DOI:
10.1039/D1CY00591J
(Communication)
Catal. Sci. Technol., 2021,
11, 4008-4011
Electrical promotion-assisted automotive exhaust catalyst: highly active and selective NO reduction to N2 at low-temperatures†
Received
5th April 2021
, Accepted 25th May 2021
First published on 2nd June 2021
Abstract
A Pd catalyst (Pd/Ce0.7Zr0.3O2) in an electric field exhibits extremely high three-way catalytic activity (TWC: NO–C3H6–CO–O2–H2O). By applying an electric field to the semiconductor catalyst, low-temperature operation of TWC can be achieved even at 473 K by virtue of the activated surface-lattice oxygen.
Introduction
With the development of global motorization, urban air pollution caused by automobile emissions and the excessive combustion of oil products have become global problems. In recent years, the development of zero emission vehicles (ZEVs) such as battery electric vehicles (BEVs) and fuel cell electric vehicles (FCVs) has been progressing. Nevertheless, from the standpoint of infrastructure facilities and cruising range, the demand for fuel-burning vehicles such as hybrid vehicles (HVs) continues. With the widespread use of “e-fuel,” a novel concept fuel made from renewable H2 and captured CO2, it is expected that these engine-powered vehicles will continue to be used in the global market in the future. That progress notwithstanding, exhaust gas purification might become more difficult because of lower exhaust gas temperatures and tighter restrictions on hazardous gas emissions. In light of this situation, many investigations have been conducted to improve exhaust gas purification at low temperatures using electrically heated catalysts (EHC),1,2 plasma catalytic systems,3–5 and highly active three-way catalysts (TWC).6,7 However, the EHC method and plasma-catalyzed reaction require higher electric power consumption.3,4,8,9 In addition, TWCs require high temperatures to achieve high purification performance.10,11 We found from an earlier study that various catalytic reactions can proceed even at low temperatures when a DC electric field is applied to a semiconductor support.12–16 Since then, we have attempted to apply this finding to TWCs. In this paper, we propose a new catalytic TWC system using a palladium catalyst supported on ceria–zirconia (Ce0.7Zr0.3O2), which can function at temperatures as low as 473 K. We prepared a mixed oxide of Ce0.7Zr0.3O2 for a catalyst support which has suitable properties (i.e. electron conductivity and surface ion conductivity) for the catalytic reaction in the electric field. Catalyst preparation and reaction procedure are described in the ESI.†
Results and discussion
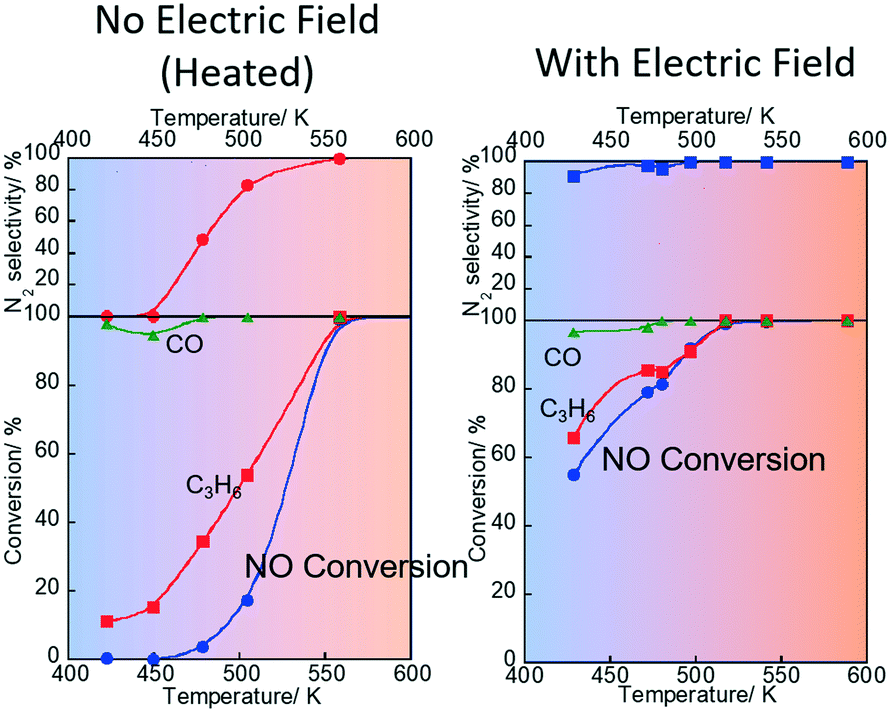
First, we conducted the three-way catalytic reaction (NO–C3H6–CO–H2O–O2) on an 0.5 wt% Pd/Ce0.7Zr0.3O2 catalyst (see the ESI† for the structural information of the prepared catalyst) with/without an electric field by 1.5 mA of direct current. The conversion of NO, C3H6, CO, and selectivity to N2 with/without the electric field are presented in Fig. 1. Application of the electric field brought very high values of NO, C3H6, and CO conversion and N2 selectivity, even at low measured temperatures (423–473 K), along with coexistence of O2 and H2O, as presented in Fig. 1(b). At 428.4 K, NO, C3H6, and CO conversion were 54.9%, 65.9%, and 96.8% respectively. Surprisingly, the selectivity to N2 was 91.3% with the electric field. In contrast, without the electric field, NO conversion and N2 selectivity were very low (almost 0%) at the low-temperature region (also see Fig. 1(a)). In these tests, the catalyst