
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural analysis of five-coordinate aluminium(salen) complexes and its relationship to their catalytic activity†
Heather
Fish
,
Sam
Hart
,
Katie J.
Lamb
 ,
Michael
North
*,
Sophie C. Z.
Quek
,
Adrian C.
Whitwood
,
Barnaby
Woods
and
Xiao
Wu
,
Michael
North
*,
Sophie C. Z.
Quek
,
Adrian C.
Whitwood
,
Barnaby
Woods
and
Xiao
Wu
Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: michael.north@york.ac.uk
First published on 15th December 2020
Abstract
The crystal structure of [Al(tBu-salen)]2O·HCl shows major changes compared to that of [Al(tBu-salen)]2O. The additional proton is localized on the bridging oxygen atom, making the aluminium atoms more electron deficient. As a result, a water molecule coordinates to one of the aluminium atoms, which becomes six-coordinate. This pushes the salen ligand associated with the six-coordinate aluminium ion closer to the other salen ligand and results in the geometry around the five-coordinate aluminium atom becoming more trigonal bipyramidal. These results experimentally mirror the predications of DFT calculations on the interaction of [Al(tBu-salen)]2O and related complexes with carbon dioxide. Variable temperature NMR studies of protonated [Al(tBu-salen)]2O complexes revealed that the structures were dynamic and could be explained on the basis of an intramolecular rearrangement in which the non-salen substituent of a five-coordinate aluminium(tBu-salen) unit migrates from one face of a square based pyramidal structure to the other via the formation of structures with trigonal bipyramidal geometries. Protonated [Al(tBu-salen)]2O complexes were shown to have enhanced Lewis acidity relative to [Al(tBu-salen)]2O, coordinating to water, dioxane and 1,2-epoxyhexane. Coordinated epoxyhexane was activated towards ring-opening, to give various species which remained coordinated to the aluminium centers. The protonated [Al(tBu-salen)]2O complexes catalysed the synthesis of cyclic carbonates from epoxides and carbon dioxide both in the presence and absence of tetrabutylammonium bromide as a nucleophilic cocatalyst. The catalytic activity was principally determined by the nature of the nucleophilic species within the catalyst structure rather than by changes to the Lewis acidity of the metal centers.
Introduction
Salen ligands have the general structure shown in Fig. 1 and are considered to be privileged ligands1 as they will complex to just about any metal in the periodic table and the resulting mono- or poly-metallic complexes can be used to catalyse a wide range of chemical reactions.2 The ligands are synthesized by reacting a diamine with two equivalents of an aldehyde or ketone and the majority of such reactions involve a 1,2-diamine to give salen ligands represented by n = 2 in Fig. 1. There are a number of stereochemical aspects of such salen ligands and their metal complexes which can have an important influence on the structure and catalytic activity of metal(salen) complexes. Metal-complexed salen ligands are not planar, but rather adopt stepped and bowl conformations, giving four potentially interconverting conformations as shown in Fig. 2A.3 Any substituents on the diamine derived part of the salen ligand (R1 in Fig. 1) can then adopt axial or equatorial positions within the 5-membered chelate ring (Fig. 2B).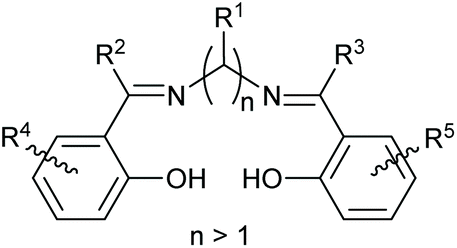 | ||
| Fig. 1 General structure of salen ligands. | ||
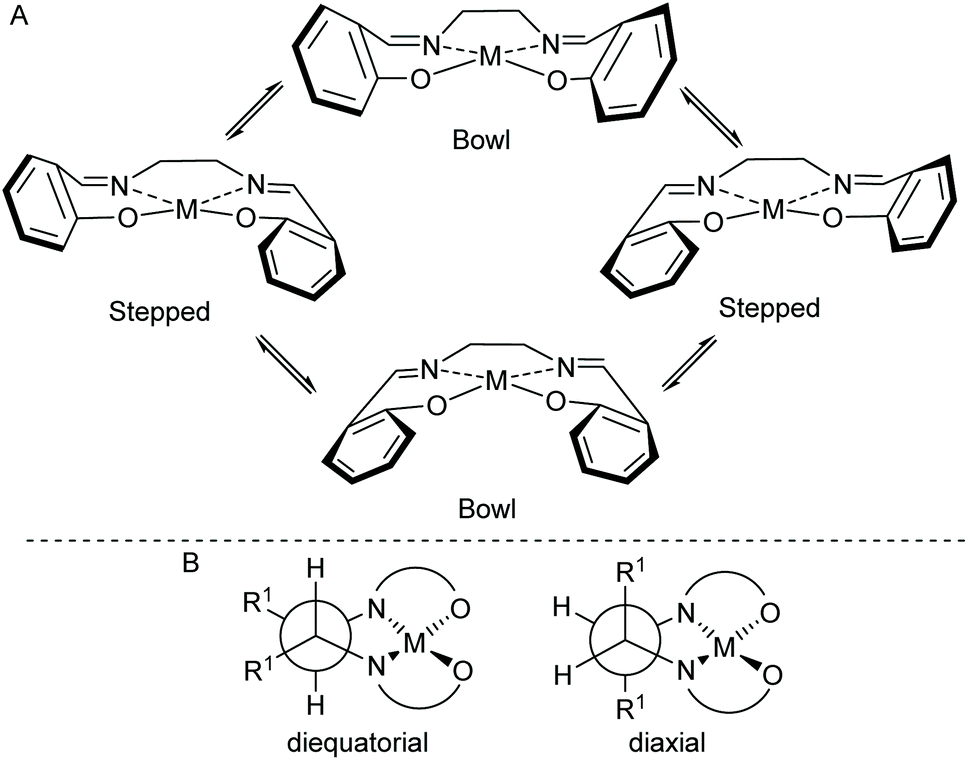 | ||
| Fig. 2 (A) Stepped and bowl conformations of a salen ligand within a metal(salen) complex. (B) Illustration of substituents (R1) in axial or equatorial positions. | ||
