
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced biocatalytic degradation of lignin using combinations of lignin-degrading enzymes and accessory enzymes†
Goran M. M.
Rashid
and
Timothy D. H.
Bugg
 *
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: T.D.Bugg@warwick.ac.uk
First published on 8th April 2021
Abstract
Methods for screening combinations of lignin-degrading enzymes and accessory enzymes for product release from polymeric lignin have been developed, using two colorimetric assays that can be applied in microtiter plate format. A set of 3 bacterial DyP-type peroxidase enzymes from Pseudomonas fluorescens, Comamonas testosteroni and Agrobacterium sp., two bacterial multi-copper oxidase enzymes CueO from Ochrobactrum sp. and CopA from Pseudomonas putida, and Sphingobacterium sp. T2 manganese superoxide dismutase have been tested in combination with one LigE β-etherase enzyme from Agrobacterium sp., two dihydrolipoamide dehydrogenase enzymes from Sphingobacterium sp. T2, Burkholderia cenocepacia peroxiredoxin, and Desulfitobacterium hafniense arylsulfotransferase. Combinations of Agrobacterium LigE with DyP-type peroxidases gave 4–10 enhancement in low molecular weight product release from technical lignins, and enhancements in product release were observed for all lignins tested, using different accessory enzymes. Analysis of products formed by reverse phase HPLC verified increases in concentrations of specific low molecular weight products.
A. Introduction
The aromatic heteropolymer lignin, found in lignocellulose plant cell walls, is the most abundant source of renewable aromatic carbon in the biosphere. There is considerable interest in the conversion of lignin into low molecular weight aromatic products using microbial conversion, biocatalysis or chemocatalysis,1,2 but also many challenges in doing so. The ether C–O and C–C bond linkages in lignin are not susceptible to cleavage under mild conditions, and the hydrophobic character of lignin often renders it insoluble as well as inert.1,2Micro-organisms that are able to break down lignin typically produce an array of extracellular oxidative enzymes. White-rot basidiomycete fungi produce extracellular lignin peroxidases, manganese peroxidases, and laccases that attack polymeric lignin.3,4 Certain soil bacteria that can degrade lignin such as Rhodococcus jostii RHA1, Amycolatopsis sp. 75iv2 and Pseudomonas fluorescens Pf-5 produce dye-decolorizing peroxidases that can oxidise lignin,5–7 while bacteria such as Streptomyces coelicolor and Ochrobactrum sp. produce lignin-oxidising multi-copper oxidases,8,9 and strains of Sphingobium, Novosphingobium and other α-Proteobacteria produce intracellular β-etherase enzymes that can utilise glutathione to attack the β-ether bond in lignin fragments.10–13 Accessory enzymes have also been identified in fungi, such as aryl alcohol oxidase that can generate hydrogen peroxide,14 and cellobiose dehydrogenase.15
Attempts to carry out the conversion of polymeric lignin in vitro using recombinant enzymes are hindered by an additional technical problem, that phenoxy radicals generated upon lignin oxidation can spontaneously repolymerise or recondense to form higher molecular weight products. In vivo it is thought that this is prevented by one-electron reduction of phenoxy radicals to phenols via flavin-dependent accessory enzymes such as cellobiose dehydrogenase in fungi,15 or dihydrolipoamide dehydrogenase in lignin-degrading bacteria.16 Therefore, in order to achieve efficient conversion of polymeric lignin into low molecular weight products, it is likely that several lignin-oxidising enzymes and accessory enzymes will be needed.
To date there are only a small number of reports of the use of enzyme combinations to attack lignin. Fungal aryl alcohol oxidase from Pleurotus eryngii has been shown to be synergistic with P. eryngii laccase to generate hydroxyl radical,17 and cellulose dehydrogenase has been reported to be synergistic with fungal manganese peroxidase for degradation of Kraft pulp lignin.18 Picart et al. have reported that treatment of beech wood lignin with Trametes versicolor laccase lcc2 M3, followed by Sphingobium SYK-6 LigE and LigG, and Novosphingobium aromaticivorans LigF, gives enhanced yields of a bio-oil containing low molecular weight aromatic compounds.19 Gall et al. have also shown that applying in vitro a combination of β-etherase enzymes LigD, LigE, LigF, LigN and NaGSTNU, together with cofactor recycling enzymes, is effective in releasing G, S and tricin units from polymeric lignin substrates.20 However, a more systematic study of enzyme combinations has not been reported, and there is no method to identify which enzyme combinations might be more effective for different lignin substrates.
The aim of this study is to examine the use of multiple combinations of lignin-degrading enzymes, by first developing high throughput assays to screen for effective enzyme combinations. We report here the use of two high throughput assays for testing enzyme combinations, and the observation of enhanced generation of specific products using different enzyme combinations for soda, organosolv, and industrial lignins.
B. Experimental section
Materials
Green Value Protobind 1000 soda lignin was purchased from Green Value SA (Orbe, Switzerland); it is a soda lignin prepared from wheat straw and sarkanda bagasse, previously characterised21 as an S/G/H lignin containing predominantly β-O-4 units, MW 3270 Mn 620 g mol−1. Poplar alkali organosolv lignin (AO) and oak dioxasolv lignin (DL) are both S/G lignins containing predominantly β-O-4 units prepared and characterised as described previously;22 wheat straw organosolv lignin was a gift from CIMV (Levallois Perret, France) (OL); it has been previously characterised21 as an S/G/H lignin containing predominantly β-O-4 units, MW 1960 Mn 450 g mol−1; miscanthus ionic liquid lignin is an S/G/H lignins containing predominantly β-O-4 units prepared and characterised as described previously.22 Dawn Technology lignin from pine wood was supplied by Avantium N. V. from their laboratory in Amsterdam, The Netherlands. Humins were supplied by Avantium N. V. and produced in their pilot plant in Geleen, The Netherlands, by conversion of fructose and glucose. All chemicals were purchased form Sigma Aldrich unless otherwise stated. O-Benzylguaiacol was prepared using the procedure of Himmel et al.23Gene cloning
The genes for Comamonas testosteroni CNB-2 dypB (NC_013446.2) and Desulfitobacterium hafniense arylsulfotransferase ast (CDX02668.1) were codon optimised for E. coli and synthesised (GenScript), then cloned into a pUC57 vector with cloning sites NdeI-EcoRV. For protein overexpression, genes were re-cloned into a pET151/D-TOPO vector using forward and reverse primers 5′CACC ATG ACA AAA GCA CAA GCA GGC ATT CTG3′ (CtDyP forward), 5′CAG GCC TGG AGT CCC AGC3′ (reverse), 5′CACC ATG AAC CCG ATC AAG AGCG 3′ (AST forward), 5′ TTA CGC GGT AAT GCT AAC GCC GGT3′ (reverse).Genes encoding DyP (Agro-DyP) and LigE were amplified from Agrobacterium sp. genomic DNA, while DHLDH1 and DHLDH2 were amplified from Sphingobacterium sp. T2 genomic DNA using forward and reverse primers as follows: Agro Dyp forward 5′CACC ATG GCG ACG TCT CTG GAG CTG3′, reverse 5′TTA CTT TGC ATC TTC GTC GCC GAC3′, Agro LigE forward 5′CACC ATG ACG ACT TCC AGA ACG CTT TAT TC3′, reverse 5′TCA CGC CGG TGT CAC ACT GC3′, DHLDH1 forward 5′CACC ATG AAC TAC GAC ATC ATT GTG ATT GG3′, reverse 5′TTA TAA GTG GAT TAC TTC ACC ATA GG3′ and DHLDH2 forward 5′CACC ATG CAA TAC GAT GTC ATA GTT ATA GG3′, reverse 5′TTA AGC GTG AAT AGC TCT GTT TGC3′. The amplicons were cloned using the Champion pET Directional TOPO Expression Kit (Invitrogen) into expression vector pET151 and transformed into E. coli TOP10 competent cells (Invitrogen). The recombinant plasmids were then transformed into BL21 E. coli BL21(DE3)pLysS (Invitrogen), for protein expression.
