
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Particle size-controlled synthesis of high-performance MnCo-based materials for alkaline OER at fluctuating potentials†
Cornelia
Broicher‡
a,
Malte
Klingenhof‡
b,
Marvin
Frisch
 b,
Sören
Dresp
b,
Nikolas Mao
Kubo
a,
Jens
Artz
a,
Jörg
Radnik
c,
Stefan
Palkovits
a,
Anna Katharina
Beine
*d,
Peter
Strasser
*b and
Regina
Palkovits
*ad
b,
Sören
Dresp
b,
Nikolas Mao
Kubo
a,
Jens
Artz
a,
Jörg
Radnik
c,
Stefan
Palkovits
a,
Anna Katharina
Beine
*d,
Peter
Strasser
*b and
Regina
Palkovits
*ad
aInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
bDepartment of Chemistry, Chemical and Materials Engineering Division, Technical University Berlin, Straße des 17. Juni 124, 10623 Berlin, Germany
cBundesanstalt für Materialforschung und -prüfung, BAM, Unter den Eichen 44-46, 12203 Berlin, Germany
dMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: katharina.beine@cec.mpg.de
First published on 4th October 2021
Abstract
For the large-scale generation of hydrogen via water electrolysis the design of long term stable and active catalysts for the oxygen evolution reaction (OER) remains a key challenge. Most catalysts suffer from severe structural corrosion that becomes even more pronounced at fluctuating potentials. Herein, MnCo based cubic particles were prepared via a hydrothermal approach, in which the edge length of the micron-sized particles can be controlled by changing the pH value of the precursor solution. The cubes are composed of varying amounts of MnCo2O4, CoCO3 and a mixed (Mn/Co)CO3 phase. Structure–activity relationships were deduced revealing a volcano-type behavior for the intrinsic OER activity and fraction of spinel oxide phase. A low overpotential of 0.37 V at 10 mA cm−2 and a stability of more than 25 h was achieved in 1.0 M KOH using a rotating disc electrode (RDE) setup. The best performing catalyst material was successfully tested under dynamic process conditions for 9.5 h and shows a superior catalytic activity as anode for the overall water splitting in an electrolyser setup in 1.0 M KOH at 333 K compared to a reference NiCo-spinel catalyst.
Introduction
Water electrolysis to generate hydrogen based on renewable energies is evolving to a core technology of modern society, which faces increasing concerns of environmental pollution and climate change. Electrochemical water splitting combined with renewable energy sources has the potential to provide large amounts of hydrogen with a significantly reduced carbon footprint (Scheme 1). Most countries aim at reducing CO2 emissions by up to 80% in the next decades. These climate targets are expected to be only met using next-generation energy production, e.g. by a commercialisation of water electrolysis. Molecular hydrogen is considered one of the most promising candidates for chemical energy storage. The efficiency of the water electrolysis is kinetically limited by the OER at the anode. The cell potential of the water splitting reaction is given by the thermodynamic standard potential E0 of 1.23 V plus an additional overpotential η, which mainly results from limitations of electron and mass transfer.1,2 Most of these losses result from the OER because of the sluggish four electron transfer reaction (eqn (1)).| 4OH− → O2 + 2H2O + 4e− | (1) |
 | ||
| Scheme 1 Commercialisation of alkaline water electrolysis for a CO2 free mobility, electricity and heat. | ||
Mn–Co spinels were prepared by several researchers using electrospinning,30 oxidative thermal scission31 and hydrothermal synthesis.32 The synthesis of Gao et al. yielded spherical instead of cubic particles resulting from a variation in the Mn/Co ratio. The catalysts showed high energy efficiency under continuous operation (1 M KOH, 100 ml min−1, T = 337 K, 1.4–2.2 V cell potential at 0.1–0.5 A cm−2).33,34 In the near future, the design of catalyst systems, which are stable dynamic operation and temporally fluctuating power supply, are needed. Until now, only few studies were performed that show catalysts with sufficient lifetime and durability at fluctuating potentials due to severe structural corrosion and degradation.35 Herein, a cheap and easy way to synthesise MnCo electrocatalysts of cubic morphology and adjustable particle size is presented. The materials were synthesised at 573 K, where the transition from carbonate to spinel occurs. The electrochemical surface area ECSA and the turnover frequency TOF of the MnCo-materials are studied to gain deeper insights into the catalytically active sites. Finally, a study on load flexibility and continuous flow-cell performance using the most stable MnCo material is presented to demonstrate industrial applicability.
Experimental section
Materials and reagents
Chemicals were purchased from Sigma-Aldrich (CoCl2, MnCl2, Nafion®, NH3(aq), NH4Cl, urea), Chemsolute (ethanol, HCl) and Merck (KOH). If not stated otherwise, chemicals were used as delivered.Synthesis of electrocatalysts
The MnCo electrode materials were prepared via hydrothermal synthesis using MnCl2 and CoCl2 as precursors in an aqueous urea solution. The metal precursors were mixed with water (30 mL, deionized) and urea (1.08 g, 18 mmol) resulting in a pH value of 5.5. Materials were also prepared at a pH value of 3 and 7 by the addition of 1 M HCl or 0.1 M NH4Cl solution, respectively. The solution was loaded into a stainless steel autoclave with Teflon inlet, sealed and heated to 393 K for 12 h. The metal precursors were used in the ratios n(Mn)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(Co) = 1:2 so that a total amount of n(Mn) + n(Co) of 6 mmol was reached. All materials were calcined at 573 K for 4 h. The synthesized materials are denoted by the different pH values of the precursor solution (3, 5.5 and 7) as follows: MnCo-pH. To examine the impact of each distinct phase on catalytic activity, a MnCo2O4 spinel reference catalyst was synthesised using the same hydrothermal synthesis procedure and a calcination temperature of 873 K.
:n(Co) = 1:2 so that a total amount of n(Mn) + n(Co) of 6 mmol was reached. All materials were calcined at 573 K for 4 h. The synthesized materials are denoted by the different pH values of the precursor solution (3, 5.5 and 7) as follows: MnCo-pH. To examine the impact of each distinct phase on catalytic activity, a MnCo2O4 spinel reference catalyst was synthesised using the same hydrothermal synthesis procedure and a calcination temperature of 873 K.
