
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting the performance of Ni/Al2O3 for the reverse water gas shift reaction through formation of CuNi nanoalloys†
Esteban
Gioria
 a,
Piyush
Ingale
a,
Felix
Pohl
a,
Raoul
Naumann d'Alnoncourt
*a,
Arne
Thomas
b and
Frank
Rosowski
ac
a,
Piyush
Ingale
a,
Felix
Pohl
a,
Raoul
Naumann d'Alnoncourt
*a,
Arne
Thomas
b and
Frank
Rosowski
ac
aBasCat – UniCat BASF JointLab, Technische Universität Berlin, Berlin 10623, Germany. E-mail: r.naumann@bascat.tu-berlin.de
bFunctional Materials, Department of Chemistry, Technische Universität Berlin, Berlin 10623, Germany
cBASF SE, Process Research and Chemical Engineering, Ludwigshafen 67056, Germany
First published on 6th November 2021
Abstract
Ni supported on alumina is extensively employed as a catalyst for the reverse water gas shift reaction. However, the formation of inactive Ni aluminates and the high selectivity to methane affects the performance of these catalysts, especially at low reaction temperatures. In this study, Cu is employed as an effective enhancer of the catalytic performance, promoting the reducibility of Ni, and suppressing methane production through the formation of stable CuNi nanoalloys. The synergy between both metals suppresses consecutive hydrogenation, reaching conversions close to equilibrium and 100% selectivity to carbon monoxide. At 500 °C, the CO yield of the Cu25Ni75/Al2O3 catalyst was twice that of Ni/Al2O3, with only half the hydrogen consumption. The formation of CuNi nanoalloys was confirmed by HAADF STEM–EDS, without segregation in monometallic phases even after 30 h time on stream. Similarly, Cu50Ni50/Al2O3 remained alloyed after the reaction. However, the catalytic activity decreased due to sintering, in agreement with a higher Cu content. This effect was significantly more pronounced for Cu75Ni25/Al2O3 and Cu/Al2O3, with formation of large particles and consequent loss of active surface area.
Introduction
Carbon monoxide is of great importance in the chemical and petrochemical industries since it is employed as a building block to produce higher-value products. The H2/CO mixture, known as synthesis gas or syngas, is the raw material in the production of alcohols and synthetic fuels through the Fischer–Tropsch process.1–4 At a commercial scale, the steam reforming of natural gas remains the preferred synthesis route and Ni has been employed as the key active site in supported reforming catalysts since their first industrial applications.5,6 Nevertheless, the use of carbon dioxide as a feedstock carbon-containing molecule has gained a lot of attention in the last few decades. Carbon dioxide is one of the main by-products of the chemical industry, and its hydrogenation employing hydrogen obtained by green methods is a potential pathway towards a circular economy and the development of more sustainable processes.7–11The chemical conversion of carbon dioxide to carbon monoxide via the reverse water gas shift (rWGS) reaction is an endothermic process (eqn (1)). This reaction requires temperatures higher than 700 °C in order to activate the thermodynamically stable CO2 molecule and reach economically feasible conversions.7,12 However, when the reaction takes place at lower temperatures, methane formation through the Sabatier reaction is favoured due to the exothermic nature of this side-reaction (eqn (2)):
| CO2 + H2 → CO + H2O ΔH0R = 41 kJ mol−1 | (1) |
| CO2 + 4H2 → CH4 + 2H2O ΔH0R = −165 kJ mol−1 | (2) |
A vast number of metal combinations have been tested for the rWGS reaction. Noble metals are highly active and selective; however, their scarcity and high cost make their large-scale application unfeasible. Thus, the use of non-noble transition metals such as Ni, Co, Fe, Cu and Mo is of great interest.18–21 Among them, Ni catalysts have shown remarkably high activity and stability even at relatively low reaction temperatures. However, high selectivity to methane via consecutive hydrogenation of carbon monoxide poses a challenge. In addition, Ni catalysts supported on alumina can form inactive species of low reducibility like Ni aluminates.22,23
The incorporation of a second metal is an effective way to tune the electronic properties of the active sites and several promoters have been proposed as an alternative to enhance the selectivity to carbon monoxide of Ni-based catalysts.24–26 Indeed, the benefits of bimetallic catalysts for the rWGS reaction are mostly related to higher metal dispersion, enhanced CO2 activation and improved CO desorption.27–29
Hence, one of the most promising alternatives is the use of Cu, due to its inherent high selectivity to carbon monoxide.30,31 Based on temperature programmed desorption studies, Liu et al. have shown that Cu significantly promotes the selectivity to carbon monoxide of supported Ni catalysts on alumina.32 This effect was attributed to the lower hydrogen adsorption capacity of Cu compared with Ni and thus suppression of consecutive carbon monoxide hydrogenation to methane. More recently, experimental results and theoretical simulations showed that Cu–Ni catalysts have an outstanding performance compared to their monometallic counterparts, related to the formation of alloyed nanostructures.33–37 However, low thermal stability and excessive sintering limit their application at relatively high reaction temperatures.38,39
Surprisingly, studies based on transmission electron microscopy and energy-dispersive X-ray spectroscopy that corroborate the formation of CuNi alloyed nanoparticles in the rWGS reaction are still scarce. In the present study, a set of five different mono- and bimetallic Cu–Ni catalysts supported on alumina were tested in the reverse water gas shift reaction in the range of 500–600 °C. The incorporation of Cu into Ni catalysts suppresses methane formation through the formation of bimetallic nanoalloys, enhancing carbon monoxide yield and decreasing hydrogen consumption. The use of alumina as a support improves the stability of bimetallic nanoparticles due to the strong interaction with the support through Ni aluminate species. In addition, Cu–Ni alloyed nanostructures were stable after 30 h time on stream, without segregation in monometallic phases.
Therefore, we believe that our work brings new insights in terms of copper-promoted nickel-based catalysts. The formation of stable bimetallic CuNi alloys suppresses methanation and thus enhances the performance in terms of CO yield and hydrogen consumption. To the best of our knowledge, this is the first report where CuNi/Al2O3 catalysts applied for rWGS are characterized by STEM–EDS, clarifying the role of nanoalloys without a doubt.
Experimental
Synthesis of Cu–Ni catalysts
The catalysts were prepared by incipient wetness impregnation. Bimetallic catalysts were prepared by co-impregnation. In a typical protocol, 1 g of the γ-alumina support was impregnated with 0.95 ml of Cu and/or Ni nitrate solution containing the desired amount of metallic precursors. The nominal metal loading was fixed to 5% wt. Metal nitrates were chosen as a precursor because of their low cost, good solubility in water and facile decomposition.40 The catalysts were dried at 120 °C overnight and calcined at 500 °C for 4 h employing a heating rate of 10 K min−1 and a 200 ml min−1 flow of 21% O2/N2. The catalysts were named CuxxNiyy/Al2O3, where xx/yy refers to the Cu/Ni mass ratio. Cu(NO3)2·3H2O, Ni(NO3)·6H2O (99.99%, Sigma-Aldrich, Germany) and H2O HPLC grade (Honeywell Riedel-de-Haën, Germany) were used as received without any further purification.Characterization
Thermal gravimetric analysis (TGA) was carried out using an STA 409 apparatus (Linseis) employing 25 mg of the sample at a temperature range of 25–900 °C, with a heating rate of 10 K min−1 and a flow rate of 20 mL min−1 of 20% O2/N2.Elemental analysis (carbon, nitrogen, hydrogen and sulphur) was performed on a Thermo FlashEA 1112 organic elemental analyzer by a dynamic flash combustion method operating at 1020 °C.
X-ray diffractograms of the powder samples were acquired in Bragg–Brentano geometry on a Rigaku SmartLab 3 kW with Cu Kα radiation (λ = 1.54 Å) over an angular range from 10° to 80° and a scan rate of 2° min−1.
The chemical composition of the mono- and bimetallic catalysts was determined by inductively coupled plasma–atomic emission spectroscopy (ICP–AES) on a Jobin Yvon Horiba ULTIMA 2000 spectrometer. The samples were digested in aqua regia HCl/HNO3 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v (nitric acid 69%, SUPRA-Qualität ROTIPURAN® and hydrochloric acid 30%, SUPRA-Qualität ROTIPURAN®) and the mean of three reproducible independent experiments is reported.
:1 v/v (nitric acid 69%, SUPRA-Qualität ROTIPURAN® and hydrochloric acid 30%, SUPRA-Qualität ROTIPURAN®) and the mean of three reproducible independent experiments is reported.
Temperature programmed reduction experiments were carried out using a BelCat-II equipped with a thermal conductivity detector (TCD). For each measurement, 250 mg of sample was heated up to 900 °C employing a heating rate of 6 K min−1 in 5% H2/N2 with a total flow of 50 mL min−1. Operating conditions were optimized following the recommendations reported by Monti et al.41
Nitrogen sorption analysis was carried out using a Quadrasorb SI (Quantachrome GmbH & Co. KG Odelzhausen, Germany) after degassing the samples for 12 h at 150 °C. The surface area was determined using the Brunauer–Emmett–Teller (BET) method, and the corresponding pore size distribution was obtained by non-local density functional theory (NLDFT).
X-ray photoelectron spectroscopy (XPS) was carried out using a Thermo Fisher Scientific ESCALAB 250Xi. An Al-Kα X-ray source (1486.6 eV) was employed for the analysis, with a pass energy of 200 eV for a survey, and 50 eV for high-resolution spectra. The samples were activated following a similar reduction treatment to that of the catalytic test and stored under an inert atmosphere in the glovebox (MBraun LABmaster Pro). Before measurements, the catalysts were briefly in contact with air during sample holding. The analysis of the spectra was done using CasaXPS software. The C1s peak of adventitious carbon at 284.6 eV was taken as a reference of charge-shift correction for the measured spectra.
The morphology, composition and size distribution of the supported nanoparticles were analysed by high-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDS) elemental mapping using a Thermo Scientific Talos F200X microscope operated at an accelerating voltage of 200 kV. The microscope is equipped with a SuperX EDX detector system. Samples were prepared by dipping the lacey carbon 400-mesh gold grids (Plano GmbH) into the powder sample. STEM–EDS mappings show the net intensities of the corresponding elements with a post-filtering Gaussian blur of 1.1 sigma.
