
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into carbamate formation from CO2 and amines: the role of guanidine–CO2 adducts†
Jere K.
Mannisto
 a,
Ljiljana
Pavlovic
*b,
Tony
Tiainen
a,
Martin
Nieger
a,
Aleksi
Sahari
a,
Kathrin H.
Hopmann
b and
Timo
Repo
*a
a,
Ljiljana
Pavlovic
*b,
Tony
Tiainen
a,
Martin
Nieger
a,
Aleksi
Sahari
a,
Kathrin H.
Hopmann
b and
Timo
Repo
*a
aDepartment of Chemistry, University of Helsinki, P.O. Box 55, A.I. Virtasen aukio 1, 00014 Helsinki, Finland. E-mail: timo.repo@helsinki.fi
bDepartment of Chemistry, UiT The Arctic University of Norway, N-9037 Tromsø, Norway. E-mail: ljiljana.pavlovic@uit.no
First published on 2nd September 2021
Abstract
Capture of CO2 by amines is an attractive synthetic strategy for the formation of carbamates. Such reactions can be mediated by superbases, such as 1,1,3,3-tetramethylguanidine (TMG), with previous implications that zwitterionic superbase–CO2 adducts are able to actively transfer the carboxylate group to various substrates. Here we report a detailed investigation of zwitterionic TMG–CO2, including isolation, NMR behavior, reactivity, and mechanistic consequences in carboxylation of aniline-derivatives. Our computational and experimental mechanistic analysis shows that the reversible TMG–CO2 zwitterion is not a direct carboxylation agent. Instead, CO2 dissociates from TMG–CO2 before a concerted carboxylation occurs, where the role of the TMG is to deprotonate the amine as it is attacking a free CO2. This insight is significant, as it opens a rational way to design new synthesis strategies. As shown here, nucleophiles otherwise inert towards CO2 can be carboxylated, even without a CO2 atmosphere, using TMG–CO2 as a stoichiometric source of CO2. We also show that natural abundance 15N NMR is sensitive for zwitterion formation, complementing variable-temperature NMR studies.
Introduction
Amines are highly important building blocks in the synthesis of natural products,1 fine chemicals2 and pharmaceuticals.3 A defining characteristic of amines is their strong affinity for various electrophiles, including CO2. Conventionally, amine-mediated CO2-capture has been widely used in industrial gas sorption processes.4 There is an increasing interest in applying amine CO2-capture for fine chemical synthesis, because CO2 is non-toxic, cheap and readily available.5,6 For example, the amine–CO2 adduct is attractive as a green pathway to N-formylation,7 methylation,7 and carbamates (Scheme 1).8–10 | ||
| Scheme 1 Selected utilizations of amine–CO2 adducts.5–10 | ||
Primary and secondary alkylamines (nitrogen bound to sp3-hybridized carbon) react rapidly with CO2 at ambient pressure and temperature. A carbamic acid is the dominant species in polar aprotic solvents (Scheme 2A).11 The situation is different in non-polar solvents, where two equivalents of amine react with CO2 to form a carbamate (salt) (Scheme 2B).11 Here, one amine acts as a Lewis base towards CO2, while the other one is a Brønsted base. Early reports in the 1990s found the addition of organic superbases (e.g. amidines and guanidines) to be critical for certain amine carboxylations.16,17 The authors proposed the formation of a mixed species of carbamate: the alkylamine acting as a Lewis base (CO2-acceptor) with the superbase as a Brønsted base (Scheme 2C). In this work, we will refer to the combination of amine and disparate base as a mixed carbamate. The term provides an important distinction to simple carbamate salts, where two equivalents of the same amine are needed for each CO2 (Scheme 2B).8,12,13 Alkyl-mixed carbamates are also reported to react with a second equivalent of CO2, forming a mixed bicarbamate (Scheme 2D).12,14,15 Clearly, the addition of a superbase markedly increases the reactivity of alkylamines with CO2.
 | ||
| Scheme 2 An overview of reactions involving CO2-capture by amines, including related nomenclature.8,11–15 | ||
The reactivity of aromatic amines (nitrogen bound to sp2-hybridized carbon) with CO2 is distinct from alkylamines; aromatic amines as such have no or low conversion (ca. 1%) to carbamic acids (Scheme 2A) or carbamates (Scheme 2B) at ambient pressure and temperature.18 Adequate reactivity is accessed by the addition of an equivalent of superbase, generating the mixed carbamate (Scheme 2C).8,12 There is no evidence of mixed bicarbamates of aromatic amines, even in presence of excess superbase.14 This and the above-mentioned low conversions suggest aromatic amines to be less reactive than alkylamines. Nevertheless, it is clear that superbases significantly increase the reactivity of aromatic amines with CO2.
Very little is known about the factors that govern mixed carbamate formation from aromatic amines in the presence of superbases. In our previous work, carboxylations were possible using a catalytic amount of superbase, which was regenerated using external base.8 It has been shown that superbases form adducts with CO2,19,20 and it has been proposed that these adducts are a form of “activated CO2” that mediate the carboxylation step.8,20–23 However, others have argued that superbase–CO2 adducts are not active intermediates in carboxylation reactions.12,24–28 Here, we present a detailed mechanistic study of superbase carboxylation reactivity, using in situ NMR studies, isolated reaction intermediates and computational modelling.
Results and discussion
Mixed carbamate mechanistic proposal
To gain insights into mixed carbamate formation with aromatic amines, we first investigated the reaction of 4-nitroaniline (1) with CO2 by NMR. There was no background reaction of 1 under CO2 (1 atm) in d6-DMSO. However, in presence of the superbase 1,1,3,3-tetramethylguanidine (TMG), we observed the mixed carbamate 2 in an equilibrium yield of 78% (Scheme 3).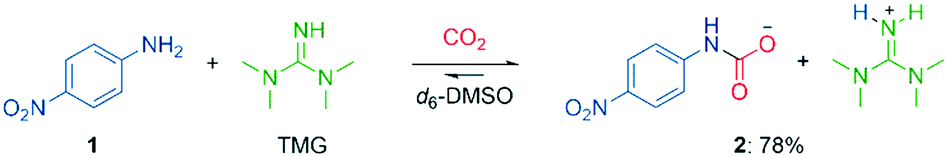 | ||
| Scheme 3 Carboxylation of 4-nitroaniline (1) in presence of TMG and CO2 in d6-DMSO. Yield determined by 1H NMR on 0.50 mmol scale. | ||
Our results prove that the superbase is essential for mixed carbamate formation from aromatic amines, but they do not give insights into the specific role of the base. We can envision at least three mechanistic possibilities for generation of mixed carbamates from aromatic amines in presence of a superbase (Scheme 4). Path 1 consists of a nucleophilic self-carboxylation. This path is based on recent reports that alkylamines can self-carboxylate with CO2, forming highly unstable zwitterions, which are deprotonated to carbamate salts.11 The analogous mechanism with aromatic amines involves two steps (Scheme 4, top): step A consists of a nucleophilic attack of the amine 1 on CO2, forming zwitterion 3. Step B consists of a rapid deprotonation by the base, forming the mixed carbamate 2. Thus in path 1, the superbase acts as a simple Brønsted base after carbamate formation has taken place.
