Lanthanum modified Fe-ZSM-5 zeolites for selective methane oxidation with H2O2
Received
10th September 2021
, Accepted 10th November 2021
First published on 10th November 2021
Abstract
Selective partial oxidation of methane to methanol under ambient conditions is a great challenge in chemistry. Iron modified ZSM-5 catalysts are shown to be effective for this reaction using H2O2 as the oxidant. However, the high consumption of H2O2 over this catalyst presents a major disadvantage. Here we report a lanthanum modified Fe-ZSM-5 (LaFe-ZSM-5) catalyst for enhanced selective methane oxidation with suppressed H2O2 consumption. Using 0.5 wt% LaFe-ZSM-5 pretreated with H2 the productivity of primary oxygenated products (CH3OH, CH3OOH, HCOOH) is 3200 mol kgLaFe−1 h−1 in 0.1 M H2O2, with a selectivity of 98.9% to primary oxygenated products. The productivity is increased to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 460 mol kgLaFe−1 h−1 in 0.5 M H2O2. Compared with Fe-ZSM-5, LaFe-ZSM-5 uses 31% less H2O2 for obtaining per mol of product under the same conditions. In situ DRIFT spectroscopy and solid state MAS NMR revealed the high H2O2 consumption in ZSM-5 based catalyst maybe closely related to the acidity of strong Brønsted acid sites (Si(OH)Al). The La modified ZSM-5 catalyst can decrease the acidity of the strong Brønsted acid sites and this suppresses the decomposition of H2O2.
460 mol kgLaFe−1 h−1 in 0.5 M H2O2. Compared with Fe-ZSM-5, LaFe-ZSM-5 uses 31% less H2O2 for obtaining per mol of product under the same conditions. In situ DRIFT spectroscopy and solid state MAS NMR revealed the high H2O2 consumption in ZSM-5 based catalyst maybe closely related to the acidity of strong Brønsted acid sites (Si(OH)Al). The La modified ZSM-5 catalyst can decrease the acidity of the strong Brønsted acid sites and this suppresses the decomposition of H2O2.
Introduction
As the principal component of natural gas, methane is a highly abundant source of hydrocarbons. An efficient conversion method of methane to liquid methanol as well as other oxygenates has captured wide interest. The current industrial route for methane conversion is indirect via the formation of synthesis gas (a mixture of CO and H2) at high temperatures and pressures. This process, however, is expensive and energy-intensive.1 The direct oxidation of methane into methanol and other oxygenates would be more attractive but remains one of the greatest challenges in catalysis today.2–4 The high C–H bond strength (104 kcal mol−1), low polarizability and negligible electron affinity of methane engenders not only extreme difficulties in the activation step, but also in avoiding over oxidation of primary oxygenates such as methanol and formaldehyde to carbon dioxide under harsh conditions necessary.5,6 Such prospects and challenges of direct methane oxidation has attracted intensive study in recent years.7–9
Cyclic gas phase oxidation of methane with Cu- or Fe metal-exchanged zeolite catalysts with O2, N2O or H2O might inhibit over-oxidation, but requires high temperatures (200 to 500 °C) to activate the oxidant and desorb the methanol product.10–13 Compared with gas-phase direct methane oxidation, liquid-phase reactions typically proceed under milder reaction conditions with less energy consumption. It has been reported electrophilic metals (such as Hg, Pd, Pt and Au complexes) can be used for C–H bond activation and achieve methane oxidation to methyl-bisulfate in the presence of strong oxidizing agents (acidic oleum and selenic acid) at 180–220 °C.14–17 However, the subsequent hydrolysis to methanol results in the formation of SO2 and it is difficult to separate methanol from the strong aqueous acidic reaction media. For the past few years, Hutchings and co-workers have reported the use of an environmentally benign oxidant H2O2 for efficiently oxidizing methane to methanol under low reaction temperatures (50 °C) in H2O:H2O2 system.3,18–22 In a series of landmark studies, gold–palladium and iron–copper modified ZSM-5 zeolites have shown the most effective activity for the low temperature oxidation of methane to methanol with aqueous H2O2.3,18 To our knowledge the highest productivity over supported Au–Pd for methane oxidation to methanol is 520 mol kgAuPd−1 h−1 over 0.13 wt% AuPd/rutile TiO2 catalyst with a primary oxygenate (CH3OH, CH3OOH, HCOOH) selectivity of 90.7% at 50 °C in 0.5 M H2O2.22 The high productivity rate was ascribed to the reduced decomposition rate of H2O2, which resulted in better propagation of the radical reaction. Using similar conditions (50 °C, 30.5 bar CH4), the productivity of 2.5 wt% Fe–Cu modified ZSM-5 for methane oxidation to methanol is 14 mol kgcat−1 h−1 in 0.5 M aqueous H2O2, corresponding to 560 mol kgFeCu−1 h−1.18 Although the iron modified ZSM-5 catalyst exhibited high activity for methane partial oxidation in aqueous H2O2, the ratio of H2O2 consumed/total product is very high. For example, this value was reported as high as 5.8 over a ZSM-5 catalyst containing 0.014 wt% Fe as an impurity.18 The relatively high cost of H2O2 used in this reaction makes it difficult to envisage an economically viable process. For both suppressing H2O2 consumption and further improving methane oxidation activity, refinement of catalyst design and in-depth understanding of methane oxidation mechanism are needed.
All selective methane oxidation pathways start with C–H bond activation, which is often the rate-limiting step. There are mainly two principle C–H activation mechanisms that have been reported for iron modified ZSM-5: (1) heterolytic and (2) homolytic reaction mechanisms.23,24 In the heterolytic mechanism, the C–H bond is activated by Lewis acid–base pairs to form an anionic alkyl group coordinated to the acid site (metal cation) and a proton coordinated to the basic part (lattice oxygen of oxide).25,26 It is well-recognized that the basicity of the heterolytic active sites plays an essential role, which allows adsorption of the weakly acidic methane.27–29 In the homolytic activation mechanism, the C–H bond is activated by charge transfer between methane and catalyst surface to form the alkyl radical and form a H radical.25,26,30 For the iron-based catalyst, it has been reported that heterolytic C–H dissociation is commonly observed over low-valence Fe sites (e.g., 2+) conjugated with a strong basic site, while the homolytic C–H cleavage occurs on high-valence Fe sites.25 Besides these two generic reaction mechanisms, a Fenton-type mechanism of C–H activation has also been reported.18,31–33 In Fenton-type reactions, free ˙OH radicals are produced from H2O2 decomposition in the presence of iron species, which initiate the radical activation of methane.25 Our previous studies showed that Fenton-type reactions only provide a small contribution to the overall methane oxidation performance with the iron modified ZSM-5 catalyst.18 Furthermore, excess ˙OH radicals in the Fenton-type reaction not only induce a rapid decomposition of H2O2 by chain reaction, but also bring about the over oxidation of the methanol product. Based on these studies, it appears that suppressing Fenton-type reactions and developing a homolytic or heterolytic activation mode may be an effective approach to both suppress H2O2 consumption and improve methane oxidation performance. In addition, it is important to determine which activation mechanism is more efficient for methane oxidation, and to date this has not been fully understood. Using iron modified zeolites as an example, the various iron clusters present may have different locations both in the framework and extra-framework of the zeolitic matrix. These can provide completely different C–H activation pathways for methane oxidation.
Herein, we choose the most potentially effective catalyst, iron-modified ZSM-5 (Fe-ZSM-5), as a model to investigate how different iron species influence the C–H activation type and what type is more efficient for methane oxidation. To study this, first a uniform Fe-ZSM-5 precursor was prepared. This precursor was then heat treated in air or an H2/Ar atmosphere separately to control the valence state of the iron species. Further studies then focus on the structure–activity relationship with H2O2 as the oxidant for methane oxidation, including the effect from the microstructure, oxidation state of the iron species and the surface acid–base sites. Based on these studies, lanthanum-modified Fe-ZSM-5 catalyst were then explored to both improve methane oxidation activity and suppress the H2O2 consumption.
