
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical investigation of the side-chain mechanism of the MTO process over H-SSZ-13 using DFT and ab initio calculations†
Michal
Fečík
 a,
Philipp N.
Plessow
*a and
Felix
Studt
ab
a,
Philipp N.
Plessow
*a and
Felix
Studt
ab
aInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: philipp.plessow@kit.edu
bInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, Engesserstrasse 18, 76131 Karlsruhe, Germany
First published on 20th April 2021
Abstract
The side-chain mechanism of the methanol-to-olefins process over the H-SSZ-13 acidic zeolite was investigated using periodic density functional theory with corrections from highly accurate ab intio calculations on large cluster models. Hexa-, penta- and tetramethylbenzene are studied as co-catalysts for the production of ethene and propene. The highest barrier, both of ethene and propene formation, is found for the methylation of the side-chain towards the formation of an ethyl or isopropyl group. All other barriers are found to be substantially lower. This leads to a clear selectivity for ethene since the elimination of ethene with a rather low barrier competes with methylation towards propene which requires a barrier that is more than 100 kJ mol−1 higher.
Introduction
The production of light olefins such as ethene and propene from methanol or dimethyl ether is experiencing growing interest1 as methanol can be produced from carbon dioxide and hydrogen as part of our future sustainable energy scenario.2 The methanol-to-olefins (MTO) process is typically catalysed by acidic zeotypes such as H-ZSM-5, H-SSZ-13 and H-SAPO-34,1,3 at temperatures in the range of 350–400 °C. The use of H-SAPO-34, for example, can lead to selectivities for light olefins (ethene and propene) exceeding 80%.3–5The mechanistic details of the MTO process have been subject to extensive experimental6–16 and theoretical studies.6,16–33 The two main concepts that have been established in recent years are the hydrocarbon pool (HCP) mechanism34–36 and the dual cycle concept.37–39 Within the olefin cycle of the HCP, olefins convert methanol to olefins autocatalytically, where olefins are repeatedly methylated to higher olefins that are subsequently cracked to lighter olefins.28,40–56 In the dual cycle concept (see Scheme 1) aromatic species also play a role in the production of light olefins, and ethene and propene in particular.6,8–14,30 This is highly interesting both from a conceptual but also practical point of view, as the selectivity towards lighter olefins might significantly differ for these two cycles.37,38 For the aromatic cycle, in addition to the methylation of aromatics,57–65 there are two main mechanistic proposals, the side-chain and the paring mechanisms shown in Scheme 1.7,17,18,66–68 In the paring mechanism, the C6 ring is contracted to a C5 ring, shaving off the olefin. The side-chain mechanism, on the other hand, involves gem-methylation of highly methylated aromatics (e.g. hexamethylbenzene) ultimately resulting in a terminal double bond that can be further methylated yielding an ethyl or isopropyl group, the ‘side-chain’. After methyl-shifts, this side-chain can be eliminated as an olefin (ethene or propene) and the aromatic co-catalyst is restored.
 | ||
| Scheme 1 Dual cycle concept inspired by mechanisms proposed in the literature.17,66 The left side shows the olefin cycle where olefins are repeatedly methylated and eventually cracked. The right side shows two proposed mechanisms of the aromatic cycle – the side-chain and the paring mechanism. In the case of the side-chain mechanism, two production cycles are distinguished by colour – ethene (blue) and propene (red). A more detailed pathway of the side-chain mechanism of the aromatic cycle is shown in Fig. 1(a). | ||
Theoretical studies of the MTO process have been extensively employed to shed light on the details of the reaction mechanism, including the HCP and dual cycle concepts.6,16–30 The production of light olefins via aromatic co-catalysts and the paring and side-chain mechanism has been investigated theoretically for H-SZM-5 and H-SAPO-34.17,20,21,27,28 For both of these zeolites, it was found that the paring mechanism is energetically less favourable than the side-chain mechanism, mostly due to high barriers for olefin elimination and the regeneration of polymethylbenzene (pMB) as the intermediate.20,21,27,28
In this work, we focus on the side-chain mechanism (see Scheme 1 and Fig. 1(a)) to produce ethene and propene, using H-SSZ-13 as the catalyst. As co-catalysts, we investigate hexamethylbenzene (HMB) and pentamethylbenzene (PMB) adsorbed at the acid site within the pore of H-SSZ-13. Additionally, the rate-determining step is also investigated for tetramethylbenzenes (TMBs) as the co-catalyst. This choice is motivated by our earlier work that identified HMB, PMB, and TMBs as the thermodynamically most stable species adsorbed in H-SSZ-13 under typical reactions of the MTO process.65 Similar observations have been made for the methylation of aromatics in H-ZSM-5.61 Methylation is studied for methanol acting as the reactant for both, the concerted and the stepwise methylation pathway.
