
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZirconium-catalysed direct substitution of alcohols: enhancing the selectivity by kinetic analysis†
Cristiana
Margarita
 ,
Piret
Villo
,
Hernando
Tuñon
,
Oscar
Dalla-Santa
,
David
Camaj‡
,
Robin
Carlsson‡
,
Malin
Lill‡
,
Anja
Ramström‡
and
Helena
Lundberg
*
,
Piret
Villo
,
Hernando
Tuñon
,
Oscar
Dalla-Santa
,
David
Camaj‡
,
Robin
Carlsson‡
,
Malin
Lill‡
,
Anja
Ramström‡
and
Helena
Lundberg
*
Department of Chemistry, KTH Royal Institute of Technology, Teknikringen 30, S-10044 Stockholm, Sweden. E-mail: hellundb@kth.se
First published on 24th September 2021
Abstract
Kinetic analysis was used as a tool for rational optimization of a catalytic, direct substitution of alcohols to enable the selective formation of unsymmetrical ethers, thioethers, and Friedel–Crafts alkylation products using a moisture-tolerant and commercially available zirconium complex (2 to 8 mol%). Operating in air and in the absence of dehydration techniques, the protocol furnished a variety of products in high yields, including glycosylated alcohols and sterically hindered ethers. In addition, the kinetic studies provided mechanistic insight into the network of parallel transformations that take place in the reaction, and helped to elucidate the nature of the operating catalyst.
Introduction
Alcohols constitute a versatile substrate class in organic synthesis. Due to their prevalence in biomass, alcohols have the potential to replace fossil-based feedstocks in fine synthesis, commodity chemicals and fuels.1–3 While the hydroxyl group is a highly useful synthetic handle for a wide variety of transformations, the synthetic toolbox largely relies on its stoichiometric derivatisation to allow for deoxygenative substitution of alcohols in one- or two-electron transformations due to the poor leaving group capacity of the OH-group. For example, alcohols can be pre-functionalised to sulfonates for direct nucleophilic displacement or transition metal-catalysed cross-couplings; to xanthate esters for single electron reduction in Barton–McCombie deoxygenation; or to oxalate esters for radical cross-couplings.4–9 Such pre-functionalization is a major drawback as it significantly reduces the process efficiencies from the perspective of atom economy, cost and labour. Direct substitution of alcohols with water as the only by-product is thus a highly attractive strategy, highlighted by the American Chemical Society (ACS) Pharmaceutical Roundtable as a key green chemistry research area.10 Catalytic nucleophilic substitution is commonly carried out by using a borrowing hydrogen strategy,11 transition metal-catalysed substitution of allylic alcohols via π-allyl intermediates12 or direct substitution using Lewis basic13–15 or Lewis acidic catalysts.2,16–22 In addition, the use of protic acids has been explored to a lesser extent.23–27 Tetravalent metal complexes with fluoroalkylsulfonate ligands constitute an interesting class of water-stable Lewis acidic catalysts for a variety of transformations.28–42 Despite this, this catalyst class is hardly represented in the context of dehydrative substitution of alcohols in contrast to its trivalent (and bivalent) counterparts.43–60In direct etherification, two alcohols are joined with a loss of water. To enable the formation of unsymmetrical ethers from two different alcohols, the transformation typically relies on one alcohol being more electrophilic than the other, e.g. by π-activation. For this reason, secondary or highly electron rich benzylic alcohols are commonly employed as electrophiles in this type of transformation,22,47,61–65 with the more challenging direct etherification of unsubstituted benzyl alcohol being less prevalent.66–68 In addition, symmetric side-products are recurrently observed for such unsymmetrical intermolecular etherification protocols and can significantly decrease the yields of these processes.21,69–72
During the last decades, user-friendly methodologies have been developed for intuitive visual kinetic analysis of organic reactions, known as reaction progress kinetic analysis (RPKA) and variable time normalization analysis (VTNA).73–78 These methodologies enable facile extraction of kinetic data by utilization of full reaction profiles for a minimal number of experiments under synthetically relevant conditions. Typically, these kinetic tools have been used for elucidation of operating mechanisms for organic transformations once a protocol has been established. Less commonly, these methods can be integrated with synthetic method development to enable streamlined reaction optimisation.79–83 Herein, we describe how the use of kinetic analysis during the early method development phase of a zirconium-catalysed protocol for direct substitution of alcohols enabled rational optimization of reaction conditions and enhanced yields by suppression of the symmetric side-product formation.
Results and discussion
In our previous work on zirconocene-catalysed esterification, small amounts of a side-product were observed in the presence of benzyl alcohol and carboxylic acid, identified as dibenzyl ether 2a.84 Interested in this zirconium-mediated direct substitution of the alcohol, an assessment of the prospects for selective formation of unsymmetrical ethers was undertaken, using benzyl alcohol 1a and 2-phenylethanol 1b as model substrates in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. To provide insight into the formation of unsymmetrical ether 2b, kinetic data was retrieved during the evaluation of reaction parameters by sampling over the course of the reaction, followed by off-line HPLC analysis.
:2 molar ratio. To provide insight into the formation of unsymmetrical ether 2b, kinetic data was retrieved during the evaluation of reaction parameters by sampling over the course of the reaction, followed by off-line HPLC analysis.
Firstly, different solvents were assessed (Fig. 1A). Similarly to the observed behaviour of Zr-catalysed esterification,84 non-polar solvents were found to furnish the unsymmetrical product 2b in higher yields compared to more polar solvents. In addition to the results in Fig. 1A, acetonitrile was assessed as solvent and resulted in <5% yield 2b. The formation of 2b in sulfolane proceeded with a high rate, but the yield was limited by a mixture of unidentified by-products. The highest yields for 2b were achieved in heptane and benzotrifluoride (BTF) with similar reaction rates. While the former solvent may present benefits in terms of environmental impact,85,86 the latter was selected for further studies due to its ability to dissolve a wide range of organic substrates. An assessment of reaction temperature indicated that heating above 80 °C is required to achieve reasonable reaction rates (Fig. 1B). For all solvents, the symmetric side-product 2a was formed along with the desired unsymmetrical product 2b, thereby limiting the yield of the latter.