Catalytic activity: CO2 hydrogenation
The catalytic experiments were carried out at atmospheric pressure and in a set-up designed by Integrated Lab Solutions (ILS, Berlin, Germany). 25 mg of each sample (pressed and sieved between 100 and 200 μm) was loaded into a quartz-tube reactor. The activation of each sample was performed in situ for 60 min at 500 °C in 100 mL min−1 of 10% H2/He with a heating rate of 10 K min−1. The reactor was purged with a 100 mL min−1 He flow after activation treatment for 10 min. Afterwards the reactant mixture (40 mL min−1 CO2:40 mL min−1 H2:20 mL min−1 N2) was introduced into the reactor, and the reaction temperature was modified in a ramping up–ramping down cycle at 500–550–600 °C. The resulting gas hourly space velocity (GHSV) was 240000 mL g−1 h−1.
The effluent gas stream was analysed using an online gas chromatograph (Agilent 7890A) equipped with a flame ionization detector and a thermal conductivity detector. The carbon dioxide conversion (XCO2), carbon monoxide selectivity (SCO), methane selectivity (SCH4) and space–time yield of carbon monoxide (STYCO) were defined as:
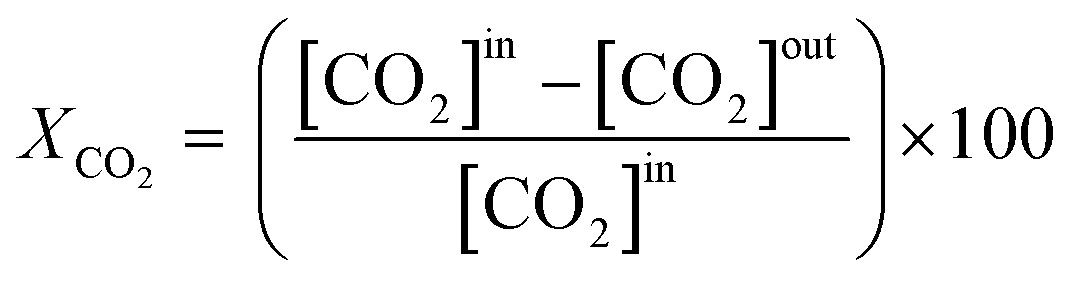
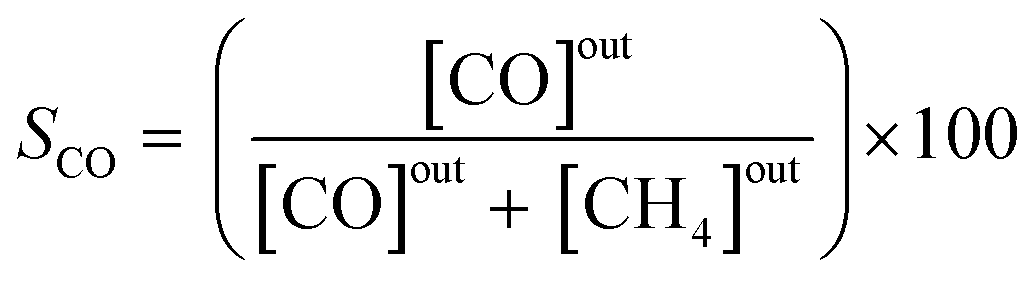
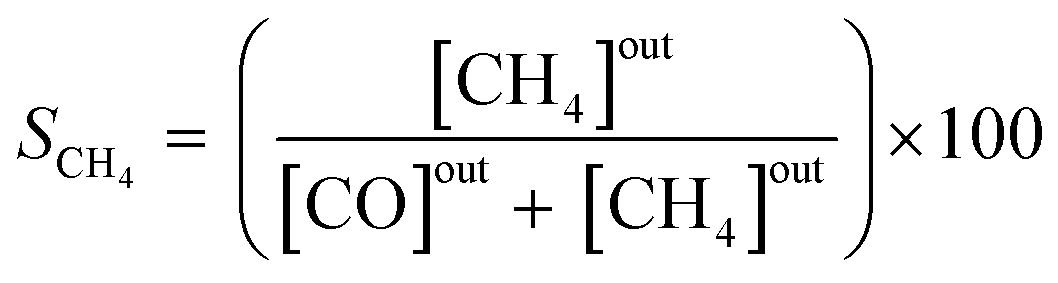
where FCO2 is the molar flow of carbon dioxide and wCuNi is the mass amount of Cu and/or Ni in the sample.
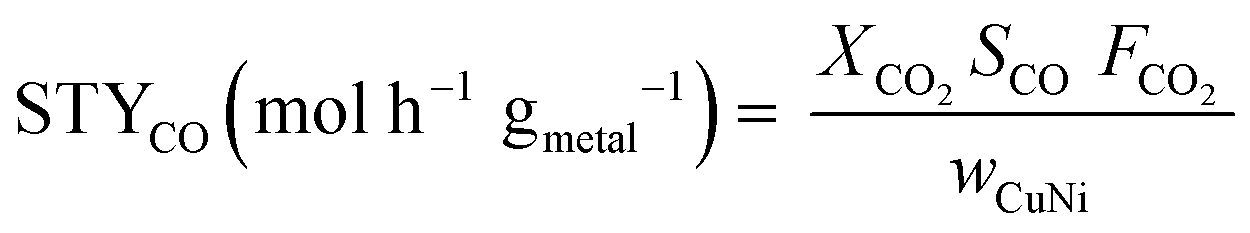
In addition, hydrogen consumption was calculated based on the CO and CH4 formation for each catalyst.
Results and discussion
Calcination process
Thermogravimetric analysis (TGA) of the impregnated catalysts was performed under analogous conditions to the calcination treatment as described in the Experimental section and is shown in Fig. S1.† Three different stages could be observed: the first one at 100 °C is related to the dehydration through evaporation of physisorbed water; the second and third one in the range of 215–260 °C and 360–410 °C respectively, are related to the decomposition of the nitrates by loss of hydration water and formation of gas products.42–45 The TGA of the second stage suggested that the temperature decomposition of the mixed salts follows a linear correlation with the Cu/Ni ratio, without the influence of the second metal (Fig. S2†).To have a clear comprehension of the decomposition process of the metallic nitrates on alumina, TG-MS analysis was performed for Cu50Ni50/Al2O3 (Fig. S3†). The dehydration process (m/z = 18; H2O) follows a two step process: the first one around 100 °C is related to physisorbed water; and the second one at 200 °C is related to the loss of structural water of the nitrates. Regarding the nitrate decomposition, only NO was detected (m/z = 30), with the first decomposition step at 180 °C, followed by the second one at 280 °C and the third one at 350 °C.46–48
In addition, elemental analysis was performed for the impregnated (before calcination) and calcined samples. In all cases, the impregnated catalysts showed a nitrogen content close to the theoretical, corresponding to the metallic nitrates deposited on alumina (Table S1†). In contrast, there was no evidence of nitrogen content for the calcined samples. Thus, the complete thermal decomposition of the metallic precursors after the calcination treatment could be confirmed.
Crystalline phases before and after activation
The crystalline phases in both calcined and reduced catalysts were characterized by powder X-ray diffraction (XRD) as described above. For the sake of comparison, the alumina support was also included. As seen from Fig. 1, all the samples present a broad peak at 37.5°, together with the peaks at 46° and 67° which are typical of the spinel-type phase of γ-alumina.49,50 Since Cu and Ni aluminates are also of spinel-type structure, it is difficult to distinguish them between the crystalline phases of the support.51–53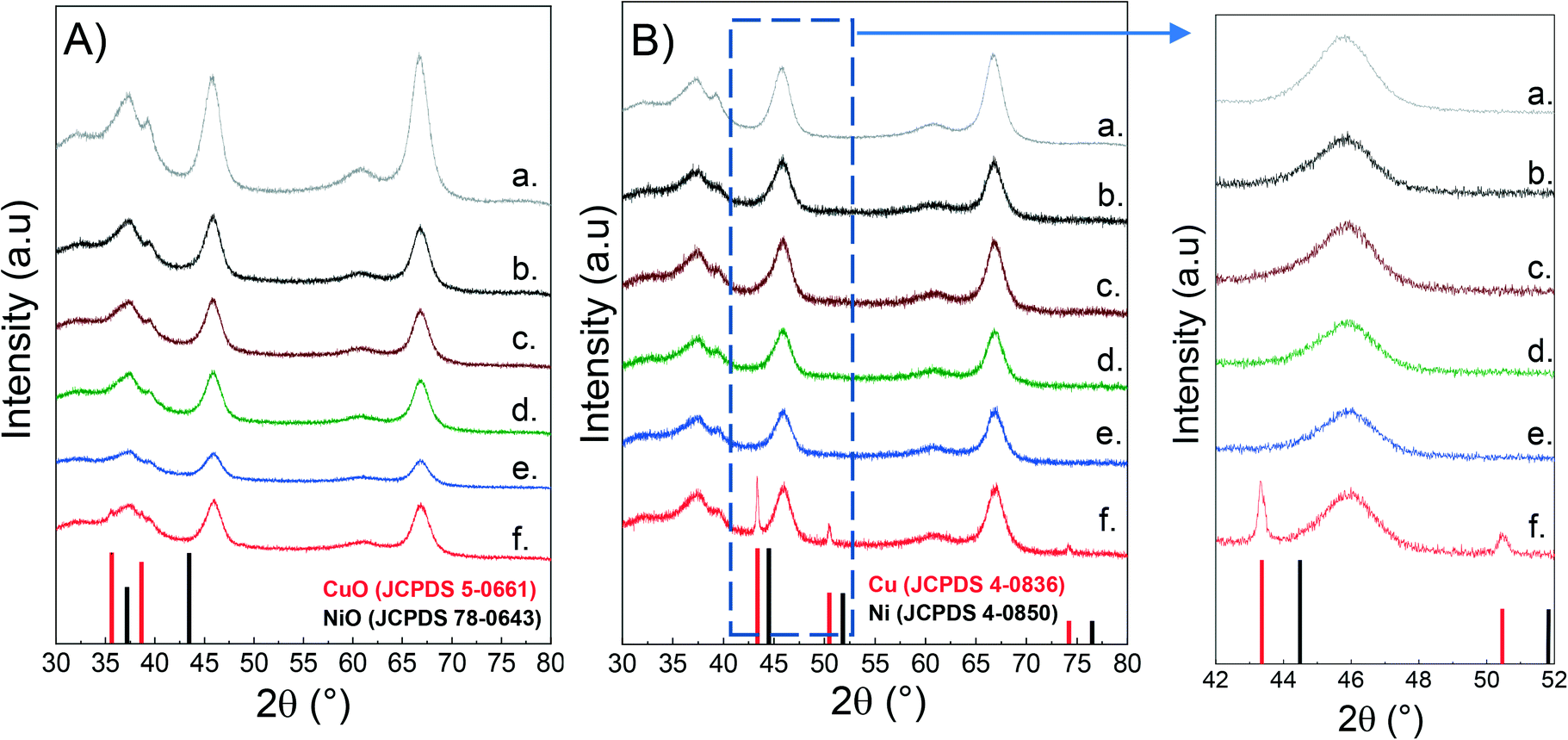 | ||
| Fig. 1 XRD of CuNi/Al2O3 catalysts: A) calcined; B) reduced; a) Al2O3; b) Ni/Al2O3; c) Cu25Ni75/Al2O3; d) Cu50Ni50/Al2O3; e) Cu75Ni25/Al2O3; f) Cu/Al2O3. | ||
Fig. 1a shows the diffractograms of the calcined catalysts. There is no evidence of either Cu or Ni oxidized species. This could be related to the presence of finely dispersed nanometric metallic oxides that cannot be detected, but also to metallic spinel species that overlap the crystalline support.54–57 However, for the Cu/Al2O3 catalyst, two peaks at 2θ 35.6° and 38.7° were observed, related to the diffractions of the (111) and (200) planes of the CuO monoclinic structure (JCPDS 5-0661).58
Fig. 1b shows the diffractograms of the reduced catalysts after activation in 10% H2 at 500 °C for 1 hour. In the monometallic Cu/Al2O3 catalyst, it can be clearly seen that Cu oxide was successfully reduced to metallic Cu (2θ: 43.29° (111), 50.43° (200) and 74.13° (220) (JCPDS 04-0836)).59,60 The absence of diffraction peaks for Ni/Al2O3 can be rationalised either by the formation of nanosized Ni nanoparticles well below the XRD detection limit and/or incomplete reduction of Ni aluminates.