A metal ion coordinated to a salen ligand is most commonly six-coordinate with two additional ligands (X and Y) also coordinated to the metal to give an octahedral geometry. However, depending on the metal and its oxidation state, four-coordinate4 and five-coordinate5 complexes can also be formed and for larger metals, higher coordination-numbers are possible.6 Within an octahedral, six-coordinate salen complex, there are three theoretically possible configurations for the salen ligand: trans, cis-α and cis-β as shown in Fig. 3.7 The trans-configuration is by far the most common, but in complexes composed of a salen ligand and a bidentate ligand, the salen ligand adopts the cis-β configuration.8 Complexes with a trans-configuration are achiral whilst complexes with cis-α or cis-β configurations are inherently chiral, giving rise to Δ and Λ stereoisomers.7
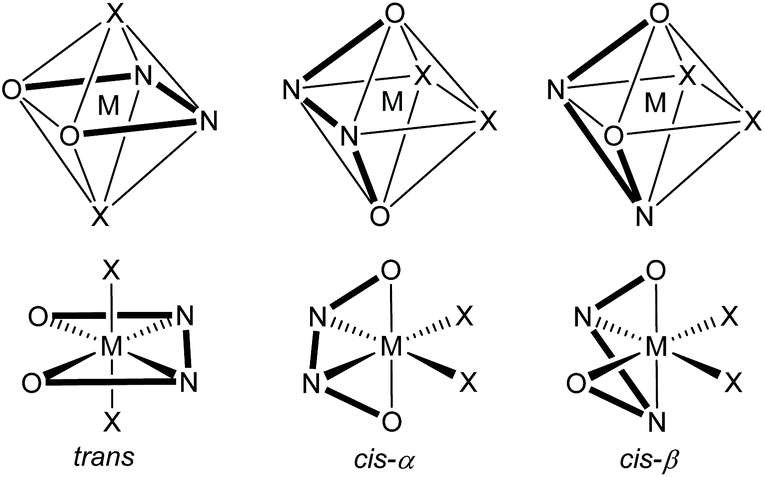 | ||
| Fig. 3 Potential configurations of six-coordinate, octahedral metal(salen) complexes. | ||
In recent years, we have worked extensively on five-coordinate aluminium complexes of salen and related ligands, showing that complexes 1–5 (Fig. 4) formed active catalysts for: the synthesis of cyclic carbonates from epoxides and carbon dioxide9–12 (Scheme 1A); the synthesis of oxazolidinones from epoxides and isocyanates13 (Scheme 1B) or from aziridines and carbon dioxide14 (Scheme 1C); the synthesis of di- and tri-thiocarbonates from epoxides and carbon disulfide15 (Scheme 1D) and asymmetric cyanohydrin synthesis16 (Scheme 1E). Complex 1 has also been used by other researchers to catalyse Michael additions3 (Scheme 1F) and Passerini-type reactions17 (Scheme 1G). Aluminium(salen) complexes in general have been widely used to initiate ring-opening polymerisation of cyclic esters.18
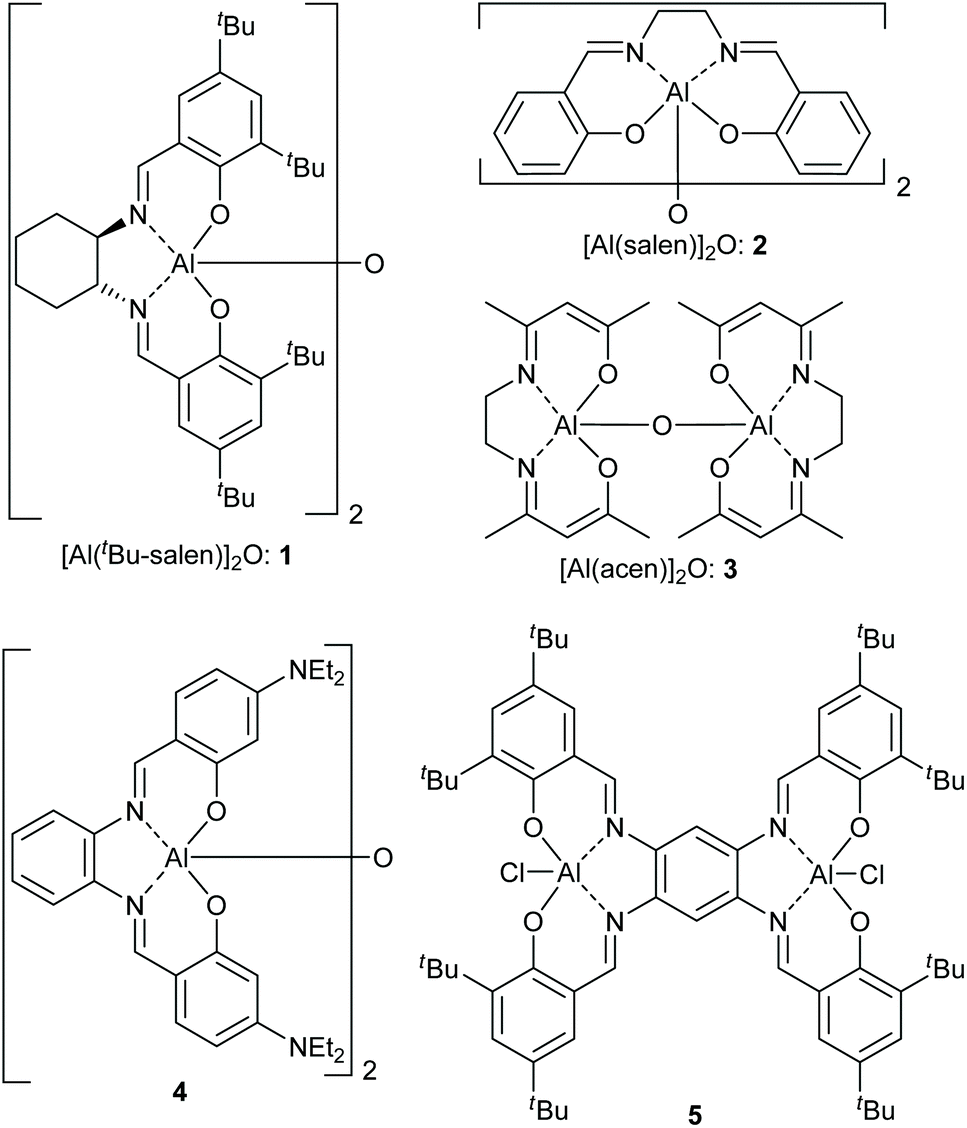 | ||
| Fig. 4 Structures of five-coordinate aluminium complexes 1–5. | ||
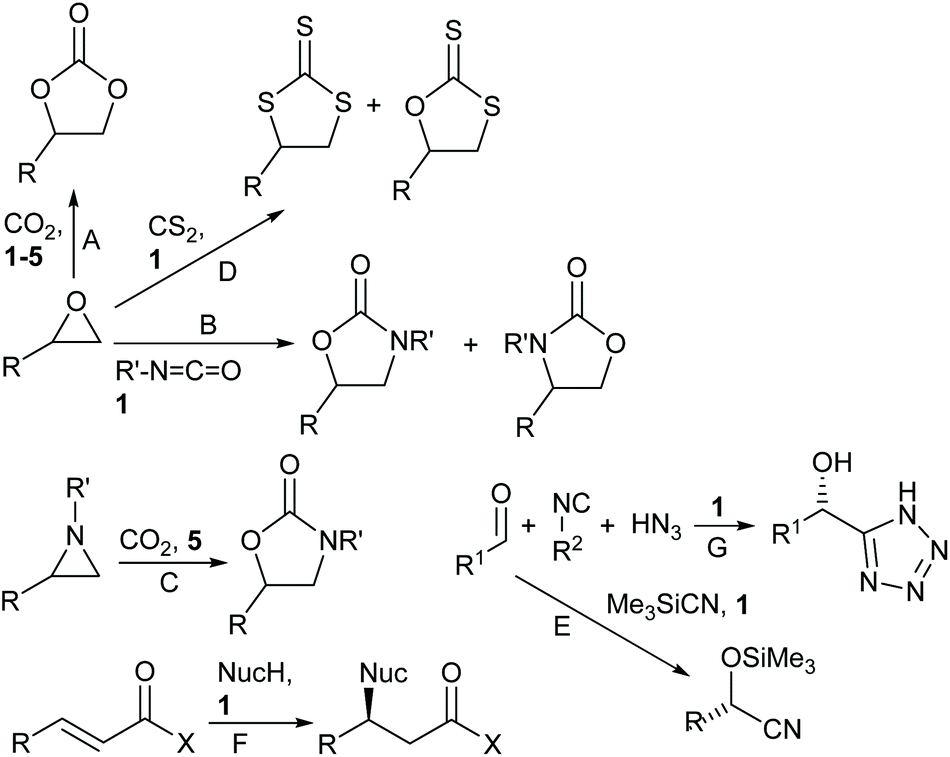 | ||
| Scheme 1 Reactions catalysed by complexes 1–5. | ||
In contrast to six-coordinate metal(salen) complexes, the structures and structural dynamics of five-coordinate metal(salen) complexes have not previously been analysed in detail. This is crucial information needed to facilitate the design of highly active catalysts based on five-coordinate complexes. Therefore, in this paper we analyse the solid and solution state structures of bimetallic aluminium(salen) complex 1 and related complexes and show that the complexes possess dynamic structures, interconverting between square-based pyramidal and trigonal bipyramidal geometries. The ability of the complexes to catalyse the synthesis of styrene carbonate from styrene oxide and carbon dioxide is also compared.
Results and discussion
Solid state structural analysis
In 2010, we reported the single-crystal X-ray structure of complex 1 (Fig. 5a).19 That sample was prepared by treatment of the corresponding salen ligand (tBu-salenH2) with aluminium triethoxide followed by aqueous work-up (Scheme 2). Both aluminium atoms within the structure were five-coordinate and the structure had an almost linear Al–O–Al unit (bond angle 175.52(15)° and 170.47(15)° for the two crystallographically unique molecules in the unit cell). Recently, we attempted to prepare complex 1 by treatment of tBu-salenH2 with diethylaluminium chloride followed by hydrolysis of the intermediate aluminium chloride complex (Scheme 2) and obtained the hydrochloride salt of complex 1.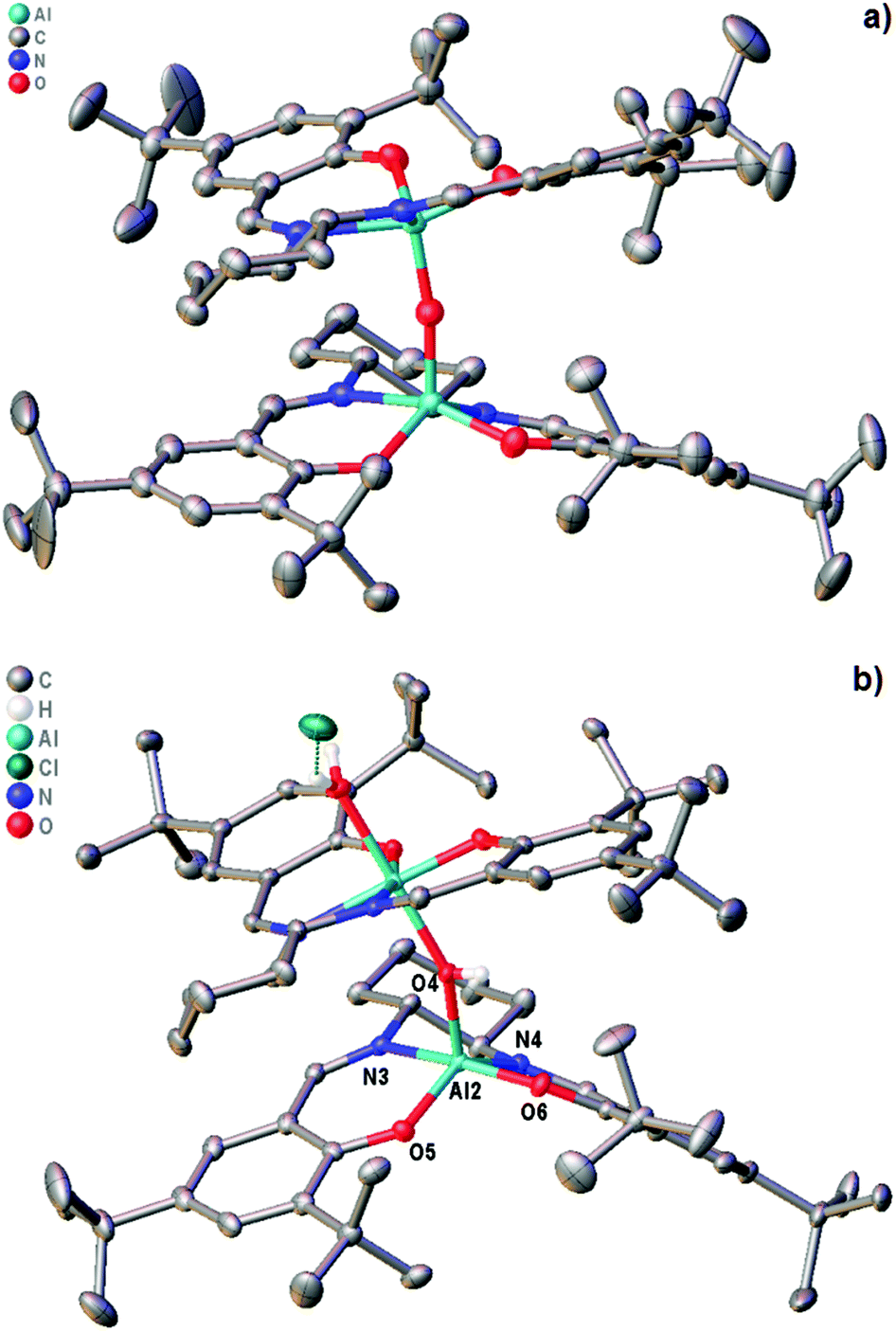 | ||
| Fig. 5 (a) structure of complex 1.19 (b) structure of complex 1·HCl. | ||
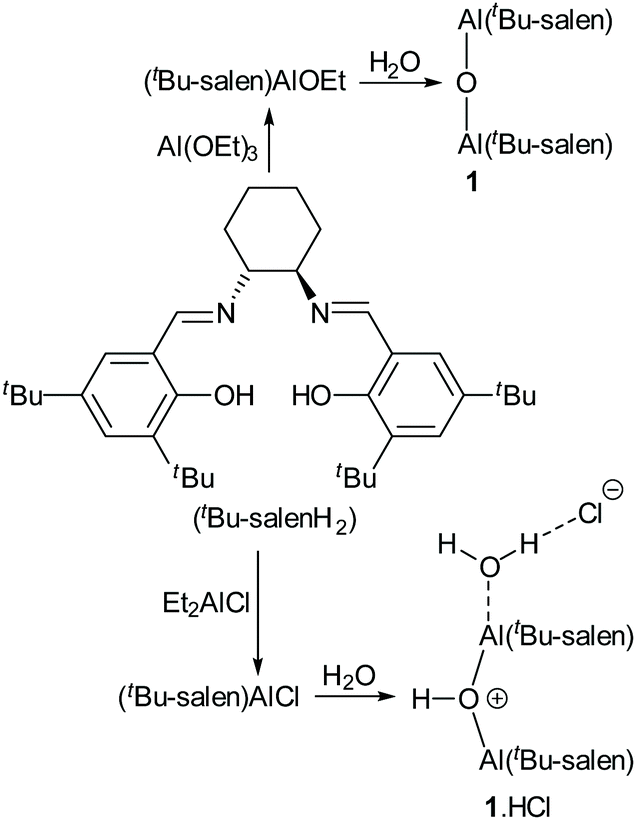 | ||
| Scheme 2 Synthesis of complexes 1 and 1·HCl. | ||
The single-crystal X-ray structure‡ of 1·HCl (Fig. 5b and ESI†) showed major differences compared to that of unprotonated 1. The additional proton is localised on the bridging oxygen, resulting in the Al–O–Al bond angle reducing to 153.17(11)°. One aluminium atom is six-coordinate (with a water occupying the sixth coordination site hydrogen-bonded to the chloride anion) whilst the other aluminium atom remains five-coordinate. There is also a marked lengthening in the aluminium to bridging oxygen distances. In complex 1 these are 1.6827(6) and 1.6842(6), whereas for complex 1·HCl the distances are 1.8112(17) to the five-coordinate aluminium and 1.9407(18) to the six-coordinate aluminium.