Physicochemical characterization
N2-Physisorption was performed on an Asap 2000 from Micromeritics. The samples were dried for 24 h at 573 K under vacuum prior to measurement. Scanning electron microscopy (SEM) images were obtained using a JEOL 7401F instrument with an accelerating voltage of 10 kV and analysed using the software ImageJ. XPS analysis was performed using a Kratos Axis Ultra DLD device. Spectra were referenced to C 1s at 285.0 eV. The X-ray diffraction (XRD) measurements were performed in Bragg–Brentano geometry on a Bruker AXS D8 Advance II theta/theta diffractometer, using Ni filtered Cu Kα1+2 radiation and a position sensitive energy dispersive LynxEye silicon strip detector. XRD patterns were recorded in continuous scanning mode in the range of 6–140° 2θ with an increment of 0.02° and a counting time of 1 s per step, resulting in a total accumulation time of 185 s. X-ray fluorescence (XRF) was performed under vacuum on an Eagle II spectrometer.Electrochemical characterization
Catalyst inks were prepared by dispersing the catalyst (2.2 mg) in a mixture of MilliQ water (49 μL), ethanol (49 μL) and Nafion (2 μL). The ink was shaken and sonicated for 15 min. 2.5 μL of catalyst ink was pipetted onto the pre-cleaned glassy carbon RDE surface with a geometric area of 0.1396 cm2 and let dry for 30 min to yield a catalyst film with a loading of 393 μg cm−2.Electrochemical measurements were performed in a glass cell using a three-electrode setup at room temperature. The modified RDE was used as working electrode. A glassy carbon stick and a Ag/AgCl (3 M KCl) electrode were used as counter and reference electrode, respectively. Measurements were carried out in 1 M KOH as electrolyte. The electrocatalytic activity was investigated by linear sweep voltammetry (LSV) using an Autolab potentiostat by Metrohm. Potentials were referenced to the RHE. Before recording any data, the catalysts were subjected to continuous potential cycling (CV) until steady voltammograms were obtained. Electrochemical impedance spectroscopy (EIS), measured in a frequency range from 50 kHz to 10 Hz at open circuit potential (OCP) with an AC perturbation of 10 mV, was used to determine the uncompensated resistance of the electrolyte. Long-term performance was investigated applying 10 mA cm−2 in 1 M KOH solution at a rotation speed of 1600 rpm.
Fabrication of catalyst coated substrates (CCS), membrane electrode assemblies (MEA) and measurement
The CCS were prepared by using a spray-coating technique for cathode and anode. For the preparation of the anode CCS, typically 50 mg of catalyst, 3 mL i-PrOH and 716 mg ionomer (3 wt-% in i-PrOH, FAA3, Fumatech) were sonicated for 15 min. The porous transport layer (PTL, 5 cm2, 10 BC, Sigratec) was fixed on a commercial vacuum-heat-table (Carbon and FuelCell) set to 328 K equipped with a pump/compressor (Welch, WOB-L 2511). By spray-coating 2 mL of the MnCo-catalyst ink onto the CCS, a homogeneous loading of 2 mg cm−2 was achieved using a Gun Piece SP-2 spray Gun (Rich) applying a 1 bar N2 flow. The cathode CCS was prepared using an ExactaCoat OP-3 (Sonotek). For the preparation of the ink, 120 mg Pt–C (Umicore, 48.5 wt% at Carbon) were mixed with 50 μL Milli-Q water, 1.72 g ionomer (see above) and 10.3 g i-PrOH achieving an ink dispersion containing 1 wt% catalyst. A mass loading with a homogeneous distribution of 0.1 mgPt cm−2 was achieved by applying ten loops with a speed of 80 mm s−1, a shaping gas flow of 3 L min−1, a nozzle power of 4 W and a liquid flow of 0.1 mL min−1. Mass loadings of anode and cathode were determined by weighing the dried (10 min at 328 K) anode and cathode electrodes before and after spray-coating. The electrolysis measurement was conducted using a commercial full cell (Carbon and FuelCell) equipped with a titanium anode and carbon cathode with a fluorinated FAS-50 (Fumatech) membrane and 1 M KOH electrolyte. Measurements were taken at 333 K. The used setup is depicted in Fig. S1 and details on the measurement protocol are given in Table S1 in the ESI.†Results and discussion
Catalyst characterization
MnCo-Materials were prepared using hydrothermal synthesis.36 Here metal precursors (in our case chlorides) are mixed with an aqueous urea solution. The pH value of the urea solution (pH = 5.5) was additionally adjusted to 3 (by addition of HCl) and 7 (by addition of NH4Cl). At elevated temperatures, urea decomposes to ammonium and carbonate ions.37 As a result the metal cations precipitate as carbonates, hydroxides, hydroxycarbonates and oxides.37,38 The solution is kept at reaction conditions for 12 h to ensure a proper aging of the precipitate. The obtained solids were filtered, dried and calcined to enable the formation of a normal spinel, which crystallises in cubic form (if for MnxCo3−xO4x lays between 0 and 1.4).39 The temperature of calcination was set to 573 K so that the phase transition to the spinel phase might be incomplete. For comparison, a MnCo2O4 spinel reference catalyst was prepared at a pH value of 5.5 during the hydrothermal synthesis and calcined at 873 K.All materials were characterised thoroughly: SEM confirms the formation of cubic particles independent of pH (Fig. 1a, c and e). More images are shown in Fig. S2 in the ESI.† Importantly, the pH value affects the particle size, i.e. the obtained average particle size decreases from 4.1 μm to 1.9 μm with increasing pH value from 3 to 7. A narrow particle size distribution is obtained (Fig. 1b, d and f). An influence of pH on the particle size and crystallinity of metal oxide materials obtained from hydrothermal synthesis was already observed by other research groups.38,40–42
 | ||
| Fig. 1 SEM images and particle size distributions of a and b) MnCo-pH 3, c and d) MnCo-pH 5.5 and e and f) MnCo-pH 7. | ||
N2-Physisorption measurements reveal a type II isotherm after IUPAC with a small hysteresis for MnCo-pH 7 (Fig. S3 in the ESI†). This classifies the materials as mainly non-porous, while MnCo-pH 7 additionally possesses a few mesopores. While for pH 3 and 5.5 a small specific surface area below 50 m2 g−1 was obtained, for pH 7, a significantly enhanced specific surface area of 150 m2 g−1 is found resulting from the decreased particle size and the increased porosity (Table 1). The amount of manganese and cobalt in the cubes was determined by XRF revealing similar atomic ratios of around 1:2 (Mn:Co) independent of pH (Table 1). XRD was performed to examine the crystal structure and phase composition of the prepared cubic particles (Fig. 2 and Table 1). Rietveld refinement was used for a quantitative analysis of the particular phases (see also Fig. S4 in the ESI†).
