of transfer of the 6-Me-2-py groups to CoII (see ESI†).
Reactions with softer transition metal centres were more successful, although under these circumstances metal coordination is observed to occur exclusively by the bridgehead Sb atom (reflecting the softness of the atoms involved). The room-temperature, 1:1 stoichiometric reactions of the arsenic ligand 1a and the antimony ligand 1b with [(Cymene)RuCl2]2 in CH2Cl2 result in quantitative formation of [(1a)(Cymene)RuCl2] (9a) and [(1b)(Cymene)RuCl2] (9b) within 5 minutes, as monitored via in situ1H NMR spectroscopy. Layering of the concentrated reaction mixtures with n-pentane yielded red crystalline 9a and 9b in 64% and 66% yield, respectively. The complexes have similar structures in the solid-state, in which the As and Sb bridgehead atoms coordinate the RuII centres (Fig. 10). There are only minor structural changes in the ligands upon coordination, with the pnictogen–ruthenium bond lengths increasing from 2.450(7) to 2.597(2) Å in line with the increase in the atomic radii of the heavier group 15 elements. The As–Ru and Sb–Ru bond lengths in both of the complexes are similar to those reported previously.32,33 In both cases, room-temperature 1H and 13C NMR spectra in MeCN confirm that the local C3 ligand symmetry is retained in solution, showing only one pyridyl environment. The aromatic cymene proton resonances in 9a are found in the region δ 5.92–5.58 ppm whereas those in 9b are in the range between δ 6.15–6.00 ppm. This indicates that the Sb atom in 9b donates more electron density to the Ru atom in 9b than the As atom does in 9a, resulting in less π-donation in the former. This somewhat counterintuitive observation presumably reflects the relative softness of the SbIII centre of 1b in respect to the soft RuII centre.
 | ||
| Fig. 10 Molecular structure of [(1a)(Cymene)RuCl2] (9a); [(1b)(Cymene)RuCl2] (9b) has a similar structure in the solid state, showing displacement ellipsoids at 50% probability; with H-atoms omitted. Selected bond lengths (Å) and angles (°): 9a, As–Cpyridyl range 1.953(4)–1.961(4), Ru–As 2.4509(4), Ru–Cl range 2.4232(9)–2.4233(9), Ru–C range 2.169(4)–2.222(3), Cpyridyl–As–Cpyridyl range 99.9(2)–100.6(2), As–Cpyridyl–N range 113.3(3)–114.9(3), Ru–As–C range 112.2(1)–121.6(1), As–Ru–Cl range 83.86(3)–88.52(2), Cl–Ru–Cl 89.13(3). 10b, Sb–Cpyridyl range 2.144(3)–2.154(3), Ru–Sb 2.5976(3), Ru–Cl range 2.4061(9)–2.4115(8), Ru–C range 2.171(4)–2.208(4), Cpyridyl–Sb–Cpyridyl range 99.4(1)–102.4(1), Sb–Cpyridyl–N range 113.1(2)–114.3(3), Ru–Sb–C range 113.4(1)–121.44(9), Sb–Ru–Cl range 84.44(2)–85.91(2), Cl–Ru–Cl 87.38(3). Colour key, As (pink), N (light blue), Ru (dark green), Cl (light green). | ||