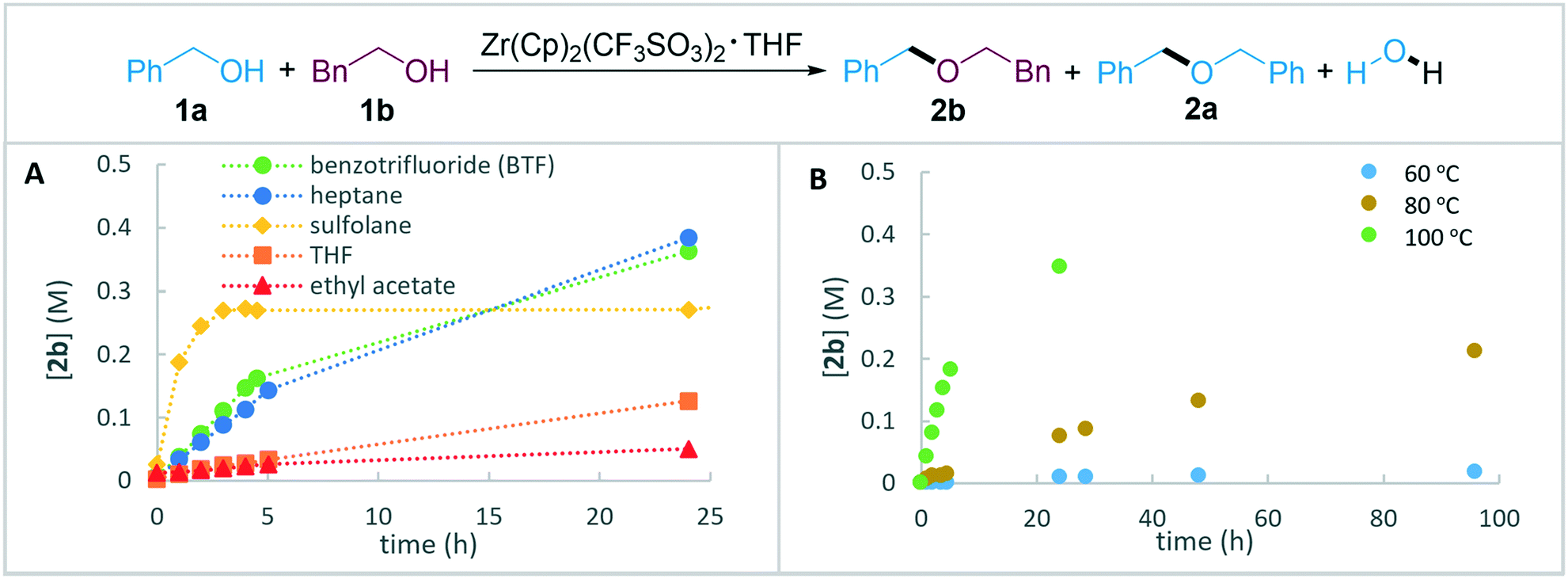 | ||
| Fig. 1 Screening of parameters for formation of unsymmetrical product 2b. A. Solvent evaluation at 100 °C. B. Temperature evaluation in BTF. Standard conditions: 1a (0.5 M), 1b (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF (1 mL), 100 °C. Yields were determined by HPLC analysis after 24 hours using 4,4′-di-tert-butylbiphenyl as internal standard. | ||
To increase the reaction selectivity, a more efficient incorporation of benzyl alcohol (1a) into product 2b was required, and a kinetic study was undertaken using the tools of RPKA and VTNA.73–78 To assess the driving forces for the formation of the two ethers, a set of different excess experiments were conducted to estimate the order in a given component. This was done by comparison of reactions where the initial concentration of the given component was varied while the others were kept constant.73,74 As shown in Fig. 2A and B the formation of 2a and 2b demonstrated a near first order rate dependence on [Zr]. The average order77 of [1a] for the formation of 2b was experimentally found to be 0.8 at low concentrations of 1a, whereas it was reduced to 0.35 at higher concentrations (Fig. 2C and D). In contrast, the rate dependence on [1a] for the formation of side-product 2a was found to be of first order for the whole concentration range examined (Fig. 2E). A slight positive rate dependence on [1b] was observed in the low concentration range for 2b formation (Fig. 2F). At higher [1b] a negative average order of −0.35 was observed for d[2b]/dt (Fig. 2G), whereas a more pronounced negative average order in [1b] was observed for the formation of side-product 2a for the whole concentration range.
 | ||
| Fig. 2 Variable time normalization analysis of different concentration experiments. A. Order in [Zr] for formation of 2b. B. Order in [Zr] for formation of 2a. C. Order in [1a] for formation of 2b at low concentrations of 1a. D. Order in [1a] for formation of 2b at high concentrations of 1a. E. Order in [1a] for formation of 2a. F. Order in [1b] for formation of 2b at low concentrations of 1b. G. Order in [1b] for formation of 2b at high concentrations of 1b. H. Order in [1b] for formation of 2a. Standard conditions: 1a (0.5 M), 1b (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF (1 mL), 100 °C. | ||
As a result of the different responses to concentration changes for 2a and 2b formation, the combined data in Fig. 2 suggested that higher selectivity for the unsymmetrical product 2b could be obtained if the concentration of 1a was kept low and 1b high throughout the reaction. Indeed, sequential addition of 1a in five portions over four hours to a BTF-solution of the catalyst and 1 M 1b resulted in maintained yield even with an increase in [1a] to a 1:1 molar ratio to [1b], all other concentrations kept constant (Fig. 3). Slow addition of 1a with a syringe pump (rate 10 μl h−1) in the equimolar reaction gave slightly lower yield for 2b compared with sequential addition, and was not explored further. Sequential addition of 1a using a 2:1 molar ratio of 1b to 1a resulted in 84% isolated yield of 2b after 24 hours, representing an increase in yield with 14 percent units compared to a setup where all material was added at the outset of the reaction (“batch addition”) using the same reactant stoichiometries (Fig. 3). As expected, the same yield of 84% of 2b was also obtained by dilution of [1a] while the concentrations of catalyst and 1b were kept constant (Fig. 3). Under these optimized “batch” conditions, the ratio of desired mixed ether 2b to symmetrical side-product 2a was measured to be 12:1 by 1H NMR spectroscopy of the crude reaction mixture.