:Co determined via XRF and composition of MnCo-materials obtained via Rietveld refinement of powder diffraction data
| Material | S BET [m2 g−1] | Mn:Co ratio [%] |
CoCO3 [%] | (Mn/Co)CO3 [%] | MnCo2O4 [%] |
|---|---|---|---|---|---|
| MnCo-pH 3 | 46 | 35/65 | — | 50.5 | 49.5 |
| MnCo-pH 5.5 | 12 | 33/67 | 17.3 | 43.9 | 38.8 |
| MnCo-pH 7 | 150 | 36/64 | 40.0 | 53.6 | 6.4 |
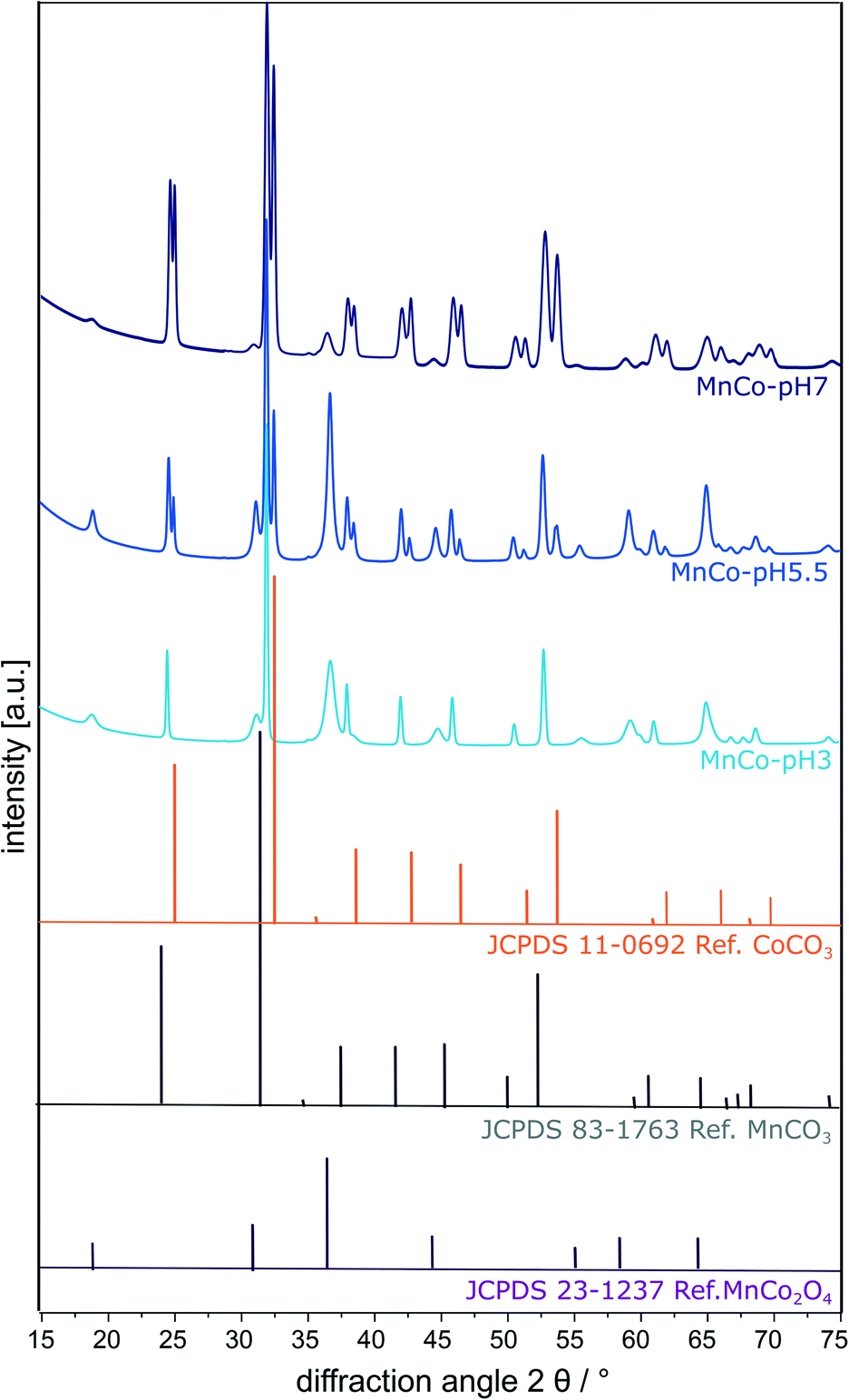 | ||
| Fig. 2 Rietveld refinements of the powder-XRD patterns of the MnCo-materials synthesised at pH 3, 5.5 and 7. In addition, three reference patterns for MnCo2O4, MnCO3 and CoCO3 are shown. | ||
A MnCo2O4 spinel fraction can be identified (JCPDS database 23-1237),27 showing reflexes at 18.5° [111], 30.5° [220], 36.0° [311], 43.8° [400], 54.3° [422], 57.9° [511], and 63.6° [440]. The amount of MnCo2O4 spinel decreases with increasing pH value. Additionally, a mixed carbonate phase (Mn/Co)CO3 with reflexes at 24.4° [012], 31.5° [104], 37.7 [100], 41.6° [113], 45.4° [202], 49.9° [024], 51.9° [116], 60.4° [122], 64.2° [214] and 68.0° [300] is observed.27 Almost the same relative amount of this mixed carbonate phase is present in all three materials, proving that an incomplete phase transition to the spinel phase occurred. A segregated crystalline CoCO3 phase (JCPDS database 011-0692)43 showing reflexes at 25.1° [012], 32.6° [104], 38.6° [110], 42.8° [113], 46.6° [202], 51.4° [024], 53.8° [116], 54.0° [018], 62.0° [122], 66.1° [214] and 69.9° [300] is formed at elevated synthesis pH values of 5.5 and 7. Hence, the cubic particles prepared at pH 5.5 and 7 consist of three segregated phases (MnCo2O4, (Mn/Co)CO3 and CoCO3). The CoCO3 fraction of the MnCo-materials increases with increasing pH, as indicated by the higher ratio of the corresponding [012] reflexes at 25.1°. The relative amount of the components is given in Table 2 and the differences can be explained by the changing pH values in the hydrothermal synthesis. If the pH value decreases, the concentration of H+ ions in the solution increases, leading to a decrease in carbonate concentration due to the protonation to hydrogen carbonate, carbonic acid and finally decomposition to CO2 and H2O.38,42 Therefore, the amount of metal carbonates in the material increases with increasing pH value. The differences in the cube size most likely also result from this varying chemical composition as the crystallisation rate of metal carbonates, hydroxides, hydroxycarbonates and oxides differs. As the smallest cubes were obtained for the carbonate richest material, it is proposed that carbonates crystallise slower, which leads to the formation of more nucleation seeds and therefore smaller crystals.
