
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural insight into an atomic layer deposition (ALD) grown Al2O3 layer on Ni/SiO2: impact on catalytic activity and stability in dry reforming of methane†
Sung Min
Kim‡
 a,
Andac
Armutlulu
a,
Wei-Chih
Liao§
b,
Davood
Hosseini
a,
Dragos
Stoian
c,
Zixuan
Chen
a,
Paula M.
Abdala
a,
Christophe
Copéret
b and
Christoph
Müller
*a
a,
Andac
Armutlulu
a,
Wei-Chih
Liao§
b,
Davood
Hosseini
a,
Dragos
Stoian
c,
Zixuan
Chen
a,
Paula M.
Abdala
a,
Christophe
Copéret
b and
Christoph
Müller
*a
aDepartment of Mechanical and Process Engineering, ETH Zurich, Leonhardstrasse 27, 8092 Zurich, Switzerland. E-mail: muelchri@ethz.ch
bDepartment of Chemistry and Applied Sciences, ETH Zurich, Vladimir Prelog Weg 1-5, 8093 Zurich, Switzerland
cSwiss–Norwegian Beamlines, ESRF, BP 220, Grenoble, 38043, France
First published on 25th October 2021
Abstract
The development of stable Ni-based dry reforming of methane (DRM) catalysts is a key challenge owing to the high operating temperatures of the process and the propensity of Ni for promoting carbon deposition. In this work, Al2O3-coated Ni/SiO2 catalysts have been developed by employing atomic layer deposition (ALD). The structure of the catalyst at each individual preparation step was characterized in detail through a combination of in situ XAS–XRD, ex situ27Al NMR and Raman spectroscopy. Specifically, in the calcination step, the ALD-grown Al2O3 layer reacts with the SiO2 support and Ni, forming aluminosilicate and NiAl2O4. The Al2O3-coated Ni/SiO2 catalyst exhibits an improved stability for DRM when compared to the benchmark Ni/SiO2 and Ni/Al2O3 catalysts. In situ XAS–XRD during DRM together with ex situ Raman spectroscopy and TEM of the spent catalysts confirm that the ALD-grown Al2O3 layer suppresses the sintering of Ni, in turn reducing also coke formation significantly. In addition, the formation of an amorphous aluminosilicate phase by the reaction of the ALD-grown Al2O3 layer with the SiO2 support inhibited catalysts deactivation via NiAl2O4 formation, in contrast to the reference Ni/Al2O3 system. The in-depth structural characterization of the catalysts provided an insight into the structural dynamics of the ALD-grown Al2O3 layer, which reacts both with the support and the active metal, allowing to rationalize the high stability of the catalyst under the harsh DRM conditions.
1. Introduction
To mitigate anthropogenic climate change, carbon dioxide capture, storage and utilization (CCSU) is a promising technology.1–5 In this context the dry reforming of methane (DRM) has emerged as a process to convert two major greenhouse gases, CH4 and CO2, into a synthesis gas (CH4 + CO2 → 2CO + 2H2 ΔH0298K = +247 kJ mol−1). Synthesis gas is a versatile chemical feedstock that can be converted further into synthetic liquid fuels and chemicals, e.g., via the Fischer–Tropsch process.6,7 In DRM the development of Ni-based catalyst has received considerable attention, owing to its high activity combined with its relatively low cost when compared to noble metals.8Ni0 is proposed to be the catalytically active state for DRM, and a linear relationship between the DRM activity and the quantity of surface Ni has been reported.5,9–12 However, under DRM conditions, Ni-based catalysts are prone to rapid deactivation due to a combination of various factors including coke formation,13,14 metal oxidation followed by the migration of Ni into the support,11,15–17 and sintering of the Ni particles.18,19 The fact that the Tammann temperature of Ni (≈700 °C) is below the envisioned operating temperatures of the DRM process (≈900 °C) makes sintering a serious issue. Coke formation during DRM can proceed through methane decomposition (CH4 → C + 2H2, ΔH0298K = +75 kJ mol−1) and CO disproportionation, i.e., Boudouard reaction (2CO → C + CO2, ΔH0298K = −171 kJ mol−1). Carbon species accumulate on the catalyst surface thereby blocking the active sites for DRM or leading to the detachment of Ni particles from the support. Coke formation is also linked to sintering since the nature and the rate of coke deposition is sensitive to the size of the Ni nanoparticles.20,21 When Ni (111) facets and step edges are too small, nucleation of C* does not proceed, restricting the formation of a graphene layer.20 Thus, the formation and the stabilization of small Ni particles (<10 nm) is a key requirement towards the development of highly active and stable Ni-based DRM catalysts.21–24 A further challenge with regards to Ni-based DRM catalysts that has to be overcome is the inhibition of the reaction of Ni with the (metal oxide) support. Although Al2O3 support is known to stabilize Ni particles more effectively against sintering than SiO2, the reaction of Ni with Al2O3 can lead to its oxidation and formation of inactive NiAl2O4.11,15,16 Hence conceiving and generating highly active and stable Ni-based DRM catalysts, several strategies have been proposed, including (i) controlling the Ni particle size with the aim of minimizing the fraction of edge and step sites on the nickel surface;11,22,25 (ii) coating Ni-based DRM catalyst by metal oxide layers (e.g., Al2O3, MgO, ZrO2 and SiO2)13,26–32 to alleviate sintering and coke formation; (iii) deposition of Ni particles on basic supports (e.g., MgO and La2O3) to reduce coke formation;11,16 (iv) promotion with alkaline (e.g., K and Na),33,34 alkaline earth (e.g., Mg, Ca and Ba)33,35 or lanthanide (e.g. La)16,36 metals to facilitate CO2 activation to gasify carbon; and (v) the alloying of Ni with a noble (e.g., Pt, Rh, Pd, and Ru)37–41 or non-noble (e.g., Fe, Cu, Mn, and Co)9,10,37,42–44 transition metal to improve its activity and stability for DRM.
SiO2-Supported Ni DRM catalysts have been plagued by a lower metal dispersion and poor sintering resistance under DRM conditions owing to a weak metal–support interaction.45 To stabilize Ni nanoparticles on a SiO2 support, several strategies have been employed. For example, nickel-silicide colloids have been deposited on SiO2 and CeO2 supports. Treatment in H2 leads to the formation of small, supported Ni nanoparticles (1–2 nm).25 Further, the addition of metal oxide overcoats (SiO2, Al2O3, MgO, ZrO2 and TiO2)13,26,27 on Ni/SiO2 has shown to reduce sintering and coke formation.
In order to precisely control the thickness of the metal oxide overcoat with atomic scale precision, atomic layer deposition (ALD) has been employed.46 ALD-grown TiO2,28 Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30,31 and ZrO229 coatings demonstrated an enhanced catalytic activity and stability. Although the deposition of Al2O3 coating onto Ni/Al2O326 alleviated the sintering of Ni under DRM conditions, Ni was found to interact with the overcoat to form NiAl2O4. Currently, the structural and electronic interaction of Ni with an Al2O3 overcoat is only poorly understood, hindering the further development of this class of material systems.
30,31 and ZrO229 coatings demonstrated an enhanced catalytic activity and stability. Although the deposition of Al2O3 coating onto Ni/Al2O326 alleviated the sintering of Ni under DRM conditions, Ni was found to interact with the overcoat to form NiAl2O4. Currently, the structural and electronic interaction of Ni with an Al2O3 overcoat is only poorly understood, hindering the further development of this class of material systems.
This work aims at obtaining an atomic-level understanding of how an ALD-grown Al2O3 overcoat reacts with Ni and the SiO2 support during pretreatment and DRM. To this end, Al2O3-coated Ni/SiO2 DRM catalysts have been obtained by ALD. The structure of the catalysts were interrogated in detail by in situ X-ray absorption spectroscopy (XAS) and X-ray powder diffraction (XRD) to probe the local structure of Ni, the average crystalline structure and changes therein with time-on-stream (TOS) during catalyst pretreatment and DRM. Further spectroscopic (Raman, FTIR and 27Al magic-angle spinning (MAS) solid-state NMR) and electron microscopy (TEM) characterizations were applied to scrutinize the structural dynamics of the Al2O3 coating during synthesis and pretreatment and to elucidate the driving forces behind sintering and coking under DRM conditions.
2. Experimental
2.1. Preparation of bare Ni/SiO2 and Ni/Al2O3 catalysts
5 wt% Ni/SiO2 and Ni/Al2O3 catalysts were prepared by incipient wetness impregnation using Ni(NO3)2 as the Ni precursor and, respectively, Aerosil 300 SiO2 (SBET = 275 m2 g−1) and γ-Al2O3 (SBET = 220 m2 g−1) supports. The catalysts were calcined at 300 °C under static air to yield NiO.2.2. Atomic layer deposition of Al2O3
An Al2O3 overcoat was deposited onto calcined catalyst at 300 °C (NiO/SiO2 and NiO/Al2O3) and onto the supports (SiO2 and Al2O3) via ALD (Picosun R-200, equipped with a powder coating system). Electronic grade trimethylaluminum (TMA) and deionized (DI) water (or ozone) were used as the Al2O3 precursor and oxidants, respectively. High-purity N2 served as both the carrier and purge gas. In a typical ALD deposition cycle the following pulse and purge (N2) times were used: 0.1 s TMA – 15 s N2 purge – 0.1 s oxidant (H2O or O3) – 15 s N2 purge. The deposition was carried out at 300 °C. High-resolution TEM allowed us to determine the deposition rate, i.e., ≈1.0 Å per ALD cycle, which is in good agreement with the thickness of an Al2O3 layer, grown simultaneously on a Si wafer and measured by ellipsometry (SENTECH SE850). To prepare the Al2O3-coated catalysts, approximately 300 mg of the catalyst (pre-calcined at 300 °C) were loaded into the reactor via a powder cartridge and then heated to 300 °C under vacuum, followed by repeated ALD cycles. The as-synthesized catalyst was subsequently calcined at 800 °C in static air in a muffle furnace.2.3. Characterization
000, 20000, and 80000 scans for calcined Al2O3-coated SiO2, Al2O3-coated NiO/SiO2, and reduced Al2O3-coated Ni/SiO2, respectively). The raw FID was shifted by 142 points towards the start of FID acquisition. All spectra were fitted with the CzSimple model using the DMFit software,51 simulating the 27Al central transitions with a Gaussian distribution of sites (d = 5).
