
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and geometric structure effects on one-electron oxidation of first-row transition metals in the same ligand framework†
Manuel
Boniolo
 a,
Petko
Chernev
a,
Mun Hon
Cheah
a,
Philipp A.
Heizmann
b,
Ping
Huang
a,
Sergii I.
Shylin
a,
Nessima
Salhi
ac,
Md Kamal
Hossain
b,
Arvind K.
Gupta
b,
Johannes
Messinger
*ad,
Anders
Thapper
*b and
Marcus
Lundberg
*ac
a,
Petko
Chernev
a,
Mun Hon
Cheah
a,
Philipp A.
Heizmann
b,
Ping
Huang
a,
Sergii I.
Shylin
a,
Nessima
Salhi
ac,
Md Kamal
Hossain
b,
Arvind K.
Gupta
b,
Johannes
Messinger
*ad,
Anders
Thapper
*b and
Marcus
Lundberg
*ac
aMolecular Biomimetics, Department of Chemistry – Ångström Laboratory, Uppsala University, 75120 Uppsala, Sweden. E-mail: johannes.messinger@kemi.uu.se; marcus.lundberg@kemi.uu.se
bSynthetic Molecular Chemistry, Department of Chemistry – Ångström Laboratory, Uppsala University, 75120 Uppsala, Sweden. E-mail: anders.thapper@kemi.uu.se
cTheoretical Chemistry, Department of Chemistry – Ångström Laboratory, Uppsala University, 75120 Uppsala, Sweden
dDepartment of Chemistry, Chemical Biological Centre, Umeå University, 90187 Umeå, Sweden
First published on 9th December 2020
Abstract
Developing new transition metal catalysts requires understanding of how both metal and ligand properties determine reactivity. Since metal complexes bearing ligands of the Py5 family (2,6-bis-[(2-pyridyl)methyl]pyridine) have been employed in many fields in the past 20 years, we set out here to understand their redox properties by studying a series of base metal ions (M = Mn, Fe, Co, and Ni) within the Py5OH (pyridine-2,6-diylbis[di-(pyridin-2-yl)methanol]) variant. Both reduced (MII) and the one-electron oxidized (MIII) species were carefully characterized using a combination of X-ray crystallography, X-ray absorption spectroscopy, cyclic voltammetry, and density-functional theory calculations. The observed metal–ligand interactions and electrochemical properties do not always follow consistent trends along the periodic table. We demonstrate that this observation cannot be explained by only considering orbital and geometric relaxation, and that spin multiplicity changes needed to be included into the DFT calculations to reproduce and understand these trends. In addition, exchange reactions of the sixth ligand coordinated to the metal, were analysed. Finally, by including published data of the extensively characterised Py5OMe (pyridine-2,6-diylbis[di-(pyridin-2-yl)methoxymethane])complexes, the special characteristics of the less common Py5OH ligand were extracted. This comparison highlights the non-innocent effect of the distal OH functionalization on the geometry, and consequently on the electronic structure of the metal complexes. Together, this gives a complete analysis of metal and ligand degrees of freedom for these base metal complexes, while also providing general insights into how to control electrochemical processes of transition metal complexes.
1 Introduction
The optimization of multi-step redox catalysis performed by molecular complexes requires a fundamental understanding of the factors that determine performance. Redox potentials of individual one-electron processes, the coupling of electron and proton transfer and the binding affinities of reactants and products are often intertwined properties that only a systematic experimental and theoretical study can disentangle.1 As the properties of coordination complexes are determined by the metal–ligand interactions, such analysis should describe their influence in a systematic manner.2 Presently available studies where the metal site is varied within the same coordination sphere mainly focus on a phenomenological description without developing deep insight into the reasons for different reactivities.3,4 Here we begin to fill this gap by a detailed study of one-electron redox reactions in a series of first-row transition metal complexes with the same pentapyridyl ligand framework (Fig. 1). This fundamental approach makes it possible to focus initially on the role of the metal in a well-defined reaction step, followed by a separate analysis of the ligand dimension.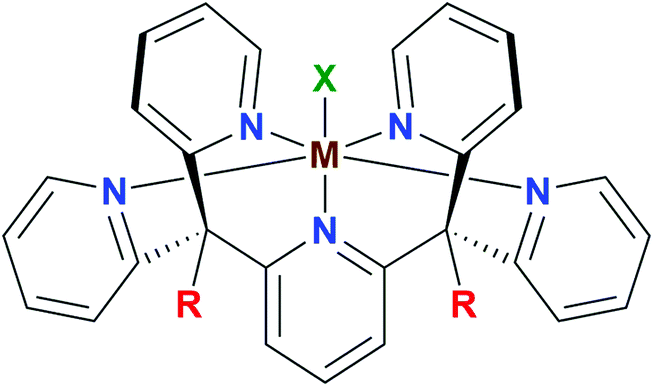 | ||
| Fig. 1 General structure of the pentapyridyl family of metal complexes (M = metal centre, R = ligand substituent, X = apical ligand). | ||
The family of pentapyridyl ligands stemming from Py5, (2,6-bis-[(2-pyridyl)methyl]pyridine) has been extensively adopted in the past two decades to complex different first-row transition metals.5 Feringa et al. reported the first example of FeII and MnII complexes with the methoxy-substituted ligand (Py5OMe), highlighting how the apical coordination site is particularly prone to ligand substitution and therefore can reversibly bind possible substrate candidates for catalytic reactions.6 Simultaneously, Stack et al. studied catalytic applications of the same complexes for the oxidation of organic substrates as they mimic the cofactor of lipoxygenases.7–9 This team also reported structural parameters, magnetic susceptibility, electrochemical behaviour, and optical properties of the series of [M(Py5OMe)Cl]+ (M = Mn, Fe, Co, Ni, Cu and Zn) complexes, but only in their reduced MII oxidation state.4 During the following two decades, Py5 complexes have also been applied as anti-tumour agents,10,11 redox mediators for dye-sensitized solar cells,12,13 and materials with tunable magnetic properties.14–16 In the field of the artificial photosynthesis, Py5-type ligands have been reported to be a suitable scaffold for synthesizing both proton reduction17–20 and water oxidation catalysts.21–24
With the idea of creating a starting point for further functionalization, our group introduced the Py5OH ligand and employed it in homogeneous water oxidation catalysis using either CoII or FeII as the metal centre.25,26 Our recent study on the [FeII(Py5OH)Cl]+ complex showed that, unlike [FeII(Py5OMe)Cl]+, it undergoes a spin-crossover from a high spin (HS) to low spin (LS) configuration, even in the presence of a weak-field ligand like Cl.15 This infers that the peripheral R-groups of the ligand can affect the electronic structure of the metal centre.
Using the Py5OH ligand framework (Fig. 1; R = OH) we synthesized here a series of metal complexes with the general formula [MII(Py5OH)Cl](PF6) (abbreviated as [MII–Cl]), where M = Mn, Fe, Co and Ni. Their one-electron redox potentials were then determined by cyclic voltammetry, and the geometric and electronic structures of both the reduced [MII–Cl] and oxidized [MIII–Cl] species were investigated in powder and/or dissolved form by employing a combination of single-crystal X-ray diffraction (XRD), synchrotron X-ray absorption spectroscopy (XAS) and density-functional theory (DFT) calculations.
The effect of the ligand sphere on the redox potential was then studied by exchange of the apical chloride ligand. This is especially interesting with regard to catalytic reactions as its exchangeability is likely important for substrate activation. To facilitate this, we also prepared the Cl-free [FeII(Py5OH)MeOH](ClO4)2 complex, which upon dissolving in dimethylformamide or acetonitrile resulted in two additional set of complexes: [FeII–DMF] and [FeII–MeCN]. Finally, the electrochemical properties of the [MII–Cl] complexes were compared to those reported previously for [MII(Py5OMe)Cl]+, abbreviated [MII–Cl]OMe. Together, this work represents a consistent set of modifications for the pentapyridyl complexes in Fig. 1, varying metal (M), apical ligand (X) and pentapyridyl substituent (R). Combined with the careful experimental and theoretical analysis of a well-defined one-electron redox event, this provides fundamental insight into the electrochemical processes of transition metal complexes.
2 Results and discussion
2.1 Structures of divalent metal complexes
Single-crystal XRD structures were obtained for all four [MII–Cl] complexes, out of which the [MnII–Cl] (Fig. S1†) and [NiII–Cl] (Fig. 2) structures are reported for the first time. The structures for [FeII–Cl] and [CoII–Cl] from the present study showed good agreement with those published previously.25,26 In all the complexes, the transition metal was embedded in a pocket composed of the five pyridines with a sixth-axial chloride ligand. The metal environment had approximate local C4v symmetry with the Fe–Cl vector along the C4-proper axis.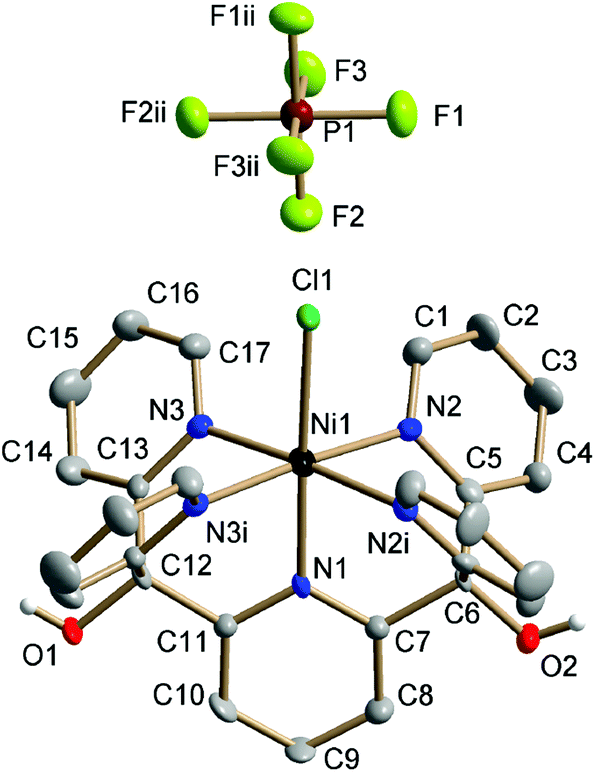 | ||
| Fig. 2 Crystal structure of [NiII–Cl] collected at 150 K. Thermal ellipsoids are drawn at the 50% probability level. Non-hydroxyl H atoms are omitted for clarity. Symmetry codes: (i) x, −1.5 − y, z; (ii) −x, −1 − y, z. | ||
Fig. 3 shows that all the metal–nitrogen bond distances (average of the equatorial M–Neq., blue; axial M–Naxial, red) decreased following the order of the periodic table, for M–Naxial from 2.249 Å in [MnII–Cl] to 2.065 Å in [NiII–Cl]. This was not the case for the metal–chlorine bond distance: although the Mn–Cl bond (2.458 Å) was longer than the Fe–Cl bond (2.419 Å), it increased again for the subsequent Co (2.432 Å) and Ni (2.431 Å) complexes, see Fig. 3 (green). These comparatively long metal–ligand distances are consistent with a high-spin (HS) configuration for all four [MII–Cl] complexes.