Experimental
Preparation
La-, Fe-, FeCu- and LaFeCu-ZSM-5 catalysts were prepared by the following method. ZSM-5 (0.5 g, Zeolyst and used as received) was stirred in a solution of the metal nitrate, ethanol (8 mL), acetonitrile (4 mL), water (2 mL). An example procedure for 0.5 wt% La-ZSM-5 (H2) catalyst is exemplified below.
Commercial ZSM-5 (Si/Al = 30) (0.5 g, Zeolyst) was stirred in a solution containing La(NO3)3·6H2O (200 μL aliquot from a 0.09 M solution of the nitrate, Aldrich) dissolved in ethanol (8 mL, Fisher), acetonitrile (4 mL, Fisher) and water (2 mL, HPLC grade, Aldrich). The suspension was stirred (12 h, 25 °C) until all the solvent had evaporated. Typically, the powder was then heated in 5% H2/Ar (550 °C, 3 h, BOC), where stated other catalysts were prepared in a similar manner and air was substituted for H2 for this heat treatment step.
Oxidation of methane using H2O2
The oxidation of methane was carried out using a 50 mL Parr stainless steel autoclave reactor with a removable Teflon liner (35 mL volume). Typically, reactions were carried out using a 10 mL reaction mixture comprising an aqueous solution of H2O2 (10 mL, 0.1 M, 1000 μmol) and the catalyst (27 mg). Prior to use, the reactor was purged with N2 (5 bar) and methane successively for three times to remove residual air. The pressure was adjusted to 30.5 bar with methane before reaction. The autoclave was then heated to the desired reaction temperature (50 °C) using the preset program. During the reaction, the solution was vigorously stirred at 1200 rpm for 30 min. After that, the stirring was stopped, and the temperature was reduced to 4 °C using an ice bath to minimize the loss of volatile products. Gaseous samples were removed by extraction into a gas bag for analysis via gas chromatography. The Varian-GC was equipped with a CPSIL5CB column (50 m, 0.33 mm internal diameter) fitted with a methaniser and analyzed by a flame ionization detector (FID). The solution reaction mixture was filtered and analyzed by 1H NMR, using a Bruker 500 MHz Ultrashield NMR spectrometer. All 1H NMR samples were analyzed against a calibrated insert containing tetramethylsilane (TMS) in deuterated chloroform (99.9% D). The remaining H2O2 was determined by titration with acidified Ce(SO4)2.
Catalyst characterization
The transmission electron microscope (TEM) and scanning transmission electron microscope (STEM) analyses were performed by a Hitachi HF5000 spherical aberration corrected transmission electron microscope. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Axis Ultra DLD spectrometer. Samples were mounted using double-sided adhesive tape and binding energies were referenced to the C 1s binding energy of adventitious carbon contamination, which taken to be 284.9 eV. Monochromatic AlKα radiation was used for all measurements; an analyser pass energy of 160 eV was used for survey scans while 40 eV was employed for detailed regional scans. UV-vis absorption spectrum of the sample was measured using an Agilent Cary 60 UV-vis spectrophotometer. The in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and pyridine Fourier transform infrared spectroscopy (Py-FTIRS) were respectively recorded on a powder sample with Bruker Tensor 27 spectrometer fitted with a HgCdTe (MCT) detector and a Harrick Praying Mantis HVC-DRP-4 cell equipped with KBr windows. In the Py-FTIR experiments, the sample were activated under vacuum and a N2 flow (10 mL min−1) at 400 °C for 2 h and then cooled to room temperature. Subsequently, N2 flow saturated with pyridine was introduced into the FTIR cell at 25 °C for 1 h to ensure that all acid sites were covered. Before measuring the Py adsorption spectra at 25 °C, the samples were purged by 10 mL min−1 N2 flow and evacuated at 25 °C for 1 h. After that, the samples were respectively heated to 200 °C and 350 °C with the rate of 4 °C min−1, and Py FTIR adsorption spectra were measured at these temperatures. Before collecting the spectra, the sample was purged by 10 mL min−1 N2 flow and evacuated at the temperature for 1 h to remove the desorbed pyridine. 27Al solid-state NMR experiments were carried out at 7.05 T on a Varian Infinity-plus 300 spectrometer. The resonance frequency was 78.11 MHz for 27Al. A 4 mm double resonance probe was employed to acquire 27NMR spectra. The 27Al MAS spectra were acquired using a one-pulse sequence with a short radio frequency (rf) pulse of 0.25 μs and a pulse delay of 0.8 s. The magic angle spinning rate was set to 10 kHz. The chemical shift was referenced to a solution of 1 M Al(NO3)3. Inductively coupled plasma optical emission spectrometry (ICP-OES) analyses were performed by Leeman Labs Prodigy7.
Results and discussions
Commercial ZSM-5 zeolites with Si/Al molar ratio of 30 was chosen as the catalyst matrix for this study. Elemental analysis by ICP of the commercial ZSM-5 showed it contained trace amounts of Fe (0.01 wt%). The commercial ZSM-5 was activated by treatment at 550 °C for 3 h in air or 5% H2/Ar prior to use, named as H-ZSM-5 (air) and H-ZSM-5 (H2) sample, respectively. Before the investigation of the different C–H activation mechanism in Fe-ZSM-5, the catalytic mechanism of H-ZSM-5 (air) and H-ZSM-5 (H2) were studied in advance. As shown in Table 1 (entry 1, 2), both of the air and H2 activated H-ZSM-5 samples exhibit catalytic activity for selective methane oxidation to partially oxygenated products (CH3OH, CH3OOH, HCOOH). However, the H-ZSM-5 (H2) sample exhibited a higher methane oxidation performance. In particular, the H2O2 consumed/partially oxygenated products ratio is sharply decreased from 21.32 to 3.12 over the H-ZSM-5 (H2) sample. To illustrate why H2 calcination can suppress H2O2 consumption and increase methane oxidation performance over the H-ZSM-5 catalyst, the active sites in H-ZSM-5 catalyst was considered.
Table 1 Catalytic studies of La, Fe, Cu containing zeolites for the oxidation of methane under mild conditionsa
| Entry |
Catalyst |
Amount of product (μmol) |
Primary oxygenate selectivity (%) |
Oxygenate productivity (mol kgcatalyst−1 per hour) |
H2O2 consumed/product |
| MeOH |
MeOOH |
FDb |
FAc |
CO2 |
|
Standard reaction conditions: 10 mL reaction media, 30 min, 50 °C, P(CH4) 30.5 bar, [H2O2] 0.1 M (1000 μmol) or 0.5 M (entry 7) (5000 μmol), catalyst 27 mg, 1200 rpm. Typical catalyst pre-treatment: 550 °C, 3 h, static air or 5% H2/Ar flow.
FD = formaldehyde.
FA = formic acid.