2.4. DRM catalytic test
The DRM reaction was carried out in a fixed-bed quartz reactor (400 mm length, 12.6 mm internal diameter). The calcined catalyst (≈20 mg) was loaded into the reactor and reduced at the temperature determined by H2-TPR in 10 vol% H2/N2 (100 ml min−1) for 2 h. Subsequently, the bed was cooled down to 700 °C to perform the DRM activity test. The total flow rate of the feed gas was 100 ml min−1 (270 L g−1 h−1; 45 ml min−1 CH4, 45 ml min−1 CO2 and 10 ml min−1 N2). The composition of the off-gas was analyzed by an online micro-GC equipped (CV-200, Thermo Scientific). N2 was used as internal standard.3. Results and discussion
3.1. Deposition of an Al2O3 overcoat onto Ni/SiO2 by ALD
Fig. 1a schematically describes the surface reactions occurring during a half and full ALD cycle. To follow the surface reactions occurring during ALD, FTIR spectra were collected after one half (i.e., after injection of a TMA pulse) and a full (i.e., TMA and subsequent injection of a steam pulse) ALD cycle on dehydroxylated SiO2 (Fig. 1b). Due to high reactivity of TMA with air and moisture, the FTIR measurements were carried out in an inert atmosphere (glovebox). SiO2 dehydroxylated at 500 °C in high vacuum (10−6 mbar) shows isolated silanols at 3746 cm−1. After one half cycle, i.e., after pulsing TMA, the intensity of the peak from isolated silanols is significantly reduced owing to their reaction with TMA. In addition, peaks associated with C–H stretching vibrations (2916 and 2966 cm−1) appear owing to methyl groups of surface-adsorbed (or chemisorbed) TMA. After injection of a pulse of steam, i.e., after the completion of a full ALD cycle, the intensity of the peak due to isolated silanols restores, albeit with a reduced intensity. In addition, a new broad band between 3461–3498 cm−1 appears, which is ascribed to a O–H interacting with Al and Si on the surface (or vicinal silanols).52 The regeneration of surface O–H groups provides the reactive sites for subsequent ALD cycles.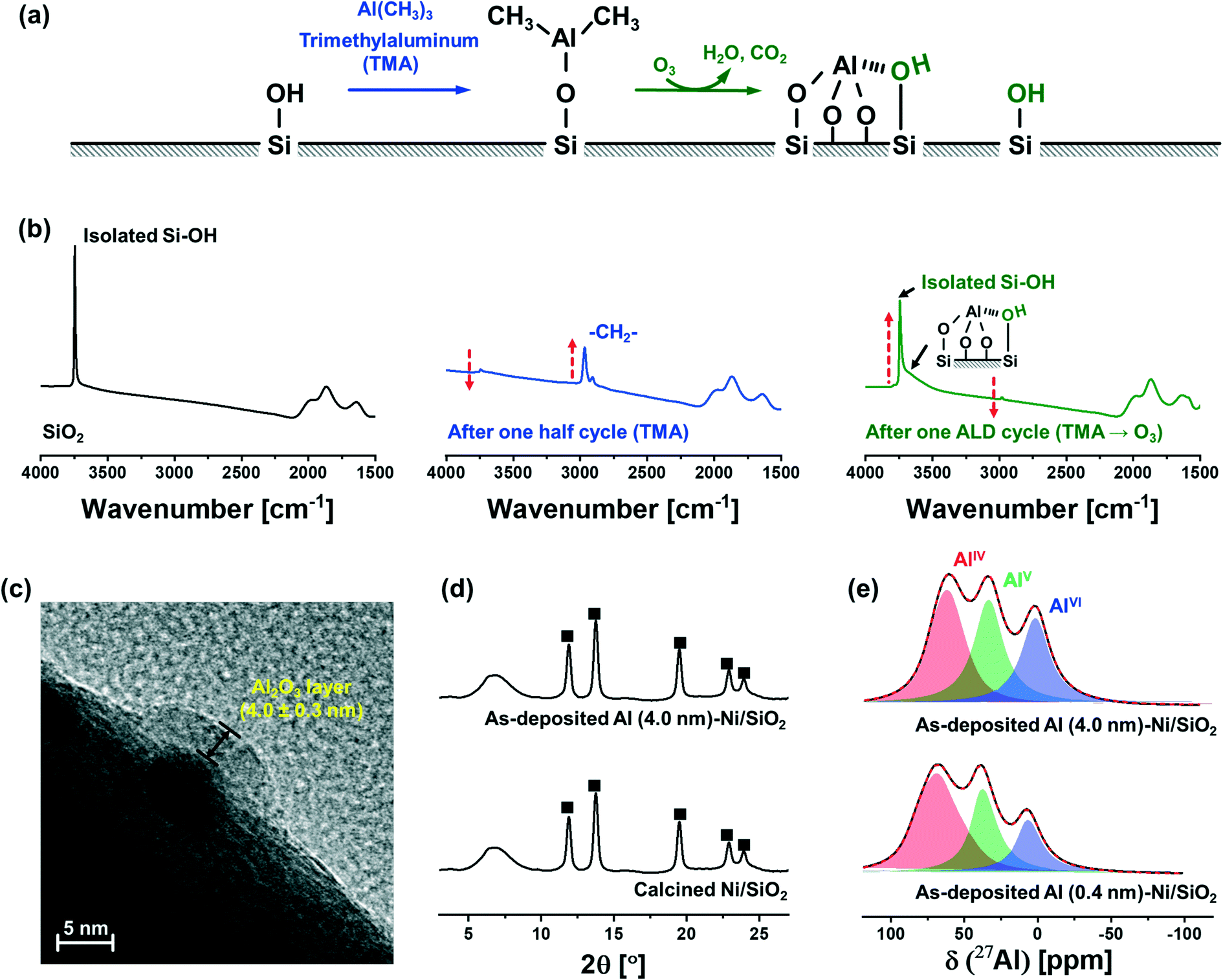 | ||
| Fig. 1 Characterization of as-synthesized Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2: (a) schematic illustrating the surface reactions occurring over a half and a full ALD cycle. (b) FTIR spectra obtained after a half and a full ALD cycle. The arrows indicate the direction of change during a full ALD cycle. (c) TEM image of Al (4.0 nm)–Ni/SiO2, showing an amorphous Al2O3 layer on top of a Ni nanoparticle dispersed on a SiO2 support. (d) XRD diffractograms of as-synthesized Al (4.0 nm)–Ni/SiO2 and calcined Ni/SiO2: (■) NiO. (e) Solid-state 27Al NMR spectra of as-synthesized Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2. | ||
The growth rate of the Al2O3 layer was estimated by HR-TEM (Fig. 1c). The thickness of the Al2O3 layer on Ni nanoparticle (5.0 ± 0.3 nm) supported on SiO2 after 40 ALD cycles (TMA/steam) was determined as 4.0 ± 0.3 nm, denoted as Al (4.0 nm)–Ni/SiO2, corresponding to a deposition rate of ∼0.1 nm/ALD cycle.
XRD characterization of Ni/SiO2 calcined at 300 °C and as-synthesized Al (4.0 nm)–Ni/SiO2 (ALD temperature 300 °C) revealed the presence of cubic NiO (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group) in Ni/SiO2 (Fig. 1d). A broad halo at 4.5–9.5° is ascribed to the amorphous SiO2 support. Similarly, in the case of as-synthesized Al (4.0 nm)–Ni/SiO2, only Bragg peaks due to NiO are present, indicating an amorphous nature of the as-deposited Al2O3 layer.
m space group) in Ni/SiO2 (Fig. 1d). A broad halo at 4.5–9.5° is ascribed to the amorphous SiO2 support. Similarly, in the case of as-synthesized Al (4.0 nm)–Ni/SiO2, only Bragg peaks due to NiO are present, indicating an amorphous nature of the as-deposited Al2O3 layer.
As-synthesized, Al2O3-coated Ni/SiO2 catalysts with different thicknesses of the Al2O3 layer (0.4 nm and 4.0 nm), i.e., Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2, were characterized further by 27Al MAS NMR (Fig. 1e). The fitting of the 27Al MAS NMR spectra was performed using the Czjzek model.51 The 27Al MAS spectra of as-synthesized Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2 contains tetra- (AlIV, NMR shift δ = 75.9, 35.2, 8.6 ppm and quadrupolar coupling constant CQ = 8.4 MHz, penta- (AlV, δ = 35.2 ppm and CQ = 6.3 MHz) and hexa- (AlVI, δ = 8.6 ppm and CQ = 5.7 MHz) coordinated Al sites, and no significant differences are observed between the spectra of as-synthesized Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2. The broad and featureless 27Al NMR line feature is characteristic of amorphous Al2O3,53–55 in line with the absence of diffraction peaks due to Al2O3 in as-synthesized Al (4.0 nm)–Ni/SiO2.
3.2. Structural characterization of the materials after calcination at 800 °C: probing the interaction of the Al2O3 layer with Ni and SiO2
The elemental composition of the materials, as determined by ICP-OES, is summarized in Table 1. The deposition of an Al2O3 overcoat onto Ni/SiO2 results in a reduction of the weight fractions of Ni (and SiO2). Elemental analysis showed that the weight percent of Ni in the materials was reduced from 5 wt% in Ni/SiO2 to 4.8 wt% and 3.7 wt% in Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2, respectively (accompanied by an increase in the Al content from 0 wt%, to 1.1 wt% and 10.2 wt%).
| Catalyst | Elemental compositiona | N2 physisorptionb | Chemisorption | Ni particle sized [nm] | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ni [wt%] | Al [wt%] | S BET [m2 g−1] | V pore,BJH [cm3 g−1] | D pore [nm] | H2 uptakec [μmol g−1] | Dispersion [%] | H2-Chem. | TEM | |
| a The weight percentage of Ni and Al in the calcined materials was determined by ICP-OES. b The specific surface area, pore volume, and pore radius were calculated using the BET and BJH models for the calcined materials. c The quantity of surface Ni in the reduced materials was determined by H2 chemisorption using a stoichiometry factor of 1.0 for H/Ni. d The average Ni particle size of the reduced materials was determined by HR-TEM. | |||||||||
| Ni/SiO2 | 5.0 | — | 197 | 1.33 | 1.9 | 102 | 12 | 8.5 | 7.1 ± 1.8 |
| Al (0.4 nm)–Ni/SiO2 | 4.8 | 1.1 | 201 | 1.40 | 1.9 | 91 | 11 | 9.1 | 7.3 ± 1.5 |
| Al (4.0 nm)–Ni/SiO2 | 3.7 | 10.2 | 204 | 1.44 | 2.0 | 70 | 11 | 9.1 | 8.2 ± 1.1 |
The N2 isotherms and BJH pore size distributions of Ni/SiO2 calcined at 300 °C, as-synthesized Al (4.0 nm)–Ni/SiO2, and Al (4.0 nm)–Ni/SiO2 calcined at 800 °C are shown in Fig. S3.† The specific surface area and BJH pore volume of Ni/SiO2 are 198 m2 g−1 and 1.33 cm3 g−1, respectively. After the deposition of a 4 nm-thick layer of Al2O3 onto Ni/SiO2, i.e., as-synthesized Al (4.0 nm)–Ni/SiO2, the pore volume in pores with a diameter dpore = 10–100 nm is significantly reduced (Fig. S3b†), resulting in a BET surface area and BJH pore volume of, respectively, 91 m2 g−1 and 0.64 cm3 g−1. This reduction in surface area is ascribed to the filling of voids by the ALD-grown Al2O3. On the other hand, Al (4.0 nm)–Ni/SiO2 after calcination (800 °C), shows a significant increase of the volume in small pores (dpore = 1–10 nm) (Fig. S3b†). Such an increase in the specific surface area and BJH pore volume was also observed in calcined Al (0.4 nm)–Ni/SiO2 (Table 1). We hypothesize that the local restructuring of the ALD-grown Al2O3 layer during calcination (including the reaction of the Al2O3 layer with NiO and SiO2 as discussed in more detail in the following section) is responsible for the formation of additional small pores.