Surprisingly, for the bimetallic catalysts there is no evidence of either metallic Cu or Ni. Thus, it can be inferred that Ni addition to Cu catalysts promotes the formation of small and dispersed CuNi nanoparticles, non-detectable by XRD.61 This was later confirmed by H2-TPR and STEM–EDS studies.
Reducibility of supported catalysts
The reducibility of supported metal catalysts strongly depends on the particle size, oxidation state, and metal–support and metal–metal interactions.62–65Fig. 2 shows the temperature programmed reduction (H2-TPR) profiles of both monometallic and bimetallic CuNi/Al2O3 catalysts. The Cu/Al2O3 catalyst exhibits a sharp reduction event at 244 °C with a shoulder at 219 °C, characteristic of the reduction of dispersed CuO nanoparticles weakly bonded to the surface.66,67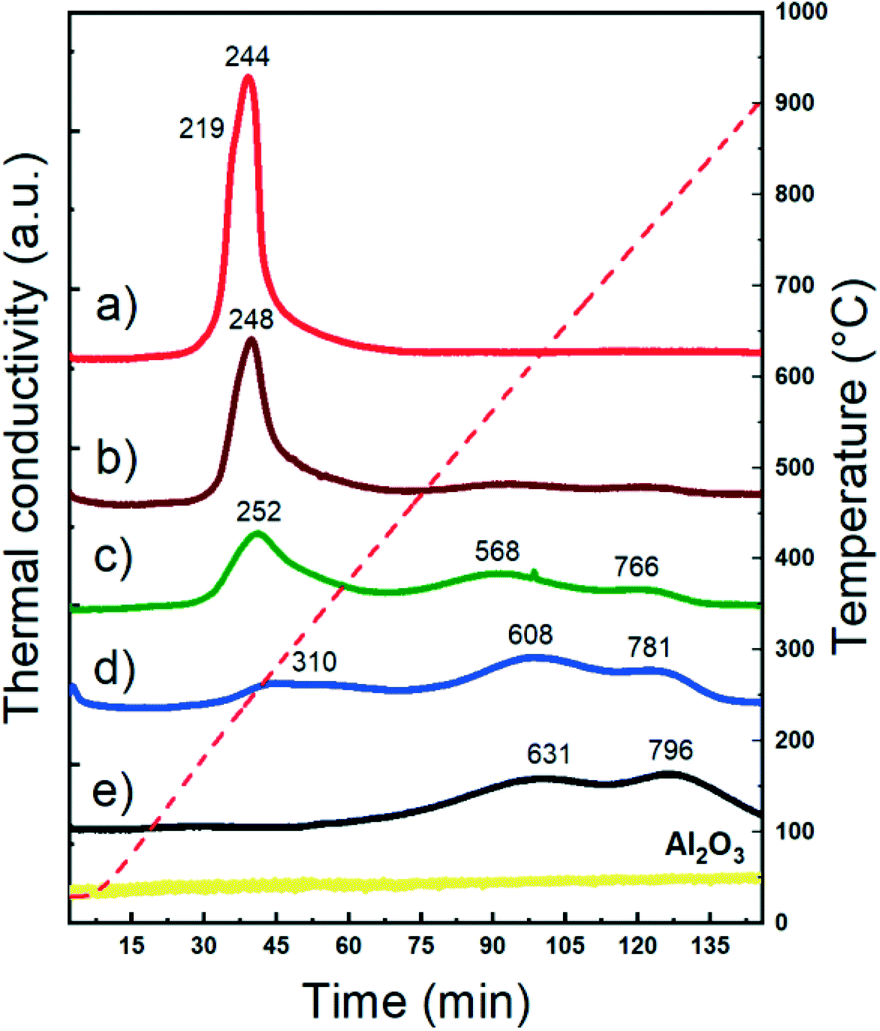 | ||
| Fig. 2 H2-TPR studies of CuNi/Al2O3 catalysts: a) Cu/Al2O3; b) Cu75Ni25/Al2O3; c) Cu50Ni50/Al2O3; d) Cu25Ni75/Al2O3; e) Ni/Al2O3. | ||
The Ni/Al2O3 catalyst shows broad reduction events at higher temperatures. It is reported that under certain synthesis conditions, e.g. calcination temperatures higher that 500 °C and metal loading below 10%, Ni nitrate decomposes forming a spinel structure with alumina.68–71 The absence of a reduction peak at 400 °C related to NiO and the reduction at high temperature indicates the presence of NiAl2O4 spinel-type species.72–74
Whether with Al2O3 or within the NiAl2O4 structure, Ni2+ strongly interacts within the framework. The first reduction event located at 631 °C is related to Ni2+oc (Ni2+ in an octahedral geometry, defined as Ni2+ ions that are not completely integrated as a spinel-type structure but in strong interaction with the support). Meanwhile, the second one at 796 °C is related to Ni2+T (Ni2+ in a tetrahedral geometry, forming nickel aluminate of spinel-type structure).72,75–77 Therefore, in agreement with the H2-TPR profiles, Ni is present as Ni2+ in strong interaction with the support in the calcined samples containing Ni.
Bimetallic catalysts exhibited different reduction profiles than their monometallic counterparts. The reduction events are located between the temperatures of the monometallic ones, which clearly suggests the synergy between CuO and Ni2+ species and thus particles in close proximity.70,78 It is already reported that Cu promotes the reducibility of Ni-based catalysts. This can be ascribed to two main contributions: i) the enhancement of the Ni reducibility due to H2 spillover produced from Cu and ii) a cooperative effect between both metals through the introduction of Cu to the Ni phase, reducing the interaction between Ni2+ and the support, and enhancing its reducibility. Indeed, from TPR it is seen that when the Cu content increased, the effect in promoting the reducibility of Ni become more pronounced.32,33,36,79,80
Table S2† shows the hydrogen consumption of the different catalysts after reduction at 500 °C. For Ni/Al2O3, 500 °C leads to a reduction degree of 13%. As expected, the reduction degree increases with the Cu content. A H2-TPR study of the calcined alumina was also performed. As expected for non-reducible supports, no hydrogen consumption was detected.
Specific surface area and porosity
Table 1 shows the surface area, total pore volume and metal composition of the different catalysts after activation in 10% H2 at 500 °C for 1 hour. The alumina support (untreated and after calcination) was included for comparative purposes. The chemical analysis performed by ICP showed that the desired Cu/Ni ratio and nominal metal loading were achieved.| Sample code | BET surfacea area (m2 g−1) | Total pore volumeb (cm3 g−1) | Cu contentc (%) | Ni contentc (%) | Total metal content (%) | Cu/Ni ratio (%) |
|---|---|---|---|---|---|---|
| a Determined by BET equation. b Obtained by non-local density functional theory (NLDF). c Determined by ICP. | ||||||
| Cu/Al2O3 | 181 | 0.397 | 4.96 | — | 4.96 | 100/0 |
| Cu75Ni25/Al2O3 | 185 | 0.411 | 3.54 | 1.17 | 4.71 | 75/25 |
| Cu50Ni50/Al2O3 | 184 | 0.400 | 2.24 | 2.25 | 4.49 | 49/51 |
| Cu25Ni75/Al2O3 | 181 | 0.401 | 1.10 | 3.21 | 4.31 | 26/74 |
| Ni/Al2O3 | 182 | 0.401 | — | 4.94 | 4.94 | 0/100 |
| Al2O3 (calcined) | 199 | 0.453 | — | — | — | — |
| Al2O3 (untreated) | 222 | 0.950 | — | — | — | — |
Fig. S4† shows the nitrogen sorption isotherms and the pore size distribution. For both alumina and supported CuNi catalysts, the adsorption–desorption branches present the characteristic H1 type hysteresis of a mesoporous type IV isotherm, according to the IUPAC classification.81 The pore size distribution showed a bimodal distribution with the main peak at 4.78 nm and a second one centred at 7.47 nm. The total pore volume decreased after impregnation, calcination and activation from 0.950 cm3 g−1 to a mean value of 0.402 cm3 g−1. However, the calcined support showed a pore volume of 0.453 cm3 g−1. Thus, the deposition of metallic species by incipient wetness impregnation does not significantly affect the textural properties of the support, without pore blockage.