|
| 1 |
H-ZSM-5 (air) |
2.8 |
3.6 |
2.4 |
1.6 |
0 |
100 |
0.8 |
21.3 |
| 2 |
H-ZSM-5 (H2) |
1.1 |
3.0 |
4.2 |
4.6 |
0.25 |
98.1 |
1.0 |
3.1 |
| 3 |
1 wt% La-ZSM-5 (H2) |
1.3 |
2.7 |
3.1 |
5.7 |
0 |
100 |
0.9 |
0 |
| 4 |
0.5 wt% Fe-ZSM-5 (air) |
14.7 |
1.5 |
12.2 |
98.7 |
14.3 |
90 |
9.4 |
3.7 |
| 5 |
0.5 wt% Fe-ZSM-5 (H2) |
33.8 |
0 |
0 |
117 |
16.9 |
90 |
11.1 |
4.5 |
| 6 |
0.5 wt% LaFe-ZSM-5 (H2) |
33.2 |
0 |
0.35 |
182.7 |
2.5 |
98.9 |
16 |
3.1 |
| 7 |
0.5 wt% LaFe-ZSM-5 (H2)a |
45.6 |
2.8 |
0 |
725.8 |
29.7 |
96.3 |
57.3 |
4.3 |
| 8 |
0.5 wt% LaFeCu-ZSM-5 (H2) |
87.6 |
0.8 |
5.6 |
73.3 |
2.9 |
98.3 |
12.4 |
4.1 |
| 9 |
0.5 wt% FeCu-ZSM-5 (H2) |
79.1 |
0.4 |
4.3 |
73.2 |
3.7 |
97.7 |
11.5 |
4.8 |
| 10 |
0.5 wt% LaFeCu-ZSM-5 (air) |
39.3 |
1.9 |
9 |
11.3 |
0 |
100 |
4.5 |
2.6 |
| 11 |
0.5 wt% FeCu-ZSM-5 (air) |
39.9 |
1.4 |
3.9 |
19.2 |
1.5 |
97.8 |
4.8 |
4.0 |
It has been reported that diiron–oxo complexes which have a binuclear Fe cluster coordinated by framework oxygen atoms and extra-framework μ-oxo are the active sites for methane oxidation over iron modified zeolite.18,25,34–37 Our previous studies found thermal pre-treatment can extract Fe from the zeolite framework to extra-framework positions. An increased concentration of extra-framework Fe–oxo complexes is accompanied by significant increases in catalytic activity.18 Compared with H-ZSM-5 (air) sample, the increased catalytic activity of H-ZSM-5 (H2) sample (Table 1, entry 1 and 2) may be ascribed to the higher concentration of extra-framework Fe–oxo complexes, which will be further discussed in the following section upon UV-vis absorption spectrum. The markedly different H2O2 consumption between the H-ZSM-5 (air) and the H-ZSM-5 (H2) samples may originate from the different C–H activation mechanism over the different Fe species in the H-ZSM-5 (H2) and the H-ZSM-5 (air) samples. The H-ZSM-5 (air) sample is prepared in an oxygen-enriched environment. This condition gives rise to the formation of trivalent Fe(III)–Fe(III)–oxo active sites which can be oxidized into Fe(IV)–Fe(IV)–oxo species by H2O2,18 and then facilitate homolytic C–H cleavage.25 Based on the above analysis, the possible reaction pathway for methane oxidation over H-ZSM-5 (air) sample by homolytic C–H cleavage mechanism are summarized in reactions ((1)–(4)).
| | | Fe(III)–Fe(III)–oxo + H2O2 → Fe(IV)–Fe(IV)–oxo + H2O | (1) |
| | | Fe(IV)–Fe(IV)–oxo + CH4 → Fe(IV)–Fe(IV)–oxo + ˙CH3 + ˙H → Fe(IV)–Fe(III)–oxo(H) + ˙CH3 → Fe(III)–Fe(III)–oxo + CH3OH | (2) |
| | | Fe(IV)–Fe(IV)–oxo + H2O2 → Fe(IV)–Fe(III)–oxo(H) + ˙OOH | (3) |
At first, the Fe(III)–Fe(III)–oxo species are oxidized by H2O2 into Fe(IV)–Fe(IV)–oxo (reaction (1)). The high valence Fe(IV)–oxo can active C–H bond by homolytic cleavage to generate ˙CH3 and ˙H radicals (reaction (2)).25 These radicals are transient species, which can supply electrons to Fe(IV)–oxo and then form CH3OH as well as hydroxyl groups on the surface, respectively (reaction (2)). In this reaction approach, the oxygen atom in CH3OH comes from the extra-framework μ-oxo. From Table 1, CH3OOH is the primary oxygenated product over H-ZSM-5 (air) sample. Fe(IV)–oxo species have been reported are the active species for both homolytic dissociation of methane (reaction (2)) and oxidation of H2O2 (reaction (3)).25 The CH3OOH species are produced by the reaction of ˙CH3 with ˙OOH which is produced from H2O2 decomposition (reaction (4)). However, the H2O2 decomposition rate is much higher than the C–H activation rate over the H-ZSM-5 (air) sample where the ratio for H2O2 consumed/partially oxygenated products is over 21.3. In H-ZSM-5 (H2) sample, this ratio decreased to 3.1, indicating a suppressed H2O2 consumption. The H-ZSM-5 (H2) sample is calcined in H2/Ar gas, giving rise to the formation of some low valence Fe(II)–oxo species besides the Fe(III)–oxo species. These low valence Fe active sites appear to be in favour of heterolytic C–H activation for methane oxidation by H2O2,25 which may suppress the undesirable H2O2 consumption over the H-ZSM-5 (H2) sample. The possible reaction pathways for methane oxidation over H-ZSM-5 (H2) sample are summarized in reactions ((5)–(8)).
| |  | (5) |
| |  | (6) |
| | | Fe(II)–oxo(H) + H2O2 → Fe(III)–oxo(H) + ˙OH + OH− | (7) |
| | | Fe(III)–oxo(H) + H2O2 → Fe(II)–oxo(H) + ˙OOH + H+ | (8) |
In the heterolytic C–H activation mode (as shown in reactions (5) and (6)), the C–H bond is activated over a Fe(II)–oxo acid–base pair to form anionic alkyl group stabilized by the Lewis acid (Fe2+) and a proton is accepted by the base part of the active site (lattice oxygen). Fenton-type decomposition of H2O2 by adjacent Fe(II) or Fe(III) species give rise to ˙OH and ˙OOH (reactions (7) and (8)) radicals which attack the Fe(II)–oxo activated Hδ+–CH3δ−, generating CH3OH and CH3OOH products, respectively. For binuclear Fe(II)–Fe(II)–oxo and Fe(II)–Fe(III)–oxo active sites, the simultaneous H2O2 decomposition and C–H bond activation by adjacent Fe centers decreased the undesirable H2O2 consumption for methane oxidation. However, the binuclear active sites also facilitate the over oxidation of methanol to formic acid, once excess ˙OH generated on the adjacent Fe(II) center.18
To investigate whether the H2O2 consumption can be further suppressed in the H2/Ar heated samples, our study found loading lanthanum (La) can further decrease the H2O2 consumption to negligible levels (Table 1, entry 3). After four successive methane oxidation reaction over 1 wt% La-ZSM-5 (Table 2, entry 1–4), only 6% of the added H2O2 (60 μmol) was decomposed to generate a total amount of 41.5 μmol primary oxygenated products (CH3OH, CH3OOH, HCHO, HCOOH). In this reaction, the average ratio for H2O2 consumed/partially oxygenated products in all of the four successive reaction cycles is only 1.45. It is worth noting that no H2O2 degradation is observed in the first and second reaction cycle over the La-ZSM-5 (H2) sample. The obvious decrease of H2O2 concentration is observed after the third cycle, which accounts for about 3% of the total amount (0.1 M in 10 mL) to generate 31.2 μmol oxygenated products (Table 2, entry 3). Without the La-ZSM-5 catalyst, approximately 10.8% of 0.1 M H2O2 was decomposed during methane oxidation under the same conditions (Table 2, entry 6). These studies revealed that the La-ZSM-5 catalyst can stabilize and inhibit the decomposition of H2O2 during methane oxidation.
Table 2 Cyclic catalytic studies of 1 wt% La-ZSM (H2) sample for the oxidation of methane under mild conditionsa
| Entry |
Catalyst |
Gas |
H2O2 amount (μmol) |
Amount of product (μmol) |
Primary oxygenate selectivity (%) |
Oxygenate product amount (μmol) |
H2O2 consumed (%) |
| MeOH |
MeOOH |
FDb |
FAc |
CO2 |
|
Standard reaction conditions: 10 mL, 30 min, 50 °C, P(CH4) 30.5 bar, [H2O2] 0.1 M (1000 μmol), catalyst 27 mg, 1200 rpm. Typical catalyst pre-treatment: calcination (550 °C, 3 h, 5% H2/Ar flow).
FD = formaldehyde.
