
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient depolymerization of lignins to alkylphenols using phosphided NiMo catalysts†
Jessi
Osorio Velasco
,
Ilse
van der Linden
,
Peter J.
Deuss
 and
Hero J.
Heeres
*
and
Hero J.
Heeres
*
Chemical Engineering Department, ENTEG, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: h.j.heeres@rug.nl
First published on 15th June 2021
Abstract
Greening up the chemical industry by using waste biomass streams as feed is a topic of high relevance. Residual lignins from for example the pulp and paper industry and second-generation bioethanol plants are interesting resources for the synthesis of biobased aromatics and alkylphenols. We here report experimental studies on the catalytic hydrotreatment of Kraft lignin to alkylphenols using non-precious metal, sulfur tolerant catalysts in the form of phosphided NiMo catalysts on different supports (AC, SiO2Al2O3, SiO2, MgO–Al2O3, and TiO2) in the absence of an external solvent. The catalysts were prepared by an incipient wetness impregnation method and characterized in detail (BET surface area, SEM, TEM, X-ray diffraction, and temperature-programmed desorption of NH3/CO2). Hydrotreatment experiments were carried out in a batch autoclave at a temperature of 400 °C, for 2 h and 100 bar initial H2 pressure. The lignin oils were analyzed extensively by GPC, GC-MS, GC×GC-FID, and elemental analysis. The highest monomer yield (51.8 wt% on lignin intake) was obtained with the NiMoP catalyst on SiO2 (5.6 wt% Ni, 9.1 wt% Mo and 5.9 wt% P), which is among the best reported in the literature so far. Of the monomers, alkylphenols are the dominant component group (30.6%), followed by aliphatics (8.1%) and aromatics (5.7%). Clear relations between support characteristics and performance were absent. The only exception is the support acidity, and apparently, intermediate acidity is required for best performance. The SiO2-supported NiMoP catalyst was also applied for the hydrotreatment of Lignoboost and Alcell lignin under the same reaction conditions. Whereas Lignoboost gave highly comparable results to Kraft lignin in terms of oil and monomer yield, Alcell lignin gave a considerably lower monomer yield (34.4 wt% on lignin intake). These results are rationalized by considering P/S exchange in the catalyst formulation during the reaction.
Introduction
Energy, transportation fuels, and chemicals obtained from fossil resources play an important role in our society. However, increasing levels of atmospheric CO2 combined with an anticipated decline in fossil resources has led to the realization that alternatives are required. Renewable energy is available from many sources (solar, wind, tidal, geothermal) whereas biobased chemicals to be used as input for the chemical industry are only accessible using a carbon-based source such as biomass.1 For the development of techno-economically viable value chains for biobased chemicals from biomass, the use of cheap biomass feeds is of pivotal importance. In this respect, lignin is considered an attractive possibility. It is a major component in lignocellulosic biomass and contains up to 40% of the biomass energy content.2 Major sources of lignin are the pulp and paper industry, processes that are typically designed to valorize the carbohydrate fraction of the lignocellulosic biomass. However, the lignin fraction is typically considered a by-product with only fuel value and is typically used for heat and/or power generation.2,3 Well-known lignins are Kraft and lignosulfonates, which contain about 1–2 wt% of S due to the use of sulfur-based reagents in the process. Approximately 55 million tons of such sulfur-containing lignins are produced annually. It is estimated that about 8 to 11 Mt per year of these lignins can be used to produce high-value aromatic platform chemicals like phenols or aromatics (benzene, toluene, xylenes) without affecting the operation of the paper mills. This requires the development of efficient technologies to depolymerize the condensed lignin structure and to selectively modify the substituent pattern (e.g. demethoxylation) of the aromatic units of the low molecular weight fragments. For this purpose, a broad spectrum of conversion routes has been developed on a lab scale and these can be classified based on temperature and pressure.4,5 Well-known examples are solvolysis, hydrothermal processing, pyrolysis, and catalytic hydrotreatments.6 The latter requires the use of catalysts and a hydrogen donor (e.g. hydrogen or hydrogen donor solvents like isopropanol). It is typically applied at elevated temperatures and when using molecular hydrogen, also at elevated pressures.Our interest is particularly focused on the catalytic hydrotreatment of lignins, as the use of catalysts allows steering of the product composition and maximizing the amount of low molecular weight alkylphenols and aromatics by rational catalyst design. Besides, an approach without the use of an external solvent is preferred to avoid the use of an (expensive) solvent recycling step in the process. As such, when using batch set-ups for preliminary catalyst studies, the lignin feed acts as the solvent as it is known to melt around 200 °C7 in the initial stage of the process, whereas, the low molecular weight products also act as solvent at prolonged batch times. In the case of continuous operation, the lignin can be dissolved in the product oil and fed to the reactor.8 Several heterogeneous catalysts have been used for the catalytic hydrotreatment of (Kraft) lignin using molecular hydrogen in the absence of an external solvent (Table 1). Typical conditions are hydrogen pressures between 30 and 200 bar and temperatures between 350 and 450 °C.
| Lignin source | Catalyst | T (°C) | Hydrogen pressure (bar) | Reaction time (h) | Oil yield (on lignin intake, %) | Monomer yield (on lignin intake, %) | Ref. |
|---|---|---|---|---|---|---|---|
| Kraft | Limonite, Geothite, FeS2, CoMo | 350–450 | 100 (initial) | 4 | 27–49 | 31 | 21 |
| Kraft | S–CoMo/S–NiMo on various supports | 400 | 100 (initial) | 4 | 28–55 (DCM soluble) | 26.4 | 3 |
| Kraft | NiMo/aluminosilica: Cr2O3, Pd/C, RANEY® Ni | 395–430 | 90–100 | 0.3–1.0 | 49–71 | 9.4 | 9 |
| Kraft/organosolv | Pd/C, RANEY® Ni, NiO–MoO3 | 350–420 | 30–120 (initial) | 0.25 | 15–81 | 23 | 8 |
| Technical lignins | Limonite | 400 | 100 (initial) | 4 | 22–41 | 29 | 22 |
| Kraft | (Ru, Pt, Pd, Rh) on AC and Al2O3 | 450 | 100 (initial) | 4 | 26–41 | 30 | 10 |
| Kraft | NiMoP/AC | 400 | 100 (initial) | 2 | 64.3 | 45.7 | 23 |
| Alcell lignin fractions | Ru/C | 400 | 100 (initial) | 4 | 70 | 18.3 | 11 |
| Alcell | Ru/C, Ru/Al2O3, Ru/TiO2, Pd/C, Pd/Al2O3, Pd/ZrO2, Cu/ZrO2 | 400 | 100 | 4 | 64–78 | 9.1 | 12 |
Noble metal catalysts such as supported Ru or Pd are active for the hydrotreatment of lignin.8–12 The highest monomer yields for Kraft lignin with these catalysts were about 30% on lignin intake (Table 1, entry 6) using Rh/Al2O3 as the catalyst. However, there is doubt about the stability of such catalysts when using sulfur-rich feeds like Kraft lignins.13 Sulfided NiMo and CoMo catalysts, often supported on Al2O3, are well-known hydrotreatment catalysts and are commonly used in the oil industry for hydrodesulfurization.14–17 They have also been tested for the catalytic hydrotreatment for Kraft lignin as sulfur tolerant catalysts and monomer yields of 26% were obtained (Table 1, entry 2) using sulfided NiMo/MgO–La2O3 catalyst at 350 °C during 4 h of reaction time and 100 bar of initial H2 pressure. Other examples of sulfur tolerant catalysts used for the reaction are Fe-based catalysts like limonite, goethite, and FeS2. The best results for Kraft lignin were a monomer yield of 31%, obtained when using limonite catalyst at 450 °C for a reaction time of 4 h. Recently, transition metal phosphide catalysts18,19 have been introduced for the catalytic hydrotreatment of Kraft lignin by our group3,20 and have shown promising catalyst performance. A series of mono and bimetallic Ni-, Mo-, and W-phosphides supported on activated carbon was tested and showed that the Mo containing phosphide catalysts exhibit better performance in terms of oil, char, and monomer yield than the W containing ones. At optimized reaction conditions for the NiMoP/AC (400 °C, 2 h batch time and a 10 wt% of catalyst loading) the monomer yield was 45.7% on lignin intake, showing the potential of this class of metal phosphides for the hydrotreatment of sulfur-rich lignins.