m space group) started to appear at ca. 500 °C (Fig. 2a), which grew in intensity as the temperature increased to 800 °C; this was accompanied by a simultaneous decrease of the intensity of the NiO peaks. The diffraction pattern of NiAl2O4 is similar to that of γ-Al2O3, yet we can clearly assign the observed peaks to NiAl2O4 when taking into account the results of the analysis of the XANES spectra. For the in situ XANES spectra at the Ni K-edge during calcination, the white line decreased in intensity and shifted from 8352 eV to 8354 eV (Fig. 2b). In addition, changes in the features of the post-edge region were observed, in line with the formation of NiAl2O4. NiO and NiAl2O4 exhibit very distinct XANES features as shown in Fig. 2b. Thus, applying linear combination fitting (LCF) analysis, using references for NiO and NiAl2O4 (Fig. 2b and c) revealed the onset of NiAl2O4 formation at ca. 500 °C. The fraction of NiAl2O4 reached 0.68 at 800 °C, whereas the fraction of NiO was reduced correspondingly. Hence in situ XRD–XAS experimentation confirms the reaction of NiO with the ALD-grown Al2O3 layer forming NiAl2O4 (NiO + Al2O3 → NiAl2O4).57 However it is worth noting that in situ XRD and Ni K-edge XANES only allows to elucidate the reaction between the ALD-grown Al2O3 and NiO and cannot probe the interaction between Al2O3 and the SiO2 support. This is due to the fact that XAS is an element specific technique and Ni K-edge XAS can only probe the environment around Ni, while XRD is unable to provide any information on amorphous phases (such as amorphous aluminosilicates).
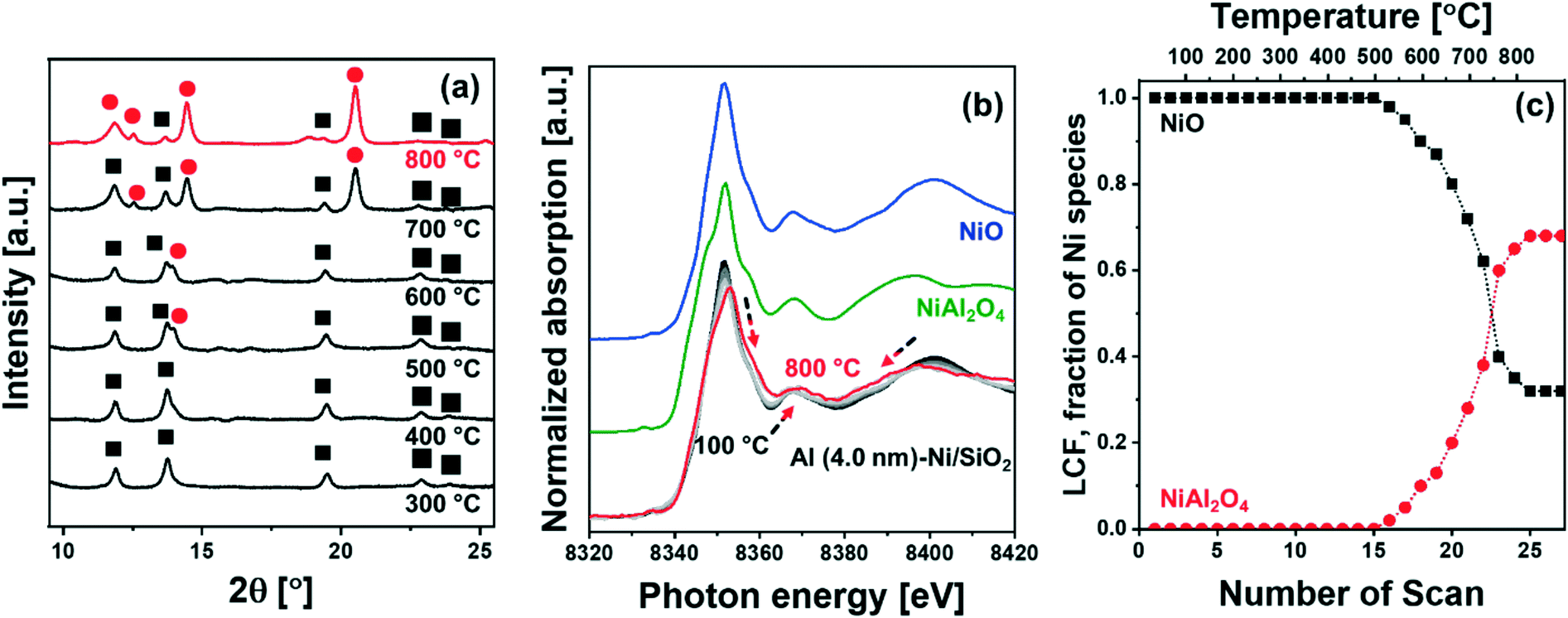 | ||
Fig. 2 Combined in situ XRD–XAS characterization: (a) in situ XRD (λ = 0.5060 Å), (b) in situ Ni K-edge XANES with NiO and NiAl2O4 references, and (c) corresponding LCF analysis results for Al (4.0 nm)–Ni/SiO2 during calcination in 20 vol% O2/N2. The symbols in the diffractogram mark the peaks due to NiO (ICSD 9866, ■) and NiAl2O4 (ICSD 9554,  ). The arrows indicate the direction of change in the XANES spectra with increasing calcination temperature. ). The arrows indicate the direction of change in the XANES spectra with increasing calcination temperature. | ||
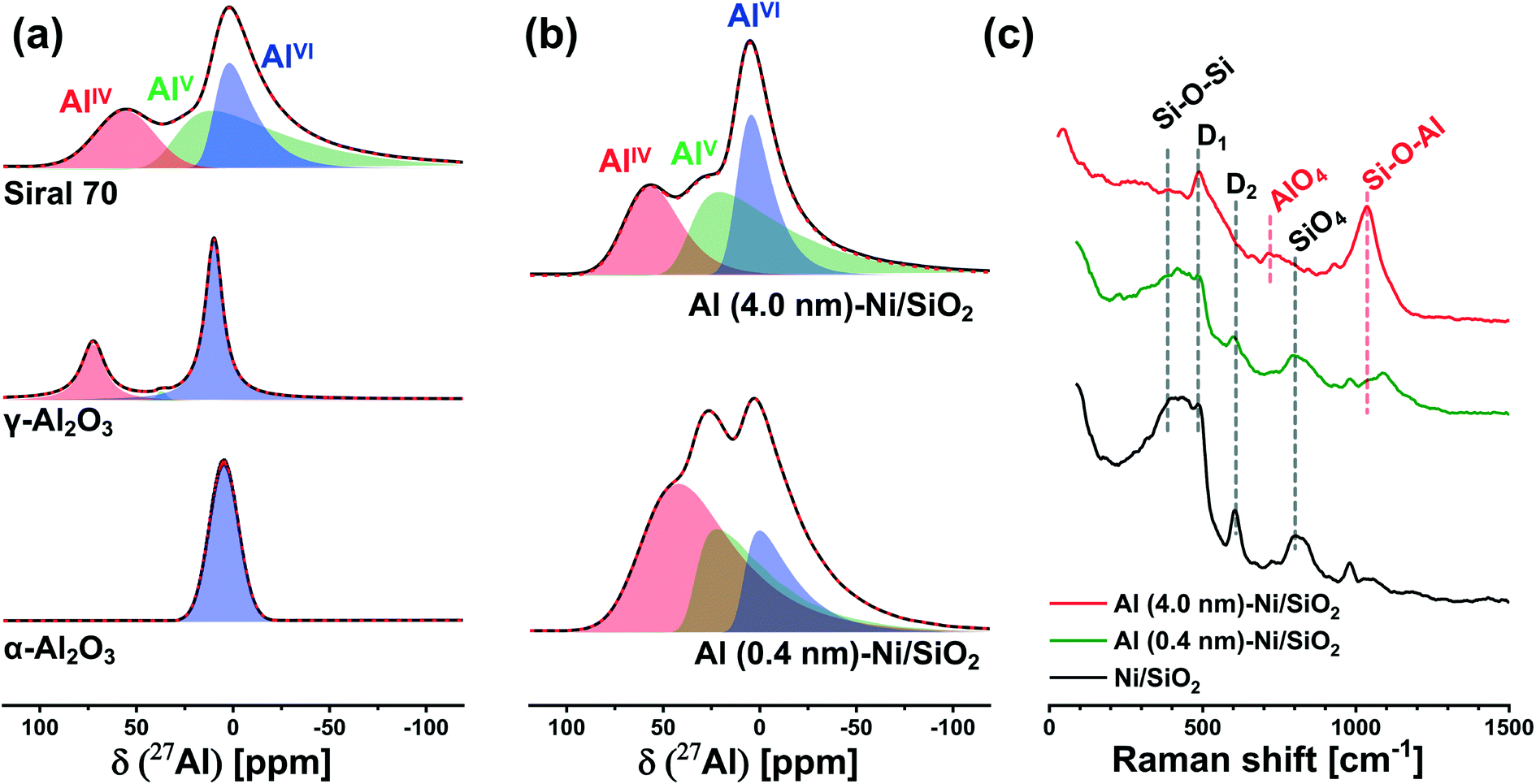 | ||
| Fig. 3 Characterization of the interaction between silica and alumina: solid-state 27Al-NMR of (a) α-Al2O3, γ-Al2O3, and aluminosilicate Siral 70 references; and (b) calcined Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2. (c) Raman spectra of calcined Ni/SiO2, Al (0.4 nm)–Ni/SiO2, and Al (4.0 nm)–Ni/SiO2. | ||
To probe more quantitatively the differences in the Al environment in calcined Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2 the respective samples were exposed to an ammonia pretreatment. Previous reports have shown that ammonia treatment of AAS leads to the transformation of its AlVI sites into AlIV sites, whereas such a transformation (upon ammonia treatment) has not been observed for γ-Al2O3.59,60,62,63 In this context, we applied ammonia treatment to calcined Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2, in order to probe the AAS content, following the hypothesis that only AlVI sites in AAS will selectively transform into AlIV sites by NH3 treatment. Indeed, a comparison of the 27Al NMR spectra of γ-Al2O3 before and after ammonia treatment (5 vol% NH3/He at 120 °C for 5 h) features a negligible difference between the two spectra (Table S1 and Fig. S6a†), confirming previously reported observations.59,60,62,63 On the other hand, for calcined Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2, a transformation of AlVI sites to AlIV sites was observed after ammonia treatment (Fig. S6b† compared to Fig. 3b). The evaluated ratios of the AlIV, AlV, and AlVI sites in Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2 before and after ammonia treatment are summarized in Table S1.† After ammonia treatment the relative abundance of AlVI sites in Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2 decreased, while the relative abundance of AlIV sites increased, providing further evidence of the presence of AAS in both materials.