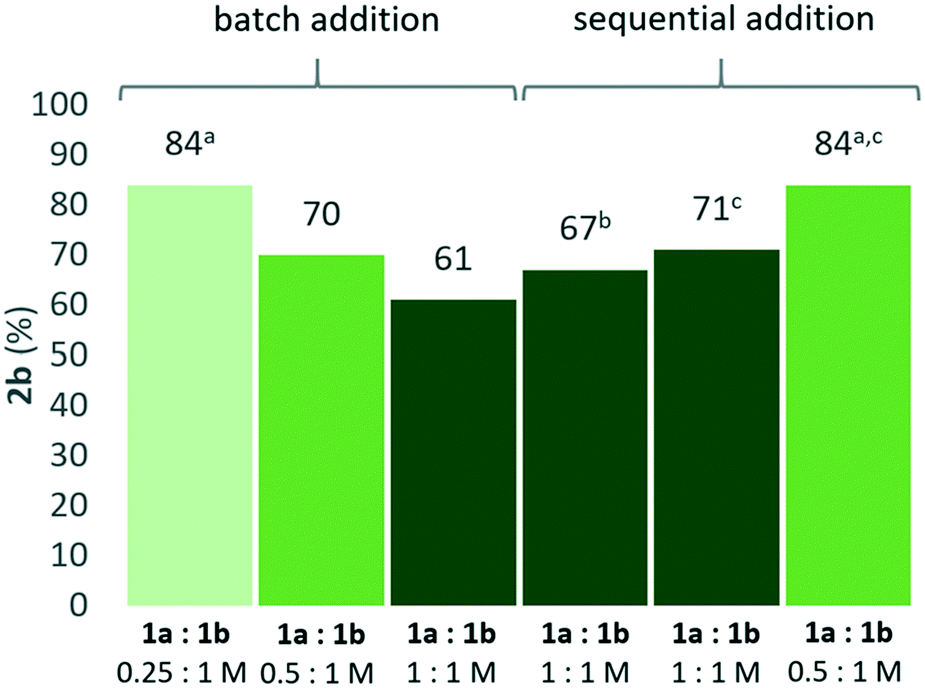 | ||
| Fig. 3 Effect of reactant concentration and mode of addition on yield of 2b. Standard conditions (batch addition): 1a (0.5 M), 1b (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF (1 mL), 100 °C. Yields were determined by HPLC analysis after 24 hours using 4,4′-di-tert-butylbiphenyl as internal standard. aIsolated yield. b1a added with syringe pump (rate 10 μl h−1) c1a added in five portions over 4 hours. | ||
While the sequential addition enables the use of less reagents, and effectively a lower catalyst loading with respect to the limiting substrate, batch addition using low [1a] offers operational simplicity. From a synthetic standpoint, the two strategies for improving the selectivity of the reaction are complementary. Interestingly, a good isolated yield (69%) could still be obtained when lowering the catalyst loading (0.01 M, 2 mol%) in neat conditions, using a 2:1 molar ratio of 1b to 1a. However, the ratio for the resulting ethers 2b:2a was 4:1 in this case. Higher concentrations of catalyst and reactants did not improve yields further after 24 hours. No reaction took place in the absence of catalyst.
A set of same excess experiments, mimicking 25% conversion in the absence and presence of the corresponding amount of water indicated that insignificant catalyst deactivation or product inhibition occurs during the reaction (Fig. 4).
 | ||
| Fig. 4 Same excess experiments, mimicking 25% conversion of 1a. Standard conditions: 1a (0.5 M), 1b (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF, 100 °C. Same excess conditions: 1a (0.375 M), 1b (0.875 M), (H2O 0.125 M, added for “same excess + H2O” experiment), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF, 100 °C. Concentrations determined by HPLC analysis after 24 hours using 4,4′-di-tert-butylbiphenyl as internal standard. | ||
Apparent Lewis acid catalysis has occasionally been reported to be due to a Brønsted acid, formed by hydrolysis of the former.87,88 In the context of metallocene triflates, Hollis et al. reported that the titanium analogue of the herein employed catalyst may undergo hydrolytic decomposition to release trifluoromethanesulfonic (triflic) acid.89 To probe whether this hydrolysis product was the operating catalyst in our protocol, a set of experiments were carried out. While addition of di-tert-butyl-4-methylpyridine to the reaction mixture containing either zirconocene triflate or triflic acid as catalyst proved inconclusive (see ESI†),87,88 an assessment of the two catalysts in the presence of different amounts of water made it clear that the two catalysts were not displaying identical behaviours (Fig. 5A). While both catalysts appear to require a sufficient amount of water, as is evident from the negligible yield in the presence of molecular sieves, the yield of 2b in the triflic acid-catalysed reaction drops gradually as a function of water addition. This behaviour is not observed for the zirconocene complex, where the 24 h yields stayed around 70% for the reactions with ratios up to 25:1 H2O:catalyst (1:1 ratio H2O:1a). In the 125:1 H2O:catalyst experiment (5:1 ratio H2O:1a) the yield of 2b did not exceed 2% in the presence of the Zr catalyst, whereas triflic acid catalysis resulted in inconsistent yields between 0 and 63%, 10% being the median value (Fig. 5A and ESI†). These observations suggest that while the zirconium complex is irreversibly deactivated at high concentrations of water, the inhibition of triflic acid is reversible. Furthermore, a set of triflic acid catalysed experiments with different [1b] were carried out, indicating an order of zero in [1b] for both d[2b]/dt and d[2a]/dt in the concentration range 1–2 M 1b (Fig. 5B and ESI†). These results stand in stark contrast to the observed negative orders in [1b] for both product and side-product formation in the zirconium-catalysed reactions (Fig. 2G and H). In addition, the observed selectivities 2b:2a were lower for reactions carried out in the presence of triflic acid. Thus, the different reaction behaviours observed when using either triflic acid or the zirconocene complex suggest that different active catalysts are operating in the two sets of reactions. By NMR-spectroscopy, a downfield shift was observed for 13C-labelled 1a in the presence of the Zr complex in d8-toluene, suggesting interaction between the metal and substrate (see ESI†). These combined observations support involvement of the zirconium centre in the mechanism.
 | ||
| Fig. 5 Comparison between Zr and CF3SO3H catalysis. A. Results from additions of water to the reactions. B. Order in [1b] for product and side-product formation under triflic acid catalysis. Standard conditions: 1a (0.5 M), 2a (1 M), Zr(Cp)2(CF3SO3)2·THF or CF3SO3H (0.02 M), BTF, 100 °C. Yields in A are determined by HPLC analysis after 24 hours using 4,4′-di-tert-butylbiphenyl as internal standard. | ||
The relative stability of the zirconium catalyst towards water, and the inhibition of catalytic activity in the presence of molecular sieves have previously been observed for zirconocene triflate.84 While the titanium analogue was reported to readily form aqua complexes with labile ligand structure,28 zirconocene with fluorinated sulfonate ligands can form dimers in the presence of water.90 Hence, the observed decelerating effect of molecular sieves may suggest that aqua complexes and/or multinuclear complexes are the catalytically active species in this system, where the former may or may not act as Brønsted acids.91 The need for a sufficient amount of water for efficient catalysis to occur has previously been reported for different types of Lewis acidic catalysts.92–95
A faster reaction rate, compared to benzyl alcohol 1a, was observed when the para-position was substituted with an electron-donating methoxy group, whereas an electron-withdrawing para-trifluoromethyl group displayed a considerably slower rate (Fig. 6). This behaviour is consistent with a mechanism involving a carbocationic intermediate, a notion further supported by observed racemization in ether 2c, formed from enantiopure (S)-1-phenylethanol and 1b (Scheme 1).