FA = formic acid.
|
| 1 |
La-ZSM-5 |
CH4 |
1000 |
1.3 |
2.7 |
3.1 |
5.7 |
0 |
100 |
12.8 |
0 |
| 2 |
La-ZSM-5 |
CH4 |
Residual from entry 1 |
1.9 |
2.2 |
3.0 |
15.2 |
0 |
100 |
22.3 |
0 |
| 3 |
La-ZSM-5 |
CH4 |
Residual from entry 2 |
2.5 |
1.9 |
2.6 |
24.2 |
0 |
100 |
31.2 |
3 |
| 4 |
La-ZSM-5 |
CH4 |
Residual from entry 3 |
3.1 |
1.8 |
2.2 |
34.4 |
0 |
100 |
41.5 |
6.1 |
| 5 |
La-ZSM-5 |
N2 |
1000 |
0 |
0 |
0 |
0 |
0 |
— |
— |
0 |
| 6 |
None |
CH4 |
1000 |
0.4 |
0.2 |
0 |
0 |
0 |
100 |
0.6 |
10.8 |
| 7 |
La-ZSM-5 |
CH4 |
0 |
Trace |
0 |
0 |
0 |
0 |
100 |
— |
— |
A comparative study on La-ZSM-5 sample showed that the concentration of La has a small effect on the methane oxidation performance (Table 3). The activities of H-ZSM-5 (H2), 0.5 wt% La-ZSM-5, 1 wt% La-ZSM-5 and 2 wt% La-ZSM-5 are very similar, indicating La could not provide the active site for methane activation. However, over the 0.5 wt% La and 0.5 wt% Fe co-modified ZSM-5 sample (0.5 wt% LaFe-ZSM-5), it was found loading La can both suppress the H2O2 consumption and increase methane oxidation performance over the H2/Ar treated LaFe-ZSM-5 (Table 1, entry 6). The productivity of primary oxygenated products from methane oxidation (30.5 bar CH4, 50 °C) over the 0.5 wt% LaFe-ZSM-5 (H2) sample is up to 16 mol kgcatalyst−1 h−1 in 0.1 M H2O2, corresponding to 3200 mol kgLaFe−1 h−1. Under the same conditions, the productivity over 0.5 wt% Fe-ZSM-5 (H2) is 11.1 mol kgcatalyst−1 h−1, corresponding to 2220 mol kgLaFe−1 h−1. The ratio of H2O2 consumed/total product in 0.5 wt% LaFe-ZSM-5 (H2) is also decreased to 3.1, as is that using 0.5 wt% Fe-ZSM-5 (H2) is 4.5. Compared with Fe-ZSM-5, LaFe-ZSM-5 uses 31% less H2O2 for obtaining per mol of product under the same conditions. The productivity of primary oxygenated products over the 0.5 wt% LaFe-ZSM-5 (H2) sample is increased to 57.3 mol kgcatalyst−1 h−1 in 0.5 M H2O2, corresponding to 11460 mol kgLaFe−1 h−1. This productivity is not only higher than the previously reported FeCu-ZSM-5 catalyst (560 mol kgFeCu−1 h−1),18 but also superior to the widely studied AuPd catalysts which exhibited the highest productivity of 520 mol kgAuPd−1 h−1 over 0.13 wt% AuPd/rutile TiO2 catalyst in 0.5 M H2O2.22 The low AuPd loading of 0.13 wt% was a contributing factor in the enhanced rate due to the low H2O2 decomposition, facilitating oxygenate production albeit at a low oxygenate yield.
Table 3 Catalytic studies of different La-ZSM (H2) samples for the oxidation of methane under mild conditionsa
| Entry |
Catalyst (La-ZSM-5) |
Amount of product (μmol) |
Primary oxygenate selectivity (%) |
Oxygenate product amount (μmol) |
H2O2 consumed |
| MeOH |
MeOOH |
FDb |
FAc |
CO2 |
|
Standard reaction conditions: 10 mL, 30 min, 50 °C, P(CH4) 30.5 bar, [H2O2] 0.1 M (1000 μmol), catalyst 27 mg, 1200 rpm. Typical catalyst pre-treatment: calcination (550 °C, 3 h, 5% H2/Ar flow).
FD = formaldehyde.
FA = formic acid.
|
| 1 |
0.5 wt% |
1.2 |
2.5 |
3 |
5.1 |
0 |
100 |
11.8 |
0 |
| 2 |
1 wt% |
1.3 |
2.7 |
3.1 |
5.7 |
0 |
100 |
12.8 |
0 |
| 3 |
2 wt% |
1.7 |
1.9 |
2.4 |
4.6 |
0 |
100 |
10.6 |
0 |
The effect of La for suppressing H2O2 consumption is also observed when comparing the methane oxidation performance of 0.5 wt% LaFeCu-ZSM-5 (H2), 0.5 wt% FeCu-ZSM-5 (H2), 0.5 wt% LaFeCu-ZSM-5 (air) and 0.5 wt% FeCu-ZSM-5 (air) samples. In the H2/Ar heated FeCu-ZSM-5 samples, La loading reduces H2O2 consumption by 14.5%, leading to a H2O2 to product ratio of 4.1 (Table 1, entry 8, 9). Over the air calcined samples, La loading reduces H2O2 consumption by 36% (Table 1, entry 10, 11), but it is accompanied by a decreased productivity of primary oxygenated products because of the lower concentration of extra-framework Fe–oxo complexes (supported by a series of UV-vis spectrum in the following section) which are active species for methane oxidation.
To further investigate why loading La can suppress H2O2 consumption and why the catalyst heated in H2/Ar exhibited higher activity, a series of characterization techniques were used to reveal the state of the catalysts. In situ pyridine-FTIR spectroscopy was employed to study the acidic properties of H-ZSM-5 and the metal loaded samples. After pyridine adsorption on H-ZSM-5 (air), H-SM-5 (H2) and La-ZSM-5 (H2) at 25 °C (Fig. 1a), absorption bands were observed in the IR spectrum at 1442, 1490 and 1550 cm−1. The IR band at 1442 cm−1 was attributed to the adsorption of pyridine coordinated on metal ions (Lewis (L) acid sites, such as Al3+).38,39 IR absorption bands around 1550 cm−1 