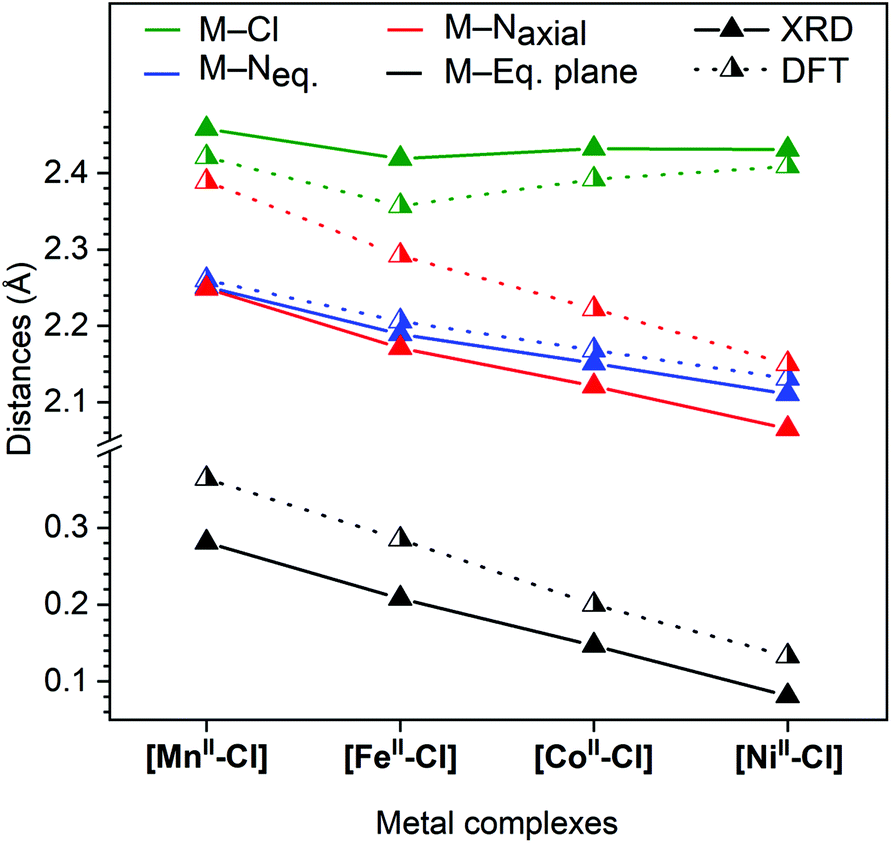 | ||
| Fig. 3 Comparison of distances from single-crystal X-ray diffraction collected at 150 K (full triangle, solid line) and calculated distances using DFT; (half-filled triangle, dotted line). M–Eq. plane, metal displacement from the equatorial plane, which is defined by the four nitrogen atoms: N2, N2i, N3 and N3i (see Fig. 2). | ||
Finally, the metal displacement from the equatorial plane followed a linear trend from 0.281 Å to 0.081 Å in the order Mn > Fe > Co > Ni (Fig. 3, black).
The solid [MII–Cl] complexes were further characterized in powder form (see Experimental section for elemental analyses) by X-ray absorption spectroscopy (XAS) at 20 K. All the complexes exhibited K-edge XANES energy positions that matched the values of divalent reference metal complexes, see Fig. S3–S6.† The simulated distances, obtained from the EXAFS measurements at 20 K of the solid [MnII–Cl], [CoII–Cl] and [NiII–Cl] samples, did not deviate significantly from those determined by XRD at 150 K. However, the EXAFS spectrum of [FeII–Cl] indicated a significantly shorter Fe–Neq. distances, by approximately 0.2 Å. As we reported recently, this is due to a spin-state change (spin-crossover, SCO) from quintet 5[FeII–Cl] to singlet 1[FeII–Cl] that occurs at a temperature below 80 K in the microcrystalline powder sample.15
To get closer to the conditions of the CV experiments, the XAS parameters were also obtained after dissolving the four samples in acetonitrile with electrolyte. The XAS spectra of the [MnII–Cl], [CoII–Cl] and [NiII–Cl] complexes remained unchanged as compared to the spectra recorded for the powder samples (see Fig. S3–S6†). By contrast, large differences in the XAS spectra were observed for [FeII–Cl] under the two conditions. A reasonable XAS fit of the dissolved Fe-complex at 20 K could be obtained with three components: (i) 40% of the chloride-bound LS form that was also observed for the powder, (ii) 40% of a HS form with long metal–ligand bonds, and (iii) 20% of a LS structure where the chloride ligand had exchanged with a solvent molecule. This was supported by a good EXAFS fit at 150 K where only two components, 80% of (ii) and 20% (iii), were necessary (see Fig. S8 and S9†). Dissolving the sample in an electrolyte solution thus led to partial ligand exchange and incomplete SCO down to 20 K.
The electronic and geometric structures of the [MII–Cl] complexes were calculated for all spin multiplicities consistent with 3dn configurations (n = 5–8). Correct spin-state energetics are in general challenging to calculate with DFT due to strong functional dependence.27–30 The results are especially sensitive to the amount of HF exchange, but as the functional dependence varies with the type of bonding it is difficult to get accurate results for a wide range of complexes.29 To address this challenge, we used the SCO in [FeII–Cl] as a reference point for selecting the functional.15
For [FeII–Cl] the B3LYP* functional (15% HF exchange) gave good results. It favoured the quintet over the singlet by 5.3 kcal mol−1 at room temperature, see Fig. 4. This value was 2.2 kcal mol−1 higher than what can be expected from the determined spin-transition temperature of the powder sample at 80 K.15 As the solvated chloride-coordinated sample had remaining HS species down to 20 K, the B3LYP* calculations with 15% HF exchange should thus represent the complex in solution with relatively high accuracy. Calculations of the other complexes showed that they all favour high-spin states, even more so than iron, see Fig. 4 (blue bars) and Table S2.† At RT, the spin-state energetics for [CoII–Cl] was very close to that of [FeII–Cl], but compared to [FeII–Cl] the LS form of the Co-complex was less well-stabilized at low temperatures, so that the calculations did not predict this to be a spin-crossover complex, see Table S5.†
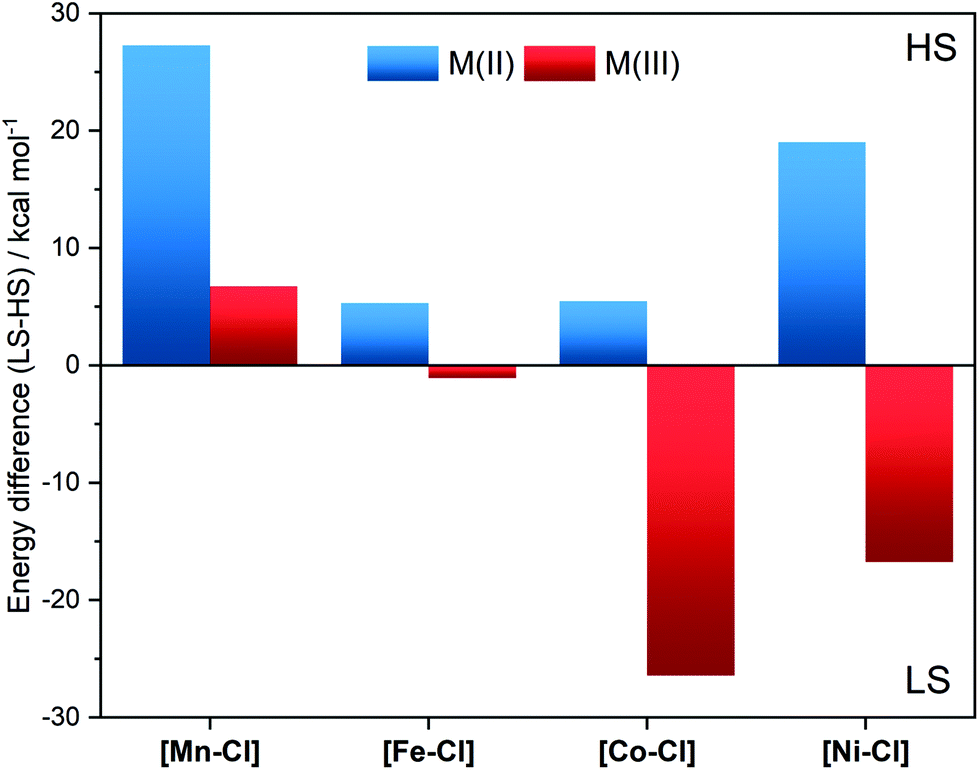 | ||
| Fig. 4 Relative free energies at 298 K of the high-spin states compared to the low-spin states for [MII–Cl] and [MIII–Cl] complexes calculated using the B3LYP* functional and the SMD solvation model for acetonitrile. Note that the species used as the LS form of the d8[NiII–Cl] requires pairing of electrons in one of the two upper d orbitals. | ||
The corresponding DFT geometric structures were, in general, in good agreement with XRD data, see Fig. 3 and Table 1. The Fe–Neq. distances had absolute deviations of less than 0.02 Å, which meant that trends in bond distances were also well reproduced. The agreement is poorer for the axial bonds, with too short M–Cl bonds (−0.1 Å on average) and much too long for M–Naxial bonds (+0.2 Å on average). This led to calculated metal centre positions that were further displaced from the equatorial plane than measured by XRD (by 0.1 Å). However, the structural trends were still very well reproduced, with all M–N distances (M–Neq. and M–Naxial) decreasing following the order in the periodic table, while the M–Cl distances showed the above-described deviation for Co and Ni.
| Complex | Ox. state | M–Neq. | M–Naxial | M–Xa | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| XRD | EXAFSb | DFT | XRD | EXAFSb | DFT | XRD | EXAFS | DFT | ||
| a For XRD, the apical ligand X is chloride for all [MII–Cl] complexes and the oxygen of DMF for the [FeII–Solv] complex. For EXAFS and DFT of [FeII–Solv] the apical ligand is the nitrogen of an acetonitrile molecule. b Average of all M–N distances modelled as one shell. c Distorted 5[MnIII–Cl] after a least-squares EXAFS fit that allows for the distances to the three closest N atoms and the Cl− to be optimized (5[MnIII–Cl]′). d Calculated distances for 5[MnIII–Cl]′ structure. e Fe–N distance: single shell Fe–N distance obtained in the best EXAFS fits employing a sample composition of 40% HS, 40% LS, and 20% chloride substituted LS complex; Fe–Cl distance: average of 50% HS and 50% LS complexes. | ||||||||||
| [Mn–Cl] | II | 2.251 | 2.25 | 2.260 | 2.249 | 2.25 | 2.389 | 2.458 | 2.43 | 2.421 |
| III | — | 2.14c | 2.158d | — | 2.14c | 2.116d | — | 2.22c | 2.264d | |
| [Fe–Cl] | II | 2.189 | 1.99e | 2.206 | 2.171 | 1.99e | 2.292 | 2.419 | 2.38e | 2.357 |
| III | — | 1.99 | 2.034 | — | 1.99 | 2.049 | — | 2.22 | 2.249 | |
| [Co–Cl] | II | 2.151 | 2.14 | 2.168 | 2.121 | 2.14 | 2.222 | 2.432 | 2.40 | 2.392 |
| III | — | 1.97 | 2.009 | — | 1.97 | 2.020 | — | 2.20 | 2.263 | |
| [Ni–Cl] | II | 2.111 | 2.09 | 2.131 | 2.065 | 2.09 | 2.150 | 2.431 | 2.42 | 2.409 |
| III | — | — | 1.993 | — | — | 2.193 | — | — | 2.420 | |
| [Fe–Solv] | II | 2.017 | 1.98 | 2.040 | 1.952 | 1.98 | 2.032 | 1.989 | 1.98 | 1.933 |
| III | — | 1.97 | 2.015 | — | 1.97 | 2.026 | — | 1.97 | 1.945 | |
The choice of functional and basis set size did not affect the calculated distances by more than 0.02 Å, see Table S3,† and the very long Fe–Naxial bond is present also with the local BP86 functional. Despite the deviations of the axial bond distances, the HS structures still give the best agreement with experiment. Structures with lower spin multiplicities all have significantly shorter Fe–Neq. bonds, which led to an underestimation by 0.2 Å, with no significant improvement in the Fe–Naxial distance, see Table S2.†
2.2 Electrochemistry
