
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric azidohydroxylation of styrene derivatives mediated by a biomimetic styrene monooxygenase enzymatic cascade†
Lía
Martínez-Montero
a,
Dirk
Tischler
b,
Philipp
Süss
c,
Anett
Schallmey
d,
Maurice C. R.
Franssen
 e,
Frank
Hollmann
a and
Caroline E.
Paul
*a
e,
Frank
Hollmann
a and
Caroline E.
Paul
*a
aDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: c.e.paul@tudelft.nl
bMicrobial Biotechnology, Ruhr-Universität Bochum, Universitätsstr. 150, 44780 Bochum, Germany
cEnzymicals AG, Walther-Rathenau-Straße 49a, 17489 Greifswald, Germany
dInstitute for Biochemistry, Biotechnology and Bioinformatics, Technische Universität Braunschweig, Spielmannstraße 7, 38106 Braunschweig, Germany
eLaboratory of Organic Chemistry, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands
First published on 14th June 2021
Abstract
Enantioenriched azido alcohols are precursors for valuable chiral aziridines and 1,2-amino alcohols, however their chiral substituted analogues are difficult to access. We established a cascade for the asymmetric azidohydroxylation of styrene derivatives leading to chiral substituted 1,2-azido alcohols via enzymatic asymmetric epoxidation, followed by regioselective azidolysis, affording the azido alcohols with up to two contiguous stereogenic centers. A newly isolated two-component flavoprotein styrene monooxygenase StyA proved to be highly selective for epoxidation with a nicotinamide coenzyme biomimetic as a practical reductant. Coupled with azide as a nucleophile for regioselective ring opening, this chemo-enzymatic cascade produced highly enantioenriched aromatic α-azido alcohols with up to >99% conversion. A bi-enzymatic counterpart with halohydrin dehalogenase-catalyzed azidolysis afforded the alternative β-azido alcohol isomers with up to 94% diastereomeric excess. We anticipate our biocatalytic cascade to be a starting point for more practical production of these chiral compounds with two-component flavoprotein monooxygenases.
Chiral epoxides are highly sought after to access optically active molecules via ring opening by nucleophiles such as azide, which affords 1,2-azido alcohols.1 These compounds are intermediates for chiral aziridines, diamines,2 and 1,2-amino alcohols found in bioactive compounds, chiral auxiliaries for asymmetric synthesis,3 and give access to click chemistry.4 Besides azidolysis,5 the synthesis of 1,2-azido alcohols traditionally involves conversion of β-diols via sulfites,6 or carbonyl reduction of α-azido ketones.7,8 The challenge is to allow for a wide array of substituents and reach enantiopurity. Enzymatic aminohydroxylation of styrenes was recently described with an engineered cytochrome c,9 and through enzymatic cascades using epoxide hydrolases, ADHs and transaminases.10–14 On the other hand, enzymatic azidohydroxylation of aromatic alkenes remains to be investigated and applied in vitro.7,15
We envisioned to produce chiral 1,2-azido alcohols from aromatic alkenes via enzymatic epoxidation with a styrene monooxygenase (SMO).16 SMO can activate and incorporate oxygen in alkenes to produce chiral epoxides with high stereo- and regioselectivity.17–19 SMOs are two-component flavoprotein monooxygenases: the reductase component StyB (EC 1.5.1.36) uses nicotinamide adenine dinucleotide (NADH) to reduce flavin adenine dinucleotide (FAD), which freely diffuses to the oxygenase component StyA (EC 1.14.14.11) and reduces molecular oxygen, forming a hydroperoxyflavin that oxidizes styrene to its chiral epoxide (Fig. 1A).20,21 StyA enzymes accept a range of aromatic17,19 and aliphatic22,23 alkene substrates, and can be easily produced recombinantly in E. coli.
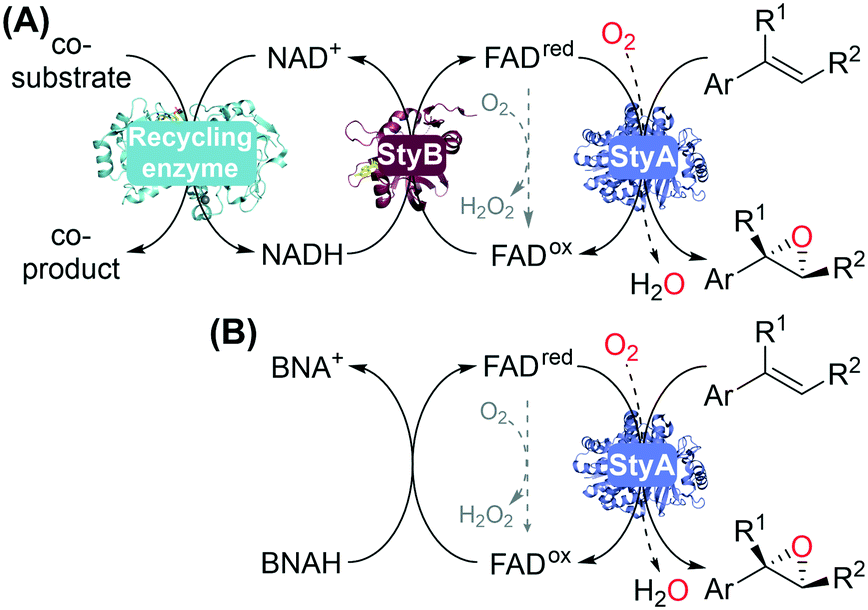 | ||
| Fig. 1 SMO-Catalyzed asymmetric epoxidation of aromatic alkenes: (A) two-component flavoprotein monooxygenase StyA and reductase StyB with the required recycled NADH cofactor; (B) oxygenase StyA with 1-benzyl-1,4-dihydronicotinamide (BNAH) as reductant for direct FAD reduction. | ||
Enzymatic recycling of the required NADH generally involves a dehydrogenase and a co-substrate (Fig. 1A), typically added in large excess. Non-enzymatic approaches have been investigated by shortcutting NADH/StyB using various photo- or electrochemical methods to reduce FAD.24–29 In particular, 1-benzyl-1,4-dihydronicotinamide (BNAH) was shown to be effective and simple to use as a reductant with StyA16,30 and other two-component flavoprotein monooxygenases such as halogenases,31 a bacterial luciferase,32 and an FMN-dependent type II Baeyer–Villiger monooxygenase.33 BNAH thus circumvents the use of two enzymes (StyB, dehydrogenase) and NADH, for using oxygenase StyA in biocatalytic reactions (Fig. 1B).