Oxidation states in the near-surface region
X-ray photoelectron spectroscopy (XPS) analysis was performed to study the chemical state and the surface composition of the mono- and bimetallic catalysts after activation in 10% H2 at 500 °C for 1 hour. The samples were stored under an inert atmosphere in the glovebox and briefly in contact with air during sample holding prior to measurements.Fig. 3a shows the spectra for the Cu 2p region. The two major peaks corresponding to Cu0 2p1/2 and 2p3/2 are located at 952.6 eV and 932.7 eV, respectively. In addition, Cu2+ was detected with peaks at 956.4 eV and 935.8 eV corresponding to Cu2+ 2p1/2 and 2p3/2, respectively.82,83 The satellite components at 943 eV are generated through the O 2p to Cu 3d charge transfer effect in CuO.84,85 The presence of CuO is related to the air contact during sample preparation. As expected, the intensity decreases with decreasing the Cu content.
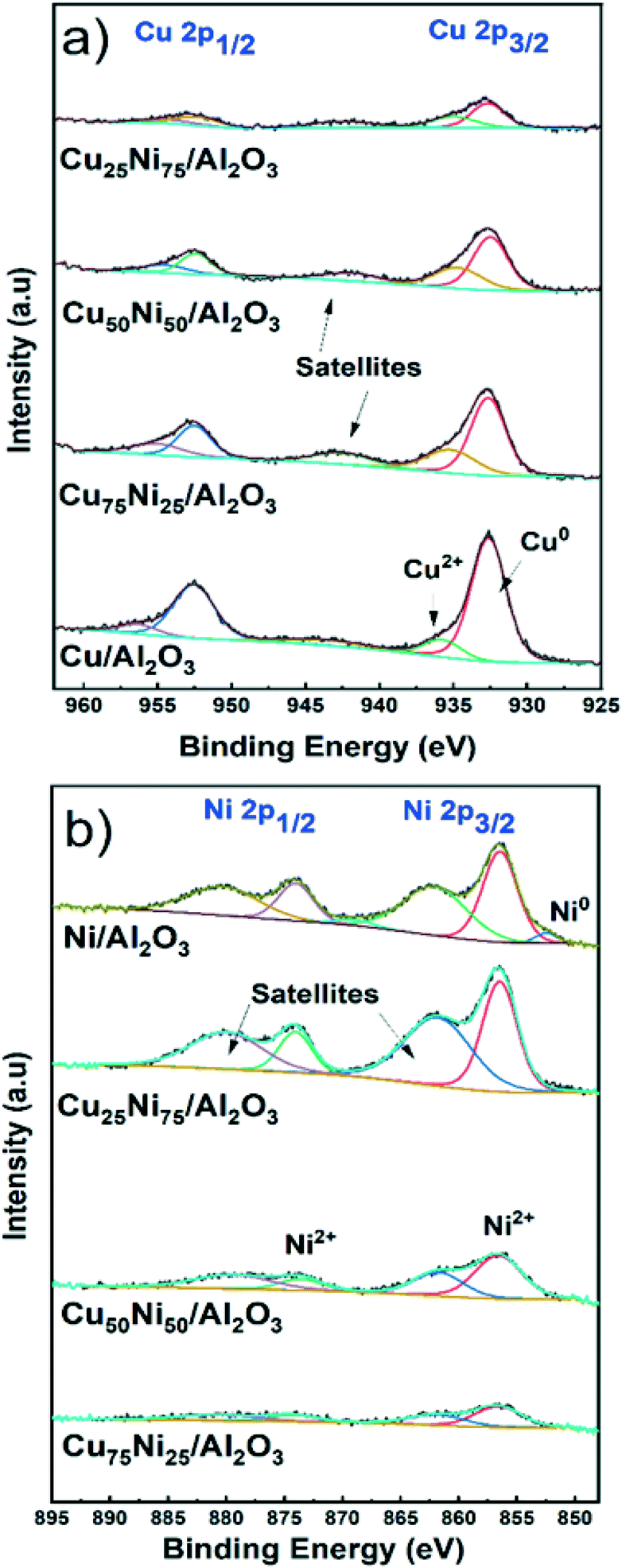 | ||
| Fig. 3 XPS spectra of the reduced catalysts. a) Cu 2p and b) Ni 2p regions. | ||
By XRD, metallic Cu could be detected only for the Cu/Al2O3 catalyst. Thus, it is inferred that small and dispersed nanoparticles were formed in bimetallic CuNi catalysts, not detectable by XRD. This was later confirmed by STEM–EDS, showing the formation of CuNi alloyed structures.
Fig. 3b shows the spectra for the Ni 2p region. Only for Ni/Al2O3, corresponding to the catalyst with the higher Ni content, a small peak at 852.5 eV related to Ni0 2p3/2 could be detected.86 Furthermore, XPS showed that only 9% of the Ni content is reduced, in agreement with the reduction degree of 13% at 500 °C determined by H2-TPR in Ni/Al2O3.
In the case of the bimetallic CuNi/Al2O3 catalysts, reduced Ni could not be detected. This is related to the low Ni content (less than 3.5%) and the small fraction of Ni0 in comparison with Ni2+ species, making the detection by XPS difficult.34,87 In addition, the effect of passivation during sample holding prior to measurements should be considered.71,88,89
Fig. 3b shows the two major peaks corresponding to Ni2+ 2p1/2 and 2p3/2, located at 873.9 eV and 856.6 eV, respectively. In addition, satellites features are located at 880.4 eV and 862.3 eV. Similarly, the satellite features are generated through the O 2p to Ni 3d charge transfer effect in Ni2+ species.87,90 The absence of peak splitting in the range 854–858 eV characteristic of NiO, and the high binding energy value are indicative of Ni2+ species in strong interaction with the support.91–95 Thus, Ni2+ is well dispersed forming spinel structures of type NiAl2O4 in all Ni-containing catalysts. Therefore, the partial reduction at 500 °C formed well distributed and small metallic nanoparticles supported on highly dispersed NiAl2O4. This is in agreement with the XRD and H2-TPR results and later confirmed by STEM–EDS.
Table S3† shows the Cu/Ni atomic ratio determined by XPS analysis. In all cases, the atomic ratio is close to the bulk composition determined by ICP. However, for both Cu75Ni25/Al2O3 and Cu50Ni50/Al2O3, the surface Cu content is slightly higher. Meanwhile, Cu25Ni75/Al2O3 showed a slight Ni enrichment, in comparison with the bulk composition.
Catalytic performance in CO2 hydrogenation
Fig. 4 shows the initial catalytic performance of the mono- and bimetallic catalysts. The evolution of the carbon dioxide conversion, selectivity to carbon monoxide and space–time yield during the 30 h time on stream can be seen in Fig. S5.† During the catalytic tests, only methane and carbon monoxide were detected as products. Hydrogen consumption was calculated based on the CO and CH4 formation for each catalyst (Fig. S6†).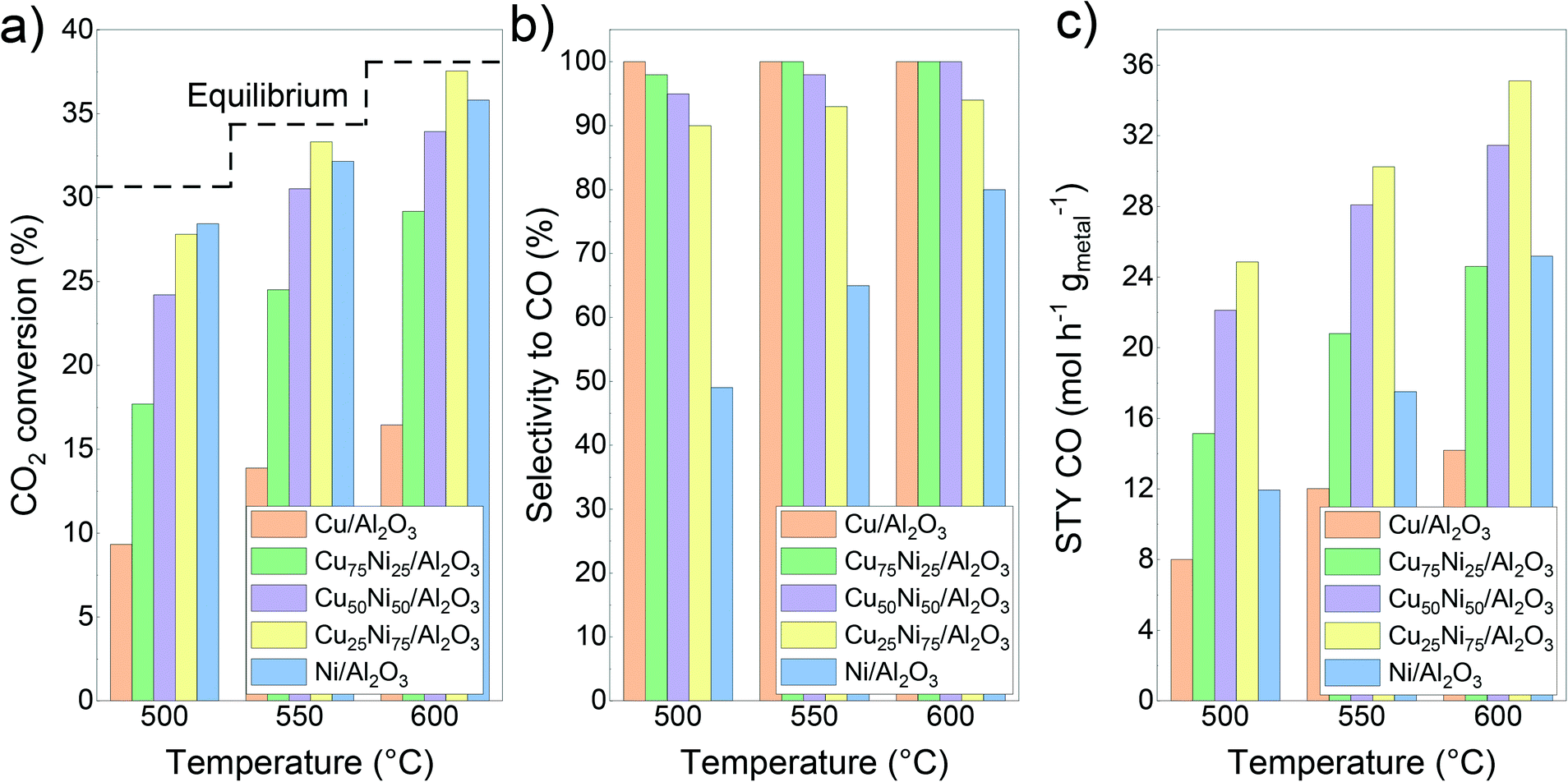 | ||
| Fig. 4 Catalytic performance of the CuNi/Al2O3 catalysts: a) CO2 conversion; b) CO selectivity; c) space–time yield of carbon monoxide. | ||
Considering the thermodynamic equilibrium and the reaction conditions (P = 1 atm; CO2:H2 = 1:1), an equimolar CO:CH4 product composition is reached at 530 °C.96 A higher temperature favours the reverse water gas shift reaction and promotes carbon monoxide production. Meanwhile, a lower temperature favours methane formation.19,22
For Ni/Al2O3, CO2 conversion reached values of 28%, 32% and 36% at 500 °C, 550 °C and 600 °C, respectively. Meanwhile, the selectivity to carbon monoxide reached values of 49%, 65% and 81% at 500 °C, 550 °C and 600 °C, respectively. In addition, no deactivation process was observed during the time on stream (Fig. S5a†). Notably, it could be observed that during the ramping down, the selectivity to carbon monoxide is increased compared with the initial one (57% vs. 49% at 500 °C and 67% vs. 65% at 550 °C, respectively, Fig. S5b†). This can be related to the continuous formation of metallic Ni sites from the reduction of the NiAl2O4 spinel, as can be inferred from the H2-TPR results. As a drawback, Ni/Al2O3 showed a higher hydrogen consumption, related to its inherent high activity and trend to form not only carbon monoxide but also methane (Fig. S6†).