In cyclic voltammetry (CV) experiments, sweeping from the equilibrium potential towards the positive direction, reversible oxidation processes were observed for all [MII–Cl] complexes (Fig. 5). This was tentatively assigned to a metal-centred one-electron oxidation from [MII–Cl] → [MIII–Cl]. The experimentally determined order of the reduction potentials of the redox couple [MIII–Cl]/[MII–Cl] was: Co (0.08 V) < Fe (0.33 V) < Mn (0.58 V) < Ni (1.17 V), which deviates from the trend of increasing potentials along the periodic table that may be expected on the basis of increasing nuclear charge density. The vertical lines in Fig. 5 indicate the calculated redox potentials, which will be discussed in detail after describing the characterization of the oxidized complexes. In the case of [FeII–Cl], a second reversible redox wave at E1/2 = 0.83 V was observed, which had an overlapping shoulder feature at Ep = 0.72 V in the oxidative scan (marked with * in Fig. 5). This shoulder feature was assigned to the quasi-reversible response of the redox-active couple Cl2/Cl− by the observed increase of this feature after addition of tetrabutylammonium chloride (TBACl) to [FeII–Cl] (Fig. S10†) and by a separate voltammogram of TBACl (Fig. S11†). The second reversible redox wave was likely not an additional one-electron oxidation of FeIII to FeIV, since there was a significant difference in current intensity between the two waves. Instead, the higher redox potential feature likely arose from a secondary species that, based on the cathodic peak analysis, made up about 25% of the total compound.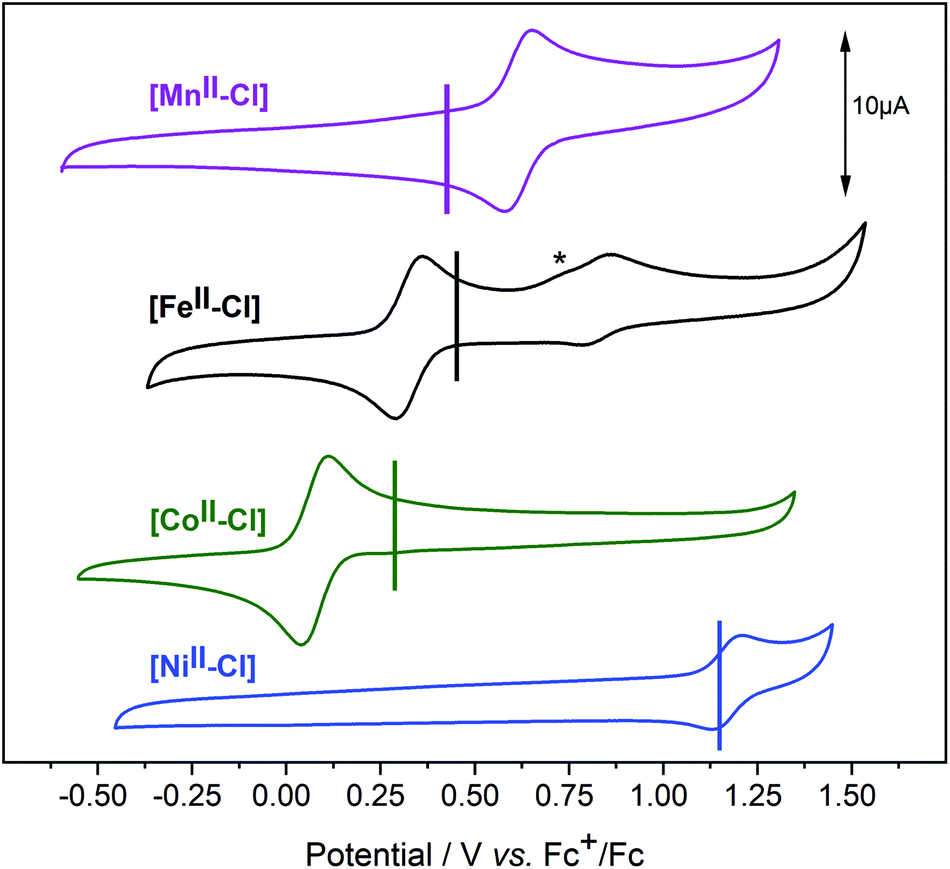 | ||
| Fig. 5 Cyclic voltammograms of the [MII–Cl] complexes in acetonitrile. The CVs were recorded under argon with a sample concentration of 0.5 mM and 0.1 M of TBAPF6 as supporting electrolyte. Scan rate 100 mV s−1. Vertical lines indicate the calculated redox potentials for the III/II oxidation, using the B3LYP* functional and the SMD solvation model for acetonitrile. The anodic current marked as * is assigned to the Cl− oxidation. | ||
Given the observation of the irreversible Cl2/Cl− redox wave, the most likely candidate was the complex where the Cl− ligand was replaced by a solvent molecule, which was also observed in the EXAFS data. This possibility will be analysed in detail in a later section.
We note that due to the difference in temperature between the experiments, the LS 1[FeII–Cl] species observed with EXAFS at 20 K was not present in the room temperature CV experiment. Thus, we estimate that the room temperature composition was 75% HS 5[FeII–Cl] and 25% 1[FeII–MeCN], in good agreement with the CV peak analysis.
2.3 Structures of oxidized complexes
To understand the redox potentials, also the products of the one-electron oxidation need to be analysed. Thus, the metal complexes were electrochemically oxidized in acetonitrile and analysed with XAS. The K-edge positions of the oxidized materials displayed a shift to higher energy compatible with a complete one-electron metal oxidation, see Fig. S3–6.† The only exception was the [NiII–Cl] complex, for which its high redox potential did not allow us to yield a significant amount of the oxidized form with our continuous flow electrosynthesis cell. Oxidation of the three remaining [MnII–Cl], [FeII–Cl] and [CoII–Cl] complexes resulted for [MnII–Cl] and [CoII–Cl] in the expected shortening of the metal–ligand bonds as visible by significant changes in the k-space data, as well as by the shift of the first peak in the Fourier transform (Fig. 7a and b; Table 1). These structural changes observed by EXAFS spectroscopy are compared below with distances obtained by DFT calculations.Oxidation of the [MnII–Cl] complex shortened the average Mn–N distance by about 0.11 Å and the Mn–Cl distance by 0.21 Å (Table 1). A direct EXAFS fit of the oxidized form requires a large Debye–Waller factor when a single Mn–N shell is used (0.085 Å, compared to around 0.065 Å for the other complexes, see Table S4†), indicating a large Jahn–Teller distortion. DFT calculations of the oxidized Mn complex give the lowest energy for a quintet 5[MnIII–Cl] structure (Fig. 4), with the triplet 3[MnIII–Cl] 6.7 kcal mol−1 higher in energy (Table S2†). Compared to the reduced complex, the calculations on the 5[MnIII–Cl] reproduced qualitatively the shorter average Mn–N distances, but maintained a long Mn–Cl distance (2.40 Å, Table S2†), in poor agreement with EXAFS data, see Fig. S12.† The 3[MnIII–Cl] has similar calculated average Mn–N distances and a much shorter Mn–Cl bond length (2.24 Å, Table S2†) as compared to the quintet, which gives better agreement with EXAFS distances (Fig. 6). However, the fit to the EXAFS spectrum was not significantly improved (Fig. S12†). Instead, an alternative quintet structure 5[MnIII–Cl]′ with a shorter Mn–Cl bond of 2.26 Å (Table 1 and Fig. 6) and elongated bonds in the equatorial plane, lies only 0.6 kcal mol−1 above the most stable quintet and gives a much better fit to EXAFS (Fig. S12†). Considering the small energy difference and the good structural match, this alternative quintet structure will also be considered in further comparisons and analyses.
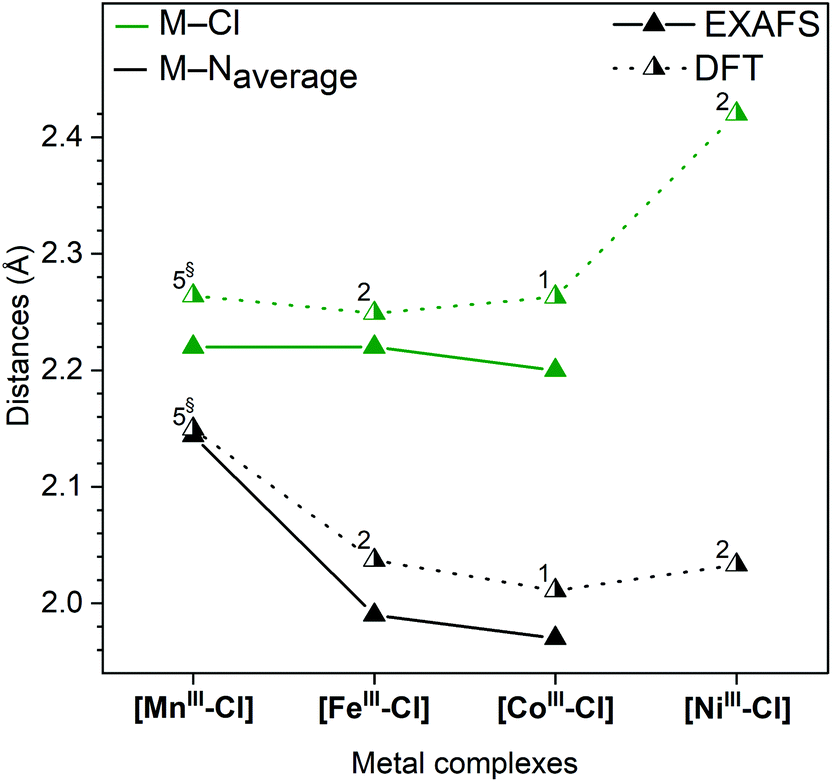 | ||
| Fig. 6 Metal–ligand distances of the oxidized n[MIII–Cl] complexes (dissolved in MeCN) obtained by EXAFS spectroscopy at 20 K (full triangles, solid line) and DFT calculations (half-filled triangles, dotted line). The numbers at the symbols indicate the spin multiplicity of the metal centres. § Calculated distorted 5[MnIII–Cl]′ structure. | ||
For [CoIII–Cl], EXAFS fitting suggests that upon oxidation the average Co–N distance shortens from 2.14 Å to 1.97 Å, and the average Co–Cl distance from 2.40 Å to 2.20 Å. The majority of reported CoIII complexes exist in a LS singlet configuration,31 and the distances found here for [CoIII–Cl] were also consistent with such an assignment. DFT calculations also prefer 1[CoIII–Cl] over 5[CoIII–Cl] by a large margin (26.4 kcal mol−1), see Fig. 4, and 3[CoIII–Cl] is also much higher in energy, see Table S2.† The calculations reproduce the short metal–ligand bonds of the EXAFS structure, although they are overestimated by approx. 0.04 Å.
In contrast to the Mn and Co complexes, the oxidation of [FeII–Cl] to [FeIII–Cl] did not lead to a similar shortening of the bonds and connected EXAFS changes. While the EXAFS spectrum of [FeII–Cl] required three components to be fitted at 20 K, the EXAFS data obtained with the oxidized complex, [FeIII–Cl], could be simulated well with a single species that had a short Fe–Cl distance (2.22 Å) and short average Fe–N distance (1.99 Å), see Table 1. This suggests the oxidation to a LS 2[FeIII–Cl] structure. Unlike the reduced complexes, there was thus no equilibrium between different spin multiplicities or axial ligands (Cl−vs. solvent) after oxidation. Energetically, DFT calculations favour the low-spin over the high-spin 6[FeIII–Cl] structure by 1.1 kcal mol−1 already at 298 K, and this tendency increases with decreasing temperature (Table S5†). The calculated average Fe–N bonds (2.04 Å) in the 2[FeIII–Cl] structure are 0.05 Å longer than predicted by experiment (Fig. 6), but still a much better fit than the 2.17 Å of the average Fe–N bond distance obtained for the 6[FeIII–Cl] complex, see Table S2.†
While no oxidized [NiIII–Cl] suitable for EXAFS analysis could be obtained, DFT calculations indicate a preference for LS 2[NiIII–Cl] over the alternative HS 4[NiIII–Cl] by 16.7 kcal mol−1, see Fig. 4. The LS structure gave short equatorial Ni–N bonds of 1.99 Å, and a much longer axial bond of 2.19 Å, for an average Ni–N bond of 2.03 Å. The calculated NiIII–Cl distance is 2.42 Å, see Fig. 6. The HS structure had much longer Ni–N distances (2.15 Å) and a shorter Ni–Cl distance (2.23 Å).