Similar observations are made in regard to coordination of PtCl2. Reactions of 1a or 1b with (benzonitrile)2PtCl2 in DCM for 5 minutes and subsequent layering of the concentrated reaction mixtures with n-pentane give the platinum complexes [(1a)2PtCl2] (10a) and [(1b)2PtCl2] (10b) as yellow crystals in 59% and 30% yields, respectively. The complexes are isostructural in the solid state (Fig. 11), in which the square-planar PtII atoms are bonded to two As- or Sb-atoms of 1a or 1b. The formation of the cis isomer in both cases is the result of the greater trans effect of the Cl ligands. Again, there are only minor changes in the structural parameters of the ligands compared to the isolated ligands themselves. The observed elongation of the pnictogen–Pt bonds going from 10a to 10b is consistent with the increase in the atomic radius of the group 15 atoms involved (2.3415(6)–2.3501(5) in 10a to 2.4945(6)–2.4977(7) in 10b). The As–Pt and Sb–Pt bond lengths in both of the complexes are similar to those reported previously.34,35
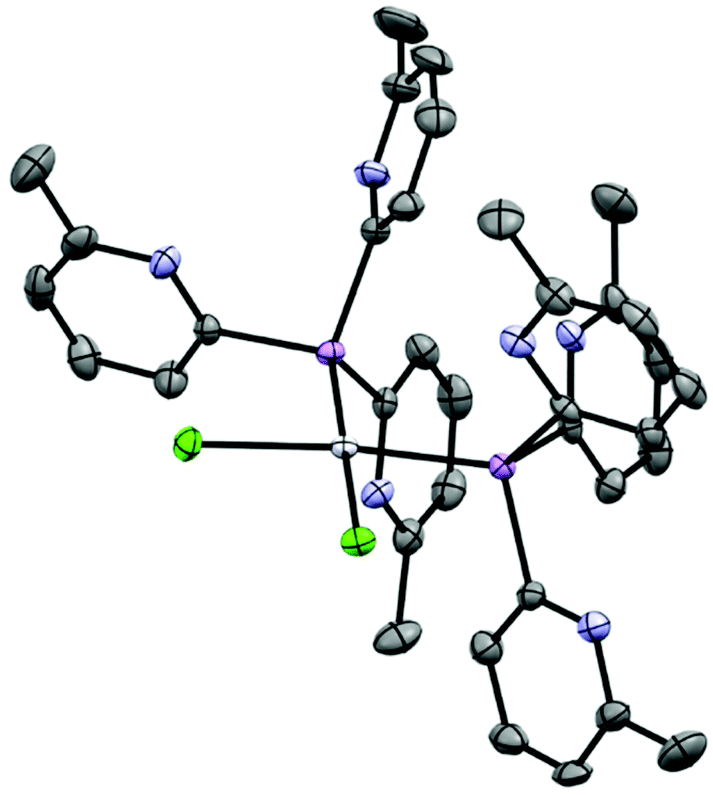 | ||
| Fig. 11 Molecular structure of [(1a)2PtCl2] (10a); [(1b)2PtCl2] (10b) has a similar structure in the solid state, showing displacement ellipsoids at 50% probability; with H-atoms omitted. Selected bond lengths (Å) and angles (°): 10a, As–Cpyridyl range 1.948(4)–1.964(4), Pt–As range 2.3415(4)–2.3503(4), Pt–Cl range 2.3326(9)–2.3523(9), Cpyridyl–As–Cpyridyl range 100.7(2)–109.4(2), As–Cpyridyl–N range 111.6(3)–119.5(3), Pt–As–C range 109.6(1)–121.4(1) As–Pt–Cl range 83.74(3)–89.81(3) (on same side), As–Pt–As 96.25(2), Cl–Pt–Cl 89.91(3). 10b, Sb–Cpyridyl range 2.126(8)–2.153(8), Pt–Sb range 2.4944(6)–2.4976(6), Pt–Cl range 2.336(2)–2.354(2), Cpyridyl–Sb–Cpyridyl range 98.2(3)–109.1(3), Sb–Cpyridyl–N range 111.3(6)–114.8(6), Pt–Sb–C range 112.0(2)–121.2(2), Sb–Pt–Cl range 84.02(5)–88.49(6) (on same side), Sb–Pt–Sb 96.89(2), Cl–Pt–Cl 90.59(7). Colour key, As (pink), N (light blue), Pt (white), Cl (light green). | ||
Conclusions
In contrast to the more well studied phosphine ligands of the type [P(2-py′)3] (where py′ is a substituted or unsubstituted pyridyl group) the 6-Me-2-pyridyl ligands 1a and 1b and the As(V) ligand 1a-O are all indefinitely air- and moisture-stable, making them not only easy to make but also easy to use. Ligands 1a and 1b are robust enough to transfer to a range of transition metal centres. Oxidation of the bridgehead atom of the arsenic ligand 1a can drastically change its coordination preferences, leading to a greater tendency for N,O-coordination involving the bridgehead O-atom and pyridyl groups. This tendency may well also be influenced by the presence of substituents on the 2-pyridyl groups in our case and can be compared to the related unsubstituted [OP(2-py)] ligand, which is observed to bond almost exclusively using a chelating N-coordination mode. We are continuing our studies of the synthesis and coordination chemistry of main group pyridyl ligands of this type, with the aim of applying these ligands in catalysis and supramolecular chemistry.