bed temperature was measured directly using a thermocouple attached to the catalyst to confirm the effects of Joule heating by the applied direct current on the catalytic activity. A great promotion with the electric field on the catalytic activity was confirmed. Therefore, the high activity/selectivity is not attributable to Joule heating. Additionally, the electric power consumption for applying the electric field to the catalyst bed was only up to 1.2 W at 428.4 K (see Table S1 in the ESI†). Therefore, the catalysis with the electric field enables a highly efficient three-way catalyst system, even at temperatures much lower than those of conventional systems.
 |
| | Fig. 1 NO, CO and C3H6 conversion and N2 selectivity over 0.5 wt% Pd/Ce0.7Zr0.3O2 under NO–C3H6–CO–O2–H2O reaction (NO: 2500 ppm, C3H6: 500 ppm CO: 3000 ppm, O2: 2500 ppm, H2O: 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm, Ar balance, SV: 72000 h−1) applying 1.5 mA direct current. 000 ppm, Ar balance, SV: 72000 h−1) applying 1.5 mA direct current. | |
This promotional effect by the electric field differs completely from electrically heated catalyst (EHC) and plasma reactions. It has been confirmed by in situ XAFS measurements that the local heating of the catalyst particles due to the Joule-heating by the application of electric field is negligible. To confirm whether the plasma effect can be ignored in this system, we conducted the plasma reaction on the same TWC gas composition. At 375 and 468 K, NO conversions using plasma reaction were only 5.6% and 7.4%, respectively. These values are very low compared to the catalytic activities in the electric field presented in Table 1. Therefore, the promotive effect for the TWC reaction using the electric field is much greater than the plasma reaction. Additionally, to confirm whether the high catalytic activity of TWC in the electric field derived from a direct NO decomposition, or not, NO decomposition activity tests were conducted while supplying only NO gas. The NO conversion values with no reductant (i.e. CO and C3H6 in this case) with the electric field were around 2.5% at each temperature. These results suggest that NO direct decomposition is unlikely to occur in this system, and suggest that the reaction between oxidants (NO, O2) and reductants (CO, C3H6), instead of NO direct decomposition, was promoted by application of the electric field: not by plasma, and not by the heat. We investigated the contribution of reductants (C3H6 and CO) to NO reduction with the electric field at low temperatures. For this purpose, we conducted NO–C3H6–O2–H2O and NO–CO–O2–H2O reactions with and without the electric field at high space velocity (SV = ca. 180000 h−1). Table 2 shows NO conversion with and without the electric field on each condition at a low-temperature region (catalyst-bed temperature: 420–500 K). On the NO–CO–O2–H2O condition, NO conversion was nearly 100% with and without the electric field. No marked difference was observed on the NO conversion. A slight increase in selectivity to N2 was observed by application of the electric field at 420 K. In contrast, a clear difference in NO conversion was apparent with and without electric field on the NO–C3H6–O2–H2O condition at 420–500 K. Moreover, drastic promotion of N2 selectivity was observed with application of the electric field in the NO–C3H6–O2–H2O condition: almost identically to the TWC condition. This result suggests that the NO–C3H6 reaction and CO oxidation are dominant reactions on the TWC condition with the electric field at low temperatures (420–500 K).