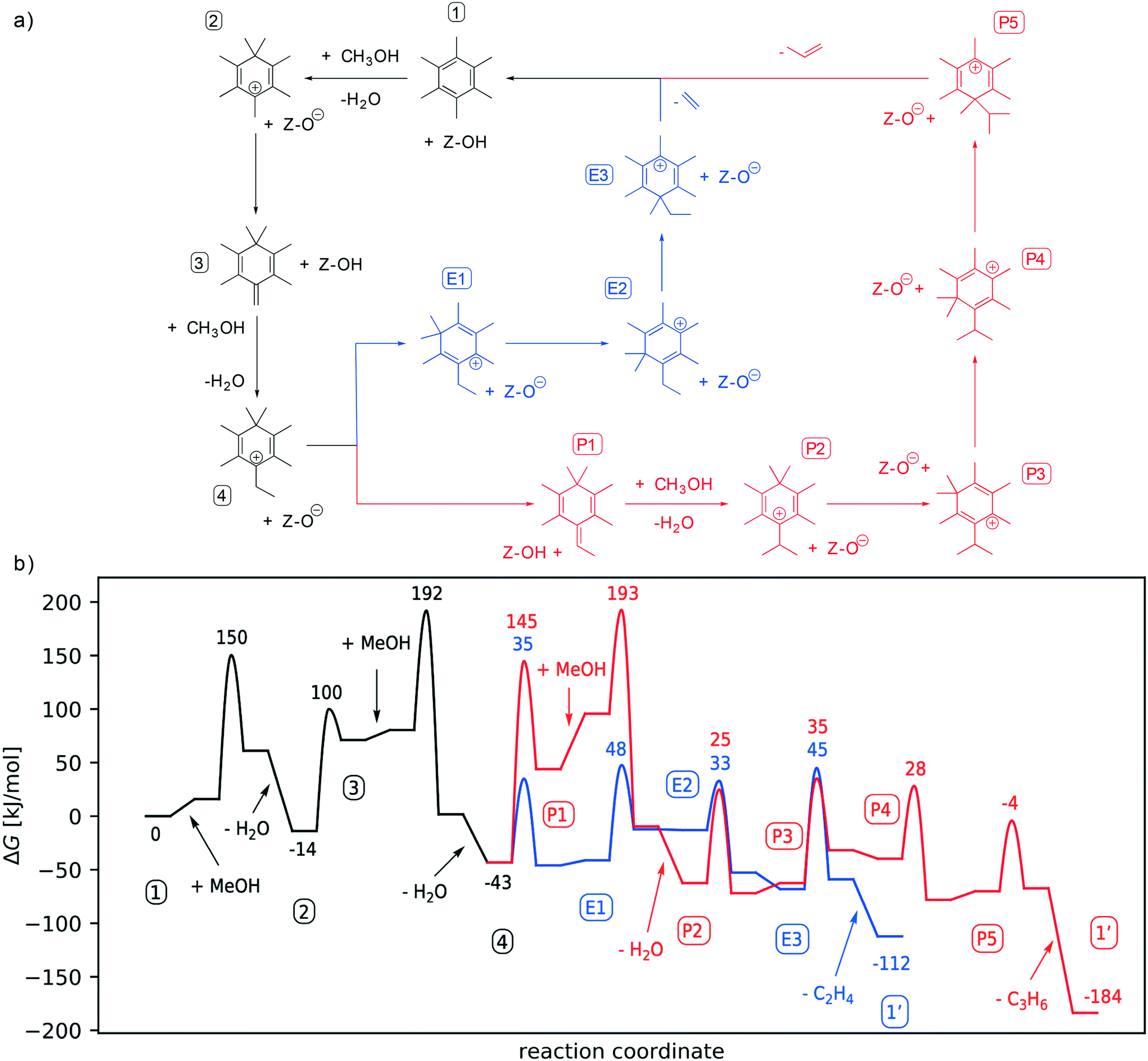 | ||
| Fig. 1 (a) Schematics of the various reaction steps of the studied side-chain methylation of the MTO process with HMB acting as the co-catalyst. After intermediate (4) the pathway can proceed to produce ethene (E, marked in blue) or propene (P, marked in red). (b) The Gibbs free energy profile of the side-chain mechanisms using HMB adsorbed at the acid site of H-SSZ-13 as the co-catalyst and methylation by methanol via the concerted mechanism. All free energies are in kJ mol−1, relative to adsorbed hexamethylbenzene (1) and are shown at 400 °C and a reference pressure of 1 bar. All indices (numbers inside boxes) and colours are the same as in (a). | ||
Computational details
As in previous work,65 all periodic DFT calculations were carried out using the projector-augmented-wave (PAW) method69 with the PBE-D3 functional70–72 with zero damping for dispersion corrections as implemented in the Vienna ab initio simulation package (VASP) in version 5.4.1.73,74 To perform the calculations, the atomic simulation environment (ASE)75 was used. The plane-wave kinetic energy cutoff was set to 400 eV. Transition state searches were performed using automated relaxed potential energy surface scans (ARPESS).76 Harmonic force constants were calculated from a central finite difference scheme. For the vibrational analysis, only a part of the zeolite – the involved oxygen atom with the Brønsted proton and the adjacent Si and Al atoms – together with the reactants inside the pore were considered. All transition states were verified to contain only a single imaginary frequency corresponding to the transition vector of the according reaction. Thermal corrections were obtained via the harmonic oscillator and the rigid rotor approximation at a temperature of 400 °C and a reference pressure of 1 bar.As described in previous studies, higher level methods are necessary for an accurate description of reaction barriers.77,78 As in the previous work,65 a hierarchical cluster approach77,79–84 is used to remedy limitations of DFT calculations. For this, highly accurate domain-based local pair natural orbital coupled cluster (DLPNO-CCSD(T))85,86 calculations together with complete basis set (CBS) extrapolation based on DLPNO-MP2 calculations were used for the correction of the periodic DFT energies obtained with a plane-wave basis set (EPBCPBE-D3) using the following formula:
| E = EPBCPBE-D3 + E46TDLPNO-CCSD(T)/DZ + ΔEMP2/CBS − E46TPBE-D3, |
| ΔEMP2/CBS = EDLPNO-MP2/CBS − E46TDLPNO-MP2/DZ. |
The lattice parameters of the unit cell of H-SSZ-13 (CHA framework) were kept fixed at a = 13.625 Å, b = 13.625 Å, and c = 15.067 Å as optimized in the previous work.78 This same unit cell was used for the H-SSZ-13 zeolite containing one Brønsted acid site that was introduced to the framework by substituting one Si atom by Al per the unit cell resulting in the Si/Al ratio of 35/1.
Results
As shown in Scheme 1, and also discussed in the literature,8,10–13,103 highly methylated aromatics are thought to play the dominant role for the side-chain mechanism. This finding is also supported by recent work showing that heavily methylated aromatics are the most stable species under relevant reaction conditions.61,65 While the detailed mechanism of the formation of aromatics from olefins is still controversially discussed in the literature,104–106 we note that in H-SSZ-13 aromatics are effectively trapped in the cavities