The salen ligand associated with the six-coordinate aluminium atom is pushed into a more equatorial plane relative to that in unprotonated complex 1 and this in turn makes the coordination geometry of the salen ligand attached to the five-coordinate aluminium more trigonal bipyramidal in complex 1·HCl (τ5 = 0.63) than in complex 1 (τ5 = 0.03 and 0.46 for the two crystallographically unique molecules).20 O6 and N3 form the two apices (N3–Al2–O6 angle 168.39(8)°) and O4, O5 and N4 form the central triangle (O4–Al2–N4 angle 118.14(8)°, O4–Al2–O5 angle 110.80(8)° and N4–Al2–O5 angle 130.62(9)°). This effectively blocks the potential sixth coordination site associated with this aluminium atom and prevents it from becoming six-coordinate.
The structural features of the X-ray structure of complex 1·HCl correlate very well with the previously reported results of DFT calculations on the interaction of complexes 2 and 3 (Fig. 4) with carbon dioxide to give adducts 6 and 7 respectively (Fig. 6).11 In complexes 6 and 7, the carbon dioxide was found to interact with the μ-oxo-bridging oxygen, giving Al–O–Al bond angles of 138–145° compared to an Al–O–Al bond angle of 152° in the X-ray structure of complex 2.21 It was also calculated that the aluminium atoms in complex 7 had increased Lewis acidity compared to those in complex 3, and would form a mono-adduct with ethylene oxide. These calculations and associated experimental results allowed us to propose a mechanism by which complexes 1–3 can catalyse the formation of cyclic carbonates from epoxides and carbon dioxide in the absence of a nucleophilic cocatalyst.11
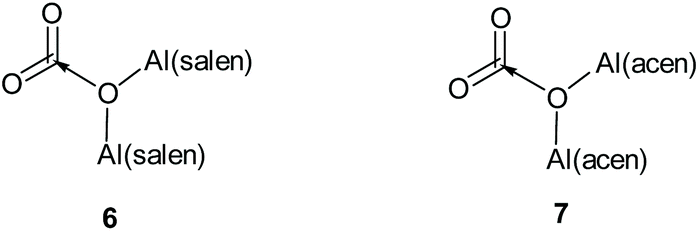 | ||
| Fig. 6 Structures of CO2 adducts 6 and 7. | ||
Solution state structural analysis
To investigate the effect of protonation of complex 1 on the solution state structure and catalytic activity of the resulting complexes, the protonation of complex 1 by trifluoracetic acid or anhydrous hydrogen chloride was investigated. Treatment of complex 1 with one equivalent of trifluoroacetic acid (to form 1·TFA) resulted in significant broadening of the 1H and 13C NMR spectra recorded at 298 K along with changes in chemical shifts. Fig. 7 shows the aromatic/imine region of the 1H NMR spectrum (6.6–8.6 ppm) and the full 1H, 13C and 19F spectra are given in the ESI.† To better understand the structure of 1·TFA and explain the line broadening, a variable temperature 1H NMR study was carried out in deuterated chloroform between 233 and 328 K. Fig. 8 shows the tert-butyl region of the resulting spectra and the full spectra are given in the ESI.† At temperatures of 273 K and below, the 1H NMR spectra of 1·TFA show four separate tert-butyl signals (labelled a–d in Fig. 8) as would be expected for a complex in which both tBu-salen units were in the same, non-C2-symmetric, environment. At 283 K however, the two central tert-butyl signals (c and d) merge to give a single peak and at 293 K the two outer signals (a and b) also merge to give an initially very broad peak centred at the same chemical shift as the merged peak derived from signals c and d. As the temperature increases from 293 to 328 K, both merged peaks sharpen and the spectrum exhibits a single peak corresponding to all four tert-butyl groups present on the salen ligand.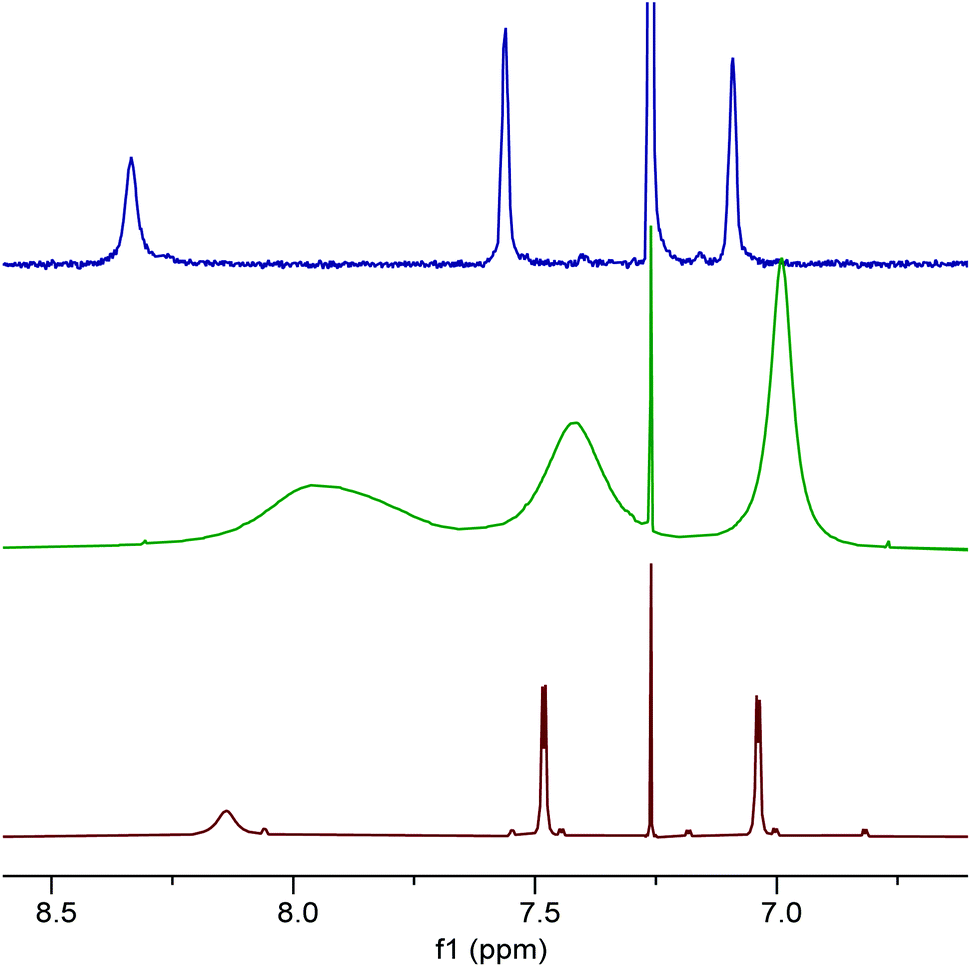 | ||
| Fig. 7 The aromatic and imine region of the 1H NMR spectra of compounds 1 (red), 1·TFA (green) and 8 (blue) recorded at 400 MHz in CDCl3 at 298 K. | ||
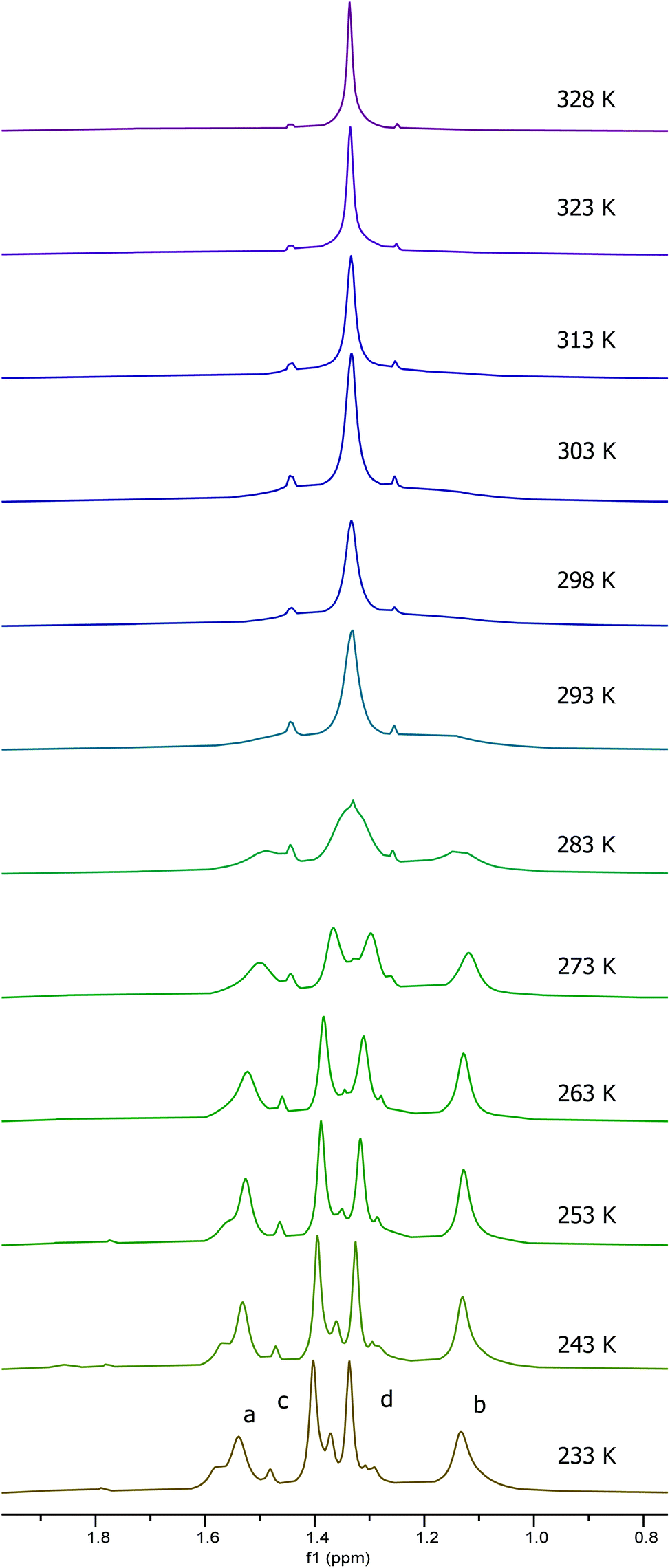 | ||
| Fig. 8 The tert-butyl group region of the 1H NMR spectra of 1·TFA recorded at 500 MHz in CDCl3 at 233–328 K. | ||
The temperature dependent changes in the tert-butyl region of the NMR spectra of 1·TFA are mirrored in the signals present in other regions of the spectra. Thus, the four aromatic and two imine signals present at temperatures below 273 K merge into two aromatic signals and one imine signal at temperatures of 303 K and above. The spectra also show a highly temperature dependent OH signal which appears at 5.7 ppm at 233 K, shifting and broadening to 4.1 ppm at 303 K before becoming too broad to detect at higher temperatures.
