
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphitic carbon nitride photocatalysis: the hydroperoxyl radical role revealed by kinetic modelling†
Inmaculada
Velo-Gala
 *a,
André
Torres-Pinto
a,
Cláudia G.
Silva
a,
Bunsho
Ohtani
b,
Adrián M. T.
Silva
a and
Joaquim L.
Faria
a
*a,
André
Torres-Pinto
a,
Cláudia G.
Silva
a,
Bunsho
Ohtani
b,
Adrián M. T.
Silva
a and
Joaquim L.
Faria
a
aLaboratory of Separation and Reaction Engineering – Laboratory of Catalysis and Materials (LSRE-LCM), Faculdade de Engenharia, Universidade do Porto, Porto 4200-465, Portugal. E-mail: invega@fe.up.pt
bInstitute for Catalysis, Hokkaido University, Sapporo 001-0021, Japan
First published on 21st October 2021
Abstract
Metal-free graphitic carbon nitride (GCN) is an optical semiconductor with the advantage of in situ H2O2 generation parallel to pollutant removal. The photocatalytic degradation mechanism using GCN is attributed to a series of reactions with reactive oxygen species and photogenerated holes. However, the role of each species in the photocatalytic process kinetics remains a challenge. This work provides novel photodegradation kinetic data through a modelling approach using the Kintecus software for the first time in heterogeneous photocatalytic reactions. The results were validated by double fitting of simultaneous phenol removal and H2O2 formation. The role of dissolved oxygen and intermediate products are also investigated. Sophisticated techniques, as photoacoustic spectroscopy for energy-resolved distribution of electron traps and diffuse reflectance spectroscopy for determination of band energies, aid to clarify the behaviour of GCN. This paper identifies the oxidation routes contributors and carefully describes the GCN photocatalytic simultaneous phenol degradation and H2O2 generation.
1 Introduction
Graphitic carbon nitride (GCN) is a polymeric photo-responsive semiconductor capable of stimulating several reactions in different fields of application. The optoelectronic properties of GCN endow this material with excellent photoactivity due to its intrinsic mild bandgap, which allows for its excitation under visible light irradiation.1–6 GCN can be easily fabricated through fast polymerisation stages under an air atmosphere with inexpensive and earth-abundant precursors, i.e. a cost-effective synthetic route.6–9 Furthermore, GCN possesses high stability under a range of operating conditions, easily manoeuvred at neutral pH and aerated conditions.5–7,10GCN has been thoroughly investigated for the photocatalytic removal of recalcitrant chemical compounds from different waters and wastewaters, including those from industry or wastewater treatment plants (e.g., organic dyes, phenols, pharmaceuticals and personal care products).11–15 In addition, GCN has attracted significant attention as a non-metallic material for eco-friendly H2O2 production.16–19 For this, heterogeneous photocatalysis takes the benefit of generating reactive oxygen species (ROS), such as the case of oxy-radicals (hydroxyl – HO˙, superoxide – O2˙−, among others). These radicals can be generated by oxidation reactions in the valence band (VB) and by reduction reactions in the conduction band (CB). The oxidation reaction between the photogenerated positive hole (h+) and aromatic molecules has been shown that is an additional advantage in the photocatalytic application of this material due to the simultaneous H2O2 generation.20,21 The latter also is of particular interest for water treatment because H2O2 is an oxidant that can generate more ROS, namely the HO˙ radicals (Fig. 1).
 | ||
| Fig. 1 General accepted mechanism for GCN photocatalysis in water solution. | ||
In the literature, many efforts to improve the efficiency of the GCN photocatalyst response are focused on reducing the fast rates of electron/hole (e−/h+) pairs recombination and slow charge carrier mobility.1,6–8,21–23 Some of these strategies are chemical doping, fabrication of heterostructures and several post-treatments. Thermal-induced exfoliation is a favourable technique to improve photoactivity by significantly increasing the specific surface area of GCN by creating higher spacing between quasi-two-dimensional layers and higher degrees of surface defects.24–26 These characteristics can potentially aid in charge transfer and hinder exciton recombination. Moreover, with the increased surface area, there is a higher probability of the GCN material absorbing light with more ease and contacting the reactive media more efficiently.27,28 To understand the complete mechanisms of GCN reactions, it is necessary to investigate the ROS that are involved in the photocatalytic system.29–31 Novel approaches, such as chemiluminescence probes or electron spin resonance spectroscopy, are being used, but the application of these techniques is limited to their availability.32–36 The most common technique to detect these active species is performing quenching experiments with relatively selective scavenging substances that react primarily with a specific oxidation agent. These techniques are considered indirect methods because although they allow a relative inactivation of some principal species, they (i) do not consider the role of by-products generated and consumed concurrently to the primary reaction, (ii) are not able to quantify the radicals themselves nor allow proper kinetics assessment and (iii) can induce misleading information as some scavengers can react with different oxidant species.37–40 Regarding this, the development of theoretical modelling that describes the kinetic behaviour of all chemical components of a photocatalytic system may be considered a very relevant tool to understand the GCN actuation mechanism definitively.
The Kintecus software has proven to be a helpful tool both for estimating kinetic data41–43 and establishing the degradation pathways taking place in different advanced oxidation processes (AOP) conducted in homogeneous systems, with or without radiation.44–46 Kintecus allows modelling the kinetics of chemical reactions along the experiment contemplating all the reactions that compose the AOP with their respective reaction rate constants (k, M−1 s−1) and infer on the species contribution.47 To the best of our knowledge, this is the first study employing the Kintecus software as a tool to entirely describe the heterogeneous photocatalytic mechanisms of pollutant degradation and H2O2 formation. As it is usually assumed in the description of a heterogeneous system in an ideal situation, it was necessary to consider that the GCN load and the media absorbance remained constant in the photocatalytic reaction modelling. The commonly accepted mechanism for the oxidising reaction by the h+ in the valence band of GCN (i.e., recombination of e−/h+ and the interactions between pollutant and H2O2 with h+)2,16,48 and the usually discarded reactions (H2O/h+)49–51 were included in this modelling. The debated theories of one-electron and two-electron reduction reactions to produce O2˙− radicals were likewise investigated.50–53 In addition, the great advantage offered by the model is the detection of secondary and tertiary reactions co-occurring in the aqueous phase and taking into account the radical species that usually are disregarded, as is the case of the hydroperoxyl radical (HO2˙). The kinetical description of all involved chemicals will facilitate understanding the influence of operation parameters and the role of each species. The exact definition of the actuation mechanism of each species involved during GCN photocatalysis will enable improvement of the efficiency and the development of novel routes for practical applications.
Phenol (PhOH) was chosen as a model compound to develop the modelling of the GCN photocatalytic system since its oxidation mechanism and its decomposition by-products are well documented in the literature,54–57 which is essential for the Kintecus model formulation. Therefore, in this study, kinetic modelling was developed for GCN photocatalytic activity under LED irradiation at 417 nm (LED-417/GCN). The aims of this study are: (i) to model the photocatalytic processes in terms of the elementary reactions and to quantitatively evaluate their contributions; (ii) to explain the observed compromise between PhOH degradation, H2O2 formation, and oxygen effect; (iii) to elucidate the mechanism for the GCN photocatalytic application both for the degradation of pollutants and for the H2O2 production; (iv) to provide novel kinetic data of photocatalytic reactions; and (v) to illustrate the kinetic model as a helpful tool to the best practical application in a heterogeneous photo-assisted system.