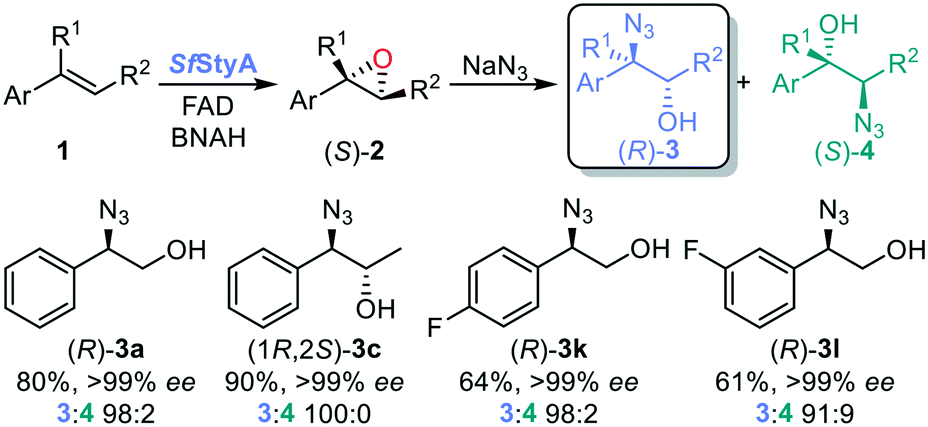
Previous enzymatic cascades involving two-component SMOs have led to the synthesis of amino acids and other valuable chiral compounds.10,34–37 Herein we establish the proof of principle for a one-pot biocatalytic cascade via StyA-catalyzed epoxidation, mediated by a nicotinamide coenzyme biomimetic (NCB), followed by either chemical or enzymatic azidolysis with halohydrin dehalogenases (HHDHs), towards the desired product isomer (Fig. 2). Our linear artificial cascade combines two (bio)chemical steps in one pot without isolation of intermediates,38 and allows for substituents on the α- or β-carbon to produce tertiary alcohols, an advantage over other cascades limited from aromatic haloketones.8,39,40 The regioselective ring opening depends on the nucleophile and epoxide, among other parameters.41
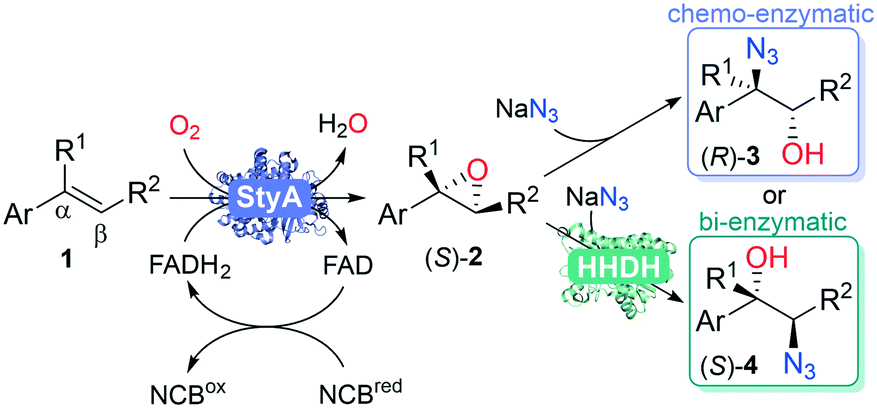 | ||
| Fig. 2 Linear one-pot chemo- or bi-enzymatic cascade from aromatic alkenes 1 towards chiral 1,2-azido alcohols. First step: StyA, FAD, NCB to enantiopure epoxides 2; second step: with NaN3 to α-azido alcohol products 3, or with NaN3 and HHDH to β-azido alcohol products 4. | ||
To establish the first step of the cascade, we explored three available StyA enzymes, recombinantly produced in E. coli and purified, for the selective asymmetric oxidation of aromatic alkenes: StyA from Pseudomonas sp. VLB120,42 StyA1 from Rhodococcus opacus 1CP,43 and SfStyA from Sphingopyxis fribergensis Kp5.2.
Compared with using reductase StyB, NADH and its recycling system (Fig. 1A), the oxygenase StyA had previously displayed similar catalytic activity using BNAH as hydride donor to reduce FAD (Fig. 1B).16 Initially, we screened a panel of aromatic alkenes 1a–s -mostly styrene derivatives- with the recently isolated SfStyA (see ESI†) to establish its substrate scope (Table 1). The aromatic alkene substrates explored bore different substitution patterns: a methyl substituent at the α- or β-position (1b–d, 1q–r), a ring substituent in para-, meta- and ortho-positions (halogen atoms and methyl groups 1e–o), a non-conjugated alkene (allylbenzene 1p) and a heterocyclic ring (2-vinylpyridine 1s).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Product 2 | [2] (mM) | TOFb (h−1) | eec (%) | |
| a Reaction conditions: [BNAH] = 15 mM, buffer (50 mM tris-SO4 pH 7.0), catalase = 650 U mL−1, [FAD] = 50 μM, [SfStyA] = 3 μM, [alkene 1] = 5 mM, 0.2% v/v dimethyl sulfoxide (DMSO), final volume 1 mL in a 2 mL plastic tube shaken on a thermomixer at 900 rpm and 30 °C, for 1 h. b Turnover frequency (TOF) = [product]/[enzyme] per hour. c Enantiomeric excess of epoxides determined by chiral GC-FID (see ESI†). Average of duplicates, standard deviation <10%. | |||||
| 1 | 2a |
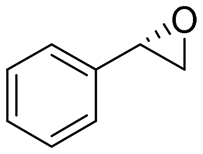
|
3.9 | 1300 | >99 (S) |
| 2 | 2b |
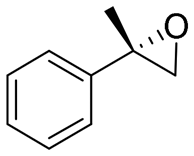
|
4.7 | 1565 | 89 (S) |
| 3 | 2c |
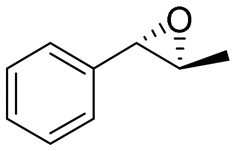
|
4.7 | 1565 | >99 (1S,2S) |
| 4 | 2d |
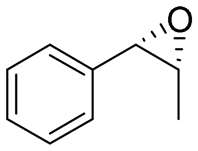
|
4.7 | 1565 | >99 (1S,2R) |
| 5 | 2e |
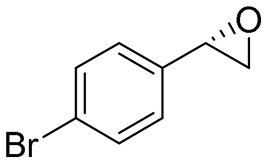
|
4.3 | 1435 | 96 (S) |
| 6 | 2f |
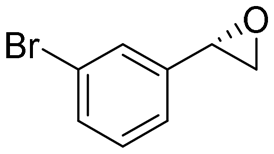
|
4.1 | 1365 | 81 (S) |
| 7 | 2g |
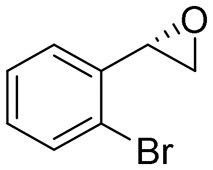
|
3.3 | 1100 | 70 (S) |
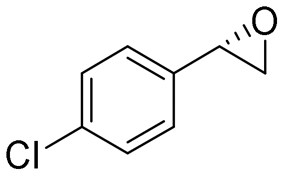
| 8 | 2h |
|
4.7 | 1565 | 97 (S) |
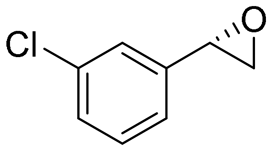
| 9 | 2i |
|
4.7 | 1565 | >99 (S) |
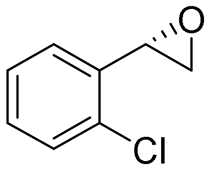