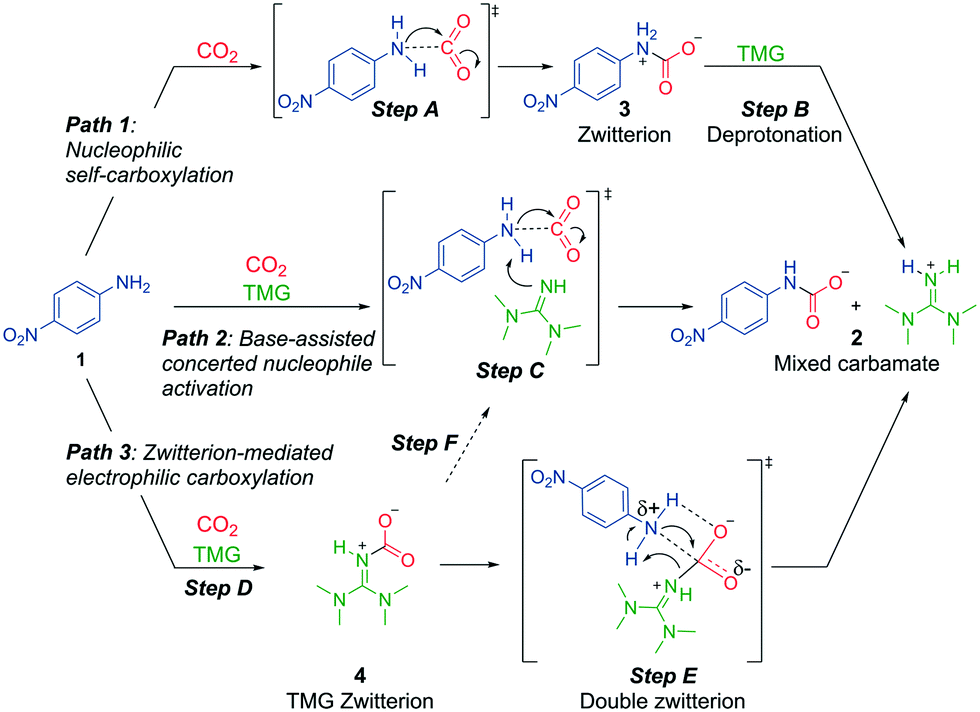 | ||
| Scheme 4 Three possible mechanistic pathways for mixed carbamate formation from aromatic amines and superbases (in this case 4-nitroaniline and TMG). | ||
Alternatively, in path 2 (Scheme 4, middle), the superbase may pre-associate with amine 1, activating it through a concerted event of deprotonation and carboxylation (step C). The concerted mode of activation implies that the nucleophile heteroatom not only must be protic, but also have an available lone pair for interaction with CO2. In this path, the superbase acts as Brønsted base and as a nucleophile activator. Similar bimolecular Brønsted base-assisted carboxylations have been proposed.26,29
In path 3 (Scheme 4, bottom), we assume that CO2 and the superbase form zwitterionic adduct 4 (step D).19,20,30 Mixed carbamate formation may then occur through concerted electrophilic carboxylation (step E). At the TS, the zwitterion may be stabilized by hydrogen bonding of the carboxylate and the ammonium protons. Eventually, the zwitterion collapses, simultaneously transferring the carboxylate to 1 and protonating TMG. In this putative mechanism, TMG acts as a transfer-carboxylation agent (as previously proposed in the literature),20,30 and as a Brønsted base. We note that there also exists the possibility that any formed TMG-zwitterion dissociates prior to carboxylation, which effectively would lead back to path 2 (step F, Scheme 4). The different mechanistic possibilities are investigated in detail below.
The effect of organic base
As part of the mechanistic analysis, we first investigated the effect of various organic bases in the carboxylation of 4-nitroaniline (Table 1). We began by testing alkylamines, which are known to form carbamate salts (Scheme 2B); hence, they may also be able to promote the reaction of 4-nitroaniline and CO2. Alkylamine DIPA did produce a low amount of mixed carbamate (9%, Table 1, entry 2), in line with previous results (7–17%).12 Assuming path 1 or 2 are active (Scheme 4), non-nucleophilic bases should produce mixed carbamate via simple deprotonation. In path 3 (Scheme 4), the base must first form a CO2-zwitterion for carboxylation to take place. As DIPA is a secondary amine, it can form a transient CO2-zwitterion,11 satisfying the criteria of path 3. Therefore, we wanted to examine a non-nucleophilic base of similar basicity (relevant for path 1 and 2), but unable to form a CO2-zwitterion, eliminating path 3. In this regard, we used the more bulky DIPEA, which readily scavenges protons, but does not react with most electrophiles,31 including CO2.29,32 No mixed carbamate was observed with DIPEA (Table 1, entry 3). Given that DIPA and DIPEA have very similar basicity, DIPEA should be a strong enough base to deprotonate the nucleophile after N–CO2 bond formation in path 1, or at the TS of path 2. Since no mixed carbamate was observed, it follows that the formation of a CO2-zwitterion could be essential (path 3 is active), or that there are other factors than basicity that influence the reaction outcome (such as sterics of the base).29|
|
|||
|---|---|---|---|
| Entry | Base | Yielda (%) | pKab (CH3CN) |
| a Yield of carboxylated 4-nitroaniline determined by 1H NMR on 0.50 mmol scale. b pKa values in CH3CN are provided, because data in DMSO is not available for all compounds. c No carbamic acid nor carbamate salt. Diisopropylamine = DIPA, N-ethyldiisopropylamine = DIPEA, 1,8-diazabicyclo[5.4.0]undec-7-ene = DBU, 1,5,7-triazabicyclo[4.4.0]dec-5-ene = TBD, 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene = MTBD. | |||
| 1 | 4-Nitroaniline | 0c | 6.2 (ref. 36) |
| 2 | DIPA | 9 | 18.8 (ref. 37) |
| 3 | DIPEA | 0c | 18.6 (ref. 38) |
| 4 | TMG | 78 | 23.4 (ref. 39 and 40) |
| 5 | tBuTMG | 92 | 26.5 (ref. 41) |
| 6 | DBU | 82 | 24.3 (ref. 39) |
| 7 | TBD | 68 | 26.0 (ref. 39) |
| 8 | MTBD | 85 | 25.5 (ref. 39) |
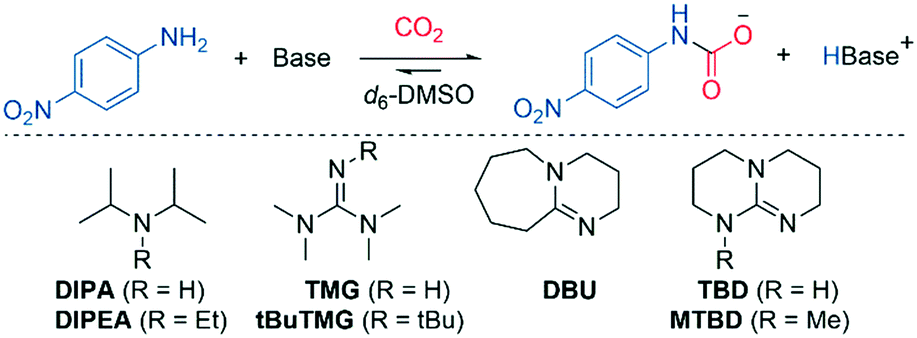
We proceeded to investigate superbases, which are more basic than alkylamines (Table 1, entries 4–8). In general, the yields for these were quite similar; however, bases with a higher Brønsted basicity than TMG did produce more of the mixed carbamate (entries 5, 6 and 8), displaying a linear relationship (R2 = 0.96, see ESI,† Fig. S1). An exception was TBD, which provided plenty of precipitate and a low amount of mixed carbamate despite high basicity (68%, entry 7). The experiment was repeated at a smaller scale (0.2 mmol) with identical precipitation and yields. Presumably, the low yields are due to unreacted TBD hydrogen-bonding to the mixed carbamate, and the resulting adduct precipitates out.33 The methylated analogue MTBD was unable to act as a hydrogen bond donor, eliminating competing adduct formation; hence, more carbamate was observed (85%, entry 8, Table 1). Superbase nucleophilicity did not correlate with mixed carbamate yield (R2 = 0.09, TBD excluded from plot, Fig. S2).34,35 Overall, the combined superbase results do not provide an unambiguous preference between the mechanistic pathways (Scheme 4).