| Material | c DL [mF cm−2] | R f [−] | ECSA [cm2] | j 1.6V [mA cm−2] | j int [mA cm−2] | b ECSA [dec−1] | R CT [Ω] | TOF [s−1] |
|---|---|---|---|---|---|---|---|---|
| MnCo-pH 3 | 0.2 | 5 | 0.68 | 1.0 | 0.20 | 62.3 | 150 | 7.9 |
| MnCo-pH 5.5 | 1.28 | 32 | 4.3 | 5.4 | 0.17 | 53.8 | 51 | 6.8 |
| MnCo-pH 7 | 4.7 | 75 | 16.1 | 10.0 | 0.09 | 53.0 | 19 | 3.4 |
For the analysis of the surface composition, XPS analysis was performed (Fig. S5 in the ESI†). The survey spectra confirm the presence of Mn, O, C and Co at the surface of the cubic particles. The carbon content at the surface increases with increasing pH value corresponding to an increasing carbonate fraction found by XRD analysis. The high-resolution spectra of Mn 2p reveal a satellite feature independent of pH. As the determination of oxidation states is rather difficult from the Mn 2p signal, a deeper look is taken into the Mn 3s high-resolution spectrum, as oxidation state can be determined from the peak splitting of the two Mn 3s multiplet split components. For pH 3 and pH 5.5, a peak splitting of ΔE = 5.3 eV and ΔE = 5.5 eV can be observed, which suggests Mn3+ species (as in Mn2O3).44 For pH 7, the largest peak splitting of ΔE = 6.5 eV can be assigned to Mn2+ (as in MnO).45 The Co 2p spectrum exhibits two spin–orbit coupling peaks and two satellite peaks. For materials prepared at pH 3 and 5.5 the binding energy of the Co 2p3/2 signal lies at 779.8 and 780.1 eV, respectively, indicating the co-existence of Co2+/Co3+ oxide phases.27,28,46 For pH 7, the Co 2p3/2 signal is shifted to a higher binding energy of 782.2 eV. This hints towards the presence of Co2+ in form of hydroxide or, more likely, carbonate phase.47 Correspondingly, the C 1s spectrum shows the largest CO32− content at a binding energy of 288.5 eV for MnCo-pH 7 (37 at%), followed by MnCo-pH 3 (30 at%) and MnCo-pH 5.5 (23 at%). In addition, C–O species at 286.1 eV and C–C species at 284.8 eV are found for the carbonate and oxide materials.48 Presumably, an increased fraction of carbonate species at the surface contributes to the stabilization of smaller particles by repulsive Coulomb forces.
Electrochemical performance
The electrocatalytic performance of the prepared MnCo-materials and reference catalysts (IrO2, RuO2, MnCo2O4 and Ni1.4Co1.6O4) was evaluated by LSV in 1.0 M KOH electrolyte. No special precautions were taken to ensure that no Fe-impurities are present in the electrolyte. Even though observed trends remain unchanged, an enhancement of the catalytic activity is therefore possible. The materials were dispersed in a water, ethanol and Nafion containing solution and drop-coated onto a glassy carbon RDE. For the reference Ni1.4Co1.6O4 material, a high catalytic activity with a low η of 0.35 V at a current density of 10 mA cm−2 was obtained at 25 °C (Fig. 3a). The Mn-containing reference material, MnCo2O4, shows a low catalytic activity with an η of 0.42 V. All synthesised MnCo-materials show η values between 0.37 V and 0.43 V. The best performing material MnCo-pH 7 even outperforms the literature known benchmark catalysts RuO2 and IrO2, which reach an η of 0.41 and 0.47 V at a current density of 10 mA cm−2, respectively (Fig. S6 in the ESI†). For the differently sized cubes prepared at different pH values, a clear trend can be observed, i.e. larger cubes show a higher η compared to smaller ones. As this trend might be referred to the larger surface area of smaller cubes and therefore a higher number of active sites, the ECSA was calculated from the double layer capacitance cDL which was extracted from cyclovoltammetry (CV) in the non-faradaic potential range (1.0–1.1 V vs. RHE) at varying scan rates (Fig. S7 in the ESI†).49,50 The cDL of MnCo-materials ranges between 0.2 and 4.7 mF cm−2 (Table 2). Dividing cDL by the literature reported specific capacitance (cS) results in the roughness factor rF. A multiplication of rF with the geometric surface area Sg of the RDE yields the ECSA. The calculation is given in eqn (S1) and (S2) in the ESI† and the ECSA values are displayed in Table 2.51 As expected, the ECSA increases with a decreasing cube size. Normalized current density plots (Fig. 3b) reveal a lower η and therefore a higher intrinsic activity for MnCo-pH 3 compared to MnCo-pH 5.5 and MnCo-pH 7. The Ni1.4Co1.6O4 reference catalyst shows a higher ECSA-normalised OER activity compared to the herein developed Mn–Co materials. From the intrinsic activity of the prepared MnCo-catalysts an influence of the chemical composition can be seen: the activity increases with increasing MnCo2O4 content and decreasing CoCO3-amount. This relationship is illustrated in Fig. 3c. Notably, for the 100% spinel-type MnCo2O4 declined catalytic activity was found. Consequently, there is an optimal composition regarding high OER activity, as indicated by MnCo-pH 3 comprised of similar amounts of (Mn/Co)CO3 and MnCo2O4.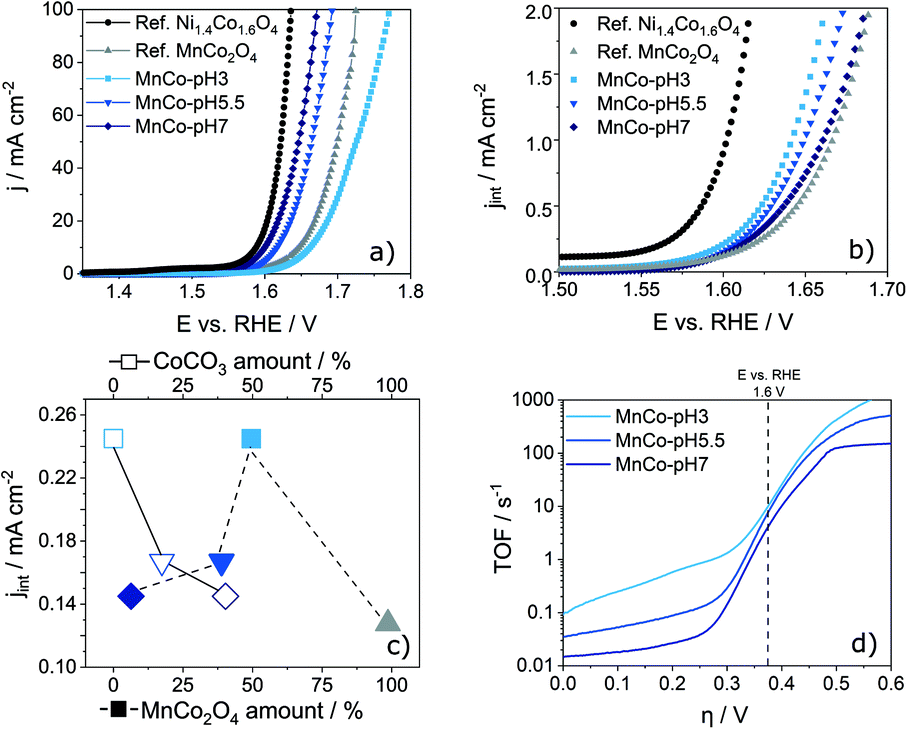 | ||