| 10 | 2j |
|
4.3 | 1435 | 83 (S) |
| 11 | 2k |
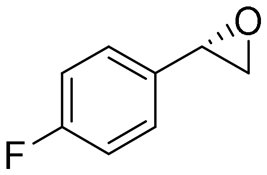
|
4.5 | 1500 | >99 (S) |
| 12 | 2l |
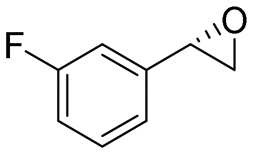
|
4.3 | 1435 | 95 (S) |
| 13 | 2m |
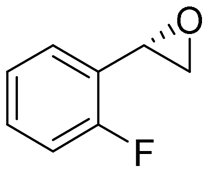
|
4.7 | 1565 | 94 (S) |
| 14 | 2n |
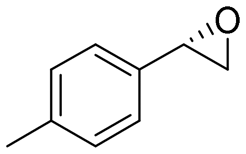
|
4.2 | 1400 | 95 (S) |
| 15 | 2o |
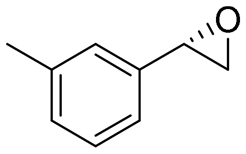
|
4.8 | 1600 | 99 (S) |
| 16 | 2p |
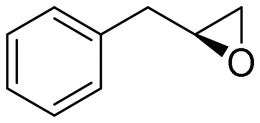
|
3.8 | 1265 | 68 (S) |
| 17 | 2q |
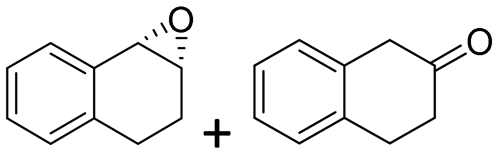
|
4.0 + 0.2 | 1400 | >99 (1S,2R) |
| 18 | 2r |
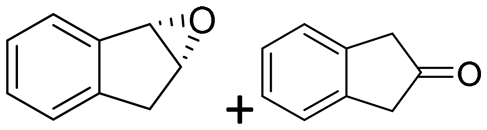
|
1.9 + 2.2 | 1365 | >99 (1S,2R) |
| 19 | 2s |
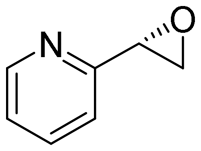
|
2.5 | 835 | >99 (S) |
Reaction mixtures were extracted after one hour for conversion comparison. All alkenes except 2-vinylpyridine 1s were converted to over 3.3 mM epoxide product within one hour (Table 1, >66% conversion, turnover frequency (TOF) >1100 h−1). Full conversion was achieved when the reaction was left to completion (Fig. 3A) and no diol was detected (see ESI†). The formation of a ketone side product was observed in two cases, with 1,2-dihydronaphthalene 1q giving 5% 2-tetralone (Table 1 entry 17), and indene 1r leading to 54% 2-indanone (entry 18). The enantiomeric excess (ee) obtained for oxide products bearing different substituents were mostly excellent (94 to >99% ee, entry 1, 3–5, 8–9, 11–15, 17–19), very good (89% ee for α-methylstyrene oxide 2b entry 2) and moderate for the non-conjugated alkene in allylbenzene 1p, affording epoxide 2p with 68% ee (entry 16).
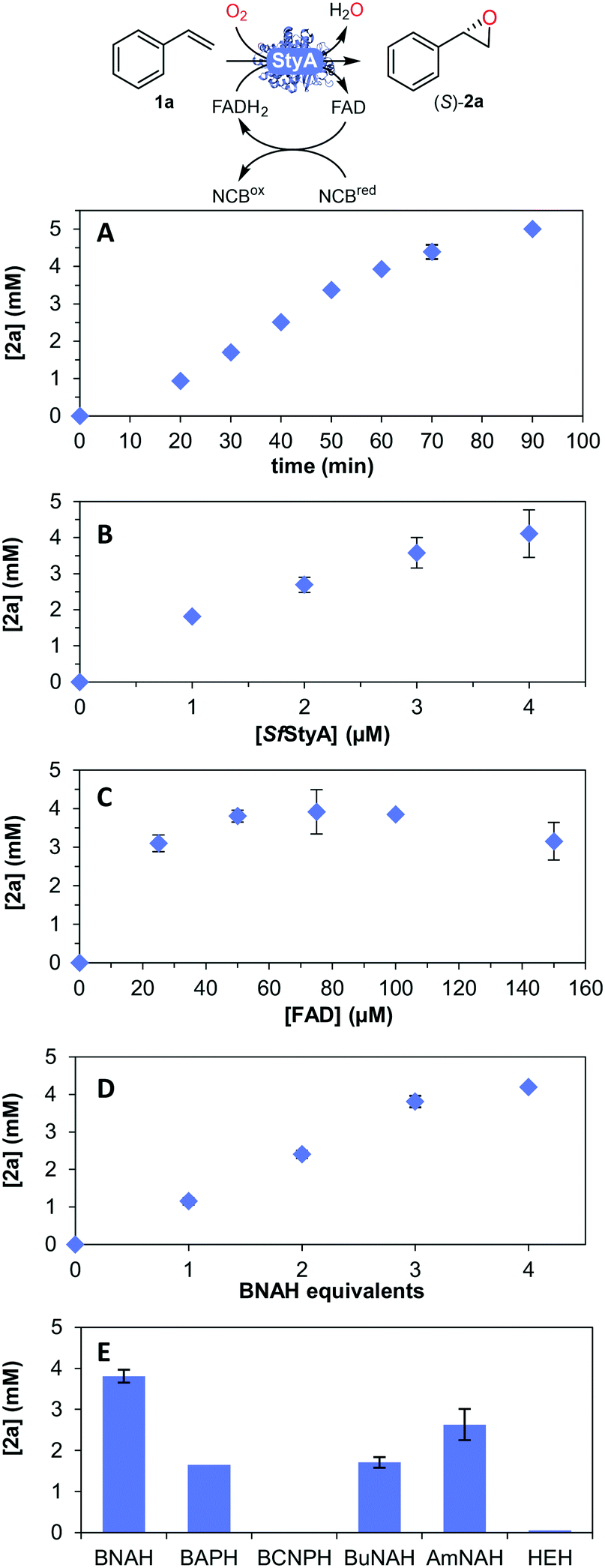 | ||
| Fig. 3 Parameters for the SfStyA-catalyzed asymmetric oxidation of styrene to its oxide 2a: (A) time course; (B) [SfStyA] = 0 to 4 μM; (C) [FAD] = 0 to 200 μM; (D) BNAH equivalents (1 equiv. = 5 mM); (E) type of NCBs hydride donor (see Fig. 4 for structures). General reaction conditions unless otherwise specified: [BNAH] = 15 mM, buffer (50 mM tris-SO4 pH 7.0), catalase = 650 U mL−1, [FAD] = 50 μM, [SfStyA] = 3 μM, [styrene] = 5 mM, 0.2% v/v DMSO, final volume 1 mL in a 2 mL plastic tube shaken on a thermomixer at 900 rpm and 30 °C, for 1 h. Average of duplicates. | ||
This substrate scope shows substituent-position effects on SfStyA, observed with other characterized StyA enzymes.44 The position of the substituent affected the ee, especially in the case of bromostyrene: the para-substituted product 2e has an ee of 96% (S), compared with 81% and 70% ee (S) obtained for meta- 2f and ortho-substituted 2g, respectively (Table 1 entry 5–7). This effect was less pronounced with chlorostyrenes 2h–j (83 to 95% ee, entry 8–10) and even less with fluorostyrenes 2k–m (94 to >99% ee, entry 11–13). Methylstyrenes 2n–o (95 to 99% ee, entry 14 and 15) showed a similar pattern as chlorostyrenes (entry 8 and 9).
The high conversions and enantioselectivity obtained with SfStyA led to its selection for further reactions, as StyA1 is less stable and gives lower TOFs,16 and StyA affords slightly lower ee for desired chiral epoxides in vitro (2a 98.0% ee, 2c 97.6% ee,24 compared with >99% ee with SfStyA in both cases). We were especially interested in substrates leading to products with two chiral centers: (S)-α-methylstyrene oxide 2b (Table 1, entry 2), (1S,2S)-1-phenylpropylene oxide 2c and (1S,2R)-1-phenylpropylene oxide 2d (entry 3 and 4) were obtained with excellent ee.