To obtain more insights into the mechanistic details, we modelled the carboxylation of 4-nitroaniline employing DFT methods (PBE0-D3BJ[IEFPCM] with DMSO as the implicit solvent model, see ESI† section 4). Initially we evaluated the attack of 4-nitroaniline at a free CO2, corresponding to path 1 (Scheme 4). A TS for this transformation could only be located if an explicit DMSO solvent molecule was added to the molecular model to stabilize the emerging positive charge on nitrogen (Fig. S3†). Although the computed barrier of around 20 kcal mol−1 appears surmountable at 298 K (Table 2, entry 1), the resulting protonated carbamate intermediate is highly endergonic, making its formation (and hence path 1) unlikely. Addition of TMG to the model allowed us to compute path 2 (Scheme 4), where the role of the base is to activate the nucleophile.26 In this model, 4-nitroaniline and TMG form a pre-associated complex, which is slightly more stable than the separated fragments (Fig. S4†), before attack on CO2 occurs. Pre-association is supported by 1H NMR studies, which show a downfield shift of the –NH2 of 4-nitroaniline by 0.07 ppm in the presence of TMG under argon. The concerted deprotonation by TMG and attack of the amine on CO2 (Fig. 1, left) has a barrier of 16.9 kcal mol−1 (Table S1,† entry 3), which decreases by 2 kcal mol−1, if an explicit DMSO molecule is included to further stabilize the emerging charges (Table 2, entry 4). The mixed carbamate formation with TMG is computed to be significantly exergonic (Table 2, entry 4), qualitatively supporting the experimentally observed product formation. The computed barrier for the formation of mixed carbamate with TMG is lower by 3.1 kcal mol−1 and 3.4 kcal mol−1 relative to the computed barriers for DIPA and DIPEA, respectively.
| Entry | Base | ΔG≠b | ΔGr |
|---|---|---|---|
| 1 atm [1 M] | 1 atm [1 M] | ||
| a PBE0-D3BJ/IEFPCM, see ESI† section 4 for details. Energies are relative to the pre-associated complex 4-nitroaniline-DMSO (entry 1) or 4-nitroaniline-base-DMSO (entries 2–5) and are given as computed (1 atm) and with a standard state correction to 1 M. b Barrier for path 2 (Scheme 4). c Barrier for path 1 (Scheme 4). | |||
| 1 | 4-Nitroaniline | 20.9 [19.0]c | 21.0 [19.1] |
| 2 | DIPEA | 20.2 [18.3] | 2.6 [0.7] |
| 3 | DIPA | 19.9 [18.0] | 1.7 [−0.2] |
| 4 | TMG | 16.8 [14.9] | −2.2 [−4.1] |
| 5 | tBuTMG | 15.4 [13.5] | −6.2 [−8.1] |
 | ||
| Fig. 1 Computed Gibbs free energies (1 M energies, kcal mol−1, 298 K, PBE0-D3BJ/IEFPCM) and schematic TS structures for carboxylation of 4-NO2–aniline via path 2 (TSC) compared to formation of the TMG–CO2 zwitterion (TSD). | ||
The alternative path 3 involves formation of a TMG–CO2 zwitterion (Scheme 4). In our calculations, formation of this complex has a low barrier and is slightly endergonic (Fig. 1, right side), indicating that TMG–CO2 may be present in the reaction mixture in low concentrations. However, a transition state for CO2 transfer from TMG–CO2 to 4-nitroaniline could not be located, as the zwitterion always dissociates prior to formation of the aniline–CO2 bond. From this, we conclude that a TMG–CO2 zwitterion is unlikely to be an active carboxylation agent and that aniline carboxylation most likely occurs through path 2, where TMG activates the nucleophile through concerted deprotonation. In this scenario, any formed TMG–CO2 zwitterion would simply be a reversible side product in the reaction mixture.
Calculations on other organic bases (Table 2, entries 2,3,5) show good agreement with the experimental results (Table 1), further supporting that path 2 is the operative mechanism. The computed reaction energy for tBuTMG is more exergonic than that for TMG, whereas with DIPEA and DIPA, the carboxylation barrier increases and the reaction is less exergonic. We also performed additional calculations with B3LYP-D3 as an alternative method. The same trend is observed, where the computed barriers and reaction energies with DIPA and DIPEA are higher relative to TMG (Tables S1 and S2†). A direct quantitative agreement between the computed reaction energies (Table 2) with the observed reaction yields (Table 1) cannot be expected for the simple computational model used here (which treats the bulk solvent as a homogeneous medium). However, we note that the general trend is reproduced well, indicating that the computational model is able to capture the relative effects that govern the reactivities.