due the small size of the pores connecting the cavities. Diffusion barriers of aromatics within H-SSZ-13 have been reported to be significantly higher than 200 kJ mol−1.107 We therefore assume that the present aromatic co-catalysts can neither enter nor leave the pores, but remain adsorbed at the acid site. The methylation of benzene in H-SSZ-13 with both methanol and DME has been extensively studied in earlier work.65 Gibbs free energy barriers for methylation have been calculated to be on the order of 150 kJ mol−1 with the thermodynamics favouring tetra- to hexamethylbenzene. Due to steric repulsion with larger aromatics, the thermodynamically most stable species have been identified to be tetramethyl-, pentamethyl-, and hexamethylbenzene. We chose hexamethylbenzene as our starting point for the investigation of the side-chain mechanism, due to its high symmetry, which reduces the number of possible isomers.Fig. 1 shows the detailed Gibbs free energy profile of the side-chain mechanism with co-catalyst HMB adsorbed at the acid site of H-SSZ-13 (HMB*). HMB* and methanol in the gas-phase are the starting point of the catalytic cycle and all free energies, including those of transition states, are given relative to this. Only the concerted pathway is depicted here, as the stepwise mechanism exhibits higher free energy barriers (see ESI†). Fig. 2 depicts the most relevant corresponding transition states of the calculated reaction mechanism. Note that not all methyl shift TS are shown as they are similar in nature.
 | ||
| Fig. 2 Relevant transition states (TS) of the side-chain mechanism of the MTO process catalyzed by HMB adsorbed on the acid site of H-SSZ-13. Frame colours and indices correspond to those shown in Fig. 1(a). Only a single methyl shift is shown due to their similarity. All distances are given in pm. The active site of H-SSZ-13 and the reactants are highlighted in the following manner: Al = blue, O = red, Si = yellow, C = brown, H = black. | ||
Starting from HMB, the first step is gem-methylation, for which – in the particular case of HMB – there is only one isomer (structure 2). Deprotonation of structure 2 at one of the methyl groups leads to the cross-conjugated intermediate 3. The deprotonation can take place in ortho- or para position (deprotonation in meta-position would lead to a biradical). We consider the para-position here since the other isomers turned out to be less favourable (see ESI†). Intermediate 3 can now be methylated at the terminal double bond to generate an ethyl group. This methylation has an overall barrier height of 206 kJ mol−1 when referenced to gas-phase methanol and the heptamethylbenzenium cation and is the rate-limiting step in ethene production. To eliminate ethene, first the additional methyl group in the gem-methylated position needs to be shifted to the carbon atom which is substituted with the ethyl group. Since the ethyl group is initially in para-position to the gem-methylated position (structure 4), this requires three successive methyl shifts, from E1, over E2 to E3. From E3, ethene formation occurs through dissociation of an ethyl cation with simultaneous deprotonation of the ethyl cation (see Fig. 2 for the atomic structure). This completes the catalytic cycle, yielding HMB*, the neutral acid site and ethene. The barriers for methyl shifts are all below 50 kJ mol−1 and the final barrier for elimination of ethene is 45 kJ mol−1 when referenced to HMB*.