2.4 Simulated redox potentials
Redox potentials of the [MIII–Cl]/[MII–Cl] couples were simulated using the optimized DFT structures of reduced and oxidized complexes. Calculations of absolute redox potentials of transition metal complexes often show significant errors, up to 0.5 eV.32–37 Highly charged complexes are in general challenging due to large absolute solvation effects.38 Techniques to reduce errors include explicit modelling of the solvent,39 and multi-step calculations to reach a neutral reference complex.40 Here, the main target was to obtain relative potentials between structurally similar complexes, rather than absolute values, and thus a standard approach was selected.The results of our DFT calculations are shown as vertical lines in the CVs plotted in Fig. 5, and are also reported in Table S6.† In agreement with our experiments, the lowest redox potential was found for [CoII–Cl]. The theoretical value of 0.18 V, is +0.10 V higher than the experimental value (0.08 V). This deviation is well within the expected accuracy of the calculations, and is likely due to error cancellation. For [FeII–Cl], the calculated value was 0.37 V, which is in even better agreement with the experiment (0.33 V). Experimentally, [MnII–Cl] had a higher redox potential (0.58 V) than both [FeII–Cl] and [CoII–Cl]. Calculations with the lowest energy structure, 5[MnIII–Cl], gave a value of 0.34 V (−0.24 V vs. exp.), which placed it at the same level as that of the iron complex. Using the alternative 5[MnIII–Cl]′ structure would instead give a redox potential of 0.37 V, which is a minor improvement compared to the experiment. Finally, the calculated redox potential of [NiII–Cl] was 1.19 V, much higher than the other complexes, and in excellent agreement with the experiment (1.17 V).
2.5 Exchange reactions at the sixth ligand position
For [FeII–Cl], the changes in EXAFS data when dissolved in acetonitrile, together with the presence of a second oxidation event in the CV, suggested that the Cl− ligand can be exchanged. Therefore, chloride-free iron complexes, where the sixth ligand position was occupied by a solvent molecule, were prepared (Scheme 1). Crystals suitable for XRD diffraction were obtained from a DMF solution, and the crystal structure of [FeII–DMF] (Fig. S2†) showed that the oxygen of the amide group in DMF coordinated to the iron. For this material, the XRD data showed that the average Fe–N bond length was, at 150 K, shorter by approximately 0.2 Å than in the [FeII–Cl] complex discussed above, indicating singlet configuration (Table 1).41 DFT calculations of the similar [FeII–MeCN] complex gave very good agreement with the experimental (XRD) distances, with a maximum deviation of 0.02 Å.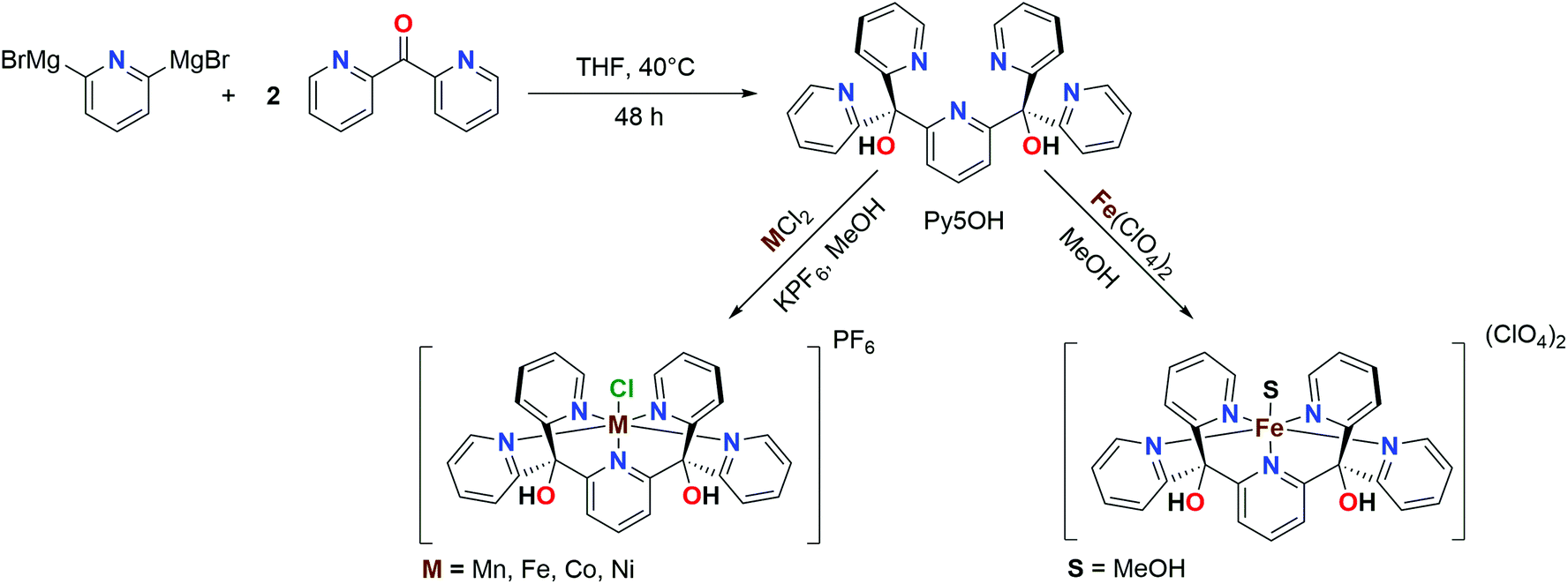 | ||
| Scheme 1 Synthetic route for the ligand and complexes investigated in this work. | ||
Preparing the chloride-free complex in methanol yielded a solid material where MeOH is in the apical coordination site. When this product was dissolved in acetonitrile, methanol was replaced by an acetonitrile molecule ([FeII–MeCN]), as observed in a HR-MS conducted at low ionization energy (Fig. S13†). Taken together, these data suggest that the solvent molecule at the apical coordination site was weakly bound and could be easily exchanged. The EXAFS data of [FeII–MeCN] shown in Fig. 7 could be fitted using a short metal–nitrogen bond length of 1.98 Å (Table 1), consistent with a LS FeII centre. The LS configuration was further supported by a 1H-NMR spectrum of [FeII–MeCN] that showed the presence of a diamagnetic metal complex (Fig. S14†). DFT calculations also favoured the LS form (by 3.2 kcal mol−1) at 298 K and gave metal–ligand distances within 0.02 Å of the EXAFS analysis.
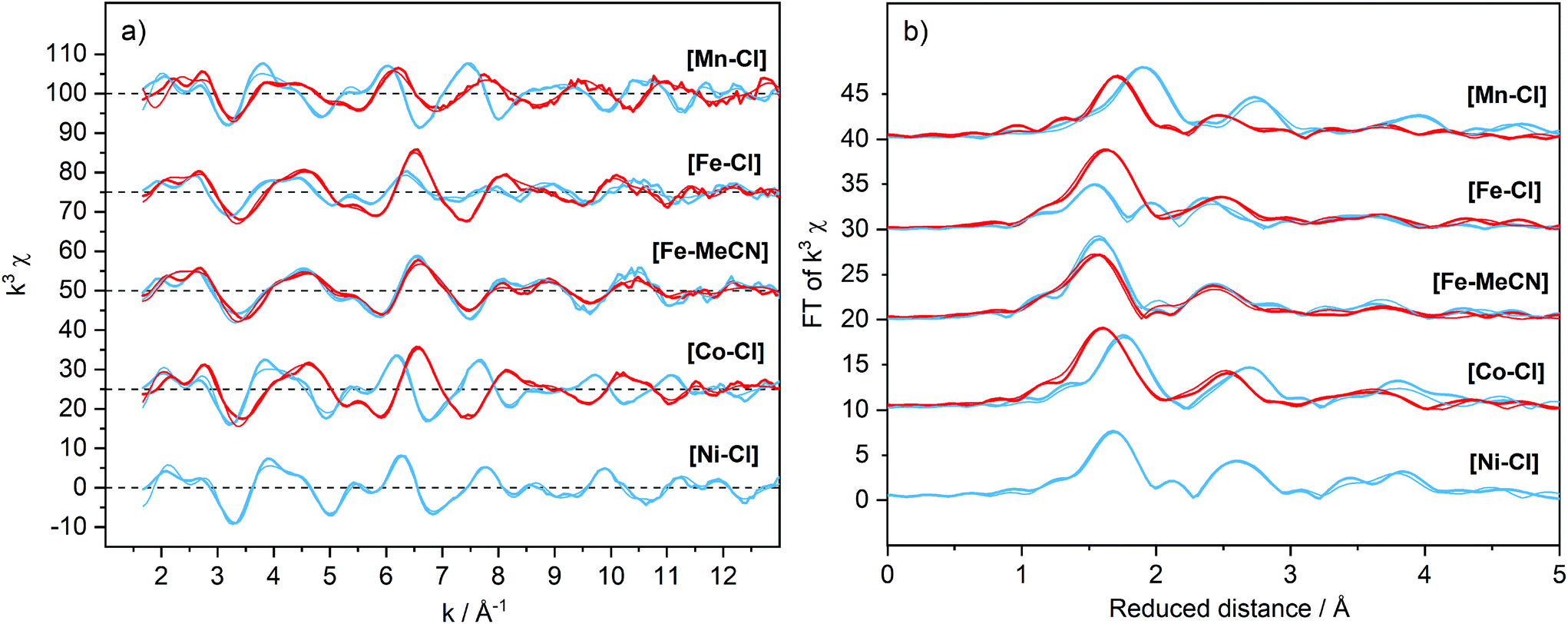 | ||
| Fig. 7 EXAFS spectra weighted by k3 (a) and their Fourier transforms (b) of the [M–X] complexes (1 mM) dissolved in acetonitrile with the addition of TBAPF6 (0.1 M). Spectra were recorded at 20 K and are offset for clarity. Divalent [MII–X], blue; oxidized [MIII–X] samples, red. Simulations of the experimental data are shown as thin lines and the parameters are given in Table S4.† | ||
In contrast to the cyclic voltammogram of [FeII–Cl] in acetonitrile that showed two reversible redox waves (E1/2 = 0.33 V and 0.83 V), the CV of [FeII–MeCN] showed only one reversible redox wave with a potential that matched the position of the second wave of the CV labelled [FeII–Cl] (Fig. 5 and S15†). Fig. 8 shows that addition of Cl− into a solution of [FeII–MeCN] in electrolyte led to the appearance of a reversible redox wave at +0.33 V that coincided with the first reversible couple of [FeII–Cl], suggesting that [FeII–Cl] and [FeII–MeCN] interchanged easily in solution. An electrochemical contribution to this ligand exchange can be excluded as we observed the same shift in the equilibrium by monitoring the Cl-titration by UV-Vis spectroscopy in pure acetonitrile (Fig. S16†). The UV-Vis data obtained in presence of TBAPF6 revealed that the supporting electrolyte partially shifted this equilibrium in favour of the [FeII–MeCN] species (Fig. S17†). This effect was probably due to the increased ionic strength of the medium, which stabilized free chloride in the solvent. This explains, why even after addition of two equivalents of Cl− a mixture of [FeII–Cl] and [FeII–MeCN] was still observed (Fig. 8 and S16a†). In addition, the Cl2/Cl− redox couple was also observed at Ep = 0.72 V in the oxidative scan in Fig. 8.