Substituent effects
The effect of aniline substitution was investigated through a Hammett study on the product equilibria of various 4-substituted anilines (Fig. 2). The conditions were similar as in Table 1, employing TMG as a base. Under these conditions, unsubstituted aniline8 and 4-methoxyaniline converted fully to the mixed carbamate (ESI† section 16). Since electron-rich anilines carboxylated quantitatively, we excluded them from the Hammett study.42 The Hammett equation is usually used in the context of reaction rates, but the equation also applies to equilibria (eqn (1)), and can be rearranged into a linear form (eqn (2)).43 | (1) |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = σρ + logK0 K = σρ + logK0 | (2) |
:1 manner (eqn (3), R ≠ H).14 Our experiments were performed with an equimolar amount of the individual aniline and TMG. Therefore, the equilibrium constant K (eqn (4)) could be simplified (eqn (5)). Equilibrium contribution of zwitterion 4 is considered negligible. Next, we defined F as the fraction of mixed carbamate formed in relation to the total amount of aniline (eqn (6)). We were able to measure F conveniently by 1H NMR spectroscopy. Eqn (5) and (6) allowed us to define the equilibrium constant K as a function of F (eqn (7)). Next, by substituting K in eqn (2), we were able to express the Hammett relationship as a function of F (eqn (8)). For derivation details, see ESI† section 5.
 | ||
| Fig. 2 Left: Experimental Hammett study of 4-substituted anilines, showing correlation between log[(F/1 − F)2] and σ at 25 °C. Fraction of mixed carbamate (F) in parenthesis. Middle: Computed Hammett plot, showing correlation between σ and the rate constant k (calculated from computed ΔG≠, 1 atm, Table 3), and right: computed Hammett plot, showing correlation between σ and the equilibrium constant K (calculated from computed ΔGr, 1 atm, Table 3). | ||
In our experiments, mixed carbamate formation was assumed to be in rapid equilibrium, and stable at the time of the measurement by NMR, as confirmed by performing a second NMR experiment 8 or 12 h later with identical results (ESI† section 17). In addition, all measurements were done at the same CO2 pressure (1 atm), thus we assumed the concentration of CO2 was constant in all samples.
| RC6H4NH2 + TMG + CO2 ⇌ RC6H4NHCO2− + TMGH+ | (3) |
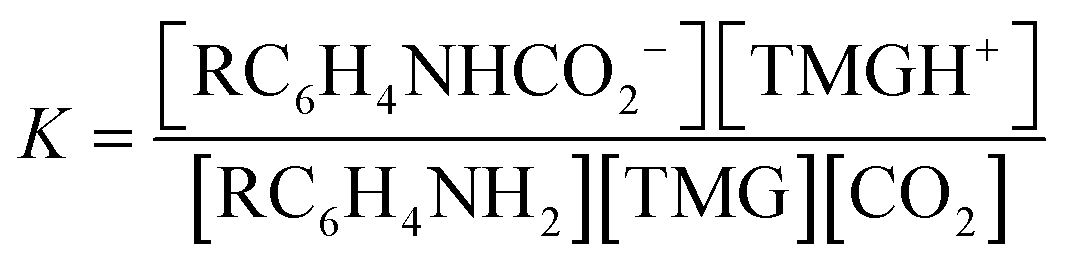 | (4) |
 | (5) |
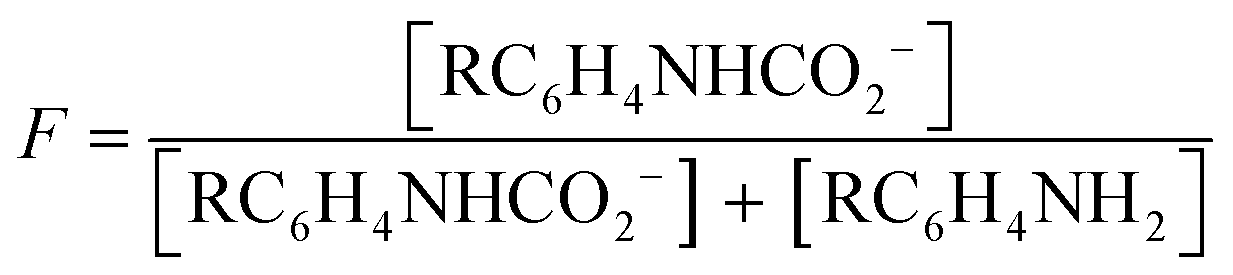 | (6) |
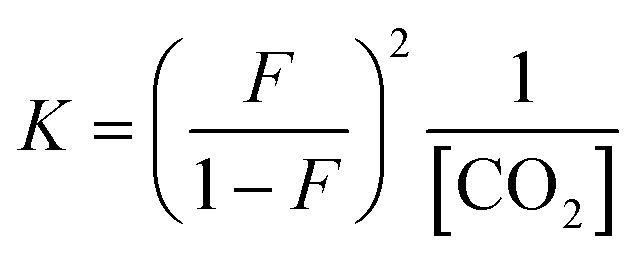 | (7) |
 | (8) |
In order to evaluate the reliability of our computational approach, we computed the Hammett plot based on a molecular model composed of TMG, an aromatic amine and CO2. Gratifyingly, the correlation between the Hammett constants and the computed reaction energies (Table 3) is very good (R2 = 0.97, Fig. 2, right) and is consistent with the experimental product yields (Fig. 2, left). We note that the computed slope is the same regardless if 1 atm or 1 M standard state energies are employed. The results indicate that the computational model is able to capture the trend of these systems well. However, the computed exergonicity for carboxylation of the less electron-deficient substrates appears somewhat too large (Table 3) compared to experiment (Table 1). Interestingly, the computed barriers for path 2 with the different anilines (Table 3) also provide a relatively good correlation with σ (R2 = 0.93, Fig. 2, middle).
| Substituent | Base | ΔG≠b 1 atm [1 M] | ΔGr 1 atm [1 M] |
|---|---|---|---|
| a PBE0-D3BJ/IEFPCM, see ESI† section 4 for details. Energies are relative to the 4-substituted aniline–TMG pre-associated complex and are given as computed (1 atm) and with a standard state correction to 1 M. b For path 2 (Scheme 4). | |||
| –Cl | TMG | 13.2 [11.3] | −4.5 [−6.4] |
| –OCF3 | TMG | 14.0 [12.1] | −3.4 [−5.3] |
| –CF3 | TMG | 14.6 [12.7] | −2.7 [−4.6] |
| –CN | TMG | 16.9 [15.1] | −2.2 [−4.1] |
| –NO2 | TMG | 18.8 [16.9] | −0.8 [−2.7] |
| –SO2CF3 | TMG | 19.3 [17.4] | −0.3 [−2.2] |
The combined experimental and computational results indicate that the most likely mechanistic pathway for mixed carbamate formation is the concerted path 2 (Scheme 4), where the role of the superbase is to activate and deprotonate the nucleophile at the carboxylation TS. However, the computations also clearly indicate that a TMG–CO2 zwitterion is kinetically and thermodynamically accessible. We thus turned towards exploring this species further.
Isolated zwitterion of TMG and CO2
The existence of zwitterionic CO2 adducts of superbases has been debated for nearly two decades. Many studies have tried to capture this elusive species, yet only bicarbonate salts have been isolated due to the presence of residual water.25,30,45 In a major breakthrough, Villiers and co-workers isolated the adduct of CO2 and TBD, stabilized by an internal hydrogen bond.19 Since then, CO2 capture has been observed for cyclic superbases TBD and MTBD in solution, but no reactivity was observed for acylic TMG.11,24 However, TMG–CO2 zwitterion 4 has been observed as a minor component in a solid mixture, but its isolation in pure form was not reported.20We reasoned that 4 is transient at room temperature (RT) in solution, as indicated by the computational results (Fig. 1), yet 4 could be more stable at lower temperatures. Low temperature experiments on superbases have been suggested,25 but to the best of our knowledge, the only successful application has been made with N-heterocyclic imines (NHI),46 and none with more common superbases, such as TMG or DBU.