It is therefore clear from the Gibbs free energy diagram that methylation of the cross-conjugated intermediate 2 requires by far the highest barrier. We have therefore investigated this transition state in more detail, looking at different orientations of the adsorbate and at different isomers. The transition state for the ortho-isomers was found to be higher by 30 kJ mol−1 at the PBE-D3 level of theory (see ESI†). We also calculated this barrier for the stepwise mechanism, where methylation proceeds via a surface-methoxy-species (SMS) that is formed upon methylation of the active site. The free energy barrier for the para isomer is calculated to be 214 kJ mol−1 when referenced to HMB*, and thus is 22 kJ mol−1 higher than the barrier for concerted methylation (see ESI†). We attribute this to steric repulsion since formation of the SMS is spatially more demanding compared to MeOH directly reacting with the intermediate.
We will now discuss the pathway for the production of propene. Starting from intermediate 4, deprotonation at the ethyl group gives the cross conjugated intermediate P1, differing from intermediate 3 only in the additional methyl group. Similar to the methylation of the double bond of 2, the analogous methylation of P1 gives an isopropyl group. Liberation of propene now proceeds in the same manner as for ethene, through three subsequent methyl shifts (P2–P5) followed by dissociation and deprotonation of the isopropyl cation which yields HMB*, propene and the neutral acid site.
Based on the free energy diagram in Fig. 1, one can clearly deduce a high kinetic selectivity for ethene formation over propene formation since the barriers for ethene formation are about 100 kJ mol−1 lower. This is partially due to the higher free energy of transition state TS(P1–P2) relative to 4 (236 kJ mol−1) compared to the 206 kJ mol−1 for TS(3–4) relative to the heptamethylbenzenium cation (structure 2) for ethene formation. The main reason, however, is that the additional methylation required for propene formation requires intrinsically a higher barrier when compared to the methyl shifts and the elimination of the olefin.
Having identified the main characteristics of the side-chain mechanism and the most relevant reaction steps, we will now turn our attention to the influence of the aromatic co-catalyst. As mentioned earlier, thermodynamics strongly favour HMB, PMB and TMB over their less methylated counterparts under relevant reaction conditions.65 We hence considered the most relevant reaction barriers with pentamethylbenzene (PMB) and tetramethylbenzene as the co-catalyst, see Fig. 3 for an illustration.
 | ||
| Fig. 3 Simplified Gibbs free energy diagram comparing barriers for forward methylation (black) increasing the total number of methyl groups attached to a given pMB, barriers for geminal methylation (red) of the considered pMBs leading towards initiation of the side-chain mechanism, and the rate-determining step of the corresponding side-chain mechanisms (blue) as identified above. The corresponding structures are shown schematically below the diagram. All energies are referenced to adsorbed HMB and are given in kJ mol−1 and were obtained for 400 °C. Barriers for forward methylations were taken from our previous study.65 | ||
For PMB, there are four possible isomers as there are four distinguishable positions where the methyl group is replaced by a hydrogen. Out of these four, only the most stable structure for the first rate-determining step was further considered. In this case, the methylation of the cross-conjugated structure 3 is accompanied by a barrier of 198 kJ mol−1 (referenced to gem-methylated PMB, the equivalent of structure 2) and the methylation P1 requires 217 kJ mol−1 (referenced to the equivalent of structure 4). Hence, HMB and PMB mediated side-chain mechanisms differ by 2 kJ mol−1 and 29 kJ mol−1 for these relevant reaction steps, respectively. For tetramethylbenzene as the co-catalyst, we find for the TS(3–4) a barrier height of 192 kJ mol−1 for durene, 210 kJ mol−1 for iso-durene and 202 kJ mol−1 for prehnitene, all referenced to the corresponding adsorbed tetramethylbenzenes. This suggests that both the activity and the selectivity for ethene is very similar for the tetra-, penta- and hexamethyl benzenes in H-SSZ-13.