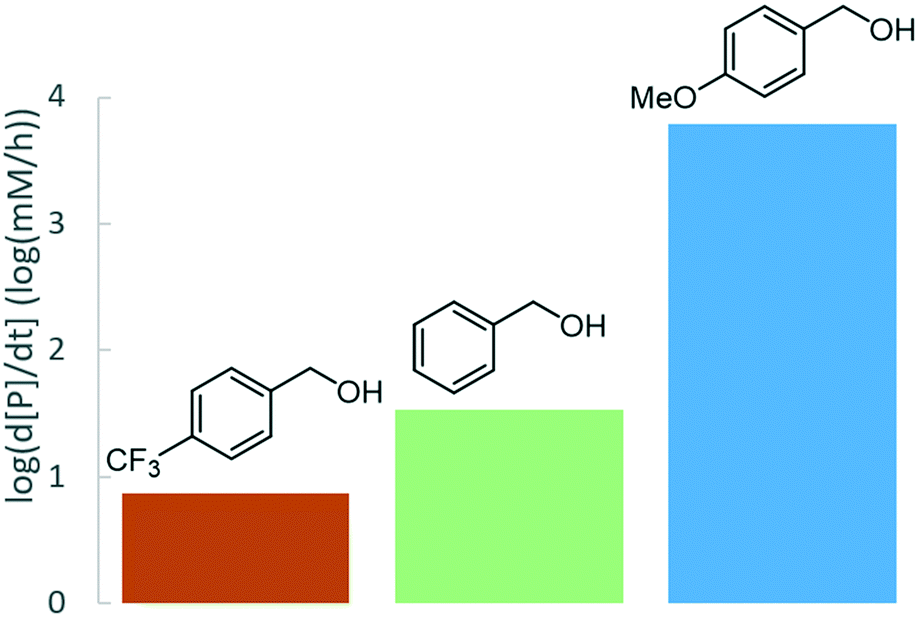 | ||
| Fig. 6 Reaction rate as a function of electrophile substitution. Standard conditions: electrophile (0.5 M), 1b (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF, 100 °C. | ||
 | ||
| Scheme 1 Racemisation of (S)-1-phenylethanol in etherification with 1b. | ||
The observed first order rate dependence on [Zr] and the positive orders on [1a] for the formation of both product 2b and side-product 2a (Fig. 2) are compatible with a mechanism in which the two ethers are formed via a single carbocationic intermediate, with C–OH activation constituting a turnover limiting step of the catalytic cycle. The average order in [1a] for the formation of [2b] was reduced from 0.8 to 0.35 at higher concentrations of 1a, whereas the average order in [1a] for the formation of 2a remained at 1. This finding explains the observed selectivity enhancement with respect to the unsymmetrical product 2b as [1a] is decreased, since the rate of 2b formation will be less disfavoured compared to that of the symmetric side-product. It is interesting to note that the strategy of maintaining reagent concentrations low for enhanced selectivity has been successful for various transformations, despite having quite different mechanistic origins.96–99 The average order in [1b] was increased from −0.35 to −0.2 at higher [1a] (see ESI†). These findings may be interpreted as if the contribution of the nucleophilic addition to the carbocationic intermediate to the rate is more pronounced under these conditions. The negative orders in [1b] for both product and side-product formation (Fig. 2) suggest that this compound is involved in the formation of off-cycle species that reduces the concentration of active catalyst in the reaction and, in turn, the reaction rate. Inhibition of catalytic activity in the presence of alcohols has previously been observed for zirconium chloride complexes, and the corresponding zirconocene dichloride complex did not yield the desired product 2b under our conditions (see ESI†).100,101 A tentative mechanism for the direct etherification and the rate laws for product and side-product formation under synthetically relevant conditions are shown in Fig. 7. The viability of the proposed mechanism was supported by kinetic modelling (see ESI†). Depending on the nature of the substrate it can be assumed that a carbocationic intermediate may form in solution as a more or less tight ion pair.102 Consequently, the degree of SN1-character of the mechanism may vary between substrates.
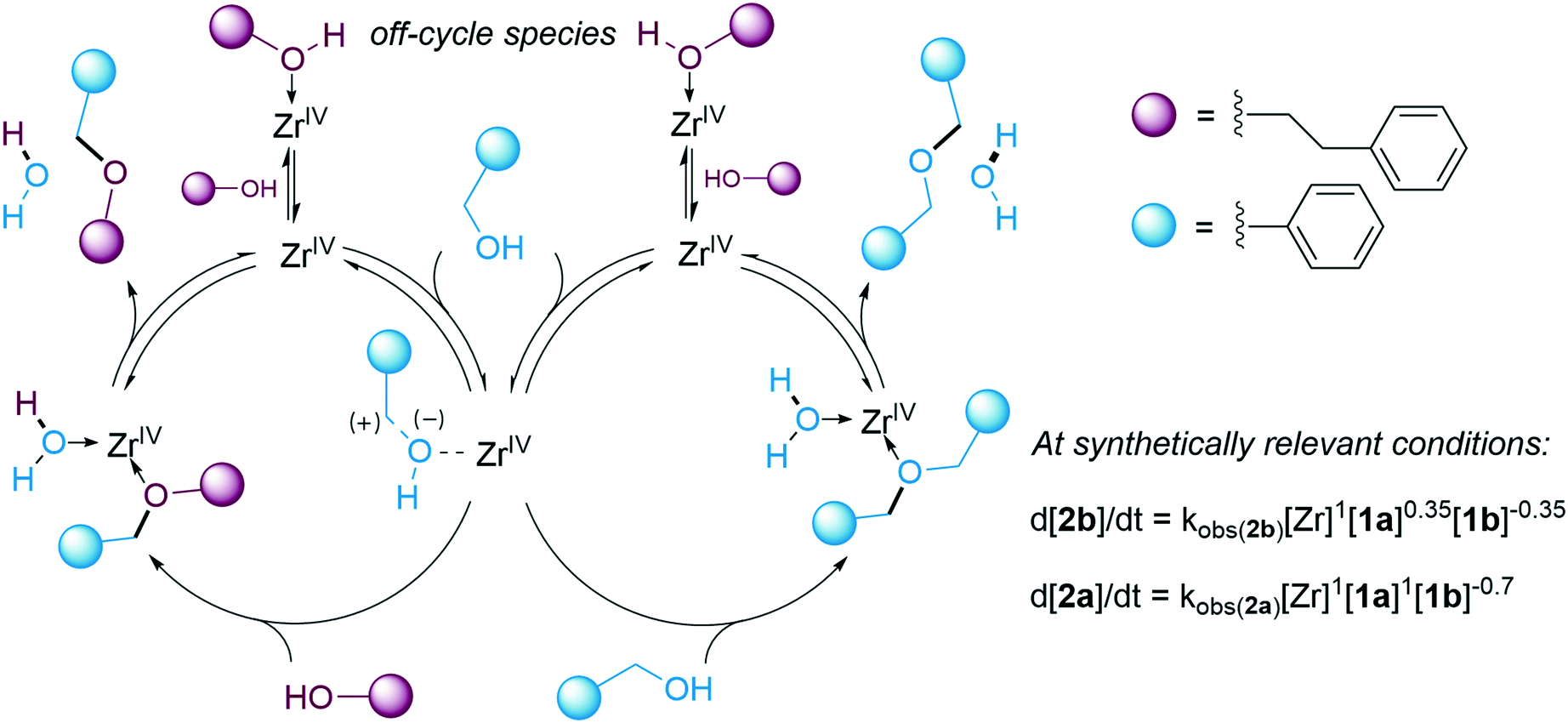 | ||
| Fig. 7 Tentative catalytic cycles for formation of 2a and 2bvia direct etherification. | ||
In addition to direct etherification, 2b can form by transetherification of 2a with 1b under the catalytic conditions (see ESI†).47 The rate of this process was found to be positively correlated to [2a] and [Zr] with a negative rate dependence on [1b]. A comparison between direct etherification and transetherification demonstrated that the latter was considerably slower than the former even at high concentrations of 2a. Thus, transetherification of 2a was judged to constitute a minor pathway for the formation of 2b, mainly operating towards the end of the reaction when [1a] has been depleted and [2a] has reached its highest levels. It is to be noted that the integrated use of RPKA and VTNA in the synthetic optimization provided this mechanistic information already at the early stages of the project, thus obviating the need for a separate work-intensive and time-consuming study using initial rate kinetics.68
Using the optimized conditions, a substrate evaluation was carried out to form a range of differently substituted ethers (Scheme 2). Higher yields for the unsymmetrical ethers were observed for the majority of the substrates using low electrophile concentration, either by sequential addition of the electrophile or by dilution under batch conditions. For operational simplicity, dilute batch conditions were chosen as standard procedure for the rest of substrate evaluation, unless otherwise indicated. Remarkable selectivity towards the formation of unsymmetrical products was observed for most benzylic substrates (ratios for formed ethers were determined by 1H NMR spectroscopy of the crude reaction mixtures, and are reported in ESI† where applicable). Whereas benzyl alcohol 1a required a reaction temperature of 100 °C to efficiently convert to the product, benzylic alcohols with electron donating groups in para position were amenable to lower reaction temperatures and/or reduced reaction times (2bvs.2j and 2k). This observation is likely due to stabilization of the proposed carbocationic intermediate. Similarly, increased degree of substitution at the benzylic position (2d–f, 2i) or extension of the aromatic system (2l, 2p–s) enabled good to excellent yields of the unsymmetrical ethers at lower reaction temperatures and/or shorter reaction times compared to the formation of 2b. Gratifyingly, sterically hindered ethers 2d–e that can be synthetically challenging, formed in good yields, indicating that the current methodology can serve as a complementary strategy to decarboxylative routes and C–H functionalization strategies.103,104 In this context, in can be noted that compound 2i was isolated in good yield, while it was previously described to undergo elimination in the presence of TfOH.105