We saturated an acetonitrile solution of liquid TMG with CO2 (1 atm) at RT under strictly anhydrous conditions. The clear solution was stored in a freezer overnight, depositing a white amorphous solid. Filtration of the cold solution using Schlenk technique under CO2 yielded TMG–CO2 zwitterion 4 as a white solid (62% yield). When placed in d6-DMSO, the solid produced small bubbles of CO2. Analysis of the solution by NMR showed significant 1H downfield shift of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH to 6.58 ppm (freebase 5.34 ppm), and the carbonyl shifted upfield to 165.4 ppm 13C (freebase 166.2 ppm). A long 13C NMR experiment revealed the carboxylate at 126.8 ppm as a broad signal (Fig. 3). Moreover, identical 13C spectra were obtained when the zwitterion was generated in situ from TMG under CO2 (1 atm). Known DBN–CO2 zwitterion displayed a similar broad carboxylate signal at 136.3 ppm in d6-DMSO.46 Note that free CO2 appears as a sharp signal at 124.2 ppm in d6-DMSO.47
NH to 6.58 ppm (freebase 5.34 ppm), and the carbonyl shifted upfield to 165.4 ppm 13C (freebase 166.2 ppm). A long 13C NMR experiment revealed the carboxylate at 126.8 ppm as a broad signal (Fig. 3). Moreover, identical 13C spectra were obtained when the zwitterion was generated in situ from TMG under CO2 (1 atm). Known DBN–CO2 zwitterion displayed a similar broad carboxylate signal at 136.3 ppm in d6-DMSO.46 Note that free CO2 appears as a sharp signal at 124.2 ppm in d6-DMSO.47
 | ||
| Fig. 3 13C{1H} NMR spectrum of zwitterion 4, dissolved in d6-DMSO in air. | ||
Adduct formation between a Lewis base and a Lewis acid is routinely assessed within frustrated Lewis pair (FLP) chemistry using NMR.48–51 It is generally accepted that a lack of apparent change in chemical shift (at room temperature) does not exclude adduct formation.48–51 The absence of any observable change in chemical shift simply indicates any hypothetical adduct equilibrium must lie far to the dissociation side.48 A reliable way to determine if an equilibrium process is present or not, is to perform a variable-temperature (VT) NMR study.49–51 In this regard, we studied TMG in d7-DMF, similar in properties to d6-DMSO but melting at lower temperatures (Fig. 4). Upon applying CO2 (1 atm) at 25 °C, the broad carboxylate signal appeared at 125.9 ppm. The 1H signal of NH shifted downfield to 5.56 ppm, while the corresponding 15N signal shifted upfield by 1.0 ppm to −207.4 ppm. These changes indicate NH is attaining increased sp3-character through quaternarization, supporting the formation of TMG–CO24. The general behavior of 1H and 15N signals of NH continued as the temperature was lowered, reaching 5.91 ppm and −209.5 ppm at 0 °C. Likewise, the carboxylate 13C signal was sensitive to temperature, becoming sharper and moving downfield to 128.7 ppm at 0 °C. The thermal behavior of 1H, 15N and 13C signals shows that 4 is stabilized by lower temperatures, thereby being present in larger concentrations. At subzero temperatures, 4 began to precipitate out, and the carboxylate signal was lost. The 1H signal of NH continued the previous trend, reaching 6.42 ppm and becoming very broad. In contrast, the 15N signal moved downfield to −205.8 ppm, which likely corresponds to the decreased concentration of 4 in solution. Overall, 15N NMR appears to be highly sensitive for changes at the CO2-bearing nitrogen (imine-like NH), reflecting the subtle changes observed by 1H and 13C NMR. Although we obtained good quality spectra generally in ca. 15 h, 15N-labelling of the CO2-bearing nitrogen could be advantageous if shorter measurements are desired.
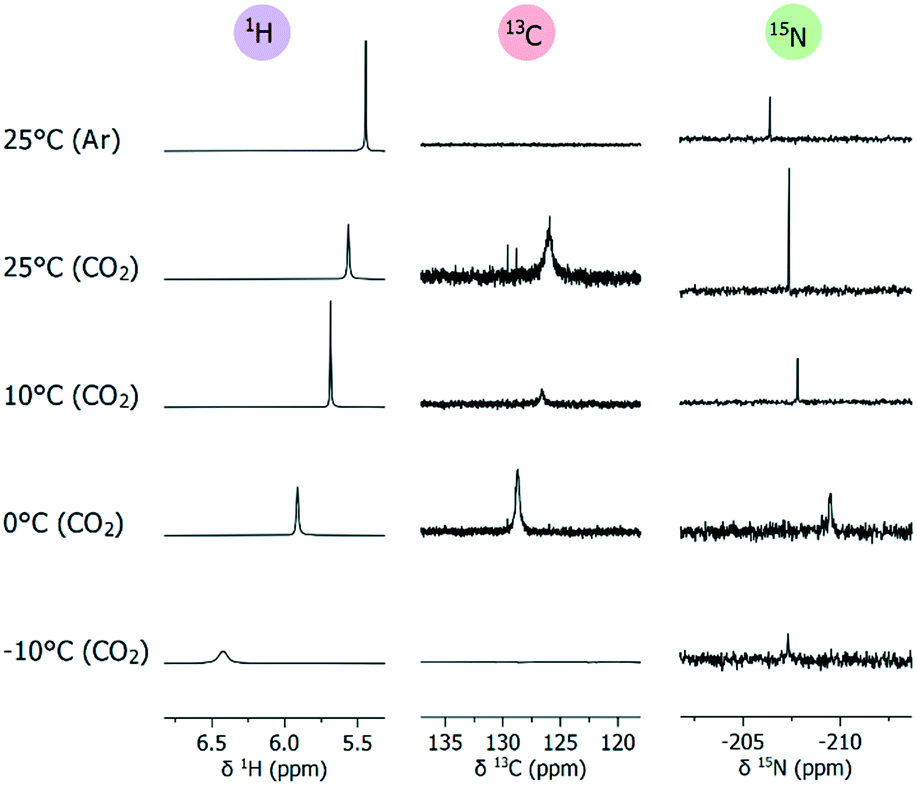 | ||
| Fig. 4 Variable-temperature NMR spectra (selected regions) of in situ generated zwitterion 4 in d7-DMF. 15N NMR (1H-coupled) obtained at natural abundance. For complete spectra, see ESI† section 9. | ||
To demonstrate the general usefulness of VT and 15N NMR in studying superbase–CO2 adducts, we investigated formation of DBU–CO2, which has been debated.25,30,46 We were able to observe the characteristic broad carboxylate 13C signal at 125.6 ppm in d7-DMF at 25 °C under CO2 (1 atm, Fig. S28†). Although changes in 1H and 13C NMR were minimal at 25 °C, 15N NMR revealed a 2.7 ppm upfield shift for the imine-like nitrogen, indicating zwitterion formation. As the temperature was decreased, the carboxylate 13C signal intensified and sharpened, reaching 126.7 ppm at −70 °C, which was accompanied by significant line broadening in 1H NMR. Our results indicate zwitterionic DBU–CO2 can form, but it is significantly more unstable than TMG–CO2 or DBN–CO2. Calculations predict DBU–CO2 formation to be exergonic at −50 °C and below (ESI† section 4.6). Furthermore, calculations suggest DBN–CO2 is stabilized by C–H⋯O interactions between the carboxylate and adjacent methylene groups (ESI† section 4.7), as is observed in the X-ray structure.46 In contrast, the C–H⋯O interactions of DBU–CO2 are weaker, likely due to geometrical restraints of the larger ring size, and not due to sterics as has been proposed.46 For detailed discussion and spectra, see ESI† section 11.