To complement the 27Al NMR studies, Raman spectroscopy was performed on calcined Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–N/SiO2 (Fig. 3c). For bare Ni/SiO2, a dominant band at 395 cm−1, which is ascribed to Si–O–Si linkages in the tetrahedral unit of SiO4, is observed.64–66 In addition, the Si–O stretching band in the Si–O–Si plane is detected at 802 cm−1. The bands at 483 cm−1 and 603 cm−1 are associate with the breathing modes of oxygen in, respectively, four (D1) and three (D2) membered rings of tetrahedral SiO4.67,68 An increase in the thickness of the ALD-grown Al2O3 layer leads to a reduction of the intensity of the Si–O–Si and SiO4 bands, whereas new characteristic features appeared at 707 cm−1 and 1043 cm−1. The band at 707 cm−1 is related to AlO4 units and the band at 1043 cm−1 is attributed to Si–O–Al,65,66,69 indicative of the presence of AAS in calcined Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2. These findings are in good agreement with 27Al NMR results. The intensified Si–O–Al band in Al (4.0 nm)–Ni/SiO2 is due to its higher loading of Al, leading to a higher signal of the Raman band at 1043 cm−1.
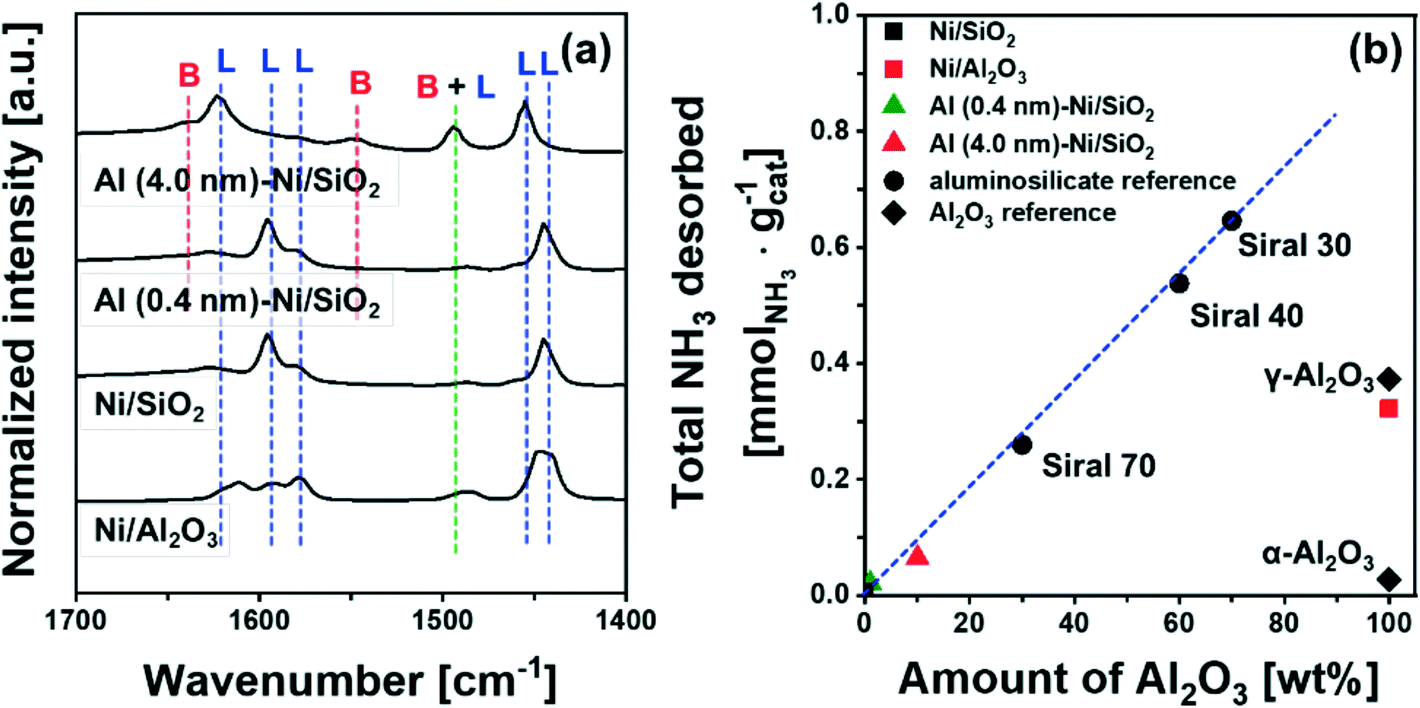 | ||
| Fig. 4 Acidic properties: (a) difference FT-IR spectra of pyridine adsorbed on calcined Ni/SiO2, Ni/Al2O3, Al (4.0 nm)–Ni/SiO2 and the references α-Al2O3, γ-Al2O3, and Siral 70. The labels L and B mark Lewis and Brønsted acid sites, respectively. (b) The quantity of NH3 desorbed during TPD as a function of the mass fraction of Al2O3. | ||
To quantify the amount of acid sites, we performed NH3-TPD (Fig. S7†). Bare Ni/SiO2 and α-Al2O3 showed a negligible desorption of NH3, i.e. 0.007 mmol NH3 per gcat and 0.027 mmol NH3 per gcat, respectively. On the other hand, 0.373 mmol NH3 per gcat and 0.322 mmol NH3 per gcat were desorbed from γ-Al2O3 and bare Ni/Al2O3, attributed largely to Lewis acid sites. For commercial AAS, the quantity of desorbed NH3 increases from 0.26 mmol NH3 per gcat to 0.65 mmol NH3 per gcat with an increase in the quantity of Al2O3 from 30 wt% (Siral 70) to 70 wt% (Siral 30). For the catalysts Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2, the amount of NH3 desorbed was 0.021 mmol NH3 per gcat and 0.065 mmol NH3 per gcat, respectively. Overall, the amount of NH3 desorbed showed a linear correlation with the amount Al2O3 in the AAS materials (Fig. 4b). Nevertheless, the amount of NH3 desorbed from Al2O3-coated Ni/SiO2 is small compared to γ-Al2O3 likely due to the amorphous nature of the Al2O3 layer.72
3.3. Catalyst activation: reduction of calcined Al2O3-coated Ni/SiO2 catalysts
The reduction behavior of the calcined Al2O3-coated Ni/SiO2 catalysts was assessed by H2-TPR (Fig. 5a). The reducible Ni2+ species can be classified as: (i) α-type (weak interaction with support, 290–475 °C), (ii) β-type (mid-interaction with support, 475–753 °C), and (iii) γ-type (strong interaction with support, 753–894 °C).73–76 As references we used commercial NiO, NiAl2O4, Ni impregnated on Siral 70 and Ni impregnated on Al2O3-coated SiO2 (Ni/Al (4.0 nm)–SiO2). The TPR profiles of the NiO and NiAl2O4 references showed a reduction peak at 415 °C and 803 °C, respectively, corresponding to α- and γ-type reduction behaviors. Ni/Siral 70 and Ni/Al (4.0 nm)–SiO2 calcined at 800 °C revealed a reduction peak at 611 °C and 617 °C, respectively, indicative of a β-type reduction. The β-type reduction is ascribed to an interaction of NiO with alumina (NiO–Al2O3). This observation suggests that the formation of AAS mitigates the further reaction of NiO with Al2O3 (forming NiAl2O4); however, the interaction of NiO with Al2O3 leads to a higher reduction temperature compared to bulk NiO.73,77–79 H2-TPR of Ni/SiO2 revealed a low reduction temperature of 450 °C indicative of a weak Ni–silica interaction, in agreement with literature.80,81 On the other hand, two reduction peaks were observed in the H2-TPR of Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2. In Al (0.4 nm)–Ni/SiO2 the reduction peaks at 450 °C and 600 °C are attributed to the reduction of NiO interacting with SiO2 and free Al2O3. Increasing the thickness of the ALD-grown Al2O3 layer, i.e., Al (4.0 nm)–Ni/SiO2, leads to reduction temperatures of 600 °C and 800 °C owing to the interaction of NiO with free Al2O3 on Al (4.0 nm)–Ni/SiO2, and the reaction of NiO with Al2O3 forming NiAl2O4, respectively.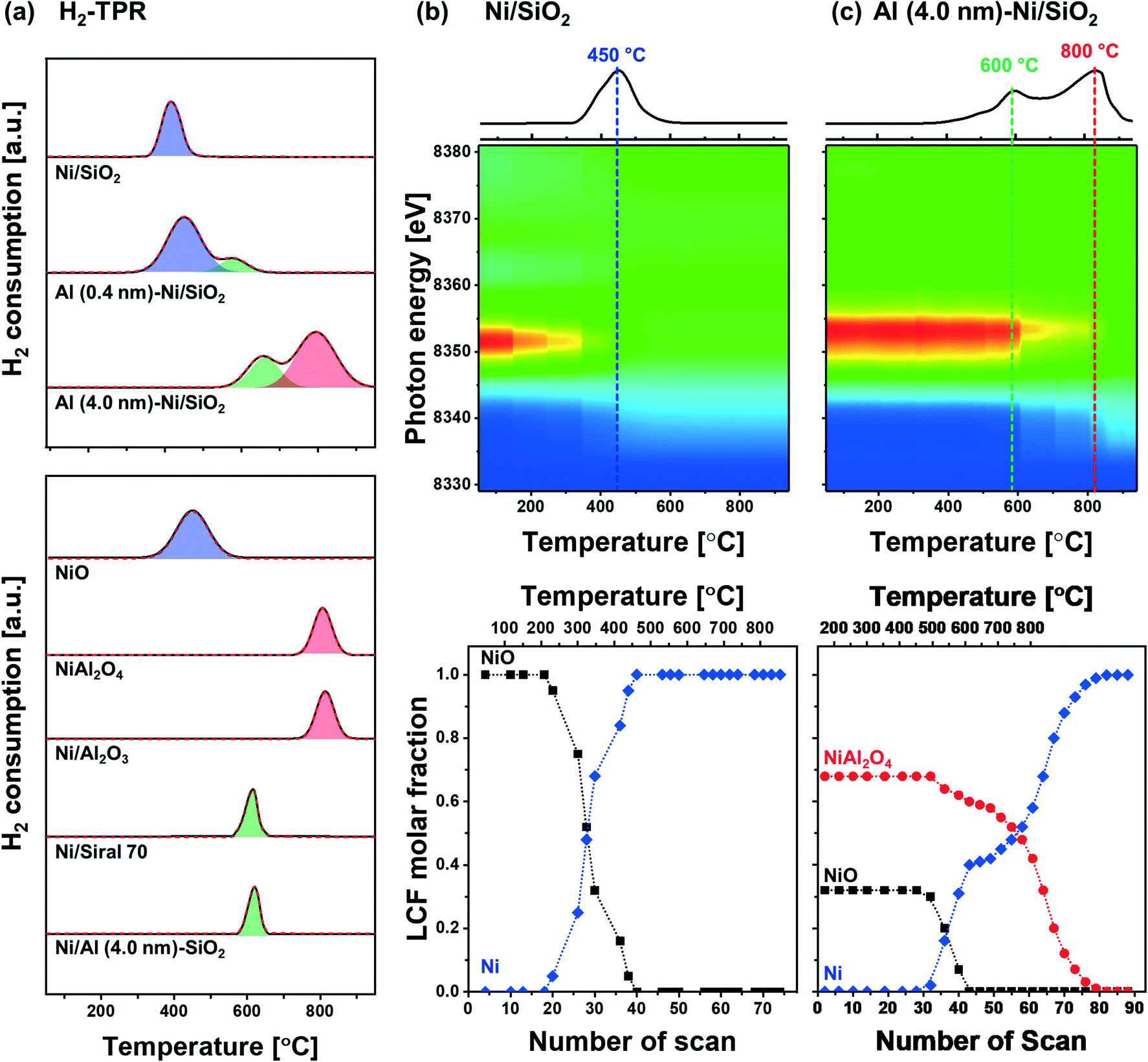 | ||
| Fig. 5 Characterization of the reduced materials: (a) H2-TPR of the references (NiO, Ni/Al2O4, Ni/Al2O3 and Ni/Siral 70) and calcined Ni/SiO2, Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2. Contour plots of the acquired in situ Ni K-edge XANES data and the corresponding LCF analysis of (b) Ni/SiO2 and (c) Al (4.0 nm)–Ni/SiO2 during reduction. | ||