Line shape analysis§ of the tert-butyl signals in the variable temperature NMR spectra of 1·TFA was carried out to allow the rate constant of the exchange process and the Gibbs energies of activation (ΔG‡) to be determined at each temperature (see ESI†). The two sets of tert-butyl signals (a, b and c, d) were modelled separately as non-coupled two spin systems and gave mutually consistent data. The rate constants for the exchange process were found to increase from 0.5–1.5 s−1 at 233 K to 4300–9000 s−1 at 328 K, but the corresponding Gibbs energies of activation were temperature independent with a value of 56.9 ± 1.3 kJ mol−1 at all twelve temperatures and for both sets of data. Eyring plots of ln(k/T) against 1/T (Fig. 9) were linear for both data sets and allowed the enthalpy (ΔH‡) and entropy (ΔS‡) of activation to be determined as 59.0 ± 3.2 kJ mol−1 and 0.0 ± 0.1 J mol−1 K−1 respectively. The zero value for ΔS‡ is consistent with the lack of temperature dependency of ΔG‡ and indicates that the process which causes the signals of the tert-butyl groups within 1·TFA to exchange position occurs without a significant change in entropy and so is likely to be an intramolecular rearrangement.
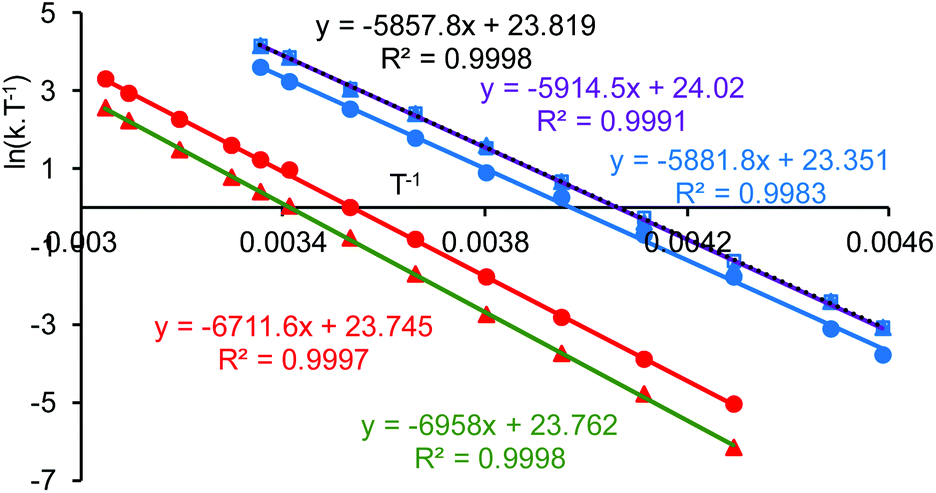 | ||
| Fig. 9 Eyring plots based on simulations of the variable temperature NMR data for compounds 1·TFA (red points) and 8 (blue points). The two data sets for 1·TFA (red and green trend lines) correspond to analysis of the two separate pairs of tert-butyl signals. The three data sets for 8 (blue, purple and black trend lines) correspond to analysis of the two separate pairs of aromatic signals and the pair of imine signals. | ||
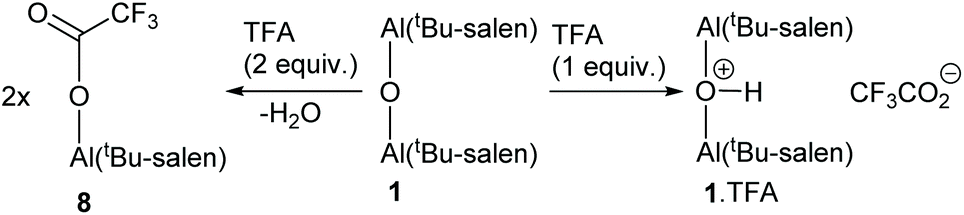
Addition of two equivalents of trifluoroacetic acid to complex 1 resulted in a resharpening of the 1H NMR peaks to positions that differed from those of compound 1 (Fig. 7). The structure of the species formed on treatment of compound 1 with two equivalents of trifluoroacetic acid was shown to be mononuclear aluminium(tBu-salen) trifluoroacetate 8 (Scheme 3) by high resolution field desorption mass spectrometry. However, the room temperature 1H NMR spectrum of complex 8 suggested a C2-symmetric structure. Therefore, a VT NMR study of complex 8 was carried out in deuterated chloroform between 218 and 328 K (see ESI†). At the lowest temperatures, the imine, aromatic and tert-butyl signals all resolved into two separate peaks; confirming that the complex actually had C1-symmetry. For compound 8, line shape analysis§ of the imine and aromatic signals in the variable temperature NMR spectra was carried out (see ESI†) as these underwent more changes than the tert-butyl signals. The imine signals and aromatic hydrogens ortho- and para-to the imine were modelled separately to give three sets of data, all of which were mutually consistent. The Gibbs energies of activation were again temperature independent with a value of 49.0 ± 1.0 kJ mol−1 for all 30 data points. Eyring plots of ln(k/T) against 1/T (Fig. 9) were linear and allowed the enthalpy (ΔH‡) and entropy (ΔS‡) of activation to be determined as 48.9 ± 0.3 kJ mol−1 and −0.2 ± 3.2 J mol−1 K−1 respectively. The approximately zero value for ΔS‡ again indicates that the process which causes the aromatic and imine signals within 8 to exchange position is likely to be an intramolecular rearrangement. The chemistry occurring on treatment of complex 1 with trifluoroacetic acid is therefore summarised in Scheme 3.
 | ||
| Scheme 3 Synthesis of complexes 1·TFA and 8. | ||
The exchange process which results in complexes 1·TFA and 8 appearing to be C2-symmetrical at higher temperatures can be explained by the process shown in Scheme 4. Borrowing terminology used in octahedral, six-coordinate complexes,7 a square pyramidal complex I has a salen ligand in the trans-configuration and a vacant coordinate site opposite the X-ligand. Movement of one of the salen ligand's oxygen atoms into this vacant site generates trigonal bipyramidal complex II. The X-ray structures of complexes 1 and 1·HCl provide good evidence for the existence of both of these coordination geometries. Subsequent migration of the X ligand to the free coordination site within complex II generates a new square pyramidal complex in which the salen ligand has a cis-β configuration. There are many examples of octahedral metal(salen) complexes with a salen ligand in the cis-β configuration.8 Movement of the remaining equatorial oxygen atom of the salen ligand into the vacant apical coordination site of complex III generates a new trigonal bipyramidal complex IV. Subsequent migration of the other apically coordinated oxygen of the salen ligand to an equatorial position generates square based pyramidal complex V in which the salen ligand again has a cis-β configuration, but with a different oxygen atom in the apical position compared to complex III. Migration of the X ligand into the free apical coordination site of complex V generates trigonal bipyramidal complex VI and finally migration of the remaining apically coordinated oxygen of the salen ligand in complex VI to an equatorial position generates square based pyramidal complex VII with a trans-configuration of the salen ligand. In complex VII, the X ligand is coordinated to the opposite face of the square planar aluminium(salen) unit compared to complex I, so that a time-averaged structure would appear to be C2-symmetric.
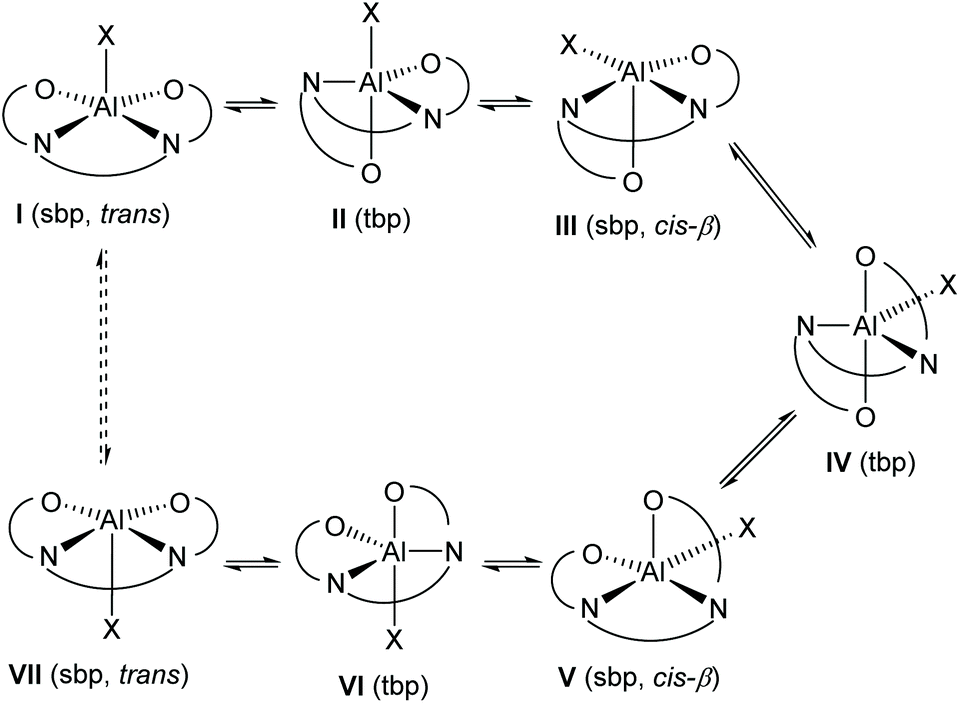 | ||
| Scheme 4 Salen ligand rearrangement within five-coordinate aluminium(salen) complexes; sbp = square based pyramid, tbp = trigonal bipyramid; trans and cis-β refer to the configuration of the salen ligand within square based pyramidal complexes. | ||
The pathway shown in Scheme 4 is fully consistent with the activation parameters determined by line-shape analysis of the variable temperature NMR data. No ligand dissociation or association occurs, so the entropy of activation would be expected to be close to zero. The X group in compound 8 is a trifluoroacetyl group, whilst for 1·TFA the X group is a much larger OAl(salen) unit. The rearrangement shown in Scheme 4 involves the X group moving into the equatorial plane of square based pyramidal complexes III and V where steric interactions with the salen ligand will be greater than for structures I and VII where the X group is in the apical position of a square based pyramid. This accounts for the higher values of the enthalpy and Gibbs energy of activation associated with the rearrangement of complex 1·TFA compared to complex 8.
Close examination of the 1H NMR spectrum of compound 1 (Fig. 7 and ESI†) showed that in addition to the major, apparently C2-symmetrical, species, a second, lower intensity set of signals corresponding to a C1-symmetrical complex were present. This is apparent in Fig. 7, where in addition to the three major aromatic/imine peaks, six other signals are present. In this case, all peaks are sharp and well resolved suggesting that at room temperature they exchange only very slowly if at all. A variable temperature NMR study (298–328 K in deuterated chloroform) revealed that the spectra of both the major and minor species were temperature dependent. In this case, although the peaks did undergo changes in chemical shift, they did not coalesce in the accessible temperature range. However, the relative amount of the minor species increased from 9% at 298 K to 14% at 328 K. Thus, the minor peaks present in the 1H NMR spectrum are consistent with complex 1 in which one of the salen ligands is in a trigonal bipyramidal geometry as observed for one of the two independent molecules in the crystal structure of complex 1.19 This interpretation was supported by a DOSY experiment (in CDCl3) which showed that the major and minor species had very similar diffusion coefficients of 5.4 × 10−10 m2 s−1 and 5.1 × 10−10 m2 s−1 respectively (see ESI†). These compare with a value of 2.0 × 10−9 m2 s−1 for the chloroform present in the NMR solvent.