corresponded to the chemisorption of pyridine on Brønsted (B) acid sites. IR absorption bands around 1490 cm−1 assigned to pyridine adsorption on both B and L acid sites.38,39 From Fig. 1a, it was found the intensity of the pyridine absorption bands from H-ZSM-5 (air), H-ZSM-5 (H2) and La-ZSM-5 (H2) samples are almost the same after saturated absorption at initial, indicating the amount of L and B acids sites are almost the same in the three different samples. After degassing at 200 °C and 350 °C, the pyridine absorption bands corresponding to B acid sites from La-ZSM-5 (H2) are obviously lower than that from H-ZSM-5 (air) and H-ZSM-5 (H2) samples. The B and L acid sites detected at 200 °C belong to weak/medium acid sites, while those observed at 350 °C are attributed to strong acid sites. The results from Fig. 1a–c indicate La-loading can obviously decrease the acid strength of strong B acid sites, which may be the main reason for the further suppression of H2O2 decomposition over La-ZSM-5 sample. The inference that La-loading decreases the acidity of strong B acid sites and then decreases the undesirable H2O2 decomposition is further demonstrated by the pyridine adsorption for the LaFeCu-ZSM-5 (air) and FeCu-ZSM-5 (air) catalysts. As shown in Fig. 1d–f, the 0.5 wt% FeCu-ZSM-5 (air) and 0.5 wt% LaFeCu-ZSM-5 (air) samples have similar B and L acid sites (Fig. 1d). After degassing at 200 °C (Fig. 1e) and 350 °C (Fig. 1f), the pyridine adsorption intensity from B acid sites for the 0.5 wt% LaFeCu-ZSM-5 (air) sample is gradually lower than that from 0.5 wt% FeCu-ZSM-5 (air), indicating the acidity of B acid sites in LaFeCu-ZSM-5 (air) is also decreased after loading La. Especially, the intensity of strong B acid sites around 1543 cm−1 in 0.5 wt% LaFeCu-ZSM-5 (air) is obviously much lower than that in 0.5 wt% FeCu-ZSM-5 (air) sample after degassing at 350 °C (Fig. 1f), indicating the large decrease in acidity of strong B aid sites. From Table 1 (entry 10, 11), the methane oxidation performance of 0.5 wt% LaFeCu-ZSM-5 (air) is very close to that over the 0.5 wt% FeCu-ZSM-5 (air) sample. However, the ratio of H2O2 consumed/partially oxygenated products in 0.5 wt% LaFeCu-ZSM-5 (air) sample is only 2.6, while that ratio in 0.5 wt% FeCu-ZSM-5 (air) is 4.0, indicating the excellent performance of La for suppressing H2O2 consumption. Combing the pyridine adsorption behavior and the methane oxidation performance of La modified H-ZSM-5 and FeCu-ZSM-5 catalysts, it can be concluded that the H2O2 decomposition rate is closely related to the acidity of the strong B acid sites in ZSM-5 based catalysts. The introduction of La species into H-ZSM-5 and Fe-ZSM-5 brings about a clear decrease in the acidity of strong B acid sites, which can then suppress the H2O2 decomposition. It is well known that the B acid sites are mainly provided by Si(OH)Al and Si(OH) in ZSM-5. Among them, OH groups from Si(OH) are classed as weak and medium B acid sites, while OH groups from Si(OH)Al provide strong B acid sites.40
 |
| | Fig. 1 Py-FTIR spectra of various catalysts degassed at 25, 200 and 350 °C: (a) HZSM-5 (H2), HZSM-5 (air) and 0.5 wt% La-ZSM-5 (H2) at 25 °C; (b) HZSM-5 (H2), HZSM-5 (air) and 0.5 wt% La-ZSM-5 (H2) at 200 °C; (c) HZSM-5 (H2), HZSM-5 (air) and 0.5 wt% La-ZSM-5 (H2) at 350 °C; (d) 0.5 wt% LaFeCu-ZSM-5 (air) and 0.5 wt% FeCu-ZSM-5 (air) sample at 25 °C; (e) 0.5 wt% LaFeCu-ZSM-5 (air) and 0.5 wt% FeCu-ZSM-5 (air) sample at 200 °C; (f) 0.5 wt% LaFeCu-ZSM-5 (air) and 0.5 wt% FeCu-ZSM-5 (air) sample at 350 °C. | |
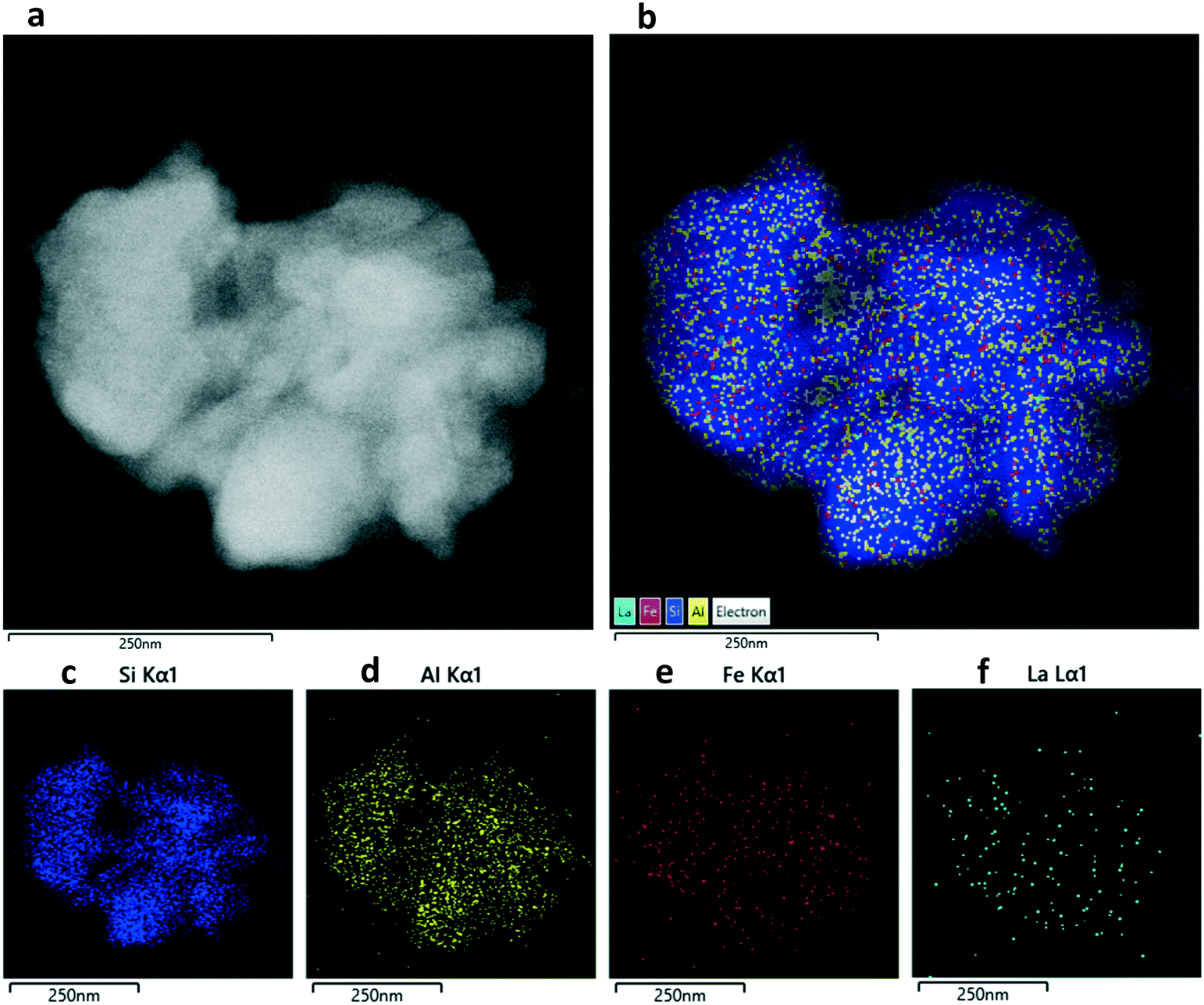
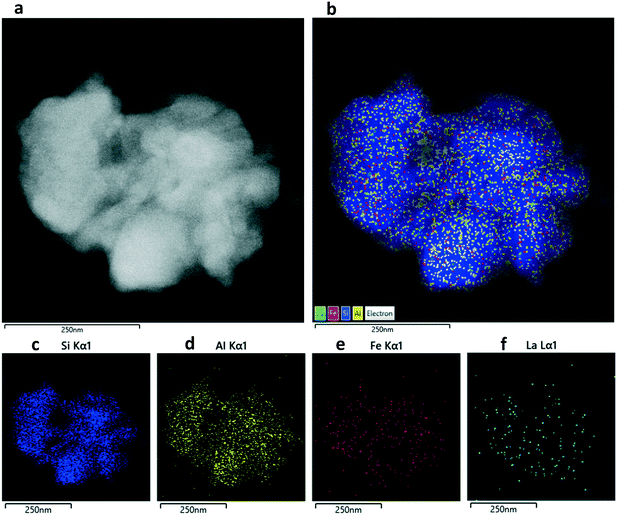
The micro-structures and element distributions of the 0.5 wt% La-ZSM-5 (H2), 0.5 wt% LaFe-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (H2) samples were investigated by high-resolution scanning transmission electron microscopy (STEM) and STEM-EDS mapping, as shown in Fig. 2–4. The ZSM-5 zeolite phases are clearly distinguished by having a high concentration in silicon and aluminum EDS signals (Fig. 2–4). After metal modification, it can be seen the La, Fe, Cu elements are found to be highly distributed in the ZSM-5 matrix of La-ZSM-5 (H2), LaFe-ZSM-5 (H2) and LaFeCu-ZSM-5 (H2) samples (Fig. 2–4). Besides the framework and extra-framework metal sites within the micropores, some larger metal oxide species were also observed from high magnified STEM images (Fig. 5). However, these larger metal oxide crystallites on the external ZSM-5 crystal surface are not the dominant species, as shown in Fig. 5.