Enzyme overexpression
Multicopper oxidase CueO from Ochrobactrum sp.,8 multicopper oxidase CopA from Pseudomonas putida,24 DyP1B from Pseudomonas fluorescens,6 peroxiredoxin from Burkholderia cenocepacia25 and MnSOD1 from Sphingobacterium sp. T2 (ref. 26) were purified as reported. Recombinant Sphingobium SYK-6 LigD27 was also expressed as a His6 fusion protein using the pET151/D-TOPO expression vector.Cultures of each recombinant strain were grown at 37 °C in 1 L of Luria–Bertani media containing 100 μg mL−1 ampicillin, induced with 0.5–1 mM IPTG (isopropyl-βD-thiogalactopyranoside) at OD600 = 0.6, then incubated overnight at 15 °C with shaking at 180 rpm. The cell pellet was harvested by centrifugation (6000g, 15 min). The cells were resuspended in 20 mM Tris pH 8.0 containing 10 mM imidazole, 0.5 M NaCl, and 1 mM PMSF, passed through a cell disruptor, centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 35 min), and the supernatant was filtered with a Whatman 0.2 μM syringe filter. The soluble protein fraction was loaded on to a 5 mL pre-equilibrated Ni-NTA column (GE Healthcare) with 20 mM Tris pH 8.0 buffer containing 20 mM imidazole, 0.5 M NaCl, and eluted with 20 mM Tris pH 7.8 containing 300 mM imidazole, 0.5 M NaCl. The purified enzyme was concentrated with an Amicon centrifugal unit (10 kDa cut off) and desalted with a PD-10 column, eluting with 50 mM sodium phosphate buffer pH 7.8.
000g, 35 min), and the supernatant was filtered with a Whatman 0.2 μM syringe filter. The soluble protein fraction was loaded on to a 5 mL pre-equilibrated Ni-NTA column (GE Healthcare) with 20 mM Tris pH 8.0 buffer containing 20 mM imidazole, 0.5 M NaCl, and eluted with 20 mM Tris pH 7.8 containing 300 mM imidazole, 0.5 M NaCl. The purified enzyme was concentrated with an Amicon centrifugal unit (10 kDa cut off) and desalted with a PD-10 column, eluting with 50 mM sodium phosphate buffer pH 7.8.
Assays for combination of oxidising enzymes with accessory enzymes
The following assays were carried out in 96-well microtiter plate format. In a deep-well microtiter plate, lignins (20–40 μL of 25 mg mL−1 DMSO or H2O in case of Kraft lignin) were added to 1 mL solutions as follows: 1) 50 μg CopA or CueO (100 μl of 0.5 mg mL−1 stock) and 15 μL of 20 mM CuSO4 in 1 mL 50 mM phosphate buffer pH 7.4; 2) 100 μg SpMnSOD1 (10 μL of 10 mg mL−1 stock) in 1 mL 50 mM Tris-HCl buffer pH 8.0, to which was added 10 μL of 100 mM H2O2; 3) 100 μg DyP enzyme (DyP1B, Ct-DyP or Agro DyP, 10 μL of 10 mg mL−1 stock) in 1 mL 50 mM acetate buffer pH 6.0 containing 1 mM MnSO4, to which was added 10 μL of 100 mM H2O2. Samples were then incubated for 1–24 h at 30 °C. Control experiment for each lignin-oxidising enzyme and lignin preparation were conducted in which enzyme was omitted.The above assays were carried out in the presence of one accessory enzyme (10 μL of 10 mg mL−1 protein stock). The accessory enzymes added were: Sphingobacterium sp. T2 DHLDH1 or DHLDH2, with cofactor NADH (20 μL of 20 mM stock); Burkholderia cenocepacia peroxiredoxin, with co-substrates H2O2 (10 μL of 100 mM stock) and reduced glutathione (10 μL of 100 mM stock); Agrobacterium sp. LigE, with co-substrate reduced glutathione (10 μL of 100 mM stock). In the case of arylsulfotransferase AST, lignins were pre-treated with AST by addition of AST (10 μL of 10 mg mL−1 stock) in 0.5 mL 10 mM Tris-glycine buffer pH 9.0 with p-nitrophenyl sulfate (5 mM final concentration), and incubated for 1 h at 30 °C, then oxidising enzyme was added as described above to make a 1 mL volume. After incubation for 1–24 h, 20 μL aliquots were removed for the following assays.
DNP assay was adapted from ref. 28. For detection of aldehyde and ketones, samples (20 μL) were mixed with 0.1 M HCl (30 μL) and 2,4-dinitrophenylhydrazine (DNP) (50 μL, 1 mM in 100 mM HCl), then incubated at room temperature for 5 min. 0.1 M NaOH (100 μL) was added, and absorbance was measured at 450 nm using a HIDEX sense microplate reader.
FCA assay was adapted from ref. 29. For detection of released phenolic compounds, samples (20 μL) were mixed with distilled water (100 μL) and Folin–Ciocalteu reagent29 (FCR) (10 μL), and NaCO3 (100 μL, 4%) was added after 4 min, then incubated in the dark for 30 min at room temperature. Absorbance was then measured at 750 nm with a HIDEX sense microplate reader.
Purpald assay was adapted from ref. 30. For detection of aldehyde products, samples (20 μL) were mixed with distilled water (80 μL) and fresh Purpald reagent (100 μL, 5 mg mL−1 in 0.5 N NaOH), and vortexed for 1 min, then incubated in the dark for 30 min at 30 °C. Absorbance was then measured at 550 nm with a HIDEX sense microplate reader.
AST assay was adapted from ref. 31. Enzyme assays were carried out in a 1.0 mL volume at 20 °C. Assays contained 5 mM p-nitrophenyl sulfate (pNS) as sulfate donor and 5 mM phenol as acceptor, using 50 mM Tris-glycine buffer (pH 9.0). After addition of AST enzyme (2–5 μL, 6 mg mL−1), the release of p-nitrophenol was monitored at 405 nm on a Varian Cary50 spectrophotometer.