A comparison of the DFT-computed (PBE-D3) free energy diagram with the coupled cluster corrected data is shown in Fig. S4.† Relative to neutral states, barriers are underestimated significantly by PBE-D3, as discussed previously.77 However, the barriers relative to cationic precursors are much less affected. Since the most stable state before side-chain methylation in Fig. 1 is the heptamethyl benzenium cation (structure 2), this means that the overall barrier is untypically accurate with PBE-D3 (197 vs. 206 kJ mol−1).
While the intrinsic selectivity for ethene production is expected to be high for the side-chain mechanism, it is important to realize that ethene can be methylated to propene thus changing the ethene-to-propene ratio. The side-chain mechanism has some similarities with the olefin cycle that we have studied previously:40 After a few methylation steps, of the olefin (or the aromatic), an olefin is split off, leaving behind an olefin (or an aromatic). The general conceptual difference is that the olefin cycle is autocatalytic, while the aromatic cycle is not. In terms of the energetics, we have found that methylation barriers are similar for olefins and aromatics.40,65 For the olefin cycle, favorable cracking reactions (involving only tertiary cations) require barriers that are lower than the methylation barriers of light olefins. On the contrary, the highest barrier for the side-chain mechanism, which is the methylation of the cross-conjugated intermediate, requires a barrier that is clearly higher than that for methylation of either aromatics or olefins. Our results are in agreement with previous DFT-calculations for H-SSZ-13, H-SAPO-34 and H-ZSM-5, which predict the same rate-limiting step and very high overall barriers for side-chain methylation (182 to 279 kJ mol−1).21,26 Similarly, comparatively lower barriers were reported for the olefin cycle.21,26,28
A comparison of the olefin and aromatic cycle in terms of reaction rates is rather difficult as this would require kinetic modeling. This is complicated by the autocatalytic nature of the olefin cycle and the fact that the side-chain mechanism requires the presence of an aromatic, which, in the case of H-SSZ-13, cannot leave the zeolite cavity due the small pore size and will therefore also influence the olefin cycle in the occupied cavity. We therefore stress that the above analysis is only qualitative and that a quantitative analysis requires advanced beyond steady-state kinetics108–114 that take the spatial and temporal changes in the reactor into account, including the formation and retention of aromatics. For a complete picture, additionally, the full aromatic cycle including the paring mechanism needs to be considered.
Conclusions
We have investigated the side-chain mechanism of the aromatic cycle of the hydrocarbon pool for H-SSZ-13 using DFT and highly accurate DLPNO-CCSD(T) calculations. For hexamethylbenzene, the entire cycle was investigated and the highest barrier (206 kJ mol−1) for ethene formation was found for the methylation of a cross-conjugated intermediate, leading to the formation of an ethyl group. The subsequent steps, methyl shifts and the eventual elimination of ethene were found to have significantly lower barriers. The side-chain mechanism is predicted to have a very high selectivity for ethene, because the additional methylation step required for the formation of an isopropyl side-chain is more than 100 kJ mol−1 higher than that for ethene elimination. It is important to note the ratio of ethene and propene is not only determined by the outcome of the side-chain mechanism. Instead, ethene also can be directly methylated to propene and both olefins may also be formed from the paring mechanism of the aromatic cycle as well as through the olefin cycle.In addition to hexamethylbenzene, we have investigated pentamethylbenzene and the tetramethylbenzenes – durene, isodurene and prehnitene. For these co-catalysts, only the rate limiting step identified for hexamethylbenzene was investigated. The corresponding barriers were generally found to be relatively high, on the order of 200 kJ mol−1 and show only small differences in reactivity between the investigated aromatics in H-SSZ-13.