In this article, the effect of catalyst support types (AC, SiO2, TiO2, SiO2/Al2O3, MgO–La2O3) on the performance of NiMo/P catalysts for Kraft lignin hydrotreatment was evaluated. The NiMo/P catalysts were prepared using an incipient wetness impregnation method, characterized by XRD, TPD, TEM, and SEM, and tested for the hydrotreatment of Kraft lignin. The lignin oils and solid residue were analyzed by GC-MS, GC×GC, GPC, and elemental analysis. The results were rationalized by establishing catalyst support structure (BET surface area, acidity/basicity, etc.)-product yield relations. The best catalysts in terms of monomer yield were also tested for other lignin sources (Lignoboost and Alcell) to determine the scope of the approach. It was shown that the choice of the support has a significant impact on catalyst performance and that by using the right support, the non-precious NiMo/P catalyst can compete and even outperform the best catalysts reported in terms of monomer and alkylphenols yield.
Experimental section
Chemicals
All chemicals used in this study were of analytical grade and used without purification. Activated carbon (AC) support was purchased from Merck KGaA, Darmstadt. SiO2 (nanopowder 10–20 nm, ≥99.5%), TiO2 (nanopowder 21 nm, mixture of anatase and rutile phase ≥99.5%) and silica-alumina (grade 135, 6.5% Al) were obtained from Sigma Aldrich. Mg(NO3)2·6H2O (≥98%), La(NO3)3·6H2O (99.999%), Ni(NO3)2·6H2O (≥98.5%), (NH4)6Mo7O24·4H2O (≥99.0%), NH4H2PO4 (99.5%), K2CO3 (≥99.0%), KOH (≥85%), HNO3, (70%) and dimethyldisulfide (DMDS) were obtained from Sigma Aldrich. Dichloromethane (DCM), di-n-butylether, acetone, and THF were obtained from Boom B.V. Hydrogen (>99.99%) and 2% O2/Ar was purchased from Hoekloos.Indulin-AT (Kraft lignin) was from MWV specialty chemicals and was provided by the Wageningen University and Research Center, The Netherlands (Dr. R. Gosselink). Indulin-AT is a purified form of Kraft pine lignin. It is free of hemicellulosic material and has an ash content of 3 wt%. Lignoboost lignin was obtained from Innventia, kindly provided by the Biomass Technology Group BV, the Netherlands. It is a purified form of Kraft lignin and contains less ash (0.42 wt%) than conventional Kraft lignin. Alcell® lignin, produced by Repap, Canada from mixed hardwood, was kindly supplied by the Wageningen University and Research Center (WUR), The Netherlands (see Gosselink24 for details). Relevant properties of the various lignins are given in Table 2.
| Kraft | Lignoboost | Alcell | ||
|---|---|---|---|---|
| a Determined by elemental analyses at the University of Groningen. b Reported by Constant et al.26 c Reported by Mattsson et al.27 d Determined by GPC using THF as the solvent at the University of Groningen, samples are not fully soluble. e Determined by HSQC NMR, procedure reported previously,25 f Data from Hita et al.22 g Number of linkages per 100 aromatic C9 units. h Data from the provider's specification sheet. | ||||
| Elemental compositiona [wt%] | C | 61.61 | 64.08 | 64.30 |
| H | 5.91 | 5.91 | 5.94 | |
| N | 0.74 | 0.03 | 0.09 | |
| S | 1.54 | 1.54 | 0.10 | |
| M W [g mol−1] | 4290b | 1670c | 2580b | |
| (812)d | (933)d | (1228)d | ||
| S/G/H ratioe | 0/100/0 | 0/100/0 | 77/23/0e and 63/37/0f | |
| Linkage typesg | β-O-4 | 8–11 | 7 | 5–18 |
| β–β | 2–4 | 4 | 4–10 | |
| β-5 | 1–3 | 2 | 2–4 | |
| Ash contenth [wt%] | 3.00 | 0.42 | N/A | |
The different lignins were analyzed by NMR to obtain information on the S/G/H ratio (syringyl alcohol (S), guaiacyl/coniferyl alcohol (G), and hydroxyphenyl/coumaryl alcohol (H)) and type and number of aromatic linkages. The lignin was dissolved in d6-acetone and 2D proton heteronuclear single quantum coherence spectra (HSQC) were recorded on a Bruker Ascend 600 NMR spectrometer following a protocol reported previously.25
Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo:P = 1:1:2). The NiMoP catalysts with the SiO, Al2O3, TiO2, and MgO–La2O3 supports were prepared using a similar matter.
:Mo:P = 1:1:2). The NiMoP catalysts with the SiO, Al2O3, TiO2, and MgO–La2O3 supports were prepared using a similar matter.
Catalyst characterization
Scanning electron microscopy (SEM) images of the catalyst were obtained using a Philips XL30 ESEM with a point-source cathode of tungsten with a surface layer of zirconia (ZrO2). Electron micrographs were recorded at a working distance of 8.8–9.3 mm at a scanning voltage of 10 kV.Transmission electron microscopy (TEM) images of the catalysts were obtained using a Tecnai T20 electron microscope (FEI) with a high-angle annular dark-field (HAADF) scanning TEM (STEM) detector operated at 200 kV. Samples for TEM measurements were ultrasonically dispersed in ethanol and subsequently deposited on a mica grid coated with carbon.
BET surface area of the catalysts and the supports were determined from the nitrogen adsorption/desorption isotherms at 77 K, recorded using a Micrometrics ASAP 2020. The samples were degassed under a vacuum at 450 °C for 4 h. The surface area was measured using the Brunauer–Emmett–Teller (BET) method in the P/Po range of 0.05–0.25. Pore size distributions (PSD) were calculated from the desorption branch of the isotherms according to the Barrett–Joyner–Halenda (BJH) method. The total pore volume was estimated by single point desorption at P/P0 = 0.25. The micropore area and volume were calculated using the t-plot method.
Temperature-programmed desorption (TPD) of NH3 and CO2 was performed using a Micrometrics AutoChem II Chemisorption Analyzer. Approximately 100 mg of sample was placed in a quartz tube. This was first heated to 650 °C at 10 °C min−1 under a He flow. Then the sample was maintained at 650 °C under a He flow for 30 minutes for degassing, after which it was cooled to 100 °C. Subsequently, the sample was saturated for 60 min with NH3 or CO2 (10 vol% in He) at 50 ml min−1. The sample was purged with a He flow to remove excess NH3 or CO2. TPD measurements were performed by heating the samples from 100 to 600 °C at a heating rate of 10 °C min−1 using He as a carrier flow at 50 ml min−1. The NH3 or CO2 desorbed from the catalyst surface was detected by an online thermal conductivity detector, calibrated by known pulses of NH3 or CO2.