Biotransformations
Lignin (5 mg, 200 μL of 25 mg mL−1 stock in DMSO) was added to 5 mL buffer (50 mM succinate pH 6.0 for DyPs, 50 mM phosphate pH 7.4 for CueO/CopA, or Tris-HCl pH 8.0 for SpMnSOD), and the mixture was heated to 40 °C for 30 min, then cooled to room temperature and 0.5 mg lignin-oxidising enzyme (50 μL of 10 mg mL−1 stock) was added. Co-substrates and cofactors were then added: 1 mM H2O2 and 1 mM MnSO4 for DyPs; 1 mM H2O2 for SpMnSOD; 0.3 mM CuSO4 for CueO/CopA. Accessory enzymes were then added: 0.5 mg DHLDH2 with 0.4 mM NADH; or 0.5 mg Agro LigE with 1 mM reduced glutathione; or 0.5 mg Prx with 1 mM reduced glutathione. Then the reactions were incubated at 30 °C for 12 h.In the case of use of AST accessory enzyme, lignins were pre-treated with AST by addition of 0.5 mg AST in 2 mL of 10 mM Tris-glycine buffer pH 9.0 with p-nitrophenyl sulfate (5 mM final concentration), and incubated for 1 h at 30 °C, then oxidising enzyme was added as described above to make 5 mL reaction with buffer (100 mM succinate pH 6.0 for DyPs, or phosphate pH 7.4 for CueO/CopA, or Tris-HCl pH 8.0 for SpMnSOD). Control experiments were carried out using no accessory enzyme, and no enzyme. 200 mg biotransformations used the same method, scaled up 20-fold (100 mL buffer, 1 mL of each enzyme from 10 mg mL−1 stock), and were incubated for 24 h at 30 °C.
The yield of biotransformed lignin was determined by two methods: 1) extraction of low molecular weight products into EtOAc, followed by evaporation; 2) the amount of solubilised lignin was estimated after biotransformation using the Coomassie blue method described below.
Method to estimate soluble lignin concentration
The amount of solubilised lignin was estimated adding 2–5 μL of sample with 200 μL Coomassie brilliant blue G-250 Bradford reagent (Sigma-Aldrich) in a 96-well microtiter plate. Absorbance was measured between 630–660 nm (GVPL, 631 nm, AV humin, 655 nm, OL 633 nm and KL 650 nm). Lignin standard solutions were prepared by dissolving 25 mg of samples with 1 mL DMSO, then 0.2–2.5 mg mL−1 standards prepared by diluting with water.HPLC analysis
HPLC and LC-MS analysis was conducted using a Phenomenex Luna 5 μm C18 reverse phase column (100 Å, 250 mm, 4.6 mm) on an Agilent 1260 analyzer and an Amazon X mass spectrometer at a flow rate of 0.5 mL min−1, monitoring at 270 nm. The gradient for HPLC and LC-MS was as follows: water/0.1% formic acid was used as solvent A, and methanol/0.1% formic acid as solvent B; method starts with 10% solvent B for 5 min; then 10–30% B from 5–10 min; 30–70% B from 10–25 min; 70–100% B from 25–30 min; 100% B from 30–35 min and 100–10% B from 35–40 min.Results
Assays for lignin degradation products
Three colorimetric assay methods were tested on several lignin preparations using DyP-type peroxidase enzyme Dyp1B from Pseudomonas fluorescens6 for measurement of the release of low molecular weight oxidised lignin degradation products (see Fig. 1A). The first method uses the Folin–Ciocalteu assay (FCA) as a colorimetric phenol detection assay.29 The second method uses 2,4-dinitrophenylhydrazine (DNP) for measuring the formation of aldehyde and ketone products formed by oxidative cleavage of lignin, which has been reported by Tonina et al.28 The third method uses the reagent Purpald to detect the formation of aldehyde products.31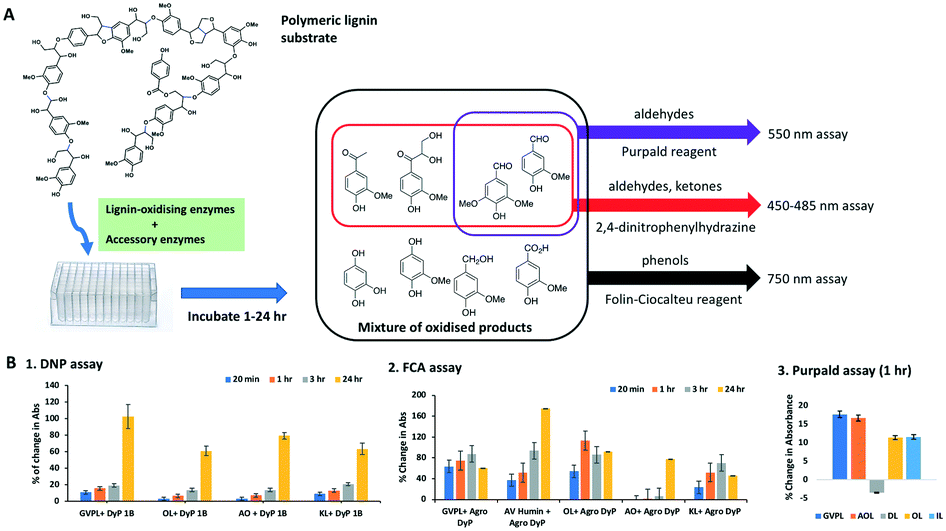 | ||
| Fig. 1 A. Scheme of FCA, DNP and Purpald assays. B. Product formation using DNP assay at 1–24 h using DyP1B; FCA assay at 1–24 h using Agro DyP; and Purpald assay at 1 h using DyP1B. Data are mean absorbance values (n = 4), after subtraction of control lacking enzyme. | ||
Five different types of lignin preparations were tested as substrate for enzymatic biotransformation, for which structural characterisation has been previously reported: a commercially available soda lignin Green Value Protobind lignin (GVPL) from wheat straw;21 poplar alkali organosolv lignin (AO);22 oak dioxasolv lignin (DL);19 wheat straw organosolv (from CIMV, France) (OL); and a miscanthus ionic liquid lignin.22 Each lignin was dissolved in DMSO, then added to 50 mM succinate buffer pH 5.5 containing 0.1 mg Dyp 1B, 1 mM MnSO4 and 1 mM H2O2, then incubated for 1 h and 24 h. Samples were taken for DNP, FCA and Purpald assays, and analysed by UV-visible spectroscopy. The results are shown in Fig. 1B.
Using the DNP assay, 2–9% increase in absorbance was observed after 1 h, and 50–120% increase in absorbance was observed after 24 h incubation (see Fig. 1B, panel 1). Using the Purpald assay, an 11–17% increase of aldehyde content was observed, compared to a control containing no enzyme, for all lignins except for dioxasolv lignin, which showed a 3.5% decrease (see Fig. 1B, panel 3).