Next, we determined the time course (Fig. 3A) and various reaction parameters for the SfStyA-catalyzed epoxidation of styrene to demonstrate the practical use of the SfStyA/BNAH system with this oxygenase component: enzyme concentration (Fig. 3B), FAD concentration (Fig. 3C), the number of BNAH equivalents (Fig. 3D), and hydride donor (Fig. 3E), which influenced product formation.
Higher enzyme concentration at 50 μM FAD led to higher product formation (Fig. 3B). FAD concentration was explored to ensure optimal product formation (Fig. 3C). FAD concentrations of up to 100 μM led to increased oxide formation, which translates to FAD reduction as the rate-limiting step. A plateau was then reached, hence the limiting step most likely became the SfStyA-catalyzed epoxidation. Less product was observed with >100 μM FAD, as previously reported with other StyA enzymes.16,24,45 The lower product formation can be ascribed to FAD competing with FADH2 for the flavin-binding site, although the affinity for FADH2 is approximately 8000-fold higher.18,43,45,46 Additionally, FAD and FADH2 can disproportionate to give FAD radicals, reacting with molecular oxygen to form hydrogen peroxide,47 leading to a futile loss of electrons.
Concerning the source of electrons, several NADH regeneration systems have been established with formate (FDH) or glucose (GDH) dehydrogenases (Fig. 1A). For our cascade design however, sodium azide can be an inhibitor for FDH, whereas high excess of glucose is typically used for GDH (see ESI†), and we aimed to achieve a more efficient and practical system. As we previously showed that the use of BNAH allows for cost-effective direct FAD reduction with up to 85% efficiency,16 we explored other NCBs. Flavin reduction by NCBs such as BNAH is known to occur through direct hydride transfer in solution.48,49 Different substituents were previously shown to affect the overall NCB redox potential and lead to different FAD reduction rates.31 Therefore, we screened six NCBs bearing substituents varying from amide, acetyl, carboxylic acid to nitrile, with either a benzyl, butyl or alkyl amide chain on the nitrogen, and the classical Hantzsch ester (HEH, Fig. 3E and 4), as a hydride donor for the reduction of FAD without interfering with the epoxidation.50 Within one-hour reactions, BNAH displayed the best overall conversion (3.9 mM), followed by AmNAH (2.6 mM). BCNPH and HEH proved to be the worst reductants. No product was observed in the absence of FAD, or enzyme, or BNAH.
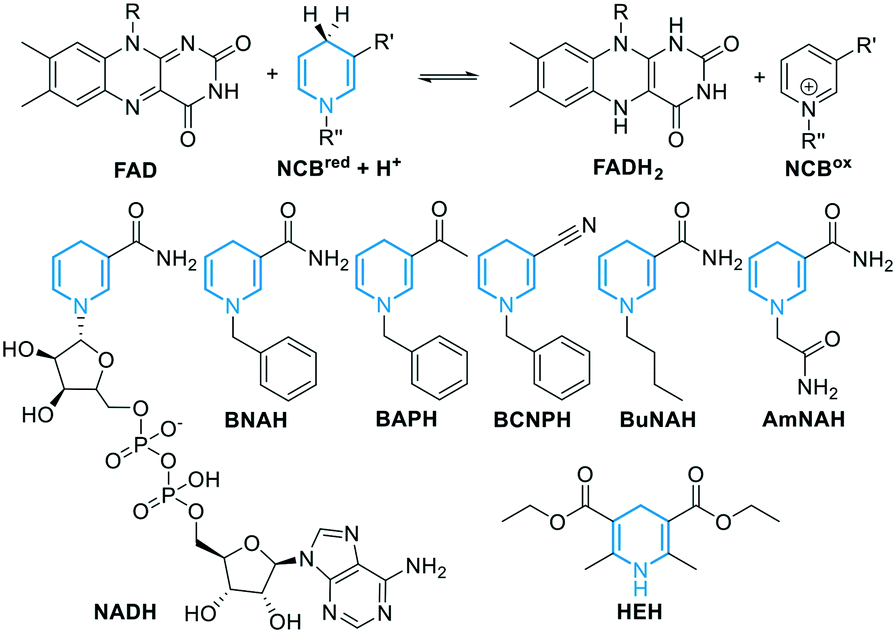 | ||
| Fig. 4 Top: schematic FAD reduction by NCBs. Bottom: chemical structures of NADH and NCBs (1-benzyl-3-acetyl-1,4-dihydropyridine BAPH, 1-benzyl-3-cyano-1,4-dihydropyridine BCNPH, 1-butyl-1,4-dihydronicotinamide BuNAH, 1-carbamoyl-1,4-dihydronicotinamide AmNAH, Hantzsch ester HEH). | ||
Once the biocatalytic asymmetric oxidation process was established, we proceeded to investigate the use of the chiral epoxides in a chemo-enzymatic cascade. Azide can cleave epoxides under mild conditions. With aliphatic epoxides, ring opening occurs through a bimolecular reaction, an SN2-type backside attack of the azide on the least substituted carbon of the protonated epoxide, forming a trans-1,2-azido alcohol as product. The rate of chemical epoxide ring opening is pH dependent, the more acidic, the faster the reaction, and also higher temperatures increase the reaction rate (see ESI†). Previous studies have shown that azidolysis of aryl-substituted epoxides show different regioselectivity, affording α-azido alcohols as the major products.51,52 The nucleophilic attack occurs at the α-carbon of the aromatic substituted epoxide, with complete inversion of configuration (see ESI†).41,53 The regioselectivity was explained by stabilization of positive charge formed through delocalization,53 but recent studies revealed that electrostatic interaction between the phenyl ring and the incoming nucleophile play a dominant role.54
We chose sodium azide salt for its high solubility in water and determined that the amount needed for fast azidolysis was seven equivalents with respect to the substrate (see ESI†). The reactivity and regioselectivity of the azidolysis in water is controlled by a neutral pH value, 30 °C temperature and low buffer concentration, thus avoiding diol formation stemming from the competition of hydroxide or water with the azide ion (see ESI†).52 Under these conditions, we achieved full conversion and highly regioselective ring opening of styrene oxide: the desired α-azido alcohol (2-azido-2-phenyl-1-ethanol 3a) was formed as the main product with only a trace amount of regioisomer 4a, determined by gas chromatography with flame ionization detection (GC-FID, ratio of 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 3a:4a). The linearly increasing product formation with higher azide concentrations can be explained by the SN2 type mechanism.
:2 3a:4a). The linearly increasing product formation with higher azide concentrations can be explained by the SN2 type mechanism.