X-ray diffraction data of the catalysts were recorded on a Bruker D8 advance diffractometer using Cu Kα radiation (λ = 0.1544 nm) at 40 kV. XRD patterns were measured in reflection geometry in the 2θ range between 5° and 80° with a step size of 0.02°.
The elemental composition of fresh and spent catalyst was determined by inductively coupled plasma (ICP) on a PerkinElmer ICP-OES Optima 7000 DV apparatus using a solid-state CCD array detector. Before analysis, the samples were dissolved by boiling them for 10 minutes in a solution containing 75% (v/v) HNO3 (3 mM) and 25% (v/v) HCl (10 mM). Yttrium (10 ppm) and scandium (10 ppm) were used as internal standards, and Ar was used as the purge gas.
Catalytic hydrotreatment of lignins
The catalytic hydrotreatment reactions were performed in a 100 ml batch autoclave (Parr Instruments Co., stainless steel type 316). The reactor was equipped with an aluminum block containing electrical heating elements as well as channels allowing the flow of cooling water. The reactor content was stirred mechanically by a Rushton-type turbine with a gas-induced impeller. The temperature was monitored via a T-controller and the pressure during the reaction was monitored using a pressure indicator. The reactor was loaded with lignin (15 g) and catalyst (1.5 g, 10 wt% on lignin) and subsequently flushed with hydrogen at 50 bar three times, after which the reactor was checked for leakage at 150 bar. The pressure was subsequently reduced to 100 bar. The reactor content was stirred at 600 rpm and heated to 400 °C with a heating rate of about 10 °C min−1. When the reactor content reached a temperature of 100 °C, the stirring rate was increased from 600 to 1200 rpm. The reaction time was set to zero when the reactor content was heated to 400 °C. After a 2 h reaction, the cooling water was activated and the reactor was cooled to room temperature with a cooling rate of approximately 10 °C min−1. Then the pressure and temperature were recorded to determine the amount of gas present in the reactor. The reactor was depressurized and the gas phase was collected in a 3 L Tedlar gas bag to determine its composition. All reactions were carried out in duplicate. The final product in the reactor always consisted of three phases (oil, water, and solids), which were separated via a centrifugation-based work-up procedure (Fig. 1). The product slurry was transferred to a centrifugation tube and the product was centrifuged at 4500 rpm for 20 min. After centrifugation, the liquids were easily separated from the solids by decanting. The oil (abbreviated PO)/water mixture was allowed to settle for 2 h, which led to phase separation. Both layers were separated and weighed. The residual slurry in the reactor was washed off using DCM and acetone, the solids were collected by filtration using a filter paper. The filter paper was dried overnight at room temperature. The filtrate was concentrated by solvent removal overnight at room temperature. The resulting lignin oil from the filtrate is marked as reactor residue oil (RO). The solid residue at the bottom of the centrifugation tube was washed about 3 times with DCM (9 mL for each wash) using an ultrasound bath-centrifugation procedure. Then, the solids were removed from the tube by rinsing with acetone and filtered on filter paper. The filtrate and filter paper were left open overnight to evaporate the solvent. The resulting lignin oil is marked as centrifugation residue oil (ResO). | ||
| Fig. 1 Schematic overview of the experimental procedure for the catalytic hydrotreatment of lignin including product work-up. | ||
Oil, water, solid, gas yield, and mass balance closure were calculated based on lignin intake (wet basis), using eqn (1)–(5)
 | (1) |
 | (2) |
 | (3) |
 | (4) |
 | (5) |
Product analysis
The composition of the gas phase was analyzed using a GC-TCD Hewlett-Packard 5890 Series II GC equipped with a Poraplot Q Al2O3/Na2SO4 column and a molecular sieve (5 Å) column. The injector temperature was set at 150 °C and the detector temperature at 90 °C. The oven temperature was kept at 40 °C for 2 min, then heated up to 90 °C at 20 °C min−1 and kept at this temperature for 2 min. A reference gas was used to quantify the results (55.19% H2, 19.70% CH4, 3.00% CO, 18.10% CO2, 0.51% ethylene, 1.49% ethane, 0.51% propylene, and 1.5% propane).GC-MS analyses of the lignin oils were performed using a Hewlett Packard 5973 MSX attached to a Hewlett Packard 6890 GC equipped with a Restek Rxi-5Sil MS column (30 m × 0.25 mm × 0.25 μm), the injection volume was 1 μl and the injector temperature was set to 280 °C. The oven temperature was kept at 45 °C for 2 min, then heated to 280 °C with a heating rate of 10 °C min−1 and then kept at 280 °C for 5 min.
GC×GC-FID analysis was used for quantitative analysis of the lignin oils. It was performed using a trace GC×GC from Interscience equipped with a cryogenic trap system and two columns (a 30 m × 0.25 mm i.d. and a 0.25 μm film of RTX-1701 capillary column connected by a meltfit to a 120 cm × 0.15 mm i.d. and a 0.15 μm film Rxi-5Sil MS column). A dual-jet modulator was applied using carbon dioxide to trap the samples. Helium was used as the carrier gas (continuous flow of 0.8 ml min−1). The injector temperature and FID temperature were set at 280 °C. The oven temperature was kept at 40 °C for 5 min and then heated up to 280 °C at a rate of 3 °C min−1. The pressure was set at 70 kPa at 40 °C. The modulation time was 6 s. The oil samples for GC-MS and GC×GC-FID were diluted 30 times with THF and 530 ppm di-n-butyl ether as an internal standard. Detailed information about the quantification of the main groups is given in Boccichini et al.31 and Tables S1 and S2 in the ESI.†
GPC analysis of the samples (lignin and lignin oils) was performed using an HP1100 equipped with three MIXED-E columns (300 × 7.5 mm PL gel 3 μm) in series using a GBC LC 1240 RI detector. The following conditions were used: THF as the eluent at a flow rate of 1 ml min−1, a pressure of 140 bar, a column temperature of 40 °C, 20 μl injection volume with a 0.2 wt% sample concentration. Toluene was used as a flow marker. Polystyrene samples of different molecular weights were used as the calibration standards. Average molecular weight calculations were performed using the PSS WinGPC Unity software from Polymer Standards Service.
Elemental analyses (C, H, N, and S) were performed using a Euro Vector 3400 CHN–S analyzer. The oxygen content was determined by difference. All experiments were carried out in duplicate, and the average value is provided.
Results and discussion
Catalyst synthesis and characterization
The phosphided NiMo catalysts on different supports (AC, SiO2–Al2O3, SiO2, MgO–La2O3, and TiO2) were prepared using an incipient wetness impregnation procedure. The amounts of Ni, Mo, and P in the catalyst formulations were determined using ICP-OES and the results are given in Table 3. The actual values are slightly lower than based on catalyst component intakes (5.6 wt% Ni, 9.1 wt% Mo, and 5.9 wt% P (molar ratio of Ni:Mo:P of 1:1:2). Before use, the catalysts were reduced in an H2 atmosphere at 650 °C and passivated in an O2/Ar mixture at room temperature.
| Elemental composition of active species | Physisorption data for supports | Physisorption data for catalysts | Chemisorption data | |||||
|---|---|---|---|---|---|---|---|---|
| Ni | Mo | P | S BET | S pores | S BET | S pores | [μmol g−1] | |
| [wt%] | [m2 g−1]a | [m2 g−1]a | ||||||
| a Cumulative pore volume as determined using the BJH method. | ||||||||
| NiMoP/AC | 4.94 | 7.86 | 5.27 | 513 | 262 | 419 | 134 | 386 (NH3) |
| NiMoP/SiO2–Al2O3 | 5.11 | 8.24 | 6.04 | 497 | 555 | 269 | 250 | 733 (NH3) |
| NiMoP/SiO2 | 542 | 152 | 174 | 133 | 411 (NH3) | |||
| NiMoP/MgO–La2O3 | 4.92 | 8.14 | 5.99 | 85 | 90 | 58 | 58 | 304 (CO2) |
| NiMoP/TiO2 | 5.01 | 7.24 | 5.29 | 54 | 49 | 35 | 32 | 145 (NH3) |
The catalysts were characterized using N2 physisorption, SEM, TEM, XRD, and NH3/CO2 chemisorption. N2 physisorption data show that the surface area of the catalysts is considerably lower than the surface area of the original support, likely due to support coverage with metal phases.