Using the FCA assay, after 1 h four lignins showed a 40–100% increase in absorbance, and after 24 h all five lignins showed 40–180% increase in absorbance (see Fig. 1B, panel 2). In some cases absorbance after 24 h in the FCA assay was slightly less than after 1–3 h, which we believe is an indication of some repolymerisation of phenolic products taking place.2
These results indicate that all three assays show time-dependent increases in absorbance in the presence of a lignin-oxidising DyP-type peroxidase, consistent with the formation of low molecular weight carbonyl and phenolic products. While there is background absorbance due to carbonyl and phenolic content in each lignin, time-dependent changes are in most cases >10 fold in excess of standard error for assay replicates (n = 4). The FCA and DNP assays were chosen for carrying out screening of enzyme combinations, since the DNP assay detects both aldehyde and ketone products.
Panel of bacterial lignin-degrading enzymes and accessory enzymes
To test different combinations of lignin oxidising enzymes and potential accessory enzymes on lignin degradation, a panel of 6 lignin-oxidizing and 5 accessory enzyme enzymes was used. Dye-decolorizing peroxidase Dyp1B from P. fluorescens has been previously reported to possess lignin-oxidising activity.6 We also identified DyP-type peroxidases in two bacterial lignin-degrading strains isolated from municipal waste:29 the genome of lignin-degrading Comamonas testosteroni TK102 contained a class B DyP peroxidase (Uniprot A0A076PUS2), and the genome of lignin-degrading Agrobacterium sp. B1 (NCBI accession PRJNA561791) contained a class C DyP peroxidase (NCBI accession WP_149145853.1).Recombinant Ct-Dyp and Agro-Dyp proteins were expressed in E. coli as His6 fusion proteins (see ESI† Fig. S1), and were characterised kinetically. Both enzymes were active in oxidation of peroxidase substrates ABTS, 2,4-dichlorophenol (DCP) and guaiacol, and both enzymes oxidised Mn2+. The KM and kcat values are shown in Table 1. The pH optima of Ct-DyP and Agro-Dyp were 5.0 and 5.5 respectively (see ESI† Fig. S2). We also tested bacterial multi-copper oxidases CueO from Ochrobactrum sp.9 and CopA from Pseudomonas putida KT2440,24 and lignin-oxidising manganese superoxide dismutase MnSOD1 from Sphingobacterium sp. T2.26
| Substrate | Ct DyP | Agro DyP | Pfl DyP1B | |||
|---|---|---|---|---|---|---|
| K m (mM) | k cat (s−1) | K m (mM) | k cat (s−1) | K m (mM) | k cat (s−1) | |
| DCP | 4.9 | 0.3 | 2.8 | 0.6 | 1.2 | 0.66 |
| Mn2+ | 11.7 | 0.3 | 9.5 | 5.4 | 7.3 | 2.4 |
| ABTS | 3.2 | 0.2 | 0.9 | 16.6 | 1.1 | 13.5 |
The genome of lignin-degrading Agrobacterium sp. B1 recently reported by our group32 also contained one putative β-etherase LigE enzyme (NCBI accession WP_149146641.1), but no other annotated LigF or LigD sequences. Recombinant Agro LigE was expressed in E. coli as a His6 fusion protein and purified (see ESI† Fig. S1). The activity of Agro LigE was tested by treatment of β-aryl ether lignin dimer GGE with recombinant Sphingobium SYK-6 LigD27 and 1 mM NAD+, generating the oxidised dimer MPHPV, however, addition of Agro LigE and 1 mM reduced glutathione gave no further reaction (see ESI† Fig. S3), indicating that Agro LigE catalyses a different reaction. Agro LigE was found to transform O-benzylguaiacol in the presence of 1 mM glutathione (see ESI† Fig. S4).
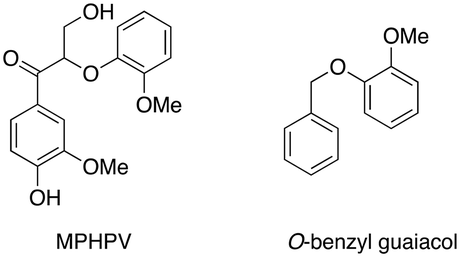
These data imply that Agro LigE catalyses a different reaction to Sphingobium SYK-6 LigE,10–13 perhaps at the benzylic α position of lignin fragments. Dihydrolipoamide dehydrogenase from Thermobifida fusca has been found to act as an accessory enzyme for lignin degradation, by preventing re-polymerisation of phenoxy radicals formed during lignin oxidation.16 We identified two dihydrolipoamide dehydrogenase enzymes in the genome of lignin-degrading Sphingobacterium sp. T2, which were expressed as His6 fusion proteins in E. coli and purified (see ESI† Fig. S1). Redox protein peroxiredoxin has been observed in proteomic analysis of lignin-degrading Pandoraea,33Bacillus ligniniphilus L1,34 and Sphingobacterium sp. T2.24 This protein reacts with hydrogen peroxide through an active site cysteine nucleophile.25 Hence we included recombinant Burkholderia cenocepacia peroxiredoxin25 in the panel of enzymes. We also included arylsulfotransferase from Desulfitobacterium hafniense, an enzyme which has been reported to transfer sulphate groups from p-nitrophenyl sulfate to phenol and polyphenol substrates, and thereby improve the water solubility of hydrophobic lignins.31,35
Testing of enzyme combinations
The combined effect of each oxidising enzymes with and without accessory enzymes was tested using the FCA and DNP assays against different lignin substrates in microtiter plate format. The lignins studied were as follows: 1) a commercially available Green Value Protobind soda lignin;21 2) commercially available alkali-Kraft lignin (KL, Sigma-Aldrich); 3) wheat straw organosolv lignin;21 4) and a Dawn Technology lignin from Avantium,36 obtained from an improved Bergius–Rheinau process which uses concentrated HCl to hydrolyse biomass.37 Characterisation data for these lignins is described in the Experimental section. We also tested an industrially produced humin, a material formed by dehydration of C5 and C6 biorefinery sugars via polymerisation of hydroxymethylfurfural.38Lignin and humin preparations were incubated with oxidising enzymes in 96 deep-well microtiter-plates in the presence of their cofactor or coenzymes for 1 h with and without accessory enzymes, then samples were taken for FCA and DNP assays (see Experimental section). The accessory enzymes were added to the reaction with the oxidising enzyme. In the case of arylsulfotransferase AST, lignins were treated with AST first for 60 min, then oxidising enzymes added.