We then carried out the full azidohydroxylation cascade with the established SfStyA-catalyzed epoxidation of alkenes 1a–o, q–s, affording the corresponding (2R)-α-azido alcohols 3 with good to excellent conversions and regioselectivity (Table 2). The ee from the SfStyA reaction was retained in all cases.
|
|
||||
|---|---|---|---|---|
| Entry | Major product | Conv.b (%) |
3:4 (%) |
|
| a Reaction conditions (i): [BNAH] = 15 mM, buffer (50 mM tris-SO4 pH 7.0), catalase 650 U mL−1, [FAD] = 50 μM, sodium azide (7 equiv.), [SfStyA] = 3 μM, [alkene] = 5 mM, 0.2% v/v DMSO, final volume 1 mL in a 2 mL plastic tube shaken on a thermomixer at 900 rpm and 30 °C, for 24 h. b Determined by chiral GC-FID. Average of duplicates, standard deviation <10%. c 5% diol product observed. d Assignment of isomers ratio not determined. | ||||
| 1 | 3a |
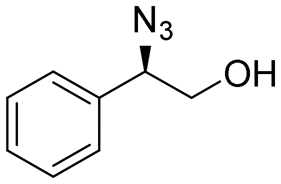
|
>99 | 98:2 |
| 2 | 3b |
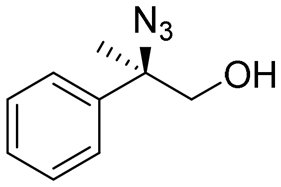
|
98c | 97:3d |
| 3 | 3c |
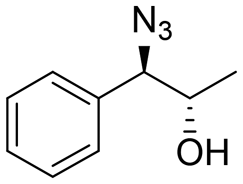
|
>99 | 100:0 |
| 4 | 3d |
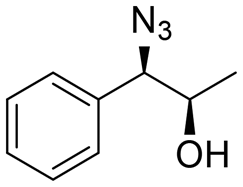
|
27 | 68:32 |
| 5 | 3e |
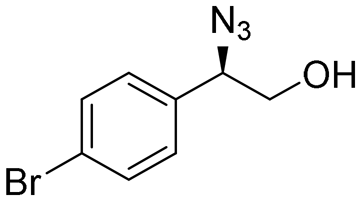
|
99 | 96:4 |
| 6 | 3f |
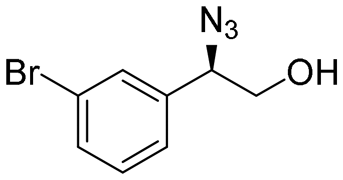
|
99 | 96:4 |
| 7 | 3g |
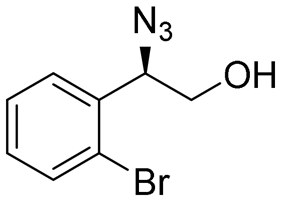
|
82 | 98:2 |
| 8 | 3h |
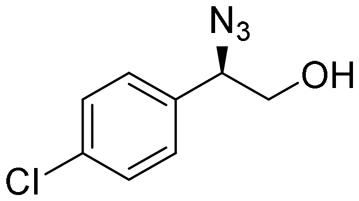
|
96 | 97:3 |
| 9 | 3i |
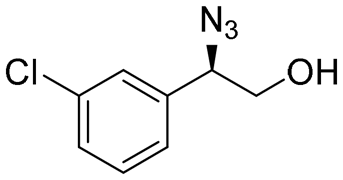
|
92 | 93:7 |
| 10 | 3j |
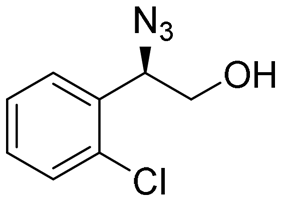
|
90 | 95:5 |
| 11 | 3k |
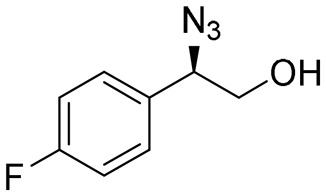
|
98 | 98:2 |
| 12 | 3l |
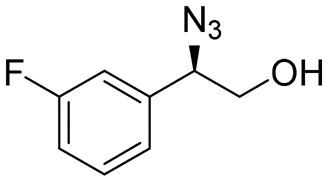
|
97 | 91:9 |
| 13 | 3m |
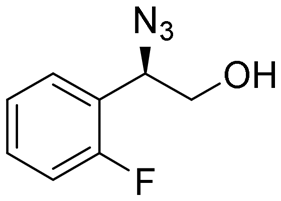
|
98 | 94:6 |
| 14 | 3n |
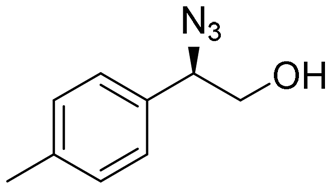
|
98 | 98:2 |
| 15 | 3o |
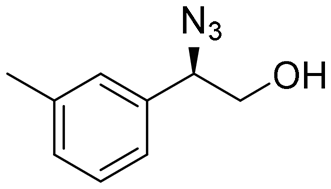
|
99 | 98:2 |
| 16 | 3q |
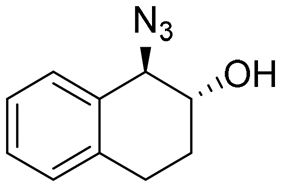
|
>99 | 94:6 |
| 17 | 3r |
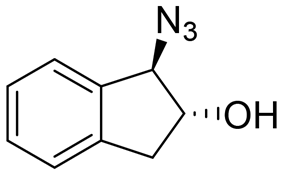
|
79 | 96:4 |
| 18 | 3s |
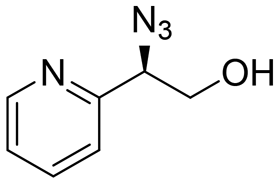
|
62 | 53:47 |
The difference in rate and degree of regioselectivity between azidolysis of trans- and cis-β-methylstyrene oxide (2cversus2d) could be explained by the orientation of the phenyl ring with respect to the epoxide (see ESI†). A further advantage of this chemo-enzymatic cascade, besides the ability to carry out the reactions in one pot via chiral epoxides, is the high reactivity of the epoxide intermediates: without the need to isolate the epoxide, the aldehyde and ketone previously observed with the simple biocatalytic epoxidation (Table 1, entries 17–18) do not have time to form. Therefore, we obtained full conversion with 1,2-dihydronaphthalene 1q and 79% conversion with indene 1r (Table 2, entry 16 and 17).
Encouraged by the enantioenriched 2-azido-2-phenyl-1-ethanol derivatives obtained, several reactions were carried out on a scale of 15 mg of substrate, increasing the substrate concentration to 10 mM. The corresponding azido alcohols obtained were simply extracted with ethyl acetate without further purification and identified and characterized by NMR spectroscopy (Fig. 5, ESI†). A higher scale reaction was performed with trans-β-methylstyrene 1c (56 mg, 10 mM) as substrate. Using SfStyA, BNAH and FAD, a 92% crude yield was obtained with >99% ee of (1R,2S)-1-azido-1-phenylpropan-2-ol 3c. 1H NMR showed a high purity product after extraction with ethyl acetate (see ESI†).
 | ||
| Fig. 5 One-pot chemo-enzymatic cascade from styrenes to chiral α-azido alcohols, 15 mg scale, % isolated yield of product. Reaction conditions: [BNAH] = 30 mM, buffer (50 mM tris-SO4, pH 7.0), catalase (650 U mL−1), [FAD] = 50 μM, [SfStyA] = 3 μM, [alkene] = 10 mM, [NaN3] = 35 mM, final volume 10 mL, shaken in Erlenmeyer flasks on an incubator shaker at 180 rpm and 30 °C for 22 h. | ||
It should be noted that sodium azide may interfere with the steady-state kinetics of substrate epoxidation catalysed by the SMO.55–57 Our one-pot cascade may be more efficient as a two-step approach, adding the sodium azide in the second step.