In contrast, the Cu/Al2O3 catalyst showed the lowest conversion compared with the other catalysts, and a quick deactivation during the time on stream, especially at 600 °C (Fig. S5a†). This could be related to its low thermal stability and thus, rapid deactivation due to sintering.39,97,98 Therefore, the active surface area decreases over time, as was later confirmed by STEM–EDS. Regarding the selectivity, the Cu catalyst formed carbon monoxide as the only product.
Bimetallic Cu75Ni25/Al2O3 and Cu50Ni50/Al2O3 showed an intermediate catalytic performance. For both catalysts, carbon dioxide conversion values led between the corresponding to the monometallic ones, as can be seen in Fig. 4a. Moreover, the addition of Cu to the Ni-based catalyst drastically improved the selectivity to carbon monoxide, suppressing methane formation (Fig. 4b). As the main disadvantage, a clear deactivation could be observed during the time on stream, especially at 600 °C (Fig. S5a†).
In comparison to Ni/Al2O3, both Cu75Ni25/Al2O3 and Cu50Ni50/Al2O3 showed a higher space–time yield of carbon monoxide, as can be seen in Fig. 4c. However, the clear deactivation at 600 °C leads to lower values during the ramping down of temperature (Fig. S5†).
In comparison with the other mono- and bimetallic formulations, Cu25Ni75/Al2O3 showed the best performance (Fig. 4). In fact, the carbon dioxide conversion reached values close to the equilibrium. However, the conversion decreases during the ramping down (Fig. S5†), probably attributed to partial sintering of the active sites. Likewise for Cu75Ni25/Al2O3 and Cu50Ni50/Al2O3, the presence of Cu inhibited methane formation. Thus, the Cu25Ni75/Al2O3 catalyst with high conversion and selectivity reached superior space–time yields of carbon monoxide. At 500 °C, carbon monoxide production was twice that of Ni/Al2O3 with only half the hydrogen consumption.
The reaction mechanism involved in the rWGS reaction is still unclear. However, based on experimental and theoretical studies, two main routes were proposed: (i) via a redox mechanism99–102 and (ii) via an associative route with formate as an intermediate.103–106
The surface redox mechanism involves the adsorption of CO2 on the metallic active site and the oxidation of the active centres, leading to CO formation. In the following step, the metal sites are regenerated by reduction with H2, forming water as a product.107–109
The associative mechanism involves hydrogen spillover on the metallic active site. The adsorption and activation of CO2 are strongly support-dependent and can take place at the metallic site or the metal–support interface. After the formation of formate species as an intermediate, the reaction with the dissociated hydrogen leads to carbon monoxide and water as products. Therefore, in contrast to the redox mechanism, CO is derived from the association of CO2 with H2.110–113
Even though CO2 hydrogenation over nickel-supported catalysts have been extensively studied, there is not yet a broad consensus regarding the reaction mechanism. The major difference is related to the pathway of CO2 activation, where the metal–support interface plays a key role.7,114–118 For Ni/Al2O3 catalysts, it is assumed that CO and CH4 are formed through formate species adsorbed on the support. Several authors proposed that CO2 adsorbs and dissociates on metallic Ni0 forming *CO species. Then, the migration onto alumina is followed by the formation of carbonates species. In the following step, carbonates react with the dissociated hydrogen leading to formate species as an intermediate. This mechanism was evidenced by in situ FT-IR experiments. The formates can be further hydrogenated forming CH4, or desorbed leading to CO and H2O.23,119–121 Similarly, it was demonstrated for Cu/Al2O3 catalysts that the CO2 hydrogenation reaction occurs via formates as an intermediate.122–125
Moreover, it is possible to rationalize the catalytic activity and selectivity of the different catalysts taking into account the different adsorption energies of the educts and products. By temperature programmed desorption studies, it is reported that carbon dioxide preferably adsorbs on Cu sites. Thus, the addition of Cu to Ni catalysts could promote the activity in CuNi alloys. As can be seen in Fig. 4a, carbon dioxide conversion in Cu25Ni75/Al2O3 is slightly superior to that in Ni/Al2O3. However, Cu-rich catalysts showed a lower performance, related to their low thermal stability and sintering of copper.97,126
On the other hand, the hydrogen adsorption capacity is higher on Ni catalysts; meanwhile Cu presents a low hydrogen adsorption capacity. In addition, carbon monoxide is preferentially adsorbed on Ni rather than on Cu sites.127,128 Thus, Ni catalysts promote methanation due to the consecutive hydrogenation of carbon monoxide to methane.32,34,129 Therefore, Cu maximizes the synergy between both metals, promoting CO desorption and suppressing methane formation.24,36,130 Thus, the catalytic performance can be improved by promoting the close interaction between both metals, enhancing the carbon dioxide conversion and selectivity to carbon monoxide.
Particle size and elemental distribution of alloyed structures
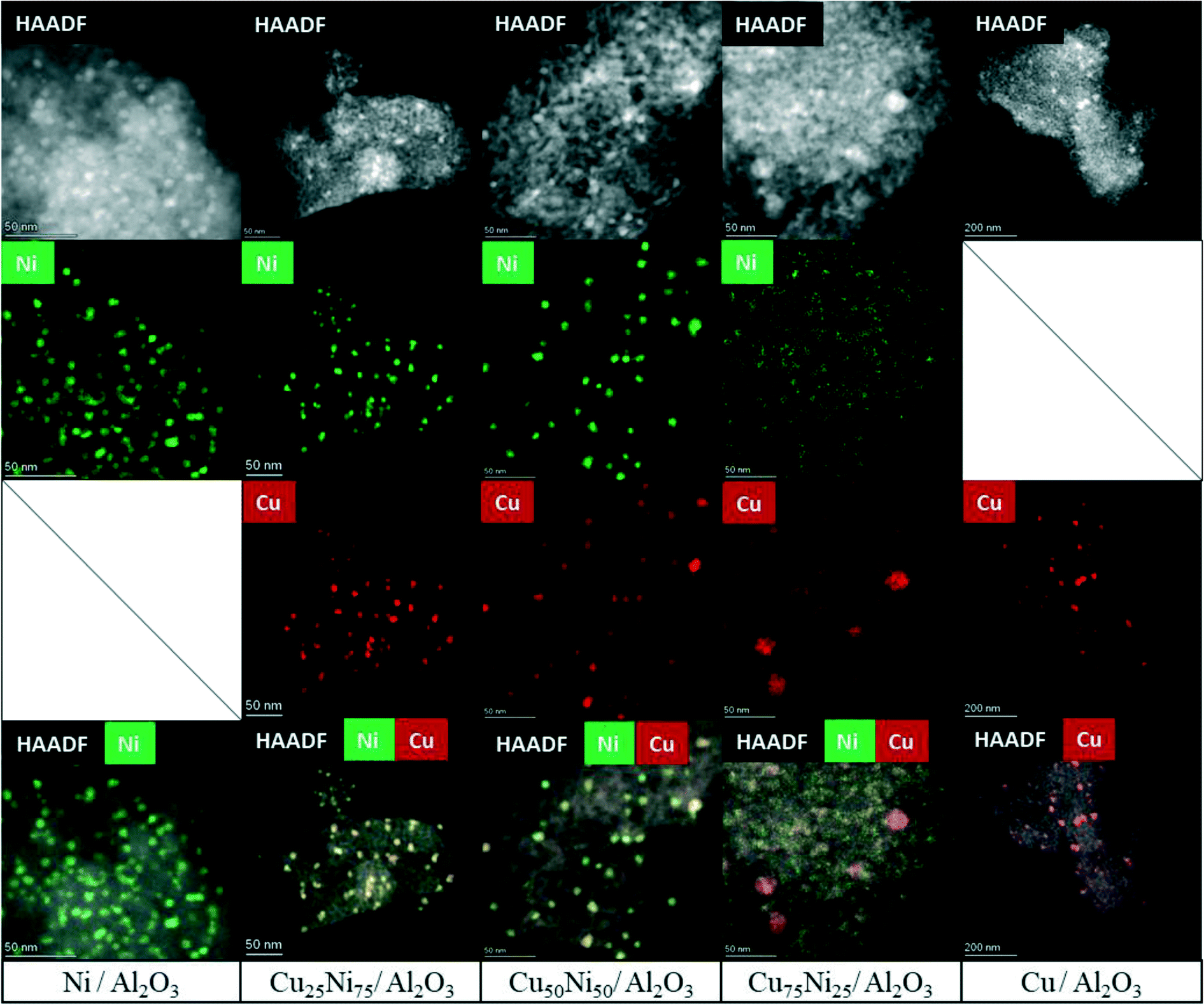
HAADF-STEM combined with EDS mapping is a powerful technique for investigating the morphology and composition of the active sites in supported multimetallic nanostructured catalysts.Before reaction, the fresh Ni/Al2O3 catalyst showed small and dispersed Ni nanoparticles of 5.2 ± 2.7 nm (Fig. 5 and S7†). After 30 h time on stream, the particle size just slightly increased showing a particle size distribution of 5.5 ± 1.9 nm. Thus, Ni nanoparticles were preserved, in agreement with the high activity and stability of the catalyst (Fig. 6 and S8†). As stated by the XPS and H2-TPR results, Ni nanoparticles are stabilized through the strong interaction with the Ni aluminate spinel phase, homogeneously dispersed on the support. In addition, small particles between 2 and 3 nm were detected on the spent catalyst (Fig. S8†). Therefore, a continuous reduction of the NiAl2O4 structure during the time on stream can be assumed for Ni/Al2O3, in agreement with the increment of the carbon monoxide selectivity. Indeed, it has been reported that the particle size of Ni nanoparticles has a prominent influence on the selectivity of the reaction. Small nanoparticles lead to CO formation, while the larger ones lead to the formation of CH4.113,131
 | ||
| Fig. 5 HAADF-STEM and EDS mapping of the fresh CuNi/Al2O3 catalysts. | ||
 | ||
| Fig. 6 HAADF-STEM and EDS mapping of the spent CuNi/Al2O3 catalysts. | ||
The fresh Cu/Al2O3 catalyst showed larger nanoparticles of 6.7 ± 2.1 nm (Fig. 5 and S9†). However, a clear sintering process took place after the reaction, forming large particles with a broad size distribution of 22.8 ± 12.9 nm (Fig. 6 and S10a†). In addition, particles up to 100 nm could be detected, in agreement with the lack of stability of supported Cu catalysts (Fig. S10b†). Thus, the lower catalytic performance of Cu/Al2O3 is related to the migration and coalescence of the nanoparticles that significantly reduces the number of available active sites.97
The HAADF-STEM–EDS images of the fresh Cu25Ni75/Al2O3 catalyst are shown in Fig. 5. It can be clearly seen that Cu and Ni form small and dispersed alloyed nanoparticles of 4.5 ± 1.2 nm, with different Cu/Ni ratios (Fig. S11†). Notably, neither Ni nor Cu was detected as segregated particles. After 30 h time on stream, a slight sintering process led to the formation of particles of 8.4 ± 2.9 nm. Some isolated agglomerates up to 20 nm could be observed (Fig. S12†). Nevertheless, the nanoparticles remained alloyed instead of forming segregated Cu and Ni phases. The tendency of the nanoparticles to stay in the alloyed state can be rationalized by the similarity of Cu and Ni (same crystal structure fcc, similar atomic radii of 117 pm and 115 pm for Cu and Ni, respectively, valence of +1 and electronegativity of 1.90 and 1.91 by Pauling scale for Cu and Ni, respectively) which favours solid solutions of bot metals in the reduced state.132,133 Thus, the outstanding performance of Cu25Ni75/Al2O3 is related to the formation of stable CuNi nanostructures (Fig. 7).