 | ||
| Scheme 2 Substrate evaluation. Standard conditions: electrophile (0.25 M, 0.25 mmol scale unless otherwise specified in ESI†), nucleophile (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF. Standard conditions (sequential addition): electrophile (0.5 mmol, added in five portions of 0.1 mmol over 4 hours), nucleophile (1 mmol, 1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF. aNMR yield (1,3,5-trimethoxybenzene as internal standard) belectrophile:nucleophile 0.5:1 M cinitial ee of secondary alcohol: 98.5% dstarting from 2,3,4,6-tetra-O-benzyl-D-glucopyranose (1:1 α:β) eelectrophile:nucleophile 0.25:0.5 M felectrophile:nucleophile 0.5:0.5 M. | ||
Various functional groups were found to be compatible with the catalytic conditions, including alkynes (2f–g), alkenes (2h, 2l), aryl bromides (2m), and carbocycles of various size (2d, 2r–s). While thiophene was tolerated under the reaction conditions (2o), substrates with basic nitrogen-containing heterocycles were not (see ESI†). Such groups have previously been shown to inhibit catalysis using the same zirconium complex in direct esterification.84 The selectivity of 2g:2a and 2h:2a was slightly lower compared to the benchmark substrate 2b (see ESI†), which may be attributed to increased steric hindrance by the longer aliphatic chain that affects the selectivity determining step. In addition, the reduced yields of 2g and 2o can be explained by observed unidentified by-product formation from the nucleophilic alcohol under the catalytic conditions.
Secondary aliphatic alcohols performed well as nucleophiles (2n, 2q), albeit with a drop in ee between 2.5 and 14.5 percent units depending on reaction temperature and time. Glycosylation of 1b at 60 °C with 2,3,4,6-tetra-O-benzyl-D-glucopyranose was efficiently carried out to form 2t in good yield, demonstrating that benzyl ethers are stable as O-protecting groups under the applied conditions. Similarly, disaccharide 2u was formed from the same glucopyranose in good yield, highlighting the compatibility of cyclic acetals with the protocol. The reaction between β-cholestanol and p- methoxybenzyl (PMB) alcohol proceeded smoothly to form the PMB-ether 2w. Similarly, PMB-protected Boc-Ser-OMe 2v formed under the catalytic conditions, albeit in moderate yield. The use of secondary and tertiary allylic alcohols as electrophiles afforded the corresponding 2-phenethyl ethers 2l and 2x in good yields with isomerization of the double bond (Scheme 2B), while using primary allylic alcohol prenol as electrophile furnished 2x only in a moderate yield of 34% due to competing elimination. Phenols have been reported to serve as both O- and C-nucleophiles in Lewis acid catalysed direct substitution of alcohols.52,106 With our conditions, phenolic OH-groups were unreactive as O-nucleophiles, which allowed for mono-etherification of diols with full selectivity for the aliphatic alcohol (Scheme 2C). Due to low solubility of the diols in BTF, these reactions were carried out in THF.
With the phenolic OH-group in para position, ether 2y was isolated in 78% whereas the ortho substituted analogue 2y′ was isolated in 34%, likely due to the increased steric hindrance and/or increased degree of catalyst chelation. The meta-substituted analogue 2y′′ was not detected. These observations are in accordance with expected substituent effects of Hammett-type and are as such consistent with the proposed carbocationic intermediate.107
While the inductively electron-withdrawing character of the hydroxyl group governs the reactivity of the meta-substituted substrate, the benzylic carbon can be expected to benefit from resonance stabilization by the functionality when situated in ortho and para positions. Similarly, other electron withdrawing substituents on the aromatic ring were found to reduce reaction rates and yields of ether products (see ESI†). The catalytic methodology was smoothly extended to include thiols as nucleophiles, affording linear as well as sterically hindered thioethers in good to excellent yields (Scheme 3).