To access the β-azido alcohol isomers, several halohydrin dehalogenases (HHDHs, EC 3.8.1.2, Enzymicals screening kit, see ESI†)58,59 were screened towards racemic styrene oxide rac-2a (Table 3), using sodium azide for the selective epoxide ring opening,58 giving two possible products, 1-azido-2-phenylethanol 3 or 2-azido-1-phenylethanol 4. To minimize the uncatalyzed background reaction of sodium azide that leads to the α-azido alcohols, the reactions with HHDHs were performed using only one equivalence of sodium azide. In general, the most active HHDH towards the formation of β-azido alcohols 4 gives a higher ratio towards the desired product.
|
|
||||
|---|---|---|---|---|
| Entry | HHDH | Ratio 3:4 |
(R)-3 eeb (%) | (R)-4 eeb (%) |
| a Reaction conditions: buffer (50 mM tris-SO4, pH 7.0), [rac-styrene oxide] = 5 mM, [NaN3] = 5 mM, lyophilized cell-free extract HHDH (10 mg mL−1), final volume 1 mL in a 2 mL plastic tube shaken on a thermomixer at 900 rpm and 30 °C for 15 h 30 min. Full conversion was observed. b Determined by chiral GC-FID. c Diol side product observed in trace amounts (<2%). | ||||
| 1c | HheA3 | 87:13 |
8 | 42 |
| 2 | HheA5 | 54:46 |
43 | 67 |
| 3c | HheB5 | 33:67 |
44 | 27 |
| 4 | HheD3 | 82:18 |
15 | 68 |
| 5c | HheD5 | 37:63 |
60 | 49 |
| 6 | HheD6 | 95:5 |
<5 | <5 |
| 7 | HheE5 | 16:84 |
22 | <5 |
| 8 | None | 98:2 |
<5 | <5 |
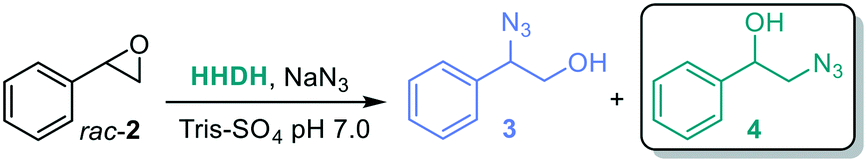
The results of the HHDH screening to obtain the β-azido alcohol were variable (Table 3): the enzymatic reaction was clearly outrun by the chemical one with HheD6 (entry 6), likely due to the low activity of the enzyme, as was the case with HheA3 (entry 1) and HheD3 (entry 4). The other HHDHs also showed mixed ratios (entries 2–3 and 6). HheE5 from Gammaproteobacterium strain IMCC3088 gave the best ratio of α:β 16:84 (entry 7).
The screened HHDHs lacked high selectivity for either the S- or R-enantiomer with racemic styrene oxide, the highest ee being reached with HheD3 (68% ee of (R)-4, entry 5), hence the importance of using a selective StyA in the asymmetric epoxidation step. In our case, the use of SfStyA provided very good to excellent ee (Table 1), but other StyA enzymes could be used to obtain the best ee depending on the substituted substrate.
We set out to further screen selected HHDHs (Enzymicals screening kit, see ESI†) with (S)-styrene oxide as starting material (Table 4). HheD4 and HheF led to trace amounts of diol side product 5 in addition to low ratios (entry 1 and 4), and once again HheE5 stood out as the most active towards β-ring opening (entry 3, the higher ratio with respect to Table 3 is ascribed to the use of a different batch of lyophilized enzyme) and was selected for further reactions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | HHDH | Ratio 3:4 |
(R)-3 eeb (%) | (S)-4 eeb (%) | (R)-5 (%) |
| a Reaction conditions: buffer (50 mM tris-SO4 pH 7.0), HHDH (10 mg), [NaN3] = 5 mM, [(S)-styrene oxide] = 5 mM, final volume 1 mL in a 10 mL glass vial shaken on an incubator shaker at 180 rpm and 30 °C for 15 h. Full conversion was observed. b Determined by chiral GC-FID. | |||||
| 1 | HheD4 | 63:37 |
>99 | >99 | 8 |
| 2 | HheE4 | 26:74 |
>99 | >99 | <1 |
| 3 | HheE5 | 6:94 |
>99 | >99 | <1 |
| 4 | HheF | 60:40 |
>99 | >99 | 5 |
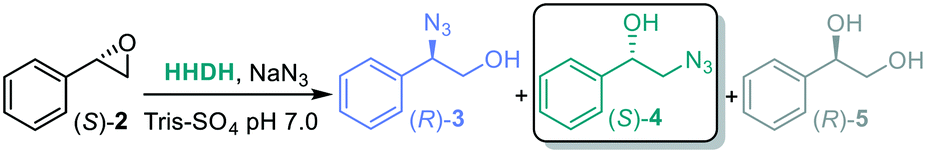
The substrates that gave highest conversion, ee and regioselectivity for the synthesis of α-azido alcohols were chosen for subsequent experiments. The bi-enzymatic cascade with SfStyA and HheE5 was thus carried out for styrene 1a, trans-β-methylstyrene 1c and 4-fluorostyrene 1k. The trans-β-methylstyrene 1c led to a mixture of α- and β-azido alcohol 4c, which can be ascribed to the poor activity of HheE5 towards the epoxide 3c.60 Further screening efforts with HHDHs did not display a more active enzyme, therefore there is room for enhancing HHDH enzymes with this type of substrate.
Pleasingly, substrates 1a and 1k afforded the corresponding β-azido alcohols 4a and 4k with good to excellent conversion, enantio- and regioselectivity (Fig. 6). With the discovery and engineering of HHDHs, we can expect variants with higher selectivity in the near future.