Table 1 NO conversion over 0.5 wt% Pd/Ce0.7Zr0.3O2 under NO–C3H6–CO–O2–H2O reaction (NO: 2500 ppm, C3H6: 500 ppm CO: 3000 ppm, O2: 2500 ppm, H2O: 7 vol%, Ar balance, total flow: 200 cc min−1) for various reactions
|
|
NO conversion/% |
| Plasma reaction |
5.6 (at 375 K) |
7.4 (at 468 K) |
|
| NO decomposition in the electric field |
2.7 (at 451 K) |
2.3 (at 532 K) |
2.5 (at 625 K) |
| Heated catalytic reaction |
0.0 (at 420 K) |
0.3 (at 449 K) |
0.26 (at 478 K) |
| Electric field reaction with reductants (3 mA) |
19.1 (at 414 K) |
16.5 (at 433 K) |
20.5 (at 457 K) |
Table 2 NO conversion and N2 selectivity over 0.5 wt% Pd/Ce0.7Zr0.3O2 under NO–C3H6–CO–O2–H2O reaction (NO: 2500 ppm, C3H6: 500 ppm CO: 3000 ppm, O2: 2500 ppm, H2O: 70000 ppm Ar balance, total flow: 200 cc min−1), NO–CO–O2–H2O reaction (NO: 2500 ppm, CO: 3000 ppm O2: 250 ppm, H2O: 70000 ppm Ar balance, total flow: 200 cc min−1) and NO–C3H6–O2–H2O reaction (NO: 2500 ppm, C3H6: 500 ppm, O2: 2500 ppm, H2O: 70000 ppm Ar balance, total flow: 200 cc min−1) with/without an electric field at 3 mA (denoted as EF)
| –Without EF– |
| Condition |
NO conversion (N2 selectivity)/% |
| 420 K |
475 K |
500 K |
| NO–CO–O2–H2O |
97.2 (5.2) |
100 (30.4) |
100 (96.2) |
| NO–C3H6–O2–H2O |
3.6 (0.9) |
3.4 (0.3) |
6.2 (5.4) |
| NO–C3H6–CO–O2–H2O (TWC) |
0.1 (0.0) |
0.27 (0.0) |
3.5 (13.4) |
| –With EF– |
| Condition |
NO conversion (N2 selectivity)/% |
| 420 K |
475 K |
500 K |
| NO–CO–O2–H2O |
95.8 (22.9) |
99.5 (32.0) |
100 (71.7) |
| NO–C3H6–O2–H2O |
14.4 (92.4) |
13.7 (100) |
17.7 (100) |
| NO–C3H6–CO–O2–H2O (TWC) |
13.7 (100) |
14.4 (100) |
23.0 (100) |
To elucidate the effect of coexistence O2 for TWC reaction, O2 partial-pressure-dependent tests were conducted under the NO–CO–O2–H2O and NO–C3H6–O2–H2O conditions with and without the electric field. As shown in Fig. S2 (ESI†), in the NO–CO–O2–H2O condition, the behavior of NO, CO and O2 conversion rates with changing O2 concentration was the same with and without the electric field. The NO conversion rate decreases and the CO reaction rate increases with increasing O2 concentration. This trend suggests that CO was consumed through the CO–O2 reaction rather than NO–CO reaction on Pd/Ce0.7Zr0.3O2. These results demonstrate that the electric field does not contribute to the NO–CO and CO–O2 reactions in the low-temperature region (420–500 K). In contrast, in the NO–C3H6–O2–H2O condition, a drastic difference is apparent in the dependence of the NO reaction rate on O2 concentration, as depicted in Fig. 2. Without the electric field, a negative dependence on the O2 concentration was observed for the NO reaction rate (Fig. 2 left). However, the NO reaction rate with the electric field positively depends on the O2 concentration of 0–1000 ppm. At 1000–1500 ppm O2, the NO reaction rate decreases considerably, which suggests that the NO reduction by C3H6 was prevented by the competitive adsorption of O2. It caused excess adsorption of O2 on Pd metal, which is the active site of the NO–C3H6 reaction. In contrast, O2 contributes to the positive effect on NO–C3H6 reaction at 0–1000 ppm O2. Burch et al. reported that O2 contributes to the NOx selective catalytic reduction by hydrocarbons (HC-SCR). NOx was reduced by hydrocarbons through these pathways (eqn (1)–(4)).10
| | | NO (g) + O2 (g) → NOx (ads) | (1) |
| | | CxHy (g) + O2 (g) → CxHyOz (ads) | (2) |
| | | NOx (ads) + CxHyOz (ads) → R–NCO (ads) | (3) |
| | | NOx (ads) + R–NCO (ads) → N2 (g) + CO2 (g) | (4) |
As those reactions show, NO and hydrocarbons were oxidized by O
2 (g) to form NO
2−, NO
3−, and C
xH
yO
z (partially oxidized species of hydrocarbons), which are the key intermediate species for HC-SCR. Subsequently, NO
x is reduced by C
xH
yO
z to N
2via R–NCO species. Higo
et al. also reported that the partially oxidized species of hydrocarbons (C
xH
yO
z) are an extremely important intermediate species for HC-SCR over Pd/perovskite catalyst.