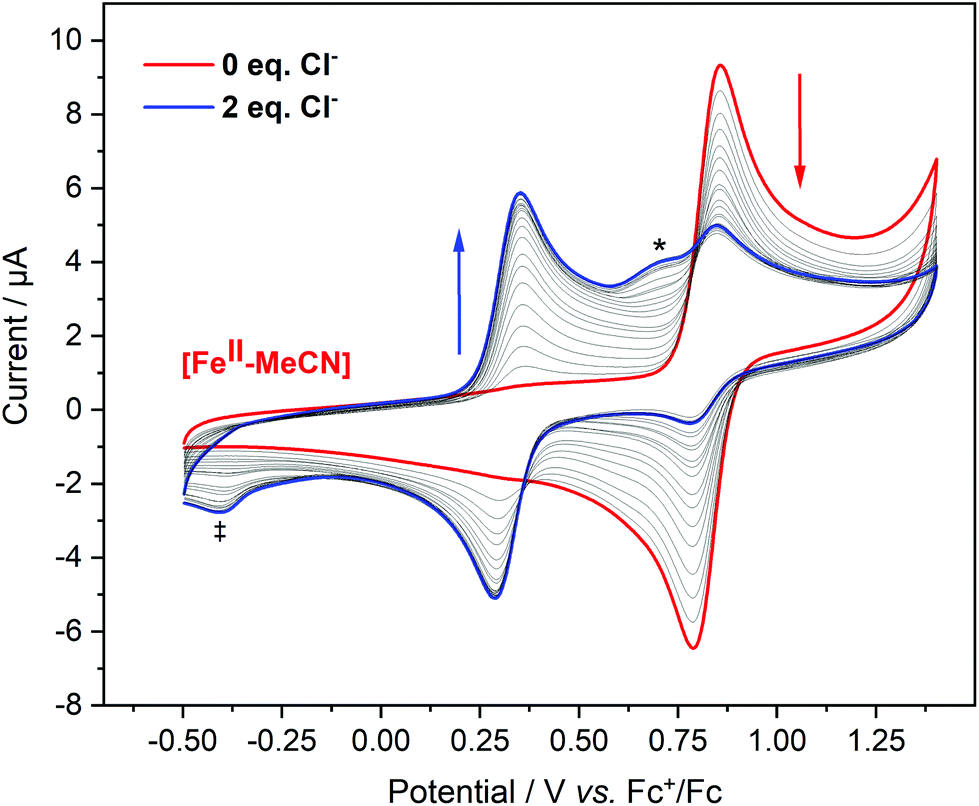 | ||
| Fig. 8 Cyclic Voltammograms of [FeII–MeCN] (0.5 mM) in 0.1 M TBAPF6 in acetonitrile with addition of 0–2 equivalent (in 0.1 equivalent steps; thin lines) of Cl− (as a MeCN solution of TBACl with 0.1 M TBAPF6 as electrolyte) with respect to the metal complex. Curves were corrected according to the dilution factors. The arrows indicate how the reversible peaks are changing upon the additions. Scan rate, 100 mV s−1. The shoulder at 0.72 V marked with (*) was assigned to the oxidation of Cl−. The reductive feature at −0.4 V marked with (‡) was tentatively assigned to a further reaction between Cl2 and the Fe complex. | ||
In contrast to the [MII–Cl] complexes, the oxidation of the [FeII–MeCN] complex occurred without significant structural changes (Fig. 7 and Table 1), although the oxidation was witnessed by the edge shift in the XANES region (Fig. S7†). The near-constant metal–ligand distances (Table 1) suggested a transition between two LS species, i.e. from 1[FeII–MeCN] to 2[FeIII–MeCN], which was supported by DFT energy and structure calculations (Table S2†). DFT calculations of the redox potential of [FeII–MeCN] gave a value of 0.76 V, which was in good agreement with the experimental value of 0.83 V (Fig. S15†).
2.6 Orbital energetics
The above experiments and DFT analysis established that the MII to MIII oxidations can be assigned to clean one-electron oxidations with the expected metal–ligand bond shortenings for the Mn- and Co complexes, while a more complex situation was identified for the Fe-complex, where the apical position was exchangeable for [FeII–Cl] and different spin states can be stabilized depending on temperature, while only one structure was observed after oxidation to FeIII. To rationalize these results and the observed spin multiplicities within simple models, we discuss the electronic structure of our complexes using a molecular orbital framework.The introducing an apical ligand (X) to a hypothetical complex with six equivalent ligands (Oh point group) decreases its symmetry. As shown by our experiments and DFT calculations, this includes for the [MII–Cl] complexes a displacement of the central metal ion from the equatorial plane along the C4-axis. Thus. a C4v symmetry is a fair assignment of the local metal environment. In reality, the symmetry deviates from C4v to a certain extent since there is no authentic C4 rotational axis giving the Py5OH ligand. This deviation, however, does not change the principles in the following section.
The calculated electronic structure of the Py5 complexes are consistent with a previous analysis that described the pyridine rings as being predominantly σ-donor and π-acceptor ligands,5 while Cl− as σ and π donor. In a C4v symmetry, the orbital degeneracy from the well-known cubic environment is lifted. Consequently, the metal 3d-dominated t2g and eg orbitals in a virtual Oh symmetry transform to b2 + e and a1 + b1 respectively, see Fig. 9. The lowest b2 level is related to a dxy orbital (equatorial pyridines π-bonding), above which is two-fold degenerated e level related to dxz and dyz orbitals (pyridine π-bonding and Cl− π-antibonding). These three orbitals will be labelled as π-type orbitals. Higher in the energy are a1 and b1 levels, both σ-type orbitals related to dz2 (Cl−/pyridine σ-antibonding) and dx2−y2 (pyridine σ-antibonding), respectively. The ordering of these two levels will be discussed in more detail below.
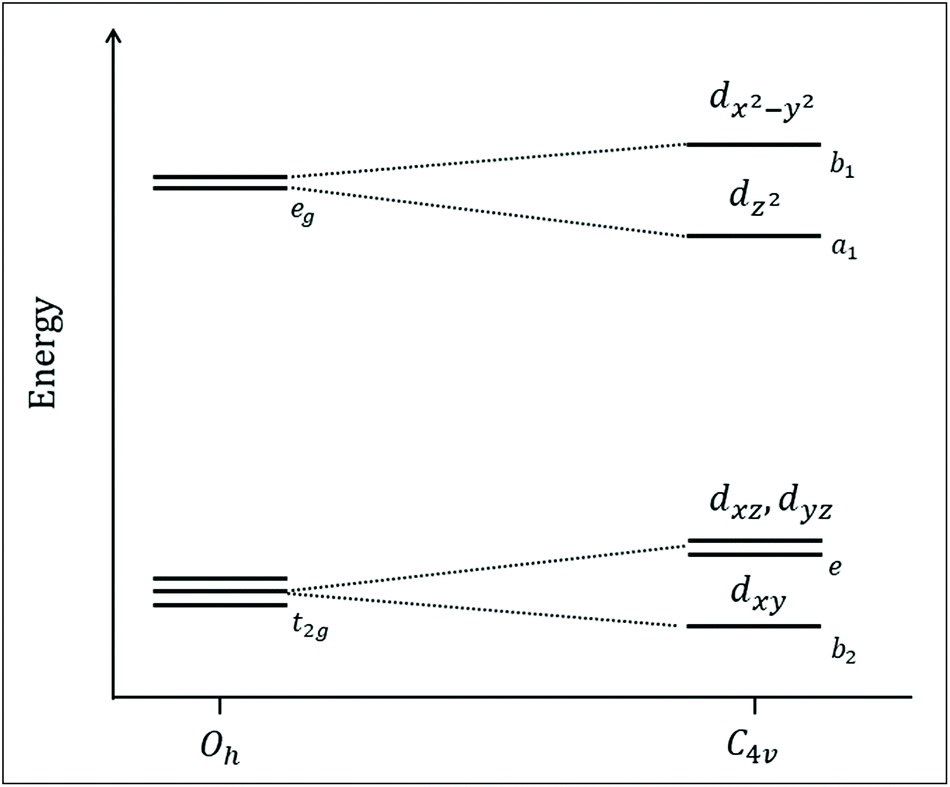 | ||
| Fig. 9 Schematic orbital energy diagram of the [MII–Cl] complexes investigated in this study, including a descent in symmetry from a hypothetical complex with six equivalent ligands (Oh point group) to local C4v symmetry at the metal. The relative position of the dx2−y2 and dz2 orbitals will be discussed further below. | ||
Going from left to right in the periodic table lowers the energy level of the metal orbitals because the additional nuclear charge is only partially screened by the extra electron. This lowering is even more pronounced when increasing the formal oxidation state. For ligand-donor bonding, lowering the metal level leads to smaller energy differences between metal and ligand orbitals, and thus a stronger interaction. For ligand-acceptor binding, it instead leads to a larger energy difference between metal and empty ligand energy levels and weaker interactions. In general, the energy difference between the π-levels and the antibonding σ-levels increases, which corresponds to a larger ligand-field splitting. These general principles can now be used to rationalize trends in geometric structure, spin-state energetics, and electrochemical behaviour.
2.7 Trends in geometric and electronic structure for different metals
Starting with the geometric structures of the reduced [MII–Cl] complexes, there is a clear trend of decreasing metal–ligand bond distances from Mn to Ni, see Fig. 3. This is consistent with a decrease in ionic radius with higher nuclear charge Z. However, the change in ionic radius from Mn to Ni is only 0.03 Å, while the average M–N distances decrease by 0.15 Å. At the same time, the M–Cl distance decreases when going from [MnII–Cl] to [FeII–Cl], but then increases slightly for later elements. As all reduced complexes are predicted to be HS, the number of electrons in the π-type orbitals increases from three to six along with the periodic table, while the number of electrons in σ-type orbitals is constant at two. Going from 6[MnII–Cl], which has one unpaired electron in each 3d orbital, to 5[FeII–Cl] adds a spin-down electron in the lowest orbital dxy. This orbital is weakly bonding with respect to the equatorial Fe–N and the effect of an extra electron is a strengthening of the Fe–N bonds and a slight decrease in metal–ligand bond lengths. At the same time, going from Mn to Fe lowers the energy of the metal 3d orbitals, which leads to stronger σ-bonding and a reduction in all bond distances. As both effects work in the same direction, all bond distances contract.The results are more interesting when going from Fe to Co. In 4[CoII–Cl] the extra electron goes into the dxz/dyz level, which is bonding with respect to the pyridines and anti-bonding with respect to Cl−. The added electron leads to shorter M–N bonds and longer M–Cl bonds. At the same time, increased donor bonding should have an overall contracting effect. Still, for the metal–chlorine bond, the anti-bonding effect dominates, and the bond length increases. Finally, going from 4[CoII–Cl] to 3[NiII–Cl] puts another electron into the dxz/dyz orbitals, which leads to similar changes as seen when going from Fe to Co. The molecular orbital analysis can thus fully explain the bond length trends for the reduced complexes.
The metal–ligand distances of the oxidized complexes show a complex behaviour upon exchange of the metal (Fig. 6). The computational analysis of the [MnIII–Cl] compound is complicated because of the deviations between the experiment and the lowest-energy 5[MnIII–Cl] (π3σ1) structure. The short Mn–Cl bond from EXAFS (2.22 Å) suggests that an electron is removed from the anti-bonding σ-type dz2 orbital upon oxidation, but the calculations instead give for 5[MnIII–Cl] a long Mn–Cl and shorter Mn–Neq. bonds, see Table S2,† consistent with an electron taken from the dx2−y2 orbital. The alternative 5[MnIII–Cl]′ structure, which with shorter axial and longer Mn–Neq. bonds, gives a better EXAFS match (Fig. 6), is more consistent with an empty dz2 orbital. It is thus possible that the calculations remove the electron from the wrong σ orbitals in this oxidation process.
As the octahedral HS MnIII complex shows a Jahn–Teller distortion, the difference between the two calculated structures can also be viewed as a change in distortion axis.
As the calculations consistently overestimate the axial Mn–N bond distances, it is possible that they artificially favour the 5[MnIII–Cl] structure with a Jahn–Teller-like elongation in this direction. Overestimations of the MnIII JT distortion have also been previously observed in DFT calculations.42
For 2[FeIII–Cl] theory and experiment largely agree, see Fig. 6. The 2[FeIII–Cl] complex has a LS (π5σ0) electron configuration and compared to 5[MnIII–Cl] this means extra electrons in dxy and one of the dxz, dyz orbitals while removing the final σ electron. This leads to short axial as well as equatorial bonds. Going further from 2[FeIII–Cl] to LS 1[CoIII–Cl] (π6σ0) adds an electron to dxz/dyz, and as these are antibonding with respect to Cl−, together with the general contraction with larger Z there should be small changes in the M–Cl distance while the M–N distances decrease when going from Fe to Co (Fig. 6). Incidentally, the extra electron is here added to the same orbital level as for the comparison of the reduced iron and cobalt complexes. Finally, the trend when going from 1[CoIII–Cl] to 2[NiIII–Cl] (π6σ1) can only be derived from calculations as the oxidized nickel complex could not be obtained. They show a significant increase in M–Cl bond length and a slight increase in the average M–N bond length. The increase in bond distances is expected as the added electron is placed in an σ antibonding orbital. As the bond distances increase more along the C4-axis, the calculations suggest that the electron ends up in the dz2 rather than the dx2−y2 orbital. LS NiIII complex is expected to show a similar Jahn–Teller-like distortion as HS MnIII, and the long M–Cl distance is similar to that of the 5[MnIII–Cl] structure. However, as seen from the [MnIII–Cl] calculations, it is possible that the structure instead would have a distortion along the equatorial M–N plane.