 | ||
| Fig. 7 HAADF-STEM and EDS mapping of Cu25Ni75/Al2O3 after 30 h time on stream. | ||
Considering the hydrogen consumption after reduction at 500 °C (Table S2†), Cu25Ni75/Al2O3 showed a reduction degree of 35%. Thus, the presence of the unreduced Ni spinel structure on the surface of the catalyst could be responsible for stabilizing the formed CuNi nanoalloys. A decrease in the carbon dioxide conversion during the ramping down is in agreement with the loss of the active surface area due to changes in the particle size. However, the preservation of the nanoalloy structure enhances the carbon monoxide selectivity, suppressing methane formation and leading to high CO yield during the 30 h of time on stream.
The fresh Cu50Ni50/Al2O3 catalyst showed an initial particle size distribution of 3.5 ± 1.6 nm, and similar to Cu25Ni75/Al2O3, Cu and Ni form alloyed structures with different Cu/Ni ratios (Fig. 5 and S13†). The small particle size is in agreement with the high initial catalytic activity. However, after the reaction the particle size increases to 10.3 ± 5.1 nm (Fig. 6 and S14a†). Moreover, alloyed particles up to 80 nm and a Cu/Ni ratio of 1:1 could be observed (Fig. S14b†). Thus, the sintering of the bimetallic CuNi alloys explains the deactivation. However, similar to Cu25Ni75/Al2O3 the particles remained alloyed after the reaction.
The corresponding STEM–EDS images of fresh Cu75Ni25/Al2O3 can be seen in Fig. 5 and S15.† In contrast to Cu25Ni75/Al2O3 and Cu50Ni50/Al2O3, bimetallic CuNi alloyed nanoparticles could not be detected. In fact, the catalyst is formed by well-defined Cu nanoparticles of 5.5 ± 1.9 nm, while Ni species are uniformly dispersed on the support. After the reaction, Cu75Ni25/Al2O3 showed the formation of both alloyed Cu–Ni and large monometallic Cu nanoparticles up to 20–30 nm (Fig. 6 and S16a and b†). Despite the formation of bimetallic alloys, the high Cu content tends to form sintered Cu nanoparticles, with the consequent reduction of the active surface area. This is in agreement with the low catalytic performance and clear deactivation of Cu75Ni25/Al2O3.
Fig. S17† shows the particle size distribution of both fresh and spent CuNi/Al2O3 catalysts. In addition, elemental analysis of both fresh and spent catalysts was performed (Table S4†). As can be seen, the carbon content was very low and close to the quantification limit. Thus, the deactivation process of the catalysts is attributed to sintering of the active sites and loss of surface area, instead of deposition of carbonaceous species and coke formation.
In summary, small and dispersed Ni particles were formed after activation of the Ni/Al2O3 catalyst. The nanoparticles were highly stable after 30 h time on stream with no significant changes in the particle size, in close interaction with the unreduced Ni aluminate phase on alumina. Cu75Ni25/Al2O3 as the catalyst with the lowest Ni content is composed of defined Cu nanoparticles supported on Ni aluminate homogeneously distributed on the support. The close interaction between Cu and Ni promotes the reducibility of Ni species and the thermal stability of Cu nanoparticles, in comparison with the monometallic Cu/Al2O3. This is in agreement with the catalytic performance. However, the relatively high Cu content tends to form large agglomerates.
Cu25Ni75/Al2O3 and Cu50Ni50/Al2O3 evidenced the formation of dispersed alloyed nanoparticles. Despite the sintering after the reaction, the supported particles remained alloyed even after 30 h time on stream. Thus, Cu was incorporated in the Ni catalysts rather than forming separate phases, improving the thermal stability. The sintering was significantly larger for Cu50Ni50/Al2O3 than Cu25Ni75/Al2O3, in agreement with a higher Cu content. Regarding Ni-rich catalysts, the incorporation of Cu significantly enhances the catalytic performance through the formation of well-dispersed bimetallic CuNi alloyed structures. In addition, the formation of CuNi alloys in strong interaction with the support promotes thermal stability, especially for Cu25Ni75/Al2O3.36,134
Conclusions
Supported CuNi/Al2O3 catalysts were tested in the reverse water gas shift reaction. It was found that the reducibility of Ni is increased by the addition of Cu, and the nature of the formed bimetallic nanoparticles depends on the nominal Cu/Ni ratio.Using Ni supported catalysts leads to methane formation through consecutive carbon monoxide hydrogenation. Meanwhile, Cu catalysts showed complete selectivity to carbon monoxide but low performance due to sintering and loss of active surface area.
For Cu25Ni75/Al2O3 the synergism between the thermal stability and bimetallic composition of the nanoparticles leads to better catalytic performance in terms of conversion and selectivity. CuNi alloyed nanoparticles maximize the synergy between both metals, promote the selectivity to carbon monoxide and suppress methane formation. Cu was incorporated in the Ni catalysts rather than forming separate phases, as was verified by HAADF-STEM EDS. In addition, Cu25Ni75 particles remained alloyed even after 30 h time on stream.
The incorporation of Cu into supported Ni catalysts is an excellent strategy to tune the electronic properties of the active metal, improving the selectivity to carbon monoxide through the formation of stable alloys and enhancing the performance in the reverse water gas shift reaction. Further studies should be conducted to improve the long-term stability of bimetallic nanoparticles under harsher reaction conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are given to Ina Remy-Speckmann (TU Berlin), Sophie Hund (TU Berlin), Christina Eichenauer (TU Berlin), Stephen Lohr (BasCat), Harald Link (TU Berlin), Stefanie Markstein (HU, Berlin), Dr. Matus Stredansky (Fritz Haber Institute of the Max Planck Society, Berlin) for the assistance in the characterisation of materials; and Dr. Benjamin Frank (BasCat) for thermodynamics calculations. Thanks are also given to Dr. Thomas Lunkenbein, Dr. Walid Hetaba and Dr. Christian Rohner (Fritz Haber Institute of the Max Planck Society, Berlin) for the training received through the ChemiTEM project. Funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat is also acknowledged.Notes and references
- Y. Lu, P. Zhou, J. Han and F. Yu, RSC Adv., 2015, 5, 59792–59803 RSC.
- D. X. Martínez-Vargas, L. Sandoval-Rangel, O. Campuzano-Calderon, M. Romero-Flores, F. J. Lozano, K. D. P. Nigam, A. Mendoza and A. Montesinos-Castellanos, Ind. Eng. Chem. Res., 2019, 58, 15872–15901 CrossRef.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- K. Wittich, M. Krämer, N. Bottke and S. A. Schunk, ChemCatChem, 2020, 12, 2130–2147 CrossRef CAS.
- E. Schwab, A. Milanov, S. A. Schunk, A. Behrens and N. Schödel, Chem. Ing. Tech., 2015, 87, 347–353 CrossRef CAS.
- P. Ingale, C. Guan, R. Kraehnert, R. Naumann d'Alnoncourt, A. Thomas and F. Rosowski, Catal. Today, 2021, 362, 47–54 CrossRef CAS.
- Y. A. Daza and J. N. Kuhn, RSC Adv., 2016, 6, 49675–49691 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS.
- R. Schlögl, Top. Catal., 2016, 59, 772–786 CrossRef.
- L. Pastor-Pérez, F. Baibars, E. Le Sache, H. Arellano-García, S. Gu and T. R. Reina, J. CO2 Util., 2017, 21, 423–428 CrossRef.
- E. Frei, A. Gaur, H. Lichtenberg, C. Heine, M. Friedrich, M. Greiner, T. Lunkenbein, J. Grunwaldt and R. Schlögl, ChemCatChem, 2019, 11, 1587–1592 CrossRef CAS.