11 The catalyst shows high NO
x reduction activity by virtue of the partially oxidized C
xH
yO
z which is produced by high-mobility lattice oxygen in the catalyst-support material. The oxygen species is expected to contribute to the formation of intermediates and to accelerate NO reduction by application of an electric field. We conducted
in situ DRIFTS measurements to elucidate details of the contribution of surface oxygen on the reaction in the electric field. The DRIFT spectra for Pd/Ce
0.7Zr
0.3O
2 catalyst in a C
3H
6 flow (3000 ppm C
3H
6 balanced Ar gas) are portrayed in
Fig. 3. Three peaks at 1596, 1440, and 1272 cm
−1 observed without the electric field (spectrum b) can be assigned to acetate and carbonate species.
17–20 This result demonstrates that C
3H
6 was oxidized by the active oxygen species on the surface of Pd/Ce
0.7Zr
0.3O
2 to form these oxygenate adsorbates. Then, after applying the electric field in the C
3H
6 flow, absorbance at 1950–1200 cm
−1 increased. New notable band observed at 1950–1775 cm
−1 were assigned to the vibration of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif)
O
21–24 (spectrum a). The change of spectrum shows that the oxygenate species are formed on the catalyst surface by application of the electric field. These findings suggest that the propylene is oxidized by the surface lattice oxygen of catalyst because no oxygen source exists in the atmosphere. The same IR measurements at 420–473 K were performed to elucidate the influence of Joule heating by the EF application (spectra c and d). No peak attributable to C
O vibration is found in these spectra, which demonstrates that C
3H
6 oxidation below 473 K proceeds only slightly on the Pd/Ce
0.7Zr
0.3O
2 catalyst without the electric field.
 |
| | Fig. 2 NO, C3H6 and O2 reaction rate, O2 conversion and N2 selectivity over 0.5 wt% Pd/Ce0.7Zr0.3O2 under NO–C3H6–O2–H2O reaction (NO: 2500 ppm, C3H6: 500 ppm O2: 0, 250, 500, 750, 1000 and 1500 ppm, H2O: 7 vol%, balance gas: Ar, total flow: 200 cc min−1) with/without the electric field (3 mA) at each temperatures (with: 373 K, without: 503 K). | |
 |
| | Fig. 3 DRIFT spectra of Pd/Ce0.7Zr0.3O2 during Ar + 3000 ppm C3H6 flow with (a) and without (b–d) electric field (3 mA) at each temperature. | |
Conclusions
This study investigated catalytic NO reduction with an electric field at low temperatures. Electric field promotion over 0.5 wt% Pd/Ce0.7Zr0.3O2 catalyst enables high activity, even at low temperatures (420–473 K) and even with coexisting O2 and H2O. For surface reactions with the electric field, the NO–C3H6 reaction is the dominant reaction on the three-way catalyst conditions in low-temperature regions (420–473 K). Furthermore, the NO reaction rate depends positively on O2 concentrations of 0–1000 ppm under a NO–C3H6–O2–H2O atmosphere. Application of the electric field promoted the formation of partially oxidized hydrocarbon (CxHyOz) using lattice oxygen in the catalyst support, as confirmed by in situ DRIFTS measurements. The activated surface lattice oxygen contributes to the formation of intermediates and accelerates NO reduction by the application of the electric field, even at low temperatures. This finding is expected to engender the development of highly efficient three-way catalyst systems.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- S. R. Khan, M. Zeeshan and S. Iqbal, Chem. Eng. Commun., 2018, 205, 680–688 CrossRef CAS.