The spin-state energetics are governed by two major factors, the ligand-field strengths and the differences in exchange stabilization between electron configurations. If the former is sufficiently large, the benefit of being at the lower orbital levels competes with and overcomes the loss of exchange stabilization and the electrons fill lower b2 and e orbitals, resulting in LS configurations. The exception is NiII where both spin configurations have two electrons in the upper two levels. In general, both ligand-field strengths and the exchange interactions increase along the periodic table. However, with changing d-electron count, the difference in the number of ligand-field excitations and exchange interactions change between LS and HS states. The resulting spin-state energetics in Fig. 4, therefore, do not follow any linear trend but are rather consistent with the spectrochemical series of metals.
Taking the reduced complexes as an example, [MnII–Cl], it has a relatively weak ligand field and the sextet has all electrons with the same spin orientation, leading to a highly stable configuration as seen in Fig. 4. Going to d6 FeII and d7 CoIII, all electrons can no longer have the same spin orientation, and the ligand-field splitting increases, resulting in the lower relative stability of the HS configurations. As mentioned above, for d8[NiII–Cl] b2 and e orbitals (b22e4) are filled in all configurations, and the high spin is favoured because it allows for a1 and b1 to be half-filled with electrons in the same spin-orientation.
2.8 Trends in redox potential for different metals
An important target of this study has been to understand the factors that determine the redox potentials of different base metals in the Py5OH complexes. Starting from a basic atomic picture, the redox potential increases when going from left to right in the periodic table as the energy of the 3d levels decrease with increasing nuclear charge. For the Py5OH complexes, the electrochemical experiments instead gave the trend Co < Fe < Mn < Ni. To analyse the effect of the ligand environment, we consider electronic, geometric, and spin degrees of freedom.From Fig. 5 it is clear that the redox potentials of all complexes except [MnII–Cl] are well described by the calculations, not only in terms of relative but also absolute potentials. The main discrepancy is that manganese is too easy to oxidize according to the calculations. However, the deviation, −0.24 eV, is within the expected accuracy of the method, and the calculations reproduce the overall experimental trend well enough to be used to rationalize the trends in redox potential.
The oxidation process will be analysed in three schematic steps. The first step is the removal of an electron from the highest occupied molecular orbital, which in all cases is the σ-type dx2−y2 orbital, and the subsequent orbital relaxation. This is followed by geometry relaxation to the minimum energy geometry of the oxidized species. Finally, in cases where a more stable spin multiplicity existed, spin change was allowed.
The description of [MnII–Cl] oxidation is straightforward: removing an electron is equivalent to going from 6[MnII–Cl] (π3σ2) to 5[MnIII–Cl] (π3σ1). Including only orbital relaxation would lead to a hypothetical redox potential of 1.11 V, see Fig. 10. This is followed by geometric relaxation of the oxidized complex to its minimum energy, which leads to a drop in the potential to 0.34 V. As the quintet is also the lowest energy of [MnIII–Cl], this is also the final calculated redox potential. Using instead the alternative 5[MnIII–Cl]′ structure, which would be consistent with the removal of a dz2 electron, would give a very similar analysis, although with a slightly higher final potential (0.37 V). The relative order of dz2 and dx2−y2 might thus not be correctly predicted, but this does not seem to have a significant effect on the analysis.
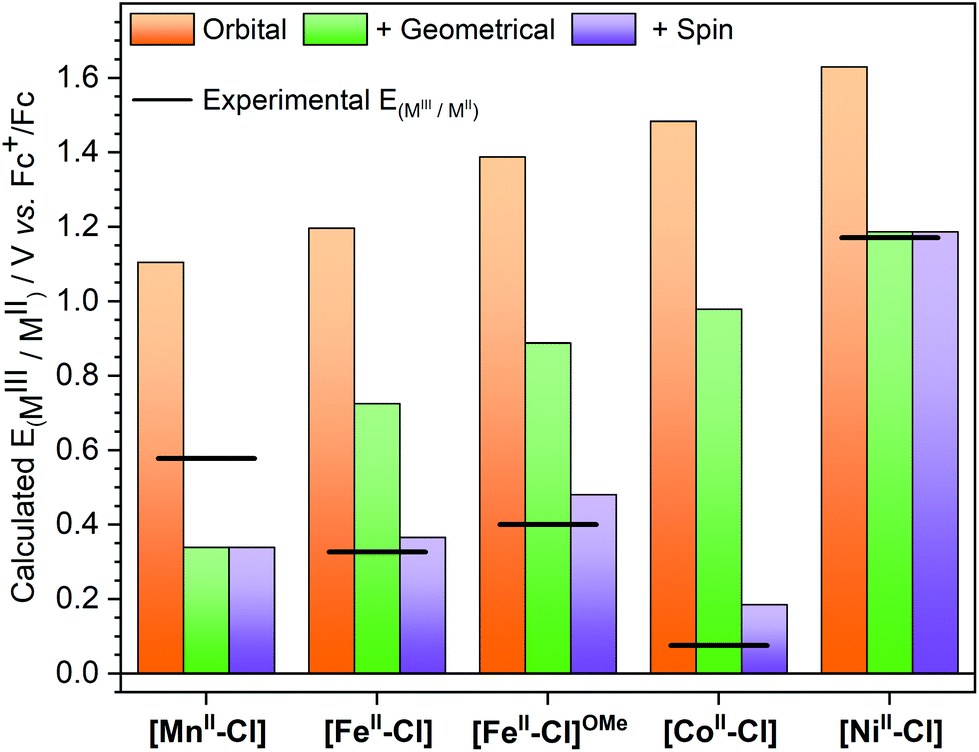 | ||
| Fig. 10 Theoretically calculated half potentials for the [MIII–Cl]/[MII–Cl] redox couples. Allowing only for orbital relaxation after removal of an electron from the same orbital in all complexes, orange bars; allowing for geometric relaxation, green bars; allowing for spin multiplicity change, purple bars. The experimental redox potentials are shown as a line. [FeII–Cl]OMe represents the [FeII(Py5OMe)Cl]+ complex with the experimental value reported by Stack et al.4 | ||
For [FeII–Cl], the oxidation is a 5[FeII–Cl] (π4σ2) to 2[FeIII–Cl] (π5σ0) transition. Taking an electron from a σ-type orbital leads initially to a 4[FeIII–Cl] π4σ1 configuration at 1.20 V, which then drops to 0.73 V upon geometry relaxation, see Fig. 10. However, for iron, there is an additional change in spin multiplicity from a quartet to a doublet, which gives the final calculated potential of 0.37 V. The analysis for the 4[CoII–Cl] (π5σ2) to 1[CoIII–Cl] (π6σ0) transition is similar to the case for iron. The oxidation to 3[CoIII–Cl] (π5σ1) gives potentials of 1.48 and 0.98 V after orbital and geometric relaxation, respectively. For [CoIII–Cl], the singlet is much more stable than the triplet, which leads to a drop in the potential down to 0.19 V. Finally, for [NiII–Cl] the electron is also taken from a σ-type orbital as it goes from 3[NiII–Cl] (π6σ2) to 2[NiIII–Cl] (π6σ1). As expected, the removal of a σ electron is more difficult in [NiII–Cl] than in the other complexes, leading to a potential of 1.19 V after geometric relaxation. As there is no further change in spin multiplicity, this is also the final redox potential, which is then much higher than for any other complex in the series. From the trends in Fig. 10 it is thus clear that if only orbital and geometry relaxation would occur, the potentials of all our Py5OH complexes would follow the order in the periodic table, i.e. show a higher redox potentials with higher Z. It is thus the difference in energy stabilization from potential changes in spin multiplicity that determines the final order of the redox potentials. The calculations can thus be used to rationalize the changes in electrochemical properties between different base metals.
2.9 Exchange reactions at the sixth ligand position
Exchange reactions at the sixth ligand position are likely required for the activation of substrates in catalytic reactions involving the [MII–Cl] complexes. From the experimental data, it is clear that [FeII–Cl] can release Cl− in the electrolyte acetonitrile solution. To get insight into the process, the reaction energy for chloride substitution by the solvent acetonitrile was calculated for the four metal complexes using DFT. The calculations show a trend in Cl− binding energies consistent with the distance analysis (Fig. 11), where binding in [FeII–Cl] is stronger than in [MnII–Cl] due to the better overlap between metal and ligand donor orbitals. The Fe–Cl bond is also stronger than in [CoII–Cl], because the latter has additional electrons in the anti-bonding dxz/dyz orbitals as described above. Finally, the binding energy to [NiII–Cl] is the highest in the series, likely due to further metal–ligand mixing with higher Z.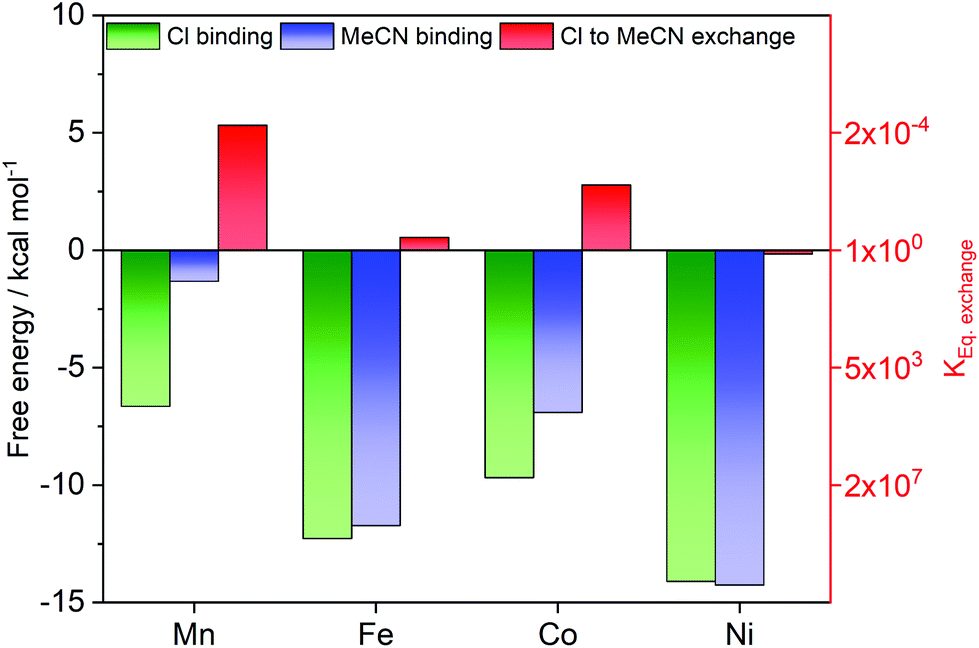 | ||
| Fig. 11 Calculated ligand binding energies for chloride [MII–Cl] (green bars) and acetonitrile [MII–MeCN] (blue bars) complexes in acetonitrile as solvent. The difference in binding energy is the Gibbs free energy required for ligand exchange ([MII–Cl]+ + MeCN ⇌ [MII–MeCN]2+ + Cl−) (red bars; right red axis for the equilibrium constant). All values are calculated at 298 K with the B3LYP* functional and the SMD solvation model. | ||
The ligand exchange equilibrium is instead determined by the energy costs for solvating the Cl− ion and the binding energy of the acetonitrile ligand, which binds stronger to Fe and Ni compared to Mn and Co. Compared to the chloride complex, acetonitrile favours the singlet state by 8.5 kcal mol−1, which makes [FeII–MeCN] a low-spin complex.
However, the rest of the [MII–MeCN] complexes remain HS, even though the energy difference is only 1.8 kcal mol−1 for [CoII–MeCN]. That means all complexes except iron have electrons in the anti-bonding dz2 orbitals. The reason for stronger bonding in Ni compared to Co could be a combination of improved overlap and the fact that the extra electron now appears in a dxz/dyz orbital that is bonding between metal and MeCN.