To our surprise, solid TMG–CO24 showed no signs of decomposition visually or by NMR when stored under CO2 at RT for six months. Unfortunately, our attempts to grow X-ray quality crystals of zwitterion 4 were unsuccessful. Therefore, we had to rule out any possibility of the corresponding hydrolysis product, bicarbonate 5 [TMGH+][HCO3−]. We decided to independently synthesize 5, and perform a comparative characterization with 4. Repeating the synthesis in the presence of water (1 equiv. to TMG) resulted in the immediate precipitation of 5 at RT. We were able to characterize bicarbonate 5 by NMR (HCO3− at 158.3 ppm) and X-ray (Fig. 5).52
 | ||
| Fig. 5 Structure of the dimer with Ci-symmetry of 5 [TMGH+][HCO3−] (symmetry operator −x + 1, −y + 1, −z + 1, displacement parameters are drawn at 50% probability level). | ||
We assessed the thermal stability of zwitterion 4 and bicarbonate 5 by thermogravimetric and differential thermal analysis under a flow of dry N2 (Fig. 6). At 25 °C, zwitterion 4 slowly degraded under the N2-flow, consistent with previous reports of CO2-adduct lability.12 The onset of thermal degradation (CO2 release) began at 44 °C, reaching a maximum at 51 °C. The major peak at 153 °C corresponds to evaporation of TMG (bp. 160 °C).53 In contrast, bicarbonate 5 showed significantly improved thermal stability. Onset of degradation occurred at 80 °C, and reaching maxima at 103 °C and 112 °C, corresponding to decomposition of the bicarbonate ion.
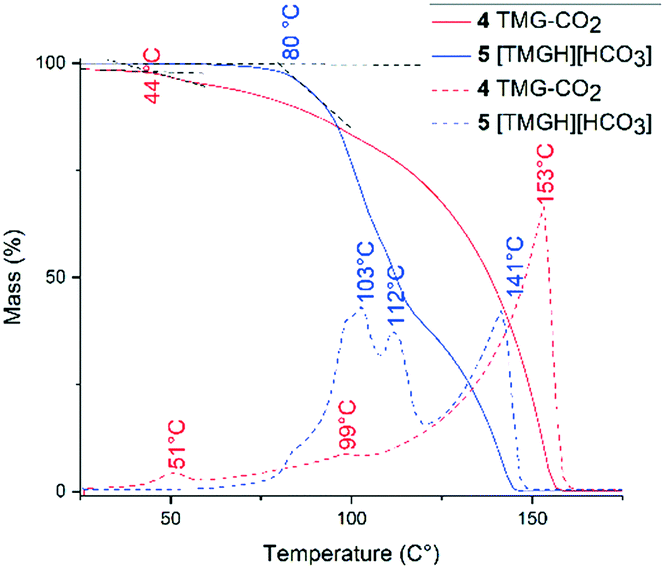 | ||
| Fig. 6 Thermogravimetric (full line) and differential thermal analysis (dashed line) of zwitterion 4 and bicarbonate 5. Intersection of black dashed lines indicates onset of thermal degradation. | ||
The IR spectra of zwitterion 4 and bicarbonate 5 was compared to known compounds (Table 4). The asymmetric CO2 stretching band in zwitterion 4 (entry 1) is similar in value to reported CO2 adducts of N-heterocyclic carbene (NHC, entry 3)54 and NHI (entry 4).46 Bicyclic superbase–CO2 zwitterions appear at higher frequencies (entries 5 and 6), suggesting the steric strain of the carboxylate in zwitterion 4 is similar to NHC and NHI.54 In contrast, bicarbonate 5 has a significantly lowered frequency (entry 2). The IR spectra of zwitterion 4 and bicarbonate 5 show significant differences (Fig. S50 and S51†). For example, zwitterion 4 shows relatively strong and sharp peaks at 3035 and 3019 cm−1, indicative of polymeric or dimeric association.55 In contrast, bicarbonate 5 has low to medium intensity broad bands in the regions of 2600–3100 cm−1, indicative of dimeric –OH⋯OC interaction,55 as is seen in the X-ray structure (Fig. 5).

When decarboxylation temperatures are compared, it is observed that zwitterion 4 falls in line with other Lewis base–CO2 adducts, with the exception of significantly more stable NHC–CO2 (Table 4, entry 3). As seen in Table 4, zwitterion 4 displays physical attributes very similar to known Lewis base–CO2 adducts, while bicarbonate 5 deviates from most CO2-adducts by being thermally more robust.
We further investigated the identity of 4 and 5 by performing computational studies on the monomers and dimers of both species (Fig. 7). It can be noted that dimeric 4 with our computational protocol is several kcal mol−1 more stable than the separated monomers, possibly due to hydrogen bonding (ΔGr = −3.1 kcal mol−1 [1 atm], −4.9 kcal mol−1 [1 M]). Further, the computed asymmetric CO2 stretching band of dimeric 4 is 1659 cm−1 (Fig. 7), in excellent agreement with the experimental frequency of 1661 cm−1 (Table 4). The monomeric 4 has a significantly higher computed frequency (1690 cm−1), whereas the monomer and dimer of 5 both have lower frequencies, in agreement with experiment (Table 4).