| Fig. 3 a) Activity plot: current density vs. potential (vs. RHE), b) ECSA normalized current density vs. potential (vs. RHE), c) correlation of the current density at a potential of 1.6 V with the amount of CoCO3 and MnCO2O4 for the synthesised MnCo-materials (conditions: scan rate 10 mV s−1, 1600 rpm, 1 M KOH) and d) plot of the results from the TOF calculations for prepared MnCo-materials. | ||
To determine a TOF, the surface area density of catalytically active metal centres has to be calculated (eqn (S3)–(S5) in the ESI†). Herein, a geometric approach for the calculation based on the cubic morphology was developed. Plotted as a function of η, the respective TOF values can be used to determine the intrinsic activity of a catalyst material at 1.6 V vs. RHE. With decreasing pH, the calculated TOF values increase. MnCo-pH 3 reaches the highest TOF of 7.9 s−1, followed by MnCo-pH 5.5 with 6.8 s−1 and MnCo-pH 7 with 3.4 s−1. The calculated TOF values are comparable to previous results reported in literature.52–54 For example, Cao et al.54 stated TOF values between 0.05 and 4.0 s−1 at 1.6 V vs. RHE for CoOx/PCN and RuOx catalysts. The presented method to determine the TOF based on a geometric approach has a great potential for the evaluation of electrocatalysts and aims for the elucidation of structure–activity relationships. The TOF values enable an evaluation of electrocatalysts regarding design, active surface, improved electrolyte and gas diffusion. Despite the comparably lower active surface area, MnCo-pH 3 shows the highest TOF values and therefore the highest intrinsic activity in alkaline OER. Accordingly, there is a correlation between the composition of spinel-type MnCo2O4 and (Mn/Co)CO3 phase in MnCo-pH 3 and the enhanced catalytic activity. Considering the rather low ECSA, the presence of few highly active metal centres is suggested. This finding is in good agreement with the acquired XPS and Rietveld refinement results showing an increasing relative fraction of Mn3+ species for decreasing pH, thereby enhancing the catalytic activity for alkaline OER, which is in accordance with earlier reports by Nakamura et al.16
Tafel plots allow insights into the reaction kinetics (Fig. S8a in the ESI†).55 In general, the OER proceeds over four steps, each of them involves an electron transfer based on the Krasil'shchikov mechanism.56 The ECSA-normalised Tafel slopes (bECSA) for the MnCo-materials vary between 53.0 and 62.3 mV dec−1. For MnCo-pH 7 and MnCo-pH 5.5 the second electron transfer step is found to be rate-determining.51 The calculated Tafel slopes are well in line with reported values for the best NiCo catalysts to date.6,20,25 The higher Tafel slope for MnCo-pH 3 indicates the transformation of unstable M–OH* towards more stable M–OH species to be rate-determining and relates most likely to the lower number of active sites.57 To further understand the electronic behaviour of the MnCo-materials, electrochemical impedance spectroscopy (EIS) was performed (Fig. S8b in the ESI†). The impedance spectra show a semicircle for all materials. Therefore, an equivalent circuit with two time constants (Fig. S8c in the ESI†) was used to analyse the impedance data and to obtain the charge transfer resistance RCT (Table 2). The Nyquist diagram shows the lowest RCT of 19 Ω for MnCo-pH 7, followed by MnCo-pH 5.5 (51 Ω) and MnCo-pH 3 (150 Ω). In comparison with LSV (Fig. 3a), a correlation between the onset potential of the three materials and the corresponding RCT can be observed. Accordingly, the lower RCT the higher the current density j at a specific η. However, ECSA-normalised plots reveal an opposite trend, as the onset potentials increase for decreasing RCT values. This peculiar behaviour can be rationalised by the different compositions of the cubic particles depending on the pH value of catalyst preparation. In this context, higher fractions of MnCo2O4 spinel in a mixed (Mn/Co)CO3 catalyst system are beneficial for high activity and improved TOF values. In terms of industrial application, not only a high activity is mandatory for a high performance catalyst, but also a sufficient stability is needed. To evaluate the electrochemical stability of the materials, chronoamperometric measurements at 10 mA cm−2 were performed (Fig. 4a). All MnCo-materials show a good catalytic activity and stability over 20 h reaching an η below 0.44 V at 10 mA cm−2. The synthesised MnCo-materials clearly outperform the reference catalysts, which deactivate after 5 h (Ni1.4Co1.6O4) and 20 h (MnCo2O4). For IrO2 and RuO2, no stability measurements were possible due to fast deactivation.