Experimental section
General experimental techniques
Where indicated, experiments were carried out on a Schlenk-line under nitrogen atmosphere or with the aid of a N2-filled glove box (Saffron type α). Although the ligands 1a, 1b and 1a-O are all indefinitely air- and -moisture stable, anaerobic conditions were employed in reactions to avoid adventitious coordination of water to metal centres in the coordination compounds or potential metal oxidation during reactions. MeCN used for reactions was dried over CaH2. n-Pentane, hexane and diethyl ether were dried over sodium or sodium/benzophenone. 6-Methyl-2-bromo-pyridine was distilled over CaH2 and stored over 4 Å molecular sieves. If not specified other compounds were acquired from Aldrich Chemical Company. 1H, 13C {1H} and 31P{1H} NMR spectra were recorded on a Bruker Avance 400 QNP or Bruker Avance 500 MHz cryo spectrometer. All spectra were recorded in CDCl3, CD2Cl2 or CD3CN, with SiMe4 (1H) or the solvent peaks as external and internal standards. Unambiguous assignments of NMR resonances were made on the basis of 2D NMR experiments (1H–1H COSY, 1H–1H NOESY, 1H–13C HMQC and 1H–13C HMBC). Fig. 12 shows the labelling scheme for NMR assignments used throughout the Experimental section. Elemental analysis was obtained using a PerkinElmer 240 elemental analyser. | ||
| Fig. 12 The numbering schemes used for (a) the 6-Me-2-py groups and (b) the cymene groups in 9a and 9b. | ||
Synthesis of new compounds
Conflicts of interest
There are no conflicts of interest.Acknowledgements
We thank the Leverhulme Trust (Grant for DSW and RG-R), The Spanish Ministry of Science, Innovation and Universities (MCIU) (grant for RG-R, project number PGC2018-096880-A-I00), the Spanish MINECO-AEI and The EU (ESF) for a Ramon y Cajal contract (RG-R, RYC-2015-19035) and the Cambridge Trust (Vice Chancellor Scholarship for AJP), The EPSRC and the Royal Dutch Shell co. Ltd. (I-Case studentship for RBJ, grant EP/R511870/1) and The University of Valladolid and Santander Bank (Fellowship for AG-R).References
- L. H. Gade and P. Hofmann, Molecular Catalysts: Structure and Functional Design, John Wiley & Sons, 2014 Search PubMed.
- I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836–5865 RSC.
- A. Caballero, M. M. Díaz-Requejo, M. R. Fructos, J. Urbano and P. J. Pérez, in Ligand Design in Metal Chemistry, John Wiley & Sons, Ltd, 2016, pp. 308–329 Search PubMed.
- S. Trofimenko, Chem. Rev., 1972, 72, 497–509 CrossRef CAS.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- H. R. Simmonds and D. S. Wright, Chem. Commun., 2012, 48, 8617–8624 RSC.
- G. M. Pawar, J. B. Sheridan and F. Jäkle, Eur. J. Inorg. Chem., 2016, 2016, 2227–2235 CrossRef CAS.
- L. F. Szczepura, L. M. Witham and K. J. Takeuchi, Coord. Chem. Rev., 1998, 174, 5–32 CrossRef CAS.
- C. S. Alvarez, F. García, S. M. Humphrey, A. D. Hopkins, R. A. Kowenicki, M. McPartlin, R. A. Layfield, R. Raja, M. C. Rogers, A. D. Woods and D. S. Wright, Chem. Commun., 2005, 198–200 RSC.
- Á. García-Romero, A. J. Plajer, L. Álvarez-Miguel, A. D. Bond, D. S. Wright and R. García-Rodríguez, Chem. – Eur. J., 2018, 24, 17019–17026 CrossRef.
- E. Yang, A. J. Plajer, Á. García-Romero, A. D. Bond, T. K. Ronson, C. M. Álvarez, R. García-Rodríguez, A. L. Colebatch and D. S. Wright, Chem. – Eur. J., 2019, 25, 14003 CrossRef CAS.
- A. J. Plajer, S. Kopf, A. L. Colebatch, A. D. Bond, D. S. Wright and R. García-Rodríguez, Dalton Trans., 2019, 48, 5692–5697 RSC.
- M. A. Beswick, M. K. Davies, P. R. Raithby, A. Steiner and D. S. Wright, Organometallics, 1997, 16, 1109–1110 CrossRef CAS.
- F. García, A. D. Hopkins, R. A. Kowenicki, M. McPartlin, M. C. Rogers and D. S. Wright, Organometallics, 2004, 23, 3884–3890 CrossRef.