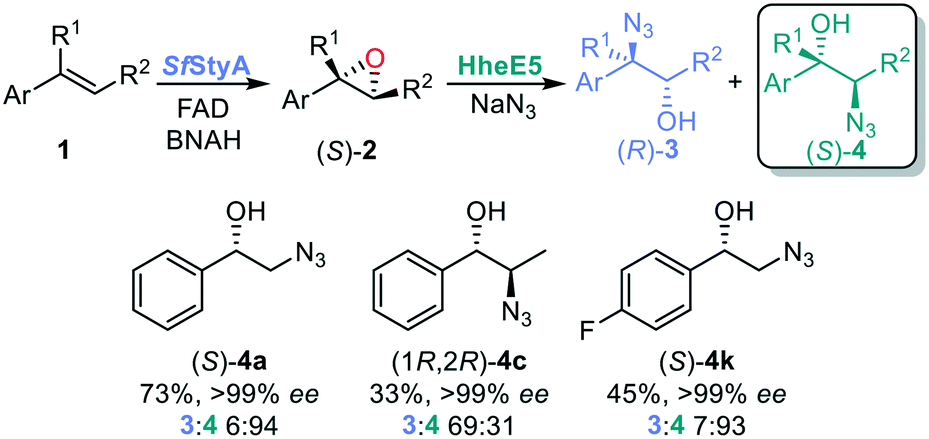 | ||
| Fig. 6 One-pot bi-enzymatic cascade to β-azido alcohols. % conversions. Reaction conditions: [BNAH] = 15 mM, buffer (50 mM tris-SO4 pH 7.0), catalase = 650 U mL−1, [FAD] = 50 μM, [alkene] = 5 mM, [SfStyA] = 3 μM, [NaN3] = 5 mM, HheE5 (20 mg), final volume 1 mL in a 10 mL glass vial shaken on an incubator shaker at 180 rpm and 30 °C for 22 h. | ||
Conclusions
In conclusion, we explored the substrate scope of a new two-component flavoprotein styrene monooxygenase SfStyA for the asymmetric oxidation of styrene derivatives, leading mostly to high conversions and ee. This study further demonstrates the use of the oxygenase component of two-component flavoprotein monooxygenases with a practical flavin reduction system. Through this biocatalytic asymmetric epoxidation with SfStyA, we established a linear artificial one-pot chemo- and bi-enzymatic cascade to achieve the asymmetric azidohydroxylation of styrenes. The epoxide ring opening was achieved with sodium azide either chemically, or catalyzed by a halohydrin dehalogenase, leading to high purity of either azidoalcohol isomers with retention of the StyA selectivity. The latest discovery of (R)-selective StyA oxygenases allows access to the other isomers.23,61,62We anticipate our biocatalytic cascade to expand the use of oxygenase StyA for the production of amino alcohols63 and styrenyl aziridines. Furthermore, the use of alternative nucleophiles, such as halides, cyanate, etc.,64 can expand the portfolio of chiral enantiopure products obtained. In the future we expect the use of peroxygenases will be attractive once evolved to achieve high enantioselectivity.65
Experimental details
All biocatalytic reactions were performed on an Eppendorf ThermoMixer C (Eppendorf, Hamburg, Germany) and followed by gas chromatography with flame ionization detection (GC-FID). Analyses were carried out on a Shimadzu GC-2010 GC-FID. Products were confirmed by reference standards and GC-MS. Product concentrations were obtained with a calibration curve equation using 5 mM dodecane as an internal standard.StyA-Catalyzed epoxidations
Stock solutions were made fresh in buffer: catalase from bovine liver (6500 U mL−1), FAD (5 mM), alkene 1 (2.5 M in DMSO). Reaction conditions: 2 mL microcentrifuge plastic tube, BNAH (15 mM), buffer (50 mM tris-SO4 pH 7.0), catalase (650 U mL−1), FAD (50 μM), SfStyA (3 μM), alkene 1 (5 mM, final 2% v/v DMSO), final volume 1 mL. The reaction mixtures were agitated on a thermomixer at 30 °C and 900 rpm for 1 h. Product concentration and ee were determined by GC-FID with a chiral column.Chemo-enzymatic cascade
Chemo-enzymatic cascade reactions (1 mL in volume) were performed in buffer (50 mM tris-SO4 pH 7.0) containing the styrene derivative 1 (5 mM), SfStyA (3 μM), FAD (50 μM), BNAH (15 mM), NaN3 (35 mM) catalase (650 U mL−1). The reaction mixtures were agitated on a thermomixer at 30 °C and 900 rpm. Comparison of retention times with authentic standards and GC-MS allowed product identification. Conversion and enantiomeric excess were determined by GC-FID analysis.Bi-enzymatic cascade
Lyophilized HHDH (10 mg, Enzymicals AG) was rehydrated in buffer for 30 min before use. Bi-enzymatic cascade reactions (1 mL in volume) were performed in buffer (50 mM tris-SO4 pH 7.0) containing the styrene derivative 1 (5 mM), SfStyA (3 μM), FAD (50 μM), BNAH (15 mM), NaN3 (5 mM), catalase (650 U mL−1) and HHDH (10 mg mL−1). The reaction mixtures were agitated on a thermomixer at 30 °C and 900 rpm. Conversion and enantiomeric excess were determined by GC-FID analysis.Experimental details for the synthesis of compounds, characterization, enzyme production and reactions, GC-FID analyses and more detailed experiments are available in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. M. M. was funded through the COST Action CM13003 Systems Biocatalysis. D. T. acknowledges the Federal Ministry for Innovation, Science and Research of North Rhine-Westphalia (grant number PtJ-TRI/1141ng006). C. E. P. acknowledges a NWO VENI grant (No 722.015.011). The halohydrin dehalogenases (HHDHs) were kindly provided by Enzymicals AG. We thank Prof. A. Schmid for providing the StyA plasmid, and A. H. Westphal, R. van Oosten and E. van der Klift for technical assistance.References
- A. K. Yudin, Aziridines and epoxides in organic synthesis, John Wiley & Sons, 2006 Search PubMed.
- X. Sun, X. Li, S. Song, Y. Zhu, Y.-F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059–6066 CrossRef CAS PubMed.
- D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–875 CrossRef CAS PubMed.
- L. S. Campbell-Verduyn, W. Szymanski, C. P. Postema, R. A. Dierckx, P. H. Elsinga, D. B. Janssen and B. L. Feringa, Chem. Commun., 2010, 46, 898–900 RSC.
- C. Molinaro, A. A. Guilbault and B. Kosjek, Org. Lett., 2010, 12, 3772–3775 CrossRef CAS PubMed.
- B. B. Lohray and J. R. Ahuja, J. Chem. Soc., Chem. Commun., 1991, 95–97 RSC.
- P. Besse, H. Veschambre, R. Chenevert and M. Dickman, Tetrahedron: Asymmetry, 1994, 5, 1727–1744 CrossRef.
- J. H. Schrittwieser, F. Coccia, S. Kara, B. Grischek, W. Kroutil, N. d'Alessandro and F. Hollmann, Green Chem., 2013, 15, 3318–3331 RSC.
- I. Cho, C. K. Prier, Z.-J. Jia, R. K. Zhang, T. Görbe and F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 3138–3142 CrossRef CAS PubMed.
- S. Wu, Y. Zhou, T. Wang, H.-P. Too, D. I. C. Wang and Z. Li, Nat. Commun., 2016, 7, 11917 CrossRef CAS PubMed.
- J. W. Zhao, H. L. Wu, J. D. Zhang, W. C. Gao, X. J. Fan, H. H. Chang, W. L. Wei and J. H. Xu, Biotechnol. Lett., 2018, 40, 349–358 CrossRef CAS PubMed.
- J. D. Zhang, J. W. Zhao, L. L. Gao, H. H. Chang, W. L. Wei and J. H. Xu, J. Biotechnol., 2019, 290, 24–32 CrossRef CAS PubMed.
- J. D. Zhang, X. X. Yang, Q. Jia, J. W. Zhao, L. L. Gao, W. C. Gao, H. H. Chang, W. L. Wei and J. H. Xu, Catal. Sci. Technol., 2019, 9, 70–74 RSC.
- M. L. Corrado, T. Knaus and F. G. Mutti, Green Chem., 2019, 21, 6246–6251 RSC.
- G. Sello, F. Orsini, S. Bernasconi and P. Di Gennaro, Tetrahedron: Asymmetry, 2006, 17, 372–376 CrossRef CAS.
- C. E. Paul, D. Tischler, A. Riedel, T. Heine, N. Itoh and F. Hollmann, ACS Catal., 2015, 5, 2961–2965 CrossRef CAS.
- T. Heine, W. J. H. van Berkel, G. Gassner, K.-H. van Pée and D. Tischler, Biology, 2018, 7, 42 CrossRef CAS PubMed.
- S. Montersino, D. Tischler, G. T. Gassner and W. J. H. van Berkel, Adv. Synth. Catal., 2011, 353, 2301–2319 CrossRef CAS.
- D. Tischler, A. Kumpf, D. Eggerichs and T. Heine, in The Enzymes, ed. P. Chaiyen and F. Tamanoi, Academic Press, 2020, ch. 13th, vol. 47, pp. 399–425 Search PubMed.
- J. Sucharitakul, R. Tinikul and P. Chaiyen, Arch. Biochem. Biophys., 2014, 555, 33–46 CrossRef PubMed.
- E. Morrison, A. Kantz, G. T. Gassner and M. H. Sazinsky, Biochemistry, 2013, 52, 6063–6075 CrossRef CAS PubMed.