2 Materials and methods
2.1 Chemicals
Dicyandiamide (C2H4N4, 99%), phenol (C6H5OH, ≥99%), titanium(IV) oxysulphate (TiO5S, ∼15 wt% in sulphuric acid), H2O2 (30 wt% in water), dimethyl sulfoxide and 2,4-dinitrophenylhydrazine were obtained from Sigma-Aldrich. Methanol (CH4O, 99.99%) was obtained from Fisher Chemical. Sulphuric acid (H2SO4, 95.0–97.0%) was obtained from Honeywell Fluka. Ultrapure water was produced in a Direct-Q Millipore system (Merck Millipore, Billerica, MA, USA).2.2 Catalyst preparation
GCN was synthesised by a two-step thermal treatment procedure in a muffle furnace, as reported elsewhere.24 Briefly, on the first stage, a certain amount of dicyandiamide was placed in a semi-closed crucible under static air, heated at 2 °C min−1, up to 450 °C remaining for 2 h at that temperature, and raised to 550 °C remaining at this temperature for 4 h. Then, after grounding, washing to remove any polymerisation intermediates, and filtering and drying at 100 °C overnight, the material was spread in open crucibles and heated up to 500 °C remaining at that temperature for 2 h.2.3 Catalyst characterisation
Our group has previously reported thorough characterisation of GCN by different advanced techniques, such as scanning electron microscopy, diffuse reflectance spectroscopy, X-ray diffraction, X-ray photoelectron spectroscopy, photoluminescence, point of zero charge, porosity and specific surface area.24,58,59An important measurement was the energy-resolved distribution of electron traps (ERDT ETs) detected by double-beam photoacoustic spectroscopy (RDB-PAS). This novel analytical technique has been applied in some recent works.60–63 The method follows the direct photoexcitation of valence-band electrons to ETs, which have a negligible photoabsorption coefficient due to the negligible density of ETs. In this study, electronic-states analysis was performed using a laboratory-made RDB-PAS instrument (code name BK1) composed of a grating monochromator with a xenon lamp (Bunkokeiki M10-RP/BXL-150), an LED (Luxeon LXHL-ND98; 625 nm) intensity-modulated at 35 Hz by a digital function generator (NF Corporation DF-1906), a UV quartz combiner light guide (Moritex MXS5-1000S-UV3), a digital lock-in amplifier (NF Corporation LI5630) and a laboratory-made photoacoustic (PA) cell equipped with a quartz window, gas-tight bulbs, a MEMS (micro-electro-mechanical system) microphone module (SparkFun MEMS Microphone Breakout, INMP401 (ADMP401)) and a stainless-steel sample holder. In typical RDB-PAS measurements,60–62 a sample powder of GCN (ca. 50–100 mg) was loaded on a stainless sample holder and placed in a PA cell, and the cell was transferred to a cubic acrylic box. Methanol-saturated nitrogen was introduced in the cell for ca. 30 min to remove oxygen, and then the cell was sealed tightly. Two light beams were introduced to the cell through a quartz window. One is continuous monochromatic light wavelength-scanned from longer wavelength to shorter wavelength (650–300 nm) with a 5 nm step. The other is an intensity-modulated (35 Hz) 625 nm LED light. PA signal was amplified with a lock-in amplifier and recorded as RDB-PA spectrum showing the accumulation of electrons in electron traps. The spectrum was differentiated from the lower energy side to obtain an ERDT pattern followed by calibration with experimentally determined conversion coefficient for titania (titanium(IV) oxide)60 to the absolute density of ETs as in titania. Therefore, the density shown in ERDT patterns is a relative value for the samples other than titania. The energy of ETs is shown in reference to valence-band top (VBT) energy for convenience, and thus obtained ERDT pattern was replotted as a bar graph with a pitch of 0.05 eV.
Conduction-band bottom (CBB) energy was estimated by single beam PAS using the same setup. The sample in a PA cell was irradiated by modulated (80 Hz) monochromatic light wavelength scanned from longer wavelength to shorter wavelength to obtain the PA spectrum, which was then calibrated with a spectrum of carbon black. To determine the absorption-edge wavelength, the bandgap energy (in eV) was calculated and plotted as CBB with reference to VBT.
2.4 Photocatalytic reactions
The photocatalytic experiments were conducted in a cylindrical borosilicate glass reactor with a total volume of 50 mL. The reactions were carried out with vigorous stirring and under the incidence of visible light by four high-intensity 10 W light-emitting diodes located symmetrically at 4.0 cm from the reactor wall with a maximum emission at 417 nm and an average irradiance of ca. 450 W m−2 each. The irradiance measurements were performed with an OceanOptics USB2000+ UV-vis spectrometer.A schematic representation of the photocatalytic setup is displayed on Fig. S1.† The reactions were conducted under gas bubbling, with 30 min of gas saturation in dark conditions to achieve an adsorption–desorption equilibrium. The gas consisted of air, argon or mixtures of air![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :argon in different proportions controlled by Bronkhorst High-Tech EF-FLOW Select F 201 CV thermal mass flow meters. The amounts of dissolved oxygen (DO) studied were 0, 5, 10, 21 and 100%.
:argon in different proportions controlled by Bronkhorst High-Tech EF-FLOW Select F 201 CV thermal mass flow meters. The amounts of dissolved oxygen (DO) studied were 0, 5, 10, 21 and 100%.
Solutions of PhOH were prepared by selecting an initial concentration of [PhOH]0 = 6.38 × 10−4 M (or 60 mg L−1), a typical concentration found in olive mill wastewaters;14 and when H2O2 was initially present, it was added dropwise to a certain amount to the PhOH solution. It has been proven that H2O2 only attacks PhOH in the presence of a specific activator (e.g., dissolved iron, photocatalyst) but not with the employed radiation source.14
The GCN powder was sonicated in the aqueous matrices for 2 min to disperse it appropriately. The GCN load was kept at 0.50 g L−1 since it was found to be the optimal amount for a compromise between the degradation of aromatic compounds and H2O2 production.58
2.5 Analytical techniques
Samples were periodically withdrawn, filtered and analysed by high-performance liquid chromatography (HPLC) using a Hitachi Elite LaChrom instrument (Hitachi, Ibaraki, Japan) equipped with both a diode array detector (DAD) and an ultraviolet detector (UV-D), a Purospher STAR RP-18 column (250 mm × 4.6 mm, 5 μm particles), and a solvent delivery pump. An optimised gradient elution method was prepared, starting with an equilibrated mixture of water (A):methanol (B) (70:30) followed by a linear gradient step to A:B (37:63) in 20 min; then, the initial conditions are re-established in a 1 min gradient step and the A:B (70:30) mixture is isocratically eluted for 7 min at a fixed flow rate of 1.0 mL min−1. PhOH was detected with the DAD at its maximum absorption wavelength (λmax) measured by ultraviolet-visible spectroscopy in a JASCO V-560 spectrophotometer (λmax,PhOH = 270 nm).
The determination of the amount of H2O2 produced was followed by a simplified colourimetric method based on the literature.64 Briefly, 300 μL of the filtered sample, 300 μL of 0.5 M H2SO4 and 10 μL of titanium(IV) oxysulphate were mixed in a polystyrene cuvette, and the absorbance read at a wavelength of 405 nm by an OceanOptics USB2000+ UV-vis spectrometer.
2.6 Kinetic modelling
The chemical kinetic model was fitted to the experimental data using Kintecus Version 6.80.47 This simulation was used to determine the reactive system mechanism of GCN photocatalysis and predict ROS concentration along this process. As a result, the photocatalytic mechanism was elucidated. The knowledge about this mechanism was extended by exploring different possible reactions that are commonly involved during GCN photocatalytic processes. As shown in Table S1,† the model was developed based on a set of 62 reactions. The associated kinetic rate constants (k) used in this study are obtained from the literature or estimated by fitting the model to the experimental data if they are unknown. This is the case of the reactions of the positive hole with PhOH, water molecules, H2O2, and its recombination with the photogenerated electron (R2–R7). The reaction rate constants were estimated by incorporating different combinations of k values into the Kintecus program to accurately simulate the kinetics of PhOH degradation and H2O2 generation in each experiment. To make the estimated k values even more precise, triplicates of these experiments were considered in the fitting simulation. As a result, a range of k values for the reactions R5–R7 was obtained. Considering this range, the fitting between the model and the triplicate experimental data were more suitable. The optimised k values were chosen by this double fit in five different experimental conditions with oxygen contents between 0% and 100%. The experiments of H2O2 removal (LED-417/GCN system without PhOH, 0% and 21% of DO) were used to estimate rate constants between H2O or H2O2 and h+. The rate constant of the photocatalytic reaction was calculated as described more in-depth in Text S1.†The precision of the obtained model was assessed by analysing the residuals sum of squares (RSS) between the experimental data and the model results explained in Text S2.† In this case, the RSS values represent the average deviation of the model result from the experimental data and assess the precision of the estimated k values. The RSS was, in turn, used to perform an analysis of the sensitivity of the model to changes in a given rate constant. This assessment test has been done in previous works.41,65,66 Likewise, for establishing the relative importance of estimated reactions in the kinetics, the RSS between the control model output and the altered model output was calculated (Text S2†).
3 Results and discussion
3.1 Phenol degradation by GCN photocatalysis
The photocatalytic system was investigated using PhOH as a model compound. The PhOH removal is attributed to the reactive species generated by the photocatalytic process since PhOH showed negligible photodegradation under visible radiation (i.e. without a photocatalyst) and is not removed by adsorption on the catalyst surface.58 Furthermore, the PhOH molecule can behave as a proton donor, a property that has been widely exposed as being able to H2O2 generation.67Previous studies of our research group evidenced the relationship between PhOH degradation and H2O2 formation in the LED-417/GCN system.58 The process of PhOH removal in aqueous solutions during photocatalysis follows a pseudo-first-order reaction rate and, under aerated conditions (i.e. in the presence of 21% dissolved oxygen (DO)) with photo-excited GCN, can promote the formation of H2O2. Interestingly, the concentration of H2O2 increases until PhOH is completely removed (Fig. 2). This behaviour has been explained owing to the presence of an aromatic compound acting as a proton donor for the H2O2 generation.49,68,69
 | ||
| Fig. 2 PhOH degradation and H2O2 evolution under air and argon saturation conditions. Initial conditions: pH0 = 6.1; [PhOH]0 = 6.38 × 10−4 M; LED-417/GCN system. | ||
The same system, but under anoxic conditions, finds no formation of H2O2, and therefore we claim that DO is necessary for H2O2 production (Fig. 2). In this way, it was interesting to investigate the role of DO in the degradation of PhOH and the evolution of H2O2 during irradiation time. Torres-Pinto et al. analysed the effect of different DO percentages, finding an evident influence of the DO and the H2O2 generation and PhOH degradation (Table S2†).58 In this study, an important input to consider is the decline of H2O2 concentration coincident with the complete removal of PhOH. This phenomenon (i.e. decline of H2O2) was observed in different studies, being ascribed to different theories: (i) H2O2 acts as a photochemical oxidation agent of the parent molecule and its intermediates;70–73 (ii) H2O2 can react directly with the photoactivated exciton pairs, through redox reactions, generating ROS;14,74 (iii) H2O2 is reduced by photogenerated electrons;75–77 or (iv) H2O2 is being transformed into water after the removal of the primary proton donor.17,50,53 In the presence of these different theories, it is also necessary to establish the correct reaction path that occurs to explain these results, as well as correlate with what was observed during PhOH degradation with the generation of H2O2, along with the impact of DO concentration.
The Kintecus program was extremely useful for modelling the kinetic reactions that define this chemical process,47 being its application simpler in homogeneous than in heterogeneous catalytic systems. In the absence of all probe compounds, this simulation is a great tool for demonstrating all different theories by the chemical species evolution throughout the system. This is the first time that photocatalytic reactions with a solid semiconductor material (GCN) are modelled, setting the stage for more comprehensively integrative approaches to research in heterogeneous photocatalysis. In this regard, a new tool is proposed to identify all chemical species role and their evolution along the reaction time in photocatalytic systems.
3.2 Modelling the kinetics of GCN photocatalytic system
For reactions with unknown rate constants, their values were obtained by a numerical routine iteration method combined in the Kintecus program to fit the model curve to experimental data accurately. This is the case for the positive hole reaction with species as the photoexcited electron (R2), the water molecule (R3, R4), H2O2 (R5, R6) and PhOH (R7). Lastly, to evaluate the optimal reaction rate constant values, the normalised residuals sum of squares (NRSS) between the model output and the experimental data was calculated, and the procedure is described in greater detail in Text S2.† The capability of the model to reproduce the role of each reaction according to the experimental data was analysed by the NRSS, taking into account a sensitivity analysis of the kinetic model to variations in individual rate constant values. The magnitude of the NRSS, depicted in Fig. S2 and S3,† offers an insight into the suitability of an attributed kinetic rate constant. The lowest value between the NRSS for the same reaction is associated with the minimised error for the selected kinetic rate constant. In addition, if the variation in the rate constant values leads to greater differences between the NRSS values, we can infer that the corresponding reaction has a more important role on the photocatalytic process.
The NRSS analysis set the optimal value for the rate constant for the recombination reaction in the H2O2 removal experiments because it is a less complex operational system. The k value for the recombination reaction was 1.00 × 1017 M−1 s−1 (Fig. S2-A and S3-A†).
All the possible reactions that have been previously investigated by other studies regarding GCN were considered in this study. The capacity of the model to reproduce the role of each reaction with the experimental data was analysed by the NRSS, considering the sensitivity analysis of the model to variations in the particular rate constant. Although the inefficient water oxidation capability of GCN materials has been established,49–52,93 the NRSS results demonstrated that the reaction between the positive hole and water molecule is necessary to effectively simulate the photocatalytic experiments (Fig. S2 and S3†). Therefore, despite the low k value, the reaction occurs and significantly impacts the overall system.
The NRSS for H2O2 removal experiments (Fig. S3†) evidenced that the positive hole and hydroxyl anion reaction does not occur in this system, as it did not influence the NRSS values (Fig. S3-D†). The lower NRSS value means that this reaction is not involved in the PhOH degradation experiment (Fig. S3-D†). However, an effect has been observed for reactions R3 and R4 (Fig. S2-B and C and S3-B and C†). The NRSS analysis revealed that the optimal k values for both reactions must be lower than 1.00 × 103 M−1 s−1, since higher reaction rate constants lead to a significant increase of the NRSS. However, for lower values of k for R3 (Fig. S2-B†), the NRSS has no considerable interference. When k values are lower and higher than 1.00 × 103 M−1 s−1 for the reaction between h+ and water molecules to produce oxygen (R4), the NRSS increases substantially, indicating its superior effect on the photocatalytic process.
For different DO concentrations, the k value of the recombination process varied significantly, as observed in Fig. S4.† This phenomenon will be better disclosed in the following sections when describing the mechanistic pathway and major species affecting PhOH removal and H2O2 generation.
In this context, by the Kintecus program, the participation of different reactions for PhOH removal was attested and quantified in terms of their k values (and supported by RSS analyses) to mimic the experimental data. The reaction between the h+ and PhOH (R7) and the effect of different ROS in PhOH degradation was considered in the model (R8–11).
The fittings for different DO percentages are shown in Fig. 3. The fitting was obtained by considering the k values of all reactions (R1–R62) to better model the concentrations of PhOH, H2O2, DO and protons (pH), as depicted in Table S2.† The errors bars established in the Kintecus results concern the best fitting obtained by substituting in the model the 27 possible combinations of k values for the R5–R7 reactions, considering the maximum, intermediate and minimum k values in the ranges established for these reactions.
 | ||
| Fig. 3 Experimental data (open symbols) and model outputs (full symbols) for the degradation of PhOH and the simultaneous H2O2 generation in the presence of different DO percentages: (a) 5%, (b) 10%, (c) 21% and (d) 100%. Initial conditions: pH0 = 6.1; [PhOH]0 = 6.38 × 10−4 M; LED-417/GCN system. | ||
As observed in Fig. 3, the model adjusts nicely to the experimental data after a careful iteration process. The k values obtained are portrayed in Table S3,† the associated error between experimental and modelled data was less significant than 3.1%, indicating that a good correlation was established.
The bottom line of most organic compound oxidation explanations in GCN photocatalysis is its reaction with the photogenerated h+.49,58,68,69,93 This oxidation pathway could induce the generation of the phenoxyl radical as the following reaction (R7):
| h+ + C6H5OH → C6H5O˙ + H+ |
In addition, there are other reactive species involved as the electron on the conduction band, the h+ on the valence band, and the combined activity of different ROS. The electron reduction of PhOH was studied, but its presence did not allow the fit in all the experiments since it promotes PhOH degradation regardless of the DO percentage (data not shown). The model disclosed each oxidant species' real and indistinguishable influence in PhOH removal (Fig. 4). In Table S3† can be found the impact of each oxidative agent, starting with the key contribution of h+ for PhOH removal being around 60%, the HO2˙ (R11) contributing with a significant 37.5%, and with the remaining corresponding to the nearly negligible HO˙ role by reaction R9.
 | ||
| Fig. 4 Kinetic modelling for the degradation of PhOH and the evolution of its oxidation reaction for DO = 21%. Initial conditions: pH0 = 6.1; [PhOH]0 = 6.38 × 10−4 M; LED-417/GCN system. | ||
The typical neglect of recognising HO2˙ influence can be attributed to the low reaction rate constant values that this radical possesses, as is the case for reaction R11. These results highlight the relevance of considering its possible contribution due to its much higher concentration along the process: [HO2˙] = 7.00 × 10−8 M at 21% of DO, compared with [O2˙−] = 2.10 × 10−10 M, [HO˙] = 4.95 × 10−16 M and [O˙−] = 4.78 × 10−20 M. The Kintecus model is of particular importance to determine the HO2˙ impact since it is difficult to find a selective chemical scavenger that can single out this radical from the system. The majority of studies concerning ROS-quenching investigate the radicals with higher k values, e.g. O2˙− and HO˙.97
As described, Fig. 4 shows the removal of PhOH by h+ and HO2˙; the primary actuators to generate intermediate species of degradation and lead to complete removal of the parent molecule. As a result, the principal immediate implication from these reactions is the generation of phenoxyl radical (C6H5O˙), phenolate anion (C6H5O−) and H2O2. Given its role in the PhOH removal, it is interesting to highlight the involvement of the phenoxyl radicals in the HO2˙ generation by reaction R12. This reaction implies that oxygen is necessary to form the second most influential PhOH oxidative species in this system. This finding could be one of the leading causes for the dependence on DO concentration. Without DO, the degradation of PhOH is only 8.50%, and H2O2 generation was not observed by the used colourimetric method. The most conventional explanation is the role of oxygen as an electron acceptor, which accelerates the heterogeneous photocatalytic degradation by inhibiting the recombination of e−/h+ pairs. Furthermore, the main consequence is the formation of O2˙− and/or H2O22,16,48,50,98,99 as the most relevant products, following reactions R30 and R31.
The different percentages of reactive species contribution for PhOH degradation were calculated to elucidate the effect of DO (Table S3†). For comparison purposes, the PhOH percentage degradation was normalised for 100% of removal and is shown in Fig. 5a. The evolution of PhOH concentration depending on irradiation time and DO% is shown in Fig. 5b.
 | ||
| Fig. 5 (a) Normalised PhOH oxidation percentages by different reactive species and (b) PhOH relative concentrations at different times as a function of the DO percentage. Initial conditions: pH0 = 6.1; [PhOH]0 = 60 mg L−1; LED-417/GCN system. | ||
The modelling results reveal that the DO effect is owed to the increase in HO2˙ concentration by the reaction between oxygen and phenoxyl radical (R12). However, it is clearly not only due to the charge recombination reaction as until now, but it was indicated as the most plausible theory of oxygen influence. The PhOH degradation at 5% of DO seems to follow zero-order kinetics, most probably because the reaction with h+ is the dominant oxidation pathway. This implies that, as the DO concentration increases, the PhOH degradation by HO2˙ increases the influence of DO, replacing the hole-oxidation as the primary contributor of PhOH removal. Consequently, the photocatalytic efficiency of this GCN material is enhanced by limiting the photogenerated charges recombination in two different ways: through the electron scavenger activity of DO and by the fostering of h+ for its direct usage as an oxidant of organic molecules.
On the other hand, the slight decreasing trend following the maximum H2O2 concentration was attributed to the decrease of catalytic activity, the decomposition of H2O2 by electrons and holes, or photodecomposition.16,49,68,101,102 The obtained model has proved to be a valuable tool for clarifying the reactions that play the leading roles in H2O2 production. H2O2 could be formed directly from reactions R11, R21, R22, R31, R34, R38, R39 and R47. The model clearly identifies HO2˙ as the key species in H2O2 production in this system. The reaction of PhOH degradation by HO2˙ (R11) and this radical recombination with itself (R38) have the highest contributions with obtaining maximum concentrations of H2O2 (Table S4†). The variation in oxygen concentration reveals that oxygen promotes the HO2˙ recombination reaction over the others. Although the intervention of reaction R11 decreases, it maintains a relevant role in forming H2O2 around 30% (Fig. 6). Fig. 6a summarises the described mechanism, showing the importance of each reaction with DO concentration defined by the Kintecus modelling. For a better understanding of H2O2 evolution, the concentrations as a function of the DO% are shown for different reaction times (Fig. 6b).
 | ||
| Fig. 6 (a) Contribution of the different reactions involved in H2O2 formation, and (b) H2O2 concentrations at different times as a function of DO percentage. Initial conditions: pH0 = 6.1; [PhOH]0 = 6.38 × 10−4 M; LED-417/GCN system. | ||
The strong DO influence is the presence of the reactions involved in the formation of HO2˙. In Fig. S5,† the concentrations of several compounds are shown along with PhOH removal and H2O2 evolution, depicting the generated amount of oxygen, hydroperoxyl, superoxide and phenoxyl radicals. Reactions R30 and R32 are the main production routes for HO2˙ generation. This is the first reaction of the sequential two-step single-electron reduction, where the oxygen reduction formed O2˙− (R30). For instance, the chemical composition of the surface of the GCN catalyst was analysed by XPS (Fig. S6†).24 Analysis of C1s showed that the significant contribution of carbon in the structure is from N-containing aromatic rings (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). The obtained 52.1% for N1s spectra is composed of a 15.2% for tertiary N (N–(C3) structure) and 29.7% for the triazine ring C–NC; the rest of N is for terminal forms as amino-functional groups and –NO2 groups.24 These configurations for C1s and N1s could result from fewer carbon vacancies on the surface.103 Both situations induce the two-step single-electron reduction pathway identified as the oxygen reduction route by the Kintecus modelling.
N). The obtained 52.1% for N1s spectra is composed of a 15.2% for tertiary N (N–(C3) structure) and 29.7% for the triazine ring C–NC; the rest of N is for terminal forms as amino-functional groups and –NO2 groups.24 These configurations for C1s and N1s could result from fewer carbon vacancies on the surface.103 Both situations induce the two-step single-electron reduction pathway identified as the oxygen reduction route by the Kintecus modelling.
To date, and our knowledge, there was only one research article where HO2˙ was the predominant ROS as observed in UVA/TiO2 system.104 This study identified HO2˙ generation by reducing oxygen by the photogenerated electrons followed by the O2˙− reaction with protons. For this specific mechanism, the authors explained the subsequent H2O2 formation by the reaction between the O2˙− with two protons and two electrons (R31). However, the effect of pH on this radical and its consequent conversion to HO2˙ must be considered (R32). Other authors explain the transformation of the superoxo radicals on the surface of materials to hydroperoxo species, concluding that their protonation leads to H2O2 evolution.99,105 Regardless of the pH influence, both radicals could follow the second reaction in the one-electron route, proposed by the authors for the generation of H2O2, because HO2˙ can also react with protons (R34) in the same way as O2˙− (R31).
Nevertheless, an unexpected route is occurring in this LED-417/GCN system. After single-electron oxygen reduction reaction (R30), the recombination reaction of HO2˙ (R38) represents the main reaction for H2O2 evolution, followed then by the interaction between PhOH and HO2˙ (R11), leading to PhOH degradation where PhOH is acting as a proton donor. This prevalent sequence is owed to the value of their reaction rate constants, with R30 showing a higher k value than that of R38 and R11. The concentrations reached by HO2˙ make its reaction with PhOH significant even though its reaction constant is relatively low (2.70 × 103 M−1 s−1). The concentration of HO2˙ could be not considered in previous studies because of the difficulty in its experimental detection and quantification. In addition, this radical is the least oxidant agent among all the reactive oxygen species. For instance, comparing the redox potentials of HO2˙ and HO˙, the latter is a much more powerful oxidising agent: E0(HO2˙/H2O2) = +1.44 V and E0(HO˙/H2O) = +2.38 V.106,107 This often leads to authors disregarding the effect of HO2˙ in the oxidation process of pollutants. Thus, Kintecus modelling allowed the recovery of a complex formulation of H2O2 evolution that has not been indicated as the main mechanism until now.
In previous studies, there is an explicit preference for the one-step two-electron direct oxygen reduction route instead of the sequential two-step single-electron indirect reduction, based on its greater effectiveness for H2O2 production.16,50,53,108 However, this assertion might not be so obvious because the result of a higher concentration of H2O2 could be related to the pathways indicated in this study. In this case, the dependence on the HO2˙ can be a substantial advantage because this species has a longer lifetime due to its lower reaction rate constants and its greater selectivity with other species.107,109
It is also necessary to point out the relevance of the phenoxyl radical to HO2˙ concentration, as evidenced by the shape and concentration values of the R12 kinetics similar to that of H2O2 evolution (Fig. S5†). The phenoxyl radical is the first by-product of PhOH oxidation by h+ (R7). This corroborates our previous theory on the by-products of PhOH degradation having an important role in the indirect production of H2O2. The phenoxyl radical reaction with DO (R12) is the other main route for HO2˙ production, reinforcing that DO should be present in the system.
According to the model (Table S4†), other routes with lower participation in the formation of H2O2 are the reaction between the hydroperoxyl anion with protons (R39). This anion comes from the complementary reactions to form the phenolate anion (R13) and its reaction with HO2˙ (R14).
It is also essential to investigate the slight decrease in H2O2 concentration after the complete PhOH removal. If only H2O2 is present in the LED-417/GCN system, its concentration decreases and the compound is removed regardless of air or argon purging (Fig. 7). GCN can effectively lead to the decomposition of H2O2 without any organic molecule present (i.e., lack of h+ scavenger), meaning that possibly H2O2 is being reduced by electrons or oxidised by h+.
 | ||
| Fig. 7 H2O2 normalised concentration ([H2O2]/[H2O2]0) in the presence of GCN and visible light ([H2O2]0 = 1 mmol L−1; gas flow = 50 mL min−1). | ||
If the reactions of h+ with H2O2 are considered in the model (R5, R6), the simulation fits much better with the experimental data (Fig. S2-E and F and S3-E and F†) for both estimated rate constants values of 1.70–2.50 × 109 M−1 s−1 and 2.00–3.00 × 109 M−1 s−1 to produce oxygen or HO2˙, respectively. This value is following that assumed by others authors for the reaction rate constant of h+ with H2O2 (1.00 × 109 M−1 s−1), corresponding to the diffusion limit of H2O2.110 The sensitivity analysis revealed that the direct oxidation of H2O2 by h+ is a crucial reaction in LED-417/GCN system, being more influential the reaction R5 since a variation in its k value has a higher impact on the residual values referring to the smaller RSS (Fig. S2-E and F and S3-E and F†). The well-known ROS reactions that remove H2O2 were also taken into account in the model's design (R42–R46). A negligible effect in its direct removal was detected by e−, O2˙−, HO˙ or HO2˙ because of their lower reaction rate constants. An increase in protons and dissolved oxygen concentrations have been recorded in the final solution, especially when PhOH degradation is higher (Table S2†). The reason is that both species are the products of reactions R5 and R6. Meanwhile, the percentage of DO also affects the process leading to the elimination of H2O2 (Table S5†). Therefore, with increasing DO percentages, it is more likely that the two main oxidative pathways are by the positive hole reaction because the presence of DO avoids the recombination of the photogenerated charge (Fig. S4†).
3.3 Phonon dispersion
The band potentials control electron relocation and formation of the oxidant species. The occurrence or not of these reactions depends on the valance and conduction band potential positions relative to their redox potentials. Their theoretical analysis can help to identify the behaviour of GCN in different systems. DRS allowed to construct the respective Tauc plot (Fig. S7†) and obtain the bandgap energy of this semiconductor.58 GCN shows a 2.73 eV bandgap, with a conduction band bottom (CBB) at −1.22 eV and a valence band top (VBT) at +1.51 eV.The energy-resolved distribution of electron traps (ERDT), detected by double-beam photoacoustic spectroscopy (RDP-PAS), has appeared as a possible trademark feature of optical semiconductors to explain mid-gap states and other possible energy levels for possible reduction reactions.60 Briefly, RDB-PAS leads to the delocalisation of electrons from the ground states in lower-energy levels to their excited states through specific photoexcitation. Then, the ERDT pattern is shown as a function of energy from VBT for convenience, such as is the case for density of states (DOS) studies. As reported in the literature, the RDB-PAS method is based on the direct photoexcitation of valence band electrons to electron traps that have negligible photo-absorption coefficient due to minor density of electron-traps.61–63 Therefore, the actual photoexcitation may occur from the high density of states part of VB, which may be lower than VBT, i.e., the VBT energy may be overestimated by the DRS results. The measured ERDTs are probably just below the CBB. As Fig. 8 shows, the RDB-PAS experiments detected distant ERDTs from the VBT, demonstrating more negative values than the CBB. This is concordant with DOS theory which explains that a phonon can occupy a given energy level (or state). In the case of electrons at the band edge of the valence band, they can be excited to specific states inside the bandgap or above the conduction band bottom. This ERDT pattern was similar to that of reported oxygen-doped GCN.63
 | ||
| Fig. 8 Representative ERDT pattern with CBB position for GCN. | ||
GCN is composed of a layered combination of nanosheets that are topologically similar to graphene; thus, this two-dimensional matrix can be expected a certain degree of anisotropy. This can lead to the belief that the electron-traps measured experimentally by RDB-PAS can be somewhat mirrored near the VBT. In this case, the DOS theory can be applied, and oxidation reactions necessary to occur, such as water, H2O2 and PhOH reactions with the available photogenerated h+, are thermodynamically favourable. Therefore, the ERDT characterisation of GCN corroborated the photocatalytic behaviour described in the Kintecus model.
The estimated photocatalytic reactions responsible for the pathway of PhOH degradation and H2O2 formation are represented in Fig. 9. The CBB (−1.22 V vs. NHE) and VBT (+1.51 V vs. NHE) of the GCN are suitable to facilitate oxygen reduction and the oxidation of H2O2, respectively. However, other oxidation reactions that might appear thermodynamically unfavourable (i.e., h+/PhOH with redox potential +1.76 V vs. NHE, and h+/H2O +1.97 V vs. NHE) may be occurring. The RDB PAS analysis found more negative values than the CBB that could be construed to additional energy band positions near the calculated VBT. In addition, some studies about the charge density distribution at VBM and CBM of GCN monolayer show that these materials exhibit large hole effective masses while small electron effective masses indicated by almost flat valence bands and dispersive conduction bands.111 These results support the viability of the oxidation reactions proposed in the model.