The results (see Fig. 2) show enhancements in product release by combining lignin-oxidising enzymes and accessory enzymes, but differences in behaviour between lignin types. For the DNP assay (Fig. 2B), enhancements in product release were observed for OL and KL for all oxidising enzymes using each of the accessory enzymes. Observation of higher levels of product formation with organosolv lignin is consistent with literature studies showing higher levels of product formation with lignins containing high β-O4 content.22 For GVPL, enhancements in phenol release were observed in the FCA assay (Fig. 2A) using all three DyP peroxidases in combination with each accessory enzyme, notably with Agro LigE, and with higher activity for Agro DyP. Even for the condensed AL, product release was observed for DyP peroxidases in combination with accessory enzymes, with highest activity for Agro DyP.
 | ||
| Fig. 2 Effect of enzyme combinations (lignin-oxidising enzymes with accessory enzymes) on production of phenolic compounds and oxidation of lignin preparations (GVPL, AVL, OL, KL and AV humin), A. Folin–Ciocalteu assay for estimation of phenolic products released, absorbance measured at 750 nm. B. 2,4-Dinitrophenylhydrazine assay for estimating the formation of aldehyde/ketone products. Colour coding: green indicates high activity, red indicates low activity. Lignin and humin preparation were dissolved in DMSO and added to 50 mM succinate, phosphate and Tris-glycine buffers at pH 5.5, 7.0, 8.0 and 9.0 (1 mL reactions, 4 replicates in 96 well microtiter plates). Data are mean absorbance values (n = 4), typical standard errors 2–3%, error analysis shown in Fig. S5.† Assays were also run with no oxidising enzyme (accessory enzyme only, bottom row) and no accessory enzyme (oxidising enzyme only, right hand column). | ||
In order to estimate the concentrations of low molecular weight products formed from these data, we used vanillin as a standard for the FCA assay (ESI† Fig. S6). For the highest activity of Agro DyP/LigE incubated with GVPL, after subtraction of control a value of A750 = 0.866 would correspond to approximately 0.5 mM vanillin.
Fig. 3 illustrates the fold increases in product formation for Agro DyP (panel A) and CopA (panel B) with different lignins. 4–10 fold increases were observed for Agro DyP, largest with Agro LigE and Prx accessory enzymes, as shown in Fig. 3A. For multi-copper oxidase CopA, more modest increases in activity were observed, as shown in Fig. 3B, the highest being a 3.7-fold enhancement of activity of CopA by Agro LigE, using AL.
 | ||
| Fig. 3 Enhancement in activity of (A) 100 μg Agrobacterium DyP and (B) 50 μg P. putida CopA in DNP assay upon addition of 100 μg accessory enzymes, compared with control containing no accessory enzyme. | ||
Enhancement of product formation from Avantium humin was also observed (see Fig. 2) using combinations of DyP peroxidases with Agro LigE or Prx. Sphingobacterium MnSOD1 showed highest activity with OL and KL, and its activity was enhanced with accessory enzymes, especially with AST.
Lignin solubilisation by AST
We have observed significant enhancements in combined activity using D. hafniense arylsulfotransferase (AST). In order to test whether this was due to solubilisation of the lignin by AST, we examined the sulfation of several different types of lignin preparations by AST. An increase in the soluble fraction of each lignin and a release of by-product p-nitrophenol were observed visually for each lignin (see ESI† Fig. S7), consistent with AST-catalysed sulfation, but quantification of the amount of solubilised lignin was challenging. Therefore, a new method was developed to quantify the amount of lignin solubilised by the enzyme.We have observed that soluble lignin interacts with Coomassie G-250 Brilliant Blue dye, giving an absorption maximum in the range 630–655 nm depending on type of lignin preparation (see ESI† Fig. S8). The observed absorbance change is directly proportional to lignin concentration, and the assay can be used to determine the concentration of Kraft lignin between 0.2 to 2.5 mg mL−1 and Green value lignin 0.5 to 8 mg mL−1 (see ESI† Fig. S9). The possible interference of low molecular weight aromatic compounds was examined, and it was found that monomeric or dimeric lignin model compounds do not interfere in the assay up to 10 mg mL−1 concentration. This assay has been used to investigate the effect of sulfate donor compound (pNS). Increasing pNS concentration 2-fold leads to an increase in the soluble fraction of GVPL by 1.4 fold. Increasing the amount of AST enzyme by 2-fold gave increases of 2-fold and 1.25 fold for IL and GVPL respectively (Table 2).
| Lignin type | Absorbance maximum for G250-lignin complex (nm) | % increase in soluble lignin after treatment with AST |
|---|---|---|
| Alkali Kraft lignin | 650 | 41% |
| Avantium lignin | 603 | 11% |
| Green Value Protobind | 631 | 23% |
| Organosolv lignin | 633 | 33% |
| Ionic liquid lignin | 635 | 137% |
For the condensed Avantium lignin, the percentage of solid lignin solubilised by AST was also estimated by gravimetric analysis. 200 mg scale biotransformations of Avantium lignin by AST were pelleted by centrifugation, and the pellets dried and weighed. Comparison of samples with and without the PNS substrate revealed mass losses of 7–16 mg/200 mg sample, implying that 4–8% of the original lignin had been solubilised.
Metabolite formation using enzyme combinations with specific lignin substrates
Biotransformations of enzymes and lignins showing increased activity in Fig. 2 were carried out at 5 mg scale, and low molecular weight products extracted into ethyl acetate after 12 h, and examined by reverse phase C18 HPLC and LC-MS. New product peaks were observed, as shown in Fig. 4.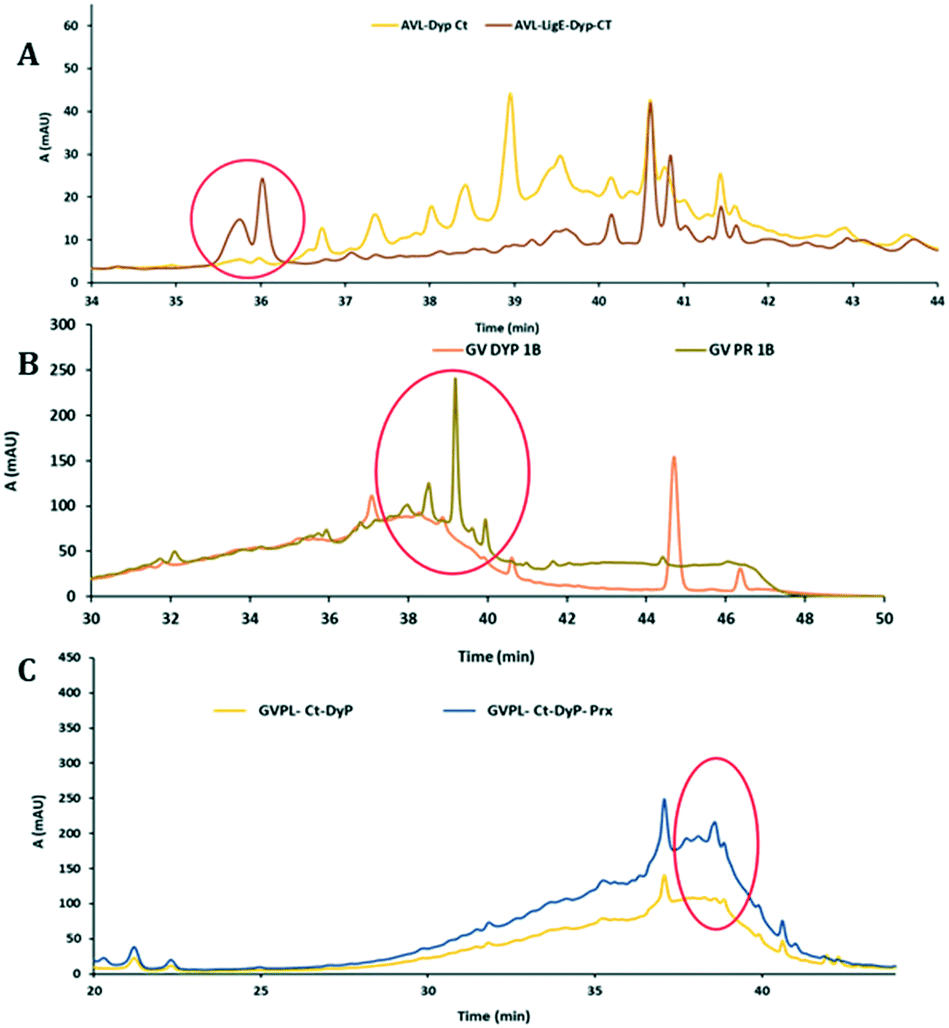 | ||
| Fig. 4 Formation of new or enhanced product peaks from lignin biotransformation by enzyme combinations, observed by reverse phase HPLC analysis. A. Addition of Agro LigE to CT DyP biotransformation of AL. B. Addition of Prx to DyP1B biotransformation of GVL. C. Addition of Prx to CT DyP biotransformation of GVL. | ||
In the presence of accessory enzymes, sometimes completely new product peaks were observed (see Fig. 4A and B), or increase in size of peaks observed using a single oxidative enzyme (see Fig. 4C). HPLC analyses for other enzyme combinations are shown in ESI† (Fig. S10–S19).
