DOI:
10.1039/D0DT03316B
(Paper)
Dalton Trans., 2021,
50, 1901-1912
Tuning the stereoelectronic factors of iron(II)-2-aminophenolate complexes for the reaction with dioxygen: oxygenolytic C–C bond cleavage vs. oxidation of complex†
Received
23rd September 2020
, Accepted 19th December 2020
First published on 23rd December 2020
Abstract
Oxidative C–C bond cleavage of 2-aminophenols mediated by transition metals and dioxygen is a topic of great interest. While the oxygenolytic C–C bond cleavage reaction relies on the inherent redox non-innocent property of 2-aminophenols, the metal complexes of 2-aminophenolates often undergo 1e−/2e− oxidation events (metal or ligand oxidation), instead of the direct addition of O2 for subsequent C–C bond cleavage. In this work, we report the isolation, characterization and dioxygen reactivity of a series of ternary iron(II)-2-aminophenolate complexes [(TpPh,Me)FeII(X)], where X = 2-amino-4-tert-butylphenolate (4-tBu-HAP) (1); X = 2-amino-4,6-di-tert-butylphenolate (4,6-di-tBu-HAP) (2); X = 2-amino-4-nitrophenolate (4-NO2-HAP)(3); and X = 2-anilino-4,6-di-tert-butylphenolate (NH-Ph-4,6-di-tBu-HAP) (4) supported by a facial tridentate nitrogen donor ligand (TpPh,Me = hydrotris(3-phenyl-5-methylpyrazol-1-yl)borate). Another facial N3 ligand (TpPh2 = hydrotris(3,5-diphenyl-pyrazol-1-yl)borate) has been used to isolate an iron(II)-2-anilino-4,6-di-tert-butylphenolate complex (5) for comparison. Both [(TpPh,Me)FeII(4-tBu-HAP)] (1) and [(TpPh,Me)FeII(4,6-di-tBu-HAP)] (2) undergo regioselective oxidative aromatic ring fission reaction of the coordinated 2-aminophenols to the corresponding 2-picolinic acids in the reaction with dioxygen. In contrast, complex [(TpPh,Me)FeII(4-NO2-HAP)] (3) displays metal based oxidation to form an iron(III)-2-amidophenolate complex. Complexes [(TpPh,Me)FeII(NH-Ph-4,6-di-tBu-HAP)] (4) and [(TpPh2)FeII(NH-Ph-4,6-di-tBu-HAP)] (5) react with dioxygen to undergo 2e− oxidation with the formation of the corresponding iron(III)-2-iminobenzosemiquinonato radical species implicating the importance of the –NH2 group in directing the C–C bond cleavage reactivity of 2-aminophenols. The systematic study presented in this work unravels the effect of the electronic and structural properties of the redox non-innocent 2-aminophenolate ring and the supporting ligand on the C–C bond cleavage reactivity vs. the metal/ligand oxidation of the complexes. The study further reveals that proper modulation of the stereoelectronic factors enables us to design a well synchronised proton transfer (PT) and dioxygen binding events for complexes 1 and 2 that mimic the structure and function of the nonheme enzyme 2-aminophenol-1,6-dioxygenase (APD).
Introduction
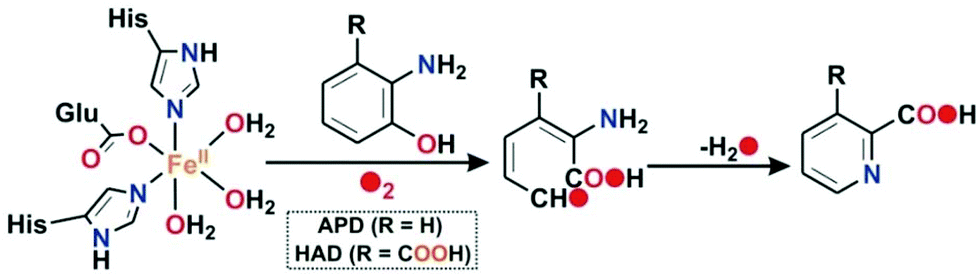
Oxidative C–C bond cleavage reaction is a key metabolic step in the biodegradation of toxic and xenobiotic compounds.1–3 The resulting ring cleavage products are used by soil bacteria as the only source of energy and carbon in nature, which in turn is critical to maintaining the global carbon cycle.1 Ring cleaving dioxygenases constitute a large family of mononuclear nonheme enzymes which are involved in the aerobic degradation of aromatic compounds, such as ortho-hydroxy phenols (catechols) or o-aminophenols or gentisic acid to aliphatic products in the soil with the incorporation of both atoms of dioxygen into the cleavage products.1,4,5 In general, for the aromatic diols (catechols), depending upon the position of the C–C bond cleavage, these enzymes are classified into two categories: intradiol catechol dioxygenases and extradiol catechol dioxygenases.6,7 2-Aminophenol-1,6-dioxygenase (APD), isolated and purified from Pseudomonas pseudoalcaligenes JS45, is a ring cleaving enzyme in a small group of nonheme extradiol type dioxygenases and is responsible for the C–C bond cleavage of 2-aminophenols (Scheme 1).8
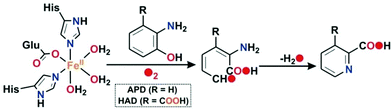 |
| | Scheme 1 Reactions catalyzed by nonheme enzyme APD and HAD. | |
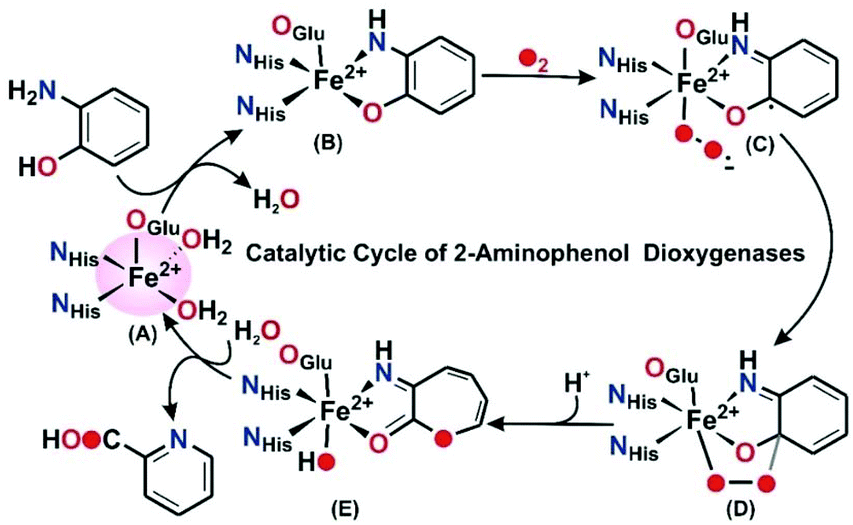
In the catalytic cycle of the C–C bond cleavage pathway of the enzyme 2-aminophenol-1,6-dioxygenase (APD), the C1–C6 bond adjacent to the –OH group is cleaved9,10 in the presence of dioxygen.11,12 The C–C cleavage product, 2-aminomuconic acid semialdehyde, spontaneously loses a water molecule to form 2-picolinic acid (Scheme 1).13 A related enzyme, 3-hydroxyanthranilate-3,4-dioxygenase (HAD), isolated from Saccharomyces cerevisiae, catalyzes the ring opening of 3-hydroxyanthranilate to quinolinic acid in the tryptophan catabolism pathway.9 Structural studies reveal that the active sites of HAD14–16 and APD17 contain an iron(II) center coordinated by the “2-His-1-Glu facial triad” motif.6,18 2-Aminophenol/3-hydroxyanthranilic acid substrates bind to the iron(II) center in a bidentate mode to initiate the aromatic ring cleavage reaction by activating dioxygen. On the basis of the structural and biochemical studies on HAD and APD, a mechanistic proposal similar to that of extradiol catechol dioxygenases has been proposed.9 The substrate binds to the ferrous ion (A) and the enzyme–substrate complex (B) activates dioxygen to form initially an iron(III)-superoxide intermediate that receives an electron from the redox non-innocent 2-aminophenolate unit to form an iron(II)-superoxide diradical species (C) (Scheme 2). An iron(II)-peroxide intermediate (D) is subsequently generated by the attack of the iron(II)-superoxo on the ligand based radical. The peroxide moiety then undergoes O–O bond heterolysis to form a lactone intermediate (E). One oxygen atom from the peroxide unit is incorporated into the lactone ring. Hydrolysis of the lactone affords the C–C bond cleavage product, 2-amino muconic acid semialdehyde, which is then converted to 2-picolinic acid through a nonenzymatic pathway (Scheme 2).19
 |
| | Scheme 2 Proposed mechanism for the catalytic cycle of APD. | |
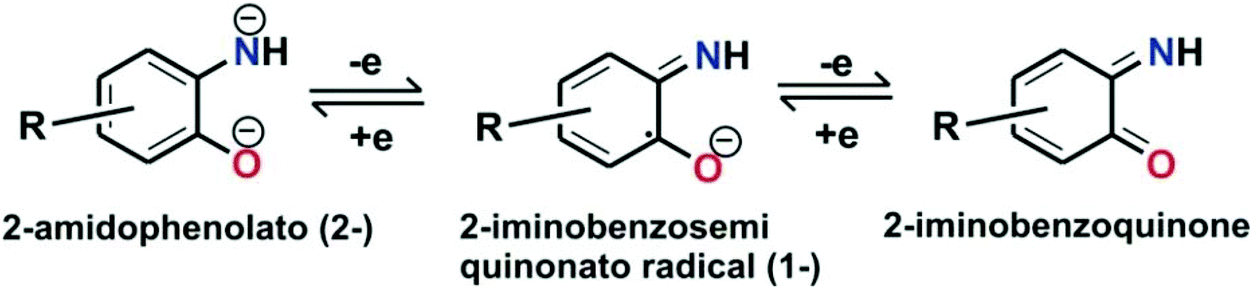
The oxidative transformation of 2-aminophenol to 2-picolinic acid by molecular oxygen has attracted the attention of biomimetic chemists to develop synthetic models of 2-aminophenol dioxygenase. Moreover, similar to catechols, o-aminophenols are well-known “redox non-innocent” ligands20–23 and are stabilized in different redox states upon binding with suitable metal ions (Scheme 3).