 | ||
| Fig. 4 a) Stability of the synthesised MnCo-materials and reference catalysts measured chronoamperometrically (conditions: scan rate 10 mV s−1, 1600 rpm, 1 M KOH); b) load flexibility stability study of MnCo-pH 7 (conditions: scan rate 10 mV s−1, 1600 rpm, 1 M KOH at changing); c) IR-free OER polarization curves for MnCo-pH 3, MnCo-pH 5.5 and MnCo-pH 7 in 1.0 M KOH at 333 K (horizontal bars represent the error of the experimental setup). | ||
Since the sustainable hydrogen production is closely related to renewable energies, which fluctuate strongly throughout the day, it is essential to measure the stability of an OER catalyst under dynamic process conditions. It was therefore evaluated for the most stable catalyst MnCo-pH 7 via chronoamperometric stability measurements at fluctuating potentials using an RDE setup (Fig. 4b). A high long-term stability of 9.5 h was achieved for potentials varying between 1.55 V and 1.64 V. To further investigate the prepared catalyst materials in a flow-cell, single-stack fuel-cell measurements were conducted mimicking industrial application. Measurements were performed in a two-electrode electrolyser for alkaline water splitting in 1.0 M KOH at 333 K. As shown in Fig. 4c and S9 in the ESI,† MnCo-pH 7 clearly outperforms the other synthesised materials and the Ni1.4Co1.6O4 reference. A high current density of 200 mA cm−2 is reached at a potential of 1.80 V. For the other catalyst materials with similar loadings of the anode and cathode, higher potentials of at least 2 V are necessary to provide similar current densities. MnCo-pH 7 shows the highest catalytic activity, as it provides a larger number of active sites compared to MnCo-pH 5.5 and MnCo-pH 3, which is in agreement with the described RDE investigations. A high stability of up to 4 h is obtained.
The spent MnCo-pH 7 catalyst was additionally investigated by XPS analysis (Fig. S10 and S11, Table S2 in the ESI†). After catalysis the materials is still composed of Co, Mn, O and C (Fig. S10 in the ESI†). Additionally, K from the electrolyte and a large amount of F are found. F results from the electrolyser setup, where a fluorinated FAS-50 membrane was applied as separator. Table S2 in the ESI† summarises the qualitative and quantitative analysis at the electrode surface. The calculated Mn:Co ratio decreased slightly from 0.21 before to 0.18 after catalysis. As the overall quantity of Mn is low, only the Mn 2p signal can be observed (Fig. S11a in the ESI†), on the basis of which it is difficult to make a statement about present Mn species. The Co 2p spectrum instead shows again two spin–orbit coupling peaks and two satellite peaks, where the Co 2p3/2 signal shows a binding energy of 779.6 eV (Fig. S11b in the ESI†). The binding energy is comparable to MnCo-pH 3 and MnCo-pH 5.5 before catalysis, indicating the presence of mixed Co2+/Co3+ oxides and thus a decrease of the CoCO3-phase. This decrease of the carbonate phase can also be observed in the O 1s element spectrum (Fig. S11c in the ESI†). The anodic treatment of the electrocatalyst thus leads to the oxidation of carbonates and molecular changes on the electrode surface.
Table 3 summarizes the performance of all herein synthesised cubic MnCo-materials in RDE experiments and in the electrolyser setup. It can be seen that our catalysts show a similar performance compared to previously investigated spinels, even though the average particle size of the herein presented electrocatalysts is significantly higher.