- S. Sourav and I. E. Wachs, Catalysts, 2020, 10, 305–321 CrossRef CAS.
- M. Behrens and R. Schlögl, Z. Anorg. Allg. Chem., 2013, 639, 2683–2695 CrossRef CAS.
- C. J. Keturakis, M. Zhu, E. K. Gibson, M. Daturi, F. Tao, A. I. Frenkel and I. E. Wachs, ACS Catal., 2016, 6, 4786–4798 CrossRef CAS.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS PubMed.
- S. S. Kim, H. H. Lee and S. C. Hong, Appl. Catal., A, 2012, 423–424, 100–107 CAS.
- F. Sun, C. Yan, Z. Wang, C. Guo and S. Huang, Int. J. Hydrogen Energy, 2015, 40, 15985–15993 CrossRef CAS.
- L. C. Wang, M. Tahvildar Khazaneh, D. Widmann and R. J. Behm, J. Catal., 2013, 302, 20–30 CrossRef CAS.
- R. Mutschler, E. Moioli, W. Luo, N. Gallandat and A. Züttel, J. Catal., 2018, 366, 139–149 CrossRef CAS.
- A. Wolf, A. Jess and C. Kern, Chem. Eng. Technol., 2016, 39, 1040–1048 CrossRef CAS.
- Z. Zhang, Y. Tian, L. Zhang, S. Hu, J. Xiang, Y. Wang, L. Xu, Q. Liu, S. Zhang and X. Hu, Int. J. Hydrogen Energy, 2019, 44, 9291–9306 CrossRef CAS.
- J. Ashok, M. H. Wai and S. Kawi, ChemCatChem, 2018, 10, 3927–3942 CrossRef CAS.
- A. I. Tsiotsias, N. D. Charisiou, I. V. Yentekakis and M. A. Goula, Nanomaterials, 2020, 11, 28–62 CrossRef.
- L. R. Winter, E. Gomez, B. Yan, S. Yao and J. G. Chen, Appl. Catal., B, 2018, 224, 442–450 CrossRef CAS.
- Y. Wang, L. R. Winter, J. G. Chen and B. Yan, Green Chem., 2021, 23, 249–267 RSC.
- S. Sengupta, A. Jha, P. Shende, R. Maskara and A. K. Das, J. Environ. Chem. Eng., 2019, 7, 102911 CrossRef CAS.
- L. Wang, H. Liu, Y. Chen and S. Yang, Int. J. Hydrogen Energy, 2017, 42, 3682–3689 CrossRef CAS.
- J.-H. Lin, P. Biswas, V. V. Guliants and S. Misture, Appl. Catal., A, 2010, 387, 87–94 CrossRef CAS.
- A. Jha, D.-W. Jeong, W.-J. Jang, C. V. Rode and H.-S. Roh, RSC Adv., 2015, 5, 1430–1437 RSC.
- Y. Liu and D. Liu, Int. J. Hydrogen Energy, 1999, 24, 351–354 CrossRef CAS.
- Y. R. Dias and O. W. Perez-Lopez, Energy Convers. Manage., 2020, 203, 112214–112223 CrossRef CAS.
- L. Deng, X. Ai, F. Xie and G. Zhou, Chem. – Asian J., 2021, 16, 949–958 CrossRef CAS.
- W. T. Figueiredo, C. Escudero, V. Pérez-Dieste, C. A. Ospina and F. Bernardi, J. Phys. Chem. C, 2020, 124, 16868–16878 CrossRef CAS.
- L. X. Wang, E. Guan, Z. Wang, L. Wang, Z. Gong, Y. Cui, Z. Yang, C. Wang, J. Zhang, X. Meng, P. Hu, X. Q. Gong, B. C. Gates and F. S. Xiao, ACS Catal., 2020, 10, 9261–9270 CrossRef CAS.
- B. Lu, Y. Ju, T. Abe and K. Kawamoto, Inorg. Chem. Front., 2015, 2, 741–748 RSC.
- D. L. Jurković, A. Pohar, V. D. B. C. Dasireddy and B. Likozar, Chem. Eng. Technol., 2017, 40, 973–980 CrossRef.
- C.-S. Chen, W.-H. Cheng and S. Lin, Appl. Catal., A, 2004, 257, 97–106 CrossRef CAS.
- J. R. A. Sietsma, A. Jos van Dillen, P. E. de Jongh and K. P. de Jong, in Studies in Surface Science and Catalysis, Elsevier Masson SAS, 2006, vol. 162, pp. 95–102 Search PubMed.
- D. Monti and A. Baiker, J. Catal., 1983, 83, 323–335 CrossRef CAS.
- J. R. A. Sietsma, H. Friedrich, A. Broersma, M. Versluijs-Helder, A. Jos van Dillen, P. E. de Jongh and K. P. de Jong, J. Catal., 2008, 260, 227–235 CrossRef CAS.
- B. C. Miranda, R. J. Chimentão, J. B. O. Santos, F. Gispert-Guirado, J. Llorca, F. Medina, F. L. Bonillo and J. E. Sueiras, Appl. Catal., B, 2014, 147, 464–480 CrossRef CAS.
- A. Kumar, E. E. Wolf and A. S. Mukasyan, AIChE J., 2011, 57, 3473–3479 CrossRef CAS.
- W. M. Keely and H. W. Maynor, J. Chem. Eng. Data, 1963, 8, 297–300 CrossRef CAS.
- P. Jeevanandam, Y. Koltypin and A. Gedanken, Mater. Sci. Eng., B, 2002, 90, 125–132 CrossRef.
- G. A. Naikoo, R. A. Dar and F. Khan, J. Mater. Chem. A, 2014, 2, 11792–11798 RSC.
- B. Małecka, A. Łącz, E. Drożdż and A. Małecki, J. Therm. Anal. Calorim., 2015, 119, 1053–1061 CrossRef.
- J. Dong, J. Wang, L. Shi, J. Yang, J. Wang, B. Shan and M. Shen, Phys. Chem. Chem. Phys., 2017, 19, 27389–27396 RSC.
- K. M. Parida, A. C. Pradhan, J. Das and N. Sahu, Mater. Chem. Phys., 2009, 113, 244–248 CrossRef CAS.
- A. Patel, P. Shukla, G. T. Pan, S. Chong, V. Rudolph and Z. Zhu, J. Environ. Chem. Eng., 2017, 5, 2350–2361 CrossRef CAS.
- W. Hu, F. Donat, S. A. Scott and J. S. Dennis, RSC Adv., 2016, 6, 113016–113024 RSC.
- P. H. Bolt, F. H. P. M. Habraken and J. W. Geus, J. Solid State Chem., 1998, 135, 59–69 CrossRef CAS.
- J. Wu, Q. Xia, H. Wang and Z. Li, Appl. Catal., B, 2014, 156–157, 265–272 CrossRef CAS.
- N. Kanjanasoontorn, T. Permsirivanich, T. Numpilai, T. Witoon, N. Chanlek, M. Niamlaem, C. Warakulwit and J. Limtrakul, Catal. Lett., 2016, 146, 1943–1955 CrossRef CAS.
- A. Devard, P. Brussino, F. A. Marchesini and M. A. Ulla, J. Environ. Chem. Eng., 2019, 7, 103201 CrossRef CAS.
- F. J. Mariño, E. G. Cerrella, S. Duhalde, M. Jobbagy and M. A. Laborde, Int. J. Hydrogen Energy, 1998, 23, 1095–1101 CrossRef.
- H. Wang, J.-Z. Xu, J.-J. Zhu and H.-Y. Chen, J. Cryst. Growth, 2002, 244, 88–94 CrossRef CAS.
- J. L. Cuya Huaman, K. Sato, S. Kurita, T. Matsumoto and B. Jeyadevan, J. Mater. Chem., 2011, 21, 7062–7070 RSC.
- W. Li, G. Fan, L. Yang and F. Li, Green Chem., 2017, 19, 4353–4363 RSC.
- Y. S. Yun, D. S. Park and J. Yi, Catal. Sci. Technol., 2014, 4, 3191–3202 RSC.
- Z. Wu, R. Ran, Y. Ma, X. Wu, Z. Si and D. Weng, Res. Chem. Intermed., 2019, 45, 1309–1325 CrossRef CAS.
- M. Romero-Sáez, A. B. Dongil, N. Benito, R. Espinoza-González, N. Escalona and F. Gracia, Appl. Catal., B, 2018, 237, 817–825 CrossRef.
- E. Gioria, F. Marchesini, A. Soldati, A. Giorello, J. Hueso and L. Gutierrez, Appl. Sci., 2019, 9, 4075–4092 CrossRef CAS.
- C. Hernández Mejía, J. E. S. van der Hoeven, P. E. de Jongh and K. P. de Jong, ACS Catal., 2020, 10, 7343–7354 CrossRef PubMed.
- Z. Wang, H. Wan, B. Liu, X. Zhao, X. Li, H. Zhu, X. Xu, F. Ji, K. Sun, L. Dong and Y. Chen, J. Colloid Interface Sci., 2008, 320, 520–526 CrossRef CAS PubMed.
- S. J. Gentry and P. T. Walsh, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 1515 RSC.
- M. Cai, J. Wen, W. Chu, X. Cheng and Z. Li, J. Nat. Gas Chem., 2011, 20, 318–324 CrossRef CAS.
- J. M. Jehng and C. M. Chen, Catal. Lett., 2001, 77, 147–154 CrossRef CAS.
- S. Srivastava, G. C. Jadeja and J. Parikh, J. Mol. Catal. A: Chem., 2017, 426, 244–256 CrossRef CAS.
- J. L. Ewbank, L. Kovarik, F. Z. Diallo and C. Sievers, Appl. Catal., A, 2015, 494, 57–67 CrossRef CAS.
- L. Yu, M. Song, P. T. Williams and Y. Wei, Ind. Eng. Chem. Res., 2019, 58, 11770–11778 CrossRef CAS.
- D. E. Rivero-Mendoza, J. N. G. Stanley, J. Scott and K. F. Aguey-Zinsou, Catalysts, 2016, 6, 170–185 CrossRef.
- C. Jiménez-González, Z. Boukha, B. de Rivas, J. J. Delgado, M. Á. Cauqui, J. R. González-Velasco, J. I. Gutiérrez-Ortiz and R. López-Fonseca, Appl. Catal., A, 2013, 466, 9–20 CrossRef.
- R. Yang, X. Li, J. Wu, X. Zhang and Z. Zhang, J. Phys. Chem. C, 2009, 113, 17787–17794 CrossRef CAS.
- S. Weber, K. L. Abel, R. T. Zimmermann, X. Huang, J. Bremer, L. K. Rihko-Struckmann, D. Batey, S. Cipiccia, J. Titus, D. Poppitz, C. Kübel, K. Sundmacher, R. Gläser and T. L. Sheppard, Catalysts, 2020, 10, 1471 CrossRef CAS.
- C. Li and Y.-W. Chen, Thermochim. Acta, 1995, 256, 457–465 CrossRef CAS.
- A. R. Naghash, T. H. Etsell and S. Xu, Chem. Mater., 2006, 18, 2480–2488 CrossRef CAS.
- F. M. Berndt and O. W. Perez-Lopez, React. Kinet., Mech. Catal., 2017, 120, 181–193 CrossRef CAS.
- M. Kang, M. W. Song, T. W. Kim and K. L. Kim, Can. J. Chem. Eng., 2002, 80, 63–70 CrossRef CAS.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS.
- M. Zhu, T. C. R. Rocha, T. Lunkenbein, A. Knop-Gericke, R. Schlögl and I. E. Wachs, ACS Catal., 2016, 6, 4455–4464 CrossRef CAS.
- G. Moretti, Surf. Interface Anal., 1991, 17, 745–750 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- L. Lyu, L. Zhang, Q. Wang, Y. Nie and C. Hu, Environ. Sci. Technol., 2015, 49, 8639–8647 CrossRef PubMed.
- M. A. Paviotti, L. A. Salazar Hoyos, V. Busilacchio, B. M. Faroldi and L. M. Cornaglia, J. CO2 Util., 2020, 42, 101328 CrossRef CAS.
- G. Garbarino, P. Riani, A. Infantes-Molina, E. Rodríguez-Castellón and G. Busca, Appl. Catal., A, 2016, 525, 180–189 CrossRef CAS.
- A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, J. A. González-Marcos, A. Bueno-López and J. R. González-Velasco, Catal. Today, 2020, 356, 419–432 CrossRef CAS.
- P. G. Savva, K. Goundani, J. Vakros, K. Bourikas, C. Fountzoula, D. Vattis, A. Lycourghiotis and C. Kordulis, Appl. Catal., B, 2008, 79, 199–207 CrossRef CAS.
- L. Xu, F. Wang, M. Chen, J. Zhang, K. Yuan, L. Wang, K. Wu, G. Xu and W. Chen, RSC Adv., 2016, 6, 28489–28499 RSC.
- M.-M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlögl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS PubMed.
- E. Heracleous, A. Lee, K. Wilson and A. Lemonidou, J. Catal., 2005, 231, 159–171 CrossRef CAS.
- S. Danaci, L. Protasova, V. Middelkoop, N. Ray, M. Jouve, A. Bengaouer and P. Marty, React. Chem. Eng., 2019, 4, 1318–1330 RSC.
- C. Jiménez-González, Z. Boukha, B. de Rivas, J. J. Delgado, M. Á. Cauqui, J. R. González-Velasco, J. I. Gutiérrez-Ortiz and R. López-Fonseca, Appl. Catal., A, 2013, 466, 9–20 CrossRef.
- L. Zhang, J. Lin and Y. Chen, J. Chem. Soc., Faraday Trans., 1992, 88, 497–502 RSC.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, React. Chem. Eng., 2021, 6, 954–976 RSC.
- M. Li, A. Borsay, M. Dakhchoune, K. Zhao, W. Luo and A. Züttel, Appl. Surf. Sci., 2020, 510, 145439–145448 CrossRef CAS.
- Y. Yu, R. Jin, J. Easa, W. Lu, M. Yang, X. Liu, Y. Xing and Z. Shi, Chem. Commun., 2019, 55, 4178–4181 RSC.
- G.-C. Wang and J. Nakamura, J. Phys. Chem. Lett., 2010, 1, 3053–3057 CrossRef CAS.
- S. Mine, T. Yamaguchi, K. W. Ting, Z. Maeno, S. M. A. H. Siddiki, K. Oshima, S. Satokawa, K. Shimizu and T. Toyao, Catal. Sci. Technol., 2021, 11, 4172–4180 RSC.
- M.-A. Serrer, A. Gaur, J. Jelic, S. Weber, C. Fritsch, A. H. Clark, E. Saraçi, F. Studt and J.-D. Grunwaldt, Catal. Sci. Technol., 2020, 10, 7542–7554 RSC.
- W. Lin, K. M. Stocker and G. C. Schatz, J. Am. Chem. Soc., 2017, 139, 4663–4666 CrossRef CAS PubMed.
- E. Vesselli, L. De Rogatis, X. Ding, A. Baraldi, L. Savio, L. Vattuone, M. Rocca, P. Fornasiero, M. Peressi, A. Baldereschi, R. Rosei and G. Comelli, J. Am. Chem. Soc., 2008, 130, 11417–11422 CrossRef CAS PubMed.
- S. Saeidi, S. Najari, F. Fazlollahi, M. K. Nikoo, F. Sefidkon, J. J. Klemeš and L. L. Baxter, Renewable Sustainable Energy Rev., 2017, 80, 1292–1311 CrossRef CAS.
- X. Chen, Y. Chen, C. Song, P. Ji, N. Wang, W. Wang and L. Cui, Front. Chem., 2020, 8, 1–21 CrossRef PubMed.
- M. I. Alam, R. Cheula, G. Moroni, L. Nardi and M. Maestri, Catal. Sci. Technol., 2021, 11, 6601–6629 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and S. M. Sidik, Appl. Catal., A, 2014, 486, 115–122 CrossRef CAS.
- M. J. L. Ginés, A. J. Marchi and C. R. Apesteguía, Appl. Catal., A, 1997, 154, 155–171 CrossRef.
- H. Jing, Q. Li, J. Wang, D. Liu and K. Wu, J. Phys. Chem. C, 2019, 123, 1235–1251 CrossRef CAS.
- L. Yang, L. Pastor-Pérez, S. Gu, A. Sepúlveda-Escribano and T. R. Reina, Appl. Catal., B, 2018, 232, 464–471 CrossRef CAS.
- Y. Zhang, L. Liang, Z. Chen, J. Wen, W. Zhong, S. Zou, M. Fu, L. Chen and D. Ye, Appl. Surf. Sci., 2020, 516, 146035 CrossRef CAS.
- A. Goguet, S. Shekhtman, R. Burch, C. Hardacre, F. Meunier and G. Yablonsky, J. Catal., 2006, 237, 102–110 CrossRef CAS.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Catal. Sci. Technol., 2015, 5, 4154–4163 RSC.
- L. Shen, J. Xu, M. Zhu and Y.-F. Han, ACS Catal., 2020, 10, 14581–14591 CrossRef CAS.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef CAS.
- N. M. Martin, F. Hemmingsson, A. Schaefer, M. Ek, L. R. Merte, U. Hejral, J. Gustafson, M. Skoglundh, A.-C. Dippel, O. Gutowski, M. Bauer and P.-A. Carlsson, Catal. Sci. Technol., 2019, 9, 1644–1653 RSC.
- S. Jantarang, E. C. Lovell, T. H. Tan, B. Xie, J. Scott and R. Amal, Catal. Sci. Technol., 2021, 11, 5297–5309 RSC.
- N. A. A. Fatah, A. A. Jalil, N. F. M. Salleh, M. Y. S. Hamid, Z. H. Hassan and M. G. M. Nawawi, Int. J. Hydrogen Energy, 2020, 45, 18562–18573 CrossRef CAS.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Catal., B, 2020, 265, 118538 CrossRef.
- R. A. Hubble, J. Y. Lim and J. S. Dennis, Faraday Discuss., 2016, 192, 529–544 RSC.
- A. M. Bahmanpour, F. Héroguel, M. Kılıç, C. J. Baranowski, L. Artiglia, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, ACS Catal., 2019, 9, 6243–6251 CrossRef CAS.
- C. S. Chen, J. H. Wu and T. W. Lai, J. Phys. Chem. C, 2010, 114, 15021–15028 CrossRef CAS.
- C.-S. Chen, W.-H. Cheng and S. Lin, Catal. Lett., 2000, 68, 45–48 CrossRef CAS.
- A. M. Bahmanpour, F. Héroguel, M. Kılıç, C. J. Baranowski, P. Schouwink, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, Appl. Catal., B, 2020, 266, 118669 CrossRef CAS.
- K. N. Papageridis, G. Siakavelas, N. D. Charisiou, D. G. Avraam, L. Tzounis, K. Kousi and M. A. Goula, Fuel Process. Technol., 2016, 152, 156–175 CrossRef CAS.
- R. Naumann d'Alnoncourt, M. Kurtz, H. Wilmer, E. Löffler, V. Hagen, J. Shen and M. Muhler, J. Catal., 2003, 220, 249–253 CrossRef.
- A. Kitla, O. V. Safonova and K. Föttinger, Catal. Lett., 2013, 143, 517–530 CrossRef CAS PubMed.
- S. Choi, B.-I. Sang, J. Hong, K. J. Yoon, J.-W. Son, J.-H. Lee, B.-K. Kim and H. Kim, Sci. Rep., 2017, 7, 41207 CrossRef CAS PubMed.
- Z. Bian, S. Das, M. H. Wai, P. Hongmanorom and S. Kawi, ChemPhysChem, 2017, 18, 3117–3134 CrossRef CAS PubMed.
- C.-S. Chen, C. S. Budi, H.-C. Wu, D. Saikia and H.-M. Kao, ACS Catal., 2017, 7, 8367–8381 CrossRef CAS.
- G. Guisbiers, S. Khanal, F. Ruiz-Zepeda, J. Roque de la Puente and M. José-Yacaman, Nanoscale, 2014, 6, 14630–14635 RSC.
- H. R. A. Ram, K. Venkatesh, K. Gopalakrishna, K. T. Kashyap and K. S. Sridhar, J. Inst. Eng.: Ser. D, 2019, 100, 243–253 CAS.
- H.-W. Chen, C.-Y. Wang, C.-H. Yu, L.-T. Tseng and P.-H. Liao, Catal. Today, 2004, 97, 173–180 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01585k |
| This journal is © The Royal Society of Chemistry 2022 |