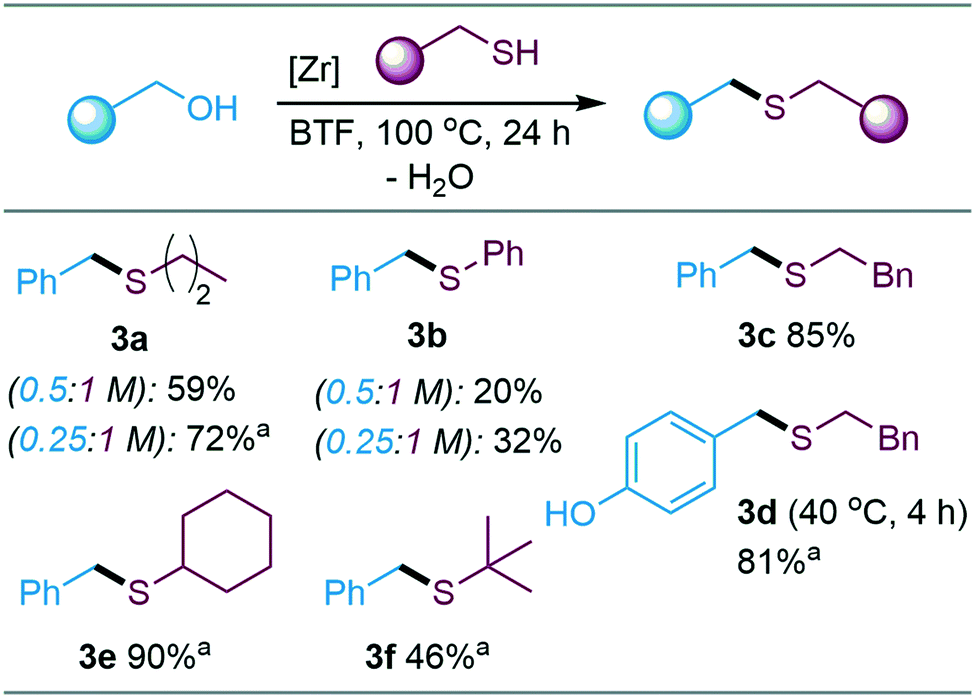 | ||
| Scheme 3 Thioetherification. Conditions: electrophile (0.25 M), nucleophile (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF, 100 °C, 24 h, unless otherwise specified. aNMR yield (1,3,5-trimethoxybenzene as internal standard). | ||
Similar to the observations made for ether formation, the yields of thioethers 3a and 3b increased when the electrophile concentration was reduced. Thiophenol acted as S-nucleophile to form thioethers (3b, Scheme 3), whereas phenols were unreactive as O-nucleophiles under the tested conditions (Scheme 2C).
However, in the absence of aliphatic O-nucleophiles, phenol was active as C-nucleophile in Friedel–Crafts alkylation (Scheme 4). When reacted with di- and triphenylmethanol, phenol selectively reacts in para position to form tertiary and quaternary carbon centres in the sterically hindered phenols 4a and 4b. The bulky 4-tritylphenol 4b and its substituted analogues constitute an interesting compound class of relevance for rotaxanes.108 The preference of phenol to react as a C- rather than O-nucleophile with electrophilic alcohols under the applied conditions stands in contrast to its reactivity under Pd catalysis.109 Friedel–Crafts alkylation was also feasible using N-methyl indole as nucleophile (4c, Scheme 4).
 | ||
| Scheme 4 Friedel–Crafts alkylation of arylmethanols. Conditions: electrophile (0.25 M), nucleophile (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), BTF, 60 °C. | ||
Conclusions
The present study describes the use of a moisture-tolerant and commercially available zirconium catalyst for direct substitution of alcohols with O-, S- and C-nucleophiles. The protocol enabled the formation of a variety of products, including sterically hindered ethers and glycosylated alcohols, in air without the need for dehydration techniques. By incorporating kinetic studies into the early method development phase, rational tuning of reaction conditions was enabled to promote selective formation of unsymmetrical products over symmetric ether side-products. High selectivity toward the unsymmetrical ethers could be obtained either with lower concentration of electrophilic alcohol at the reaction onset or via its sequential addition to the reaction mixture. In addition to synthetic benefits, the kinetic studies simultaneously provided mechanistic insights and illuminated a complex network of interdependent catalytic pathways as well as provided support for direct involvement of the zirconium centre in the catalysis with a minimal number of experiments. As such, the approach represents an attractive shift in the classic workflow of method development and mechanistic studies of organic reactions to benefit the synthetic community.Funding sources
The Swedish Research Council (2015-06466), Stiftelsen Olle Engkvist Byggmästare, Stiftelsen Lars Hiertas Minne and Magnus Bergvalls Stiftelse supported the present study.Abbreviations
| BTF | Benzotrifluoride |
| THF | Tetrahydrofuran |
| Cp | Cyclopentadienyl |
| ee | Enantiomeric excess |
| NMR | Nuclear magnetic resonance |
| HPLC | High performance liquid chromatography |
Author contributions
The study was designed by H. L. and C. M. Kinetic studies were carried out by C. M. assisted by O. D.-S., H. T., D. C., R. C., M. L. and A. R. Substrate evaluation was carried out by C. M. and P. V. assisted by H. T. The manuscript and ESI† were written by H. L. and C. M. with contribution from P. V. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Dr. Matthew Mower for valuable discussions and assistance with the kinetic modelling. Further, we thank Associate Professor Dr. Zoltán Szabó for support with the NMR-studies of the catalyst, Dr. Merle Plassman for assistance with HRMS measurements, and Dr. Johannes Röckl for input during the preparation of the manuscript. The Swedish Research Council, Stiftelsen Olle Engkvist Byggmästare, Stiftelsen Lars Hiertas Minne and Magnus Bergvalls Stiftelse are gratefully acknowledged for financially supporting the study.Notes and references
- T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Rev. Chem., 2018, 2, 35–46 CrossRef CAS.
- J. Sun, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2014, 4, 3346–3356 CrossRef CAS.
- R. J. J. Nel and A. de Klerk, Ind. Eng. Chem. Res., 2009, 48, 5230–5238 CrossRef CAS.
- D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC.
- S. C. Dolan and J. MacMillan, J. Chem. Soc., Chem. Commun., 1985, 1588–1589 RSC.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed.
- E. Bisz and M. Szostak, ChemSusChem, 2017, 10, 3964–3981 CrossRef CAS PubMed.
- B. A. Vara, N. R. Patel and G. A. Molander, ACS Catal., 2017, 7, 3955–3959 CrossRef CAS PubMed.
- K. Kang, L. Huang and D. J. Weix, J. Am. Chem. Soc., 2020, 142, 10634–10640 CrossRef CAS PubMed.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- J. M. J. Williams, in Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, John Wiley & Sons, Weinheim, 2013, ch. 7, pp. 121–137 Search PubMed.
- B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467–4483 RSC.
- P. H. Huy, Eur. J. Org. Chem., 2020, 2020, 10–27 CrossRef CAS.
- R. H. Beddoe, K. G. Andrews, V. Magné, J. D. Cuthbertson, J. Saska, A. L. Shannon-Little, S. E. Shanahan, H. F. Sneddon and R. M. Denton, Science, 2019, 365, 910 CrossRef CAS PubMed.