| Material | Loading [mg cm−2] | Particle size | j 1.8V [mA cm−2] | η [V] |
|---|---|---|---|---|
| MnCo-pH 3 | 2.0 | 4.1 μm | 70 | 0.43 |
| MnCo-pH 5.5 | 2.0 | 3.2 μm | 83 | 0.39 |
| MnCo-pH 7 | 2.0 | 1.9 μm | 200 | 0.37 |
| Ni1.4Co1.6O4 (ref. 9) | 2.0 | 10 nm | 80 | 0.36 |
| NiFeOx (ref. 58) | 2.5 | 10 nm | 520 | — |
| NiFe2O4 (ref. 58) | 2.5 | 5 nm | 120 | — |
| NiMn2O4 (ref. 59) | 3.0 | — | 210 | — |
| CMK-3-MnPc-WI18 | — | — | — | 0.49 |
| IrOx (ref. 51) | — | — | — | 0.32 |
| MnOx/NCNT60 | — | — | — | 0.52 |
Conclusion
In this study, cubic MnCo-materials were synthesised, characterised and tested as catalysts for OER in alkaline electrolyte. Controlling the pH of the precursor solution enabled to precisely tune the average edge length of the formed particles. Due to the higher surface area of smaller cubic particles, a higher ECSA and a lower η are obtained. However, after a normalisation of the electrochemical data to the ECSA, a reverse trend is observed. The combination of MnCo2O4 and mixed (Mn/Co)CO3 phases was shown to increase the OER activity, while the catalyst consisting of 100% spinel phase was found to be less active. Based on the calculation of TOFs for cubic particles using a newly developed geometric approach, the MnCo-catalyst prepared at pH 3 with a TOF of 7.9 s−1 at 1.60 V vs. RHE was identified to be most active due to its high fraction of MnCo2O4 spinel in a mixed carbonate catalyst system. A decreasing TOF with increasing synthesis pH value was found.Concerning an industrial application of a high performance electrocatalyst, MnCo-pH 7 seems more promising as it provides a significantly larger number of active sites due to its lower average particle size. Additionally a high stability in the chronoamperometry (>20 h), under fluctuating potentials (9.5 h) and in a flow-cell (4 h) is obtained for the material. The presence of CoCO3 shows a beneficial impact on stability, which is key for long-term industrial applications on a larger scale. In this context, the catalyst material is superior to other literature-known MnCo and NiCo spinels.6,20,25,27,28 Due to the excellent long-term stability, MnCo-pH 7 emerged as most promising electrocatalyst for application in future hydrogen technology, as it combines a sufficient activity with a high stability. Hence, the synergetic effects of different metal oxide and carbonate species were used to enhance not only the catalytic activity but also the stability of the cubic electrocatalysts.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. CB, MK (marked by ‡) contributed equally to the publication. CB and MF contributed to the writing of the manuscript as well as all aspects of data analysis and interpretation. With the help of NMK, CB synthesised and characterised the materials and performed electrocatalytic tests in a three-electrode RDE setup. MK performed electrocatalytic tests in a two-electrode electrolyser. MF developed an approach for the calculation of TOF values. MK and MF contributed to the editing of the manuscript. JR performed XPS analyses and contributed to the data evaluation. SD, JA and SP helped with the interpretation of the data in this work and contributed to the success of the project through a variety of scientific discussions. AKB, PS, and RP conceived the study, contributed to the writing and editing of the manuscript and to data interpretation. The research was carried out in close collaboration between the research groups in Aachen, Berlin and Mülheim an der Ruhr. Therefore, all three group leaders function as corresponding authors for the publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support by the European Regional Development Fund (ERDF) and the state of North-Rhine Westphalia, Germany under the operational program “Regional Competitiveness and Employment” Project “Sustainable Chemical Synthesis” and the Federal Ministry of Education and Research (BMBF) for funding this work within the MANGAN research cluster BMBF-PTJ FKz 03SF0508. Financial support by the German Research Foundation (DFG) through Grant Reference Number STR 596/12-1 and the Federal Ministry for Economic Affairs and Energy (Bundesministerium für Wirtschaft und Energie, BMWi) under Grant Number 03EIV041F in the collaborative research project “MethQuest” in the group “MethFuel” are gratefully acknowledged. Thanks to Frank Girgsdies from the Fritz Haber Institute of the Max Plank Society in Berlin for XRD measurements. Many thanks to Jörg M. Stockmann from the Bundesanstalt für Materialforschung und -prüfung (BAM) for assistance in XPS measurements. Open Access funding provided by the Max Planck Society.Notes and references
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- H. Shi and G. Zhao, J. Phys. Chem. C, 2014, 118, 25939–25946 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. C. Cruz, V. Baglio, S. Siracusano, R. Ornelas, L. Ortiz-Frade, L. G. Arriaga, V. Antonucci and A. S. Aricò, J. Nanopart. Res., 2011, 13, 1639–1646 CrossRef CAS.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518–9523 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. Zhang, X. Fan, J. Jian, D. Yu, Z. Zhang and L. Dai, Energy Environ. Sci., 2017, 10, 2312–2317 RSC.
- A. K. Beine, C. Broicher, Q. Hu, L. Mayerl, T. Bisswanger, H. Hartmann, A. Besmehn, S. Palkovits, A.-H. Lu and R. Palkovits, Catal. Sci. Technol., 2018, 8, 6311–6315 RSC.
- F. Zeng, C. Broicher, J. P. Hofmann, S. Palkovits and R. Palkovits, ChemCatChem, 2020, 12, 710–716 CrossRef CAS.
- C. Broicher, F. Zeng, N. Pfänder, M. Frisch, T. Bisswanger, J. Radnik, J. M. Stockmann, S. Palkovits, A. K. Beine and R. Palkovits, ChemCatChem, 2020, 12, 5378 CrossRef CAS.
- K. H. Cho, H. Seo, S. Park, Y. H. Lee, M. Y. Lee, N. H. Cho and K. T. Nam, Adv. Funct. Mater., 2020, 30, 1910424 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- C. Broicher, J. Artz, S. Palkovits, H. Antoni, M. Drogeler, D. M. Morales, C. Stampfer and R. Palkovits, Catal. Sci. Technol., 2018, 8, 1517–1521 RSC.
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Energy Environ. Sci., 2013, 6, 2745–2755 RSC.
- P. F. Smith, B. J. Deibert, S. Kaushik, G. Gardner, S. Hwang, H. Wang, J. F. Al-Sharab, E. Garfunkel, L. Fabris, J. Li and G. C. Dismukes, ACS Catal., 2016, 6, 2089–2099 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153–18156 CrossRef CAS PubMed.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- C. M. Hull, J. A. Koza and J. A. Switzer, J. Mater. Res., 2016, 31, 3324–3331 CrossRef CAS.
- Y. Huang, W. Yang, Y. Yu and S. Hao, J. Electroanal. Chem., 2019, 840, 409–414 CrossRef CAS.
- C. Broicher, F. Zeng, J. Artz, H. Hartmann, A. Besmehn, S. Palkovits and R. Palkovits, ChemCatChem, 2019, 11, 412–416 CrossRef CAS.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef PubMed.
- L. Kong, M. Zhong, W. Shuang, Y. Xu and X.-H. Bu, Chem. Soc. Rev., 2020, 49, 2378–2407 RSC.
- L. Negahdar, F. Zeng, S. Palkovits, C. Broicher and R. Palkovits, ChemElectroChem, 2019, 6, 5588–5595 CrossRef CAS.
- S. Hirai, S. Yagi, A. Seno, M. Fujioka, T. Ohno and T. Matsuda, RSC Adv., 2016, 6, 2019–2023 RSC.