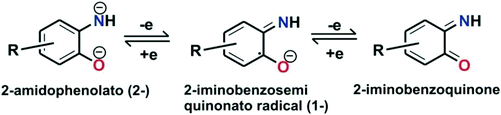 |
| | Scheme 3 Redox non-innocence behavior of 2-aminophenols. | |
Reports of several iron(II) complexes coordinated by the non-innocent o-aminophenolate ligands in different redox states have been published by Wieghardt and coworkers.21,22,24–28 In biomimetic chemistry, dioxygen reactivity of model iron(II)-2-aminophenolate is less explored compared to the catecholate complexes.6 Paine's group reported the O2-dependent C–C bond cleavage reactivity of iron(II)-2-aminophenolate complexes supported by neutral N4 donor ligands.29,30 The complexes have been shown to react with dioxygen under ambient conditions to afford substituted 2-picolinic acids mimicking the function of 2-aminophenol dioxygenases (APD and HAD).29,30 These model complexes provide insights into the mechanism of aromatic ring cleavage of 2-aminophenols. Subsequently, the Paine group developed catalytic models of 2-aminophenol dioxygenases using neutral tridentate31 and tetradentate ligands.32 The role of the second sphere residue33 as well as the pH of the reaction solution in controlling the catalytic reactivity and selectivity has been proposed. However, the reported catalytic model of 2-aminophenol dioxygenase with a tridentate ligand was not structurally characterized.31 In fact, there is no report of structurally characterized iron(II)-2-aminophenolate complex that closely mimics the active site structure of APD and at the same time displays C–C cleavage activity. Monoanionic tris(pyrazolyl)borates are often used as bioinspired N3 donor ligands to mimic the ‘2-His-1-carboxylate facial triad’ found in the active sites of many dioxygen activating nonheme mono-iron enzymes.34–43 Since APD and HAD belong to the superfamily of nonheme iron enzymes with the ‘2-His-1-carboxylate’ facial triad, tris(pyrazolyl)borates could be appropriate ligands to develop biomimetic models of these enzymes. Fiedler's group reported the synthesis and characterization of iron(II)-2-aminophenolate complexes with the monoanionic TpPh2 ligand (TpPh2 = hydrotris(3,5-diphenyl-pyrazol-1-yl)borate) and neutral Ph2TIP tris(4,5-diphenyl-1-methylimidazol-2-yl)phosphine ligand.44–46 The iron(II)-2-aminophenolate complex, supported by the TpPh2 ligand, when oxidized chemically using one electron oxidant led to the isolation of an iron(II)-2-iminobenzosemiquinone radical intermediate with S = 3/2 spin state.44 The radical species has relevance to an intermediate proposed in the catalytic cycle of APD. Although the dioxygen reactivity of the TpPh2 supported iron(II)-2-aminophenolate complex was not reported, the electronic structure of the intermediate provided mechanistic information on the catalytic cycle of the enzyme.44 On the other hand, the dioxygen reactivity of the neutral Ph2TIP ligand led to the 2e− oxidation process to form [(Ph2TIP)FeIII(4,6-di-tBu-ISQ)]+, where ISQ is 2-iminobenzosemiquinone. Similar 2e− oxidation events of 2-aminophenol in iron complexes of monoanionic N3C donor ligands have been reported by Paine and co-workers. The low-spin complexes do not exhibit aromatic C–C bond cleavage reactions.47,48 These studies reveal that the difference in the electronic structure of the biomimetic complexes with a change in ligand backbones apparently controls reactivity, whether a simple 1e−/2e− oxidation event or the direct addition of O2 to form the C–C bond cleavage reactions occurs in the biomimetic iron(II)-2-aminophenolate complexes.30,44,46 Recently, Fiedler and coworkers have reported the dioxygen reactivity of pentacoordinate cobalt(II)-2-aminophenolate complexes, [Co(TpMe2)(4,6-di-tBu-HAP)] and [Co(TpPh2)(4,6-di-tBu-HAP)].49 Both the Co(II)-4,6-di-tBu-HAP complexes exhibit hydrogen atom transfer reactivity (HAT) at room temperature upon exposure to dioxygen to form the corresponding (TpR2)CoII-2-iminobenzosemiquinone species. However, at low temperature (−70 °C) the [Co(TpR2)(4,6-di-tBu-HAP)] complex supported by the TpMe2 ligand backbone reacts with dioxygen to form an oxygen adduct which was identified as the CoIII-superoxo species. The TpPh2 supported CoII-4,6-di-tBu-HAP complex on the other hand is unreactive towards dioxygen at lower temperatures suggesting that sterics can modulate the energetics of oxygen binding. Interestingly, the cobalt(II)-2-iminosemibenzoquinone intermediate supported by the TpMe2 ligand further reacts with dioxygen at reduced temperature to form a CoIII-alkylperoxo intermediate bound to the substituted iminobenzoquinone ligand. The observed CoIII-superoxo as well as the CoIII-alkylperoxo species mimic the proposed intermediates in the enzymatic cycle.9 In fact the crystal structure of CoIII-alkylperoxo resembles the iron-alkylperoxo intermediate reported by Lipscomb in a crystal structure of catechol dioxygenase HPCD.50 Unfortunately, none of these synthetic oxygen intermediates led to the C–C bond cleavage of substituted 2-aminophenol, in contrast to the enzymatic system. Thus tuning of stereoelectronic properties of the supporting ligand and 2-aminophenol substrates is one of the key parameters that may be given due consideration while developing models of APD.
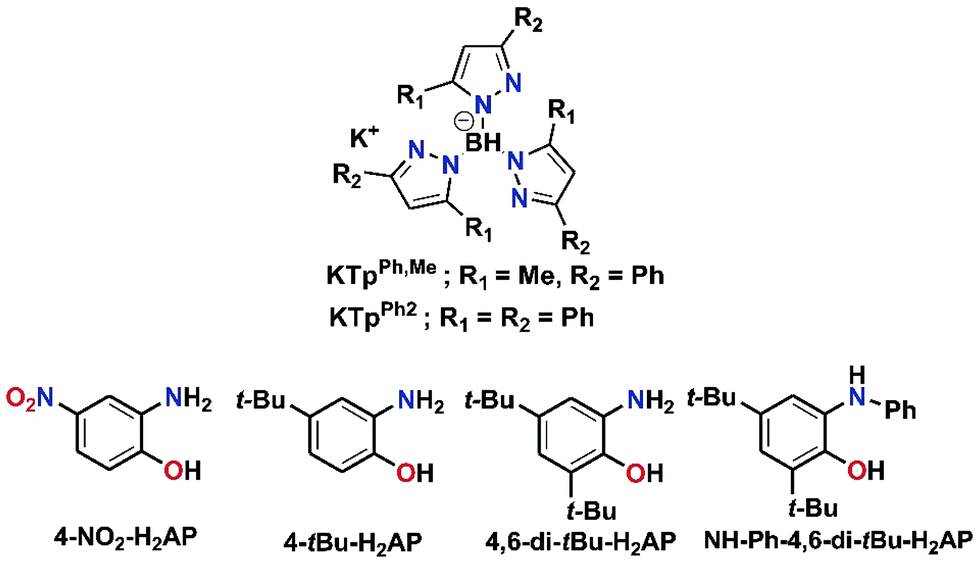
With this background, we have explored in this work the dioxygen reactivity of a series of iron(II)-2-aminophenolate complexes [(TpPh,Me)FeII(X)] (X = 2-amino-4-tert-butylphenolate (4-tBu-HAP) (1); X = 2-amino-4,6-di-tert-butylphenolate (4,6-di-tBu-HAP) (2); X = 2-amino-4-nitrophenolate (4-NO2-HAP) (3); and X = 2-anilino-4,6-di-tert-butylphenolate (NH-Ph-4,6-di-tBu-HAP) (4) of the facial tridentate nitrogen ligand (TpPh,Me = hydrotris(3-phenyl-5-methylpyrazol-1-yl)borate) (Chart 1). Another facial N3 ligand (TpPh2 = hydrotris(3,5-diphenyl-pyrazol-1-yl)borate) (Chart 1) has also been utilized to isolate the corresponding iron(II)-2-anilino-4,6-di-tert-butylphenolate complex (5). We have envisioned that the dioxygen activation and subsequent aromatic ring cleavage activity of substituted 2-aminophenols could be controlled via fine-tuning of the supporting ligand as well as controlling the electron density on the aromatic ring of 2-aminophenols. The structural tuning of the monoanionic tris(pyrazolyl)borate ligand allows us not only to design close structural mimics but also reveal the functional activity of 2-aminophenol dioxygenases. By using the TpPh,Me = hydrotris(3-phenyl-5-methylpyrazol-1-yl)borate, which is sterically less congested and at the same time has proven superior activity compared to the TpPh2 backbone in biomimetic chemistry,51 we expected a different reactivity pattern of the corresponding iron(II)-2-aminophenolate complexes toward dioxygen. A comparative study of the dioxygen reactivity of the iron(II)-2-aminophenolate complexes is presented in this work. The results of this study shed light on the effect of the supporting ligand and of the substituted 2-aminophenolate in directing the course of reactivity towards oxygenolytic C–C bond cleavage reactivity vs. oxidation of the iron(II)-2-aminophenolate complexes (1 or 2 electron metal and/or ligand oxidation).
 |
| | Chart 1 Facial N3 ligands and substituted 2-aminophenols used in this work. | |
Results and discussion
The iron(II)-2-aminophenolate complexes [(TpPh,Me)FeII(4-tBu-HAP)] (1), [(TpPh,Me)FeII(4,6-di-tBu-HAP)] (2) and [(TpPh,Me)FeII(4-NO2-HAP)] (3) were synthesized by mixing equimolar amounts of the ligand (KTpPh,Me), iron(II) chloride, and the sodium salt of the respective 2-aminophenol in a dichloromethane–methanol solvent mixture at room temperature under a nitrogen atmosphere (Scheme 4). The complexes [(TpPh,Me)FeII(NH-Ph-4,6-di-tBu-HAP)] (4) and [(TpPh2)FeII(NH-Ph-4,6-di-tBu-HAP)] (5) were isolated from the reaction of the respective ligand (TpPh,Me for 4 and TpPh2 for 5) with iron(II) salt (iron chloride for 4 and iron perchlorate for 5) and 2-anilino-4,6-di-tert-butylphenol in the presence of triethylamine as a base. Complexes 4 and 5 can also be synthesized by the reaction of the respective ligand (TpPh,Me for 4 and TpPh2 for 5) with iron(II)-chloride and the sodium salt of 2-anilino-4,6-di-tertbutylphenol in a dichloromethane–methanol solvent mixture at room temperature under a nitrogen atmosphere (Scheme 4). Room temperature magnetic moment values ranging between 4.89 and 4.86μB indicate the high-spin nature of the complexes (ESI†). The 1H NMR spectra of the complexes show relatively sharp, well-resolved, and paramagnetically shifted resonances comparable to other high-spin iron(II) complexes of TpR,R′ ligands (ESI†).40,43,51–53
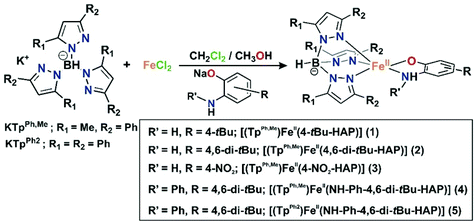 |
| | Scheme 4 Syntheses of iron(II)-2-aminophenolate complexes. | |
Crystal structures of 1, 2, and 5
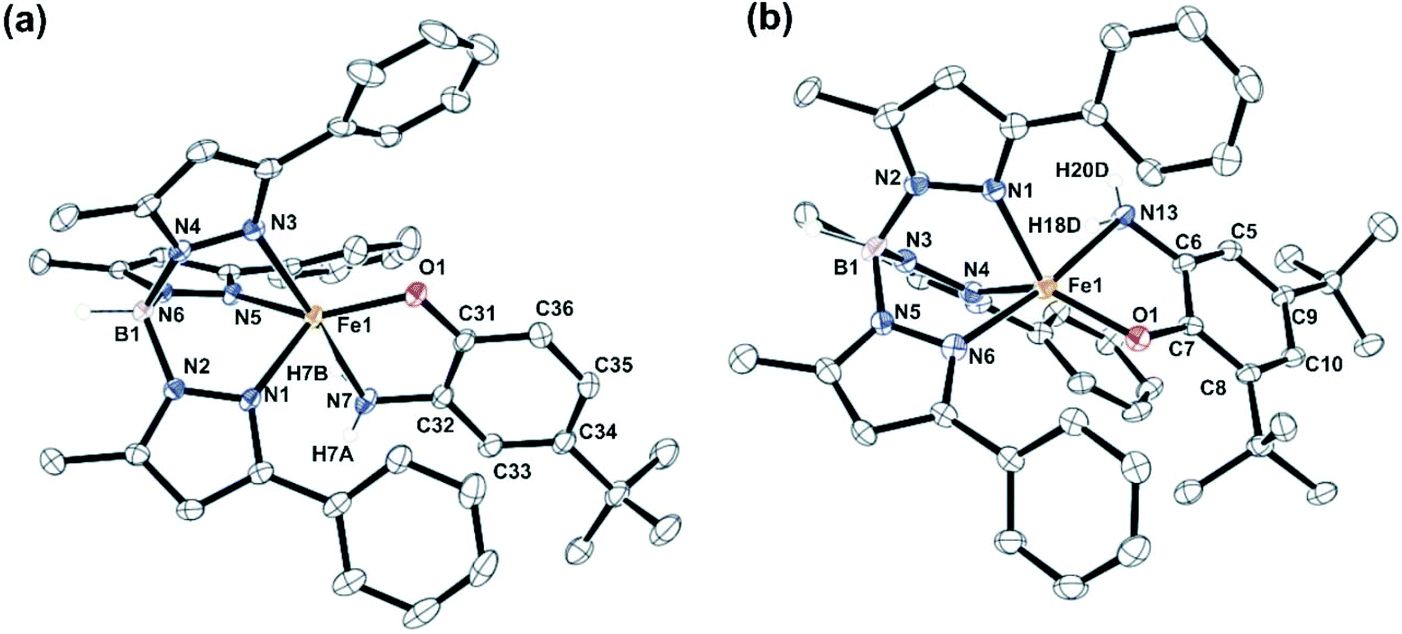
Complexes 1, 2 and 5 were characterized by single-crystal X-ray diffraction studies. The X-ray structures reveal that each of the neutral iron complexes (1 and 2) contains a five-coordinate center coordinated by one TpPh,Me ligand and one monoanionic 2-aminophenolate. Three nitrogen donors from the ligand (TpPh,Me) coordinate facially to the iron center and the remaining two sites are occupied by the bidentate 2-aminophenolates (Fig. 1). The average Fe–N distance of all the three complexes is comparable to that of the reported high-spin iron(II) complex tris(pyrazolyl borate) ligands (Tables S3 and S4, ESI†).43,44,51,54,55 In the five-coordinate complexes, two pyrazole nitrogens from the supporting ligand and the phenolate oxygen from 2-aminophenolate unit form the equatorial plane while the amine nitrogen of 2-aminophenol and one pyrazole nitrogen of the ligand occupies the axial positions giving rise to distorted trigonal bipyramidal (τ = 0.63 for 1 and τ = 0.69 for 2) coordination geometry at the iron center (Fig. 1). The C–C bond distances of the coordinated 2-aminophenolate ring in both 1 and 2 are similar to those of TpPh2FeII(4,6-di-tert-butylaminophenolate) reported by Fiedler and coworkers44 supporting the aromatic nature of the 2-aminophenolate ring (Table S3, ESI†).
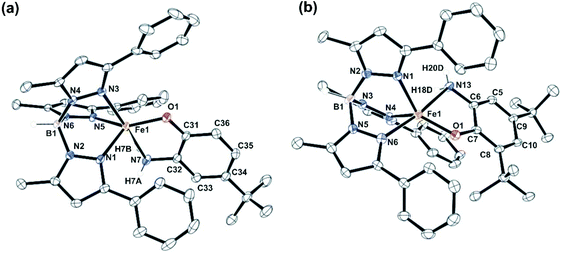 |
| | Fig. 1 Molecular structures of the neutral complexes (a) [(TpPh,Me)FeII(4-tBu-HAP)] (1) (All hydrogen atoms except those on N7 and B1 have been omitted for clarity) and (b) [(TpPh,Me)FeII(4,6-di-tBu-HAP)] (2). (All hydrogen atoms except those on N13 and B1 have been omitted for clarity). | |
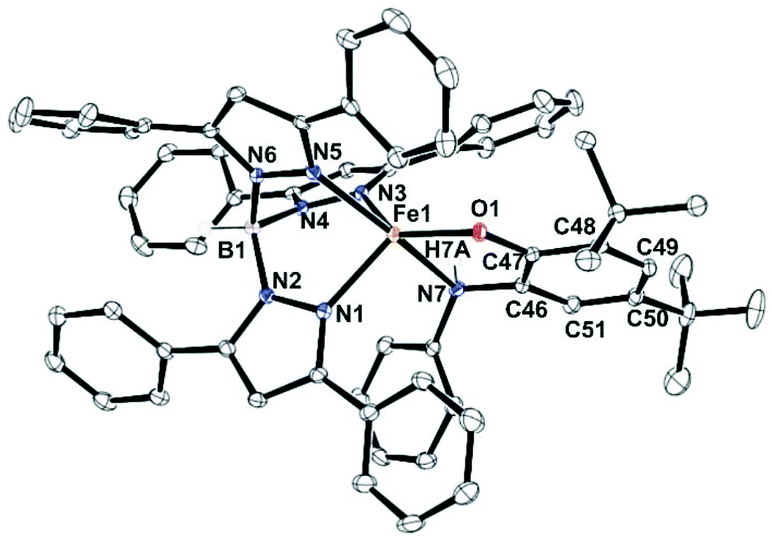
In the case of complex 5 (Fig. 2), a five-coordinate iron(II) center coordinated by one TpPh2 ligand and one monoanionic NHPh-2-aminophenolate unit is observed. The 2-aminophenolate oxygen O1 binds to the iron center trans to the pyrazole nitrogen N3 with the O1–Fe1–N3 angle of 131.46(5)° (Table S4, ESI†). The nitrogen atom N7 of the 2-aminophenolate unit occupies an axial position and is trans to the pyrazole nitrogen N5 with an N7–Fe1–N5 angle of 161.55(5)°. The oxygen atom O1 from 2-aminophenolate and two nitrogen donors (N3 and N1) from the ligand form the equatorial plane of the distorted trigonal bipyramidal complex (τ = 0.50).55 Compared to complexes 1 and 2, the distortion in 5 toward trigonal bipyramid is less. The C–C bond distances in the coordinated 2-aminophenolate moiety indicate the retention of the aromatic character without any quinonoid distortion (Table S4, ESI†). Although X-ray diffraction quality single crystals could not be isolated for [(TpPh,Me)FeII(NH-Ph-4,6-di-tBu-HAP)] (4), the structure and binding motif of 4 is expected to be very similar to that of [(TpPh2)FeII(NH-Ph-4,6-di-tBu-HAP)] (5) having a coordinated 2-anilino-4,6-di-tert-butylphenolate. The spectroscopic and analytical data of 3 suggest the complex to be mononuclear with five-coordinate geometry at the iron(II) center similar to that in 1 and 2. The binding mode of the 2-aminophenolate complexes (1) and (2) at the iron(II) centre bears similarity with the reported crystal structure of APD from Comamonas sp. strain CNB-1.14–17 However, unlike the enzymatic system which has an anionic NNO facial environment around the metal centre, the complexes (1 and 2) reported here have anionic NNN facial environment and thus serve as close structural mimics of 2-aminophenol dioxygenase.
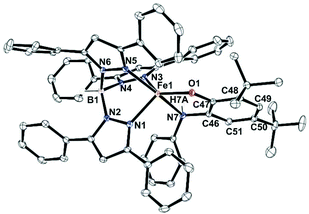 |
| | Fig. 2 Molecular structures of [(TpPh2)FeII(NH-Ph-4,6-di-tBu-HAP)] (5). All hydrogen atoms except those on N7 and B1 have been omitted for clarity. | |
Reactions of iron(II)-2-aminophenolate complexes with O2
Dioxygen reactivity of 1 and 2
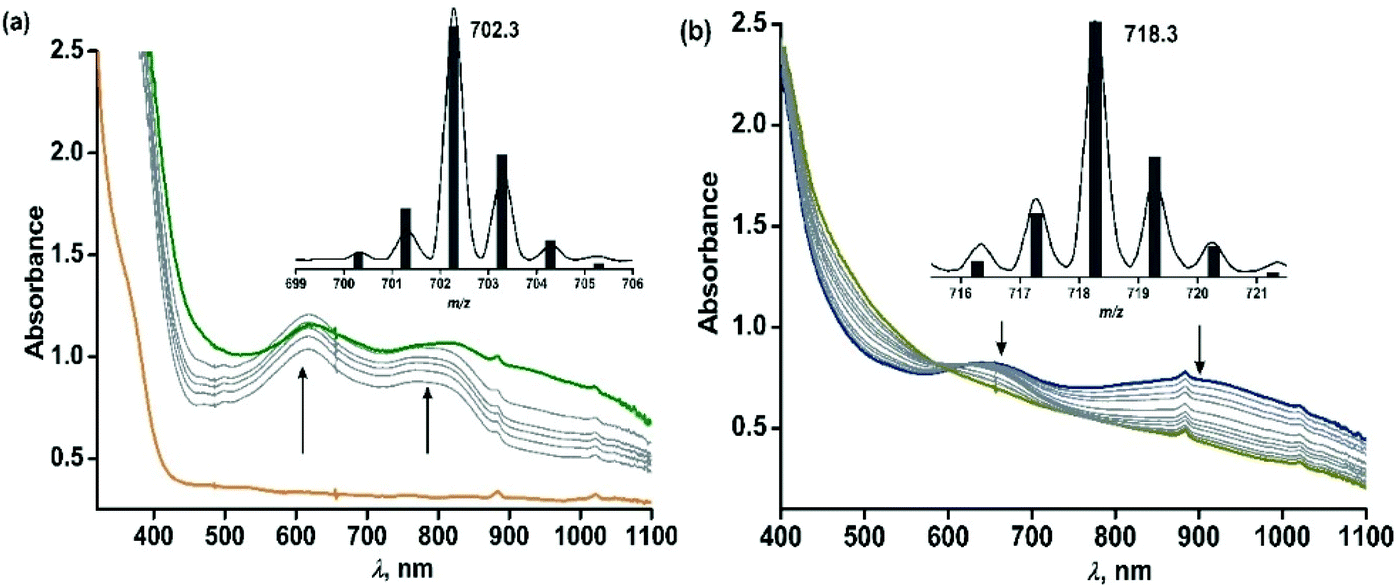
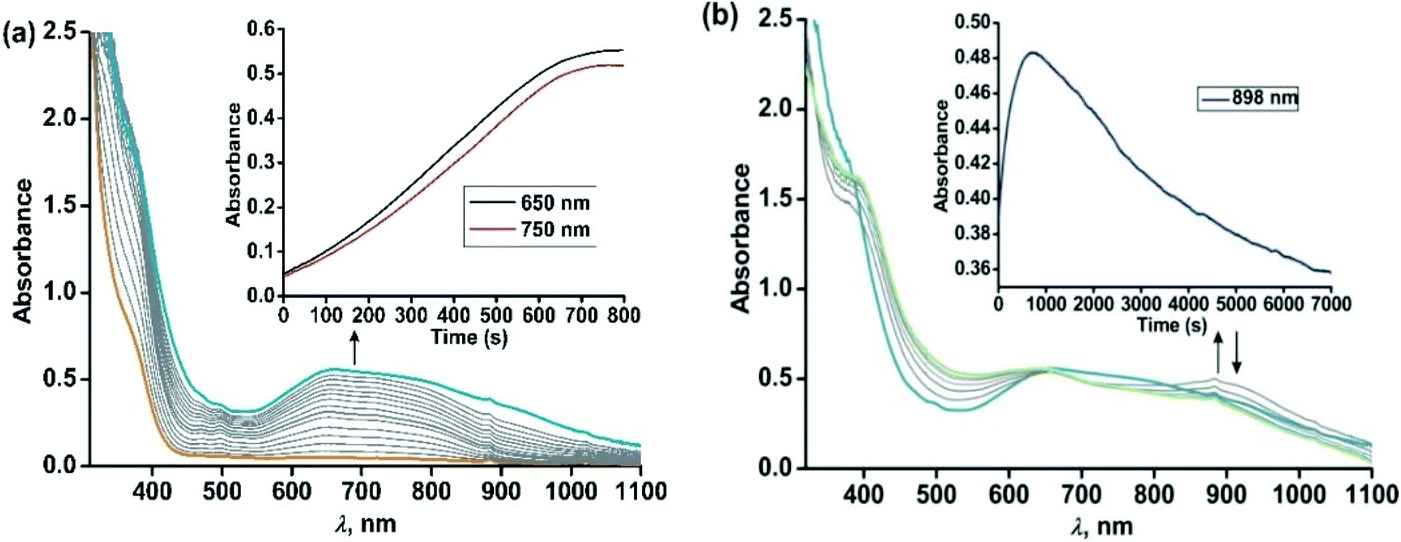
The iron(II)-2-aminophenolate complex [(TpPh,Me)FeII(4-tBu-HAP)] (1) reacts with O2 at room temperature, with the formation of two charge-transfer bands at 618 nm and 807 nm within 1 min (Fig. 3). The ESI-mass spectrum of the solution shows a molecular ion peak at m/z = 702.3 with the isotope distribution patterns calculated for [(TpPh,Me)Fe(4-tBu-AP)]+ (Fig. 3, inset). The X-band EPR spectrum of the reaction solution at this point exhibits a rhombic signal at g = 4.3, typical of a high-spin iron(III) complex (Fig. S6, ESI†).29,30 In the presence of dioxygen, complex 1 is oxidized to its iron(III)-2-amidophenolate form (1Ox). In the next step of the reaction, the band at 618 nm shifts to 660 nm and the band at 807 nm shifts to lower energy to generate a broad band at 900 nm within 2 min (Fig. 3b). The time-dependent EPR spectrum suggests the formation of a ligand-based radical (g = 1.99) along with a rhombic signal at g = 4.3 (Fig. S6, ESI†).29,30 In the final step, the bands at 660 nm and 900 nm slowly decay over a period of 2 h to form a brown-green solution (Fig. 3b). The final EPR solution exhibits no radical signature, but the rhombic signal at g = 4.3 remains (Fig. S6, ESI†). The ESI-mass spectrum of the final oxidized solution shows an ion signal at m/z = 718.3 with the isotope distribution patterns calculated for [(TpPh,Me)Fe(4-tert-butyl-2-picolinate) + H]+ (Fig. 3b, inset) which shifts by two mass units in the presence of 18O2 (Fig. S9, ESI†).
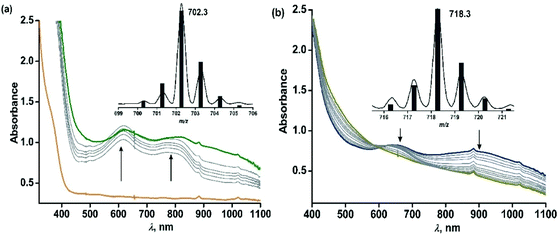 |
| | Fig. 3 Time-dependent optical spectral changes of 1 (0.5 mM in dichloromethane, 298 K) during the reaction with dioxygen (a) within 1 min. Inset: ESI-MS of the solution formed after 1 min (b) during the reaction with dioxygen after 1 min. Inset: ESI-MS of the final oxidized solution. | |
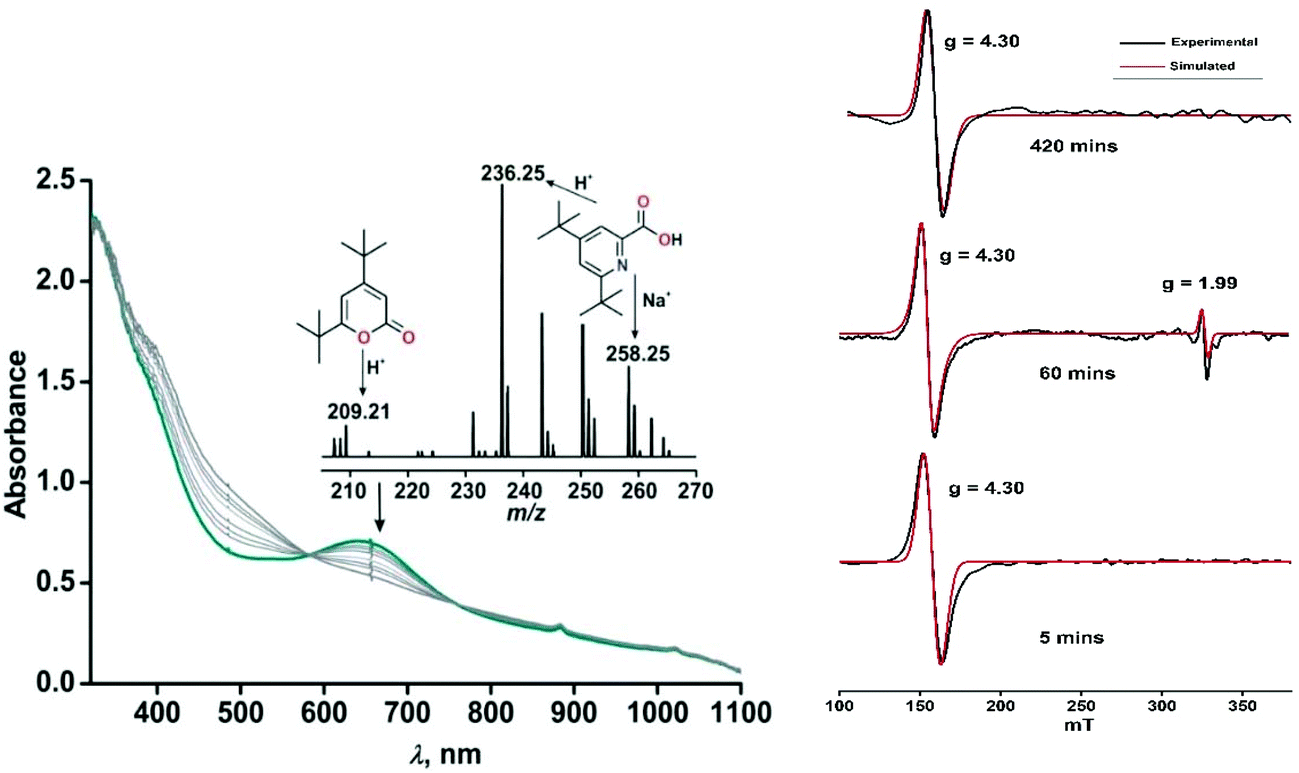
Exposure of a dichloromethane solution of [(TpPh,Me)FeII(4,6-ditBu-HAP)] 2 to dioxygen at room temperature immediately results in the formation of a greenish-blue solution. The colour change associated with the reaction of 2 with dioxygen can be monitored by optical spectral change, which indicated a multistep reaction. At first, the greenish blue solution exhibits a broad band at 650 nm typical of iron(III)-2-amidophenolate complexes (Fig. 4a).30,47 The X-band EPR spectrum of 2 at 77 K in a dichloromethane–toluene glass shows a rhombic signal at g = 4.3 typical of high-spin iron(III) complexes (Fig. 5, right).30 Thus, complex 2 directly converts to its iron(III)-2-amidophenolate (2Ox) form unlike the iron(II)-2-iminobenzosemiquinonate intermediate formed with a TpPh2 supporting ligand.44 However in a subsequent step, a new band appears near 898 nm which decays over a period of 2 h (Fig. 4b) and consequently a sharp signal at g = 1.99 is observed presumably due to the formation of a ligand-based radical species in solution (Fig. 5, right).30 Notably, the intensity of the EPR signal at g = 1.99 slowly diminishes as the reaction proceeds further. The final step is a slow process where the initial charge-transfer band at 650 nm slowly decays over a period of 5 h to form a greenish brown solution (Fig. 5, left). The final solution displays a rhombic EPR signal at g = 4.3 due to the formation of a high-spin iron(III) species (Fig. 5, right).
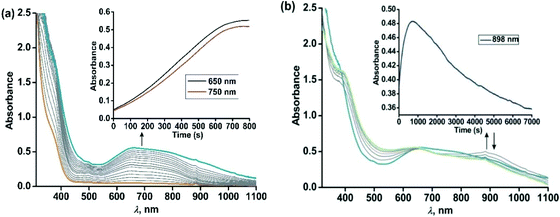 |
| | Fig. 4 Time-dependent optical spectral changes of 2 (0.5 mM in dichloromethane) during the reaction with dioxygen at room temperature. (a) Step 1 and (b) step 2. Inset: Plot of absorbance vs. time(s). | |
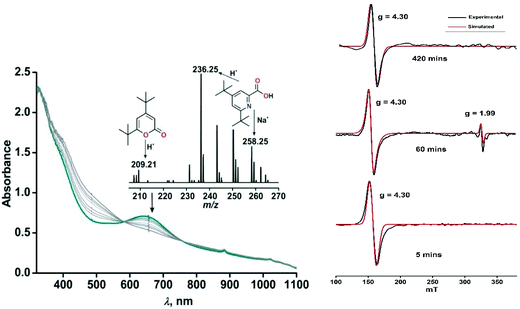 |
| | Fig. 5 (Left) Time dependent optical spectral changes of 2 (0.5 mM in dichloromethane) during the reaction with dioxygen at room temperature (step 3). Inset: ESI-MS of the final oxidized solution of 2 after reaction with dioxygen. (Right) X-band EPR spectral changes with time during the reaction of 2 with dioxygen in dichloromethane–toluene glass at 77 K. The red traces show the simulated EPR spectra. For detailed simulation parameters see Table S1.† | |
Interestingly, simulation of the EPR spectra of the time dependent dioxygen reactivity study for both complexes 1 and 2 revealed a significant change in the zero-field splitting (|D|) (see the ESI, Table S1†). For paramagnetic systems with S ≥ 1, a zero-field splitting (ZFS) can often be observed which arises from dipolar and spin–orbit couplings, causing differences among energy levels in the absence of an external magnetic field. There is still a limited understanding of how ZFS parameters relate to the geometric and electronic structures of transition metals compounds, including how metal–ligand bonding affects ZFS.56,57 However studies from Neese and coworkers have revealed a correlation between the increase of D with the spectrochemical series showing a decrease of the ligand field strength.57 Looking at our zero-field splitting values for the S = 5/2 spin system, from the simulated EPR spectra it may be qualitatively predicted that compared to the iron(III)-2-aminophenolate species (1Ox and 2Ox), the binding of dioxygen changes the coordination geometry and decreases the |D| value. In the final stage of the reaction, complexes show a significant increase in the |D| value, in line with the fact that compared to strongly bound 2-aminophenolate ligand, the C–C bond cleavage results in weakly coordinating iron(III)-2-picolinate complexes, thereby increasing the zero-field splitting.