Analysis via LC-MS led to the identification of several monocyclic aromatic products, including hydroxyquinol (benzene-1,2,4-triol), 2-methoxy-hydroquinone, vanillin and vanillic acid (see Fig. 5). For 9 of the 11 monomers, structures were identified by co-elution with commercial standards. For the two G3 monomers, the same compound or a closely related compound had been identified in an earlier study,22 with the same retention time. At later retention times were species with molecular mass 297–367 g mol−1 at retention times 35–45 min (see Fig. 4, panels A and B) that likely correspond to oxidised dimeric lignin fragments. As shown in Fig. 5, addition of accessory enzymes led to enhancement in concentration of these metabolites. LC-MS data is shown in ESI† (Fig. S20–S33).
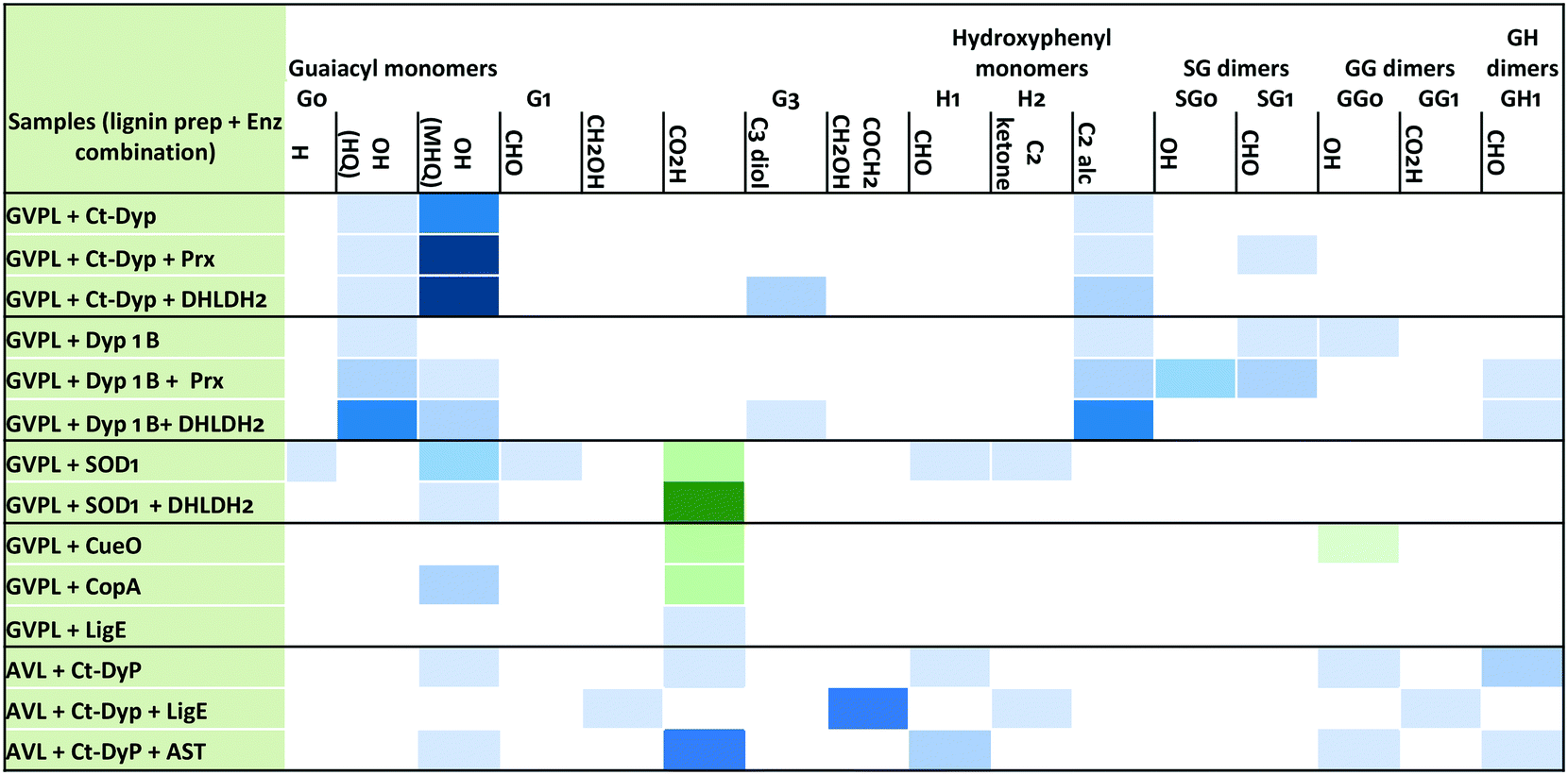 | ||
| Fig. 5 Enhancement in concentration of selected peaks by addition of accessory enzymes. Blue colour indicates the formation of a new product not found in no-enzyme control; green colour indicates enhancement in concentration of a compound observed in no-enzyme control; intensity of colour indicates the relative peak size (2, 3, 5, 10 fold enhancement). Labels: G0, G1, G3 refer to guaiacyl monomers containing 0, 1, or 3 carbon sidechains; H1, H2 refer to hydroxyphenyl monomers containing 1 or 2 carbon sidechains; SG, GG, GH dimers refer to β-O-4 dimers containing additional substituents OH, CHO or CO2H; HQ, hydroxyquinol, MHQ, methoxyhydroquinone. | ||
Larger scale biotransformations
Selected biotransformations were carried out using Green Value Protobind lignin on a 200 mg scale over 24 h, and the extent of lignin conversion was estimated via three different approaches, shown in Table 3. Firstly, using the Coomassie Blue G250 dye assay described above, the amount of soluble lignin present was estimated, and then compared to a control lacking enzyme. Using this method, 17.8% conversion of soluble lignin was estimated using Agro DyP combined with Agro LigE, compared with 13.9% conversion with LigE alone, and a 2.4% increase in soluble lignin with Agro DyP alone, thought to be due to some lignin recondensation. Secondly, the weight of total products extracted into ethyl acetate was measured. Again, the highest yield of extractable products was observed using Agro DyP and Agro LigE enzymes in combination, but with a significant background of extractable products with no enzyme added. Finally, individual product peaks observed by LC-MS were calibrated against authentic standards. Again, the highest combined yield was for Agro DyP and Agro LigE, for which the four identifiable products were observed in a total of 9.9% yield. The latter method underestimates total yield, since there are further unidentified product peaks, however, the trend observed in each method is the same, showing that percentage conversion of lignin to low molecular weight products can be enhanced by the use of enzyme combinations identified using the screening methods described above.| Enzymes | Soluble lignina (mg) | % conversion of soluble lignina | % products extracted into EtOAc (w/w) | Individual product peaks observed by LC-MS, total% |
|---|---|---|---|---|
| a Estimated by Coomassie blue assay method. b Increase in soluble lignin due to recondensation. MHQ, methoxyhydroquinone; HQ, hydroxyquinol; VA, vanillic acid; 4HB, 4-hydroxybenzaldehyde. | ||||
| No enzyme | 180 ± 2 | — | 15.9 | — |
| Agro DyP | 184 ± 2 | (−2.4)b | 17.6 | MHQ, HQ, ∑7.5% |
| LigE | 155 ± 2 | 13.9 | 18.9 | VA, 8.7% |
| Agro DyP + LigE | 148 ± 2 | 17.8 | 22.4 | MHQ, HQ, VA, 4HB, ∑9.9% |
| Agro DyP + Prx | 182 ± 2 | — | 16.5 | MHQ, HQ, ∑8.0% |
| Agro DyP + DHLDH2 | 178 ± 2 | 1.2 | 18.7 | MHQ, HQ, ∑7.1% |
Conclusions