- R. García-Rodríguez and D. S. Wright, Dalton Trans., 2014, 43, 14529–14532 RSC.
- R. García-Rodríguez, S. Hanf, A. D. Bond and D. S. Wright, Chem. Commun., 2017, 53, 1225–1228 RSC.
- A. J. Plajer, A. L. Colebatch, F. J. Rizzuto, P. Pröhm, A. D. Bond, R. García-Rodríguez and D. S. Wright, Angew. Chem., Int. Ed., 2018, 57, 6648–6652 CrossRef CAS.
- T. Gneuß, M. J. Leitl, L. H. Finger, N. Rau, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44, 8506–8520 RSC.
- A. Steiner and D. Stalke, Organometallics, 1995, 14, 2422–2429 CrossRef CAS.
- G. B. Deacon and J. H. S. Green, Spectrochim. Acta, Part A, 1969, 25, 355–364 CrossRef CAS.
- T. Osako, Y. Tachi, M. Taki, S. Fukuzumi and S. Itoh, Inorg. Chem., 2001, 40, 6604–6609 CrossRef CAS.
- R. T. Jonas and T. D. P. Stack, Inorg. Chem., 1998, 37, 6615–6629 CrossRef CAS.
- Y. Shimazaki, H. Yokoyama and O. Yamauchi, Angew. Chem., Int. Ed., 1999, 38, 2401–2403 CrossRef CAS.
- M. Kodera, Y. Kajita, Y. Tachi and K. Kano, Inorg. Chem., 2003, 42, 1193–1203 CrossRef CAS.
- A. V. Artem'ev, I. Y. Bagryanskaya, E. P. Doronina, P. M. Tolstoy, A. L. Gushchin, M. I. Rakhmanova, A. Y. Ivanov and A. O. Suturina, Dalton Trans., 2017, 46, 12425–12429 RSC.
- M. Nardelli, C. Pelizzi, G. Pelizzi and P. Tarasconi, Dalton Trans., 1985, 321 RSC; S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m1537 CrossRef CAS; G. A. Bowmaker, Effendy, R. D. Hart, J. D. Kildea, E. N. de Silva, B. W. Skelton and A. H. White, Aust. J. Chem., 1997, 50, 539 CrossRef; U. M. Tripathi, A. Bauer and H. Schmidbaur, Dalton Trans., 1997, 2865 RSC; A. Bonardi, A. Cantoni, C. Pelizzi, G. Pelizzi and P. Tarasconi, J. Organomet. Chem., 1991, 402, 281 CrossRef; A. Cingolani, Effendy, J. V. Hanna, M. Pellei, C. Pettinari, C. Santini, B. W. Skelton and A. H. White, Inorg. Chem., 2003, 42, 4938 CrossRef.
- A. V. Artem'ev, J. A. Eremina, E. V. Lider, O. V. Antonova, E. V. Vorontsova and I. Yu Bagryanskaya, Polyhedron, 2017, 138, 218–224 CrossRef.
- A. J. Plajer, A. L. Colebatch, M. Enders, Á. García-Romero, A. D. Bond, R. García-Rodríguez and D. S. Wright, Dalton Trans., 2018, 47, 7036–7043 RSC.
- P. Espinet, R. Hernando, G. Iturbe, F. Villafañe, A. G. Orpen and I. Pascual, Eur. J. Inorg. Chem., 2000, 2000, 1031–1038 CrossRef.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- Á. García-Romero, A. J. Plajer, D. Miguel, D. S. Wright, A. D. Bond, C. M. Álvarez and R. García-Rodríguez, Inorg. Chem., 2020, 59, 7103 CrossRef.
- R. Cini, C. Bellucci, G. Tamasi, M. Corsini, M. Fontani and P. Zanello, Inorg. Chim. Acta, 2002, 339, 89–103 CrossRef CAS.
- J. H. Nelson, K. Y. Ghebreyessus, V. C. Cook, A. J. Edwards, W. Wielandt, S. B. Wild and A. C. Willis, Organometallics, 2002, 21, 1727–1733 CrossRef CAS.
- S. Otto and M. H. Johansson, Inorg. Chim. Acta, 2002, 329, 135–140 CrossRef CAS.
- O. F. Wendt, A. Scodinu and L. I. Elding, Inorg. Chim. Acta, 1998, 277, 237–241 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectroscopic and X-ray characterisation. CCDC 2039592–2039603 and 2039862. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03732j |
| This journal is © The Royal Society of Chemistry 2021 |