Taken together, the calculations show that the exchange energy, which is the difference between the two binding energies, is close to zero for [FeII–Cl] and [NiII–Cl], while it is slightly positive for [MnII–Cl] and [CoII–Cl], see Fig. 11. For [MnII–Cl], [FeII–Cl] and [CoII–Cl] this reproduces the trend observed in the experiments, as we observed the exchange of the apical ligand only for the [FeII–Cl] complex. By contrast, for [NiII–Cl] the thermodynamically expected ligand exchange was not observed, which may indicate a kinetic limitation of this process. Interestingly, for the related [NiII(Py5Me)Cl]+ complex the chloride was recently reported to exchange with the solvent.23
Calculations of [FeIII–Cl] show that in the oxidized state, the corresponding exchange reaction for a neutral solvent molecule is unfavourable, which is consistent with the EXAFS observations of [FeIII–Cl] described above. The difference in calculated redox potential between [FeII–Cl] and [FeII–MeCN] can also be attributed to the difference in charge, with the anionic ligand favouring lower redox potentials.
2.10 Effects of Py5 modifications
The properties of the complexes can be further modified by synthetic changes to the Py5 ligand. Here the focus is on a comparison of the hydroxyl-substituted Py5OH and previously published results of the methoxy-substituted Py5OMe ligands. The steric hindrance of the methoxy groups in Py5OMe-ligated complexes induces a tilting of the axial pyridine ring relative to the equatorial plane and a nonsymmetrical pyridine coordination in the equatorial plane.15 In the case of Py5OH, the perpendicular pyridine is aligned with the axial axis and a less distorted octahedral geometry is observed. We previously calculated that the distortion in Py5OMe leads to a 2.9 kcal mol−1 destabilization of the LS state,15 so that [FeII(Py5OMe)Cl]+, unlike [FeII(Py5OH)Cl]+, stays HS at all temperatures and does not show a SCO behaviour.Electrochemically, the Py5OMe complexes have redox potentials that are 0.08 V higher than the Py5OH complexes, see Table S6.† The exception is Ni, where the reported redox potential of the Py5OMe complex is 0.78 V lower.4 Based on the good agreement between calculated and measured half potentials for the [NiIII–Cl]/[NiII–Cl] redox couple in this study, we assume that the reported values for [NiII(Py5OMe)Cl]+ are not reflecting the same redox couple.
To analyse the differences between the two versions of the ligand, the redox potential of the [FeII(Py5OMe)Cl]+ complex was calculated using the same protocol as for [FeII–Cl]. With this ligand, the ferric complex is also LS, although by a very small margin at 298 K (0.7 kcal mol−1). The calculated redox potential is 0.11 V higher than for the [FeII–Cl] complex, in good agreement with the experimental difference of 0.07 V. Breaking down the process into the different steps shows that the main contribution is the increased energy required to remove the σ electron (+0.18 V), while geometric relaxation and changes in spin multiplicity work in the opposite direction ([FeII–Cl]OMe in Fig. 10). In the calculations, the electron is taken from the antibonding dx2−y2 orbital, and the structural distortion with the Py5OMe ligand lowers the dx2−y2 level because of poorer overlap. This effect is also visible from the longer equatorial bond distances, by 0.08 Å, in [FeII–Cl]OMe. If the observed differences in redox potential could be directly translated into differences in barrier heights, a 0.1 eV difference corresponds to a hundred-fold effect on reaction rates. This shows the potential of modifying Py5 type ligands for tuning the reactivity of these complexes.
3 Conclusions
Herein we reported the first comprehensive analysis of the effects of substituting the central base metal, the auxiliary ligand, and distal modification to the ligand framework have been on the redox potential of Py5 coordinated complexes. This analysis was based on a careful structural characterization of both reduced and oxidized species. The trends in structure, spin-state energies, redox potentials, and ligand-exchange reactivity were then rationalized in a molecular orbital picture, supported by DFT calculations. The deviations from a continuous increase in redox potential with increasing nuclear charge were explained by significant stabilization from changes in spin multiplicity for the Fe and especially Co complexes, leading to large stabilizations of the oxidized states and correspondingly lower redox potentials. Moreover, we demonstrated that the exchange dynamics of the apical ligand, which is observed for Fe in both EXAFS and CV data, can be explained by the favourable binding of the solvent molecule, rather than a weak Fe–Cl interaction. Finally, the analysis quantifies the role of Py5 ligand modifications far from the metal site, which had, in line with previously reported spin-state energetics,4,15 also measurable effects on the electrochemical behaviour. Thereby, this study shows that even a simple one-electron oxidation reaction can entail unexpected complexity and tunability. Thus, developing efficient and stable multi-electron catalysis with base metals requires a detailed study of all steps of the catalytic cycle. We also demonstrated that DFT calculations could be employed to understand, in most cases, the trends observed. We thus expect that this systematic approach will, in the long run, facilitate deriving design principles allowing the development of efficient and stable catalysts via predictive calculations.4 Experimental
4.1 Synthesis
All starting reagents were obtained from commercial sources and used as received. All glassware were cleaned and dried overnight at 120 °C. The synthesis of the metal complexes was conducted under dry Argon atmosphere. The synthesized samples were stored in air without observing any degradation.The complexes were fully characterized to verify the purity of the desired product by means of 1H-NMR, FT-IR, UV-VIS, HR-MS and elemental analysis as described in the ESI.† The oxidised metal complexes were obtained by electrolysis in acetonitrile solution (1 mM) as described in the next section.
Caution: Perchlorate salts are potentially explosive and should be handled with care.
Py5OH was synthesized following a procedure introduced by us recently.15 To a dry THF solution (40 ml) of 2,6-dibromopyridine (0.65 g, 2.72 mmol) in a 100 ml 3-neck round bottom flask, an excess of Mg (0.25 g, 102 mmol) was added. After sonicating the mixture for 20 minutes at 35 °C using an ultrasonic bath (45 kHz frequency, model USC300TH, VWR Collection) most of the starting solid Mg was dissolved and the transparent solution turned dark. A THF solution (20 ml) of di(2-pyridyl)ketone (1.00 g, 5.44 mmol) was added dropwise to the Grignard reagent with subsequent formation of a white precipitate. The mixture was stirred for 48 hours followed by the addition of 10% HCl (30 mL). The organic solvent was evaporated and the aqueous solution was washed with CH2Cl2 (2 × 50 mL). Neutralization of the aqueous solution with saturated Na2CO3-solution was followed by extraction with CH2Cl2 (3 × 100 mL). The CH2Cl2 solvent was evaporated and the penta-pyridylcarbinol product was recrystallized from hot acetone (250 mg, 0.558 mmol, yield: 10.2%). 1H-NMR (400 MHz, CDCl3): δ = 7.17 (4 H, t of d, J1 = 5.3 Hz, J2 = 2.3 Hz, 5-H of Py arms (py-a)), 7.55–7.60 (8 H, m, 3-Hpy-a, 4-Hpy-a), 7.71 (3 H, m, 2-H and 3-H of bridging Py), 8.50 (4 H, m, 6-Hpy-a) ppm. 13C-NMR (400 MHz, CDCl3): δ = 162.1, 161.2, 147.2, 137.8, 136.5, 123.5, 122.5, 120.8, 80.9 ppm. MS (+ESI-ToF): m/z: 448.1878 [Py5OH + H+]+, 470.1689 [Py5OH + Na+]+.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and kept under vacuum overnight (112.4 mg, 0.16 mmol, yield: 49.4%). High Resolution ESI-MS shows two main molecular fragments at 537.07329 m/z, [MnII(Py5OH)Cl]+ and 501.10787 m/z [MnII(Py5OH) − H+]+. The UV-Vis spectrum presents a strong absorption below 300 nm with a shoulder at 325 nm (ε = 0.76 × 103 M−1 cm−1), see Fig. S15.† Solid FT-IR (KBr) of the complex shows the same vibration modes as the ligand with a blueshift of 7 cm−1 and the characteristic P–F stretching at 842 cm−1 from the PF6− ion.43 MnC27H21N5ClO2PF6 (682.85 g mol−1) calcd C 47.49, H 3.10, N 10.26, Cl 5.19; found C 46.76, H 4.37, N 10.41, Cl 5.14.
:1) and kept under vacuum overnight (112.4 mg, 0.16 mmol, yield: 49.4%). High Resolution ESI-MS shows two main molecular fragments at 537.07329 m/z, [MnII(Py5OH)Cl]+ and 501.10787 m/z [MnII(Py5OH) − H+]+. The UV-Vis spectrum presents a strong absorption below 300 nm with a shoulder at 325 nm (ε = 0.76 × 103 M−1 cm−1), see Fig. S15.† Solid FT-IR (KBr) of the complex shows the same vibration modes as the ligand with a blueshift of 7 cm−1 and the characteristic P–F stretching at 842 cm−1 from the PF6− ion.43 MnC27H21N5ClO2PF6 (682.85 g mol−1) calcd C 47.49, H 3.10, N 10.26, Cl 5.19; found C 46.76, H 4.37, N 10.41, Cl 5.14.
High Resolution ESI-MS shows two main molecular fragments at 541.08128 m/z, [CoII(Py5OH)Cl]+ and 505.10374 m/z, [CoII(Py5OH) − H+]+. The UV-Vis spectrum shows a strong absorption below 400 nm and a weak broad composite band between 425 nm and 550 nm (ε460 = 0.04 × 103 M−1 cm−1), see Fig. S15.† Solid FT-IR (KBr) of the complex shows the same vibration modes as the ligand with a blueshift of 5 cm−1 and the characteristic P–F stretching at 842 cm−1 from the PF6− ion.43 CoC27H21N5ClO2PF6 (686.84 g mol−1) calcd C 47.21, H 3.08, N 10.20; found C 48.64, H 4.16, N 10.43.
The UV-Vis spectrum shows a strong absorption below 300 nm with a shoulder at 320 nm (ε = 0.75 × 103 M−1 cm−1), see Fig. S15.† Solid FT-IR (KBr) of the complex shows the same vibration modes as the ligand with a blueshift of 4 cm−1 and the characteristic P–F stretching at 842 cm−1 from the PF6− ion.43 NiC27H21N5ClO2PF6·4H2O (758.66 g mol−1) calcd C 42.75, H 3.85, N 9.23, Cl 4.67; found C 42.55, H 3.90, N 9.23, Cl 4.91.
FeC28H25N5Cl2O11 (734.28 g mol−1) calcd C 45.80, H 3.43, N 9.54; found C 46.3, H 4.30, N 10.98.