- R. M. Heck and R. J. Farrauto, Appl. Catal., A, 2001, 221, 443–457 CrossRef CAS.
- S. Bröer and T. Hammer, Appl. Catal., B, 2000, 28, 101 CrossRef.
- B. M. Penetrante, R. M. Brusasco, B. T. Merritt and G. E. Vogtlin, Pure Appl. Chem., 1999, 71, 1829 CAS.
- M. A. Malik and K. H. Schoenbach, Int. J. Plasma Environ. Sci. Technol., 2011, 5(1), 50–57 Search PubMed.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- H. S. Gandhi, G. W. Graham and R. W. McCabe, J. Catal., 2003, 216, 433–442 CrossRef CAS.
- J. Gao, G. Tian and A. Sorniotti, Energy Sci. Eng., 2019, 7, 2383–2397 CrossRef CAS.
- W. Maus, R. Brück, R. Konieczny and A. Scheeder, MTZ Worldw., 2010, 71, 34–39 CrossRef.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283–303 CrossRef CAS.
- T. Higo, K. Ueno, Y. Omori, H. Tsuchiya, S. Ogo, S. Hirose, H. Mikami and Y. Sekine, RSC Adv., 2019, 9, 22721–22728 RSC.
- K. Murakami, Y. Tanaka, R. Sakai, Y. Hisai, S. Hayashi, Y. Mizutani, T. Higo, S. Ogo, J.-G. Seo, H. Tsuneki and Y. Sekine, Chem. Commun., 2020, 56, 3365–3368 RSC.
- M. Torimoto, S. Ogo, D. Harjowinoto, T. Higo, J.-G. Seo, S. Furukawa and Y. Sekine, Chem. Commun., 2019, 55, 6693–6695 RSC.
- A. Sato, S. Ogo, K. Kamata, Y. Takeno, T. Yabe, T. Yamamoto, S. Matsumura, M. Hara and Y. Sekine, Chem. Commun., 2019, 55, 4019–4022 RSC.
- K. Takise, A. Sato, K. Murakami, S. Ogo, J.-G. Seo, K. Imagawa, S. Kado and Y. Sekine, RSC Adv., 2019, 9, 5918–5924 RSC.
- R. Manabe, H. Nakatsubo, A. Gondo, K. Murakami, S. Ogo, H. Tsuneki, M. Ikeda, A. Ishikawa, H. Nakai and Y. Sekine, Chem. Sci., 2017, 8, 5434–5439 RSC.
- H. Abdulhamid, J. Dawody, E. Fridell and M. Skoglundh, J. Catal., 2006, 244, 169–182 CrossRef CAS.
- Y. Ji, T. J. Toops, J. A. Pihl and M. Crocker, Appl. Catal., B, 2009, 91, 329–338 CrossRef CAS.
- M. Haneda, Y. Kintaichi, M. Inaba and H. Hamada, Catal. Today, 1998, 42, 127–135 CrossRef CAS.
- W. S. Epling, C. H. F. Peden and J. Szanyi, J. Phys. Chem. C, 2008, 112, 10952–10959 CrossRef CAS.
- I. V. Yudanov, R. Sahnoun, K. M. Neyman, N. Rösch, J. Hoffmann, S. Schauermann, V. Johánek, H. Unterhalt, G. Rupprechter, J. Libuda and H. J. Freund, J. Phys. Chem. B, 2003, 107, 255–264 CrossRef CAS.
- H. Tiznado, S. Fuentes and F. Zaera, Langmuir, 2004, 20, 10490–10497 CrossRef CAS PubMed.
- M. Haneda, N. Bion, M. Daturi, J. Saussey, J. C. Lavalley, D. Duprez and H. Hamada, J. Catal., 2002, 206, 114–124 CrossRef CAS.
- T. Baidya, P. Bera, B. D. Mukri, S. K. Parida, O. Kröcher, M. Elsener and M. S. Hegde, J. Catal., 2013, 303, 117–129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00591j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Toru
Uenishi
b and
Yasushi
Sekine
a,
Toru
Uenishi
b and
Yasushi
Sekine