- P. H. Huy, T. Hauch and I. Filbrich, Synlett, 2016, 27, 2631–2636 CrossRef CAS.
- M. Dryzhakov, E. Richmond and J. Moran, Synthesis, 2016, 48, 935–959 CrossRef CAS.
- E. Emer, R. Sinisi, M. G. Capdevila, D. Petruzziello, F. De Vincentiis and P. G. Cozzi, Eur. J. Org. Chem., 2011, 2011, 647–666 CrossRef.
- M. Bandini and M. Tragni, Org. Biomol. Chem., 2009, 7, 1501–1507 RSC.
- A. Baeza and C. Nájera, Synthesis, 2014, 46, 25–34 CrossRef.
- R. Kumar and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 1121–1146 RSC.
- S. Biswas and J. S. M. Samec, Chem. – Asian J., 2013, 8, 974–981 CrossRef CAS PubMed.
- M. Hellal, F. C. Falk, E. Wolf, M. Dryzhakov and J. Moran, Org. Biomol. Chem., 2014, 12, 5990–5994 RSC.
- V. D. Sarca and K. K. Laali, Green Chem., 2006, 8, 615–620 RSC.
- R. Ortiz and R. P. Herrera, Molecules, 2017, 22, 574 CrossRef PubMed.
- R. Sanz, A. Martínez, D. Miguel, J. M. Álvarez-Gutiérrez and F. Rodríguez, Adv. Synth. Catal., 2006, 348, 1841–1845 CrossRef CAS.
- S. Estopiñá-Durán and J. E. Taylor, Chem. – Eur. J., 2021, 27, 106–120 CrossRef PubMed.
- M. Dryzhakov, M. Hellal, E. Wolf, F. C. Falk and J. Moran, J. Am. Chem. Soc., 2015, 137, 9555–9558 CrossRef CAS PubMed.
- T. K. Hollis, N. P. Robinson and B. Bosnich, J. Am. Chem. Soc., 1992, 114, 5464–5466 CrossRef CAS.
- S. Kobayashi, Synlett, 1994, 1994, 689–701 CrossRef.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W. L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS PubMed.
- S. Kobayashi and C. Ogawa, Chem. – Eur. J., 2006, 12, 5954–5960 CrossRef CAS PubMed.
- R. Qiu, G. Zhang, X. Xu, K. Zou, L. Shao, D. Fang, Y. Li, A. Orita, R. Saijo, H. Mineyama, T. Suenobu, S. Fukuzumi, D. An and J. Otera, J. Organomet. Chem., 2009, 694, 1524–1528 CrossRef CAS.
- L. Yang, Y. Li and P. E. Savage, Ind. Eng. Chem. Res., 2014, 53, 2633–2639 CrossRef CAS.
- R. Qiu, X. Xu, L. Peng, Y. Zhao, N. Li and S. Yin, Chem. – Eur. J., 2012, 18, 6172–6182 CrossRef CAS PubMed.
- N. Li, X. Zhang, X. Xu, Y. Chen, R. Qiu, J. Chen, X. Wang and S.-F. Yin, Adv. Synth. Catal., 2013, 355, 2430–2440 CrossRef CAS.
- H. Ishitani, H. Suzuki, Y. Saito, Y. Yamashita and S. Kobayashi, Eur. J. Org. Chem., 2015, 2015, 5485–5499 CrossRef CAS.
- N. Li, L. Wang, L. Zhang, W. Zhao, J. Qiao, X. Xu and Z. Liang, ChemCatChem, 2018, 10, 3532–3538 CrossRef CAS.
- H. Lundberg, F. Tinnis and H. Adolfsson, Appl. Organomet. Chem., 2019, 33, e5062 CrossRef.
- M. de Léséleuc and S. K. Collins, ACS Catal., 2015, 5, 1462–1467 CrossRef.
- S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287–8288 CrossRef CAS.
- X. Zhang, R. Qiu, C. Zhou, J. Yu, N. Li, S. Yin and X. Xu, Tetrahedron, 2015, 71, 1011–1017 CrossRef CAS.
- Y.-C. Wu, H.-J. Li, L. Liu, N. Demoulin, Z. Liu, D. Wang and Y.-J. Chen, Adv. Synth. Catal., 2011, 353, 907–912 CrossRef CAS.
- F. Liu, K. De Oliveira Vigier, M. Pera-Titus, Y. Pouilloux, J.-M. Clacens, F. Decampo and F. Jérôme, Green Chem., 2013, 15, 901–909 RSC.
- J. Tummatorn, C. Thongsornkleeb, S. Ruchirawat, P. Thongaram and B. Kaewmee, Synthesis, 2015, 47, 323–329 CrossRef CAS.
- S. Matsunaga and M. Shibasaki, in Bismuth-Mediated Organic Reactions, ed. T. Ollevier, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 179–197, DOI:10.1007/128_2011_169.
- R. A. Watile, A. Bunrit, J. Margalef, S. Akkarasamiyo, R. Ayub, E. Lagerspets, S. Biswas, T. Repo and J. S. M. Samec, Nat. Commun., 2019, 10, 3826 CrossRef PubMed.
- P. K. Sahoo, S. S. Gawali and C. Gunanathan, ACS Omega, 2018, 3, 124–136 CrossRef CAS PubMed.
- R. Mazzoni, M. Salmi, S. Zacchini and V. Zanotti, Eur. J. Inorg. Chem., 2013, 2013, 3710–3718 CrossRef CAS.
- T. Ohshima, J. Ipposhi, Y. Nakahara, R. Shibuya and K. Mashima, Adv. Synth. Catal., 2012, 354, 2447–2452 CrossRef CAS.
- P.-A. Payard, Q. Gu, W. Guo, Q. Wang, M. Corbet, C. Michel, P. Sautet, L. Grimaud, R. Wischert and M. Pera-Titus, Chem. – Eur. J., 2018, 24, 14146–14153 CrossRef CAS PubMed.
- M. Gohain, C. Marais and B. C. B. Bezuidenhoudt, Tetrahedron Lett., 2012, 53, 1048–1050 CrossRef CAS.
- M. Noji, T. Ohno, K. Fuji, N. Futaba, H. Tajima and K. Ishii, J. Org. Chem., 2003, 68, 9340–9347 CrossRef CAS PubMed.
- T. Tsuchimoto, K. Tobita, T. Hiyama and S.-I. Fukuzawa, Synlett, 1996, 1996, 557–559 CrossRef.
- G. V. M. Sharma and A. K. Mahalingam, J. Org. Chem., 1999, 64, 8943–8944 CrossRef CAS PubMed.
- W. Huang, Q.-S. Shen, J.-L. Wang and X.-G. Zhou, Chin. J. Chem., 2008, 26, 729–735 CrossRef CAS.
- C. C. Silveira, S. R. Mendes and G. M. Martins, Tetrahedron Lett., 2012, 53, 1567–1570 CrossRef CAS.
