
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multivalency in CXCR4 chemokine receptor targeted iron oxide nanoparticles†
Neazar E.
Baghdadi
 ab,
Benjamin P.
Burke
c,
Tahani
Alresheedi
bcg,
Shubhanchi
Nigam
bc,
Abdu
Saeed
d,
Farooq
Almutairi
bf,
Juozas
Domarkas
bc,
Abid
Khan
ce and
Stephen J.
Archibald
*bc
ab,
Benjamin P.
Burke
c,
Tahani
Alresheedi
bcg,
Shubhanchi
Nigam
bc,
Abdu
Saeed
d,
Farooq
Almutairi
bf,
Juozas
Domarkas
bc,
Abid
Khan
ce and
Stephen J.
Archibald
*bc
aCentre of Nanotechnology, King Abdul-Aziz University, Jeddah, Saudi Arabia
bDepartment of Chemistry, University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: s.j.archibald@hull.ac.uk
cDepartment of Biomedical Sciences and PET Research Centre, University of Hull Cottingham Road, Hull, HU6 7RX, UK
dDepartment of Physics, Faculty of Science, King Abdul-Aziz University, Jeddah 21589, Saudi Arabia
eDivision of Pharmacy and Optometry, School of Health Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, UK
fCollege of Applied Medical Sciences, University of Hafar Al-Batin, Hafar Al-Batin, Saudi Arabia
gDepartment of Chemistry, College of Science and Art, Qassim University, Qassim, Saudi Arabia
First published on 19th January 2021
Abstract
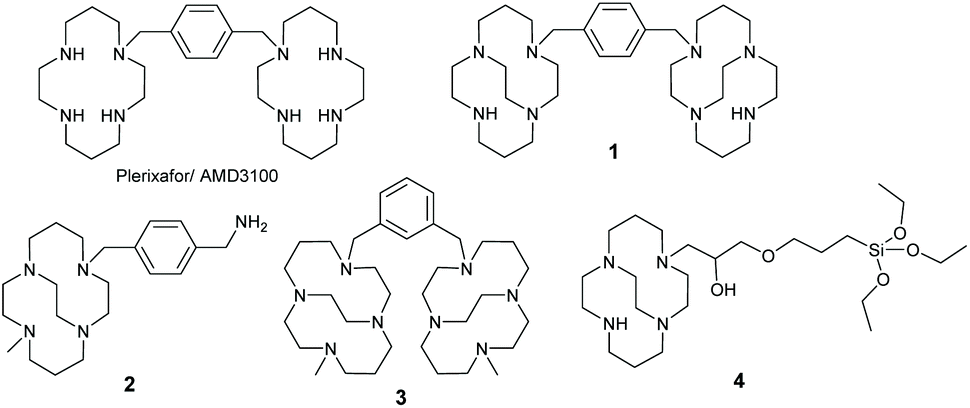
The CXCR4 chemokine receptor is an important biomolecular target in cancer diagnostics and therapeutics. In a new multivalent approach, iron oxide nanoparticles were conjugated with multiple binding units of a low affinity azamacrocylic CXCR4 antagonist. The silica coated nanostructure has good suspension stability, a mode size of 72 nm and high affinity for CXCR4, showing >98% inhibition of anti-CXCR4 mAb binding in a receptor binding competition assay on Jurkat cells.
Targeting of cell surface receptors on cancer cells with nanomaterials has offered new diagnostic and therapeutic prospects for cancer imaging and treatment by optimising tumour uptake efficiency and reducing toxicity.1,2 One of the key challenges in nanotechnology is achieving high tumour accumulation with a low background to minimise off target signal or effects. A potential mechanism to optimise this parameter is to apply a tuneable affinity for tumour overexpressed receptors and, ideally, combine it into a system where surface modification can be used to influence blood pool retention.3,4 The CXCR4 chemokine receptor is a highly relevant biological target in the development of new diagnostics and therapies for cancers.5–9 Nanoparticles targeting CXCR4 will allow the use of multimodal imaging to inform disease prognosis and improve treatment options. The use of superparamagnetic iron oxide nanoparticles allows T2 weighted MRI contrast and this can be combined with radiolabelling of the surface with radioisotopes for PET or SPECT imaging in the next phase of the research work.
 | ||
| Fig. 1 A low affinity mono-macrocycle binding unit can be used to generate high affinity CXCR4 chemokine receptor (represented as the green GPCR in the cell membrane) binding nanoparticles through multivalent receptor binding interactions. The approach could be used to optimise affinity for high CXCR4 expressing tissues and cells. | ||
The potential for targeting NPs to CXCR4 in drug delivery and imaging has been investigated previously. Various nanostructures have been created and modified for targeting CXCR4 receptors over a wide range of different cancer types for multifunctional applications, including diagnosis, chemotherapy, and gene expression control. Peptide targeting has been the main mechanism of CXCR4 recognition in the ca. 20 published CXCR4 targeting NP constructs. Unzueta et al. have described a targeting construct for CXCR4 chemokine receptors using green fluorescence protein attached to peptide T22 antagonists.18 De la Torre et al. also utilised T22 with mesoporous silica NPs loaded with doxorubicin and tested for drug release in lymphoma cells.19 Chittasupho et al. using cellulose NPs coated with poly lactic-co-glycolic acid (PLGA) for drug delivery, targeting CXCR4 with the LFC131 peptide.20 Di-Wen et al. also used LFC131 for liver uptake with PLGA/micellar NPs.21 Only three research groups have reported results on CXCR4 targeted iron oxide NPs with two groups attaching anti-CXCR4 antibodies to the NP surface.22–24 Long, Aboagye and co-workers covalently attached a CXCR4 binding cyclic peptide to the surface of the iron oxide NPs for enhanced magnetic resonance imaging. However, there are no reported studies of iron oxide NPs with azamacrocycle CXCR4 antagonists on the surface,23 and only one example of a metal NP (gold), where AMD3100 is attached to the surface.25 The multivalent approach to engineer high affinity CXCR4 binding nanoconstructs from multiple low affinity antagonists conjugated to the surface has not been investigated. Although multivalency may have occurred with other constructs they were not produced from low affinity antagonists to allow more exquisite and precise tuning of the affinity.
Our focus, in designing an attachment process to the surface of NPs, was to develop a simple protocol with relatively few steps to allow future combination with the chelator free radiolabelling approach that we have previously developed for gallium-68 with mesoporous silica coated NPs.4 In order to achieve this, a novel siloxy precursor was needed for formation of both a silica shell and incorporation of the antagonist on to the surface of the NP. Barreto et al. investigated a process for attaching azamacrocycles to the surface of iron oxide NPs by reacting them with 3-(3-(triethoxysiloxy)propoxy(propan-2-ol). In their study, the aim was to use the macrocycles including cyclam and cyclen for radiolabelling with copper-64 (although this process is unlikely to give sufficiently stable chelator labelled complexes for in vivo use).26 Modification of the procedure to use cross-bridged macrocycles for receptor targeting (rather than for radiolabelling) was investigated in our work to develop high affinity CXCR4 binding NPs.
Some of the previously reported approaches to produce nanostructures have low coating efficiency and produce polydisperse particle sizes. For example, the traditional approach of coating isolated “bare” NPs suffers from lower coating efficiency which can cause aggregation of the NPs under physiological conditions. Therefore, more complex multi-step protocols have been developed that can reduce yields or adversely affect the final physical properties. Our approach is direct and simple, coating the nanostructure surface with one or multiple siloxane components in a one-step reaction by exchanging the oleic acid on the surface.4,27 The first step was to produce iron oxide NPs, which was carried out using a co-precipitation method adapted from the work of Larsen et al.28 The preparation relies on elevated temperature and the presence of oleic acid to produce NPs of specific size via temperature control. Nucleation and growth principles were applied, where the iron oxide nuclei were created and then the rate of the growth is dependent on the temperature. In this instance, the core size of the produced NPs is less than 20 nm with a mono-disperse size distribution, see Fig. S2.2.† The prepared NPs were suspended in organic solvent (toluene), compatible with the oleic acid present on the surface, and stored for future use. These particles can then be used for the silica coating and attachment of the cross-bridged macrocycle complex on the NPs surface.
A new siloxane cross-bridged cyclam derivative 4 was designed, due to the optimised binding properties of the CB cyclam component,29,30 and prepared to allow coating of the NPs with silica and functionalisation with the potential CXCR4 antagonist coating. The aim was to provide a coating, via the ligand exchange reaction, that also rendered the NPs water-soluble due to the hydrophilicity of the silica and the metal complexes. 4 was synthesised, see Scheme 1 and S2.1,† from CB-cyclam (prepared according to the methods of Weisman and co-workers). A commercially available oxirane derivative was reacted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio resulting in a single product. The product was analysed and its identity confirmed using mass spectrometry and NMR spectroscopy.
:1 ratio resulting in a single product. The product was analysed and its identity confirmed using mass spectrometry and NMR spectroscopy.
 | ||
| Scheme 1 Formation of the siloxy macrocycle derivative and the NPs coated with siloxane/CXCR4 binding unit to give a silica shell on the NP surface (see S2† for experimental details). | ||
The coating of the NP and the macrocycle attachment was achieved in a single step reaction, see Scheme 1, forming a silica layer with the CXCR4 binding molecule displayed on the NPs surface. A biphasic system solvent system was used in the reaction with toluene as the organic solvent and a basic aqueous layer with the addition of triethylamine. The oleic acid was displaced to allow formation of the polymeric silica shell on the surface with strong coordinate bonds to the NP surface. A range of techniques were used to characterise the coating, including ICP-OES, NTA, and CHN analysis.
The TEM analysis of the NPs prior to silica coating showed mono-disperse NPs with slightly irregular roughly spherical shapes and uniform sizes of less than 20 nm, see Fig. S2.2.† After the coating process, with the siloxy ether functionalised macrocyclic complex, see Fig. 2, the TEM shows some degree of aggregation, likely due to the polymer coating linking some of the iron oxide cores together, forming slightly larger particles, which are still mostly monodisperse. The NTA analysis shows one main particle population and demonstrates size distribution has been shifted by around 40 nm with a mean diameter of 92 nm and mode of 72 nm, consistent with our previous studies.
 | ||
| Fig. 2 Nanoparticle tracking analysis of the prepared coated NPs with the silica CB-cyclam-Ni coating suspended in deionised water; the TEM image is inset. | ||
The elemental analysis of the macrocycle coated NPs (CHN combustion analysis and ICP-EOS) shows that the nanostructure consists mostly of the iron oxide core (Fe 44.8%) with a silica shell present on the surface of the NPs. Nickel(II) was selected as the metal ion to complex with the CB-cyclam on the particle surface due to the flexibility in aryl ring positioning of bi-cyclam derivatives to retain affinity. Our previous work has shown that the affinity of the para-bis-CBcyclam nickel(II) complex 2 is retained in the meta-bis-CBcyclam nickel(II) complex 3 indicating that this metal ion may have appropriate coordination properties for multivalent interactions with the CXCR4 receptor on the cell surface.14,31 The presence of carbon, hydrogen, and nitrogen indicate attachment of the macrocycles on the NPs surface. The masses of N and Ni were scaled to indicate a ratio of macrocycle (4 N, assuming no other sources of nitrogen) to nickel(II), showing that there is incomplete occupancy of the macrocyclic cavities with a ratio of ca. 0.2 nickel(II) ions per macrocycle.
The nanostructure surface charge was measured using zeta potential at pH 7.4 for physiological relevance. The nickel(II) macrocycle functionalised particles have a zeta potential of −5.5 mV indicating that they will repel one another, and that stability to aggregation and precipitation is likely at this pH. Qualitative observational studies were applied to monitor the stability of aqueous suspensions over a range of ionic strengths and pHs, in order to determine the coated NPs stability under a range of physiological conditions. At a fixed ion concentration of 7.5 mM, pH stability was tested in the range pH 1 to 12, see Fig. S2.3,† with good stability observed across the whole range after 6 hours. In a parallel study, ionic strength was varied, see Fig. S4.2.† A small amount of precipitate occurred on addition of salt solutions in some cases; however, this was easily removed by filtration and the remaining suspension was stable for several days. The NPs showed high stability at varying solution ionic strengths from 0.1 to around 200 mOsm L−1.
Fig. S2.1† shows the infra-red spectra of the different NPs (oleic acid NPs and Si-CB-cyclam-Ni NPs). The coating layer has peaks in the expected region, between 600 and 1000 cm−1, for Fe–O, Si–O–C and Si–O–Fe bond stretches. Peaks showing the presence of – OH and oleic acid (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CH2 and CH3) vibration frequencies were observed for the NPs before silica coating. In the silica-CB-cyclam-Ni coated NPs, absorption peaks were observed at 1488, 1459, and 1404 cm−1 assigned to the CB cyclam and 1117 cm−1 to the silica polymer coating.
C, CH2 and CH3) vibration frequencies were observed for the NPs before silica coating. In the silica-CB-cyclam-Ni coated NPs, absorption peaks were observed at 1488, 1459, and 1404 cm−1 assigned to the CB cyclam and 1117 cm−1 to the silica polymer coating.
To measure the binding efficiency of NPs-Si-CB-cyclam-Ni to the CXCR4 chemokine receptors displayed on the surface of cancer cells, a competition binding assay was carried out using an anti-CXCR4 monoclonal antibody known to bind to epitopes blocked by azamacrocyclic antagonists, see Fig. 3.32 Jurkat human T-lymphoblastic cancer cells were used as they overexpress CXCR4 chemokine receptors. In a flow cytometry experiment, the cell surface bound anti-CXCR4 specific mAb tagged with the PE fluorescent dye was used for competition binding with the NPs. Two concentrations of the NPs were tested, see S2.5.† A meaningful IC50 value cannot be determined as the antagonist surface concentration and nature of the multivalent interaction is unknown, however, a 10-fold dilution maintained effective antibody competition. The solid purple peak on the left represents the negative control (isotype), in which no fluorescent tagged anti-CXCR4 antibody is added and the green peak on the right indicates the addition of the dye tagged antibody with no blocking agent (i.e. the maximum signal). At both concentrations tested, the peaks almost completely overlap with the negative control showing that the NPs are bound to CXCR4 with high affinity, and that the anti-CXCR4 mAb is unable to displace them (mean fluorescence intensity calculation indicates ca. 100% of receptors are occupied by the NPs, see S2.5†).
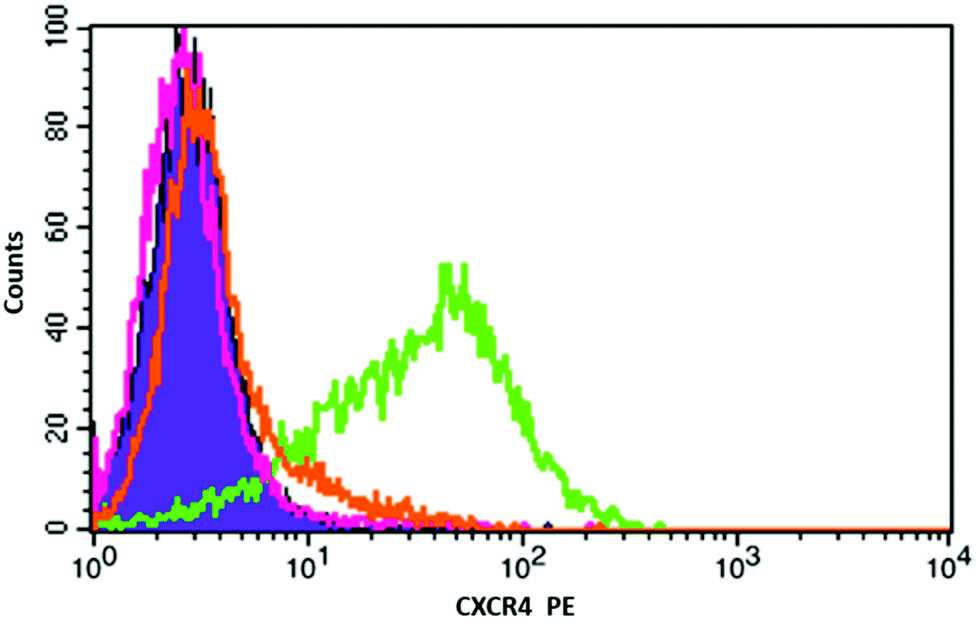 | ||
| Fig. 3 Flow cytometry histogram of the binding of anti-CXCR4-PE (12G5) on the cell surface of Jurkat cells. Negative (solid purple), anti-CXCR4-PE mAb only (green), and competition with NPs (pink & orange). | ||
To put this result in context, comparison binding studies were carried out with a nickel(II) complex of a mono-macrocyclic compound ([Ni2]2+) which was selected as an appropriate control and synthesised, see S2.4.†14 The nickel(II) complex of 4 cannot be isolated as a pure compound due to polymer formation. Nanoparticles that were not coated with an azamacrocycle were used as control for NP binding showing no affinity for the target (and showed issues with suspension stability in the cell culture medium). Mono-macrocycle compound [Ni2]2+ exhibits 12G5 antibody inhibition of ca. 38% (with an error of ±3.0%) at a saturating concentration. This clearly demonstrates a multivalent effect is present for the NPs when compared with ca. 100% inhibition exhibited by the nickel(II) macrocycle coated nanoparticles.
Conclusions
This work presents a direct and simple route to generate a stable suspension of NPs that are suitable for biomedical applications targeting CXCR4. The NPs are reacted with a siloxy derivative to give a CB-cyclam-Ni coated silica surface that forms stable suspensions of NPs, which can be used in vitro and potentially in vivo. The key feature is the demonstration (for the first time) of a metal complex CXCR4 receptor recognition via a multivalent approach indicating the potential for tuning and optimisation of receptor recognition. The opportunity to control the affinity is important, and could also be combined with a varying amount of PEG coating on the surface and also be extended to coat various NP shapes and compositions, as we have demonstrated in our previous research. The next stage of this work is to select a range of NPs with varied affinity coatings, radiolabel the silica surface (chelator free) with radiometals33 and perform in vivo studies to determine uptake and retention in CXCR4 expressing tumours. This will allow us to exploit the large library of varied affinity bridged azamacrocycle antagonists developed in the Archibald group.The simplest way to radiolabel a NP is via the direct interaction of the radioisotope with the surface of a preformed nanoconstruct.4,27 This means direct labelling can be carried out without significant alteration of surface properties, making it attractive for use with NPs that are optimised for targeting. The use of an iron oxide core also allows multimodal imaging using T2 weighted magnetic resonance imaging and will be investigated in future studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Saudi Cultural Attaché with studentships to NB and TA. We gratefully acknowledge the Daisy Appeal Charity for funding (Grant: DAhul0211) and fellowship funding for BPB, and the University of Hull (UoH) for PET infrastructure support. We thank Dr Assem Allam and his family for their generous donation to help found the PET Research Centre at the UoH. Fig. 1 was created with BioRender.com.Notes and references
- B. P. Burke, C. Cawthorne and S. J. Archibald, in Clinical Applications of Magnetic Nanoparticles: From Fabrication to Clinical Applications, ed. N. T. K. Thanh, CRC Press, 2018, ch. 23, pp. 409–428 Search PubMed.
- N. Bertrand, J. Wu, X. Y. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS.
- R. Hennig, K. Pollinger, A. Veser, M. Breunig and A. Goepferich, J. Controlled Release, 2014, 194, 20–27 CrossRef CAS.
- B. P. Burke, N. Baghdadi, A. E. Kownacka, S. Nigam, G. S. Clemente, M. M. Al-Yassiry, J. Domarkas, M. Lorch, M. Pickles, P. Gibbs, R. Tripier, C. Cawthorne and S. J. Archibald, Nanoscale, 2015, 7, 14889–14896 RSC.
- M. E. Mayerhoefer, S. J. Archibald, C. Messiou, A. Staudenherz, D. Berzaczy and H. Schöder, J. Magn. Reson. Imaging, 2020, 51, 1325–1335 CrossRef.
- S. Poty, P. Désogère, C. Goze, F. Boschetti, T. D'Huys, D. Schols, C. Cawthorne, S. J. Archibald, H. R. Maëcke and F. Denat, Dalton Trans., 2015, 44, 5004–5016 RSC.
- B. P. Burke, C. S. Miranda, R. E. Lee, I. Renard, S. Nigam, G. S. Clemente, T. D'Huys, T. Ruest, J. Domarkas, J. A. Thompson, T. J. Hubin, D. Schols, C. J. Cawthorne and S. J. Archibald, J. Nucl. Med., 2020, 61, 123–128 CrossRef CAS.
- M. Kircher, P. Herhaus, M. Schottelius, A. K. Buck, R. A. Werner, H. J. Wester, U. Keller and C. Lapa, Ann. Nucl. Med., 2018, 32, 503–511 CrossRef CAS.
- T. Liu, X. B. Li, S. You, S. S. Bhuyan and L. Dong, Exp. Hematol. Oncol., 2016, 5, 19 CrossRef.
- I. Renard and S. J. Archibald, Adv. Inorg. Chem., 2020, 75, 447–476 CrossRef.
- G. C. Valks, G. McRobbie, E. A. Lewis, T. J. Hubin, T. M. Hunter, P. J. Sadler, C. Pannecouque, E. De Clercq and S. J. Archibald, J. Med. Chem., 2006, 49, 6162–6165 CrossRef CAS.
- A. Khan, J. Greenman and S. J. Archibald, Curr. Med. Chem., 2007, 14, 2257–2277 CrossRef CAS.
- A. Khan, J. D. Silversides, L. Madden, J. Greenman and S. J. Archibald, Chem. Commun., 2007, 416–418 RSC.
- R. Smith, D. Huskens, D. Daelemans, R. E. Mewis, C. D. Garcia, A. N. Cain, T. N. C. Freeman, C. Pannecouque, E. D. Clercq, D. Schols, T. J. Hubin and S. J. Archibald, Dalton Trans., 2012, 41, 11369–11377 RSC.
- G. McRobbie, G. C. Valks, C. J. Empson, A. Khan, J. D. Silversides, C. Pannecouque, E. De Clercq, S. G. Fiddy, A. J. Bridgeman, N. A. Young and S. J. Archibald, Dalton Trans., 2007, 36, 5008–5018 RSC.
- X. Montet, M. Funovics, K. Montet-Abou, R. Weissleder and L. Josephson, J. Med. Chem., 2006, 49, 6087–6093 CrossRef CAS.
- K. Abstiens, M. Gregoritza and A. M. Goepferich, ACS Appl. Mater. Interfaces, 2019, 11, 1311–1320 CrossRef CAS.
- U. Unzueta, M. V. Cespedes, N. Ferrer-Miralles, I. Casanova, J. Cedano, J. L. Corchero, J. Domingo-Espin, A. Villaverde, R. Mangues and E. Vazquez, Int. J. Nanomed., 2012, 7, 4533–4544 CAS.
- C. de la Torre, I. Casanova, G. Acosta, C. Coll, M. J. Moreno, F. Albericio, E. Aznar, R. Mangues, M. Royo, F. Sancenon and R. Martinez-Manez, Adv. Funct. Mater., 2015, 25, 687–695 CrossRef CAS.
- C. Chittasupho, P. Kewsuwan and T. Murakami, Curr. Drug Delivery, 2017, 14, 1060–1070 CAS.
- S. Di-Wen, G. Z. Pan, L. Hao, J. Zhang, Q. Z. Xue, P. Wang and Q. Z. Yuan, Int. J. Pharm., 2016, 500, 54–61 CrossRef.
- Y. L. He, W. Song, J. Lei, Z. Li, J. Cao, S. Huang, J. Meng, H. Y. Xu, Z. Y. Jin and H. D. Xue, Acta Radiol., 2012, 53, 1049–1058 CrossRef.
- J. Gallo, N. Kamaly, I. Lavdas, E. Stevens, Q. D. Nguyen, M. Wylezinska-Arridge, E. O. Aboagye and N. J. Long, Angew. Chem., Int. Ed., 2014, 53, 9550–9554 CrossRef CAS.
- V. Vilas-Boas, B. Espina, Y. V. Kolen’ko, M. Banobre-Lopez, J. A. Duarte, V. C. Martins, D. Y. Petrovykh, P. P. Freitas and F. D. Carvalho, Biointerphases, 2018, 13, 011005 CrossRef.
- Y. F. Zhao, L. Detering, D. Sultan, M. L. Cooper, M. You, S. H. Cho, S. L. Meier, H. Luehmann, G. R. Sun, M. Rettig, F. Dehdashti, K. L. Wooley, J. F. DiPersio and Y. J. Liu, ACS Nano, 2016, 10, 5959–5970 CrossRef CAS.
- J. A. Barreto, M. Matterna, B. Graham, H. Stephan and L. Spiccia, New J. Chem., 2011, 35, 2705–2712 RSC.
- B. P. Burke, N. Baghdadi, G. S. Clemente, N. Camus, A. Guillou, A. E. Kownacka, J. Domarkas, Z. Halime, R. Tripier and S. J. Archibald, Faraday Discuss., 2014, 175, 59–71 CAS.
- B. A. Larsen, M. A. Haag, N. J. Serkova, K. R. Shroyer and C. R. Stoldt, Nanotechnology, 2008, 19, 265012 CrossRef.
- R. D. Maples, A. N. Cain, B. P. Burke, J. D. Silversides, R. E. Mewis, T. D'Huys, D. Schols, D. P. Linder, S. J. Archibald and T. J. Hubin, Chem. – Eur. J., 2016, 22, 12916–12930 CrossRef CAS.
- A. Khan, G. Nicholson, J. Greenman, L. Madden, G. McRobbie, C. Pannecouque, E. De Clercq, R. Ullom, D. L. Maples, R. D. Maples, J. D. Silversides, T. J. Hubin and S. J. Archibald, J. Am. Chem. Soc., 2009, 131, 3416–3417 CrossRef CAS.
- A. N. Cain, T. N. Carder Freeman, K. D. Roewe, D. L. Cockriel, T. R. Hasley, R. D. Maples, E. M. A. Allbritton, T. D'Huys, T. Van Loy, B. P. Burke, T. J. Prior, D. Schols, S. J. Archibald and T. J. Hubin, Dalton Trans., 2019, 48, 2785–2801 RSC.
- A. van Hout, T. D’Huys, M. Oeyen, D. Schols and T. Van Loy, PLoS One, 2017, 12, e0176057 CrossRef.
- B. P. Burke, C. Cawthorne and S. J. Archibald, Philos. Trans. R. Soc., A, 2017, 375, 20170261 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0dt02626c |
| This journal is © The Royal Society of Chemistry 2021 |