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge support by the state of Baden-Württemberg bwHPC (bwunicluster and JUSTUS, RV bw16G001 and bw17D011). Financial support from the Helmholtz Association is also gratefully acknowledged.Notes and references
- U. Olsbye, S. Svelle, M. Bjorgen, P. Beato, T. V. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed
.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS
- J. Q. Chen, A. Bozzano, B. Glover, T. Fuglerud and S. Kvisle, Catal. Today, 2005, 106, 103–107 CrossRef CAS
- J. J. Spivey, Chem. Eng. Commun., 1991, 110, 123–142 CrossRef CAS
- D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, V. Van Speybroeck, M. Waroquier and J. F. Haw, Angew. Chem., Int. Ed., 2008, 47, 5179–5182 CrossRef CAS
- T. Mole, J. A. Whiteside and D. Seddon, J. Catal., 1983, 82, 261–266 CrossRef CAS
- B. Arstad and S. Kolboe, J. Am. Chem. Soc., 2001, 123, 8137–8138 CrossRef CAS PubMed
- B. Arstad and S. Kolboe, Catal. Lett., 2001, 71, 209–212 CrossRef CAS
- A. Sassi, M. A. Wildman, H. J. Ahn, P. Prasad, J. B. Nicholas and J. F. Haw, J. Phys. Chem. B, 2002, 106, 2294–2303 CrossRef CAS
- W. Song, J. F. Haw, J. B. Nicholas and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 10726–10727 CrossRef CAS
- M. Bjørgen, F. Bonino, S. Kolboe, K.-P. Lillerud, A. Zecchina and S. Bordiga, J. Am. Chem. Soc., 2003, 125, 15863–15868 CrossRef PubMed
- M. Bjørgen, U. Olsbye, D. Petersen and S. Kolboe, J. Catal., 2004, 221, 1–10 CrossRef
- M. Bjørgen, U. Olsbye, S. Svelle and S. Kolboe, Catal. Lett., 2004, 93, 37–40 CrossRef
- W. Dai, X. Wang, G. Wu, N. Guan, M. Hunger and L. Li, ACS Catal., 2011, 1, 292–299 CrossRef CAS
- K. Hemelsoet, J. Van der Mynsbrugge, K. De Wispelaere, M. Waroquier and V. Van Speybroeck, ChemPhysChem, 2013, 14, 1526–1545 CrossRef CAS PubMed
- D. Lesthaeghe, A. Horré, M. Waroquier, G. B. Marin and V. Van Speybroeck, Chem. – Eur. J., 2009, 15, 10803–10808 CrossRef CAS PubMed
- B. Arstad, J. B. Nicholas and J. F. Haw, J. Am. Chem. Soc., 2004, 126, 2991–3001 CrossRef CAS PubMed
- C.-M. Wang, Y.-D. Wang and Z.-K. Xie, Catal. Sci. Technol., 2014, 4, 2631–2638 RSC
- C.-M. Wang, Y.-D. Wang, H.-X. Liu, Z.-K. Xie and Z.-P. Liu, Microporous Mesoporous Mater., 2012, 158, 264–271 CrossRef CAS
- C.-M. Wang, Y.-D. Wang, Y.-J. Du, G. Yang and Z.-K. Xie, Catal. Sci. Technol., 2016, 6, 3279–3288 RSC
- C.-M. Wang, Y.-D. Wang, Z.-K. Xie and Z.-P. Liu, J. Phys. Chem. C, 2009, 113, 4584–4591 CrossRef CAS
- K. De Wispelaere, K. Hemelsoet, M. Waroquier and V. Van
Speybroeck, J. Catal., 2013, 305, 76–80 CrossRef CAS
- D. Lesthaeghe, B. De Sterck, V. Van Speybroeck, G. B. Marin and M. Waroquier, Angew. Chem., Int. Ed., 2007, 46, 1311–1314 CrossRef CAS PubMed
- Y. Chu, X. Sun, X. Yi, L. Ding, A. Zheng and F. Deng, Catal. Sci. Technol., 2015, 5, 3507–3517 RSC
- C.-M. Wang, Y.-D. Wang, Y.-J. Du, G. Yang and Z.-K. Xie, Catal. Sci. Technol., 2015, 5, 4354–4364 RSC
- C.-M. Wang, Y.-D. Wang, H.-X. Liu, Z.-K. Xie and Z.-P. Liu, J. Catal., 2010, 271, 386–391 CrossRef CAS
- S. Wang, Y. Chen, Z. Wei, Z. Qin, H. Ma, M. Dong, J. Li, W. Fan and J. Wang, J. Phys. Chem. C, 2015, 119, 28482–28498 CrossRef CAS
- C.-M. Wang, Y.-D. Wang, H.-X. Liu, G. Yang, Y.-J. Du and Z.-K. Xie, Chin. J. Catal., 2015, 36, 1573–1579 CrossRef CAS
- V. Van Speybroeck, K. De Wispelaere, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet and M. Waroquier, Chem. Soc. Rev., 2014, 43, 7326–7357 RSC
- C. Chizallet, ACS Catal., 2020, 5579–5601, DOI:10.1021/acscatal.0c01136