In the following, we utilize in situ XANES at the Ni K-edge (Fig. 5b) and in situ XRD (Fig. S8b†) during reduction to probe in more detail the reduction pathway of Ni2+ species in the calcined catalysts. Calcined Ni/SiO2 (α-type Ni2+) and Al (4.0 nm)–Ni/SiO2 (β- and γ-type Ni2+) were chosen for in situ XAS–XRD experiments as they show the largest difference in the H2-TPR experiments (whereby Al (0.4 nm)–Ni/SiO2 can be considered, from a reduction behavior perspective, as a mixture of Ni/SiO2 (α-type Ni2+) and Al (4.0 nm)–Ni/SiO2 (β- and γ-type Ni2+) features). For bare Ni/SiO2, in situ XANES revealed that the reduction of NiO started at 250 °C; NiO was fully reduced to metallic Ni at 450 °C. The appearance of (111), (200) and (220) reflections due to Ni0 (fcc) in the in situ XRD data at temperatures exceeding 300 °C is indicative of the formation of metallic Ni through the direct reduction of bulk NiO (Fig. S8a†). LCF analysis of the XANES data of calcined Al (4.0 nm)–Ni/SiO2 yielded a composition of 32 mol% NiO and 68 mol% NiAl2O4. LCF of the in situ XANES and XRD showed that in calcined Al (4.0 nm)–Ni/SiO2, NiO was reduced in the temperature range 500–600 °C, whereas NiAl2O4 was reduced between 550 °C and 800 °C.
To summarize, combining our insight of the structure of calcined Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2 with its reduction behavior, we can deduce that the reduction occurring at 400 °C is ascribed to the reduction of NiO weakly interacting with SiO2, whereas NiO interacting with free Al2O3 in Al2O3-modified SiO2, i.e., AAS, reduces only at 600 °C. In addition, the availability of excessive, free Al2O3 domains in Al (4.0 nm)–Ni/SiO2 results in the formation of NiAl2O4 (NiO + Al2O3 → NiAl2O4) which reduces at 800 °C. 27Al NMR measurements of the reduced materials revealed no significant differences to the calcined material (Fig. S6c†), indicating that AAS remains as such in the catalysts after reduction. Overall, our structural characterization using XRD, XAS, Raman and 27Al NMR point to the formation of both AAS, and NiAl2O4 in Al2O3-coated Ni/SiO2 catalysts.
Electron microscopy of reduced Al (4.0 nm)–Ni/SiO2 was performed to visualize the morphology of the Ni nanoparticles and the ALD-grown Al2O3 layer (Fig. 6). HR-TEM of reduced Al (4.0 nm)–Ni/SiO2 (Fig. 6a) shows metallic Ni nanoparticles with a size of 8 ± 1 nm. The Ni nanoparticles were partly embedded in the support and partly covered by an amorphous (ALD-grown) layer. Hence, increasing the thickness of the ALD-grown Al2O3 layer from 0.4 nm to 4.0 nm, will reduce the number of active (and accessible) active Ni active sites at the surface, which is in line with a decrease in the quantity of chemisorbed H2 with increasing thickness of the ALD-grown Al2O3 layer, as tabulated in Table 1. The mean Ni particle size determined by H2 chemisorption using a hemisphere model is larger than the TEM-estimated Ni particle size. This difference is ascribed to the partial covering of the Ni surface by the Al2O3 overcoat.
 | ||
| Fig. 6 Electron microscopy-based morphological characterization: (a) HR-TEM and (b) HAADF-STEM images with (c–e) EDX mapping for reduced Al (4.0 nm)–Ni/SiO2. The arrows represent the ALD-grown layer. | ||
To probe further the morphology and composition of the Al2O3 layer, STEM images combined with elemental EDX analysis (Fig. 6b–e and S9†) of reduced Al (4.0 nm)–Ni/SiO2 were acquired. At the top of the silica spheres, an Al-containing layer forms. The overlap of the signals from Si and Al are indicative of a reaction between SiO2 and Al2O3. The particle size of Ni after reduction, as determined by TEM (Fig. S4† and Table 1) was similar for all of the reduced catalysts: Ni/SiO2 (7 ± 2 nm), Al (0.4 nm)–Ni/SiO2 (7 ± 2 nm) and Al (4.0 nm)–Ni/SiO2 (8 ± 1 nm). However, the amount of surface Ni of the reduced catalysts, quantified by H2 chemisorption using a stoichiometry factor of H/Ni = 1 (ref. 48 and 50) decreased with increasing thickness of the ALD-grown Al2O3 layer, i.e., 102 μmolNi gcat−1 (Ni/SiO2) > 91 μmolNi gcat−1 (Al (0.4 nm)–Ni/SiO2) > 70 μmolNi gcat−1 (Al (4.0 nm)–Ni/SiO2). This observation can be explained by the partial coverage of surface Ni by the ALD-grown Al2O3 layer.
3.4. Dry reforming of methane activity and selectivity
The catalytic activity tests for DRM were performed in a fixed-bed quartz reactor. In a typical experiment, prior to the activity test, the respective calcined catalysts were first reduced (based on the TPR results) at 400 °C (Ni/SiO2), 600 °C (Al (0.4 nm)–Ni/SiO2), and 800 °C (Ni/Al2O3 and Al (4.0 nm)–Ni/SiO2) in 10 vol% H2/N2 (100 ml min−1) for 2 h. The DRM performance was evaluated in the kinetic regime, i.e., far from the equilibrium conversion of methane, at 700 °C using a GHSV of 300 L gcat−1 h−1 (100 mL min−1 of 45% CH4, 45% CO2 and 10% N2). The DRM performances of the Al2O3-coated Ni/SiO2 based catalysts (Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2) were compared with the following benchmark catalysts: (i) Ni supported on SiO2 or Al2O3 (Ni/SiO2 and Ni/Al2O3), (ii) Ni supported on Al2O3-coated SiO2 (Ni/Al (4.0 nm)–SiO2) and Ni supported on Al2O3-coated Al2O3 (Ni/Al (4.0 nm)–Al2O3) and (iii) Al2O3-coated Ni/Al2O3 (Al (0.4 nm)–Al2O3).As expected, we observe severe deactivation for Ni/SiO2, exhibiting a drastic reduction of the methane consumption rate with TOS, i.e., from 33 mmolCH4 min−1 gcat−1 to 5 mmolCH4 min−1 gcat−1 after 10 h TOS (Fig. 7a). A gradual decrease of the methane consumption rate was also observed for Ni/Al2O3 (from 27 mmolCH4 min−1 gcat−1 to 20 mmolCH4 min−1 gcat−1), Ni/Al (4.0 nm)–SiO2 (from 38 mmolCH4 min−1 gcat−1 to 30 mmolCH4 min−1 gcat−1) and Ni/Al (4.0 nm)–Al2O3 (from 30 mmolCH4 min−1 gcat−1 to 26 mmolCH4 min−1 gcat−1). These results show that the deactivation tendency of Ni catalysts when impregnated onto Al2O3 (Ni/Al2O3) and Al2O3-coated supports (Ni/Al (4.0 nm)–SiO2 and Ni/Al (4.0 nm)–Al2O3) improves only partially.