 |
| | Fig. 2 Characterization of the 0.5 wt% La-ZSM-5 (H2) sample: (a) high-resolution bright-field STEM image, (b) STEM-EDS layered image, (c–f) STEM-EDS map for Si, O, Al and La. | |
 |
| | Fig. 3 Characterization of the 0.5 wt% LaFe-ZSM-5 (H2) sample: (a) high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) image, (b) STEM-EDS layered image, (c–f) STEM-EDS map for Si, Al, Fe and La. | |
 |
| | Fig. 4 Characterization of the 0.5 wt% LaFeCu-ZSM-5 (H2) sample: (a) high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) image, (b) STEM-EDS layered image, (c–g) STEM-EDS map for Si, Cu, Al, Fe and La. | |
 |
| | Fig. 5 High magnified high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) image: (a) 0.5 wt% La-ZSM-5 (H2) sample, (b) 0.5 wt% LaFe-ZSM-5 (H2) sample. | |
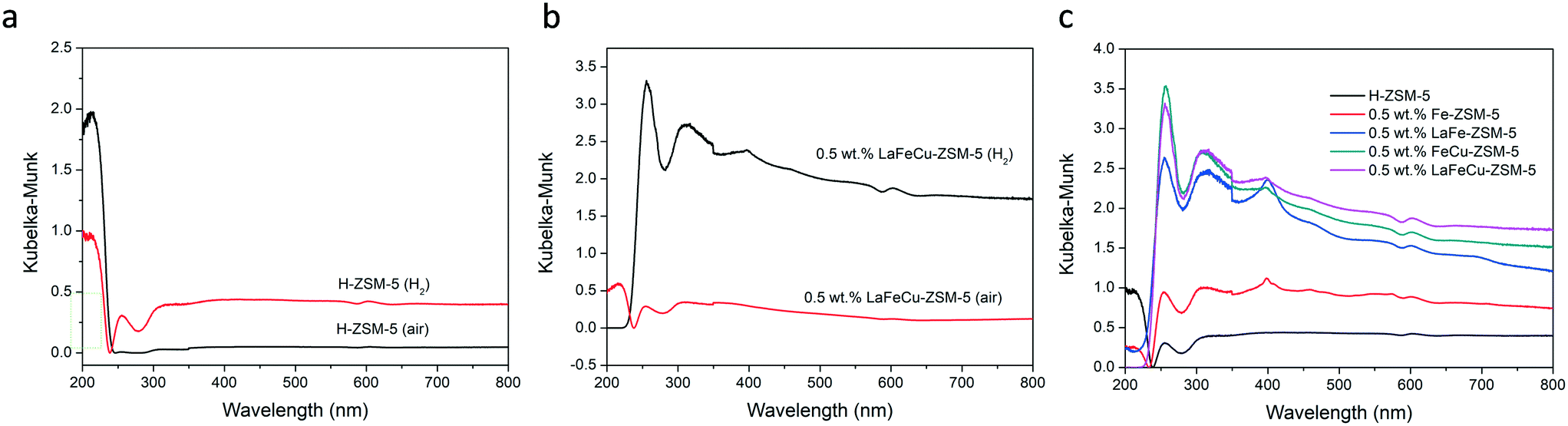
UV-vis absorption spectroscopy was used to investigate the nature of the iron metal sites in the catalysts. It has been reported that Fe species in ZSM-5 give rise to absorption bands among UV-vis region, where the absorption band between 200 to 250 nm comes from isolated Fe3+ in framework tetrahedral (Td) positions; the absorption band between 250 to 300/350 nm is attributed to isolated or oligonuclear extra-framework Fe species in square-pyramidal and distorted octahedral (Oh) environments; the absorption band between 300/350 to 450 nm is ascribed to Fe3+xOy clusters and the absorption above 450 nm is from larger Fe2O3 crystallites on the external ZSM-5 crystal surface.21,41–45Fig. 6a shows the UV-vis absorption spectrum of H-ZSM-5 (H2) and H-ZSM-5 (air) sample. One can see that isolated framework Fe3+ with the absorption band around 213 nm is the dominant iron species in H-ZSM-5 (air) sample. After heat treated by 5% H2/Ar at 550 °C, some isolated framework Fe3+ species transformed into extra-framework Fe species with the absorption band around 256 nm. Previous studies have proved extra-framework Fe–oxo complexes are the main active species for methane oxidation,18 the increased extra-framework Fe species in the H2/Ar treated H-ZSM-5 (H2) sample might be the main reason for its enhanced methane oxidation performance. This phenomenon is also observed when we compare the UV-vis absorption spectrum of 0.5 wt% LaFeCu-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (air) sample. As shown in Fig. 6b, there is an obvious absorption band at 217 nm for air treated 0.5 wt% LaFeCu-ZSM-5 (air) sample. Meanwhile, the absorption band around 256 nm is decreased when compared with H2/Ar heated sample (Fig. 6b), indicating that isolated Fe3+ in framework Td positions within the zeolite channels are the dominant Fe species in air heated LaFeCu-ZSM-5 (air) sample. These framework Fe species are not active species for methane oxidation, which may be the main reasons for the lower catalytic activity of air treated LaFeCu-ZSM-5 (air) sample. For further investigating the Fe species in H2/Ar heated samples, the UV-vis absorption spectra for 0.5 wt% Fe-ZSM-5 (H2), LaFe-ZSM-5 (H2), FeCu-ZSM-5 (H2), and LaFeCu-ZSM-5 (H2) catalysts are presented in Fig. 6c from which two strong absorption bands centered around 256 and 310 nm are observed, indicating isolated extra-framework Fe species are the dominant iron metal sites in the H2/Ar heated catalysts. Besides that, a weak absorption band around 400 nm appeared, demonstrating the presence of the larger FexOy clusters on the external zeolite surface. These larger FexOy clusters mainly contribute to Fenton type decomposition of H2O2.
 |
| | Fig. 6 UV-vis absorbance spectra of various ZSM-5 based materials heat treated at 550 °C in 5% H2/Ar or air for 3 h: (a) the comparison of H-ZSM-5 (H2) and H-ZSM-5 (air) samples; (b) the comparison of 0.5 wt% LaFeCu-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (air) samples; (c) the comparison of H-ZSM-5 (H2), 0.5 wt% Fe-ZSM-5 (H2), 0.5 wt% LaFe-ZSM-5 (H2), 0.5 wt% FeCu-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (H2) samples. | |
The chemical states of La, Fe and Cu element species in the 0.5wt% La-ZSM-5 (H2), 0.5wt% LaFe-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (H2) samples were characterized by XPS. As shown in Fig. 7a, the La 3d5/2 core level spectra in 0.5 wt% La-ZSM-5 (H2) sample were deconvoluted after Tougaard background subtraction. The main peak located around 835.9 eV was associated with the main component of La 3d5/2 XPS spectrum,40 labeled cf0 in Fig. 7a. The other two peaks located at 837.2 and 839.3 eV were attributed to satellite components,40 labeled cf1L antibonding and cf1L bonding respectively (Fig. 7a). These results are in line with the previous studies on lanthanum hydroxide species.46 Compared to the 0.5 wt% La-ZSM-5 (H2) sample, the main binding energy of La 3d5/2 in 0.5 wt% LaFe-ZSM-5 (H2) (Fig. 7b) sample shifted from 835.9 to 836.1 eV, indicating some electrons may transfer from La to Fe and then contribute to maintain much more low-valence Fe species in the H2 treated LaFe-ZSM (H2) sample. The main binding energy of La 3d5/2 XPS spectrum in 0.5 wt% LaFeCu-ZSM-5 (H2) (Fig. 7c) sample shifted −0.1 eV when compared with that in 0.5 wt% LaFe-ZSM-5 (H2) sample, indicating Cu may partly disturb the electron transfer from La to Fe in their co-modified sample. This is further proved by comparison of the XPS spectrum of Fe 2p in 0.5 wt% LaFe-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (H2) samples. As shown in Fig. 7d for the 0.5 wt% LaFe-ZSM-5 (H2) sample, the binding energies of 710.7 and 724.1 eV with a satellite signal at 715.6 and 730.6 eV are characteristic of Fe2+, while the binding energies of 712.4 and 726.6 eV with a satellite signal at 717.8 and 733.5 eV are characteristic of Fe3+.47 Compared with the LaFe-ZSM-5 (H2) sample, it was found the binding energies of Fe 2p in LaFeCu-ZSM-5 (H2) samples shifted to lower values (Fig. 7e). Besides, an additional peak around 708 eV appeared in the 0.5 wt% LaFeCu-ZSM-5 (H2) sample, which is possibly due to electron-deficient Fe2+ sites, created by the deficient oxygen coordination around it. Fig. 7f is the XPS spectrum of Cu 2p3/2 in 0.5 wt% LaFeCu-ZSM-5 (H2) sample. The spectrum is deconvoluted into two peaks. One at 932.75 eV is derived from Cu2+. The other peak at 934 eV is ascribed to Cu+. The low-valence Cu+ species are resulted from the reduction of Cu2+ by H2 treatment.