Analyses of organic products from 1 and 2 and labelling experiments
The final oxidised products obtained from the reaction of 1 with dioxygen was analysed by 1H NMR spectrum after acidic work-up, maintaining the pH and extraction in diethyl ether and quantification using 1,4-benzoquinone as the internal standard, following the literature procedure.29 A quantitative transformation of 2-amino-4-tert-butylphenol to 4-tert-butyl-2-picolinic acid was observed in 2.5 h (Scheme 5 and Fig. S10, see the ESI† for details).29 No starting material, 2-amino-4-tert-butylphenol, was detected. The ESI-MS of the reaction solution after work up shows the peak at m/z = 180.21 which corresponds to the distribution pattern calculated for [4-tert-butyl-2-picolinic acid + H+] (Fig. S7, ESI†). The GC-mass spectrum of the organic product (2-picolinic acid was esterified with diazomethane, see ESI†) shows a distinct ion peak at m/z = 193 with the expected fragmentation patterns for methyl-4-tert-butyl-2-picolinate (Fig. S8a, ESI†). The ion peak at m/z at 193 is shifted to 195 when the reaction is performed with 18O2 supporting the origin of oxygen atoms from dioxygen (Fig. S8b, ESI†).
 |
| | Scheme 5 Aromatic ring cleavage products of substituted 2-aminophenolates during the reaction of 1 and 2 with dioxygen. | |
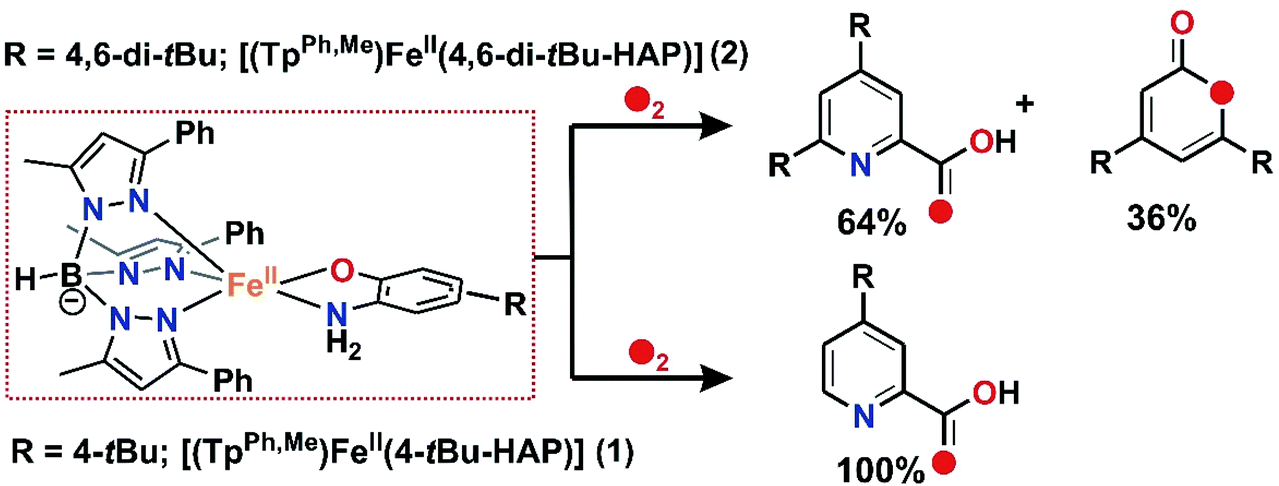
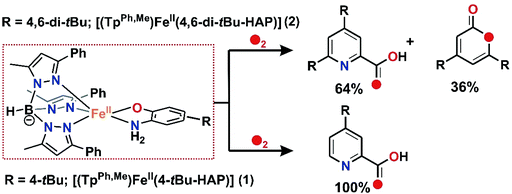
The oxidized products derived from 2 were similarly extracted into an organic solvent after acid treatment, following the literature procedure,30,31 and the organic products were analyzed by ESI-MS and GC-MS and quantified by 1H NMR spectroscopy using 1,4-benzoquinone as the internal standard (see the ESI†). The 1H NMR spectrum derived from the dioxygen reactivity of 2 reveals that 2-amino-4,6-di-tert-butylphenol quantitatively transforms to a mixture of C–C bond cleavage products, 4,6-di-tert-butyl-2H-pyran-2-one (36%) and 4,6-di-tert-butyl-2-picolinic acid (64%) (Scheme 5 and Fig. S13†).30,31 The ESI-MS shows distinct ion peaks at m/z = 209.21 which correspond to the distribution pattern calculated for [4,6-di-tert-butyl-2H-pyran-2-one + H]+ and at m/z = 236.35 and 258.25 for [4,6-di-tert-butyl-2-picolinate + H]+ and [4,6-di-tert-butyl-2-picolinate + Na]+, respectively (Fig. S11, ESI†). The GC-mass spectrum of organic products (2-picolinic acid was esterified with diazomethane) shows two distinct ion peaks at m/z = 208 and 249 with the expected fragmentation patterns of 4,6-di-tert-butyl-2H-pyran-2-one and methyl-4,6-di-tert-butyl-2-picolinate (Fig. S12a, ESI†). When the reaction is carried out with 18O2, the ion peak at m/z = 208 is shifted two mass units higher to m/z = 210 and the peak at m/z = 249 is shifted to m/z = 251 (Fig. S12b, ESI†). The labeling experiment confirms the incorporation of one 18O atom into each of the cleavage products.
Thus, both the substituted iron(II)-2-aminophenolate complexes (1 and 2) react with dioxygen to quantitatively form the C–C bond cleavage product of the corresponding 2-aminophenols. For iron(II)-2-amino-4-tert-butylphenolate complex (1), 4-tert-butyl-2-picolinic acid is observed as the sole C–C bond cleavage product after the reaction with molecular oxygen. On the other hand, the reaction of iron(II)-2-amino-4,6-di-tert-butylphenolate complex (2) with oxygen results in a mixture of 4,6-di-tert-butyl-2H-pyran-2-imine and 4,6-di-tert-butyl-2-picolinic acid. However, in both the cases, the products are found to be regioselective and result from the C1–C6 bond cleavage of the respective 2-aminophenols upon dioxygen activation (Scheme 5).
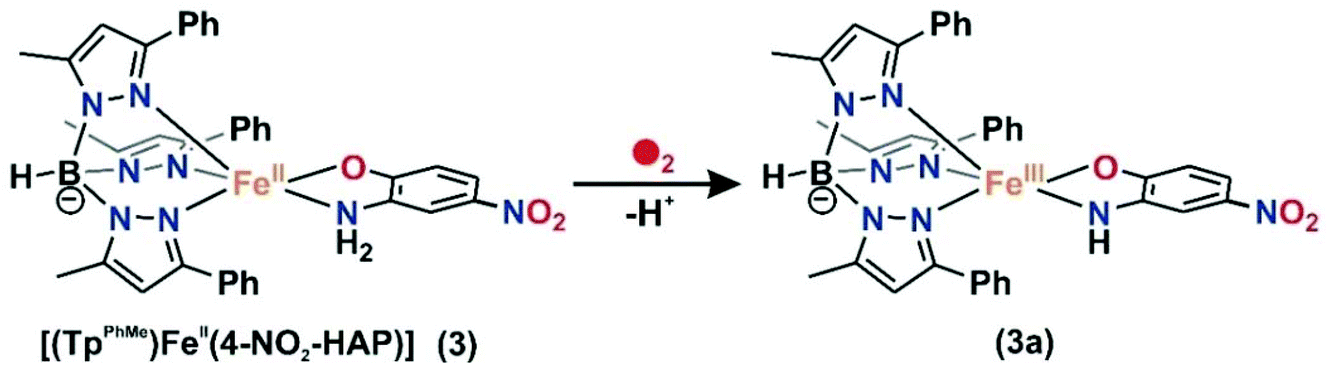
Dioxygen reactivity of 3
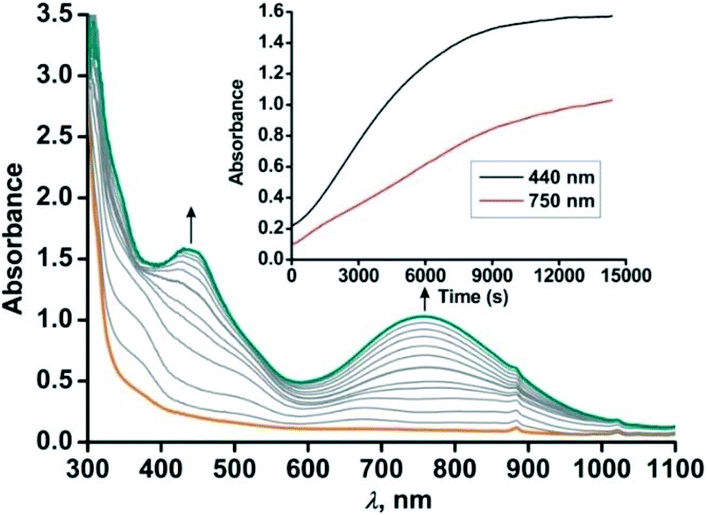
For the –NO2 substituted 2-aminophenol, complex [(TpPh,Me)FeII(4-NO2-HAP)] (3), the light yellow solution in dichloromethane rapidly transforms to a greenish-blue species (3a) upon exposure to dioxygen. During the reaction, two broad bands at 530 nm and 720 nm, characteristic of the LMCT transitions of iron(III)-2-amidophenolate species (Fig. 6), are generated over a period of 2 h. The X-band EPR spectrum of the final oxidized solution exhibits an intense rhombic signal at g = 4.3 (Fig. S14, ESI†). Similar to the iron(II)-catecholate complexes,38,55 iron(II)-2-aminophenolate complexes are converted to the corresponding iron(III)-2-amidophenolate species (Scheme 6). The intensity and position of the ligand-to-metal charge-transfer bands of 3a remain unchanged even after several hours under an oxygen atmosphere. Analyses of the organic product by 1H NMR after acidic workup of the bluish-green solution reveal that 4-NO2-aminophenol is quantitatively recovered without the formation of any C–C cleavage product.
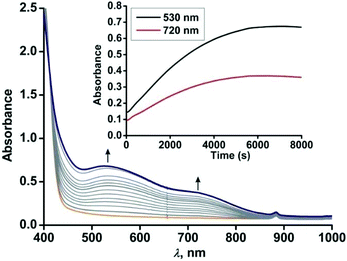 |
| | Fig. 6 Optical spectral changes with time during the reaction of 3 (0.5 mM solution in dichloromethane) with dioxygen at 298 K. Inset: Plot of absorbance vs. time. | |
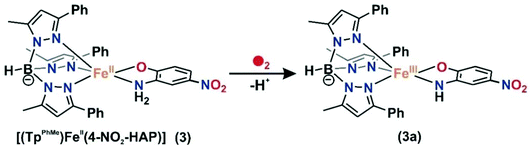 |
| | Scheme 6 Reaction of [(TpPh,Me)FeII(4-NO2-HAP)] (3) with dioxygen. | |
Role of –NH2 group in the C–C bond cleavage reactivity of iron(II)-2-aminophenolate complexes
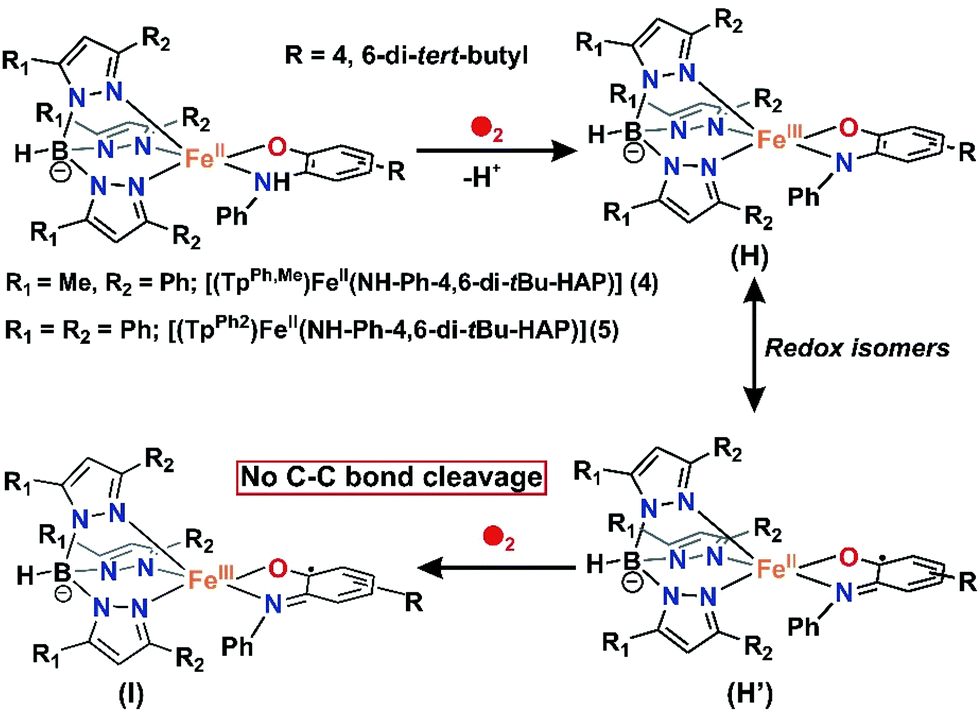
Both the iron(II)-2-aminophenolate complexes 4 and 5 with –NH–Ph groups instead of –NH2 are sensitive towards dioxygen in solution. The light yellow solution of [(TpPh,Me)FeII(NH-Ph-4,6-di-tBu-HAP)] (4) in dichloromethane rapidly transforms into greenish-blue species (4a) upon exposure to dioxygen. Initially, two broad peaks at 680 nm and 800 nm are generated rapidly whose feature and absorbance match with the reported iron(II)-2-iminobenzosemiquinonate species.44 However, the two broad peaks merge extremely fast and during the subsequent course of reaction of 4 with dioxygen, two prominent bands at 440 nm and 750 nm (Fig. 7) generate over a period of 4 h. The time dependent EPR spectrum reveals the formation of an intense rhombic signal at g = 4.3 for a high-spin iron(III) species after 5 min. However, with time, the intensity of the g = 4.3 signal decreases and the final oxidized solution after 4 h becomes EPR silent (Fig. S15, ESI†). The intensity and position of the optical transition of 4a remain unchanged even after several hours under an oxygen atmosphere. No oxidative C–C bond cleavage of 2-anilino-4,6-di-tert-butylphenol is observed upon the reaction of 4 with dioxygen. Thus, in the presence of dioxygen, complex 4 undergoes two electron oxidation on the redox non-innocent 2-aminophenol to form an EPR silent iron(III)-2-iminobenzosemiquinonate species (Scheme 7 and Fig. S15, ESI†).32,46–48 However, attempts to crystallize the final oxidized solution were unsuccessful.
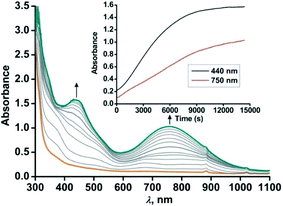 |
| | Fig. 7 Optical spectral changes of 4 (0.5 mM in dichloromethane) during the reaction with dioxygen at 298 K. Inset: Plot of absorbance vs. time. | |
 |
| | Scheme 7 Proposed mechanism for the reaction of 4 and 5 with dioxygen. | |
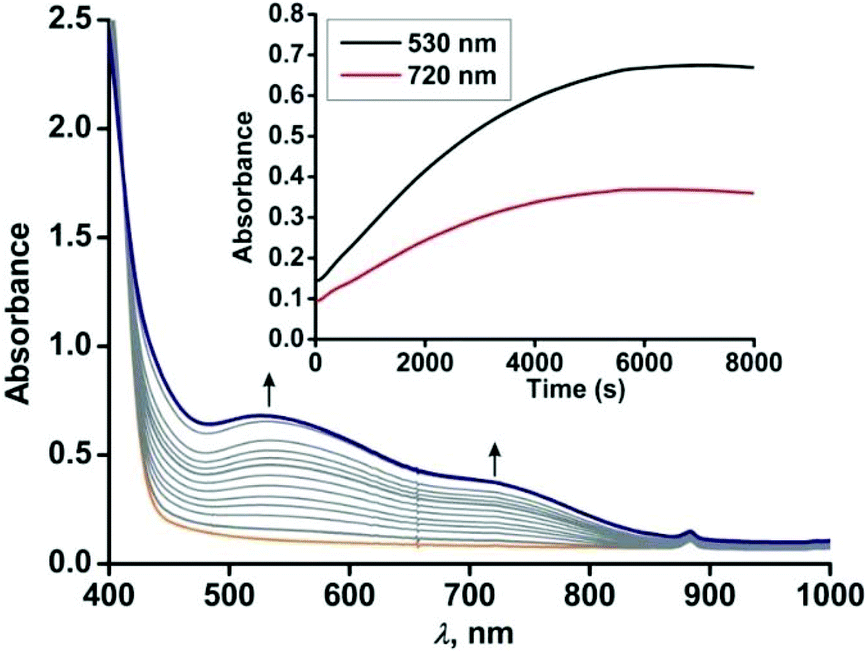
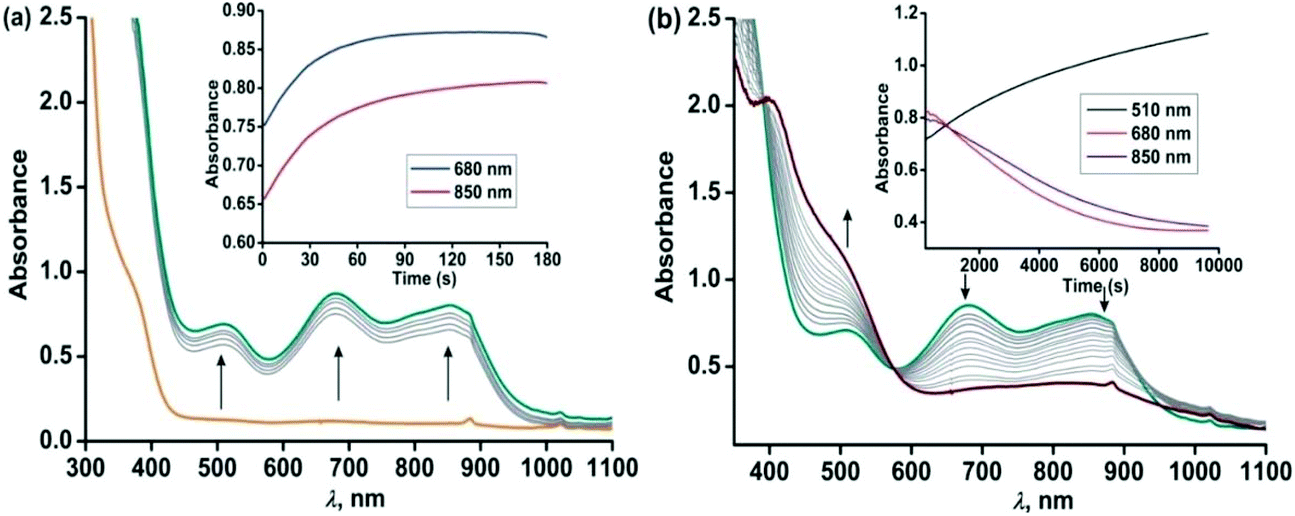
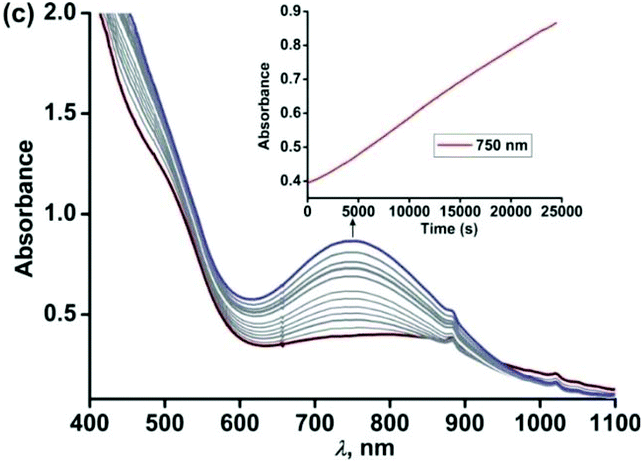
Similar to complex 4, complex [(TpPh2)FeII(NH-Ph-4,6-di-tBu-HAP)] (5) is reactive towards dioxygen. However, changing the ligand to TpPh2, from TpPh,Me, enables to monitor three distinct oxidation steps, as evident from the optical spectrum. Upon exposure to dioxygen, the yellow solution of 5 immediately turns green. Three charge transfer transitions at 510 nm, 680 nm and 850 nm arise within 3 min (Fig. 8). The X-band EPR spectrum of the oxidized solution at this point shows an intense rhombic signal at g = 4.3 (Fig. S16, ESI†). The spectral data suggest the formation of an iron(III)-2-amidophenolate species. During the next steps of the reaction under an oxygen atmosphere, the charge-transfer transitions at 680 nm and 850 nm decay over a period of 3 h to form a single broad band at around 800 nm (Fig. 8). The molar extinction coefficient and position of the optical transition after 3 h resemble that of the optical spectrum of the chemically one electron oxidized form of [TpPh2FeII(2-amino-4,6-di-tert-butylphenol)], i.e. an iron(II)-2-iminobenzosemiquinonate species reported by Fiedler et al.44
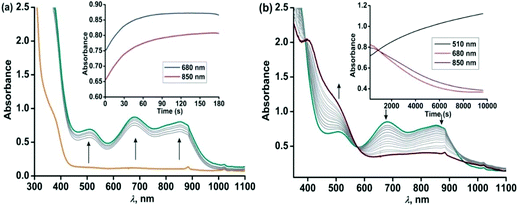 |
| | Fig. 8 Optical spectral changes of 5 (0.5 mM in dichloromethane) during the reaction with dioxygen at room temperature over a period of (a) 2 min (step 1) and (b) over a period of 3 h (step 2). Inset: Plot of absorbance vs. time. | |
However, the X-band EPR spectrum at 77 K reveals signals at g = 1.99 possibly due to the presence of a ligand-based radical species in solution along with a small peak at g = 4.3 (Fig. S16, ESI†).30 No peak corresponding to the S = 3/2 spin state was observed at 77 K. Attempts to isolate the species formed at this point, lead to the isolation of greenish blue crystals, which were of extremely poor quality for exact structure determination. Finally, the broad band at 800 nm undergoes a blue-shift to 750 nm and slowly increases in intensity over a period of 7 h (Fig. 9) and remains stable for several days. The final oxidized solution is EPR silent.32,46–48 Such a stable UV-vis peak at the higher wavelength region of around 750 nm with a molar absorption coefficient of approx. 4000 M−1 cm−1 is characteristic for the literature reported iron(III)-2-iminobenzosemiquinone species.44,46 Reports from Paine and coworkers as well as Fiedler and coworkers have revealed such iron(III)-iminobenzosemiquinone species to be EPR silent likely due to the coupling between iron(III) and iminobenzosemiquinone ligand based radicals, making it overall an integer spin EPR silent species.32,46–48 The iron(II)-2-anilino-4,6-di-tert-butylphenolate complex, thus undergoes two electron oxidation via its iron(II)-2-iminobenzosemiquinonate form. No oxidative C–C bond cleavage of 2-anilino-4,6-di-tertbutylphenol was observed in the reaction of 5 with dioxygen.