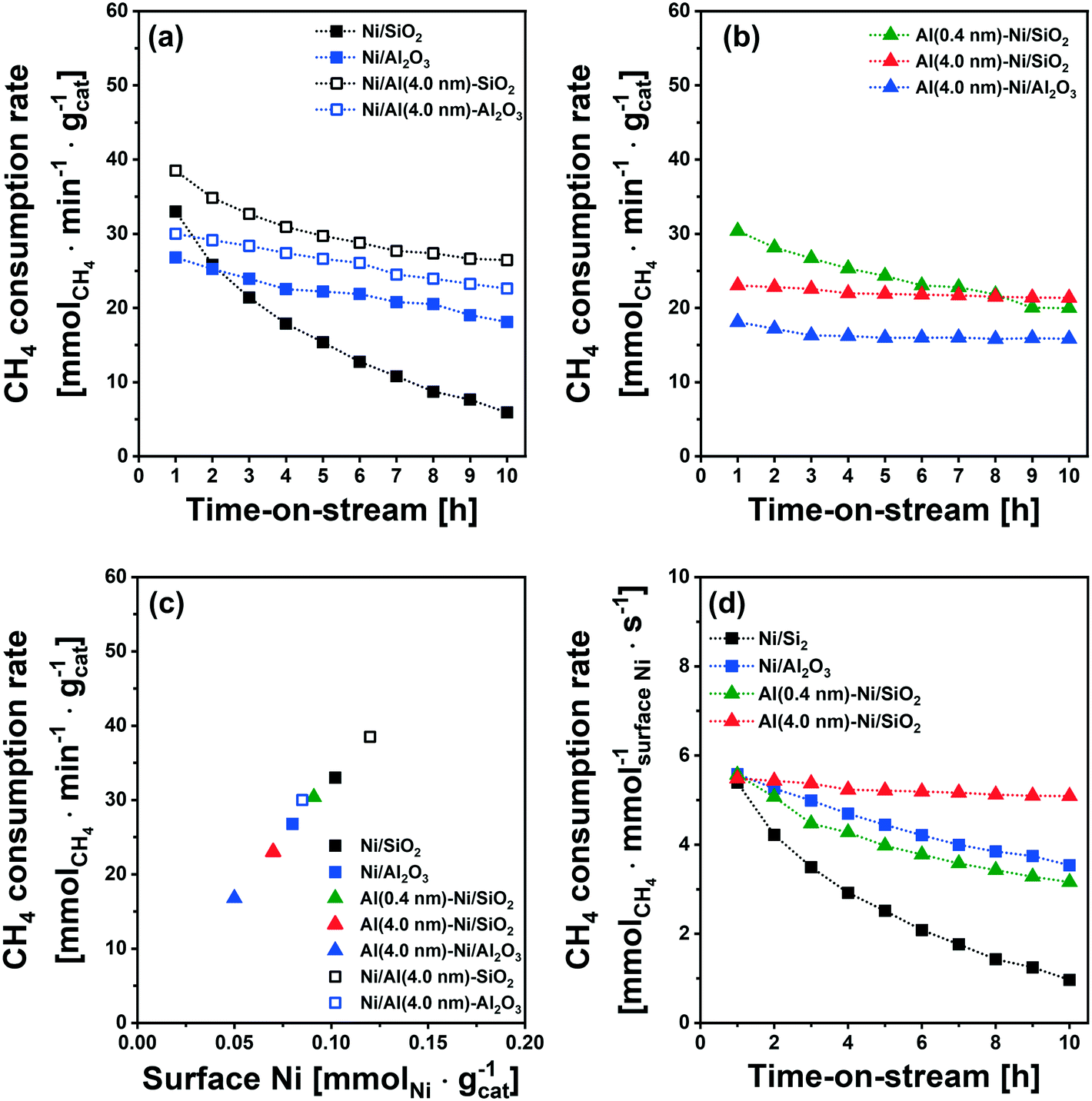 | ||
| Fig. 7 DRM performance: rate of methane consumption for (a) Ni supported on bare SiO2 and Al2O3-coated SiO2 and Al2O3, and (b) Al2O3-coated Ni/SiO2 and Ni/Al2O3 as a function of TOS, (c) rate of methane consumption at 1 h of TOS as a function of surface Ni as quantified by H2 chemisorption (H/Ni = 1.0), and (d) rate of methane consumption normalized by surface Ni of the freshly reduced catalysts, quantified by H2 chemisorption (H/Ni = 1.0), as a function of TOS. | ||
Turning now to the catalysts for which the Al2O3 overcoat was deposited after Ni impregnation onto the SiO2 support, the methane consumption rate at 1 h of TOS of Al (0.4 nm)–Ni/SiO2 (30 mmolCH4 min−1 gcat−1) Al (4.0 nm)–Ni/SiO2 (23 mmolCH4 min−1 gcat−1) and Al (4.0 nm)–Ni/Al2O3 (18 mmolCH4 min−1 gcat−1) are reduced compared to the corresponding benchmarks Ni/SiO2 and Ni/Al2O3 (Fig. 7a and b). After 10 h of TOS, the methane consumption rate of Al (0.4 nm)–Ni/SiO2 has reduced to 20 mmolCH4 min−1 gcat−1, while a very stable methane consumption rate was observed for Al (4.0 nm)–Ni/SiO2 (23 mmolCH4 min−1 gcat−1 over 10 h TOS). For Al (4.0 nm)–Ni/Al2O3 the initial methane consumption rate (1 h TOS) was reduced from 18 mmolCH4 min−1 gcat−1 to 16 mmolCH4 min−1 gcat−1 after 2 h of TOS, but remained stable at 16 mmolCH4 min−1 gcat−1) at 10 h TOS. These findings show that an Al2O3 overcoat of a thickness of 4 nm deposited onto Ni/SiO2 or Ni/Al2O3 is effective to stabilize the DRM performance of the impregnated Ni catalysts. The inverse preparation route, i.e., the impregnation of Ni onto Al2O3-coated SiO2 and Al2O3 supports did not yield stable catalysts.
It is important to note that the initial methane consumption rate (normalized by weight catalyst) decreases with increasing thickness of the Al2O3 overcoats. This is ascribed to (i) the reduced Ni content per mass catalyst and (ii) the reducing number of accessible active surface Ni sites (H2 chemisorption data, Table 1) with an increasing quantity of Al2O3 deposited. Fig. 7c confirms that there is a linear correlation between the methane consumption rate (1 h of TOS) with surface Ni, as quantified by H2 chemisorption (H/Ni = 1.0), providing further evidence that surface metallic Ni is the active site for DRM. The methane consumption rate (1 h of TOS) normalized by surface Ni (Fig. 7d) features very similar values (5.4–5.7 mmolCH4 mmolsurface Ni−1 s−1) for Ni/SiO2, Ni/Al2O3, Al (0.4 nm)–Ni/SiO2 and Al (4.0 nm)–Ni/SiO2.
3.5. Structure–stability relationship: Al2O3 overcoat stabilizing the catalyst against coking and sintering
To obtain further insight into the enhanced stability of Al (4.0 nm)–Ni/SiO2 when compared to the uncoated benchmark catalysts (i.e., Ni/SiO2 and Ni/Al2O3), additional in situ and post-reaction analyses were performed.In situ XRD data acquired during the DRM at 700 °C (Fig. 8a) revealed a noticeable (002) reflection due to graphitic carbon (space group P63/mmc) for Ni/SiO2 and Ni/Al2O3 after 5 h of TOS. On the other hand, no crystalline carbon was detected for Al (4.0 nm)–Ni/SiO2, implying that graphite C formation was at least one of the reasons leading to the deactivation of uncoated Ni/SiO2 and Ni/Al2O3. For Ni/Al2O3in situ XRD also reveals a decrease in the intensity of the Ni0 peaks (with respect to the intensity of the support), likely due to the formation of NiAl2O4. However, due to overlapping diffraction peaks the distinction between spinel γ-Al2O3 and NiAl2O4 is difficult. Thus, the in situ XRD study was complemented by XAS characterization of the post-reaction catalysts to determine the oxidation state of Ni. To examine the oxidation state of Ni in the catalyst materials, in situ and ex situ XANES spectra at the Ni K-edge after 5 h and 10 h of DRM were collected (Fig. 8b and S10†). It should be noted that all of the catalysts were completely reduced to metallic Ni0 prior to DRM, i.e., TOS = 0 h. The in situ Ni K-edge XANES data (Fig. 8b) does not show significant changes in the oxidation state of Ni for Ni/SiO2 and Al (4.0 nm)–Ni/SiO2 after 5 h of DRM. On the other hand, the XANES spectra of spent Ni/Al2O3 showed a partial oxidation of metallic Ni forming NiAl2O4. The oxidation of metallic Ni to NiAl2O4 was also observed for Ni/Al (4.0 nm)–Al2O3 and Al (4.0 nm)–Ni/Al2O3 (Fig. S10†). From a LCF analysis of the ex situ XANES data we quantified that 10, 10 and 9 mol% of Ni in Ni/Al2O3, Ni/Al (4.0 nm)–Al2O3 and Al (4.0 nm)–Ni/Al2O3 were converted into NiAl2O4 during 10 h of DRM (Table S2†). The migration of Ni0 into the Al2O3 lattice forming NiAl2O4 can explain (with carbon deposition) the deactivation of these three catalysts. Importantly, we did not observe any Ni oxidation in spent Al (4.0 nm)–Ni/SiO2 and Ni/Al (4.0 nm)–SiO2, i.e. Ni was maintained in its metallic form over 10 h of TOS. These peculiar results of the Al2O3-ALD/SiO2-based catalysts are possibly due to the presence of aluminosilicates in Al (4.0 nm)–Ni/SiO2 and Ni/Al (4.0 nm)–SiO2 that prevent the formation of NiAl2O4. Hence, the presence of aluminosilicates resulting from the interaction of the ALD-grown Al2O3 layer with SiO2 upon calcination seems to mitigate the migration (and oxidation) of Ni into the Al2O3 matrix.
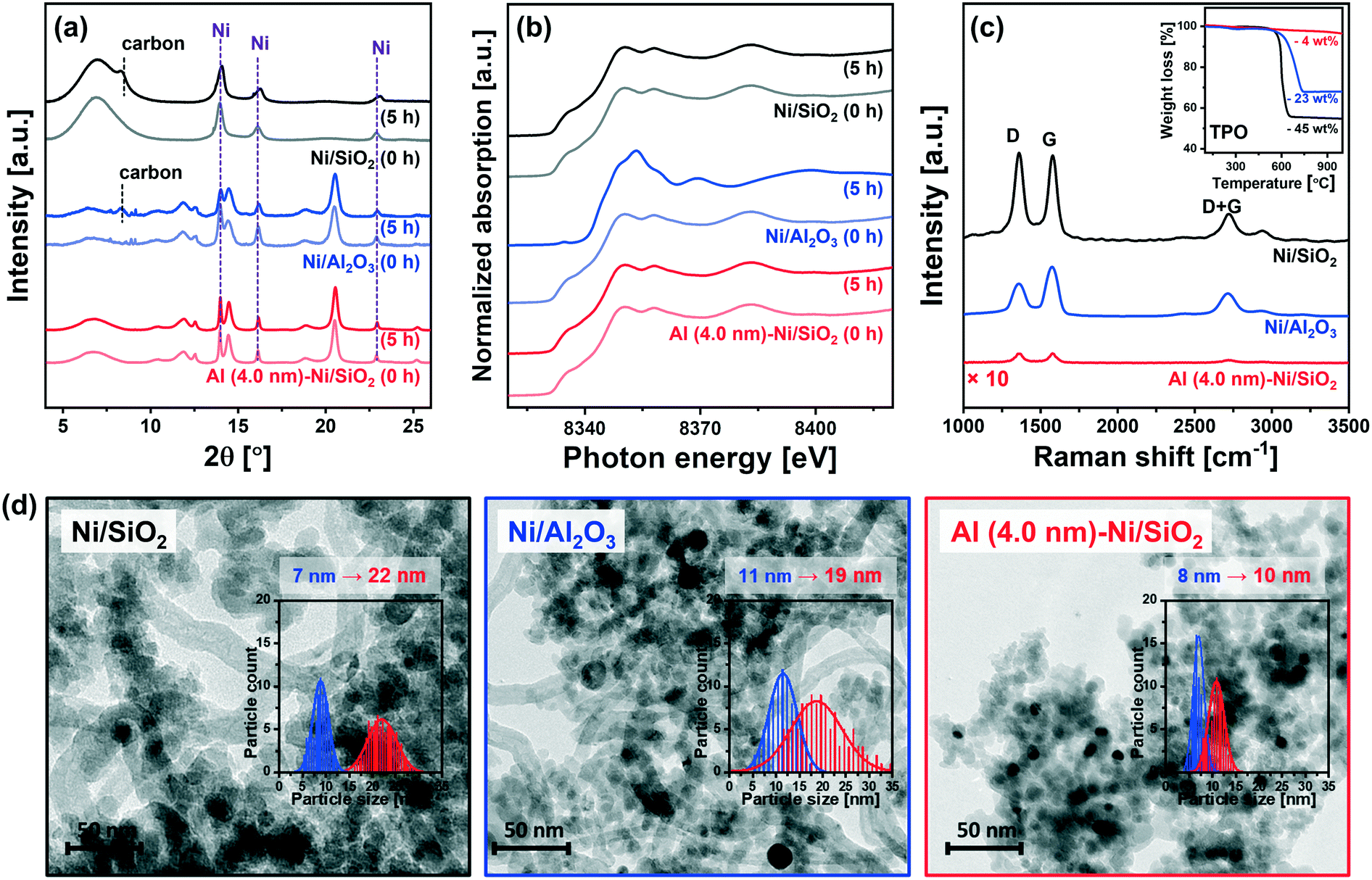 | ||
| Fig. 8 Morphological and structural characterization of the spent catalysts: Selected in situ (a) diffractograms and (b) Ni K-edge XANES acquired under DRM conditions and (c) Raman spectra of spent Ni/SiO2, Ni/Al2O3 and Al (4.0 nm)–Ni/SiO2. The asterisk in (a) represents γ-Al2O3 (ICSD 66559) or NiAl2O4 (ICSD 9554). The inset in (b) plots the weight loss of the spent catalysts as a function of temperature during a TPO experiment. (d) TEM images and Ni particle size distribution of the spent catalyst. | ||
The quantity of the carbon deposited was determined by temperature programmed oxidation (TPO) on the spent catalysts that were collected after the in situ XAS–XRD measurements under DRM conditions for 5 h (inset in Fig. 8c). The weight increase in the temperature range of 300–450 °C is mostly due to the oxidation of Ni to NiO. The weight loss due to the oxidation of deposited carbon species occurs at 600 °C for Ni/SiO2 and Al (4.0 nm)–Ni/SiO2, and at 715 °C for Ni/Al2O3. The quantity of deposited carbon followed the order: Ni/SiO2 (45 wt%) > Ni/Al2O3 (23 wt%) ≫ Al (4.0 nm)–Ni/SiO2 (4 wt%).