Protonation of complex 1 using a solution of anhydrous hydrogen chloride in dioxane gave more complex results as the dioxane acted as a Lewis base. Addition of one equivalent of hydrogen chloride to complex 1, followed by evaporation of the excess dioxane in vacuo cleanly gave complex 9 (Scheme 5). The 1H NMR spectrum of complex 9 (at room temperature) suggested that the complex was C2-symmetric, but the broadness of the signals again indicated that this was due to a dynamic process (Fig. 10 and ESI†). The residual dioxane signal integrated to eight protons, indicating that just one dioxane was coordinated within complex 9. The structure of complex 9 exactly mirrors the structure of 1·HCl determined by X-ray crystallography (Fig. 5b) with the water molecule replaced by a dioxane ligand.
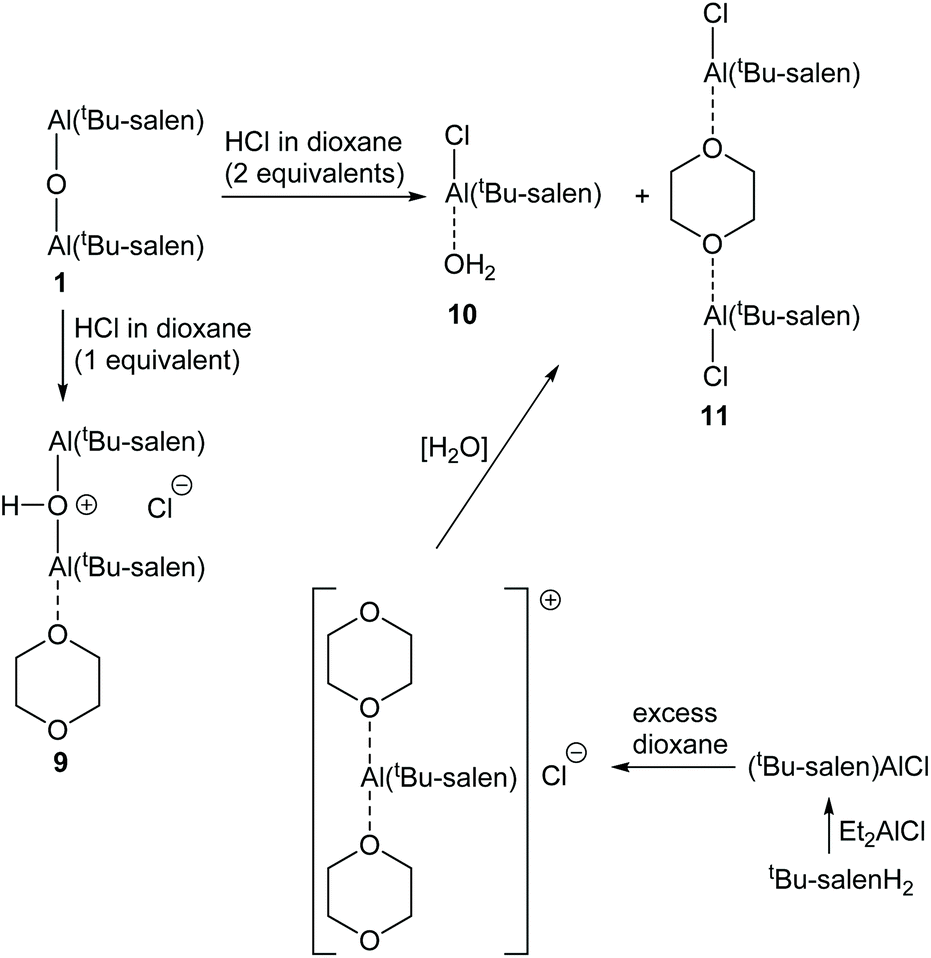 | ||
| Scheme 5 Synthesis of complexes 9–11. | ||
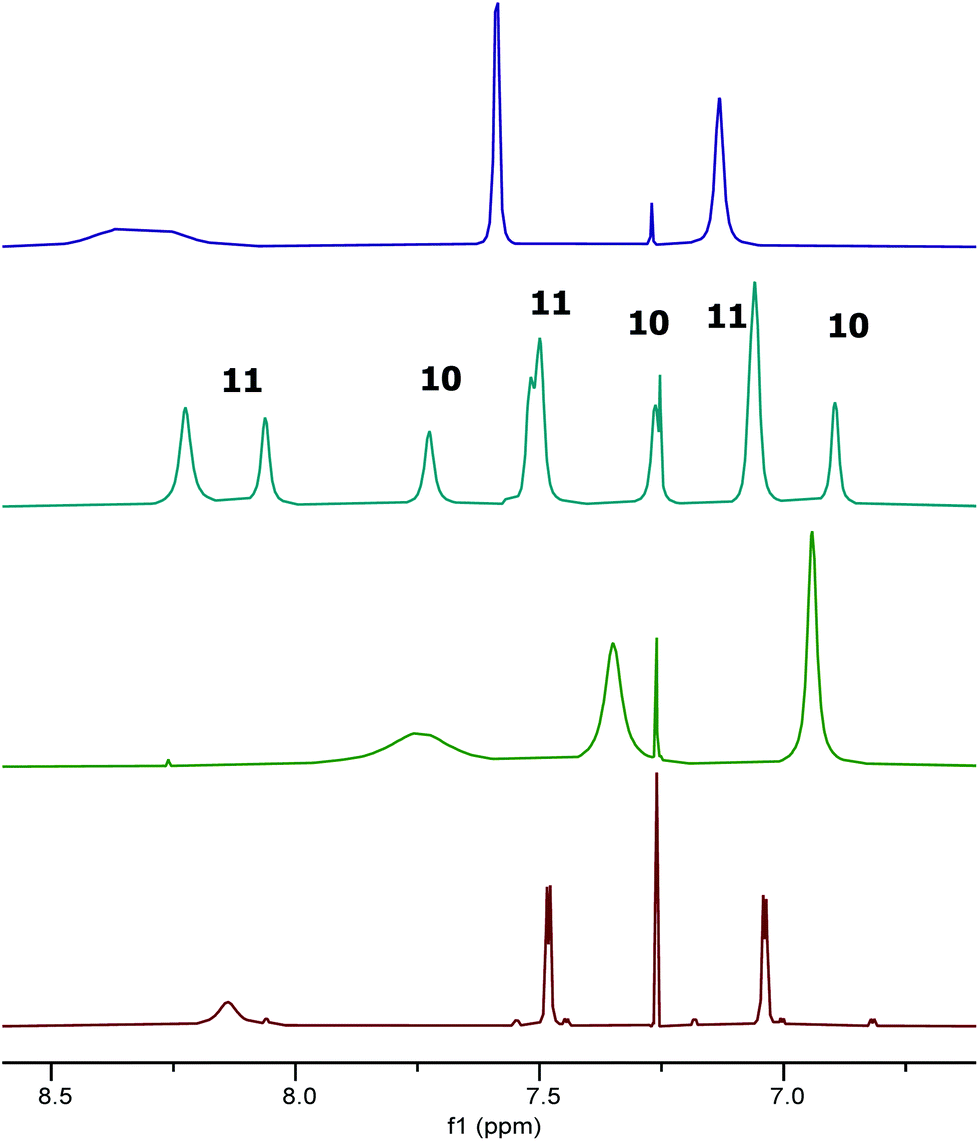 | ||
Fig. 10 Extracts of the 1H NMR spectra of compounds: 1 (red); 9 (green); 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.25 mixture of 10 and 11 (cyan); and 11 (purple) in CDCl3. :1.25 mixture of 10 and 11 (cyan); and 11 (purple) in CDCl3. | ||
In contrast to the treatment of complex 1 with two equivalents of trifluoroacetic acid, addition of two equivalents of hydrogen chloride in dioxane to complex 1, followed by removal of excess dioxane in vacuo resulted in the formation of two new species 10 and 11 in a 1:1.25 ratio (Fig. 10). In addition to the imine and aromatic signals shown in Fig. 10, the full 1H NMR spectrum (see ESI†) showed a broad OH peak at 4.8 ppm; evidence for coordinated dioxane in complex 11; and the expected cyclohexyl and tert-butyl protons.
To confirm the structures of complexes 10 and 11, an alternative synthesis of these species was undertaken. Thus, treatment of the tBu-salenH2 ligand with diethylaluminium chloride gave the known12 five-coordinate Al(tBu-salen)Cl complex (Scheme 5). Addition of excess dioxane to a sample of Al(tBu-salen)Cl formed the dioxane solvate {[Al(tBu-salen)(dioxane)2]+ Cl−} in situ. The 1H NMR spectra of this solvate did not match those of complexes 10 and/or 11 (see ESI†). However, after evaporation of excess dioxane from the solvate, the 1H NMR spectrum did match that of complexes 10 and 11 obtained from complex 1, though with a 1:2.5 ratio of 10:11 (see ESI†).
Treatment of bimetallic complex 1 with anhydrous hydrogen chloride to give products in which the μ-oxo bridge has been broken will produce half an equivalent of water relative to the concentration of aluminium. Therefore, a 1:1 ratio of complexes 10 and 11 would be expected to form when complex 1 was treated with two equivalents of anhydrous hydrogen chloride and the observed ratio was 1:1.25. In contrast, no water is generated when Al(tBu-salen)Cl is treated with dioxane. Hence, the formation of complex 10 under these conditions must be due to adventitious water in the reaction and this accounts for the lower amount of complex 10 formed by this route. By carrying out the synthesis from Al(tBu-salen)Cl under rigorously anhydrous conditions with evaporation of excess dioxane on a Schlenk line, the formation of complex 10 could be completely avoided and only complex 11 was formed (Fig. 10 and ESI†). The 1H NMR spectrum of pure complex 11 confirmed that half a dioxane molecule was present for each Al(tBu-salen) unit.
As shown in Fig. 10, the 1H NMR signals for complex 11 in the mixture of species 10 and 11 do not exactly superimpose with the signals observed when only species 11 was present. However, a NOESY spectrum recorded on a sample with a 1:2.5 ratio of complexes 10 and 11 showed cross-peaks between the signals for each species, indicating that they were in chemical exchange with each other (see ESI†). Thus, slightly different chemical shifts and line widths would be expected for complex 11 in the presence and absence of complex 10.
It was apparent from the room temperature 1H NMR spectra of compounds 10 and 11 that aspects of their structures were again not fully explained by the spectra. In particular, the aromatic signals shown in Fig. 10 were far simpler than would be expected and the imine signals in Fig. 10 appeared extremely broad or to be split into two separate peaks. Therefore a variable temperature 1H NMR study was undertaken on the 1:2.5 mixture of 10 and 11 in deuterated chloroform at 500 MHz between 218 and 328 K. Fig. 11 shows the tert-butyl region of the resulting spectra and the full spectra are given in the ESI.† At 218 K, eight tert-butyl signals (labelled a–h) are present between 1.55 and 0.95 ppm. This is the number expected for a combination of complexes 10 and 11 as within each of these complexes the aluminium(tBu-salen) units are not C2-symmetric. However, as the temperature increases, the tert-butyl signals start to merge, first to six signals at 243–253 K, then to two signals (one of which is very broad) at 313 K. Similar changes are apparent in other regions of the spectra (see ESI†). The signal for the coordinated water molecule of complex 10 undergoes a particularly large change in chemical shift: from 5.75 ppm at 218 K to 3.20 ppm at 328 K.