In this work we have assembled a collection of 6 bacterial lignin-oxidising enzymes and 5 accessory enzymes, and have carried out a systematic investigation of combining different lignin-oxidising and accessory enzymes. Included in the collection are two new DyP-type peroxidase enzymes from C. testosteroni and Agrobacterium sp., which both show activity for Mn2+ oxidation (Table 1), and the activity of Agro DyP towards polymeric lignin is somewhat higher than that of P. fluorescens DyP1B (Fig. 2). In order to test enzyme combinations with polymeric lignin substrates, we have developed colorimetric assays to monitor release of aldehyde, ketone and phenol products, which show time-dependent increases in absorbance (Fig. 1B), and function reliably in microtiter plate format (Fig. 2). These assays provide a convenient and rapid method to identify effective combinations of enzymes for bioconversion of different lignins, and could in principle be used to study the time-course of lignin conversion.The results in Fig. 2 show that the activity of lignin-oxidising enzymes is enhanced by addition of accessory enzymes. For treatment of organosolv lignin, Green Value lignin & Kraft lignin, the activity of the three DyP peroxidase enzymes is assisted by reductive accessory enzymes DHLDH2 or peroxiredoxin (Prx), which may be via trapping of phenoxy radicals to prevent repolymerisation.16
Activity was also enhanced by addition of Agrobacterium LigE, an enzyme whose homologues attack β-aryl ether linkages using glutathione as co-substrate.10–13 The Agrobacterium LigE enzyme appears to catalyse a novel ether cleavage transformation, whose precise mechanism is currently under further study, which assists in the depolymerisation of several lignins. Activity was also enhanced by addition of arylsulfotransferase AST, due to partial solubilisation of lignin in aqueous buffer.31,35 Hence there appear to be different mechanisms by which lignin oxidation can be enhanced via addition of accessory enzymes, and different effects are observed with different lignin substrates, for which the colorimetric assays are a valuable tool.
Of particular interest that enzyme combinations show increases in low molecular weight products with industrial lignins such as Kraft lignin that are hard to valorise via biocatalysis or chemocatalysis.22 Significant increases in product formation were observed for treatment of Kraft lignin by DyP peroxidase enzymes and Agro LigE or Prx, and some product release was observed for the condensed Avantium lignin, which can be solubilised to a small extent by AST. Product release was also observed for Avantium humin (see Fig. 2), although we were not able to identify specific low molecular weight products formed from humin breakdown.
The chemical structures of the products observed by LC-MS analysis are illustrated in Fig. 6. Monomeric products arise mainly from either aryl-Cα oxidative cleavage (2-methoxyhydroquinone, hydroxyquinol), or Cα–Cβ oxidative cleavage (e.g. vanillin, vanillic acid), with demethylation evident in the formation of hydroxyquinol (see Fig. 6).
 | ||
| Fig. 6 Chemical structures of products observed from enzyme biotransformations (listed in Fig. 5), indicating which products are enhanced by addition of accessory enzymes. | ||
In the presence of accessory enzymes, the yield of monomeric products is increased (see Fig. 5). In the case of GVPL, product yield is enhanced by DHLDH2 or Prx, whereas for AVL, product yield is enhanced by AST, which aids lignin solubilisation. We also observed the formation of oxidised dimeric products, whose yield was also enhanced by addition of accessory enzymes.
Concentrations of specific products are enhanced in the presence of accessory enzymes (Fig. 5), and the overall yield of lignin conversion to low molecular weight products was shown to be enhanced for the Agro DyP/Agro LigE combination (Table 3). The practical issues involved in lignin depolymerisation such as structural complexity, solubility, and repolymerisation are still challenging to address,2 but the present work has shown the potential advantage of combining lignin-oxidising enzymes with accessory enzymes for the formation of aromatic bioproducts from lignin.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the Bio-Based Industries Joint Undertaking under the European Union Horizon 2020 Research and Innovation Programme under Grant Agreement 720303. We would like to thank Dr Annelie Jongerius (Avantium) for provision of lignin and humin samples, Dr Dominic Campopiano (University of Edinburgh) for provision of an expression strain for B. cenocepacia peroxiredoxin, and Prof. Stéphanie Baumberger (INRA Versailles) for helpful comments on the manuscript.References
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- T. D. H. Bugg and R. Rahmanpour, Curr. Opin. Chem. Biol., 2015, 29, 10–17 CrossRef CAS PubMed.
- D. W. S. Wong, Appl. Biochem. Biotechnol., 2009, 157, 174–209 CrossRef CAS PubMed.