The acidity/basicity of the catalysts was evaluated by temperature-programmed desorption of NH3 and CO2, respectively (Table 3, figures are given in the ESI,† Fig. S2). The SiO2–Al2O3 supported catalyst shows the highest acidity (733 μmol g−1), in line with literature data ([760 μmol g−1]30 and [630 μmol g−1]32), which is due to the presence of Al2O3 in the formulation.3,33–36 Of all the acidic supports, the TiO2-supported catalyst showed the lowest acidity (145 μmol g−1). Intermediate acidity was found for NiMoP supported on SiO2 and AC catalysts, both with a desorption peak in the temperature range of 120–450 °C. The latter is higher than reported for NiMo/AC3 probably due to differences in the type of AC used and catalyst loading. MgO–La2O3 is the only basic support in the series with a CO2 uptake of 304 μmol g−1 in the temperature range of 150–400 °C.
X-ray diffraction (Fig. S7, ESI†) was used to study the various metal phases in the catalysts. For the NiMoP on AC, SiO2, TiO2, the presence of a clear Ni2P phase was observed with peaks at 2θ values of 40, 44, 47, and 54°.37 For NiMoP/MgO–La2O3 a NiP phase was observed with a peak at a 2θ value of 43°.40 In the case of the SiO2, a MoP phase is also present.38 XRD analysis of the TiO2 support shows that the TiO2 nanoparticles are a mixture of rutile and anatase phase,39 in line with the support data provided by the supplier.
A representative TEM image for the AC supported catalyst is given in Fig. 2, TEM images for the other catalysts are given in the ESI† (Fig. S3). The TEM image for the NiMoP/AC shows a clear contrast between the metal particles and the support. The particle size shows a broad distribution from 4 to 25 nm (Fig. 2), with an average particle size of 15.5 nm. This value is higher than found for NiMoP/SiO235 (4–10 nm with an average particle size of 8.1 nm), likely due to differences in synthesis protocols and the type of silica used. Reliable data for metal nanoparticle sizes using TEM for the other supports could not be obtained. The SiO2–Al2O3 and SiO2 show metal nano-particles in the range of 20 nm, however, reliability is limited due to overlapping support particles.
 | ||
| Fig. 2 a) Representative TEM image and b) particle size distribution for NiMoP/AC determined by TEM analysis. | ||
Catalytic hydrotreatment of Kraft lignin using the NiMoP catalysts
Hydrotreatment reactions of Kraft lignin were performed in a batch reactor set-up at 400 °C for 2 h using the NiMoP catalysts on the different supports (AC, SiO2, SiO2–AlO3, TiO2, MgO–La2O3) without an external solvent. The initial hydrogen pressure was set at 100 bar at room temperature and was at max. 200 bar at the initial stage of the batch reaction. Consumed hydrogen was not replenished. All experiments were performed in duplicate and good repeatability was observed (see ESI† Table S2).For all experiments, the major product is a dark brown lignin oil with yields in the range of 54–68 wt% on lignin intake (Fig. 3). Besides, a separate water phase (10–20%), gas-phase (∼10 wt%), and a solid residue (2–15 wt%) were formed and quantified. Relevant data for the composition of the gas phase are given in the ESI† (Table S3) and it typically contains CH4 and CO2, besides unconverted H2. Mass balance closure is very good and ranges between 88–93 wt%. Carbon balance closures range between 84–102% (see Fig. S4†). Hydrogen consumption is between 0.34 and 0.43 NL g−1 of lignin (see ESI,† Fig. S5).
 | ||
| Fig. 3 Mass balance and yields for Kraft lignin using NiMoP catalysts on different supports. Yields are given on wt% on lignin intake. Reaction conditions: 15 g Kraft lignin, 1.5 g catalyst, initial hydrogen pressure of 100 bar at RT, 2 h at 400 °C, 1200 RPM. | ||
The highest oil yield of 68.1 wt% on lignin was obtained with the silica-supported catalyst, whereas the worst result (54.5 wt%) was found for the catalyst with the basic MgO/La2O3 support (Fig. 3). The latter is due to the formation of significant amounts of solids (14.8 wt%), indicative of the occurrence of repolymerisation reactions during the hydrotreatment.
The desired oil (PO) was analyzed in detail by GC×GC-FID, GPC, and CHNS elemental analysis. The elemental compositions are given in the form of a van Krevelen plot on a dry basis (Fig. 4). Not all lignin oils are depicted in the van Krevelen diagram, due to the high viscosity of some of the oils which excluded accurate water determinations by Karl Fisher titrations.
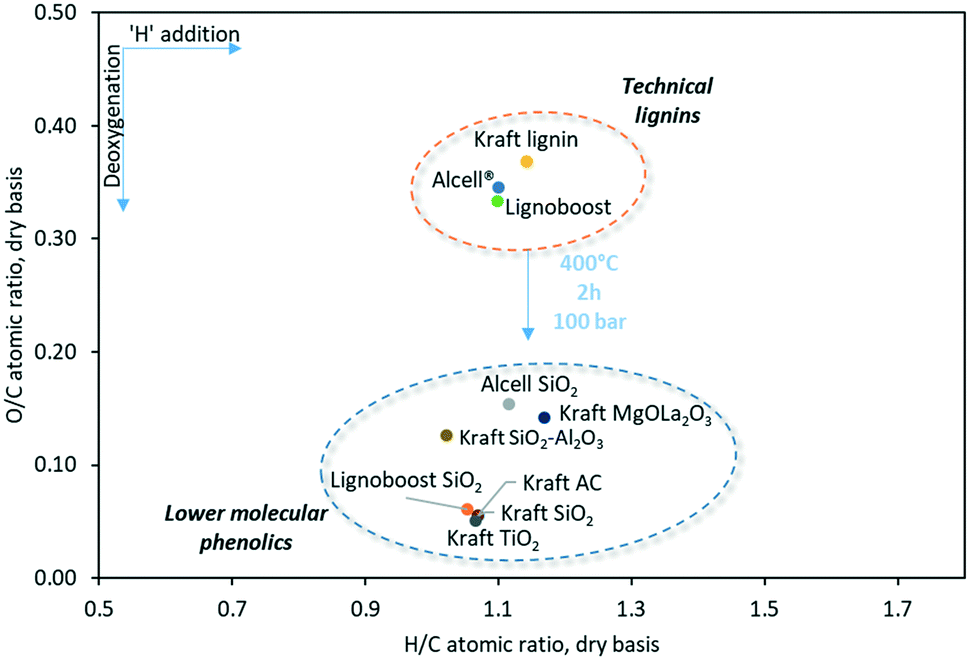 | ||
| Fig. 4 Van Krevelen plot for product oils obtained by the catalytic hydrotreatment of technical lignins using various catalysts. Reaction conditions: 15 g Kraft lignin, 1.5 g catalyst, initial hydrogen pressure of 100 bar at RT, 2 h @ 400 °C, 1200 RPM. | ||
The atomic H/C ratios of the lignin oils from Kraft lignin are all in a narrow range (1.06–1.07). Compared to the Kraft lignin feed, the oxygen content of the oils is reduced considerably, indicative of the occurrence of hydrodeoxygenation reactions. Besides, it emphasizes that quantitative oxygen removal is possible under these conditions. Moreover, the O/C and H/C ratios of the oils are in between that of typical aromatics and alkylphenols. This indicates the presence of considerable amounts of such components in the oils, supported by GC techniques (vide infra). Elemental analysis data also reveal the presence of some sulfur in the lignin oils (∼0.3 wt%), although the amounts are substantially lower than for the Kraft lignin feed (1.54 wt%). Sulfur removal can be due to hydrodesulfurization with the concomitant formation of H2S and/or selective transfer of S to the solids. The latter was shown to occur to a significant extent and it was shown that the solids contain 4–6 wt% sulfur (vide infra).