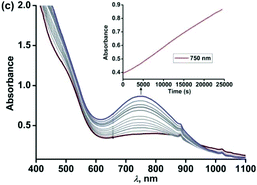 |
| | Fig. 9 Optical spectral changes of 5 (0.5 mM in dichloromethane) during the reaction with dioxygen at 298 K over a period of 7 h (step 3). Inset: Plot of absorbance vs. time. | |
Despite sufficient electron density on the aromatic ring for 2-anilino-4,6-di-tert-butylphenol in complexes 4 and 5, they undergo two electron oxidation of the redox non-innocent ligand,44,47 instead of showing C–C bond cleavage reactivity. This demonstrates the importance of the –NH2 group in substituted 2-aminophenols in displaying C–C bond cleavage reactions. The Fe(II)-2-iminobenzosemiquinonato species (H′) reacts with dioxygen and the lack of the –NH2 group directs the course of reactivity towards further oxidation on the metal center to form Fe(III)-2-iminobenzosemiquinonate complex (I)44,46 and no further reaction with dioxygen occurs (Scheme 7). However, Mukherjee and coworkers have shown an example of an iron complex of 2-aminophenol-appended supporting ligand where reaction with dioxygen resulted in a dimeric iron(III) μ2 oxo-bridged complex coordinated by a furan derivative in each metal unit, which was proposed to be formed as a result of the oxidative aromatic C–C bond cleavage product of 2-aminophenol.58
Discussion and proposed C–C bond cleavage mechanism of iron(II)-2-aminophenolate complexes
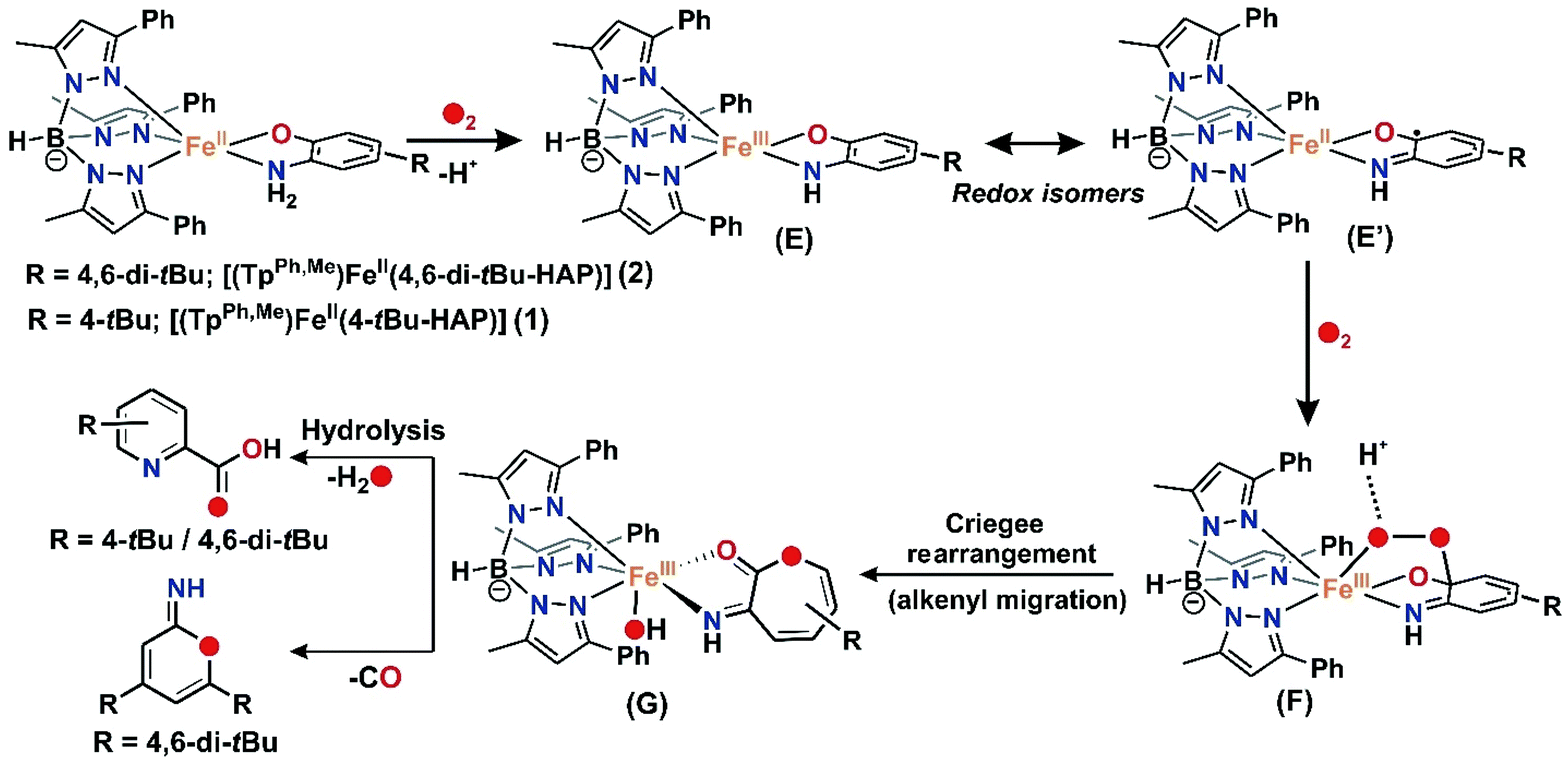
For iron(II)-catecholate model complexes, an outer sphere one electron oxidation of iron(II)-catecholate to iron(III)-catecholate species have been reported by Que and Moro-oka.6,38 The activation of dioxygen is initiated at the iron(II)-semiquinone intermediate (a redox isomer of iron(III)-catecholate) to form an iron(III)-alkylperoxo intermediate for the C–C bond cleavage of catechol. Paine and coworkers reported the first functional model of 2-aminophenol dioxygenases supported by a tetradentate tripodal ligand system that undergoes C–C bond cleavage via an iron(III)-2-amidophenolate species.29,30 The reactivity (and stability) of the intermediate, which directs the course of the aromatic ring cleavage reaction, was reported to be dependent on the nature of ring substituent on 2-aminophenols.30 Fiedler and co-workers have isolated and structurally characterized an iron(II)-2-aminophenolate complex,44–46 [(TpPh2)FeII(4,6-di-tert-butylaminophenol)], which was converted to an iron(II)-2-iminobenzosemiquinonato radical species upon one-electron chemical oxidation. On the basis of our experimental observations and literature precedents, it is proposed that complexes 1, 2 and 3 with TpPh,Me as the supporting ligand react with dioxygen to undergo one-electron oxidation to form an iron(III)-2-amidophenolate species in the first step (Schemes 6 and 8). The presence of an electron withdrawing nitro group at the 4-position of the 2-aminophenolate ring in 3 makes it unreactive towards dioxygen. Only electron transfer via an outer sphere mechanism operates and so complex 3 becomes stable in its iron(III)-2-amidophenolate (3a) form and does not react further with dioxygen (Scheme 6).30 However, the incorporation of electron donating groups on the aromatic ring of 2-aminophenol (tert-butyl) for complex 1 and 2 results in the oxygenolytic aromatic ring cleavage reaction. The proper steric geometry provided by the TpPh,Me ligand along with appropriate electron density on the aromatic ring of 2-aminophenols triggers the iron(II)-2-iminobenzosemiquinonato species (E), a redox isomer of iron(III)-2-amidophenolate species (E′) from 1 and 2 to react with dioxygen and generate an iron(III)-alkyl peroxo intermediate (F). The iron(III)-alkyl peroxo intermediate undergoes an alkenyl Criegee rearrangement, ultimately leading to the regioselective C–C bond cleavage of substituted 2-aminophenols (Scheme 8). However, the isolation and stabilization of putative species like iron(II)-2-iminobenzosemiquinonato under an oxygen environment (unlike chemical oxidation) is extremely challenging. The reported models (1 and 2) undergo regioselective C–C bond cleavage of substituted 2-aminophenols at a rate much faster than those reported for the tetradentate N4 system by Paine and coworkers.29,30 Whereas complex 1 takes only 2.5 h to undergo the C–C bond cleavage of 4-tBu-HAP upon reaction with dioxygen, the corresponding iron(II)-2-aminophenolate complex of the tetradentate ligand 6Me3TPA takes around 6 h for the reaction. Similarly, there is a three-fold increase in the rate of reaction for C–C bond cleavage of 4,6-di-tBu-HAP by complex 2 (7 h) compared to the iron(II)-4,6-di-tBu-HAP complex supported by the 6Me3TPA ligand (22 h). Though we were unable to isolate any reactive intermediate, it might perhaps not be hyperbolic to term the complexes (1 and 2) as first models simultaneously resembling the structural motif as well as emulating the functional reactivity of 2-aminophenol dioxygenases.
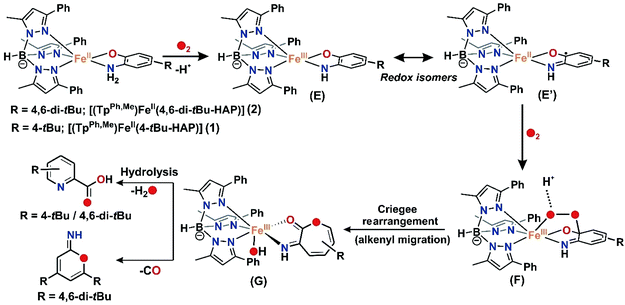 |
| | Scheme 8 Proposed mechanism for the C–C bond cleavage of 2-aminophenolates by biomimetic iron(II) complexes 1 and 2 using O2 as the oxidant. | |
So far from this study and previous literature reports (see Table S5† for a comparative analysis of dioxygen reactivity of reported 2-aminophenol model complexes), it is evident that unlike catechol cleaving dioxygenases, the development of the functional models of 2-aminophenol dioxygenase is more challenging. It is instructive from both the experimental and theoretical studies that differences in the denticity of the supporting ligand backbone, electronic factors of both the supporting ligands and the substituted 2-aminophenols as well as the proton shuttle in the reaction medium and probable second sphere residues exhibit a remarkable effect on the ring cleaving activity of model 2-aminophenol complexes.30–32,59 Whereas [(TpPh2)FeII4,6-di-tBu-HAP] or [(TIPPh2)FeII4,6-di-tBu-HAP] undergoes chemical one-electron oxidation or reaction with dioxygen and undergo the HAT reaction to form the corresponding FeII-2-iminosemibenzoquinone species, these complexes do not show the C–C bond cleavage reactivity of substituted 2-aminophenols, likely due to the steric and/or electronic factors of the supporting ligands.49 For the CoII-2-iminosemibenzoquinone species formed by HAT upon exposure to the dioxygen of TpR2CoII4,6-di-tBu-HAP (R2 = Me2 or R2 = Ph2), dioxygen adduct formation was hindered for the sterically bulky Ph2 substituted ligand compared to its Me2 congener, highlighting once again the importance of steric effect in modulating the energetics of oxygen binding. Thus, in this work, the success of the C–C bond cleavage of substituted 2-aminophenols was governed by both the steric effect and electronics of the supporting ligand TpPh,Me and also the electron density on the 2-aminophenolate ring (reactivity of 4-tBu-H2AP > 4,6-di-tBu-H2AP > 4-NO2-H2AP). Furthermore, for iron(II) complexes 4 and 5, despite having similar facial N3 coordination motif by the supporting ligands (TpPh2 and TpPh,Me) around the metal centre, the lack of a –NH2 group in coordinated 2-anilino-4,6-di-tert-butylphenolate directs the course of reactivity towards the two-electron oxidised Fe(III)-2-iminobenzosemiquinonate complex instead of undergoing C–C bond cleavage. Thus a synchronized way of both proton transfer (PT) and O2 binding along with the proper control of stereoelectronic factors are perhaps the keys for the oxygenolytic scission of C–C bond of substituted 2-aminophenols. The efficient management of these events is well executed in the enzymatic system in harmony with the active site and the conserved second sphere residues.