- V. K. Gupta, C. P. Kubicek, J. G. Berrin, D. W. Wilson, M. Couturier, A. Berlin, E. X. F. Filho and T. Ezeji, Trends Biochem. Sci., 2016, 41, 633–645 CrossRef CAS PubMed.
- M. Ahmad, J. N. Roberts, E. M. Hardiman, R. Singh, L. D. Eltis and T. D. H. Bugg, Biochemistry, 2011, 50, 5096–5107 CrossRef CAS PubMed.
- M. E. Brown, T. Barros and M. C. Y. Chang, ACS Chem. Biol., 2012, 7, 2074–2081 CrossRef CAS PubMed.
- R. Rahmanpour and T. D. H. Bugg, Arch. Biochem. Biophys., 2015, 574, 93–98 CrossRef CAS PubMed.
- S. Majumdar, T. Lukk, J. O. Solbiati, S. Bauer, S. K. Nair, J. E. Cronan and J. A. Gerlt, Biochemistry, 2014, 53, 4047–4058 CrossRef CAS PubMed.
- R. S. Granja-Travez, R. C. Wilkinson, G. F. Persinoti, F. M. Squina, V. Fülöp and T. D. H. Bugg, FEBS J., 2018, 285, 1684–1700 CrossRef CAS PubMed.
- P. Picart, C. Müller, J. Mottweiler, L. Wiermans, C. Bolm, P. Dominguez de Maria and A. Schallmey, ChemSusChem, 2014, 7, 3164–3171 CrossRef CAS PubMed.
- D. L. Gall, H. Kim, F. Lu, T. J. Donohue, D. R. Noguera and J. Ralph, J. Biol. Chem., 2014, 289, 8656–8667 CrossRef CAS PubMed.
- W. S. Kontur, C. A. Bingman, C. N. Olmsted, D. R. Wassarman, A. Ulbrich, D. L. Gall, R. W. Smith, L. M. Yusko, B. G. Fox, D. R. Noguera, J. J. Coon and T. J. Donohue, J. Biol. Chem., 2018, 293, 4955–4968 CrossRef CAS PubMed.
- H. Voβ, C. Amata Heck, M. Schallmey and A. Schallmey, Appl. Environ. Microbiol., 2020, 86, e02026 Search PubMed.
- A. Hernández-Ortega, P. Ferreira and A. T. Martínez, Appl. Environ. Microbiol., 2012, 93, 1395–1410 Search PubMed.
- M. D. Cameron and S. D. Aust, Enzyme Microb. Technol., 2001, 28, 129–138 CrossRef CAS PubMed.
- R. Rahmanpour, L. D. W. King and T. D. H. Bugg, Biochem. Biophys. Res. Commun., 2017, 482, 57–61 CrossRef CAS PubMed.
- F. Guillén, V. Gómez-Toribio, M. Jesús Martínez and A. T. Martínez, Arch. Biochem. Biophys., 2000, 383, 142–147 CrossRef PubMed.
- F. Huang, J. Fang, X. Lu, P. Gao and J. Chen, Chin. Sci. Bull., 2001, 46, 1956 CrossRef CAS.
- P. Picart, H. Liu, P. M. Grande, N. Anders, L. Zhu, J. Klankermayer, W. Leitner, P. Domínguez de María, U. Schwaneberg and A. Schallmey, Appl. Microbiol. Biotechnol., 2017, 101, 6277–6287 CrossRef CAS PubMed.
- D. L. Gall, W. S. Kontur, W. Lan, H. Kim, Y. Li, J. Ralph, T. J. Donohue and D. R. Noguera, Appl. Environ. Microbiol., 2018, 84, e02076 CrossRef PubMed.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- C. S. Lancefield, G. M. M. Rashid, F. Bouxin, A. Wasak, W.-C. Tu, J. Hallett, S. Zein, J. Rodríguez, S. D. Jackson, N. J. Westwood and T. D. H. Bugg, ACS Sustainable Chem. Eng., 2016, 4, 6921–6930 CrossRef CAS.
- M. E. Himmel, K. K. Oh, D. W. Sopher and H. L. Chum, J. Chromatogr. A, 1983, 267, 249–265 CrossRef CAS.
- R. S. Granja-Travez and T. D. H. Bugg, Arch. Biochem. Biophys., 2018, 660, 97–107 CrossRef CAS PubMed.
- D. J. Clarke, X. P. Ortega, C. L. Mackay, M. A. Valvano, J. R. W. Govan, D. J. Campopiano, P. Langridge-Smith and A. R. Brown, Biochemistry, 2010, 49, 1319–1330 CrossRef CAS PubMed.
- G. M. M. Rashid, C. R. Taylor, Y. Liu, X. Zhang, D. Rea, V. Fülöp and T. D. H. Bugg, ACS Chem. Biol., 2015, 10, 2286–2294 CrossRef CAS PubMed.
- Y. Sato, H. Moriuchi, S. Hishiyama, Y. Otsuka, K. Oshima, D. Kasai, M. Nakamura, S. Ohara, Y. Katayama, M. Fukuda and E. Masai, Appl. Environ. Microbiol., 2009, 75, 5195–5201 CrossRef CAS PubMed.
- F. Tonina, E. Vignalia, L. Pollegionia, P. D'Arrigob and E. Rosini, Enzyme Microb. Technol., 2017, 96, 143–150 CrossRef PubMed.
- G. M. M. Rashid, M. J. Duran-Pena, R. Rahmanpour, D. Sapsford and T. D. H. Bugg, J. Appl. Microbiol., 2017, 123, 159–171 CrossRef CAS PubMed.
- M. S. Quesenberry and Y. C. Lee, Anal. Biochem., 1996, 234, 50–55 CrossRef CAS PubMed.
- M. A. Van Der Horst, A. F. Hartog, R. El Morabet, A. Marais, M. Kircz and R. Wever, Eur. J. Org. Chem., 2015, 534–541 CrossRef CAS.
- R. S. Granja-Travez, G. F. Persinoti, F. M. Squina and T. D. H. Bugg, Appl. Microbiol. Biotechnol., 2020, 104, 3305–3320 CrossRef CAS PubMed.
- M. Kumar, S. Verma, R. K. Gazara, M. Kumar, A. Pandey, P. K. Verma and I. S. Thakur, Biotechnol. Biofuels, 2018, 11, 154 CrossRef PubMed.
- D. Zhu, P. Zhang, C. Xie, W. Zhang, J. Sun, W.-J. Qian and B. Yang, Biotechnol. Biofuels, 2017, 10, 44 CrossRef PubMed.
- P. Prinsen, A. Narani, A. F. Hartog, R. Wever and G. Rothenberg, ChemSusChem, 2017, 10, 2267–2273 CrossRef CAS PubMed.
- A. Smith, Avantium renewable chemistries update, International Forest Biorefining Conference, Thunder Bay, Ontario, Canada, 2017 Search PubMed.
- J. Wang, J. Xi and Y. Wang, Green Chem., 2015, 17, 737–751 RSC.
- I. van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00431j |
| This journal is © The Royal Society of Chemistry 2021 |