The composition of the product oils was determined using GC techniques. The list of identified components in the lignin oil as determined by GC-MS for a representative oil from NiMoP/SiO2 is provided in the ESI† (Table S1). The main products were alkylphenols and cyclic alkanes, examples are phenol, cresol, p-ethylphenol, 4-propyl phenol, and cyclohexane.
To quantify the amounts of the main organic compound classes (aromatics, phenolics, alkanes, etc.), the lignin oils were analyzed using GC×GC-FID. The GC detectable components were categorized into eight distinct regions, see Fig. 5 for a representative example.
 | ||
| Fig. 5 Typical GC×GC-FID chromatogram for a product oil obtained using NiMoP/SiO2 showing the various component groups: 1 = cyclic alkanes, 2 = linear/branched alkanes, 3 = aromatics, 4 = ketones/alcohols, 5 = naphthalenes, 6 = volatile fatty acids, 7 = guaiacols, 8 = alkylphenols, 9 = catechols, A = di-n-dibutylether (internal standard) and B = BHT (stabilizer in THF). | ||
The total monomer yield after the hydrotreatment reaction (as determined by GC×GC-FID) ranges from 31–52% on lignin intake (Fig. 6), implying that a large proportion of the oils is of low molecular weight and GC detectable. As such, it shows that the catalysts indeed are capable of depolymerizing Kraft lignin to a significant extent. The highest amount of monomers was obtained with the SiO2 supported catalyst, whereas the worst results were obtained using the more acidic SiO2/Al2O3 support. The major component groups in the oils are alkylphenols, followed by alkanes and aromatics (Fig. 6).
 | ||
| Fig. 6 Monomer yields (wt% on lignin intake) for Kraft lignin using NiMoP catalysts on different supports, as determined by GC×GC-FID. Reaction conditions: 15 g Kraft lignin, 1.5 g catalyst, initial hydrogen pressure of 100 bar at RT, 2 h @ 400 °C, 1200 RPM. | ||
Substantial depolymerization of the Kraft lignin was also confirmed by GPC measurements on the lignin oils (Fig. 7). Sharp peaks are observed in the region of 80–160 g mol−1 for all lignin oils, indicating the presence of substantial amounts of low molecular weight monomers, in line with the GC×GC data. The Mw values are between 360 and 450, which is considerably lower than reported for Kraft lignin (4.2–5.3 kDa).26,40
 | ||
| Fig. 7 GPC data for the Kraft lignin oils obtained with NiMoP on various supports. | ||
To determine the fate of the S in the Kraft lignin feed upon hydrotreatment, the S-content of the solid residue, water phase, and lignin oil were determined after the reaction by CHNS analysis. The percentage of S in the various phases based on the S-intake in the feed is given in Fig. 8. It can be concluded that the oils still contain small amounts of S (0.05–0.8 wt% S), likely in the form of sulfonated groups on lignin fragments. The amount of S in the water phase is typically the lowest (0.3–0.6 wt% S). When considering the solids, the S content varies considerably and is between 4–6 wt%, with the highest amount for the catalyst with the basic support (NiMoP/MgO–La2O3). XRD analysis (ESI, Fig. S8†) reveals that the S in the solids is not only S incorporated in the char but is also present in the form of metal sulfides (e.g. MoS2, and Ni7S6). The overall S-balance closure is far from quantitative and well below 60% (see Fig. 8). The most likely explanation is the formation of substantial amounts of S-containing gas phase components like H2S. Though evident from the smell, we were not able to quantify the amount accurately with our GC methods. This also explains the large amounts of S in the solids for the NiMoP/MgOLa2O3 catalysts, which is basic, and likely absorbs considerable amounts of acidic H2S.
 | ||
| Fig. 8 Sulfur distribution for the solids, oil, and water phase based on intake S in the feed. | ||
 | ||
| Fig. 9 Proposed reaction network for Kraft lignin hydrotreatment using NiMoP catalysts (adapted from Kloekhorst et al.50). | ||
 | ||
| Fig. 10 Support effect on oil and monomer yield (wt% on lignin intake) for Kraft lignin hydrotreatment using supported NiMoP catalysts. | ||
Here, the catalysts are categorized according to their acidity, with the basic MgOAl2O3 and the left and the most acidic SIO2/Al2O3 at the right. When aiming for high oil and monomer yields, it appears that the catalyst with medium acidity (SiO2) performs best, whereas basic and more acidic supports are less favourable. The latter is likely due to the formation of substantial amounts of char when using the more acidic SiO2/Al2O3 support (4.7 wt%) compared to SiO2 (2.0 wt%), implying that supports with a high acidity promote repolymerisation. This is in agreement with findings in literature data.3,32 Apparently, a too-low acidity (and even basicity) also has a negative effect on the depolymerization activity of the catalysts and result in the formation of lower amounts of monomers (Fig. 10). These findings suggest that the liquid phase yield and composition is a delicate balance between depolymerization and re-polymerization which apparently may be tuned by the acidity of the support. However, other factors may also play a role, for example metal particle sizes (not considered as proper TEM images of some of the catalyst could not be obtained) and changes in the active phases of the phosphided catalyst in combination with S-rich feeds by S-incorporation (see below).
The results in this study show that the NiMoP/SiO2 catalyst is very effective in depolymerizing lignin and gives the best performance when considering both oil and monomer yield when compared to literature data (Table 1). Additionally, the results indicate that desulfurization of the lignin occurs, giving lignin oils with a notable low sulfur content (0.2 wt%, Table S4†) and low solid yields, showing the potential of a solvent-free hydrotreatment with phosphides catalyst for depolymerization and desulfurization of Kraft lignin.