 | ||
| Fig. 7 Computed structures and asymmetric CO2 stretching band of the monomer and dimer of 4 and 5. For 5-monomer, two equally strong peaks are observed and for 5-dimer, some CO2 asymmetric stretching contribution is also observed for a peak at 1652 cm−1, besides the dominant peak at 1611 cm−1. | ||
The combined results confirm the identity of zwitterion 4 and bicarbonate 5. Given the transient existence of superbase–CO2 adducts in solution at RT, why is zwitterion 4 isolable from a cold solution? Here, it is probable TMG is rapidly carboxylated, yet the resulting zwitterion decomposes rapidly at 298 K, in agreement with the computed energies (Scheme 5), showing a low barrier for formation of 4 (8.7 kcal mol−1), but a slightly endergonic reaction (0.4 kcal mol−1). At lower temperatures, the entropic penalty is reduced and solvent induced dissociation of 4 into TMG and CO2 is slower (see NMR discussion, vide supra). Indeed, calculations indicate that formation of 4 from CO2 and TMG would be slightly exergonic at 263 K (−0.7 kcal mol−1, Scheme 5). At subzero temperatures, the higher concentration of 4 leads to aggregates, which precipitate out. According to calculations, formation of dimer 4′ is exergonic (Fig. S5†).
 | ||
| Scheme 5 Computed free energies (kcal mol−1, PBE0-D3BJ/IEFPCM[acetonitrile], 1 M standard state) for reversible formation of zwitterion 4. | ||
Having established 4 as a true zwitterion, we set out to explore its reactivity under neat conditions, in order to eliminate solvent induced dissociation of CO2. In a glovebox, we added liquid protic nucleophiles 6a–c to solid zwitterion 4, resulting in an immediate reaction of bubbling (CO2) and gel formation (slowly solidifying). Analysis by NMR showed successful carboxylation of aromatic amine 7a, pyrrole 7b, and 1-butanol 7c (Scheme 6), although 7a was obtained in somewhat lower yield compared to the in situ experiment (Fig. 2, left). These experiments support that CO2 dissociates from zwitterion 4 before carboxylation occurs, in accord with the calculations (Fig. 1). Similar stoichiometric carboxylations have been performed with TBD–CO2 in solution, but the necessity of CO2 dissociation from TBD–CO2 was not discussed.56
 | ||
| Scheme 6 Carboxylation of protic nucleophiles using solid zwitterion 4 or bicarbonate 5 on 0.5 mmol scale. Yields determined by 1H NMR. | ||
Lewis base–CO2 adducts have been reported to carboxylate amines,20,30 however, their chemical identity has been debated, as others have suggested the adducts are better described as bicarbonate salts.25 Therefore, we repeated our experiments in Scheme 6 with bicarbonate 5. No bubbles were observed, although the mixtures did solidify. Carboxylation of 1-butanol 7c was similar in yield, while bicarbonate 5 was significantly less effective at carboxylating nitrogen compounds. We recommend future superbase–CO2 zwitterion studies to include the corresponding bicarbonate as a control in carboxylation reactions. Tetraalkylammonium bicarbonate salts are able to carboxylate amines,57 but not pyrroles.58 Considering 5 readily carboxylated pyrrole 7b, this suggests the presence of TMG is critical.
In summary, zwitterionic TMG–CO24 may exist under the carboxylation conditions, in agreement with the low barrier computed for its formation (Fig. 1), however, our mechanistic analysis indicates that 4 is likely to dissociate into TMG and CO2 prior to carboxylation (see mechanistic discussion, vide supra). At low temperatures, dissociation is slowed down, allowing isolation of 4. We further note that the experiments in Scheme 6 showcase the potential of zwitterion 4 as a source of dry CO2,46 and that carboxylations are possible without an atmosphere (large excess) of CO2.
Conclusions
Previous literature has proposed zwitterionic superbase–CO2 adducts as active carboxylation agents. In this work, we were able to isolate pure TMG–CO2 as a stable solid, allowing us to explore its carboxylation reactivity. Our experimental and computational results indicate that CO2 must first dissociate from the TMG–CO2 zwitterion before carboxylation can occur. This opens a low energy pathway, where the base pre-associates to the nucleophile, activating it towards attack at a free CO2, with carboxylation and deprotonation occurring concertedly (path 2, Scheme 4).The identity of the TMG–CO2 zwitterion was confirmed by NMR and computational studies, in addition to a comparative analysis of the corresponding hydrolysis product (bicarbonate). Room temperature NMR studies were used to observe TMG–CO2 and the long-sought DBU–CO2 adduct, both displaying a broad carboxylate 13C signal, due to reversible binding of CO2. These results were complimented by VT NMR studies. Natural abundance 15N NMR was found to be sensitive for zwitterion formation, with significant upfield shifts of the CO2-bearing nitrogen, which was introduced here as a highly useful tool for studying superbase interactions with CO2.
Our experimental and computational findings are significant for further design and development of CO2-based synthesis, because they demonstrate that superbases are able to activate dormant species, such as aromatic amines, which are otherwise inert towards CO2. For example, as shown in this work, both zwitterionic TMG–CO2 and the corresponding bicarbonate can carboxylate nucleophiles without an atmosphere of CO2 (large excess). However, zwitterionic TMG–CO2 was significantly more effective at carboxylating nitrogen nucleophiles. Although the carboxylation is stoichiometric in superbase, we foresee increasing use of superbases as activating agents in many catalytic applications. In this context, a very important observation is that although superbases can transiently bind and store CO2, the resulting zwitterion is not a form of “activated CO2”.
Author contributions
JKM conceived the project. JKM, TT, and AS carried out the experiments. LP and KHH performed the computational studies. MN performed the X-ray crystallography studies. The manuscript and supplemental information was prepared jointly by all authors. TR supervised the experimental work, and KHH the computational work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. Lauri Partanen for his advice regarding equilibrium studies, Dr. Kristina Sorochkina for her insightful comments, and Dr. David Casadio and Dr. Sami Heikkinen for their help with NMR studies. This work has been supported by CHEMS – The Doctoral Programme in Chemistry and Molecular Sciences at University of Helsinki, Academy of Finland (project 310767), NordForsk (Grant No. 85378) and the members of the Nordic Consortium for CO2 Conversion (NordCO2), and Nylands Nation (JKM). KHH thanks the Tromsø Research Foundation (TFS2016KHH), the Research Council of Norway (grant No. 300769 and 262695) and Sigma2 for grants of computer time (No. nn9330k and nn4654k).Notes and references
- A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Nat. Prod. Rep., 2016, 33, 491–522 RSC.
- A. Ricci, Modern amination methods, John Wiley & Sons, Inc., 2007 Search PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- T. Niemi and T. Repo, Eur. J. Org. Chem., 2018, 1180–1188 Search PubMed.
- J. Vaitla, Y. Guttormsen, J. K. Mannisto, A. Nova, T. Repo, A. Bayer and K. H. Hopmann, ACS Catal., 2017, 7, 7231–7244 CrossRef CAS.
- F. D. Bobbink, S. Das and P. J. Dyson, Nat. Protoc., 2017, 12, 417–428 CrossRef CAS PubMed.
- J. K. Mannisto, A. Sahari, K. Lagerblom, T. Niemi, M. Nieger, G. Sztanó and T. Repo, Chem. – Eur. J., 2019, 25, 10284–10289 CrossRef CAS PubMed.
- W. Schilling and S. Das, ChemSusChem, 2020, 13, 6246–6258 CAS.
- D. Riemer, P. Hirapara and S. Das, ChemSusChem, 2016, 3, 1916–1920 CrossRef.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5940–5966 CrossRef CAS.