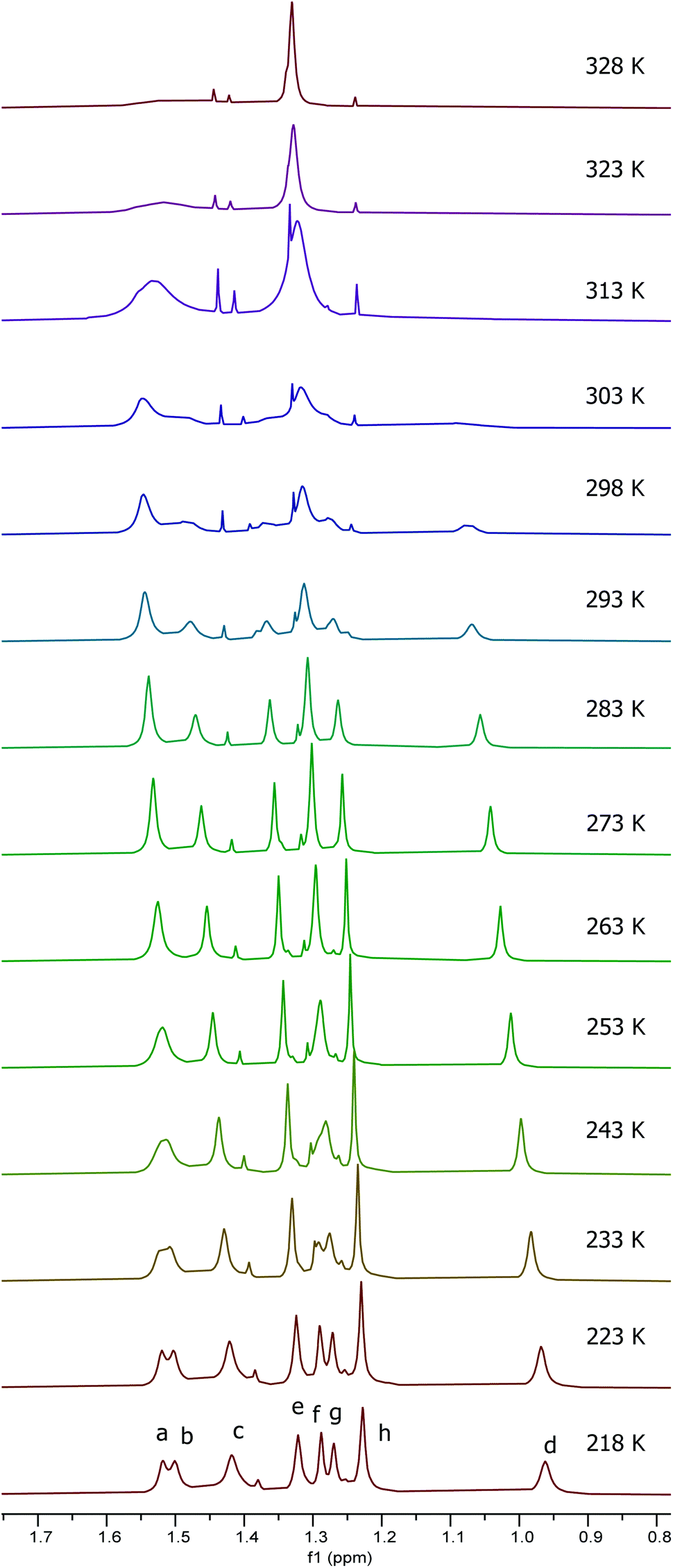 | ||
| Fig. 11 Stacked plot of extracts of the 500 MHz 1H NMR spectra of a 1:2.5 mixture of compounds 10 and 11 in CDCl3 recorded at 218 to 328 K. | ||
The eight tert-butyl signals could be divided into two groups of four (a–d and e–h) each of which merges to a single peak at the higher temperatures and which correspond to the tert-butyl groups ortho- and para-to the phenols in the salen ligand. These two sets of tert-butyl signals were separately subjected to line shape analysis§ (using a four non-coupled spin model, see ESI†) to allow rate constants and Gibbs energies of activation to be determined at each temperature. For this system, ΔG‡ was temperature dependent, increasing steadily from 50.7 kJ mol−1 at 218 K to 67.6 kJ mol−1 at 328 K based on analysis of signals a–d and from 52.8 kJ mol−1 at 218 K to 69.5 kJ mol−1 at 328 K based on analysis of signals e–f. Eyring plots for the two sets of data are given in Fig. 12 and allowed ΔH‡ to be calculated as 17.9 ± 1.1 kJ mol−1 and ΔS‡ to be determined as −155 ± 1 J mol−1 K−1.
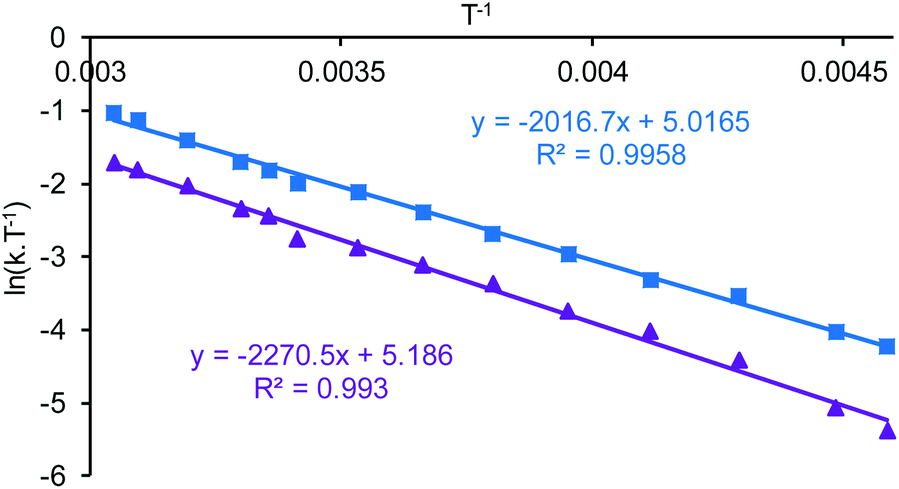 | ||
| Fig. 12 Eyring plots based on the simulations of the variable temperature NMR data a 1:2.5 mixture of compounds 10 and 11. The blue points and line correspond to analysis of peaks a–d whilst the purple points and line correspond to analysis of peaks e–h. | ||
Within the mixture of compounds 10 and 11, three separate exchange processes will occur. Both compounds can independently undergo the same salen ligand rearrangement discussed above for compounds 1·TFA and 8 and illustrated in Scheme 4. This intramolecular rearrangement does however, require the initial dissociation of a weakly bound water or dioxane ligand to generate a five-coordinate complex and hence will have a positive entropy of activation. Compounds 10 and 11 also exchange their weakly bound water and dioxane ligands as illustrated in Scheme 6 and the negative value observed for ΔS‡ suggests that this occurs through an associative mechanism such as that shown in the transition state structure in Scheme 6. The associative ligand exchange outweighs the dissociative ligand rearrangement, resulting in an overall negative entropy of activation.
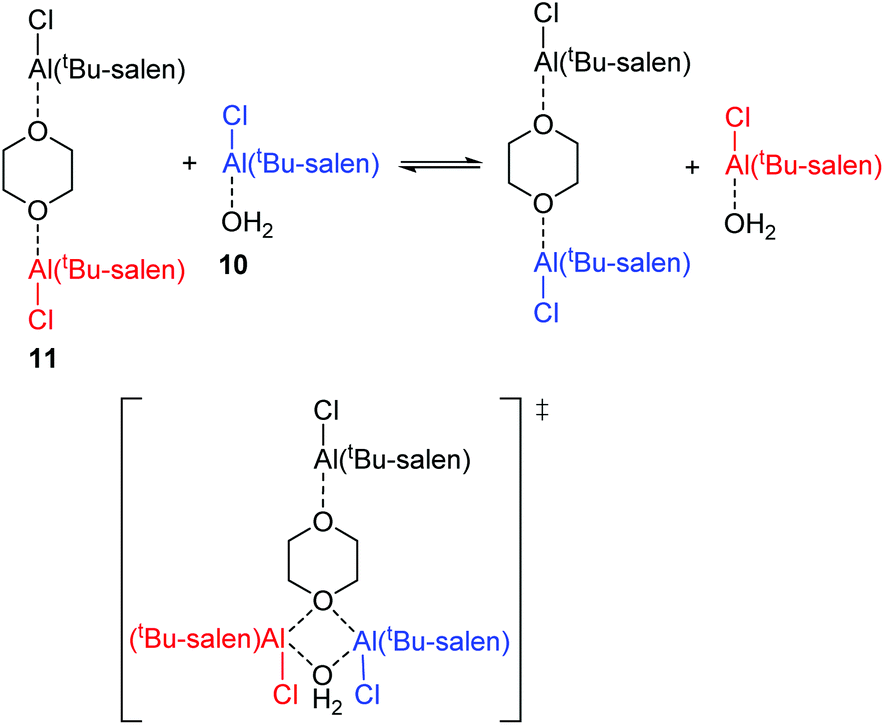 | ||
| Scheme 6 Associative mechanism for the exchange of neutral ligands between compounds 10 and 11. | ||
Investigation of the lewis basicity of aluminium(salen) complexes
Since water and dioxane were capable of acting as Lewis bases towards the Al(tBu-salen) units in complexes 10 and 11, the ability of an epoxide to coordinate to complexes 1, 1·TFA, 9 and (tBu-salen)AlCl was investigated to probe the significance of these results to the mechanisms of reactions catalysed by these and related complexes (Scheme 1). DFT calculations11 have previously shown that the Lewis acidity of complex 1 could be increased by withdrawing some of the electron density associated with the bridging oxygen. 1,2-Epoxyhexane was chosen as the Lewis base on the basis of its convenient boiling point (118–120 °C). Addition of excess 1,2-epoxyhexane to a solution of complex 1 in deuterated chloroform resulted in detectable shifts in the 1H NMR signals of both the aromatic hydrogens of complex 1 and the hydrogens of 1,2-epoxyhexane (see ESI†). Evaporation of the 1,2-epoxyhexane resulted in the aromatic hydrogen signals of complex 1 returning to their original chemical shifts. This indicates that complex 1 can act as a Lewis acid towards epoxides as required by its proposed mechanism of action in the presence of tetrabutylammonium bromide as a cocatalyst, but that the epoxide is only weakly bound to the aluminium complex.9Addition of excess 1,2-epoxyhexane to a solution of (tBu-salen)AlCl in deuterated chloroform resulted in smaller shifts to the 1H NMR aromatic and epoxide signals (see ESI†). These were accompanied by the appearance of multiple new signals in the range of 3–4 ppm and 6.5–9 ppm, indicative of the epoxide having undergone ring–opening to form diol and oligomeric ethers which compete with the 1,2-epoxyhexane for coordination to the aluminium. Consistent with this, when the excess 1,2-epoxyhexane was evaporated, the aromatic signals did not revert to their original shifts and the multiple peaks at 3–4 ppm were still present in the 1H NMR spectrum. There is literature precedent for aluminium(salen) complexes initiating the ring-opening polymerisation of epoxides.22
When excess 1,2-epoxyhexane was added to 1·TFA, no changes in the 1H NMR chemical shifts of the aromatic or epoxide hydrogens was observed, though the aromatic and imine signals did become sharper (see ESI†). On evaporation of excess 1,2-epoxyhexane, changes in the aromatic and imine signals did become apparent. In addition to the three signals at the original positions, new signals corresponding to a salen ligand in a non-C2 symmetric environment appeared as did peaks between 3 and 4 ppm in the 1H NMR spectrum and between 65 and 75 ppm in the 13C NMR spectrum corresponding to ring–opened epoxide. These results can be explained on the basis that 1,2-epoxyhexane undergoes ring–opening as the solution is being evaporated to give oligomeric ethers22 which coordinate to one of the aluminium centres of 1·TFA, forming a six-coordinated aluminium which can no longer undergo the salen ligand rearrangement shown in Scheme 4. A very similar situation was observed when 1,2-epoxyhexane was added to complex 9, though in this case a small change in the chemical shift of the epoxide signals was seen (see ESI†). On evaporation of the 1,2-epoxyhexane, the dioxane initially weakly coordinated to complex 9 was also removed. These results indicate that complexes 1, 1·TFA, 9 and (tBu-salen)AlCl are all capable of acting as Lewis acids towards 1,2-epoxyhexane and in the case of 1·TFA, 9 and (tBu-salen)AlCl this activation of the epoxide results in its ring–opening by a nucleophile present in the reaction to give oligomers22 which also coordinate to the aluminium(salen) units.