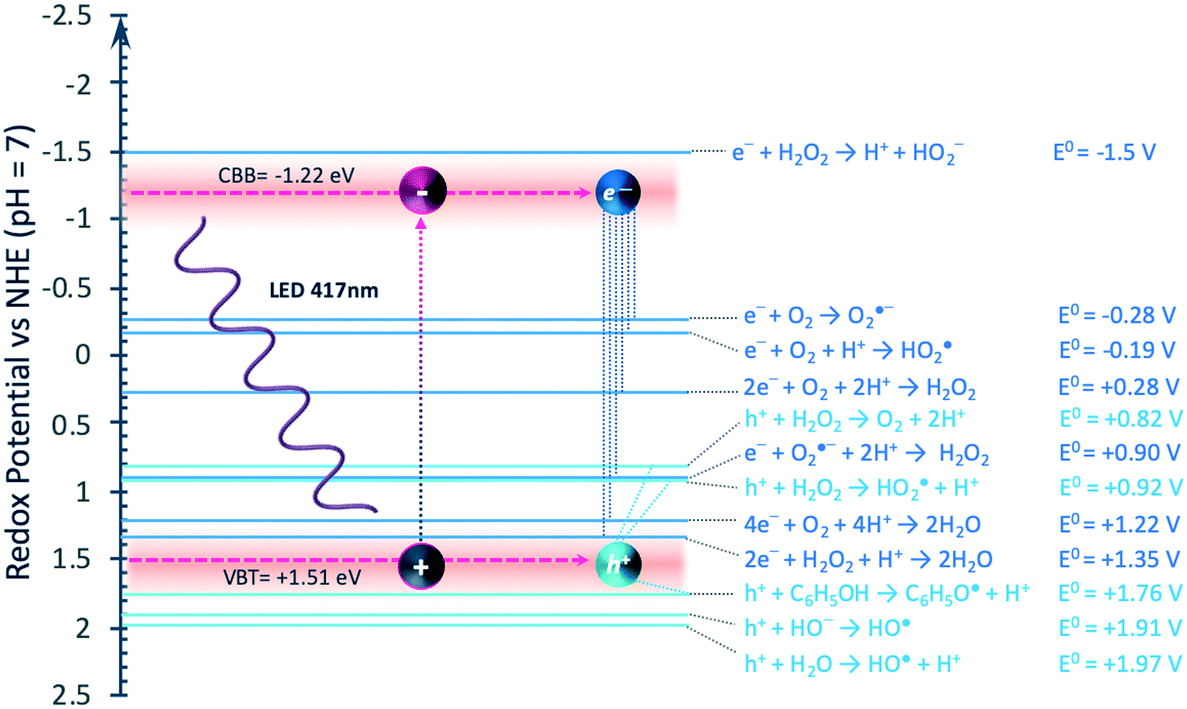 | ||
| Fig. 9 Energy levels of GCN and redox potential of reaction with photogenerated charges. Redox potential values obtained from ref. 106. | ||
3.4 Photocatalytic mechanism
This work aims to clarify the photocatalytic behaviour of GCN under LED irradiation (maximum emission at 417 nm, i.e. visible light). Therefore, it is imperative to explain the overall links detected between DO, PhOH, H2O2 and the photogenerated e−/h+ pairs by the simulation model. The proposed mechanistic pathway is shown in Fig. 10. | ||
| Fig. 10 Photocatalytic mechanism proposed for GCN under visible-LED. | ||
Higher percentages of DO in the medium reveal an increment of PhOH degradation as well as a higher H2O2 production. The PhOH oxidation by photogenerated h+ represents more than half of PhOH degradation, and the remainder being held by HO2˙ degradation. The initial by-products of these reactions are phenoxyl radicals and H2O2. The relevance of DO in this system is not only the evident avoidance of photogenerated e−/h+ pairs. The formation of O2˙− by oxygen reduction in the conduction band is vital in this system, not for its direct reaction for H2O2 generation but through its conversion into HO2˙ (R32), with k = 7.20 × 1010 M−1 s−1. This is supported by considering the value of pKa(HO2˙/O2˙−) = 4.69112 and that all main reactions generate H+ as a product. The protonation of O2˙− to yield HO2˙ induces the acidification of the aqueous medium along with the photocatalytic treatment, with final pH values of 5.14, 4.06 and 3.83, respectively for the experiments with 10%, 21% and 100% of DO (Table S2†), this lower pH leads to effective HO2˙ production and faster PhOH removal and H2O2 generation rates. The other effect of DO is its necessary reaction with the phenoxyl radical to increase HO2˙ concentration. Consequently, the production of H2O2 is primarily due to the HO2˙ recombination, following the reaction between PhOH and HO2˙. The relevance of the pathways established in this proposed mechanism is usually not taken into account, perhaps due to the lower reactivity of this radical and the difficulty of its experimental determination between other ROS.
With regards to the observed slight decrease in the H2O2 concentration, it has been induced only by the reactions with the photogenerated h+ in the VB, and neither by H2O2 reaction with by-products nor by its reduction through photogenerated e−. As a result, this reduction is slow since the products from H2O2 oxidation by h+ are HO2˙, oxygen and protons simultaneously involved in H2O2 generation.
Succinctly, with the obtained data, we can establish two main routes of PhOH degradation and H2O2 generation, addressing the major contributors for each species evolution, supported by the modelling data. In conditions with low DO concentration, there is a mainly oxidative pathway where h+ promote the conversion of PhOH into aromatic radicals that aid in HO2˙ formation, which by itself oxidises PhOH and generates low amounts of H2O2. In contrast, when air is saturated in the reactive medium (i.e., >21% O2), oxygen reduction reactions are significantly superior, giving rise to a two-step one-electron reduction of O2 to form HO2˙, which is recombined into H2O2 and also acts with around a 40% PhOH removal. Therefore, two situations are portrayed in H2O2 formation: anoxic/oxidative and oxygenated/reductive, representing a novel mechanism focus to approach GCN photocatalytic reactions for H2O2 generation.
4 Conclusions
Many studies showed the capability of GCN materials for simultaneously producing H2O2 and high degradation of organic pollutants in waters. Different mechanisms have been proposed for the heterogeneous GCN mediated photocatalytic process. These authors indicated that the photogenerated electrons are consumed by DO, forming H2O2 by the two-electron or one-electron reduction routes. For the last, protons are necessary for H2O2 generation. In addition, O2˙− and HO˙ could be formed. These ROS and the photogenerated h+ can oxidise organic pollutants.In this context, an estimation of the participation of the different reactive species has been attempted using radical scavengers, but this methodology has significant drawbacks that influence their application and limit the interpretation of results. Achieving a deep insight into the photocatalytic mechanism of GCN materials might be helpful to its practical application and contribute to the knowledge in radical pathways. However, it is not simple to isolate the role of each chemical species, especially the ROS with lower reactivity, because their scavenging effect involves the removal of other more reactive species. This is the case of HO2˙, whose role in advanced oxidation processes would be hidden by the non-selective and highest reactivity of HO˙. In this context, this study reports the principal role of HO2˙ in GCN based systems by the first kinetic simulation of this photocatalytic process.
Kintecus simulation has confirmed to be an effective tool for defining the mechanism of photocatalytic processes. This work provided considerable kinetic data of radical reactions with the GCN material; many of them were estimated for the first time. Multiple combinations of all different degradation pathways identified in the literature were investigated, deducing for the first time that HO2˙, as a secondary radical, is the main species involved in both the primary oxidation of PhOH and simultaneous H2O2 generation. The oxidation of PhOH by h+ is positioned as responsible for around 60% of PhOH oxidation, and PhOH reduction by photogenerated e− was discarded by the simulation. Although the redox potential is more positive than the VBT of GCN, the reaction between PhOH and holes is necessary for achieving the best fit between the experimental results and the simulation. This was supported by the RDB-PAS analysis of the ERDT of electron-traps that found more negative values than the CBB. These energy states could be interpreted as additional band positions near the calculated VBT. So, investigating the ERDT of electron-traps corroborated the thermodynamic feasibility of the proposed reactions. In addition, the established photocatalytic model reveals that the influence of DO has a different effect than that described in classical theories. The avoidance of recombination of the photogenerated charges is favoured by the presence of DO due to the reduction reaction of oxygen in the CB. However, this reaction is not increased by the percentage of DO in water as it had been established until now. The modelling results revealed that the DO amount increases HO2˙ concentration by the reaction between phenoxyl radical and oxygen, increasing the effect of this radical on PhOH degradation. In addition, HO2˙, oxygen and protons are identified as the products from the slight decrease in H2O2 concentration from its reaction with h+. This reaction is only favoured when PhOH is completely removed. The simulation concluded a negligible effect of the electrons, O2˙−, HO˙ or HO2˙ in H2O2 removal. In sum, we could establish the route for PhOH degradation, portraying the major contributors to its removal and the importance of DO in the system. The simultaneous evolution of H2O2 was clarified by combining the reactions taken into account in this system. Carefully planned simulation methods of experimental data can better comprehend the phenomena occurring during pollutant oxidation that we cannot rationalise experimentally.