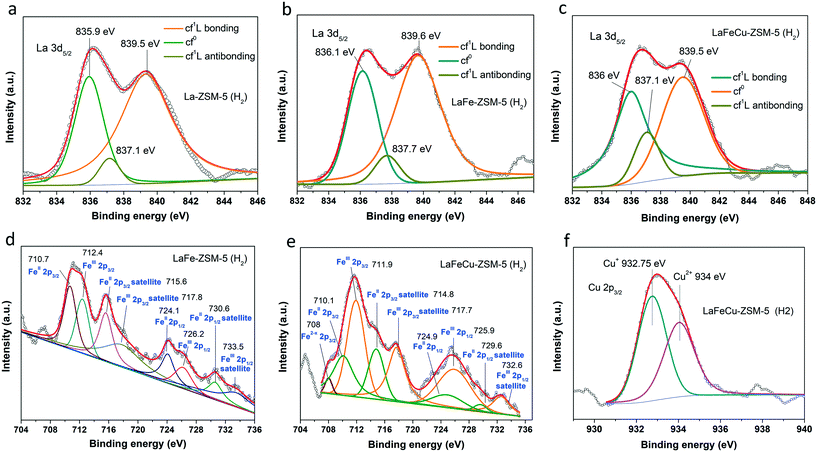 |
| | Fig. 7 XPS spectrum of La 3d5/2 (a–c), Fe 2p (d and e) and (f) Cu 2p3/2 in 0.5 wt% La-ZSM-5 (H2), 0.5 wt% LaFe-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (H2) samples. | |
To investigate whether the La modification can induce dealumination of ZSM-5 matrix and then decreased the strong B acid sites Si(OH)Al, diffuse reflectance FTIR spectra for a series of H-, La-, FeCu- and LaFeCu-ZSM-5 catalysts were obtained after pretreatment of these catalyst at 400 °C for 2 h under N2 flowing. Fig. 8a illustrates the FTIR absorption spectra of H-ZSM-5 (H2) and 0.5 wt% La-ZSM-5 (H2) sample. The absorption band around 3609, 3666, and 3734 cm−1 are attributed to the vibration of OH from Si(OH)Al (strong B acid sites), extra-framework Al(OH)x and terminal isolated Si(OH).48,49 The smaller absorption band at 3609 cm−1 in the 0.5 wt% La-ZSM-5 (H2) sample indicates the decreased concentration of Si(OH)Al site on the catalyst surface. Fig. 8b shows the FTIR absorption spectra of 0.5 wt% FeCu-ZSM-5 (air) and 0.5 wt% FeCu-ZSM-5 (H2) samples. It was found that all of the FTIR absorption bands from Si(OH)Al, Al(OH) and Si(OH) are increased when the sample is calcined in H2/Ar gas, indicating an increased concentration of B acid sites in these samples, which may be the main reason why the 0.5 wt% FeCu-ZSM-5 sample (H2) sample exhibited a higher H2O2 consumption ratio than that over 0.5 wt% FeCu-ZSM-5 (air) in the methane oxidation reaction. For the 0.5 wt% LaFeCu-ZSM-5 (H2) and 0.5 wt% LaFeCu-ZSM-5 (air) samples, the FTIR absorption band (Fig. 8c) from Si(OH)Al and Al(OH) almost disappeared when compared with the 0.5 wt% FeCu-ZSM-5 samples (Fig. 8b), demonstrating further that La modification can decrease the concentration of strong B acid sites.
 |
| | Fig. 8
In situ DRIFT spectra of different catalysts at 25 °C after treatment at 400 °C for 2 h under N2 flow. (a) Comparison of HZSM-5 (H2) and 0.5 wt% La-ZSM-5 (H2) sample. (b) Comparison of 0.5 wt% FeCu-ZSM-5 (air) and 0.5 wt% FeCu-ZSM-5 (H2) sample. (c) Comparison of 0.5 wt% LaFeCu-ZSM-5 (air) and 0.5 wt% LaFeCu-ZSM-5 (H2) sample. | |
Fig. 9 shows the 27Al MAS NMR spectra of the H-ZSM-5 (H2), 0.5 wt% La-ZSM-5 (H2), 0.5 wt% Fe-ZSM-5 (H2) and 0.5 wt% LaFe-ZSM-5 (H2) samples. All of these samples exhibit an intense and sharp signal at 54.0 ppm, which is due to an aluminum species in a tetrahedral (Td) coordination, indicating that addition of La and Fe has no effect upon the MFI framework. An additional, weak peak centered around 0 ppm is normally assigned to octahedrally (Oh) coordinated extra-framework Al species. However, compared with the H-ZSM-5 (H2) sample, the intensity of the resonances for Td27Al in framework sites and Oh27Al in extra-framework sites were gradually decreased in 0.5 wt% La-ZSM-5 (H2), 0.5 wt% Fe-ZSM-5 (H2) and 0.5 wt% LaFe-ZSM-5 (H2) samples. Besides, from the partial enlarged detail in Fig. 9, the peak positions for both of the Td- and Oh-coordinated Al species slightly moved to a lower ppm position. These phenomenon indicates that La and Fe modification may stimulate deprivation of the Al element in ZSM-5 matrix during the preparation of metal modified ZSM-5 (H2) catalysts, and then change the concentration of strong B acid sites. The Si/Al molar ratios of H-ZSM-5 (H2), 0.5 wt% La-ZSM-5 (H2), 0.5 wt% Fe-ZSM-5 (H2) and 0.5 wt% LaFe-ZSM-5 (H2) samples were investigated by ICP-OES, which is 29.5, 30.6, 31.5 and 31.7 respectively, further confirming the dealumination by La, Fe modification. Compared with Fe-ZSM-5, the suppressed H2O2 consumption in La modified Fe-ZSM-5 catalyst may be ascribed to the further decreased acidity of strong B acid sites after La modification.
 |
| | Fig. 9
27Al MAS-NMR spectra of HZSM-5 (H2), 0.5wt% La-ZSM-5 (H2), 0.5wt% Fe-ZSM-5 (H2) and 0.5wt% LaFe-ZSM-5 (H2) samples. | |
Conclusions
We have demonstrated that La modified Fe-ZSM-5 catalysts exhibit a higher activity for selective partial oxidation of methane and lower H2O2 consumption under ambient conditions. The productivity of primary oxygenated products (CH3OH, CH3OOH, HCOOH) from methane oxidation over the 0.5 wt% LaFe-ZSM-5 (H2) sample gives 3200 mol kgLaFe−1 h−1 in 0.1 M H2O2, with a selectivity of 98.9% to primary oxygenated products. With 0.5 M H2O2 the productivity is increased to 11460 mol kgLaFe−1 h−1, which is about 20 times higher than that over an Au–Pd catalyst under the similar conditions. Further studies indicate La modification can decrease the acidity of strong B acid sites and then suppress the H2O2 decomposition in the methane oxidation process. Compared with the Fe-ZSM-5 (H2) catalyst, the LaFe-ZSM-5 (H2) catalyst can save 31% of H2O2 for generating per mol of product under the same conditions. Over the La modified FeCu-ZSM-5 (air) catalyst, about 36% of H2O2 can be saved for methane oxidation when compared with FeCu-ZSM-5 (air) catalyst. This study shows a way forward towards the application of direct methane oxidation to valuable C–H products, which can stimulate other studies on refinement of catalyst design to further improve the productivity under a lower H2O2 consumption.