Analysis of the spent catalyst
To gain insights into the structure of the best catalyst (NiMoP/SiO2) after the batch reaction, the residue was analyzed using elemental analyses (P and S content) and XRD. Elemental analyses showed that the solid residues after catalytic hydrotreatment contained 4–6 wt% sulfur (Table S5, ESI†), indicating that between 6 and 38% of the sulfur intake ends up in the solid residue (Fig. 8). Elemental analysis (ICP) of the solid residue indicates that there is still 1–4 wt% phosphorus present in the spent catalyst, though it is not possible to determine leaching levels as the spent catalysts are covered with char, an inevitable reaction product. The X-ray diffractogram of fresh and spent NiMoP/SiO2 are given in the ESI† (Fig. S8). Peaks corresponding to metal phosphide phases are still present in the spent catalyst. Moreover, new peaks appear at 2θ values 14, 31, 33, 38, 51, and 53°, corresponding to molybdenum and nickel sulfides.41–44 This implies that at least part of the phosphorus in the catalyst formulation is replaced by sulfur. These findings suggest that the exchange of phosphorus and sulfur occurs in the catalyst formulation during hydrotreatment. This may result in the formation of (partly) sulphided NiMo species, which are also known to be active for catalytic hydrotreatment.17,18,45,46 An overview of hydrotreatment studies of Kraft lignin using both sulphided and phosphided catalysts is given in Fig. S10.† It clearly shows that sulphided catalysts are also active, though performance seems to be somewhat lower than for the phosphided ones. However, a comparison is hampered because not all data were obtained at similar conditions and experimental set-ups.The possibility of P–S exchange is also supported by literature data. For instance, when using Ni2P catalysts for HDS reactions (e.g. using 4,6-dimethyl dibenzothiophene as the substrate), it was found by elemental analyses that a substantial amount of S accumulates on the catalyst.45,47–49 The use of in situ X-ray absorption fine structure (XAFS) indeed showed the presence of Ni-S bonds in the catalyst formulation after the reaction, indicating P–S exchange and the formation of surface phosphosulfide (NiPS) species. XRD measurements were reported and showed a clear peak of a mixed NiPS phase at 2θ = 32.3°. XRD spectra for spent NiMoP/SiO2 indeed show a clear peak at this value (Fig. S8†), further supporting the idea that S is incorporated in the catalyst structure during the reaction. Besides, it shows that quantitative exchange of S with P does not occur, in line with the elemental analyses data. To gain further insights into the P/S exchange process on catalyst performance, and for instance, to determine the time scale of P/S exchange, model studies will be required and these are currently in progress.
Catalytic hydrotreatment of other lignin types with NiMoP on SiO2
To explore the scope for the depolymerization of lignins using phosphide catalysts, the catalyst with the highest performance (NiMoP/SiO2) regarding oil and monomer yields was used for two other types of lignins in the form of Alcell (AL), a well-known organosolv lignin, and Lignoboost® (LB), a purified form of Kraft lignin. The elemental composition of the different lignins was determined by CHN–S analyses and is given in Table 2. Kraft and Lignoboost lignin both contain 1.5 wt% sulfur, whereas the sulfur content is by far lower for Alcell lignin. Another major difference is the amount of OMe groups on the aromatic nuclei, which is by far higher for Alcell lignin (mainly S units) compared to the other ones (mainly G units).22The reaction conditions were identical to the Kraft hydrotreatment experiments, viz a 2 h batch time at 400 °C, with 100 bar initial hydrogen pressure and a stirring rate of 1200 rpm. All lignins gave lignin oil as the main product, with oil yields between 65.6–72.5 wt% on lignin intake and monomer yields between 34 and 52% (Fig. 11). Hydrodeoxygenation of the lignin feedstock leads to the formation of a water phase in a yield of ∼10 wt% on lignin intake. Additionally, some permanent gases (9–12%) were formed, with the highest gas yield for Alcell lignin. This is likely due to the higher amount of –OMe groups for Alcell lignins, which are known to be converted to gas-phase components during catalytic hydrotreatment. This finding is in line with previous research in our group.22 The amount of solids formed was very low (∼2 wt%) indicating a low extent of repolymerization of the oxygenated structures. The mass balance closures are satisfactory and range from 88 to 92%. Hydrogen consumption is between 0.33 and 0.43 Nl g−1 of lignin.
 | ||
| Fig. 11 Mass balance and product yields for selected lignins using NiMoP/SiO2 catalyst. Yields are given on wt% on lignin intake. Reaction conditions: 15 g lignin, 1.5 g catalyst, initial hydrogen pressure of 100 bar at RT, 2 h at 400 °C, 1200 RPM. | ||
The lignin oil was analyzed by GC×GC-FID, GPC, and elemental analysis. Analysis by GC×GC-FID showed a comparable monomer yield for the hydrotreated Kraft and Lignoboost lignin (51.8 and 51.5%, respectively, Fig. 12). Surprisingly, the monomer yield is considerably lower for Alcell lignin, with a 34.4 wt% monomer yield on lignin intake. In each case, alkylphenols are the predominant class of monomers present in the lignin oil, accompanied by linear alkanes, aromatics, naphthalenes, and dihydroxybenzenes.
 | ||
| Fig. 12 Monomer yields (wt% on lignin intake) for selected lignins using NiMoP catalyst on SiO2, as determined by GC×GC-FID. Reaction conditions: 15 g lignin, 1.5 g catalyst, initial hydrogen pressure of 100 bar at RT, 2 h @ 400 °C, 1200 RPM. | ||
The elemental composition of the oils was determined by CHN–S elemental analysis and the results are given in a van Krevelen diagram in Fig. 4. The oxygen of the oils (7–11%) is by far lower than the feed (28–30%), indicative for substantial though not quantitative deoxygenation. As such, all lignin oils show a considerably reduced O/C ratio. GPC analysis of the lignin oils shows a sharp peak in the monomer region (80–160 g mol−1) for all lignin oils (Fig. S6†). The molecular weight of the oil after hydrotreatment is the highest for Alcell lignin.
All in all, Kraft and Lignoboost lignin show very comparable results regarding yields and oil composition. This is not surprising, as Lignoboost is a purified version of Kraft lignin and thus contains lower amounts of impurities (sugar residues) and ash. However, according to the results provided here, these impurities have little or no influence on the hydrotreatment reaction with the NiMoP/SiO2 catalyst.
The hydrotreatment reaction using Alcell lignin gave a by far lower monomer yield (34.4%) than the reactions with Kraft or Lignoboost lignin. This is rather surprising as the molecular weight of Alcell lignin is lower than that of Kraft/Lignoboost (Table 2), and thus less depolymerization is required for a substantial reduction in molecular weight. So far we do not have a sound explanation for these differences in yields when comparing Alcell and Kraft/Lignoboost lignin. A possible explanation is the absence of S in the Alcell lignin, which may affect catalyst performance, particularly when considering that P/S exchange occurs on the catalyst (vide supra). Indeed, the X-ray diffractogram of NiMoP/SiO2 after Alcell hydrotreatment (Fig. S9†) is similar to the fresh one and shows no peaks corresponding to metal sulfides. Very preliminary, these differences in performance between Alcell on the one hand and Kraft/Lignoboost on the other could indicate that mixed sulphided/phosphided catalysts are more active than the phosphided ones. Additional experiments were carried out using Alcell lignin in the presence of DMDS to prove that S has a beneficial effect on the activity of phosphided NiMo catalysts. Unfortunately, inconclusive results were obtained. However, the experiments with a sulfur free Alcell lignin feed using the NiMoP/SiO2 catalyst clearly reveals that the phosphided catalysts are active for the depolymerization of lignins and that the presence of S is beneficial though not essential.