Conclusion
In conclusion, we have synthesized a series of iron(II)-2-aminophenolate complexes supported by tridentate monoanionic facial ligands which are all reactive towards dioxygen. The works presented herein show how the electronic and structural properties of 2-aminophenolate rings as well as the supporting ligand influence the C–C bond cleavage of 2-aminophenols and also highlight the role of –NH2 group in controlling the oxidative C–C bond cleavage reactivity. This work represents the first closest structural mimic of 2-aminophenol dioxygenases which exhibits excellent functional selectivity of the enzyme to regioselectively cleave the C1–C6 bond of substituted 2-aminophenols. Future directions would aim to explore detailed spectroscopic and theoretical calculations to evaluate the subtle changes in the geometric and electronic factors that modulate the dioxygen reactivity pathway from one or two electron oxidation events to regioselective C–C bond cleavage of 2-aminophenols in model complexes.
Experimental section
See ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the School of Chemical Sciences, Indian Association for the Cultivation of Science, India. R. D. J thanks the Council of Scientific and Industrial Research (CSIR), India for a research fellowship. Special thanks to Prof. Tapan Kanti Paine for sharing his laboratory space, chemicals and analytical equipment and also for the insightful suggestions and critical proofreading of the manuscript.
References
- F. H. Vaillancourt, J. T. Bolin and L. D. Eltis, Crit. Rev. Biochem. Mol. Biol., 2006, 41, 241–267 CrossRef CAS.
- G. Fuchs, M. Boll and J. Heider, Nat. Rev. Microbiol., 2011, 9, 803–816 CrossRef CAS.
- S. Fetzner, Appl. Environ. Microbiol., 2012, 78, 2505–2514 CrossRef CAS.
- T. D. H. Bugg and G. Lin, Chem. Commun., 2001, 941–952 RSC.
- S. Harayama, M. Kok and E. L. Neidle, Annu. Rev. Microbiol., 1992, 46, 565–601 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- T. D. H. Bugg, Tetrahedron, 2003, 59, 7075–7101 CrossRef CAS.
- U. Lendenmann and J. C. Spain, J. Bacteriol., 1996, 178, 6227–6232 CrossRef CAS.
- K. L. Colabroy, H. Zhai, T. Li, Y. Ge, Y. Zhang, A. Liu, S. E. Ealick, F. W. McLafferty and T. P. Begley, Biochemistry, 2005, 44, 7623–7631 CrossRef CAS.
- L. Que Jr., Biochem. Biophys. Res. Commun., 1978, 84, 123–129 CrossRef CAS.
- B. E. Haigler and J. C. Spain, Appl. Environ. Microbiol., 1991, 57, 3156–3162 CrossRef CAS.
- C. C. Somerville, S. F. Nishino and J. C. Spain, J. Bacteriol., 1995, 177, 3837–3842 CrossRef CAS.
- Z. He and J. C. Spain, J. Ind. Microbiol. Biotechnol., 2000, 25, 25–28 CrossRef CAS.
- I. Dilovic, F. Gliubich, G. Malpeli, G. Zanotti and D. Matkovic-Calogovic, Biopolymers, 2009, 91, 1189–1195 CrossRef CAS.
- X. Li, M. Guo, J. Fan, W. Tang, D. Wang, H. Ge, H. Rong, M. Teng, L. Niu, Q. Liu and Q. Hao, Protein Sci., 2006, 15, 761–773 CrossRef CAS.
- Y. Zhang, K. L. Colabroy, T. P. Begley and S. E. Ealick, Biochemistry, 2005, 44, 7632–7643 CrossRef CAS.
- D.-F. Li, J.-Y. Zhang, Y.-J. Hou, L. Liu, Y. Hu, S.-J. Liu, D.-C. Wang and W. Liu, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 32–43 CrossRef CAS.
- P. C. A. Bruijnincx, G. van Koten, K. Gebbink and J. M. Robertus, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC.
- K. L. Colabroy and T. P. Begley, J. Am. Chem. Soc., 2005, 127, 840–841 CrossRef CAS.
- P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermueller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213–2223 CrossRef CAS.
- A. I. Poddel'sky, V. K. Cherkasov and G. A. Abakumov, Coord. Chem. Rev., 2009, 253, 291–324 CrossRef.
- H. Chun, C. N. Verani, P. Chaudhuri, E. Bothe, E. Bill, T. Weyhermueller and K. Wieghardt, Inorg. Chem., 2001, 40, 4157–4166 CrossRef CAS.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- H. Chun, E. Bill, E. Bothe, T. Weyhermueller and K. Wieghardt, Inorg. Chem., 2002, 41, 5091–5099 CrossRef CAS.
- H. Chun, T. Weyhermuller, E. Bill and K. Wieghardt, Angew. Chem., Int. Ed., 2001, 40, 2489–2492 (
Angew. Chem.
, 2001
, 113
, 2552–2555
) CrossRef CAS.
- H. Chun, E. Bill, T. Weyhermueller and K. Wieghardt, Inorg. Chem., 2003, 42, 5612–5620 CrossRef CAS.
- K. S. Min, T. Weyhermueller and K. Wieghardt, Dalton Trans., 2003, 1126–1132 CAS.
- S. Mukherjee, T. Weyhermueller, E. Bill, K. Wieghardt and P. Chaudhuri, Inorg. Chem., 2005, 44, 7099–7108 CrossRef CAS.
- B. Chakraborty and T. K. Paine, Angew. Chem., Int. Ed., 2013, 52, 920–924 (
Angew. Chem.
, 2013
, 125
, 954–958
) CrossRef CAS.
- B. Chakraborty, S. Bhunya, A. Paul and T. K. Paine, Inorg. Chem., 2014, 53, 4899–4912 CrossRef CAS.
- S. Chatterjee and T. K. Paine, Inorg. Chem., 2015, 54, 1720–1727 CrossRef CAS.
- T. R. Lakshman, S. Chatterjee, B. Chakraborty and T. K. Paine, Dalton Trans., 2016, 45, 8835–8844 RSC.
- G. Dong, J. Lu and W. Lai, ACS Catal., 2016, 6, 3796–3803 CrossRef CAS.
- E. L. Hegg and L. Que Jr., Eur. J. Biochem., 1997, 250, 625–629 CrossRef CAS.
- N. Burzlaff, Angew. Chem., Int. Ed., 2009, 48, 5580–5582 (
Angew. Chem.
, 2009
, 121
, 5688–5690
) CrossRef CAS.
- E. H. Ha, R. Y. N. Ho, J. F. Kisiel and J. S. Valentine, Inorg. Chem., 1995, 34, 2265–2266 CrossRef CAS.
- S. Hikichi, T. Ogihara, K. Fujisawa, N. Kitajima, M. Akita and Y. Moro-oka, Inorg. Chem., 1997, 36, 4539–4547 CrossRef CAS.
- T. Ogihara, S. Hikichi, M. Akita and Y. Moro-oka, Inorg. Chem., 1998, 37, 2614–2615 CrossRef CAS.
- E. L. Hegg, R. Y. N. Ho and L. Que Jr., J. Am. Chem. Soc., 1999, 121, 1972–1973 CrossRef CAS.
- M. P. Mehn, K. Fujisawa, E. L. Hegg and L. Que Jr., J. Am. Chem. Soc., 2003, 125, 7828–7842 CrossRef CAS.
- T. K. Paine, H. Zheng and L. Que Jr., Inorg. Chem., 2005, 44, 474–476 CrossRef CAS.
- A. Mukherjee, M. A. Cranswick, M. Chakrabarti, T. K. Paine, K. Fujisawa, E. Munck and L. Que Jr., Inorg. Chem., 2010, 49, 3618–3628 CrossRef CAS.
- R. Rahaman, B. Chakraborty and T. K. Paine, Angew. Chem., Int. Ed., 2016, 55, 13838–13842 (
Angew. Chem.
, 2016
, 128
, 14042–14046
) CrossRef CAS.
- M. M. Bittner, S. V. Lindeman and A. T. Fiedler, J. Am. Chem. Soc., 2012, 134, 5460–5463 CrossRef CAS.
- M. M. Bittner, D. Kraus, S. V. Lindeman, C. V. Popescu and A. T. Fiedler, Chem. – Eur. J., 2013, 19, 9686–9698 CrossRef CAS.
- M. M. Bittner, S. V. Lindeman, C. V. Popescu and A. T. Fiedler, Inorg. Chem., 2014, 53, 4047–4061 CrossRef CAS.
- P. Halder, S. Paria and T. K. Paine, Chem. – Eur. J., 2012, 18, 11778–11787 CrossRef CAS.
- S. Banerjee, P. Halder and T. K. Paine, Z. Anorg. Allg. Chem., 2014, 640, 1168–1176 CrossRef CAS.
- P. Kumar, S. V. Lindeman and A. T. Fiedler, J. Am. Chem. Soc., 2019, 141, 10984–10987 CrossRef CAS.
- E. G. Kovaleva and J. D. Lipscomb, Science, 2007, 316, 453–457 CrossRef CAS.
- S. Chatterjee, S. Bhattacharya and T. K. Paine, Inorg. Chem., 2018, 57, 10160–10169 CrossRef CAS.
- S. Paria, L. Que and T. K. Paine, Angew. Chem., Int. Ed., 2011, 50, 11129–11132 (
Angew. Chem.
, 2011
, 123
, 11325–11328
) CrossRef CAS.
- D. Sheet, S. Bhattacharya and T. K. Paine, Chem. Commun., 2015, 51, 7681–7684 RSC.
- M. Sallmann, I. Siewert, L. Fohlmeister, C. Limberg and C. Knispel, Angew. Chem., Int. Ed., 2012, 51, 2234–2237 (
Angew. Chem.
, 2012
, 124
, 2277–2280
) CrossRef CAS.
- S. Paria, P. Halder, B. Chakraborty and T. K. Paine, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2011, 50, 420–426 Search PubMed.
- J. Nehrkorn, S. A. Bonke, A. Aliabadi, M. Schwalbe and A. Schnegg, Inorg. Chem., 2019, 58, 14228–14237 CrossRef CAS.
- S. E. Stavretis, M. Atanasov, A. A. Podlesnyak, S. C. Hunter, F. Neese and Z.-L. Xue, Inorg. Chem., 2015, 54, 9790–9801 CrossRef CAS.
- G. C. Paul, S. Banerjee and C. Mukherjee, Inorg. Chem., 2017, 56, 729–736 CrossRef CAS.
- G. J. Christian, S. Ye and F. Neese, Chem. Sci., 2012, 3, 1600–1611 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2012465–2012467. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03316b |
| ‡ Present address: Max-Planck-Institute for Chemical Energy Conversion, Stiftstr, 45470 Mülheim an der Ruhr, Germany. |
| § Present address: Department of Chemistry, Indian Institute of Technology, Hauz Khas, New Delhi 110016, India. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Sridhar
Banerjee
a,
Rahul Dev
Jana
a,
Shrabanti
Bhattacharya
a,
Biswarup
Chakraborty§
a and
Sergio Augusto Venturinelli
Jannuzzi
*a,
Sridhar
Banerjee
a,
Rahul Dev
Jana
a,
Shrabanti
Bhattacharya
a,
Biswarup
Chakraborty§
a and
Sergio Augusto Venturinelli
Jannuzzi