Author contributions
S. Sun, N. F. Dummer and G. J. Hutchings – designed the project. G. J. Hutchings – supervised the progress of the entire project. S. Sun – conducted the catalysts preparation, activity tests and sample characterizations by TEM and ESR. S. Sun and T. Bere – carried out in situ DRIFT measurements. S. Sun and A. J. Barnes – carried out NMR measurements. S. Sun and G. Shaw – carried out XRD and DRS measurements. G. Xiaoxiao and R. Lewis – contributed to other characterizations. N. Richards contributed to the analysis of XRD results. D. J. Morgan – conducted and analysed the sample by XPS. The manuscript was written through collective contributions from all authors. All authors approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22175034, 21671197) and the Shanghai Science and Technology Commission (No. 21ZR1401600). The research has also been financially supported from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Actions (Grant Agreement No. 785794).
References
- International Energy Agency (IEA), “Resources to reserves 2013” (IEA, 2013), www.iea.org/publications/freepublications/publication/Resources2013.pdf.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS PubMed.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F. S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- C. Díaz-Urrutia and T. Ott, Science, 2019, 363, 1326–1329 CrossRef PubMed.
- C. Copéret, Chem. Rev., 2010, 110, 656–680 CrossRef PubMed.
- A. Hu, J. J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- H. D. Gesser, N. R. Hunter and C. B. Prakash, Chem. Rev., 1985, 85, 235–244 CrossRef CAS.
- B. L. Conley, W. J. Tenn III, K. J. H. Young, S. K. Ganesh, S. K. Meier, V. R. Ziatdinov, O. Mironov, J. Oxgaard, J. Gonzales, W. A. Goddard III and R. A. Periana, J. Mol. Catal. A: Chem., 2006, 251, 8–23 CrossRef CAS.
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nat. Catal., 2019, 2, 485–494 CrossRef CAS.
- E. V. Starokon, M. Parfenov, S. S. Arzumanov, L. V. Pirutko, A. G. Stepanov and G. I. Panov, J. Catal., 2013, 300, 47–54 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- T. Sheppard, C. D. Hamill, A. Goguet, D. W. Rooney and J. M. Thompson, Chem. Commun., 2014, 50, 11053–11055 RSC.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Löffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340–343 CrossRef CAS PubMed.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS PubMed.
- A. Stephen, K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef CAS PubMed.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef CAS PubMed.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. Ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 1835–1844 CrossRef CAS.
- M. M. Forde, R. D. Armstrong, R. McVicker, P. P. Wells, N. Dimitratos, Q. He, L. Lu, R. L. Jenkins, C. Hammond, J. A. Lopez-Sanchez, C. J. Kielyc and G. J. Hutchings, Chem. Sci., 2014, 5, 3603–3616 RSC.
- C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- C. Coperet, Chem. Rev., 2010, 110, 656–680 CrossRef CAS PubMed.
- A. Szecsenyi, G. Li, J. Gascon and E. A. Pidko, ACS Catal., 2018, 8, 7961–7972 CrossRef CAS PubMed.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- A. Maitra, I. Campbell and R. J. Tyler, Appl. Catal., A, 1992, 85, 27–46 CrossRef CAS.
- P. Käßner and M. Baerns, Appl. Catal., A, 1996, 139, 107–129 CrossRef.
- V. R. Choudhary and V. H. Rane, J. Catal., 1991, 130, 411–422 CrossRef CAS.
- M. Eichelbaum, M. Havecker, C. Heine, A. M. Wernbacher, F. Rosowski, A. Trunschke and R. Schlogl, Angew. Chem., Int. Ed., 2015, 54, 2922–2926 CrossRef CAS PubMed.
- G. B. Shulpin and G. V. Nizova, React. Kinet. Catal. Lett., 1992, 48, 333–338 CrossRef CAS.
- G. V. Nizova, G. Suss-Fink and G. B. Shul'pin, Chem. Commun., 1997, 397–398 RSC.
- G. Suss-Fink, G. V. Nizova, S. Stanislas and G. B. Shul'pin, J. Mol. Catal. A: Chem., 1998, 130, 163–170 CrossRef CAS.
- E. Andris, R. Navrátil, J. Jašík, M. Puri, M. Costas, L. Que and J. Roithová, J. Am. Chem. Soc., 2018, 140, 14391–14400 CrossRef CAS PubMed.
- A. A. Battiston, J. H. Bitter, W. M. Heijboer, F. M. F. de Groot and D. C. Koningsberger, J. Catal., 2003, 215, 279–293 CrossRef CAS.
- J. Jia, Q. Sun, B. Wen, L. X. Chen and W. M. H. Sachtler, Catal. Lett., 2002, 82, 7–11 CrossRef CAS.
- A. A. Battiston, J. H. Bitter and D. C. Koningsberger, J. Catal., 2003, 218, 163–177 CrossRef CAS.
- L. P. Oleksenko, V. K. Yatsimirsky, G. M. Telbiz and L. V. Lutsenko, Adsorpt. Sci. Technol., 2004, 22, 535–541 CrossRef CAS.
- T. Gong, X. Zhang, T. Bai, Q. Zhang, L. Tao, M. Qi, C. Duan and L. Zhang, Ind. Eng. Chem. Res., 2012, 51, 13589–13598 CrossRef CAS.
- Z. Liu, J. P. H. Li, E. Vovk, Y. Zhu, S. Li, S. Wang, A. P. van Bavel and Y. Yang, ACS Catal., 2018, 8, 11761–11772 CrossRef CAS.
- G. Centi, S. Perathoner, F. Pino, R. Arrigo, G. Giordano, A. Katovic and V. Pedulà, Catal. Today, 2005, 110, 211–220 CrossRef CAS.
- T. Inui, H. Nagata, T. Takeguchi, S. Iwamoto, H. Matsuda and M. Inoue, J. Catal., 1993, 139, 482 CrossRef CAS.
- G. Lehmann, Z. Phys. Chem., 1970, 72, 279–297 CrossRef CAS.
- J. Pérez-Ramírez, J. C. Groen, A. Brückner, M. S. Kumar, U. Bentrup, M. N. Debbagh and L. A. Villaescusa, J. Catal., 2005, 232, 318–334 CrossRef.
- E. J. M. Hensen, Q. Zhu, M. M. R. M. Hendrix, A. R. Overweg, P. J. Kooyman, M. V. Sychev and R. A. van Santen, J. Catal., 2004, 221, 560–574 CrossRef.
- L. Zhang, Y. Liu, Y. Wang, X. Li and Y. Wang, Appl. Surf. Sci., 2021, 557, 149838 CrossRef CAS.
- J. Jiao, W. Qiu, J. Tang, L. Chen and L. Jing, Nano Res., 2016, 9, 1256–1266 CrossRef CAS.
- G. Paul, C. Bisio, I. Braschi, M. Cossi, G. Gatti, E. Gianotti and L. Marchese, Chem. Soc. Rev., 2018, 47, 5684–5739 RSC.
- S. Schallmoser, T. Ikuno, M. F. Wagenhofer, R. Kolvenbach, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, J. Catal., 2014, 316, 93–102 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abc,
Alexandra J.
Barnes
a,
Xiaoxiao
Gong
ad,
Richard J.
Lewis
*abc,
Alexandra J.
Barnes
a,
Xiaoxiao
Gong
ad,
Richard J.
Lewis