Raman spectra of all of the spent catalysts showed characteristic D and G bands of carbon at 1250–1350 cm−1 and 1500–1700 cm−1, respectively (Fig. 8c). The D bands represent disordered carbon species, e.g., amorphous or defective filamentous carbon, whereas the G band represents the stretching mode of the sp2 bonds in ordered graphite.82,83 The very low intensity of the peaks in the Raman spectra for Al (4.0 nm)–Ni/SiO2 is in line with the quantitative carbon analysis using TPO, i.e. very little carbon deposition on spent Al (4.0 nm)–Ni/SiO2 compared to spent Ni/SiO2 and Ni/Al2O3. The ratio of the intensities of the G and D bands (IG/ID) provides an indication on the degree of crystallinity of the deposited carbon,84 whereby a high ratio of IG/ID points to a high crystallinity. For spent Ni/Al2O3IG/ID = 1.3, which is higher than the values for spent Ni/SiO2 (IG/ID = 1.0) and Al (4.0 nm)–Ni/SiO2 (IG/ID = 1.0). The crystallinity of the carbon deposited affects the oxidation temperature of carbon, i.e., an increasing oxidation temperature with an increasing degree of carbon crystallinity.9 Hence our findings concerning the IG/ID values for the different catalysts are in line with the higher oxidation temperature of carbon deposited onto Ni/Al2O3 (715 °C) compared to Ni/SiO2 and Al (4.0 nm)–Ni/SiO2 (600 °C). These results confirm that the significant deposition of coke is a key reason for the deactivation of the uncoated catalysts, i.e. Ni/SiO2 and Ni/Al2O3, whereas Al (4.0 nm)–Ni/SiO2 shows a very high resistance to coke formation. An increasing degree of coke formation during DRM can be explained by the following factors: (i) surface acidity of the catalyst support (basic sites can alleviate coke deposition by promoting the oxidation of surface carbon85) and (ii) sintering of Ni particles. Considering the observed trends in coke formation, particle size and acidity, we ascribe the main driver to coke formation in our set of model catalysts to differences in the growth of the Ni particle size during DRM. Coke formation and particle growth were also visualized by TEM. The formation of filamentous carbon on spent Ni/SiO2 and Ni/Al2O3 was observed, whereas no coke formation was revealed in the TEM images of spent Al (4.0 nm)–Ni/SiO2 (Fig. 8d). Measuring the particle growth during DRM through the analysis of TEM images, a 3.1-fold (from 7 nm to 22 nm) and 1.7-fold (from 11 nm to 19 nm) increase in the Ni particle size was observed for Ni/SiO2 and Ni/Al2O3, respectively. On the other hand, a significantly reduced increase in the particle size of Ni (from 8 nm to 10 nm, i.e., 25%) was observed for Al (4.0 nm)–Ni/SiO2, demonstrating the stabilizing effect of an ALD-grown overcoat of Al2O3 on the Ni nanoparticles, preventing in turn their sintering during DRM.
To summarize our detailed characterization of the as-prepared and spent catalysts attribute the remarkable stability of Al (4.0 nm)–Ni/SiO2 during DRM to (i) the confinement of Ni nanoparticles by the Al2O3 overcoat preventing coke deposition on edge and step sites in large Ni particles, and (ii) the formation of aluminosilicates that mitigate the migration and oxidation of Ni preventing NiAl2O4 formation.
4. Conclusions
We have developed stable Ni-based DRM catalysts by employing atomic layer deposition (ALD) to ensure the deposition of a conformal layer of Al2O3 onto Ni nanoparticles supported on SiO2. Al2O3-coated Ni/SiO2 that showed a remarkable DRM stability when compared to the benchmark references Ni/SiO2 and Ni/Al2O3. An in-depth structural characterization of the Al2O3-coated catalysts and several benchmark systems via in situ Ni K-edge XAS–XRD complemented by H2-TPR and solid-state 27Al NMR revealed the reaction of the ALD-grown Al2O3 layer with the SiO2 support and Ni during calcination, i.e., forming an aluminosilicate and NiAl2O4, respectively. The deactivation of the uncoated benchmark catalysts, i.e., Ni/Al2O3 and Ni/SiO2, is ascribed to Ni particle growth and coke formation during DRM. An additional deactivation route of the Ni/Al2O3 benchmark is the migration/oxidation of Ni into the Al2O3 matrix forming (inactive) NiAl2O3. The best catalyst prepared, i.e. Al (4.0 nm)–Ni/SiO2, showed a remarkable stability (maintenance of 89% of the initial methane consumption rate after 10 h of TOS) under DRM conditions owing to the confinement of Ni nanoparticles by the Al2O3 overcoat and the inhibition of the oxidation of Ni and reaction with Al2O3 in the presence of aluminosilicates.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge ETH (ETH 57 12-2) and the Swiss National Science Foundation (200020_156015) for financial support. ESRF and the Swiss Norwegian Beamlines (SNBL at ESRF) are gratefully acknowledged for providing access to the synchrotron facility. We would like to thank the Scientific Center for Optical and Electron Microscopy (ScopeM) and the Center for Micro- and Nanoscience (FIRST) at ETH Zurich for providing training and access to electron microscopes. This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation and partially supported by the European Research Council under the European Union's Horizon 2020 research and innovation program grant agreement 819573.References
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- J. C. Abanades, E. S. Rubin, M. Mazzotti and H. J. Herzog, Energy Environ. Sci., 2017, 10, 2491–2499 RSC.
- I. Dimitriou, P. García-Gutiérrez, R. H. Elder, R. M. Cuéllar-Franca, A. Azapagic and R. W. K. Allen, Energy Environ. Sci., 2015, 8, 1775–1789 RSC.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC.
- S. M. Kim, P. M. Abdala, M. Broda, D. Hosseini, C. Copéret and C. Müller, ACS Catal., 2018, 8, 2815–2823 CrossRef CAS.
- R. L. Espinoza, A. P. Steynberg, B. Jager and A. C. Vosloo, Appl. Catal., A, 1999, 186, 13–26 CrossRef CAS.
- A. P. Steynberg, R. L. Espinoza, B. Jager and A. C. Vosloo, Appl. Catal., A, 1999, 186, 41–54 CrossRef CAS.
- J. R. Rostrupnielsen and J. H. B. Hansen, J. Catal., 1993, 144, 38–49 CrossRef CAS.
- S. M. Kim, P. M. Abdala, T. Margossian, D. Hosseini, L. Foppa, A. Armutlulu, W. van Beek, A. Comas-Vives, C. Copéret and C. Müller, J. Am. Chem. Soc., 2017, 139, 1937–1949 CrossRef CAS PubMed.
- T. Margossian, K. Larmier, S. M. Kim, F. Krumeich, C. Müller and C. Copéret, ACS Catal., 2017, 7, 6942–6948 CrossRef CAS.
- T. Margossian, K. Larmier, S. M. Kim, F. Krumeich, A. Fedorov, P. Chen, C. R. Müller and C. Copéret, J. Am. Chem. Soc., 2017, 139, 6919–6927 CrossRef CAS PubMed.
- S. M. Kim, A. Armutlulu, A. M. Kierzkowska, D. Hosseini, F. Donat and C. Müller, Sustainable Energy Fuels, 2020, 4, 713–729 RSC.
- J. W. Han, C. Kim, J. S. Park and H. Lee, ChemSusChem, 2014, 7, 451–456 CrossRef CAS PubMed.
- J.-H. Kim, D. J. Suh, T.-J. Park and K.-L. Kim, Appl. Catal., A, 2000, 197, 191–200 CrossRef CAS.
- S. Kawi, Y. Kathiraser, J. Ni, U. Oemar, Z. Li and E. T. Saw, ChemSusChem, 2015, 8, 3556–3575 CrossRef CAS PubMed.
- A. Tsoukalou, Q. Imtiaz, S. M. Kim, P. M. Abdala, S. Yoon and C. R. Müller, J. Catal., 2016, 343, 208–214 CrossRef CAS.
- D. L. Trimm, Catal. Today, 1999, 49, 3–10 CrossRef CAS.
- A. J. van Dillen, R. J. A. M. Terörde, D. J. Lensveld, J. W. Geus and K. P. de Jong, J. Catal., 2003, 216, 257–264 CrossRef CAS.
- L. Xu, H. Song and L. Chou, ACS Catal., 2012, 2, 1331–1342 CrossRef CAS.
- D. M. Argyle and H. C. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y.-T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y.-F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef PubMed.
- D. Baudouin, U. Rodemerck, F. Krumeich, A. D. Mallmann, K. C. Szeto, H. Ménard, L. Veyre, J.-P. Candy, P. B. Webb, C. Thieuleux and C. Copéret, J. Catal., 2013, 297, 27–34 CrossRef CAS.
- J. A. Lercher, J. H. Bitter, W. Hally, W. Niessen and K. Seshan, in Stud. Surf. Sci. Catal., ed. J. W. Hightower, W. Nicholas Delgass, E. Iglesia and A. T. Bell, Elsevier, 1996, vol. 101, pp. 463–472 Search PubMed.
- S. Tang, L. Ji, J. Lin, H. C. Zeng, K. L. Tan and K. Li, J. Catal., 2000, 194, 424–430 CrossRef CAS.
- D. Baudouin, K. C. Szeto, P. Laurent, A. De Mallmann, B. Fenet, L. Veyre, U. Rodemerck, C. Copéret and C. Thieuleux, J. Am. Chem. Soc., 2012, 134, 20624–20627 CrossRef CAS PubMed.
- P. Littlewood, S. Liu, E. Weitz, T. J. Marks and P. C. Stair, Catal. Today, 2020, 343, 18–25 CrossRef CAS.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Appl. Catal., B, 2017, 203, 625–632 CrossRef CAS.
- C. Wang, H. Wang, Q. Yao, H. Yan, J. Li and J. Lu, J. Phys. Chem. C, 2016, 120, 478–486 CrossRef CAS.
- T. M. Onn, S. Zhang, L. Arroyo-Ramirez, Y.-C. Chung, G. W. Graham, X. Pan and R. J. Gorte, ACS Catal., 2015, 5, 5696–5701 CrossRef CAS.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205 CrossRef CAS PubMed.
- E. Baktash, P. Littlewood, R. Schomäcker, A. Thomas and P. C. Stair, Appl. Catal., B, 2015, 179, 122–127 CrossRef CAS.
- Y. Lou, M. Steib, Q. Zhang, K. Tiefenbacher, A. Horváth, A. Jentys, Y. Liu and J. A. Lercher, J. Catal., 2017, 356, 147–156 CrossRef CAS.
- S. Wang and G. Q. Lu, J. Chem. Technol. Biotechnol., 2000, 75, 589–595 CrossRef CAS.
- J. Juan-Juan, M. C. Román-Martínez and M. J. Illán-Gómez, Appl. Catal., A, 2006, 301, 9–15 CrossRef CAS.
- D. La Rosa, A. Sin, M. L. Faro, G. Monforte, V. Antonucci and A. S. Aricò, J. Power Sources, 2009, 193, 160–164 CrossRef CAS.
- M. H. Amin, S. Putla, S. Bee Abd Hamid and S. K. Bhargava, Appl. Catal., A, 2015, 492, 160–168 CrossRef CAS.
- Z. Bian, S. Das, M. H. Wai, P. Hongmanorom and S. Kawi, ChemPhysChem, 2017, 18, 3117–3134 CrossRef CAS PubMed.
- B. Steinhauer, M. R. Kasireddy, J. Radnik and A. Martin, Appl. Catal., A, 2009, 366, 333–341 CrossRef CAS.
- B. Pawelec, S. Damyanova, K. Arishtirova, J. L. G. Fierro and L. Petrov, Appl. Catal., A, 2007, 323, 188–201 CrossRef CAS.
- Z. Hou and T. Yashima, Catal. Lett., 2003, 89, 193–197 CrossRef CAS.
- C. Crisafulli, S. Scirè, R. Maggiore, S. Minicò and S. Galvagno, Catal. Lett., 1999, 59, 21–26 CrossRef CAS.
- P. Djinović, I. G. Osojnik Črnivec, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef.
- J. Zhang, H. Wang and A. K. Dalai, J. Catal., 2007, 249, 300–310 CrossRef CAS.
- N. Rahemi, M. Haghighi, A. A. Babaluo, M. F. Jafari and S. Khorram, Int. J. Hydrogen Energy, 2013, 38, 16048–16061 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef.
- S. M. Maier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 8005–8013 CrossRef CAS.
- C. H. Bartholomew, Catal. Lett., 1990, 7, 27–51 CrossRef CAS.
- C. H. Bartholomew, R. B. Pannell and J. L. Butler, J. Catal., 1980, 65, 335–347 CrossRef CAS.
- T. Kammler, S. Wehner and J. Küppers, Surf. Sci., 1995, 339, 125–134 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- L. Baggetto, V. Sarou-Kanian, P. Florian, A. N. Gleizes, D. Massiot and C. Vahlas, Phys. Chem. Chem. Phys., 2017, 19, 8101–8110 RSC.
- S. K. Lee and C. W. Ahn, Sci. Rep., 2014, 4, 4200 CrossRef PubMed.
- V. Sarou-Kanian, A. N. Gleizes, P. Florian, D. Samélor, D. Massiot and C. Vahlas, J. Phys. Chem. C, 2013, 117, 21965–21971 CrossRef CAS.
- P. M. Abdala, O. V. Safonova, G. Wiker, W. van Beek, H. Emerich, J. A. van Bokhoven, J. Sá, J. Szlachetko and M. Nachtegaal, Chimia, 2012, 66, 699–705 CrossRef CAS PubMed.
- L. Zhou, L. Li, N. Wei, J. Li and J.-M. Basset, ChemCatChem, 2015, 7, 2508–2516 CrossRef CAS.
- E. J. M. Hensen, D. G. Poduval, P. C. M. M. Magusin, A. E. Coumans and J. A. R. V. Veen, J. Catal., 2010, 269, 201–218 CrossRef CAS.
- A. Omegna, J. A. van Bokhoven and R. Prins, J. Phys. Chem. B, 2003, 107, 8854–8860 CrossRef CAS.
- M. F. Williams, B. Fonfé, C. Sievers, A. Abraham, J. A. van Bokhoven, A. Jentys, J. A. R. van Veen and J. A. Lercher, J. Catal., 2007, 251, 485–496 CrossRef CAS.
- K. Monu, L. César, C. Zixuan, G. David, W. Elena, M. Christoph, F. Franck, M. Dominique, F. Alexey, C. Christophe, L. Anne and F. Pierre, Atomic-Scale Structure and its Impact on Chemical Properties of Aluminum Oxide Layers Prepared by Atomic Layer Deposition on Silica, 2021 Search PubMed.
- J. A. Bokhoven, D. C. Koningsberger, P. Kunkeler, H. van Bekkum and A. P. M. Kentgens, J. Am. Chem. Soc., 2000, 122, 12842–12847 CrossRef.
- E. Bourgeat-Lami, P. Massiani, F. Di Renzo, P. Espiau, F. Fajula and T. Des Courières, Appl. Catal., 1991, 72, 139–152 CrossRef CAS.
- B. H. Babu, M. Niu, X. Yang, Y. Wang, L. Feng, W. Qin and X.-T. Hao, Opt. Mater., 2017, 72, 501–507 CrossRef CAS.
- A. K. Yadav and P. Singh, RSC Adv., 2015, 5, 67583–67609 RSC.
- K. Chah, B. Boizot, B. Reynard, D. Ghaleb and G. Petite, Nucl. Instrum. Methods Phys. Res., Sect. B, 2002, 191, 337–341 CrossRef CAS.
- J. W. Chan, T. Huser, S. Risbud and D. M. Krol, Opt. Lett., 2001, 26, 1726–1728 CrossRef CAS PubMed.
- M. Sakakura, M. Terazima, Y. Shimotsuma, K. Miura and K. Hirao, J. Appl. Phys., 2011, 109, 023503 CrossRef.
- M. Okuno, N. Zotov, M. Schmücker and H. Schneider, J. Non-Cryst. Solids, 2005, 351, 1032–1038 CrossRef CAS.
- P. Du, P. Zheng, S. Song, X. Wang, M. Zhang, K. Chi, C. Xu, A. Duan and Z. Zhao, RSC Adv., 2016, 6, 1018–1026 RSC.
- R. Huirache-Acuña, T. A. Zepeda, E. M. Rivera-Muñoz, R. Nava, C. V. Loricera and B. Pawelec, Fuel, 2015, 149, 149–161 CrossRef.
- E. J. M. Hensen, D. G. Poduval, V. Degirmenci, D. A. J. M. Ligthart, W. Chen, F. Maugé, M. S. Rigutto and J. A. R. V. Veen, J. Phys. Chem. C, 2012, 116, 21416–21429 CrossRef CAS.
- C. Jiménez-González, Z. Boukha, B. de Rivas, J. R. González-Velasco, J. I. Gutiérrez-Ortiz and R. López-Fonseca, Energy Fuels, 2014, 28, 7109–7121 CrossRef.
- Q. Liu, J. Gao, M. Zhang, H. Li, F. Gu, G. Xu, Z. Zhong and F. Su, RSC Adv., 2014, 4, 16094–16103 RSC.
- J. Gao, C. Jia, M. Zhang, F. Gu, G. Xu and F. Su, Catal. Sci. Technol., 2013, 3, 2009–2015 RSC.
- C. Jia, J. Gao, J. Li, F. Gu, G. Xu, Z. Zhong and F. Su, Catal. Sci. Technol., 2013, 3, 490–499 RSC.
- N. Miletić, U. Izquierdo, I. Obregón, K. Bizkarra, I. Agirrezabal-Telleria, L. V. Bario and P. L. Arias, Catal. Sci. Technol., 2015, 5, 1704–1715 RSC.
- W. Tsai, J. A. Schwarz and C. T. Driscoll, J. Catal., 1982, 78, 88–95 CrossRef CAS.
- J. Zhang, H. Xu, X. Jin, Q. Ge and W. Li, Appl. Catal., A, 2005, 290, 87–96 CrossRef CAS.
- C. Guimon, N. El Horr, E. Romero and A. Monzon, in Stud. Surf. Sci. Catal., ed. A. Corma, F. V. Melo, S. Mendioroz and J. L. G. Fierro, Elsevier, 2000, vol. 130, pp. 3345–3350 Search PubMed.
- S. He, H. Wu, W. Yu, L. Mo, H. Lou and X. Zheng, Int. J. Hydrogen Energy, 2009, 34, 839–843 CrossRef CAS.
- J. Guo, H. Lou and X. Zheng, Carbon, 2007, 45, 1314–1321 CrossRef CAS.
- P. Wang, E. Tanabe, K. Ito, J. Jia, H. Morioka, T. Shishido and K. Takehira, Appl. Catal., A, 2002, 231, 35–44 CrossRef CAS.
- P. Mallet-Ladeira, P. Puech, C. Toulouse, M. Cazayous, N. Ratel-Ramond, P. Weisbecker, G. L. Vignoles and M. Monthioux, Carbon, 2014, 80, 629–639 CrossRef CAS.
- T. Sodesawa, A. Dobashi and F. Nozaki, React. Kinet. Catal. Lett., 1979, 12, 107–111 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01149a |
| ‡ Institute for Integrated Catalysis, Pacific Northwest National Laboratory, Richland, Washington 99352, USA. |
| § DSM Applied Science Center, P.O. Box 1066, 6160 BB Geleen, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2021 |