Author contributions
IVG: conceptualisation, methodology and formal analysis by Kintecus program, investigation, resources, data curation, writing-original draft, visualization, supervision, project administration, funding acquisition; ATP: conceptualisation, validation, investigation, data curation, writing-original draft, visualization; CGS: resources, writing – reviewing and editing; BO: formal analysis by ERDT, writing – reviewing and editing; AMTS: resources, writing – reviewing and editing; JLF: resources, writing – reviewing and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I. V.-G. acknowledges the financial support of her Marie Sklodowska Curie Action (Project SED 842988) from the European Union Framework Programme for Research and Innovation (Horizon 2020). A. T.-P. acknowledges Fundação para a Ciência e a Tecnologia (FCT) for his scholarship SFRH/BD/143487/2019. A free academic license for the Kintecus version 6.80 package for kinetic modelling is also gratefully acknowledged as well as the courteous and professional technical service of Dr James Ianni, the author of the Kintecus software. This work was partially financed by project NORTE-01-0145-FEDER-031049 (InSpeCt PTDC/EAM-AMB/31049/2017) funded by FEDER funds through NORTE 2020 – Programa Operacional Regional do NORTE – and by national funds through FCT/MCTES (PIDDAC), and by project POCI-01-0145-FEDER-031398 funded by FEDER through COMPETE2020 – Programa Operacional Competitividade e Internacionalização (POCI) – and by national funds through FCT. We would also like to thank the scientific collaboration under Base-UIDB/50020/2020 and Programmatic-UIDP/50020/2020 Funding of LSRE-LCM, funded by national funds through FCT/MCTES (PIDDAC).References
- M. Ismael, J. Alloys Compd., 2020, 846, 156446 CrossRef CAS
.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC
- Y. Yang, X. Li, C. Zhou, W. Xiong, G. Zeng, D. Huang, C. Zhang, W. Wang, B. Song, X. Tang, X. Li and H. Guo, Water Res., 2020, 184, 116200 CrossRef CAS PubMed
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- D. Mittal and D. P. Dutta, J. Mater. Sci.: Mater. Electron., 2021, 32, 18512–18543 CrossRef CAS
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed
- A. Wang, C. Wang, L. Fu, W. Wong-Ng and Y. Lan, Nano-Micro Lett., 2017, 9, 47 CrossRef PubMed
- N. F. F. Moreira, M. J. Sampaio, A. R. Ribeiro, C. G. Silva, J. L. Faria and A. M. T. Silva, Appl. Catal., B, 2019, 248, 184–192 CrossRef CAS
- Y. Cui, J. Huang, X. Fu and X. Wang, Catal. Sci. Technol., 2012, 2, 1396–1402 RSC
- S. Liu, J. Ke, H. Sun, J. Liu, M. O. Tade and S. Wang, Appl. Catal., B, 2017, 204, 358–364 CrossRef CAS
- A. Torres-Pinto, M. J. Sampaio, J. Teixo, C. G. Silva, J. L. Faria and A. M. T. Silva, J. Water Process. Eng., 2020, 37, 101467 CrossRef
- M. A. Barros, M. J. Sampaio, A. R. Ribeiro, C. G. Silva, A. M. T. Silva and J. L. Faria, J. Environ. Chem. Eng., 2021, 9, 105554 CrossRef
- Z. Haider, H.-I. Cho, G.-H. Moon and H.-I. Kim, Catal. Today, 2019, 335, 55–64 CrossRef CAS
- A. Torres-Pinto, M. J. Sampaio, C. G. Silva, J. L. Faria and A. M. T. Silva, Catalysts, 2019, 9, 990 CrossRef CAS
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC
- Y. Cao, G. Zhou, X. Chen, Q. Qiao, C. Zhao, X. Sun, X. Zhong, G. Zhuang, S. Deng, Z. Wei, Z. Yao, L. Huang and J. Wang, J. Mater. Chem. A, 2020, 8, 124–137 RSC
- H. Zhang, L. H. Guo, L. Zhao, B. Wan and Y. Yang, J. Phys. Chem. Lett., 2015, 6, 958–963 CrossRef CAS PubMed
- Q. Zheng, E. Xu, E. Park, H. Chen and D. Shuai, Appl. Catal., B, 2019, 240, 262–269 CrossRef CAS
- V. Hasija, P. Raizada, A. Sudhaik, K. Sharma, A. Kumar, P. Singh, S. B. Jonnalagadda and V. K. Thakur, Appl. Mater. Today, 2019, 15, 494–524 CrossRef
- X. Huo, H. Yi, Y. Fu, Z. An, L. Qin, X. Liu, B. Li, S. Liu, L. Li, M. Zhang, F. Xu, G. Zeng and C. Lai, Environ. Sci.: Nano, 2021, 8, 1835–1862 RSC
- M. J. Lima, A. M. T. Silva, C. G. Silva and J. L. Faria, J. Catal., 2017, 353, 44–53 CrossRef CAS
- H. Xu, J. Yan, X. She, L. Xu, J. Xia, Y. Xu, Y. Song, L. Huang and H. Li, Nanoscale, 2014, 6, 1406–1415 RSC
- H. Zhao, H. Yu, X. Quan, S. Chen, Y. Zhang, H. Zhao and H. Wang, Appl. Catal., B, 2014, 152, 46–50 CrossRef
-
G. A. Somorjai, Active Sites in Heterogeneous Catalysis, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1977, pp. 1–68 Search PubMed
- J. C. Védrine, Appl. Catal., A, 2014, 474, 40–50 CrossRef
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed
- J. Herman, Y. Zhang, V. Castranova and S. L. Neal, Anal. Bioanal. Chem., 2018, 410, 6079–6095 CrossRef CAS PubMed
- P. Fernández-Castro, M. Vallejo, M. F. San Román and I. Ortiz, J. Chem. Technol. Biotechnol., 2015, 90, 796–820 CrossRef
- K.-i. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2000, 104, 4934–4938 CrossRef CAS
- T. Hirakawa and Y. Nosaka, J. Phys. Chem. C, 2008, 112, 15818–15823 CrossRef CAS
- T. Hirakawa, K. Yawata and Y. Nosaka, Appl. Catal., A, 2007, 325, 105–111 CrossRef CAS
- D. Dvoranová, Z. Barbieriková and V. Brezová, Molecules, 2014, 19(8), 12078–12098 CrossRef PubMed
- Z. Wang, W. Ma, C. Chen, H. Ji and J. Zhao, Chem. Eng. J., 2011, 170, 353–362 CrossRef CAS
- Y. Jing and B. P. Chaplin, Environ. Sci. Technol., 2017, 51, 2355–2365 CrossRef CAS PubMed
- J. Zhang and Y. Nosaka, Appl. Catal., B, 2015, 166–167, 32–36 CAS
- C.-Y. Chen and C. T. Jafvert, Environ. Sci. Technol., 2010, 44, 6674–6679 CrossRef CAS PubMed
- O. Micic, Y. Zhang, K. R. Cromack, A. Trifunac and M. Thurnauer, J. Phys. Chem., 1993, 97, 13284–13288 CrossRef CAS
- A. L. Rose and T. D. Waite, Environ. Sci. Technol., 2002, 36, 433–444 CrossRef CAS PubMed
- Z. Wang and J. Liu, Catal. Today, 2014, 224, 244–250 CrossRef CAS
- I. Velo-Gala, M. J. Farré, J. Radjenovic and W. Gernjak, Environ. Sci. Technol. Lett., 2019, 6, 106–111 CrossRef CAS
- Z. Wu, J. Fang, Y. Xiang, C. Shang, X. Li, F. Meng and X. Yang, Water Res., 2016, 104, 272–282 CrossRef CAS PubMed
- Y. Lei, J. Lu, M. Zhu, J. Xie, S. Peng and C. Zhu, Chem. Eng. J., 2020, 379, 122339 CrossRef CAS
- L. Lian, B. Yao, S. Hou, J. Fang, S. Yan and W. Song, Environ. Sci. Technol., 2017, 51, 2954–2962 CrossRef CAS PubMed
-
J. C. Ianni, Kintecus. Windows Version 6.8, 2019 Search PubMed
- G. Mamba and A. K. Mishra, Appl. Catal., B, 2016, 198, 347–377 CrossRef CAS
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS
- Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6, 7021–7029 CrossRef CAS
- S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26–35 CrossRef CAS
- Y. Wang, D. Meng and X. Zhao, Appl. Catal., B, 2020, 273, 119064 CrossRef CAS
- Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2015, 5, 3058–3066 CrossRef CAS
-
P. Chowdhury, S. Nag and A. K. Ray, Degradation of Phenolic Compounds Through UV and Visible-Light-Driven Photocatalysis: Technical and Economic Aspects, Phenolic Compounds - Natural Sources, Importance and Applications, IntechOpen Limited, London, UK, 2016 Search PubMed
- C.-H. Chiou, C.-Y. Wu and R.-S. Juang, Chem. Eng. J., 2008, 139, 322–329 CrossRef CAS
- P. Neta and J. Grodkowski, J. Phys. Chem. Ref. Data, 2005, 34, 109–199 CrossRef CAS
- E. Mvula, M. N. Schuchmann and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 2001, 264–268 RSC
- A. Torres-Pinto, M. J. Sampaio, C. G. Silva, J. L. Faria and A. M. T. Silva, Appl. Catal., B, 2019, 252, 128–137 CrossRef CAS
- J. C. Lopes, M. J. Sampaio, R. A. Fernandes, M. J. Lima, J. L. Faria and C. G. Silva, Catal. Today, 2019, 357, 32–38 CrossRef
- A. Nitta, M. Takase, M. Takashima, N. Murakami and B. Ohtani, Chem. Commun., 2016, 52, 12096–12099 RSC
- A. Nitta, M. Takashima, M. Takase and B. Ohtani, Catal. Today, 2019, 321-322, 2–8 CrossRef CAS
- P. Unwiset, G. Chen, B. Ohtani and K. C. Chanapattharapol, Catalysts, 2019, 9(12), 1010 CrossRef CAS
- C. Chuaicham, S. Karthikeyan, R. R. Pawar, Y. Xiong, I. Dabo, B. Ohtani, Y. Kim, J. T. Song, T. Ishihara and K. Sasaki, Chem. Commun., 2020, 56, 3793–3796 RSC
- G. Eisenberg, Ind. Eng. Chem., Anal. Ed., 1943, 15, 327–328 CrossRef CAS
- A.
N. Pham, G. Xing, C. J. Miller and T. D. Waite, J. Catal., 2013, 301, 54–64 CrossRef CAS
- T. Liu, X. Li, X. Yuan, Y. Wang and F. Li, J. Mol. Catal. A: Chem., 2016, 414, 122–129 CrossRef CAS
- H. Zhang, L.-H. Guo, L. Zhao, B. Wan and Y. Yang, J. Phys. Chem. Lett., 2015, 6, 958–963 CrossRef CAS PubMed
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed
- L. Svoboda, P. Praus, M. J. Lima, M. J. Sampaio, D. Matýsek, M. Ritz, R. Dvorský, J. L. Faria and C. G. Silva, Mater. Res. Bull., 2018, 100, 322–332 CrossRef CAS
- B. Balagam and D. E. Richardson, Inorg. Chem., 2008, 47, 1173–1178 CrossRef CAS PubMed
- K. P. Mangalgiri, S. Patton, L. Wu, S. Xu, K. P. Ishida and H. Liu, Environ. Sci. Technol., 2019, 53, 13323–13331 CrossRef CAS PubMed
- J. M. Barazesh, C. Prasse, J. Wenk, S. Berg, C. K. Remucal and D. L. Sedlak, Environ. Sci. Technol., 2018, 52, 195–204 CrossRef CAS PubMed
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed
- S.-Y. Hsu, C.-L. Lee, C.-H. Kuo and W.-C. Kuo, Sens. Actuators, B, 2021, 328, 129015 CrossRef CAS
- C. M. Sánchez-Sánchez and A. J. Bard, Anal. Chem., 2009, 81, 8094–8100 CrossRef PubMed
- I. Sirés, E. Brillas, M. A. Oturan, M. A. Rodrigo and M. Panizza, Environ. Sci. Pollut. Res., 2014, 21, 8336–8367 CrossRef PubMed
- D. Fattakhova-Rohlfing, A. Zaleska and T. Bein, Chem. Rev., 2014, 114, 9487–9558 CrossRef CAS PubMed
- Q. Li and F.-T. Li, Chem. Eng. J., 2021, 421, 129915 CrossRef CAS
- J. Diaz-Angulo, J. Porras, M. Mueses, R. A. Torres-Palma, A. Hernandez-Ramirez and F. Machuca-Martinez, J. Photochem. Photobiol., A, 2019, 383, 112015 CrossRef CAS
- S. Zhao and X. Zhao, Appl. Catal., B, 2019, 250, 408–418 CrossRef CAS
- Y.-H. Chen, B.-K. Wang and W.-C. Hou, Chemosphere, 2021, 278, 130334 CrossRef CAS PubMed
- R. Shanker Sahu, A. Dubey and Y.-H. Shih, Carbon, 2021, 182, 89–99 CrossRef CAS
- T. Luo, X. Hu, Z. She, J. Wei, X. Feng and F. Chang, J. Mol. Liq., 2021, 324, 114772 CrossRef CAS
- Y. Yang and Z. Bian, Sci. Total Environ., 2021, 753, 141908 CrossRef CAS PubMed
- H. Klingshirn, B. Faltermeier, W. Hengl and M. Maier, Chem. Phys. Lett., 1982, 93, 485–489 CrossRef CAS
- R. Protz and M. Maier, J. Chem. Phys., 1980, 73, 5464–5467 CrossRef CAS
- F. E. Scully and J. Hoigné, Chemosphere, 1987, 16, 681–694 CrossRef CAS
- D. Slawinska, Photochem. Photobiol., 1978, 28, 453–458 CrossRef CAS
- P. G. Tratnyek and J. Hoigne, Environ. Sci. Technol., 1991, 25, 1596–1604 CrossRef CAS
- H. J. Guiraud and C. S. Foote, J. Am. Chem. Soc., 1976, 98, 1984–1986 CrossRef CAS
- N. Serpone, J. Photochem. Photobiol., A, 1997, 104, 1–12 CrossRef CAS
- T. Velegraki, E. Hapeshi, D. Fatta-Kassinos and I. Poulios, Appl. Catal., B, 2015, 178, 2–11 CrossRef CAS
- Y. J. O. Asencios, V. S. Lourenço and W. A. Carvalho, Catal. Today, 2020 Search PubMed
- Z. Li and X. Meng, J. Hazard. Mater., 2020, 399, 122939 CrossRef CAS PubMed
- Y. Cherifi, A. Barras, A. Addad, B. Ouddane, P. Roussel, A. Chaouchi, S. Szunerits and R. Boukherroub, Chemosphere, 2021, 268, 128798 CrossRef CAS PubMed
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS
- M. Zhang, Y. Yang, X. An and L.-A. Hou, Chem. Eng. J., 2021, 412, 128663 CrossRef CAS
- Y. Shiraishi, S. Kanazawa, D. Tsukamoto, A. Shiro, Y. Sugano and T. Hirai, ACS Catal., 2013, 3, 2222–2227 CrossRef CAS
- J. J. Guerard, P. R. Tentscher, M. Seijo and J. Samuel Arey, Phys. Chem. Chem. Phys., 2015, 17, 14811–14826 RSC
- Q. Wu, J. Cao, X. Wang, Y. Liu, Y. Zhao, H. Wang, Y. Liu, H. Huang, F. Liao, M. Shao and Z. Kang, Nat. Commun., 2021, 12, 483 CrossRef CAS PubMed
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS
- Y. Xie, Y. Li, Z. Huang, J. Zhang, X. Jia, X.-S. Wang and J. Ye, Appl. Catal., B, 2020, 265, 118581 CrossRef CAS
- G.-H. Moon, M. Fujitsuka, S. Kim, T. Majima, X. Wang and W. Choi, ACS Catal., 2017, 7, 2886–2895 CrossRef CAS
- H. Hirakawa, S. Shiota, Y. Shiraishi, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6, 4976–4982 CrossRef CAS
- L. Wang, Q. Zhang, B. Chen, Y. Bu, Y. Chen, J. Ma, F. L. Rosario-Ortiz and R. Zhu, Water Res., 2020, 174, 115605 CrossRef CAS PubMed
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS
- Y. Zhou, G. Chen and J. Zhang, J. Mater. Chem. A, 2020, 8, 20849–20869 RSC
- J. Zheng, S. R. Springston and J. Weinstein-Lloyd, Anal. Chem., 2003, 75, 4696–4700 CrossRef CAS PubMed
- T. Ma, S. Garg, C. J. Miller and T. D. Waite, J. Colloid Interface Sci., 2015, 446, 366–372 CrossRef PubMed
- B. Mortazavi, F. Shojaei, M. Shahrokhi, M. Azizi, T. Rabczuk, A. V. Shapeev and X. Zhuang, Carbon, 2020, 167, 40–50 CrossRef CAS
- B. H. J. Bielski, Photochem. Photobiol., 1978, 28, 645–649 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01657a |
| This journal is © The Royal Society of Chemistry 2021 |