- P. V. Kortunov, L. S. Baugh, M. Siskin and D. C. Calabro, Energy Fuels, 2015, 29, 5967–5989 CrossRef CAS.
- C. Chiappe, G. Pampaloni, L. Biancalana, G. Bresciani and F. Marchetti, New J. Chem., 2017, 41, 1798–1805 RSC.
- W. McGhee, D. Riley, K. Christ, Y. Pan and B. Parnas, J. Org. Chem., 1995, 60, 2820–2830 CrossRef CAS.
- A. L. Ethier, J. R. Switzer, A. C. Rumple, W. Medina-Ramos, Z. Li, J. Fisk, B. Holden, L. Gelbaum, P. Pollet, C. A. Eckert and C. L. Liotta, Processes, 2015, 3, 497–513 CrossRef CAS.
- W. D. McGhee, D. P. Riley, M. E. Christ and K. M. Christ, Organometallics, 1993, 12, 1429–1433 CrossRef CAS.
- M. Costa, G. P. Chiusoli and M. Rizzardi, Chem. Commun., 1996, 1699–1700 RSC.
- K. Masuda, Y. Ito, M. Horiguchi and H. Fujita, Tetrahedron, 2005, 61, 213–229 CrossRef CAS.
- C. Villiers, J. P. Dognon, R. Pollet, P. Thuéry and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CrossRef CAS PubMed.
- F. S. Pereira, E. R. de Azevedo, E. F. da Silva, T. J. Bonagamba, D. L. da Silva Agostíni, A. Magalhães, A. E. Job and E. R. Pérez González, Tetrahedron, 2008, 64, 10097–10106 CrossRef CAS.
- Z. Xin, C. Lescot, S. D. Friis, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2015, 54, 6862–6866 CrossRef CAS PubMed.
- H. C. Fu, F. You, H. R. Li and L. N. He, Front. Chem., 2019, 7, 1–15 CrossRef PubMed.
- H. Zhou, W. Chen, J. H. Liu, W. Z. Zhang and X. B. Lu, Green Chem., 2020, 22, 7832–7838 RSC.
- R. Nicholls, S. Kaufhold and B. N. Nguyen, Catal. Sci. Technol., 2014, 4, 3458–3462 RSC.
- D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef CAS PubMed.
- W. Li, N. Yang and Y. Lyu, Org. Chem. Front., 2016, 3, 823–835 RSC.
- M. Hulla and P. J. Dyson, Angew. Chem., 2020, 59, 1002–1017 CrossRef CAS PubMed.
- M. Hulla, S. M. A. Chamam, G. Laurenczy, S. Das and P. J. Dyson, Angew. Chem., Int. Ed., 2017, 56, 10559–10563 CrossRef CAS PubMed.
- P. V. Kortunov, M. Siskin, M. Paccagnini and H. Thomann, Energy Fuels, 2016, 30, 1223–1236 CAS.
- E. R. Pérez, R. H. A. Santos, M. T. P. Gambardella, L. G. M. De Macedo, U. P. Rodrigues-Filho, J. C. Launay and D. W. Franco, J. Org. Chem., 2004, 69, 8005–8011 CrossRef PubMed.
- M. J. Diem, D. F. Burow and J. L. Fry, J. Org. Chem., 1977, 42, 1801–1802 CrossRef CAS.
- J. E. Rainbolt, P. K. Koech, C. R. Yonker, F. Zheng, D. Main, M. L. Weaver, J. C. Linehan and D. J. Heldebrant, Energy Environ. Sci., 2011, 4, 480–484 RSC.
- N. von Wolff, C. Villiers, P. Thuéry, G. Lefèvre, M. Ephritikhine and T. Cantat, Eur. J. Org. Chem., 2017, 2017, 676–686 CrossRef CAS.
- M. Baidya and H. Mayr, Chem. Commun., 2008, 1792–1794 RSC.
- A. Gholamipour-Shirazi and C. Rolando, Org. Biomol. Chem., 2012, 10, 8059–8063 RSC.
- V. A. Bren', E. N. Maloesheva and V. I. Minkin, Org. React., 1967, 4, 534–555 Search PubMed.
- E. I. Rõõm, A. Kütt, I. Kaljurand, I. Koppel, I. Leito, I. A. Koppel, M. Mishima, K. Goto and Y. Miyahara, Chem. – Eur. J., 2007, 13, 7631–7643 CrossRef PubMed.
- D. J. Heldebrant, P. K. Koech, J. E. Rainbolt, F. Zheng, T. Smurthwaite, C. J. Freeman, M. Oss and I. Leito, Chem. Eng. J., 2011, 171, 794–800 CrossRef CAS.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- K. Kaupmees, A. Trummal and I. Leito, Croat. Chem. Acta, 2014, 87, 385–395 CrossRef.
- C. M. Zall, J. C. Linehan and A. M. Appel, ACS Catal., 2015, 5, 5301–5305 CrossRef CAS.
- Substituents with Hammett constants below a certain threshold (σ ≤ 0) would all produce the same result (100% yield), prohibiting us from constructing a linear correlation.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- I. Tiritiris, J. Mezger, E. V. Stoyanov and W. Kantlehner, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 407–418 CAS.
- L. F. B. Wilm, T. Eder, C. Mück-Lichtenfeld, P. Mehlmann, M. Wünsche, F. Buß and F. Dielmann, Green Chem., 2019, 21, 640–648 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- S. Tanaka, Y. Saito, T. Yamamoto and T. Hattori, Org. Lett., 2018, 20, 1828–1831 CrossRef CAS PubMed.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 1–6 CrossRef PubMed.
- A. C. Shaikh, J. M. Veleta, J. Moutet and T. L. Gianetti, Chem. Sci., 2021, 12, 4841–4849 RSC.
- L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164–10168 CrossRef CAS PubMed.
- X-Ray Data for 5 (CIF), CCDC 2044161, contains the Supplementary Crystallographic Data for this paper. It is a redetermination of COWGON at 123 K using Cu-Kα radiation. Refcode COWGON: CCDC-1912766 CrossRef CAS; V. Ramkumar and R. L. Gardas, J. Chem. Eng. Data, 2019, 64, 4844–4855 CrossRef CAS; A related hemihydrate has been reported. Refcode IQAPIA, CCDC-796566. See ref. 45.
- A. E. Sherr and A. A. Krupnik, J. Appl. Polym. Sci., 1965, 9, 2707–2714 CrossRef CAS.
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS PubMed.
- T. Hase, Tables for Organic Spectrometry, Otatieto, 10th edn, 2008 Search PubMed.
- C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef PubMed.
- A. Inesi, V. Mucciante and L. Rossi, J. Org. Chem., 1998, 3263, 1337–1338 CrossRef.
- S. Cesa, V. Mucciante and L. Rossi, Tetrahedron, 1999, 55, 193–200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2044161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cy01433a |
| This journal is © The Royal Society of Chemistry 2021 |