- H. Toda, R. Imae and N. Itoh, Adv. Synth. Catal., 2014, 356, 3443–3450 CrossRef CAS.
- T. Heine, A. Scholtissek, S. Hofmann, R. Koch and D. Tischler, ChemCatChem, 2020, 12, 199–209 CrossRef CAS.
- F. Hollmann, P. C. Lin, B. Witholt and A. Schmid, J. Am. Chem. Soc., 2003, 125, 8209–8217 CrossRef CAS PubMed.
- F. Hollmann, K. Hofstetter, T. Habicher, B. Hauer and A. Schmid, J. Am. Chem. Soc., 2005, 127, 6540–6541 CrossRef CAS PubMed.
- R. Ruinatscha, K. Buehler and A. Schmid, J. Mol. Catal. B: Enzym., 2014, 103, 100–105 CrossRef CAS.
- M. M. C. H. van Schie, C. E. Paul, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2019, 55, 1790–1792 RSC.
- R. Ruinatscha, C. Dusny, K. Buehler and A. Schmid, Adv. Synth. Catal., 2009, 351, 2505–2515 CrossRef CAS.
- R. Amongre and G. Gassner, Bioelectrochemistry, 2021, 137, 107679 CrossRef CAS PubMed.
- W. Y. Zhang and F. Hollmann, Chem. Commun., 2018, 54, 7281–7289 RSC.
- M. Ismail, L. Schroeder, M. Frese, T. Kottke, F. Hollmann, C. E. Paul and N. Sewald, ACS Catal., 2019, 9, 1389–1395 CrossRef CAS PubMed.
- J. Phonbuppha, R. Tinikul, T. Wongnate, P. Intasian, F. Hollmann, C. E. Paul and P. Chaiyen, ChemBioChem, 2020, 21, 2073–2079 CrossRef CAS PubMed.
- R. Röllig, C. E. Paul, M. Claeys-Bruno, K. Duquesne, S. Kara and V. Alphand, Org. Biomol. Chem., 2021, 19, 3441–3450 RSC.
- S. K. Wu, Y. Z. Chen, Y. Xu, A. T. Li, Q. S. Xu, A. Glieder and Z. Li, ACS Catal., 2014, 4, 409–420 CrossRef CAS.
- J. D. Zhang, S. K. Wu, J. C. Wu and Z. Li, ACS Catal., 2015, 5, 51–58 CrossRef CAS.
- S. K. Wu, Y. Zhou, D. Seet and Z. Li, Adv. Synth. Catal., 2017, 359, 2132–2141 CrossRef CAS.
- S. K. Wu, Y. Zhou and Z. Li, Chem. Commun., 2019, 55, 883–896 RSC.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- W. Szymanski, C. P. Postema, C. Tarabiono, F. Berthiol, L. Campbell-Verduyn, S. de Wildeman, J. G. de Vries, B. L. Feringa and D. B. Janssen, Adv. Synth. Catal., 2010, 352, 2111–2115 CrossRef CAS.
- J. H. Schrittwieser, I. Lavandera, B. Seisser, B. Mautner and W. Kroutil, Eur. J. Org. Chem., 2009, 2293–2298 CrossRef CAS.
- H.-Y. Wang, K. Huang, M. De Jesús, S. Espinosa, L. E. Piñero-Santiago, C. L. Barnes and M. Ortiz-Marciales, Tetrahedron: Asymmetry, 2016, 27, 91–100 CrossRef CAS PubMed.
- S. Panke, M. Held, M. G. Wubbolts, B. Witholt and A. Schmid, Biotechnol. Bioeng., 2002, 80, 33–41 CrossRef CAS PubMed.
- D. Tischler, R. Kermer, J. A. D. Groning, S. R. Kaschabek, W. J. H. van Berkel and M. Schlomann, J. Bacteriol., 2010, 192, 5220–5227 CrossRef CAS PubMed.
- S. Bernasconi, F. Orsini, G. Sello and P. Di Gennaro, Tetrahedron: Asymmetry, 2004, 15, 1603–1606 CrossRef CAS.
- A. Kantz, F. Chin, N. Nallamothu, T. Nguyen and G. T. Gassner, Arch. Biochem. Biophys., 2005, 442, 102–116 CrossRef CAS PubMed.
- U. E. Ukaegbu, A. Kantz, M. Beaton, G. T. Gassner and A. C. Rosenzweig, Biochemistry, 2010, 49, 1678–1688 CrossRef CAS PubMed.
- V. Massey, J. Biol. Chem., 1994, 269, 22459–22462 CrossRef CAS.
- M. F. Powell, W. H. Wong and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4604–4608 CrossRef CAS PubMed.
- R. Stewart and D. J. Norris, J. Chem. Soc., Perkin Trans. 2, 1978, 246–249 RSC.
- C. Zheng and S. L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- J. Boruwa, J. C. Borah, B. Kalita and N. C. Barua, Tetrahedron Lett., 2004, 45, 7355–7358 CrossRef CAS.
- F. Fringuelli, O. Piermatti, F. Pizzo and L. Vaccaro, J. Org. Chem., 1999, 64, 6094–6096 CrossRef CAS.
- J. J. Blumenstein, V. C. Ukachukwu, R. S. Mohan and D. L. Whalen, J. Org. Chem., 1993, 58, 924–932 CrossRef CAS.
- C.-H. Wu, B. Galabov, J. I.-C. Wu, S. Ilieva, P. R. von Schleyer and W. D. Allen, J. Am. Chem. Soc., 2014, 136, 3118–3126 CrossRef CAS PubMed.
- B. Entsch, D. P. Ballou and V. Massey, J. Biol. Chem., 1976, 251, 2550–2563 CrossRef CAS.
- K. Detmer and V. Massey, J. Biol. Chem., 1984, 259, 1265–1272 CrossRef.
- A. Kantz and G. T. Gassner, Biochemistry, 2011, 50, 523–532 CrossRef CAS PubMed.
- J. Koopmeiners, B. Halmschlag, M. Schallmey and A. Schallmey, Appl. Microbiol. Biotechnol., 2016, 100, 7517–7527 CrossRef CAS PubMed.
- A. Schallmey and M. Schallmey, Appl. Microbiol. Biotechnol., 2016, 100, 7827–7839 CrossRef CAS PubMed.
- E. Calderini, J. Wessel, P. Suss, P. Schrepfer, R. Wardenga and A. Schallmey, ChemCatChem, 2019, 11, 2099–2106 CrossRef CAS.
- C. Cui, C. Guo, H. Lin, Z.-Y. Ding, Y. Liu and Z.-L. Wu, Enzyme Microb. Technol., 2020, 132, 109391 CrossRef CAS PubMed.
- H. Xiao, S. Dong, Y. Liu, X. Q. Pei, H. Lin and Z. L. Wu, Catal. Sci. Technol., 2021, 11, 2195–2201 RSC.
- X. Ariza, O. Pineda, F. Urpi and J. Vilarrasa, Tetrahedron Lett., 2001, 42, 4995–4999 CrossRef CAS.
- G. Hasnaoui-Dijoux, M. M. Elenkov, J. H. L. Spelberg, B. Hauer and D. B. Janssen, ChemBioChem, 2008, 9, 1048–1051 CrossRef CAS PubMed.
- M. C. R. Rauch, F. Tieves, C. E. Paul, I. W. C. E. Arends, M. Alcalde and F. Hollmann, ChemCatChem, 2019, 11, 4519–4523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00855b |
| This journal is © The Royal Society of Chemistry 2021 |