4.2 X-ray diffraction
Single crystals suitable for measurements were obtained for all four [MII–Cl] complexes and the [FeII–DMF] complex by slow diffusion of toluene into a dimethylformamide (DMF) solution of the complex. All the measurements were performed using graphite-monochromatized Mo Kα radiation at 150 K using a Bruker D8 APEX-II equipped with a CCD camera. Data reduction was performed with SAINT.44 Absorption corrections for the area detector were performed using SADABS.45,46 The structure was solved by direct methods and refined by full-matrix least-squares techniques against F2 using all data (SHELX).45,46 All non-hydrogen atoms were refined with anisotropic displacement parameters if not stated otherwise. Hydrogen atoms were constrained in geometric positions to their parent atoms. The structures were refined using the OLEX2 suite of programs.47 See Table S1† for the detailed parameters.4.3 X-ray absorption spectroscopy
X-ray absorption spectroscopy measurements were performed at the KMC-3 beamline at the BESSY II synchrotron facility in Berlin, Germany. The incident X-ray energy was scanned through the Mn, Fe, Co, and Ni K-edge regions using a silicon (111) double-crystal monochromator. Measurements were performed with samples positioned at 45° with respect to the incident beam in a liquid helium-cooled cryostat (Oxford Instruments). Samples in powder form were mixed with boron nitride in a weight ratio of 1:30. Solution samples of the complexes (1 mM) were prepared in dry acetonitrile. A 10 μm Fe, Co, or Ni foil (Goodfellow Cambridge Limited) positioned behind the sample served as an energy calibration standard. For Mn, a thin layer of KMnO4 was used. Energy calibration was done by assigning the position of the maximum of the first derivative of the absorption of the Fe, Co, and Ni foils to energies 7112 eV, 7709 eV, and 8333 eV, respectively. The position of the KMnO4 pre-edge was set to 6543.3 eV. Kα fluorescence signals from the samples were recorded with a 13-element silicon drift detector (RaySpec) positioned perpendicular to the incident beam. For each sample, 8–16 scans were taken in the energy range from 120 eV before the corresponding K-edge up to 750 eV after the edge. Each scan was collected on a new sample spot, to avoid possible radiation damage; additionally, three consecutive scans at the same sample spot confirmed that there was no observable radiation damage on the time scale of the XAS measurement. E0 used for EXAFS extraction was 6539 eV (Mn), 7115 eV (Fe), 7710 eV (Co) and 8334 eV (Ni). EXAFS simulations were done with the FEFF 9.0 software48 (using settings NLEG 6, CRITERIA 12 5, RPATH 7, SCF 7 1 30 0.05). The amplitude reduction factor S02 was 0.90 for Mn, Co and Ni, and 0.85 for Fe. Least-squares fitting of k3-weighted EXAFS data by changing of interatomic distances for the first four single-scattering shells and Debye–Waller factors for all shells (with all multiple-scattering shells having the same Debye–Waller factor) was done with in-house software (SimXLite). The k3-weighted EXAFS data were fitted between k = 1.6 Å−1 and 13 Å−1. Fit parameter errors were determined as described previously.49
4.4 Bulk electrolysis
The solution samples of the reduced complexes (1 mM) in 0.1 M TBAPF6 acetonitrile were oxidized employing a custom-made continuous flow electrosynthesis cell.50 The applied potential was selected by recording a slow (10 mV s−1) cyclic voltammogram in steady condition. The extent of electrolysis was monitored by recording the current response (typically around 0.2 mA at 0.05 ml min−1 flow rate) and changes in the UV-Vis spectrum to provide an estimation of the percentage of the conversion. The oxidized sample solution was collected and frozen in liquid nitrogen for XAS measurements.4.5 Cyclic voltammetry
All solvents, chemicals, glassware, and electrochemical tools were carefully dried. A pseudo reference electrode that consisted of a AgCl coated Ag wire immersed in a 0.1 M TBAPF6 acetonitrile solution contained in a porous glass tipped tube was employed. The electrochemical stability of the Ag pseudo reference electrode was tested over 2 days by recording the cyclic voltammetry of ferrocene. A drift of 0.014 V was observed in the redox potential (E1/2 (Fc+/Fc) = 0.47 V vs. Ag pseudo reference, in MeCN). All potentials quoted in this study are relative to the Fc+/Fc couple.The glassy carbon working electrode (3 mm diameter) was polished with alumina particles (0.05 μm) immediately prior use. The counter electrode was a platinum rod polished with sandpaper before use. Unless stated otherwise, the following parameters were used to record cyclic voltammetry on our samples: scan rate: 100 mV s−1, step potential 0.002 V. The sample concentration was 0.5 mM with tetrabutylammonium hexafluorophosphate supporting electrolyte (100 mM) in acetonitrile.
4.6 Density-functional theory
Density-functional theory (DFT) calculations were performed using Gaussian09 E.01.51 Initial geometry optimization for all complexes was performed using the B3LYP functional with the Lanl2DZ basis set for transition metals, and 6-31G(p,d) for all other atoms. Thermal contributions were calculated from the subsequent Hessian calculations. Structures were then reoptimized using the B3LYP-D3 functional with the larger Lanl2TZ(f) basis set for the transition metals, and 6-311+G(2df,2pd) for all other atoms. All optimizations were performed in acetonitrile solvent using the default Polarizable Continuum Model (PCM) in Gaussian09. Final energies were calculated with the B3LYP* functional,52 using the D3 parameters from B3LYP.53 This functional was previously shown to give good results for spin-state energetics in the spin-crossover complex [FeII–Cl].15 Final energy calculations are done using the Solvation Model based on Density (SMD). This model gives good absolute solvation energies, which is important for the calculation of half potentials.54 Binding energies are calculated from the reaction [MII] + X ⇌ [MII–X] with all components in acetonitrile solvent, including the apical ligand X. For reactions with differences in concentration between components, a configurational entropy factor has been added according to Boltzmann's formula, S = kBlnW, where kB is Boltzmann's constant and W is the number of solvent molecules per complex.
Calculated redox potentials are obtained using the reaction [MII–X] ⇌ [MIII–X] + e−. The energy of the solvated electron was calculated using 4.28 V for the absolute potential of the standard hydrogen electrode (SHE).55 The choice of reference value affects the absolute potentials, but not the comparison between complexes. Calculated values are given in V vs. Fc+/Fc for comparison with experimental results, with 0.40 V for the E1/2 of Fc+/Fc vs. the SHE.56 The analysis of the different contributions to the redox potential was made by separating the calculations of the oxidized species into three steps. First, the energy for removing a spin-up σ electron was calculated using the structure of the reduced complex (orbital contribution). Second, the structure was optimized using the same spin multiplicity (geometric). Finally, the energy of the most stable spin multiplicity was calculated (spin).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Energy Agency through grant No. 45421-1. The authors thank the Helmholtz-Zentrum Berlin (HZB) for the allocation of synchrotron radiation beamtime at the KMC-3 beamline (BESSY II synchrotron, Berlin Adlershof). Calculations were performed on resources provided by SNIC through the National Supercomputer Centre at Linköping University (Tetralith) under projects snic2018-3-575 and snic2019-3-498. SIS thanks the ÅForsk Foundation for support (grant No. 19-349). AT and MKH thank the Olle Engkvist Foundation for support (grant number 198-0369).Notes and references
- K. D. Vogiatzis, M. V. Polynski, J. K. Kirkland, J. Townsend, A. Hashemi, C. Liu and E. A. Pidko, Chem. Rev., 2019, 119, 2453–2523 CrossRef CAS PubMed.
- D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- R. J. M. Klein Gebbink, R. T. Jonas, C. R. Goldsmith and T. D. P. Stack, Inorg. Chem., 2002, 41, 4633–4641 CrossRef CAS PubMed.
- E. G. Pavel, N. Kitajima and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 3949–3962 CrossRef CAS.
- M. E. de Vries, R. M. La Crois, G. Roelfes, H. Kooijman, A. L. Spek, R. Hage and B. L. Feringa, ChemComm, 1997, 1549–1550 RSC.
- R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 8566–8567 CrossRef CAS.
- C. R. Goldsmith, R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 2002, 124, 83–96 CrossRef CAS PubMed.
- C. R. Goldsmith, A. P. Cole and T. D. P. Stack, J. Am. Chem. Soc., 2005, 127, 9904–9912 CrossRef CAS PubMed.
- E. L.-M. Wong, G.-S. Fang, C.-M. Che and N. Zhu, ChemComm, 2005, 4578–4580 RSC.
- W.-L. Kwong, C.-N. Lok, C.-W. Tse, E. L.-M. Wong and C.-M. Che, Chem. – Eur. J., 2015, 21, 3062–3072 CrossRef CAS PubMed.
- M. K. Kashif, J. C. Axelson, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, J. Am. Chem. Soc., 2012, 134, 16646–16653 CrossRef CAS PubMed.
- J. Baillargeon, Y. Xie, A. L. Raithel, B. Ghaffari, R. J. Staples and T. W. Hamann, Inorg. Chem., 2018, 57, 11633–11645 CrossRef CAS PubMed.
- B. Bechlars, D. M. D'Alessandro, D. M. Jenkins, A. T. Iavarone, S. D. Glover, C. P. Kubiak and J. R. Long, Nat. Chem., 2010, 2, 362–368 CrossRef CAS PubMed.
- M. Boniolo, S. I. Shylin, P. Chernev, M. H. Cheah, P. A. Heizmann, P. Huang, N. Salhi, K. Hossain, A. Thapper, M. Lundberg and J. Messinger, ChemComm, 2020, 56, 2703–2706 RSC.
- D. J. Rudd, C. R. Goldsmith, A. P. Cole, T. D. P. Stack, K. O. Hodgson and B. Hedman, Inorg. Chem., 2005, 44, 1221–1229 CrossRef CAS PubMed.
- E. J. Sundstrom, X. Yang, V. S. Thoi, H. I. Karunadasa, C. J. Chang, J. R. Long and M. Head-Gordon, J. Am. Chem. Soc., 2012, 134, 5233–5242 CrossRef CAS PubMed.
- A. E. King, Y. Surendranath, N. A. Piro, J. P. Bigi, J. R. Long and C. J. Chang, Chem. Sci., 2013, 4, 1578–1587 RSC.
- E. Deponti, A. Luisa, M. Natali, E. Iengo and F. Scandola, Dalton Trans., 2014, 43, 16345–16353 RSC.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, ChemComm, 2011, 47, 4249–4251 RSC.
- D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, ChemComm, 2012, 48, 2107–2109 RSC.
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das, A. Thapper, J. Uhlig, P. Dinér and L. Sun, J. Catal., 2016, 335, 72–78 CrossRef CAS.
- S. Ohzu, T. Ishizuka, H. Kotani and T. Kojima, ChemComm, 2014, 50, 15018–15021 RSC.
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemComm, 2015, 51, 13074–13077 RSC.
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemSusChem, 2016, 9, 1178–1186 CrossRef CAS PubMed.
- J. N. Harvey, in Principles and Applications of Density Functional Theory in Inorganic Chemistry I, ed. N. Kaltsoyannis and J. E. McGrady, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 151–184 Search PubMed.
- M. Swart, A. R. Groenhof, A. W. Ehlers and K. Lammertsma, J. Phys. Chem. A, 2004, 108, 5479–5483 CrossRef CAS.
- M. Radoń, Phys. Chem. Chem. Phys., 2014, 16, 14479–14488 RSC.
- B. Pinter, A. Chankisjijev, P. Geerlings, J. N. Harvey and F. De Proft, Chem. – Eur. J., 2018, 24, 5281–5292 CrossRef CAS PubMed.
- W. Klaeui, W. Eberspach and P. Guetlich, Inorg. Chem., 1987, 26, 3977–3982 CrossRef CAS.
- J. Li, C. L. Fisher, J. L. Chen, D. Bashford and L. Noodleman, Inorg. Chem., 1996, 35, 4694–4702 CrossRef CAS.
- R. Gilson and M. C. Durrant, Dalton Trans., 2009, 10223–10230 RSC.
- S. V. Jerome, T. F. Hughes and R. A. Friesner, J. Phys. Chem. B, 2014, 118, 8008–8016 CrossRef CAS PubMed.
- M.-H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407–7412 CrossRef CAS.
- L. E. Roy, E. R. Batista and P. J. Hay, Inorg. Chem., 2008, 47, 9228–9237 CrossRef CAS PubMed.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- L. E. Roy, E. Jakubikova, M. G. Guthrie and E. R. Batista, J. Phys. Chem. A, 2009, 113, 6745–6750 CrossRef CAS PubMed.
- J. Cheng, X. Liu, J. VandeVondele, M. Sulpizi and M. Sprik, Acc. Chem. Res., 2014, 47, 3522–3529 CrossRef CAS PubMed.
- D. Bím, L. Rulíšek and M. Srnec, J. Phys. Chem. Lett., 2016, 7, 7–13 CrossRef PubMed.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, in Spin Crossover in Transition Metal Compounds II, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 97–128 Search PubMed.
- M. Lundberg and P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2004, 6, 4772–4780 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th edn, 1986 Search PubMed.
- B. A. I. Bruker, APEX2 and SAINT., Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- M. Risch, K. Klingan, J. Heidkamp, D. Ehrenberg, P. Chernev, I. Zaharieva and H. Dau, ChemComm, 2011, 47, 11912–11914 RSC.
- J. Yeo, M. H. Cheah, M. I. Bondin and S. P. Best, Aust. J. Chem., 2012, 65, 241–253 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. B. Bloino, G. R. Janesko, B. Gomperts, H. Mennucci, P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. S. Greeley, W. T. Smith Jr., R. W. Stoughton and M. H. Lietzke, J. Phys. Chem., 1960, 64, 652–657 CrossRef CAS.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894–7899 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2013481-2013483. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03695a |
| This journal is © The Royal Society of Chemistry 2021 |