- G. Li and E. A. Pidko, ChemCatChem, 2018, 11, 134–156 CrossRef
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma, E. Abou-Hamad, I. Vollmer, M. Goesten, B. Mezari, E. J. M. Hensen, J. S. Martinez-Espin, M. Morten, S. Mitchell, J. Perez-Ramirez, U. Olsbye, B. M. Weckhuysen, V. Van Speybroeck, F. Kapteijn and J. Gascon, Nat. Chem., 2018, 10, 804–812 CrossRef CAS PubMed
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS
- I. M. Dahl and S. Kolboe, J. Catal., 1996, 161, 304–309 CrossRef CAS
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed
-
M. Bjørgen, K.-P. Lillerud, U. Olsbye and S. Svelle, in Studies in Surface Science and Catalysis, ed. F. B. Noronha, M. Schmal and E. F. Sousa-Aguiar, Elsevier, 2007, vol. 167, pp. 463–468 Search PubMed
- P. N. Plessow and F. Studt, Catal. Sci. Technol., 2018, 8, 4420–4429 RSC
- J. Rey, C. Bignaud, P. Raybaud, T. Bucko and C. Chizallet, Angew. Chem., Int. Ed., 2020, 59, 18938–18942 CrossRef CAS PubMed
- C.-M. Wang, Y.-D. Wang and Z.-K. Xie, J. Catal., 2013, 301, 8–19 CrossRef CAS
- D. Lesthaeghe, J. Van der Mynsbrugge, M. Vandichel, M. Waroquier and V. Van Speybroeck, ChemCatChem, 2011, 3, 208–212 CrossRef CAS
- M. N. Mazar, S. Al-Hashimi, M. Cococcioni and A. Bhan, J. Phys. Chem. C, 2013, 117, 23609–23620 CrossRef CAS
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS
- C.-J. Chen, S. Rangarajan, I. M. Hill and A. Bhan, ACS Catal., 2014, 4, 2319–2327 CrossRef CAS
- O. Bortnovsky, P. Sazama and B. Wichterlova, Appl. Catal., A, 2005, 287, 203–213 CrossRef CAS
- Y.-H. Guo, M. Pu, B.-H. Chen and F. Cao, Appl. Catal., A, 2013, 455, 65–70 CrossRef CAS
- X. Zhu, S. Liu, Y. Song and L. Xu, Appl. Catal., A, 2005, 288, 134–142 CrossRef CAS
- S. Mitchell, M. Boltz, J. Liu and J. Pérez-Ramírez, Catal. Sci. Technol., 2017, 7, 64–74 RSC
- Y.-X. Sun, J. Yang, L.-F. Zhao, J.-X. Dai and H. Sun, J. Phys. Chem. C, 2010, 114, 5975–5984 CrossRef CAS
- X. Hou, Y. Qiu, X. Zhang and G. Liu, Chem. Eng. J., 2017, 307, 372–381 CrossRef CAS
- S. Standl, T. Kühlewind, M. Tonigold and O. Hinrichsen, Ind. Eng. Chem. Res., 2019, 58, 18107–18124 CrossRef CAS
- V. Blay, E. Epelde, R. Miravalles and L. A. Perea, Catal. Rev.: Sci. Eng., 2018, 60, 278–335 CrossRef CAS
- P. del Campo, M. T. Navarro, S. K. Shaikh, M. D. Khokhar, F. Aljumah, C. Martínez and A. Corma, ACS Catal., 2020, 10, 11878–11891 CrossRef CAS
- V. Blay, P. J. Miguel and A. Corma, Catal. Sci. Technol., 2017, 7, 5847–5859 RSC
- K. De Wispelaere, J. S. Martínez-Espín, M. J. Hoffmann, S. Svelle, U. Olsbye and T. Bligaard, Catal. Today, 2018, 312, 35–43 CrossRef CAS
- Z. Wen, H. Zhu and X. Zhu, Catal. Lett., 2019, 150, 21–30 CrossRef
- R. Y. Brogaard, B. M. Weckhuysen and J. K. Nørskov, J. Catal., 2013, 300, 235–241 CrossRef CAS
- S. L. C. Moors, K. De Wispelaere, J. Van der Mynsbrugge, M. Waroquier and V. Van Speybroeck, ACS Catal., 2013, 3, 2556–2567 CrossRef CAS
- M. DeLuca, P. Kravchenko, A. Hoffman and D. Hibbitts, ACS Catal., 2019, 9, 6444–6460 CrossRef CAS
- Y.-Y. Chen, Z. Wei, S. Wang, J. Li, M. Dong, Z. Qin, J. Wang, H. Jiao and W. Fan, Catal. Sci. Technol., 2016, 6, 5326–5335 RSC
- J. Van der Mynsbrugge, M. Visur, U. Olsbye, P. Beato, M. Bjørgen, V. Van Speybroeck and S. Svelle, J. Catal., 2012, 292, 201–212 CrossRef CAS
- B. Arstad, S. Kolboe and O. Swang, J. Phys. Chem. B, 2002, 106, 12722–12726 CrossRef CAS
- M. Fečík, P. N. Plessow and F. Studt, ACS Catal., 2020, 10, 8916–8925 CrossRef
- M. W. Erichsen, S. Svelle and U. Olsbye, Catal. Today, 2013, 215, 216–223 CrossRef
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS PubMed
- R. Sullivan, C. J. Egan, G. Langlois and R. P. Sieg, J. Am. Chem. Soc., 1961, 83, 1156–1160 CrossRef CAS
- P. E. Blochl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- A. Hjorth Larsen, J. Jorgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dulak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiotz, O. Schutt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef
- P. N. Plessow, J. Chem. Theory Comput., 2018, 14, 981–990 CrossRef CAS PubMed
- T. J. Goncalves, P. N. Plessow and F. Studt, ChemCatChem, 2019, 11, 4368–4376 CrossRef CAS
- P. N. Plessow and F. Studt, ACS Catal., 2017, 7, 7987–7994 CrossRef CAS
- C. Tuma and J. Sauer, Phys. Chem. Chem. Phys., 2006, 8, 3955–3965 RSC
- C. Tuma, T. Kerber and J. Sauer, Angew. Chem., Int. Ed., 2010, 49, 4678–4680 CrossRef CAS PubMed
- N. Hansen, T. Kerber, J. Sauer, A. T. Bell and F. J. Keil, J. Am. Chem. Soc., 2010, 132, 11525–11538 CrossRef CAS PubMed
- S. Svelle, C. Tuma, X. Rozanska, T. Kerber and J. Sauer, J. Am. Chem. Soc., 2009, 131, 816–825 CrossRef CAS
- M. Rybicki and J. Sauer, J. Am. Chem. Soc., 2018, 140, 18151–18161 CrossRef CAS PubMed
- Q. Ren, M. Rybicki and J. Sauer, J. Phys. Chem. C, 2020, 124, 10067–10078 CrossRef CAS
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef
- D. Feller, J. Chem. Phys., 1992, 96, 6104–6114 CrossRef CAS
- T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys., 1997, 106, 9639–9646 CrossRef CAS
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS
- TURBOMOLE V7.1 2016, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com, http://www.turbomole.com, (accessed 05.05. 2017)
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS
- F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753–12762 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- C. L. Janssen and I. M. B. Nielsen, Chem. Phys. Lett., 1998, 290, 423–430 CrossRef CAS
- W. Jiang, N. J. DeYonker and A. K. Wilson, J. Chem. Theory Comput., 2012, 8, 460–468 CrossRef CAS PubMed
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS
- Y. Minenkov, E. Chermak and L. Cavallo, J. Chem. Theory Comput., 2015, 11, 4664–4676 CrossRef CAS
- Y. Minenkov, G. Bistoni, C. Riplinger, A. A. Auer, F. Neese and L. Cavallo, Phys. Chem. Chem. Phys., 2017, 19, 9374–9391 RSC
- M. Saitow, U. Becker, C. Riplinger, E. F. Valeev and F. Neese, J. Chem. Phys., 2017, 146, 164105 CrossRef PubMed
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef PubMed
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS
- Ø. Mikkelsen, P. O. Rønning and S. Kolboe, Microporous Mesoporous Mater., 2000, 40, 95–113 CrossRef
- Y. Liu, F. M. Kirchberger, S. Muller, M. Eder, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 1462 CrossRef PubMed
- S. Wang, Y. Chen, Z. Qin, T.-S. Zhao, S. Fan, M. Dong, J. Li, W. Fan and J. Wang, J. Catal., 2019, 369, 382–395 CrossRef CAS
- J. S. Martínez-Espín, K. De Wispelaere, T. V. W. Janssens, S. Svelle, K. P. Lillerud, P. Beato, V. Van Speybroeck and U. Olsbye, ACS Catal., 2017, 7, 5773–5780 CrossRef
- A. T. Smith, P. N. Plessow and F. Studt, Chem. Phys., 2021, 541, 111033 CrossRef CAS
- S. M. Alwahabi and G. F. Froment, Ind. Eng. Chem. Res., 2004, 43, 5098–5111 CrossRef CAS
- T. V. W. Janssens, S. Svelle and U. Olsbye, J. Catal., 2013, 308, 122–130 CrossRef CAS
- P. Kumar, J. W. Thybaut, S. Svelle, U. Olsbye and G. B. Marin, Ind. Eng. Chem. Res., 2013, 52, 1491–1507 CrossRef CAS
- P. Kumar, J. W. Thybaut, S. Teketel, S. Svelle, P. Beato, U. Olsbye and G. B. Marin, Catal. Today, 2013, 215, 224–232 CrossRef CAS
- P. N. Plessow, A. Smith, S. Tischer and F. Studt, J. Am. Chem. Soc., 2019, 141, 5908–5915 CrossRef CAS PubMed
- S. Standl, F. M. Kirchberger, T. Kühlewind, M. Tonigold, M. Sanchez-Sanchez, J. A. Lercher and O. Hinrichsen, Chem. Eng. J., 2020, 402, 126023 CrossRef CAS
- J. Amsler, P. N. Plessow and F. Studt, Catal. Lett., 2021 DOI:10.1007/s10562-020-03492-6
Footnote |
| † Electronic supplementary information (ESI) available: Additional computation information and the total energies in PDF. Structure files in XYZ and CIF formats are attached in the ZIP file. See DOI: 10.1039/d1cy00433f |
| This journal is © The Royal Society of Chemistry 2021 |