Conclusions
We here show that sulfur tolerant, non-precious metal catalysts in the form of phosphided NiMo catalysts on different supports are highly active for Kraft lignin hydrotreatment in the absence of a solvent. Several NiMo/P catalysts on different supports were prepared, characterized, and tested in a batch set-up and product yield and compositions were determined. Best results when considering oil (68.1 wt%) and monomer yield (51.8 wt%) were obtained using a moderately acidic SiO2-supported catalyst. These yield/composition data are the highest reported so far in the literature for a solvent-free catalytic hydrotreatment of Kraft lignin. The scope of the reaction was extended by testing Lignoboost® and Alcell lignin. Analysis of the reaction products showed that Lignoboost lignin gave very comparable results to Kraft lignin, not surprisingly as Lignoboost lignin is a purified form of Kraft lignin. Remarkably, however, an organosolv lignin (Alcell) gave a lower monomer yield after hydrotreatment, possibly due to the absence of S in the Alcell. Catalysts characterization studies on spent catalysts reveal that phosphorus in the catalyst formulation is partially replaced by sulfur when using sulfur-containing Kraft or Lignoboost lignin as a feedstock. This finding implies that the actual active species are not only phosphided catalysts mixed P/S or even sulfided catalysts may be active and affect product yields and composition.Author contributions
I. vd. L. carried out most of the experimental work, J. O. V. supervised the project and prepared the manuscript with input from H. J. H. Both H. J. H. and P. J. D. supervised the research and contributed to the final version of the manuscript.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
We thank Dr. R. Gosselink from the Wageningen University and Research Center, The Netherlands for providing the Inulin-AT lignin. We acknowledge Qingqing Yuan from the University of Groningen for help with acquiring the TEM images for the catalysts. We also thank the Colombian Ministry of Science, Technology, and Innovation (Minciencias) for financial support with the educational grant no. 756-2016 for abroad Ph.D. funding and to the project “Strategy of transformation of the Colombian energy sector in the horizon 2030” supported with the call 788 of Minciencias Scientific Ecosystem (contract number FP44842-210-2018).References
- E. de Jong, H. Stichnothe, G. Bell, H. Jørgensen, I. de Bari, J. van Haveren and J. Lindorfer, Bio-Based Chemicals A 2020 Update Bio-Based Chemicals, 2020 Search PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Lignin Valorization: Improving Lignin Processing in the Biorefinery, Science, 2014, 344, 6185 CrossRef PubMed.
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Solvent free depolymerization of Kraft lignin to alkyl-phenolics using supported NiMo and CoMo catalysts, Green Chem., 2015, 17, 4921–4930 RSC.
- D. Knezevic, Hydrothermal Conversion of Biomass, PhD Thesis, University of Twente, 2009 Search PubMed.
- B. Joffres, D. Laurenti, N. Charon, A. Daudin, A. Quignard and C. Geantet, Conversion thermochimique de la lignine en carburants et produits chimiques: Une revue, Oil Gas Sci. Technol., 2013, 68, 753–763 CrossRef CAS.
- B. Joffres, D. Laurenti, N. Charon, A. Daudin, A. Quignard and C. Geantet, Thermochemical conversion of lignin for fuels and chemicals: a review, Oil Gas Sci. Technol., 2013, 68, 753–763 CrossRef CAS.
- A. Kloekhorst, Biobased Chemicals from Lignin, PhD thesis, University of Groningen, 2015 Search PubMed.
- D. Meier, R. Ante and O. Faix, Catalytic hydropyrolysis of lignin: Influence of reaction conditions on the formation and composition of liquid products, Bioresour. Technol., 1992, 40, 171–177 CrossRef CAS.
- A. Oasmaa, R. Alén and D. Meier, Catalytic hydrotreatment of some technical lignins, Bioresour. Technol., 1993, 45, 189–194 CrossRef CAS.
- I. Hita, P. J. Deuss, G. Bonura, F. Frusteri and H. J. Heeres, Biobased chemicals from the catalytic depolymerization of Kraft lignin using supported noble metal-based catalysts, Fuel Process. Technol., 2018, 179, 143–153 CrossRef CAS.
- A. Kloekhorst and H. J. Heeres, Catalytic hydrotreatment of Alcell lignin fractions using a Ru/C catalyst, Catal. Sci. Technol., 2016, 6, 7053–7067 RSC.
- A. Kloekhorst and H. J. Heeres, Catalytic Hydrotreatment of Alcell Lignin Using Supported Ru, Pd, and Cu Catalysts, ACS Sustainable Chem. Eng., 2015, 3, 1905–1914 CrossRef CAS.
- W. Mu, H. Ben, A. Ragauskas and Y. Deng, Lignin Pyrolysis Components and Upgrading—Technology Review, BioEnergy Res., 2013, 6, 1183–1204 CrossRef CAS.
- Y.-C. Lin, C.-L. Li, H.-P. Wan, H.-T. Lee and C.-F. Liu, Catalytic Hydrodeoxygenation of Guaiacol on Rh-Based and Sulfided CoMo and NiMo Catalysts, Energy Fuels, 2011, 25, 890–896 CrossRef CAS.
- A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, CoMo Sulfide-Catalyzed Hydrodeoxygenation of Lignin Model Compounds: An Extended Reaction Network for the Conversion of Monomeric and Dimeric Substrates, J. Catal., 2012, 285(1), 315–323 CrossRef CAS.
- S. Mukundan, L. Atanda and J. Beltramini, Thermocatalytic cleavage of C-C and C-O bonds in model compounds and kraft lignin by NiMoS2/C nanocatalysts, Sustainable Energy Fuels, 2019, 3, 1317–1328 RSC.
- S. G. Parto, J. M. Christensen, L. S. Pedersen, A. B. Hansen, F. Tjosås, C. Spiga, C. D. Damsgaard, D. B. Larsen, J. Duus and A. D. Jensen, Liquefaction of Lignosulfonate in Supercritical Ethanol Using Alumina-Supported NiMo Catalyst, Energy Fuels, 2019, 33, 1196–1209 CrossRef CAS.
- T. Wada, K. K. Bando, T. Miyamoto, S. Takakusagi, S. T. Oyama and K. Asakura, Operando QEXAFS studies of Ni2P during thiophene hydrodesulfurization: direct observation of Ni-S bond formation under reaction conditions, J. Synchrotron Radiat., 2012, 19, 205–209 CrossRef CAS.
- B. Dhandapani, S. Ramanathan, C. C. Yu, B. Frühberger, J. G. Chen and S. T. Oyama, Synthesis, characterization, and reactivity studies of supported Mo2C with phosphorus additive, J. Catal., 1998, 176, 61–67 CrossRef CAS.
- Z. Li, Q. Liu, Y. Shi, Z. Yao, W. Ding and Y. Sun, Novel synthesis of a NiMoP phosphide catalyst in a CH4–CO2 gas mixture, Dalton Trans., 2019, 48, 14256 RSC.
- S. Agarwal, R. K. Chowdari, I. Hita and H. J. Heeres, Experimental Studies on the Hydrotreatment of Kraft Lignin to Aromatics and Alkylphenolics Using Economically Viable Fe-Based Catalysts, ACS Sustainable Chem. Eng., 2017, 5, 2668–2678 CrossRef CAS PubMed.
- I. Hita, H. J. Heeres and P. J. Deuss, Insight into structure–reactivity relationships for the iron-catalyzed hydrotreatment of technical lignins, Bioresour. Technol., 2018, 267, 93–101 CrossRef CAS PubMed.
- R. K. Chowdari, S. Agarwal and H. J. Heeres, Hydrotreatment of Kraft Lignin to Alkylphenolics and Aromatics Using Ni, Mo, and W Phosphides Supported on Activated Carbon, ACS Sustainable Chem. Eng., 2019, 7(2), 2044–2055 CrossRef CAS PubMed.
- R. J. A. Gosselink, Lignin as a Renewable Aromatic Resource for the Chemical Industry, PhD thesis, Wageningen University, Wageningen, 2011 Search PubMed.