Catalytic activity of aluminium(salen) complexes
Given their abilities to coordinate epoxides, the relative abilities of complexes 1, 1·TFA, 8 and 9 to catalyse the synthesis of styrene carbonate 13 from styrene oxide 12 and carbon dioxide under two different sets of solvent-free reaction conditions were investigated. Initially, reactions were carried out at 25 °C and 1 bar pressure of carbon dioxide, using a combination of aluminium complex and tetrabutylammonium bromide as catalyst9,19 (Scheme 7, conditions A) and the results are detailed in Table 1. Entries 1–3 confirm that at 25 °C and one bar carbon dioxide pressure, neither complex 1 nor tetrabutylammonium bromide is an effective catalyst, but when used together they form an effective synergistic catalyst system. Entry 4 shows that this catalytic activity is maintained in the presence of dioxane, indicating that the coordinated dioxane present in complex 9 could be displaced by styrene oxide during the catalytic cycle. This is confirmed by entry 5 which shows that bimetallic complex 9 had similar catalytic activity to complex 1. Entries 6 and 7 show that 1·TFA also has a similar catalytic activity to complex 1 especially when generated in situ. In contrast, entries 8 and 9 show that the monometallic complex 8 has lower catalytic activity than complex 1, whether preformed complex 8 was used at 2.5 mol% loading or complex 8 was generated in situ at 5 mol% loading.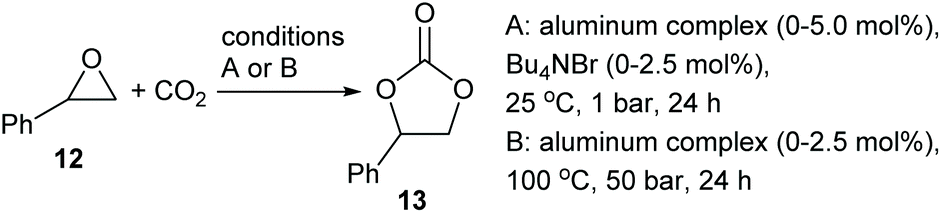 | ||
| Scheme 7 Synthesis of styrene carbonate 13 from styrene oxide 12 and CO2. | ||
| Entry | Aluminium complex (mol%) | Bu4NBr (mol%) | T (°C) | P (bar) | Conversiona or (isolated yield) |
|---|---|---|---|---|---|
|
a Determined by 1H NMR analysis of the reaction mixture.
b 5 mol% of dioxane added.
c 1·TFA was formed in situ by addition of TFA (2.5 mol%) to complex 1 (2.5 mol%) prior to addition of Bu4NBr.
d Complex 8 (5 mol%) was formed in situ by addition of TFA (5.0 mol%) to complex 1 (2.5 mol%) prior to addition of Bu4NBr.
|
|||||
| 1 | None | 2.5 | 25 | 1 | 0 |
| 2 | 1 (2.5) | 0 | 25 | 1 | 9 |
| 3 | 1 (2.5) | 2.5 | 25 | 1 | 53 |
| 4 |
1b (2.5) |
2.5 | 25 | 1 | 55 |
| 5 | 9 (2.5) | 2.5 | 25 | 1 | 46 |
| 6 | 1·TFA (2.5) | 2.5 | 25 | 1 | 36 |
| 7 |
1·TFAc (2.5) |
2.5 | 25 | 1 | 48 |
| 8 | 8 (2.5) | 2.5 | 25 | 1 | 27 |
| 9 |
8d (5.0) |
2.5 | 25 | 1 | 11 |
| 10 | None | 0 | 100 | 50 | 0 |
| 11 | 1 (2.5) | 0 | 100 | 50 | (59) |
| 13 | 1·TFA (2.5) | 0 | 100 | 50 | (29) |
| 12 | 9 (2.5) | 0 | 100 | 50 | (40) |
| 14 | 8 (2.5) | 0 | 100 | 50 | (7) |
The results presented in entries 3–7 of Table 1 suggest that it is the nucleophile (tetrabutylammonium bromide) rather than the Lewis acid that is the dominant factor in determining the effectiveness of a tetrabutylammonium bromide/bimetallic aluminium(salen) complex catalyst system. Therefore, 1, 1·TFA, 8 and 9 were tested as catalysts under alternative, tetrabutylammonium bromide free conditions at 100 °C and 50 bar carbon dioxide pressure11 (Scheme 7, conditions B). Entry 10 shows that under these conditions, no reaction occurs in the absence of a catalyst. Complex 1 shows reasonable catalytic activity under these reaction conditions (entry 11). Protonation of complex 1 with trifluoroactic acid to form 1·TFA or with HCl to form complex 9 reduced the catalytic activity (entries 12 and 13). The significantly higher catalytic activity of complex 9 compared to 1·TFA can be explained by the presence of a reasonable nucleophile (chloride) in complex 9 but not in complex 1·TFA. In the presence of chloride two catalytic cycles are possible under these reaction conditions, involving epoxide ring-opening by chloride or carboxylate as previously determined.9–11,19 The lower catalytic activity of both complexes 9 and 1·TFA compared to complex 1 is consistent with the need for the μ-oxo group of the complexes to interact with carbon dioxide (as shown in Fig. 6) during the catalytic cycle in the absence of a nucleophilic cocatalyst.10,11 Protonation of the μ-oxo group will inhibit this interaction. Finally, entry 14 shows that monometallic complex 8 is again a very poor catalyst under these conditions.
Conclusions
Five-coordinate aluminium(salen) complexes are shown to have dynamic structures with both trigonal bipyramidal and square based pyramidal conformations accessible in both the solution and solid states. These interconvert by a salen ligand rearrangement mechanism which line shape analysis of variable temperature NMR spectra show is neither associative or dissociative. The rearrangement is often rapid on the NMR timescale at ambient temperature which results in deceptively simple, apparently C2-symmetrical structures being observed.Protonation of μ-oxo bridged bimetallic aluminium(salen) complexes occurs on the bridging oxygen and results in significant changes to the coordination geometry and ligand conformations around the aluminium centres. Protonation also increases the Lewis acidity of the aluminium centres as shown by their interaction by Lewis bases such as water, dioxane and 1,2-epoxyhexane. However, the protonated complexes have lower catalytic activity than the unprotonated complex for the synthesis of styrene carbonate from styrene oxide and carbon dioxide which suggests that nucleophilicity (of a cocatalyst or the μ-oxo bridge) is more important than Lewis acidity for the catalytic activity.
Experimental
(μ2-Hydroxy)-bis(2,2′-((1R,2R)-cyclohexane-1,2-diylbis (nitrilomethylylidene)) bis(4,6-di-tert-butylphenolato))-di-aluminium chloride hydrate 1·HCl
2,2′-((1R,2R)-cyclohexane-1,2-diylbis(nitrilomethylylidene))bis (4,6-di-tert-butylphenolato)-aluminium chloride12 was dissolved in chloroform and the ensuing solution allowed to slowly evaporate to dryness at room temperature. The resulting solid was dissolved in toluene and the solution allowed to slowly evaporate at room temperature, giving crystals of 1·HCl suitable for analysis by X-ray crystallography. Diffraction data were collected at 110 K, with Cu–Kα radiation (λ = 1.54184 Å) on an Oxford Diffraction SuperNova using an EOS CCD camera. The crystal was cooled with an Oxford Instruments Cryojet. Diffractometer control, data collection, initial unit cell determination, frame integration and unit-cell refinement was carried out with “Crysalis”.23 Face-indexed absorption corrections were applied using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm.24 OLEX225 was used for overall structure solution, refinement and preparation of computer graphics and publication data. Within OLEX2, the algorithm used for structure solution was “ShelXS direct methods”,26 using direct methods. Refinement by full-matrix least-squares used the SHELXL algorithm23 within OLEX222 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed using a “riding model” and included in the refinement at calculated positions except for O–H hydrogens which were located by difference map. The difference map revealed the presence of three molecules of solvent occupying at least three orientations. All attempts to model this solvent using a discrete atom model failed and so a solvent mask was used to model this electron density.
(μ2-Hydroxy)-bis(2,2′-((1R,2R)-cyclohexane-1,2-diylbis (nitrilomethylylidene))bis(4,6-di-tert-butylphenolato))-di-aluminium trifluoroacetate 1·TFA
To a stirred solution of complex 1 (0.100 g, 0.086 mmol) in CH2Cl2 (0.5 mL) was added trifluoroacetic acid (6.6 μL, 1 equiv.). The solution was stirred for 5 minutes then evaporated in vacuo to provide 1·TFA (0.110 g, 0.086 mmol) in quantitative yield. [α]20D –548 (c 1.0, CH2Cl2); νmax(ATR) 3638, 2952, 2907, 2866, 1706 and 1622 cm−1; δH(400 MHz, 298 K): 7.92 (4H, br, 4× CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 7.42 (4H, br, 4× ArCH), 6.99 (4H, br, 4× ArCH), 3.25 (5H, br, 4× NCH + OH), 2.32 (4H, br, 2× CH2), 2.0–1.0 (12H, br, 6×CH2), 1.32 (72H, s, 4× C(CH3)3); δC(100 MHz, 298 K): 167–164 (br), 162.1, 158.4 (q 2JCF 36.3 Hz), 140.4, 139–137 (br), 130.5, 129–127 (br), 118.0, 116.1 (q 1JCF 292.1 Hz), 65–62 (br), 35.4, 33.9, 31.4, 29.4, 26.9, 23.3; δF(377 MHz, 298 K): −76.2; m/z (FD+): found 1158.76872; C72H104N4O5Al2 (M − TFA)+ requires 1158.76320; found: C 69.0, H 8.7, N 4.5%; C74H104N4O7F3Al2·H2O requires: C 68.9, H 8.3, N 4.3%.