- M. Gliech, A. Bergmann, C. Spöri and P. Strasser, J. Energy Chem., 2016, 25, 278–281 CrossRef.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2010, 3, 79 CrossRef PubMed.
- P. W. Menezes, A. Indra, N. R. Sahraie, A. Bergmann, P. Strasser and M. Driess, ChemSusChem, 2015, 8, 164–171 CrossRef CAS PubMed.
- P. W. Menezes, A. Indra, V. Gutkin and M. Driess, Chem. Commun., 2017, 53, 8018–8021 RSC.
- F. Gao, C. Chu, W. Zhu, X. Tang, H. Yi and R. Zhang, Appl. Surf. Sci., 2019, 479, 548–556 CrossRef CAS.
- Y. Jung, H. Park, J.-A. Park, J. Noh, Y. Choi, M. Jung, K. Jung, M. Pyo, K. Chen, A. Javey and G. Cho, Sci. Rep., 2015, 5, 8105 CrossRef CAS PubMed.
- A. Zhao, J. Masa, W. Xia, A. Maljusch, M.-G. Willinger, G. Clavel, K. Xie, R. Schlögl, W. Schuhmann and M. Muhler, J. Am. Chem. Soc., 2014, 136, 7551–7554 CrossRef CAS PubMed.
- R. Xu, K. Ye, D. Cao, J. Huang, T. Liu, K. Cheng, J. Yin and G. Wang, J. Power Sources, 2014, 268, 204–211 CrossRef.
- A. Manabe, M. Kashiwase, T. Hashimoto, T. Hayashida, A. Kato, K. Hirao, I. Shimomura and I. Nagashima, Electrochim. Acta, 2013, 100, 249–256 CrossRef CAS.
- S. Dresp, F. Luo, R. Schmack, S. Kühl, M. Gliech and P. Strasser, Energy Environ. Sci., 2016, 9, 2020 RSC.
- A. R. Zeradjanin, J. Masa, I. Spanos and R. Schlögl, Front. Energy Res., 2021, 8, 613092 CrossRef.
- Z. Zhao, G. Tian, V. Trouillet, L. Zhu, J. Zhu, A. Missiul, E. Welter and S. Dsoke, Inorg. Chem. Front., 2019, 6, 1861–1872 RSC.
- T. Shishido, M. Yamamoto, D. Li, Y. Tian, H. Morioka, M. Honda, T. Sano and K. Takehira, Appl. Catal., A, 2006, 303, 62–71 CrossRef CAS.
- C. Baltes, S. Vukojević and F. Schüth, J. Catal., 2008, 258, 334–344 CrossRef CAS.
- H. T. Zhang and X. H. Chen, Nanotechnology, 2006, 17, 1384–1390 CrossRef CAS.
- S. Ishii, T. Nakane, S. Uchida, M. Yoshida and T. Naka, J. Asian Ceram. Soc., 2018, 6, 7–12 CrossRef.
- H.-S. Na, S.-Y. Ahn, J.-O. Shim, K.-W. Jeon, H.-M. Kim, Y.-L. Lee, W.-J. Jang, B.-H. Jeon and H.-S. Roh, Korean J. Chem. Eng., 2019, 36, 1243–1248 CrossRef CAS.
- G. Simson, E. Prasetyo, S. Reiner and O. Hinrichsen, Appl. Catal., A, 2013, 450, 1–12 CrossRef CAS.
- R. Zhang, X. X. Huang, D. Wang, T. K. A. Hoang, Y. Yang, X. M. Duan, P. Chen, L.-C. Qin and G. W. Wen, Adv. Funct. Mater., 2018, 28, 1705817 CrossRef.
- Z. Zhao, G. Tian, V. Trouillet, L. Zhu, J. Zhu, A. Missiul, E. Welter and S. Dsoke, Inorg. Chem. Front., 2019, 6, 1861–1872 RSC.
- V. Di Castro and G. Polzonetti, J. Electron Spectrosc. Relat. Phenom., 1989, 48, 117–123 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- D. Lee, Q. X. Xia, J. M. Yun and K. H. Kim, Appl. Surf. Sci., 2018, 433, 16–26 CrossRef CAS.
- A. Shchukarev and D. Korolkov, Open Chem., 2004, 2, 347 CAS.
- S. J. Trasatti, Electroanal. Chem., 1980, 111, 125–131 CrossRef CAS.
- S. J. Trasatti and O. A. Petri, Pure Appl. Chem., 1991, 63, 711–734 CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- E. Locke, S. Jiang and S. K. Beaumont, Top. Catal., 2018, 61, 977–985 CrossRef CAS PubMed.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- L. Cao, Y. Cao, X. Liu, Q. Luo, W. Liu, W. Zhang, X. Mou, T. Yao and S. Wei, J. Mater. Chem. A, 2018, 6, 15684–15689 RSC.
- J. Y. Cheon, T. Kim, Y. Choi, H. Y. Jeong, M. G. Kim, Y. J. Sa, J. Kim, Z. Lee, T.-H. Yang, K. Kwon, O. Terasaki, G.-G. Park, R. R. Adzic and S. H. Joo, Sci. Rep., 2013, 3, 2715 CrossRef PubMed.
- R. L. Doyle and M. E. G. Lyons in Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, ed. S. Giménez and J. Bisquert, Springer International Publishing, Cham, Berline, 2016, vol. 2, pp. 41–104 Search PubMed.
- F. J. Pérez-Alonso, C. Adán, S. Rojas, M. A. Peña and J. L. G. Fierro, Int. J. Hydrogen Energy, 2014, 39, 5204–5212 CrossRef.
- S. Campagna Zignani, M. Lo Faro, S. Trocino and A. S. Aricò, Energies, 2020, 13, 1720 CrossRef.
- A. Carbone, S. Campagna Zignani, I. Gatto, S. Trocino and A. S. Aricò, Int. J. Hydrogen Energy, 2020, 45(16), 9285–9292 CrossRef CAS.
- H. Antoni, W. Xia, J. Masa, W. Schuhmann and M. Muhler, Phys. Chem. Chem. Phys., 2017, 19, 18434–18442 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00905b |
| ‡ Authors contributed equally to the publication. |
| This journal is © The Royal Society of Chemistry 2021 |