- D. S. Zijlstra, C. A. Analbers, J. de Korte, E. Wilbers and P. J. Deuss, Efficient Mild Organosolv Lignin Extraction in a Flow-Through Setup Yielding Lignin with High β-O-4 Content, Polymers, 2019, 11(12), 1913 CrossRef CAS PubMed.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. De Peinder, R. Boelens, D. S. Van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, New insights into the structure and composition of technical lignins: A comparative characterisation study, Green Chem., 2016, 18, 2651–2665 RSC.
- C. Mattsson, S. I. Andersson, T. Belkheiri, L. E. Åmand, L. Olausson, L. Vamling and H. Theliander, Using 2D NMR to characterize the structure of the low and high molecular weight fractions of bio-oil obtained from LignoBoost™ kraft lignin depolymerized in subcritical water, Biomass Bioenergy, 2016, 95, 364–377 CrossRef CAS.
- R. Kishore, M. L. Kantam, J. Yadav, M. Sudhakar, S. Laha and A. Venugopal, Pd/Mg-La mixed oxide catalyzed oxidative sp2 CH bond acylation with alcohols, J. Mol. Catal. A: Chem., 2013, 379, 213–218 CrossRef CAS.
- P. Yang, H. Kobayashi, K. Hara and A. Fukuoka, Phase Change of Nickel Phosphide Catalysts in the Conversion of Cellulose into Sorbitol, ChemSusChem, 2012, 5, 920–926 CrossRef CAS PubMed.
- R. H. Bowker, B. Ilic, B. A. Carrillo, M. A. Reynolds, B. D. Murray and M. E. Bussell, Carbazole hydrodenitrogenation over nickel phosphide and Ni-rich bimetallic phosphide catalysts, Appl. Catal., A, 2014, 482, 221–230 CrossRef CAS.
- P. Bocchini, G. Galletti, S. Camarero and A. Martinez, Absolute quantitation of lignin pyrolysis products using an internal standard, J. Chromatogr. A, 1997, 773, 227–232 CrossRef CAS.
- B. Jiang, J. Gong, J. Zhang, F. Li, J. Zhang, Y. Liu, Y. Chen and H. Song, Highly Active Ni2P Catalyst Supported on Core–Shell Structured Al2O3@TiO2 and Its Performance for Benzofuran Hydrodeoxygenation, Ind. Eng. Chem. Res., 2017, 56, 12038–12045 CrossRef CAS.
- A. S. Piskun, Catalytic Conversion of Levulinic Acid to -Valerolactone Using Supported Ru Catalysts: From Molecular to Reactor Level, PhD thesis, University of Groningen, 2016 Search PubMed.
- S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, Sustainable production of acrolein: investigation of solid acid-base catalysts for gas-phase dehydration of glycerol, Green Chem., 2007, 9(10), 1130–1136 RSC.
- Z. Pan, R. Wang, Z. Nie and J. Chen, Effect of a second metal (Co, Fe, Mo and W) on performance of Ni2P/SiO2 for hydrodeoxygenation of methyl laurate, J. Energy Chem., 2016, 25, 418–426 CrossRef.
- A. Ramesh, P. Tamizhdurai, K. Suthagar, K. Sureshkumar, S. Theres and K. Shanthi, Intrinsic role of pH in altering catalyst properties of NiMoP over alumino-silicate for the vapour phase hydrodeoxygenation of methyl heptanoate†, New J. Chem., 2019, 43, 3545 RSC.
- B. Jiang, J. Gong, J. Zhang, F. Li, J. Zhang, Y. Liu, Y. Chen and H. Song, Highly Active Ni2P Catalyst Supported on Core−Shell Structured Al2O3@TiO2 and Its Performance for Benzofuran Hydrodeoxygenation, Ind. Eng. Chem. Res., 2017, 56(42), 12038–12045 CrossRef CAS.
- X. Liang, D. Zhang, Z. Wu and D. Wang, The Fe-promoted MoP catalyst with high activity for water splitting, Appl. Catal., A, 2016, 524, 134–138 CrossRef CAS.
- K. Thamaphat, P. Limsuwan and B. Ngotawornchai, Phase Characterization of TiO 2 Powder by XRD and TEM, 2008, vol. 42 Search PubMed.
- R. Gosselink, J. Van Dam, de P. Wild, J. J. Wouter, T. Bridgewater, H. J. Heeres, E. Scott, J. Sanders, J. Van Dam, P. De Wild, W. Huijgen, T. Bridgwater, D. Nowakowski, E. Heeres, A. Kloekhorst, E. Scott and J. Sanders, Valorisation of lignin–Achievements of the LignoValue project, 2011 Search PubMed.
- E. Rodríguez-Castellón, A. Jiménez-López and D. Eliche-Quesada, Nickel and cobalt promoted tungsten and molybdenum sulfide mesoporous catalysts for hydrodesulfurization, Fuel, 2008, 87, 1195–1206 CrossRef.
- M. Grilc, G. Veryasov, B. Likozar, A. Jesih and J. Levec, Hydrodeoxygenation of solvolysed lignocellulosic biomass by unsupported MoS2, MoO2, Mo2C and WS2 catalysts, Appl. Catal., B, 2015, 163, 467–477 CrossRef CAS.
- A. Olivas, J. Cruz-Reyes, V. Petranovskii, M. Avalos and S. Fuentes, Synthesis and characterization of nickel sulfide catalysts, J. Vac. Sci. Technol., A, 1998, 16, 3515–3520 CrossRef CAS.
- R. Silva-Rodrigo, H. C. Jimenez, A. Guevara-Lara, J. A. A. Melo-Banda, H. Castillo Jimenez, A. Guevara-Lara, J. A. A. Melo-Banda, A. Olivas Sarabia, A. I. Reyes de la Torre, F. Morteo Flores and A. Castillo Mares, Synthesis, characterization and catalytic properties of NiMoP/MCM41-γAl2O3 catalysts for DBT hydrodesulfurization, Catal. Today, 2015, 250, 2–11 CrossRef CAS.
- S. T. Oyama, X. Wang, Y. K. Lee, K. Bando and F. G. Requejo, Effect of phosphorus content in nickel phosphide catalysts studied by XAFS and other techniques, J. Catal., 2002, 210, 207–217 CrossRef CAS.
- S. T. Oyama, in Journal of Catalysis, Academic Press Inc., 2003, vol. 216, pp. 343–352 Search PubMed.
- J. H. Kim, X. Ma, C. Song, Y. K. Lee and S. T. Oyama, Kinetics of two pathways for 4,6-dimethyldibenzothiophene hydrodesulfurization over NiMo, CoMo sulfide, and nickel phosphide catalysts, Energy Fuels, 2005, 19, 353–364 CrossRef CAS.
- S. J. Sawhill, K. A. Layman, D. R. Van Wyk, M. H. Engelhard, C. Wang and M. E. Bussell, Thiophene hydrodesulfurization over nickel phosphide catalysts: Effect of the precursor composition and support, J. Catal., 2005, 231, 300–313 CrossRef CAS.
- T. Wada, K. K. Bando, S. T. Oyama, T. Miyamoto, S. Takakusagi and K. Asakura, Operando Observation of Ni2P Structural Changes during Catalytic Reaction: Effect of H2S Pretreatment, Chem. Lett., 2012, 41, 1238–1240 CrossRef CAS.
- A. Kloekhorst, J. Wildschut and H. J. Heeres, Catalytic hydrotreatment of pyrolytic lignins to give alkylphenolics and aromatics using a supported Ru catalyst, Catal. Sci. Technol., 2014, 4, 2367–2377 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00588j |
| This journal is © The Royal Society of Chemistry 2021 |