N), 7.42 (4H, br, 4× ArCH), 6.99 (4H, br, 4× ArCH), 3.25 (5H, br, 4× NCH + OH), 2.32 (4H, br, 2× CH2), 2.0–1.0 (12H, br, 6×CH2), 1.32 (72H, s, 4× C(CH3)3); δC(100 MHz, 298 K): 167–164 (br), 162.1, 158.4 (q 2JCF 36.3 Hz), 140.4, 139–137 (br), 130.5, 129–127 (br), 118.0, 116.1 (q 1JCF 292.1 Hz), 65–62 (br), 35.4, 33.9, 31.4, 29.4, 26.9, 23.3; δF(377 MHz, 298 K): −76.2; m/z (FD+): found 1158.76872; C72H104N4O5Al2 (M − TFA)+ requires 1158.76320; found: C 69.0, H 8.7, N 4.5%; C74H104N4O7F3Al2·H2O requires: C 68.9, H 8.3, N 4.3%.
2,2′-((1R,2R)-Cyclohexane-1,2-diylbis(nitrilomethylylidene))bis (4,6-di-tert-butylphenolato)-aluminium trifluoroacetate 8
To a stirred solution of complex 1 (0.100 g, 0.086 mmol) in CH2Cl2 (0.5 mL) was added trifluoroacetic acid (13.2 μL, 2 equiv.). The solution was stirred for 5 minutes then evaporated in vacuo to provide complex 8 (0.118 g, 0.173 mmol) in quantitative yield. [α]20D –410 (c 1.0, CH2Cl2); νmax(ATR) 2953, 2908, 2867, 1696 and 1622 cm−1; δH(400 MHz, 298 K): 8.33 (2H, s, 2× CHN), 7.56 (2H, d 4JHH 2.1 Hz, 2× ArCH), 7.09 (2H, d 4JHH 2.2 Hz, 2× ArCH), 3.50 (2H, br, 2× NCH), 2.54 (2H, br, CH2), 2.10 (2H, br, CH2), 1.59 (2H, br, CH2), 1.51 (18H, s, 2× C(CH3)3), 1.47 (2H, br, CH2), 1.31 (18H, s, 2× C(CH3)3); δC(100 MHz, 298 K): 158.4 (q 2JCF 38.4 Hz), 141.1, 139.0, 136.4, 131.4, 127.9, 126.2, 118.2, 115.7 (q 1JCF 287.6 Hz), 65–63 (br), 35.6, 34.2, 31.5, 29.7, 27.3, 23.9; δF(377 MHz, 298 K): −76.2; m/z (FD+): found 684.37197; C38H52N2O4F3Al (M+) requires 684.36891; found: C 63.2, H 7.5, N 3.9%; C38H52N2O4F3Al·2H2O requires: C 63.2, H 7.8, N 3.9%.
(μ2-Hydroxy)-bis(2,2′-((1R,2R)-cyclohexane-1,2-diylbis (nitrilomethylylidene)) bis(4,6-di-tert-butylphenolato))-di-aluminium chloride dioxane 9
To a stirred solution of complex 1 (0.100 g, 0.086 mmol) in CH2Cl2 (0.5 mL) was added a solution of HCl in dioxane (21.6 μL of a 4.0 M solution, 1 equiv.). The solution was stirred for 5 minutes then evaporated in vacuo to provide complex 9 (0.1061 g, 0.083 mmol) in 96% yield. [α]20D –499 (c 1.0, CH2Cl2); νmax(ATR) 2952, 2906, 2885 and 1622 cm−1; δH(400 MHz, 298 K): 7.75 (4H, br, 4× CHN), 7.35 (4H, br, 4× ArCH), 6.94 (4H, br, 4× ArCH), 3.64 (8H, s, 4× CH2O), 3.03 (4H, br, 4× NCH), 1.61 (5H, br, OH + 2× CH2), 1.5–0.9 (12H, br, 6× CH2), 1.28 (36H, s, 4× C(CH3)3), 1.18 (36H, s, 4× C(CH3)3); δC(100 MHz, 298 K): 164.7 (br), 161.7, 140.5, 138.8 (br), 130.9 (br), 128.4 (br), 118.1, 67.1, 63.1 (br), 35.5 (br), 34.1, 31.4, 29.3 (br), 26.7 (br), 23.3 (br); m/z (FD+): found 1158.76381; C72H104N4O5Al2 (M − Cl-dioxane)+ requires 1158.76320.
(μ2-Dioxane)-bis(2,2′-((1R,2R)-cyclohexane-1,2-diylbis (nitrilomethylylidene)) bis(4,6-di-tert-butylphenolato))-di-aluminium chloride 11
1–4 Dioxane (1 mL) was added to 2,2′-((1R,2R)-cyclohexane-1,2-diylbis(nitrilomethylylidene))bis(4,6-di-tert-butyl phenolato)-aluminium chloride12 (0.100 g, 0.186 mmol) in a stoppered oven dried round bottomed flask and the solution left to stir until dissolution. The flask was then attached to a Schlenk line and solvent removed in vacuo to leave compound 11 (0.111 g, 0.160 mmol) as a yellow solid in 86% yield. [α]20D –567 (c 1.0, CH2Cl2); νmax(ATR) 2953, 2909, 2865 and 1616 cm−1; δH(400 MHz, 298 K): 8.36 (4H, br, 4× CHN), 7.59 (4H, br, 4× ArCH), 3.86 (3H, br, 3× NCH), 3.71 (8H, s, 4× OCH2), 3.19 (1H, br, NCH), 2.52 (4H, br, 2× CH2), 2.08 (4H, br, 2× CH2), 1.8–1.1 (8H, m, 4× CH2), 1.58 (36H, s, 4× C(CH3)3), 1.34 (36H, s, 4× C(CH3)3); δC(100 MHz, 298 K): 160.4 (br), 141.2, 138.8 (br), 131.4 (br), 130.8 (br), 127.9 (br), 118.3, 67.2, 63.0 (br), 62.2 (br), 35.7, 34.2, 31.5, 29.9, 27.5, 23.8. m/z (FD+): found 606.35561; C36H52N2O2ClAl (0.5 M-dioxane)+ requires 606.35327.
General procedure for the synthesis of styrene carbonate 13 at 1 bar pressure
A mixture of styrene oxide 12 (2 mmol, 0.23 ml) and catalyst or catalyst precursor (2.5 mol%) was stirred vigorously in a reaction vial. If the catalyst was being formed in situ, TFA (2.5 mol%, 3.83 μl or 5 mol%, 7.65 μl) was added; followed in all cases by Bu4NBr (2.5 mol%, 0.0161 g). Upon dissolution, the reaction vial was purged for 5 minutes with CO2 and then fitted with a balloon filled with CO2. The reaction was then stirred at 25 °C for 24 hours. The unpurified reaction mixture was analysed by 1H NMR spectroscopy to determine the conversion of styrene oxide 12 into styrene carbonate 13.General procedure for the synthesis of styrene carbonate 13 at 50 bar pressure
Styrene oxide 12 (2 mmol, 0.23 ml) was added to a sample vial preloaded with the desired catalyst (2.5 mol%) and a magnetic stirrer bar. The vial was sealed with a lid pierced by a needle and placed in a high-pressure reactor. The reactor was flushed with CO2, heated to 100 °C and pressurized to 50 bar with CO2. The reaction was stirred for 24 hours, then the reactor was cooled with a liquid nitrogen bath, before releasing the CO2 pressure. The reaction mixture was dissolved in dichloromethane and filtered through silica to remove catalyst before being analyzed by 1H NMR spectroscopy to determine the conversion. Pure styrene carbonate 13 was obtained by column chromatography using hexane:EtOAc mixtures (1:0, 1:6, 1:4, 1:2, 1:1 and 0:1) as eluent. νmax(CH2Cl2) 3078, 3038, 2926, 1784, 1459 and 1064 cm−1; 1H NMR δH(400 MHz, CDCl3): 7.5–7.4 (3H, m, ArH), 7.4–7.3 (2H, m, ArH), 5.67 (1H, dd 3J 8.1, 7.9 Hz, PhCHO), 4.80 (1H, dd 3J 8.3 Hz, 2J 8.6 Hz, CH2O), 4.34 (1H, 3J 7.9, 2J 8.6 Hz, CH2O); 13C NMR δC(101 MHz, CDCl3): 155.0, 135.9, 129.8, 129.3, 125.9, 78.1, 71.3; m/z (ESI+): found 187.0369; C9H8O3Na (M + Na)+ requires 187.0366.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011, ch. 7 Search PubMed
.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS
- A. Gualandi, F. Calogero, S. Potenti and P. G. Cozzi, Molecules, 2019, 24, article 1716 CrossRef
- Y. N. Belokon, J. Fuentes, M. North and J. W. Steed, Tetrahedron, 2004, 60, 3191–3204 CrossRef CAS
- G. A. Morris, H. Zhou, C. L. Stern and S. T. Nguyen, Inorg. Chem., 2001, 40, 3222–3227 CrossRef CAS
- Q. Liu and M. Ding, J. Organomet. Chem., 1998, 553, 179–181 CrossRef CAS
- A. M. Sargeson and G. H. Searle, Inorg. Chem., 1965, 4, 45–52 CrossRef CAS
- V. Vergopoulos, S. Jantzen, D. Rodewald and D. Rehder, J. Chem. Soc., Chem. Commun., 1995, 377–378 RSC
- M. North, ARKIVOC, 2012, (i), 610–628 Search PubMed
- J. A. Castro-Osma, M. North and X. Wu, Chem. – Eur. J., 2014, 20, 15005–15008 CrossRef CAS
- J. A. Castro-Osma, M. North, W. K. Offermans, W. Leitner and T. E. Müller, ChemSusChem, 2016, 9, 791–794 CrossRef CAS
- M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, ACS Catal., 2015, 5, 3398–3402 CrossRef CAS
- T. Baronsky, C. Beattie, R. W. Harrington, R. Irfan, M. North, J. G. Osende and C. Young, ACS Catal., 2013, 3, 790–797 CrossRef CAS
- M. Sengoden, M. North and A. C. Whitwood, ChemSusChem, 2019, 12, 3296–3303 CrossRef CAS
- W. Clegg, R. W. Harrington, M. North and P. Villuendas, J. Org. Chem., 2010, 75, 6201–6207 CrossRef CAS
- M. North, P. Villuendas and C. Williamson, Tetrahedron, 2010, 66, 1915–1924 CrossRef CAS
- T. Yue, M.-X. Wang, D.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2008, 47, 9454–9457 CrossRef CAS
- J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC
- W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828–6843 CrossRef CAS
- A. W. Addison, N. T. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- P. L. Gurian, L. K. Cheatham, J. W. Ziller and A. R. Barron, J. Chem. Soc., Dalton Trans., 1991, 1449–1456 RSC
- H. Sugimoto, C. Kawamura, M. Kuroki, T. Aida and S. Inoue, Macromolecules, 1994, 27, 2013–2018 CrossRef CAS
-
CrysAlisPro, Version 1.171.34.41, Oxford Diffraction Ltd Search PubMed
-
Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm within CrysAlisPro software, Version 1.171.34.40, Oxford Diffraction Ltd Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray data and copies of all recorded and simulated spectra. CCDC 2022416. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03598j |
| ‡ X-ray data for complex 1·HCl has been deposited with the CCDC 2022416.† |
| § Line shape analysis was carried out using WinDNMR (https://www.chem.wisc.edu/areas/reich/plt/windnmr.htm). |
| This journal is © The Royal Society of Chemistry 2021 |