- M. Noji, Y. Konno and K. Ishii, J. Org. Chem., 2007, 72, 5161–5167 CrossRef CAS PubMed.
- K. Chen, H. J. Chen, J. Wong, J. Yang and S. A. Pullarkat, ChemCatChem, 2013, 5, 3882–3888 CrossRef CAS.
- P. Khedar, K. Pericherla and A. Kumar, Synlett, 2014, 25, 515–518 CrossRef CAS.
- O. S. Nayal, M. S. Thakur, R. Rana, R. Upadhyay and S. K. Maurya, ChemistrySelect, 2019, 4, 1371–1374 CrossRef CAS.
- R. R. Singh, A. Whittington and R. S. Srivastava, Mol. Catal., 2020, 492, 110954 CrossRef CAS.
- S.-S. Meng, Q. Wang, G.-B. Huang, L.-R. Lin, J.-L. Zhao and A. S. C. Chan, RSC Adv., 2018, 8, 30946–30949 RSC.
- L. Zhang, A. Gonzalez-de-Castro, C. Chen, F. Li, S. Xi, L. Xu and J. Xiao, Mol. Catal., 2017, 433, 62–67 CrossRef CAS.
- Q. Xu, H. Xie, P. Chen, L. Yu, J. Chen and X. Hu, Green Chem., 2015, 17, 2774–2779 RSC.
- A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298–300 CrossRef CAS PubMed.
- Y. Yang, Z. Ye, X. Zhang, Y. Zhou, X. Ma, H. Cao, H. Li, L. Yu and Q. Xu, Org. Biomol. Chem., 2017, 15, 9638–9642 RSC.
- J. Kim, D.-H. Lee, N. Kalutharage and C. S. Yi, ACS Catal., 2014, 4, 3881–3885 CrossRef CAS.
- Y. Liu, X. Wang, Y. Wang, C. Du, H. Shi, S. Jin, C. Jiang, J. Xiao and M. Cheng, Adv. Synth. Catal., 2015, 357, 1029–1036 CrossRef CAS.
- S. Estopiñá-Durán, L. J. Donnelly, E. B. McLean, B. M. Hockin, A. M. Z. Slawin and J. E. Taylor, Chem. – Eur. J., 2019, 25, 3950–3956 CrossRef PubMed.
- A. B. Cuenca, G. Mancha, G. Asensio and M. Medio-Simón, Chem. – Eur. J., 2008, 14, 1518–1523 CrossRef CAS PubMed.
- H. T. Ang, J. P. G. Rygus and D. G. Hall, Org. Biomol. Chem., 2019, 17, 6007–6014 RSC.
- S. Estopiñá-Durán, E. B. McLean, L. J. Donnelly, B. M. Hockin and J. E. Taylor, Org. Lett., 2020, 22, 7547–7551 CrossRef PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 10852–10866 CrossRef CAS PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- J. Burés, Top. Catal., 2017, 60, 631–633 CrossRef.
- C. D. T. Nielsen and J. Burés, Chem. Sci., 2019, 10, 348–353 RSC.
- D. G. Blackmond, T. Rosner and A. Pfaltz, Org. Process Res. Dev., 1999, 3, 275–280 CrossRef CAS.
- X. Companyó and J. Burés, J. Am. Chem. Soc., 2017, 139, 8432–8435 CrossRef PubMed.
- C. Colletto, J. Burés and I. Larrosa, Chem. Commun., 2017, 53, 12890–12893 RSC.
- J. Wang, H. Lundberg, S. Asai, P. Martín-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O'Donnell, A. S. Ratnayake, P. Richardson, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404–E6410 CrossRef CAS PubMed.
- G. Hutchinson, C. Alamillo-Ferrer and J. Burés, J. Am. Chem. Soc., 2021, 143, 6805–6809 CrossRef CAS PubMed.
- P. Villo, O. Dalla-Santa, Z. Szabó and H. Lundberg, J. Org. Chem., 2020, 85, 6959–6969 CrossRef CAS PubMed.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- T. C. Wabnitz, J.-Q. Yu and J. B. Spencer, Chem. – Eur. J., 2004, 10, 484–493 CrossRef CAS PubMed.
- I. Šolić, H. X. Lin and R. W. Bates, Tetrahedron Lett., 2018, 59, 4434–4436 CrossRef.
- T. K. Hollis and B. Bosnich, J. Am. Chem. Soc., 1995, 117, 4570–4581 CrossRef CAS.
- R. Qiu, X. Xu, Y. Li, G. Zhang, L. Shao, D. An and S. Yin, Chem. Commun., 2009, 1679–1681, 10.1039/B821366F.
- R. K. Schmidt, K. Müther, C. Mück-Lichtenfeld, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2012, 134, 4421–4428 CrossRef CAS PubMed.
- R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876–2879 CrossRef CAS PubMed.
- N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed.
- H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS.
- S. Zhang, X. Zhang, X. Ling, C. He, R. Huang, J. Pan, J. Li and Y. Xiong, RSC Adv., 2014, 4, 30768–30774 RSC.
- J. S. M. Wai, I. Marko, J. S. Svendsen, M. G. Finn, E. N. Jacobsen and K. B. Sharpless, J. Am. Chem. Soc., 1989, 111, 1123–1125 CrossRef CAS.
- G. Manolikakes, M. A. Schade, C. M. Hernandez, H. Mayr and P. Knochel, Org. Lett., 2008, 10, 2765–2768 CrossRef CAS PubMed.
- N. A. Vermeulen, M. S. Chen and M. Christina White, Tetrahedron, 2009, 65, 3078–3084 CrossRef CAS.
- J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537–9542 CrossRef CAS PubMed.
- G. V. M. Sharma, K. S. Kumar, B. S. Kumar, S. V. Reddy, R. S. Prakasham and H. Hugel, Synth. Commun., 2014, 44, 3156–3164 CrossRef CAS.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chem. – Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed.
- D. Crich, J. Am. Chem. Soc., 2021, 143, 17–34 CrossRef CAS PubMed.
- J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398–402 CrossRef CAS PubMed.
- H. Wang, K. Liang, W. Xiong, S. Samanta, W. Li and A. Lei, Sci. Adv., 2020, 6, eaaz0590 CrossRef CAS PubMed.
- D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179–4182 CrossRef CAS PubMed.
- R. M. P. Veenboer and S. P. Nolan, Green Chem., 2015, 17, 3819–3825 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- A. H. G. David, R. Casares, J. M. Cuerva, A. G. Campaña and V. Blanco, J. Am. Chem. Soc., 2019, 141, 18064–18074 CrossRef CAS PubMed.
- T. Rukkijakan, S. Akkarasamiyo, S. Sawadjoon and J. S. M. Samec, J. Org. Chem., 2018, 83, 4099–4104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, kinetic data, NMR (1H and 13C), HPLC chromatograms can be found in the ESI. See DOI: 10.1039/d1cy01219c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |