
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation, dynamics, and long-term stability of Mn- and Fe-promoted Rh/SiO2 catalysts in CO hydrogenation†
Phil
Preikschas
 *a,
Julia
Bauer
a,
Kristian
Knemeyer
a,
Raoul
Naumann d'Alnoncourt
*a,
Ralph
Kraehnert
a and
Frank
Rosowski
ab
*a,
Julia
Bauer
a,
Kristian
Knemeyer
a,
Raoul
Naumann d'Alnoncourt
*a,
Ralph
Kraehnert
a and
Frank
Rosowski
ab
aBasCat, UniCat BASF JointLab, Technische Universität Berlin, 10623 Berlin, Germany. E-mail: p.preikschas@bascat.tu-berlin.de; r.naumann@bascat.tu-berlin.de
bProcess Research and Chemical Engineering, Heterogeneous Catalysis, BASF SE, 67056 Ludwigshafen, Germany
First published on 21st June 2021
Abstract
The conversion of syngas (CO/H2) to ethanol (StE) is one promising example to generate a high-value fuel and key intermediate for various base chemicals, preferably from non-fossil carbon resources. Rh-Based catalysts demonstrated the highest selectivities towards C2+ oxygenates and ethanol, in particular. However, the accomplished yields still must be increased, and the catalyst's stability must be improved for industrial application. One primary strategy to improve C2+ oxygenate yields over Rh is the addition of one or several promoters. Specifically, Mn and Fe are among the most frequently used metals to improve rhodium's catalytic performance in binary and ternary systems. To date, experimental studies primarily focused on increasing the C2+ oxygenate yields, but long-term catalytic investigations are only rarely reported. Consequently, Mn and Fe's specific role as promoter and their influence on the long-term and thermal stability of supported Rh catalysts are not clarified as yet. A holistic view of atomistic promoter effects and their impact on the stability and dynamics of Rh-based catalysts under reaction conditions is thereby highly desired. Herein, we report a comprehensive study about the stability and dynamics of Mn- and Fe-promoted Rh/SiO2 catalysts at industrially relevant high-pressure conditions (54 bar). For this purpose, unpromoted Rh/SiO2, single-promoted RhMn/SiO2 and RhFe/SiO2, and complex multi-promoted RhMnFe/SiO2 catalysts were systematically investigated in four different states: calcined, reduced, after a long-term catalytic study (>22 days on stream), and after a high temperature stability investigation (T = 243–320 °C). The thorough analysis of each catalyst in the different states with integral and local characterization methods led to specific structural models before and after long-term catalytic investigations. These structural models provide a detailed view on compositions, electronic properties, and morphologies of promoted Rh/SiO2 catalysts and serve as a basis for improved catalyst design strategies and more sophisticated computational modeling efforts.
Introduction
Depletion of fossil resources and the increasing demand of a growing world population lead to new challenges to provide industry and society with chemicals. In this manner, syngas (CO/H2) as an alternative feedstock for the production of base chemicals becomes increasingly interesting in view of growing climate and carbon management concerns.1 The production of synthetic ethanol is one promising example of this transition. To date, ethanol is mainly produced by fermentation of sugars from corn or sugarcane. This process is inefficient, energy-intensive, and directly competes with food sources raising ethical issues.2 For this reason, the direct conversion of syngas to ethanol (StE) is a promising alternative route from non-fossil carbon resources.Over the last decades, various catalyst systems have been tested for the direct conversion of StE, and Rh-based catalysts offer the most promising results.1–4 However, the accomplished yields still must be increased, and the catalyst's stability must be improved before industrial applications become viable. Despite these heavy research efforts, the entire complexity of this reaction at process-relevant conditions has not been unraveled so far.2,5 Until now, experimental studies primarily focused on increasing the C2+ oxygenate yields, but long-term investigations (>100 h on stream) are still limited.2,6 Subsequently, the stability of Rh-based catalysts and the influence of promoters on Rh's deactivation behavior are only rarely studied. Furthermore, specific promoter effects and related structure–function relationships cover mainly the initial reactivity of Rh-based catalysts.5 However, studies on supported metal catalysts have shown that a catalyst can undergo a change in activity and selectivity over time on stream during an initial formation phase.7–9
One primary strategy to improve C2+ oxygenate yields over Rh is the addition of one or several promoters. In this manner, a wide range of metallic and oxidic promoters, including transition metals and rare-earth elements, have been tested. Mn and Fe are among the most frequently used metals to improve Rh's catalytic performance in binary and ternary systems.2
Even so the reaction network and active Rh sites are still elusive, ethanol is likely formed on Rh through hydrogen-assisted dissociation of adsorbed CO, formation of CHx (x = 1–3) surface fragments, insertion of CHO/CO into Rh–CHx bonds, and subsequent hydrogenation.1–3,10,11 As Mn addition has shown high potential to increase C2+ oxygenate yields and ethanol, in particular, its role in the StE reaction has been described as promoting CO dissociation and CO insertion.2 However, the influence of Mn on Rh's morphological and electronic properties is rarely discussed. Experimental evidence of specific promoter effects has been contributed by Mao et al. that tilted adsorbed CO species at Rh–MnOx interfacial sites cause increased CO dissociation rates that lead to enhanced activity and selectivity.12 Other studies contradict this hypothesis and doubt the relevance of tilted adsorbed CO species for reactivity.13 Whereas others proposed that MnOx stabilizes Rh+ sites at their interface and ultimately promotes CO insertion.14–18 On the contrary, Yu and co-workers proposed that Rh+ sites are not stable under reaction conditions and thereby not relevant for the StE reaction.19
Similarly, the role of Fe as promoter for Rh-based catalysts is still under debate and controversially discussed. On the one hand, increased ethanol selectivities and suppressed methane formation were observed and ascribed to the formation of RhFe nanoalloy structures under reaction conditions.20–23 In contrast, the stabilization of Rh+ sites has been proposed through FeOx species in their vicinity.24–26 Similar to RhMnOx, these sites enhance molecular CO adsorption and finally CO insertion. Besides the promotional effect of Fe in enhancing ethanol formation, Mo and co-workers ascribed higher selectivities towards methane and methanol to the addition of Fe. Subsequently, only a small improvement in EtOH selectivity was observed.27
Despite these conversely discussed promoter effects, it is widely accepted that a close proximity of the promoter and Rh is needed, the so-called strong metal–promoter interactions.28–30 However, a lack of long-term catalytic investigations and thorough characterization of spent samples after catalytic reaction have limited atomistic insights into the Rh–promoter interface. Likewise, the specific role of each promoter and their influence on Rh's catalytic properties over longer periods of time are not clarified yet. For these reasons, a holistic view of promoter effects on an atomic level and their impact on Rh-based catalysts under reaction conditions is highly desired.
Herein, we report a comprehensive study about the stability and dynamics of Mn- and Fe-promoted Rh-based catalysts under industrially relevant high-pressure conditions (54 bar, 243–320 °C). Long-term catalytic investigations (>22 days on stream time) combined with an extended catalyst characterization provided simplified structural models of each catalyst before and after catalysis. We foresee that these structural models will serve as a basis for improved catalyst design strategies and more sophisticated computational modeling efforts.
Experimental section
Catalyst synthesis
The four different catalysts were synthesized according to a previously reported procedure.6,29 Aqueous solutions of the respective metal nitrates were impregnated on the silica support (Davisil Grade 636, Sigma-Aldrich) by incipient wetness impregnation method. Drying and calcination were performed in four steps under a constant flow of synthetic air (500 mL min−1) with a ramp of 5 K min−1 at 80, 100, 120, and 350 °C for 30, 30, 180, and 180 min, respectively. After calcination, the pre-catalysts were sieved to receive the target particle size for catalytic testing of 250–315 μm. Metal loadings from ICP-OES (wt%): 2.2 Rh (Rh/SiO2); 2.4 Rh, 1.6 Mn (RhMn/SiO2); 2.3 Rh, 0.5 Fe (RhFe/SiO2); 2.3 Rh, 1.5 Mn, 0.5 Fe (RhMnFe/SiO2). Atomic percent of metals (at%): 1.0 Rh (Rh/SiO2); 1.0 Rh, 1.3 Mn (RhMn/SiO2); 1.0 Rh, 0.4 Fe (RhFe/SiO2); 1.0 Rh, 1.2 Mn, 0.4 Fe (RhMnFe/SiO2).Catalyst characterization
All samples have been transferred under ambient conditions after calcination, reduction, long-term catalytic investigation, and high temperature study for sample characterization.Powder X-ray diffraction (XRD) measurements were performed in Bragg–Brentano geometry on a D8 Advance II theta/theta diffractometer (Bruker AXS), using Ni-filtered Cu Kα1,2 radiation and a position sensitive energy dispersive LynxEye silicon strip detector. The sample powder was filled into the recess of a cup-shaped sample holder, the surface of the powder bed being flush with the sample holder edge (front loading).
Scanning transmission electron microscopy (STEM), energy-dispersive X-ray spectroscopy (EDX) were conducted on a FEI Talos F200X microscope. The microscope was operated at an acceleration voltage of 200 kV. STEM-EDX elemental maps were recorded by a Super-X system including four silicon drift detectors. Background-corrected and fitted intensities were used for image visualization. All samples were prepared on holey carbon-coated copper grids (Plano GmbH, 400 mesh). Particle size distributions were determined by measuring at least 250 particles by using ImageJ software.31
X-ray photoelectron spectroscopy (XPS) was measured on K-Alpha™ + X-ray Photoelectron Spectrometer System (Thermo Scientific), with Hemispheric 180° dual-focus analyzer with 128-channel detector. This system uses a micro-focused, monochromatic Al Kα X-ray source powered at 6 mA and 12 kV. Charge compensation was performed using a dual-beam flood source of low-energy Ar+ ions and low energy electrons (less than 1 eV). For the measurement, the as-prepared samples were directly loaded on the sample holder. The data was collected with X-ray spot size of 200 μm, 20 scans for survey, and 50 scans for regions. The pass energy was set at 200 eV for survey and 50 eV for high-resolution spectra. All survey spectra (1400–0 eV, 1.0 eV step size) and high-resolution spectra (0.1 eV step size) of Si 2p (116–92 eV), C 1s (300–276 eV), Rh 3d (328–296 eV), O 1s (544–524 eV), and Fe 2p (740–700 eV) regions are provided in the ESI.† Data analysis was carried out using Thermo Scientific Avantage software. For composition analysis, a method for carbon contamination correction proposed by Smith has been applied.32
Elemental analysis (Rh, Mn, Fe, Mg) was performed via inductively coupled plasma optical emission spectrometry (ICP-OES) by the contract laboratory Mikroanalytisches Labor Kolbe, Oberhausen (Germany).
Catalytic testing for synthesis gas conversion
The catalytic testing of the syngas-to-ethanol reaction was performed in a 4-fold parallel testing unit. Catalyst amounts of 0.1–0.5 g (approx. 0.2–1.0 mL) with a particle size of 100–200 μm were loaded into each stainless-steel reactor with an effective inner diameter of 6.25 mm. The reaction temperature was monitored by temperature sensors with three thermocouples along the catalyst bed.Four mass flow controllers were used to adjust the flow rates of the inlet gases N2 (99.999%), CO (99.997%), H2 (99.999%) and Ar (99.999%, all Air Liquide). The CO feed line was equipped with a carbonyl trap to remove all metal carbonyls that might be formed by high-pressure of CO in contact with stainless steel. The carbonyl trap consisted of a U-shaped 1/2′′ stainless steel tube filled with Al2O3 and heated to 170 °C by a heating sleeve.
Compounds in the effluent gas that condense at 180 °C were removed by a coalescence filter in the downstream oven. All remaining compounds in the effluent gas were analyzed with an online gas chromatograph (Agilent 7890B) equipped with one thermal conductivity detector (TCD) and one flame ionization detector (FID) using He as the carrier gas. TCD detects the inlet gases H2, Ar, N2, and CO. The FID is used to detect a large variety of paraffins, olefins and oxygenates (alcohols, acetaldehyde, acetic acid) using a Poraplot Q column. Installation of a Polyarc® reactor allowed detection of CO and CO2 with the FID and precise quantification of all compounds. The carbon balance was between 96–102% for all measurements.
The catalysts were reduced in situ at 54 bar, 265 °C with 5% H2 in N2 for 1 h with a volume flow of 41.6 NmL min−1. Subsequently, synthesis gas feedstock mixture containing CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2:N2:Ar (20:60:10:10%, v:v) was admitted at a total pressure of 54 bar. The volume flow was kept constant to achieve a GHSV of 2500–12500 h−1. The amount of catalyst was chosen to yield approx. 5% CO conversion at standard conditions. The temperature was varied in a range of 243–320 °C. Each step was held constant for at least 15 h to allow the catalysts to equilibrate.
:H2:N2:Ar (20:60:10:10%, v:v) was admitted at a total pressure of 54 bar. The volume flow was kept constant to achieve a GHSV of 2500–12500 h−1. The amount of catalyst was chosen to yield approx. 5% CO conversion at standard conditions. The temperature was varied in a range of 243–320 °C. Each step was held constant for at least 15 h to allow the catalysts to equilibrate.
The obtained concentrations of all compounds were corrected for volume changes due to the reaction and the subsequent N2 dilution. Therefore, the mole fraction of Ar was used as inert internal standard according to eqn (1).
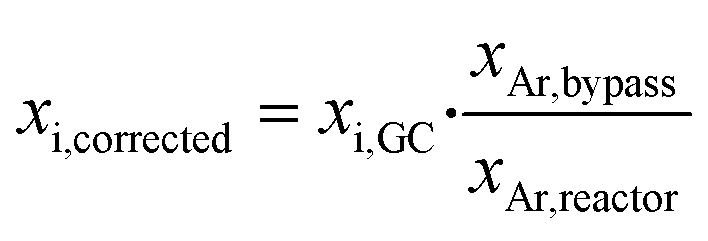 | (1) |
Carbon monoxide conversion XCO was calculated based on the sum of carbon numbers in all products (eqn (2)).
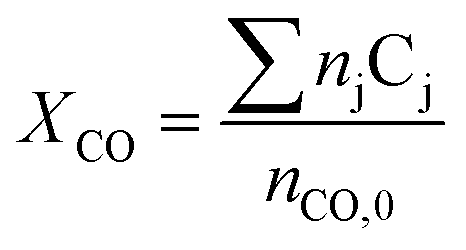 | (2) |
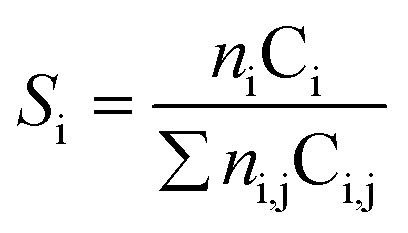 | (3) |
Results and discussion
Four different Rh-based samples were synthesized by co-impregnation of metal nitrates, drying, and calcination at 350 °C in flowing air. Besides a pure Rh/SiO2 catalyst with a nominal loading of 2.4 wt%, three modified catalysts with Mn and/or Fe as promoters were investigated with nominal loadings of 1.5 and 0.5 wt%, respectively (for exact values from ICP-OES analysis see catalyst synthesis in Experimental section). These Mn and Fe loadings were selected based on preliminary catalytic tests to identify the highest ethanol space–time yields. Especially the Fe loading has a crucial impact on methanol and ethanol formation rates.22,33 In the case of Mn, the chosen Mn:Rh ratio is in agreement with the optimal molar ratio of 1 reported by Ojeda and co-workers.34
Throughout this work, the catalysts will be referred to with the simplified expressions Rh/SiO2, RhMn/SiO2, RhFe/SiO2, and RhMnFe/SiO2 due to readability. This denotation does not contain information about oxidation states or different morphologies.
The four catalysts were tested in two independent catalytic tests: (1) a comprehensive long-term catalytic study with over 22 days on stream at 243–260 °C and (2) a high temperature study, which focused on the stability of the materials investigated, with a maximum temperature of 320 °C applied. For both studies, the as-prepared, calcined pre-catalysts were reduced in situ prior to catalytic testing at 265 °C in 5% H2/N2 and measured under industrially high-pressure conditions (54 bar). To investigate the stability and surface dynamics of the Rh/Mn/Fe/SiO2 catalysts under high-pressure conditions, every catalyst system was thoroughly characterized in the four different states: calcined (black), reduced (blue; after in situ H2 treatment), after long-term catalytic study (dark green), and after high temperature stability investigation (light green; Scheme 1).
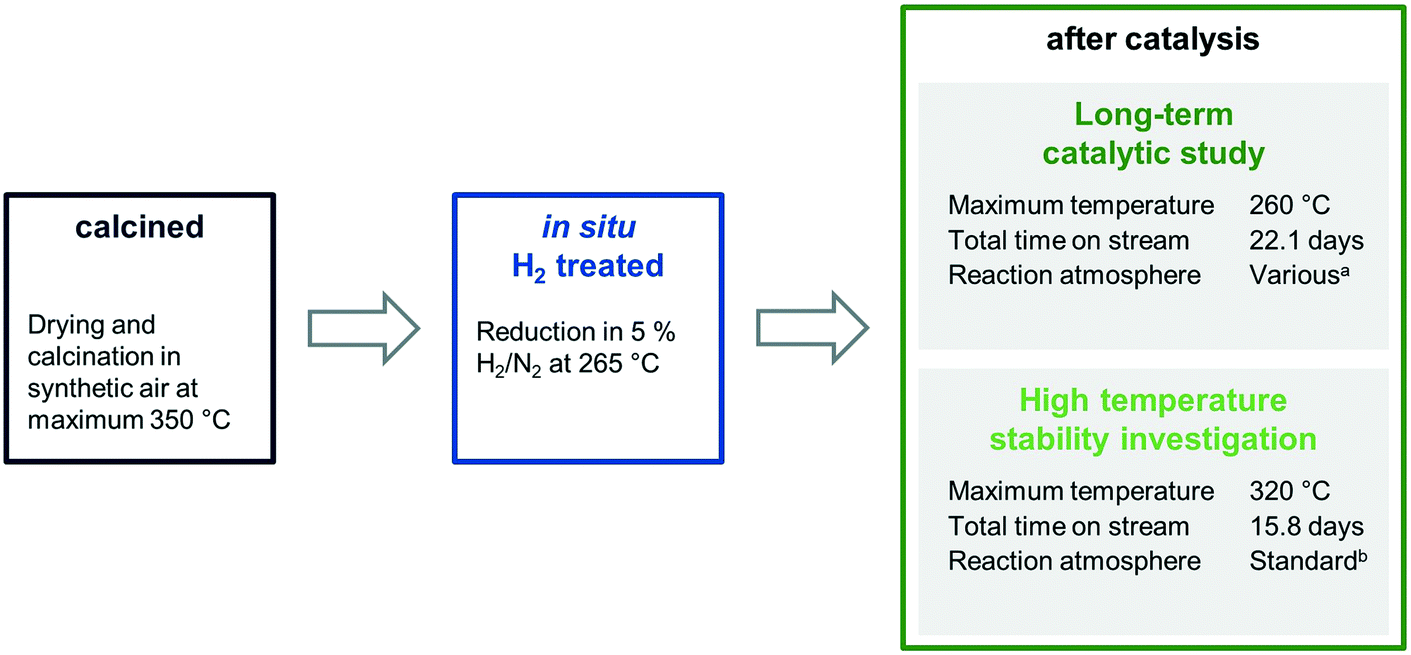 | ||
| Scheme 1 Overview of the four different states in which each sample was investigated. aReaction atmosphere was varied: CO:H2:Ar:N2 = 2.5–20:30–60:10:balance in 18 steps at 54 bar total pressure. bStandard reaction atmosphere: CO:H2:Ar:N2 = 20:60:10:10 at 54 bar total pressure. | ||
The characterization results of different states provide meaningful insights into catalyst composition, electronic properties, and morphology. Their different nanostructures before and after catalysis are represented as simplified structural models, which provide the opportunity to clarify the role of Mn and Fe in binary and ternary Rh catalysts on an atomic level and industrially relevant reaction times.
Bulk and surface compositions of Rh/Mn/Fe/SiO2 catalysts
The bulk compositions of all four catalysts remained the same before and after catalytic testing, as proved by ICP-OES. Therefore, any influence of changing bulk compositions on the reactivity during catalysis can be excluded.Moreover, stable bulk compositions are necessary to investigate surface compositions of each catalyst via XPS. These surface compositions were determined by comparing the respective elemental peak areas from high-resolution scans after carbon contamination correction (see Experimental section for more details). The Si 2p signal (103.5 eV) was used as charge reference. For all samples, the Si:O ratio is about 1:2, as expected for SiO2 as support. Si and O together make up 98–99 at% of the entire surface (Table 1).
| Entry | Sample | Treatment | Surface composition (at%) | Mn/Fe/Rh surface ratio | Mn/Fe/Rh bulk ratio from ICP | ||||
|---|---|---|---|---|---|---|---|---|---|
| Si | O | Rh | Mn | Fe | |||||
| a Surface compositions have been determined from high-resolution scans. Si 2p, O 1s, Rh 3d, and Fe 2p high-resolution spectra are provided in the ESI,† and Mn 2p spectra as Fig. 2. | |||||||||
| 1 | SiO2 | Calcination | 33 | 66 | — | — | — | — | — |
| 2 | Rh/SiO2 | Calcination | 33 | 66 | 0.38 | — | — | — | — |
| 3 | Reduction | 33 | 66 | 0.32 | — | — | — | — | |
| 4 | High temperatures | 33 | 66 | 0.34 | — | — | — | — | |
| 5 | Long-term study | 33 | 66 | 1.02 | — | — | — | — | |
| 6 | RhMn/SiO2 | Calcination | 33 | 66 | 0.42 | 0.46 | — | 1.1/—/1 | 1.2/—/1 |
| 7 | Reduction | 33 | 66 | 0.31 | 0.48 | — | 1.5/—/1 | ||
| 8 | High temperatures | 33 | 66 | 0.37 | 0.84 | — | 2.3/—/1 | ||
| 9 | Long-term study | 33 | 65 | 0.65 | 1.43 | — | 2.2/—/1 | ||
| 10 | RhFe/SiO2 | Calcination | 33 | 66 | 0.46 | — | 0.52 | —/1.1/1 | —/0.4/1 |
| 11 | Reduction | 33 | 66 | 0.54 | — | 0.78 | —/1.4/1 | ||
| 12 | High temperatures | 33 | 66 | 0.27 | — | 0.60 | —/2.2/1 | ||
| 13 | Long-term study | 33 | 66 | 0.52 | — | 0.85 | —/1.6/1 | ||
| 14 | RhMnFe/SiO2 | Calcination | 33 | 66 | 0.40 | 0.45 | 0.41 | 1.1/1.1/1 | 1.2/0.4/1 |
| 15 | Reduction | 33 | 66 | 0.33 | 0.60 | 0.48 | 1.8/1.4/1 | ||
| 16 | High temperatures | 33 | 66 | 0.29 | 0.55 | 0.49 | 1.8/1.7/1 | ||
| 17 | Long-term study | 33 | 65 | 0.58 | 1.01 | 0.79 | 1.7/1.4/1 | ||
An overall Rh surface content of around 0.38 at% was found for the unpromoted Rh/SiO2 sample, which was slightly decreased after H2 reduction. Interestingly, the Rh surface content increased by a factor of 3.2 to 1.02 at% after the long-term catalytic study (Table 1, entries 3 and 5). This finding suggests that a significantly higher Rh dispersion on the silica surface is present after catalysis.
For the calcined RhMn/SiO2 sample, the Mn:Rh ratio was about 1.1 as expected from bulk composition analysis (Table 1, entry 6). Rh and Mn show thereby a comparable distribution over the sample, and their ratio is slightly increased after H2 treatment. After catalysis, the Mn:Rh ratio was significantly increased with values of 2.3 and 2.2, respectively (Table 1, entries 8 and 9). This suggests an increased mobility of MnOx under reaction conditions.
Moreover, this increased ratio and a lower increase in Rh dispersion (factor of 2.1; Table 1, entries 7 and 9) might be an indication for migration of MnOx species to Rh nanoparticles and partial coverage of Rh surface sites. A complementary observation was also reported for similar Mn-promoted Rh/SiO2 catalysts, suggesting the formation of MnOx overlayers at the Rh–MnOx interface through strong metal–promoter interactions.28
In the case of RhFe/SiO2, a Fe:Rh ratio of 1.1 was determined after calcination (Table 1, entry 10). This ratio does not fit the bulk composition (0.33), which leads to the assumption that Fe is finely distributed over the support surface, and thereby an agglomeration of Fe can be excluded. After catalysis, the Fe surface content was further increased, but to a smaller extent compared to Mn (Table 1, entries 11 and 13). Therefore, Fe might also be mobile on the surface of silica under reaction conditions, similar to the case of Mn. In contrast to the unpromoted and Mn-promoted Rh catalyst, the Rh dispersion remained the same after long-term catalytic study and was significantly decreased after the high temperature investigation.
The complex multi-promoted RhMnFe/SiO2 catalyst shows similar trends as described for the single-promoted systems resulting in Mn/Fe/Rh ratio of 1.1/1.1/1 after calcination (Table 1, entry 14). Reduction in H2 leads to an increase in Mn surface content. In contrast, the reduction of the pre-catalyst has no significant influence on the surface content of Fe. After catalysis, further enrichment of Mn and Fe contents by factors of 1.7 and 1.6 were observed, respectively (Table 1, entries 15 and 17).
To conclude, the Rh dispersion increased in the monometallic and Mn-containing samples after the catalytic conversion of syngas to ethanol. Mn and Fe seem to be mobile under reaction conditions, and partial coverage of Rh by a MnOx phase is rather likely. Overall, the XPS analysis reveals a significant influence of the applied reaction conditions on the specific surface composition.
Oxidation states and electronic properties of Rh/Mn/Fe/SiO2 catalysts
To investigate the oxidation states, electronic properties, and phase compositions in the four states mentioned in Scheme 1, XPS and XRD were applied. In the calcined state of all pre-catalysts investigated, Rh is present as Rh2O3 as indicated by the most intense reflection of Rh2O3 at 2θ = 35° in the respective X-ray diffractograms (Fig. 1a). This reflection is clearly visible in direct comparison with the diffractogram of the pure SiO2 support (grey; Fig. 1). In the case of RhFe/SiO2, this feature is broader, which might be caused by formation of a complex mixture of Rh and Fe oxides as reported for Rh(Mn,Fe)Ox/SiO2 catalysts.28,29 The XRD data is further consistent with XPS analysis showing typical binding energy peaks of Rh3+ at 308.6–308.9 eV in corresponding Rh 3d spectra (Fig. S1a–d†).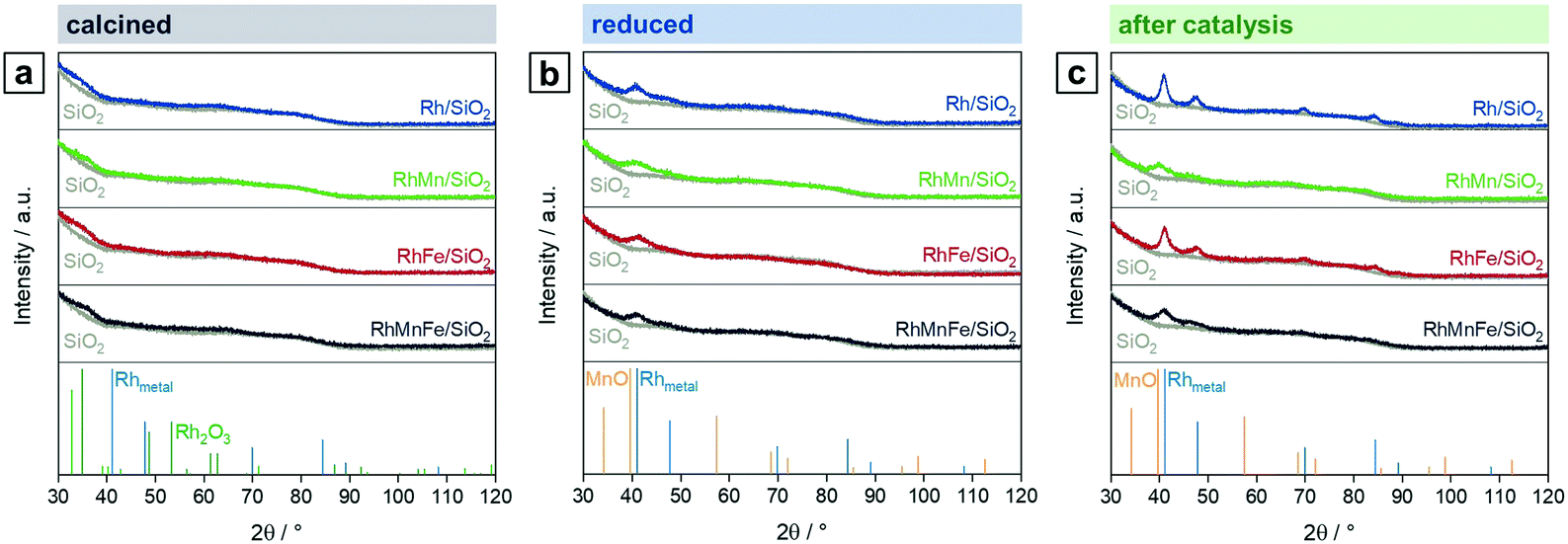 | ||
| Fig. 1 X-ray diffractograms of Rh/SiO2 (blue), RhMn/SiO2 (green), RhFe/SiO2 (red), and RhMnFe/SiO2 (black) in direct comparison with pure SiO2 (grey) after (a) calcination (350 °C, synthetic air), (b) reduction (265 °C, 5% H2/N2), and (c) after catalysis (long-term catalytic study; 54 bar, max. 260 °C; 530 h on stream). Rh (blue; C5-685), Rh2O3 (green; C41-541), and MnO (orange; 065-0638) references were taken from ICDD database. | ||
The H2 treatment leads to a full reduction of Rh reflected in binding energy shifts to the typical region for metallic Rh from 307.4–306.9 eV (Fig. S1a–d and Table S1†). Subsequent exposure to ambient air after reduction does not cause oxidation of supported Rh. The full reduction of Rh2O3 to Rh also becomes visible in XRD spectra of Rh/SiO2 and RhFe/SiO2, showing a broad peak at the position of the typical main reflection of metallic Rh (2θ = 41°; Fig. 1b). An exact assignment is not possible regarding the Mn-promoted catalysts due to a broadening of the signal and the common detection limit of XRD below crystallite sizes of 3 nm.35 Moreover, this broadening might be a result of an overlap with a MnO reflection, which usually appears in the same range (2θ = 39.7°; Fig. 1b). Hence, it can be assumed that the addition of Mn to Rh/SiO2 catalysts leads to a higher Rh dispersion after synthesis and in situ reduction.
Respective Mn 2p spectra of RhMn/SiO2 and RhMnFe/SiO2 show binding energy peaks in the distinct region of oxidized Mn at 641.6 and 641.5 eV, respectively (Fig. 2a and b). Not even the reduction in H2, or the long-term catalytic study under high-pressure syngas condition, led to a further reduction of Mn. This is in accordance with one of our previously reported studies about RhMnOx/SiO2 catalysts. In this study, we demonstrated that the oxidation of Mn to MnOx is inevitable on silica supports. Impregnation of Na[Mn(CO)]5, bearing Mn in the low oxidation state of −1, did not lead to the formation of a zero-valent Mn species.30 The presence of typical satellite peaks in all Mn 2p spectra after reduction or catalysis indicates that Mn is presumably in a formal oxidation state of +2 (Fig. 2a and b). It is consequently assumed that most likely MnO is the prevailing phase.
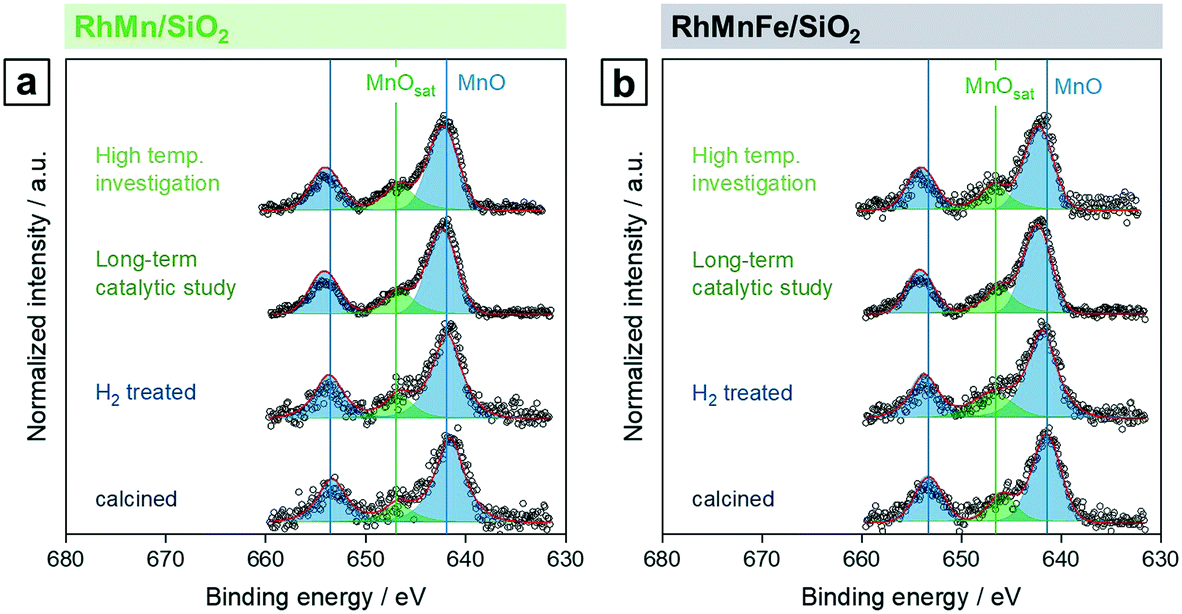 | ||
| Fig. 2 Mn 2p high-resolution XP spectra (660–630 eV, 0.1 eV step size) of (a) RhMn/SiO2 and (b) RhMnFe/SiO2 in four different states: calcined (350 °C, synthetic air), reduced (265 °C, 5% H2/N2), after long-term catalytic study (54 bar, max. 260 °C; 530 h on stream), and after high temperature stability investigation (54 bar, max. 320 °C; 350 h on stream). For corresponding Rh 3d and Fe 2p spectra see Fig. S1 and S2.† | ||
After long-term catalysis, significant binding energy shifts of 0.7 eV to higher binding energies (642.3 and 642.2 eV, respectively) were observed in respective spectra of RhMn/SiO2 and RhMnFe/SiO2 (Fig. 2a and b and Table S1†). These shifts are probably caused by forming Mn acetates and/or carbonates under the reductive syngas conditions. In a recent study, we reported the formation of surface carbonates on MnOx-supported Rh catalysts during the conversion of syngas based on XRD analysis of respective spent catalysts.36 Due to the low Mn loading, the formation of acetate or carbonate species on RhMn/SiO2 and RhMnFe/SiO2 could not be verified by XRD. Therefore, further investigations are required for a clear statement.
On the contrary, no significant changes in the high-resolution Fe 2p spectra have been observed for the Fe-containing samples, RhFe/SiO2 and RhMnFe/SiO2, after catalytic reactions. Subsequently, the Fe 2p XPS data indicate that Fe is mainly in an oxidized state, probably in the form of Fe2O3 (Fig. S2a and b†). However, a shift to lower binding energies indicating that Fe got more reduced under reaction conditions or a contribution of metallic Fe states have been expected as observed for similar RhFe/SiO2 catalysts due to RhFe nanoalloy formation.36 Concerning the low Fe content and thereby low intensities of corresponding Fe 2p signals, no reliable information regarding a metallic Fe phase was available from XPS. Furthermore, the formation of a surface passivation layer upon sample handling under ambient conditions might explain the apparently high amounts of Fe oxides present after catalytic reaction. However, additional investigations might be required to clarify the Rh and Fe speciation, e.g., in the form of in situ experimentation.
Influence of reaction conditions on particle sizes of Rh/Mn/Fe/SiO2 catalysts
For all catalysts in each specific state, particle size distributions were determined by systematically measuring at least 250 different particles per sample (Fig. S3†). For this purpose, bright-field scanning transmission electron microscopy (BF-STEM) images of three different areas were examined. The calcined samples have not been investigated due to their high beam-sensitivity and consequent insignificance.28 All particle diameters of metallic Rh particles and crystallite sizes of Rh2O3 are summarized in Table 2.| Treatment | Rh | RhMn | RhFe | RhMnFe |
|---|---|---|---|---|
| a Crystallite sizes were estimated with Langford and Wilson's representation of the Scherrer equation.37 | ||||
| Calcination | 1.3 (XRD) | 1.4 (XRD) | 1.5 (XRD) | 1.6 (XRD) |
| Reduction | 2.9 ± 0.9 | 2.4 ± 0.7 | 2.9 ± 0.8 | 2.2 ± 0.5 |
| High temperature | 3.9 ± 0.9 | 2.5 ± 0.7 | 2.8 ± 0.7 | 2.2 ± 0.5 |
| Long-term study | 4.4 ± 1.4 | 2.9 ± 1.0 | 3.3 ± 1.2 | 3.1 ± 0.9 |
The four catalysts show relatively small mean Rh2O3 crystallite sizes after calcination ranging from 1.3–1.6 nm from XRD (Table 2). In situ reduction in H2 led to metallic Rh particles with the smallest mean particle sizes of 2.4 and 2.2 nm for RhMn/SiO2 and RhMnFe/SiO2, respectively. The mean particle sizes of Rh/SiO2 and RhFe/SiO2 are significantly larger with a value of 2.9 nm. It is assumed that Mn addition leads to a higher Rh dispersion already after in situ reduction. All catalysts show a similar, narrow particle size distribution reflected in small standard deviations of 0.5–0.8 nm (Table 2 and Fig. S3†).
After long-term catalytic study, a slight particle growth was observed for all catalysts, which is further consistent with XRD results. The unpromoted Rh/SiO2 catalyst shows the largest increase in particle size with a factor of 1.5 and a value of 4.4 nm. The particles on the promoted catalysts are about 25–35% smaller in size than on Rh/SiO2. Interestingly, the addition of Mn leads to the smallest particles with mean sizes of 2.9 nm for RhMn/SiO2 and 3.1 nm for RhMnFe/SiO2 (Table 2).
However, these findings are in contrast to the aforementioned surface compositions obtained from XPS data, showing a higher Rh dispersion after catalysis for all catalysts investigated. Suzuki et al. reported that metallic Rh clusters on Al2O3 could disintegrate into isolated Rh+ sites through CO chemisorption.38 A similar Rh particle disintegration on silica under the influence of high CO partial pressures could explain the increased Rh dispersion besides a slight particle growth. It should be noted that those Rh+ sites are not detectable by STEM or XRD due to common detection limits. However, further investigations about the disintegration of Rh particles on SiO2 are part of further studies and will be required for a clear proof.
Although the RhMnFe/SiO2 catalyst shows the smallest mean particle size after reduction, its particles are slightly larger after catalysis in comparison to RhMn/SiO2, which is further consistent with respective surface compositions from XPS analysis (Table 1). Considering the indication of RhFe nanoalloys formation from XPS in this context, the alloy formation probably follows incorporation of Fe into Rh nanoparticles. This incorporation would then lead to larger particles and would explain the more pronounced particle growth. This assumption is in accordance with our previous study about the in situ formation of RhFe nanoalloy structures through reduction of FeOx at Rh–FeOx interfacial sites via hydrogen spillover.36
To summarize, Mn addition to silica-supported Rh catalysts leads to a stabilization of the Rh particles and prevents agglomeration by reducing the mobility of Rh on the support. Consequently, Rh particles on MnO/SiO2 almost retained their size after high temperature investigation and are slightly increased after long-term catalytic study. Therefore, an expected particle growth has been reduced. The addition of Fe does not suppress particle growth to the same extent and leads to larger particles on RhMnFe/SiO2 compared to RhMn/SiO2. This increase in particle size suggests RhFe nanoalloy formation through the incorporation of Fe into Rh nanoparticles.
Influence of reaction conditions on the morphology of Rh/Mn/Fe/SiO2 catalysts
STEM in combination with EDX spectroscopy was performed on the freshly reduced and spent samples after long-term catalytic study and high temperature investigation. The morphology and general structural features of the four different Rh-based catalysts were evaluated from images taken with dark-field (DF), bright-field (BF), and high-angle annular dark-field (HAADF) detectors. STEM-EDX mappings were used to investigate the elemental distribution over the silica support, and line profile scans were performed to analyze the elemental composition of the nanoparticles.Representative overview STEM images of the unpromoted Rh/SiO2 catalyst show well-dispersed particles over the support after in situ reduction (Fig. 3a). The corresponding EDX mapping clearly demonstrates that only pure Rh nanoparticles are present, indicated by the overlap of the HAADF and Rh EDX signals (Fig. S4a†). The respective EDX spectrum further proved that no contaminations such as Fe are present (Fig. S4b†). Those contaminations are often correlated with the reactivity of Rh/SiO2 catalysts in syngas conversion.39 After long-term catalysis, the Rh nanoparticles are unevenly distributed over the support and structured mostly in agglomerates of a few particles (<10 particles; Fig. 3b).
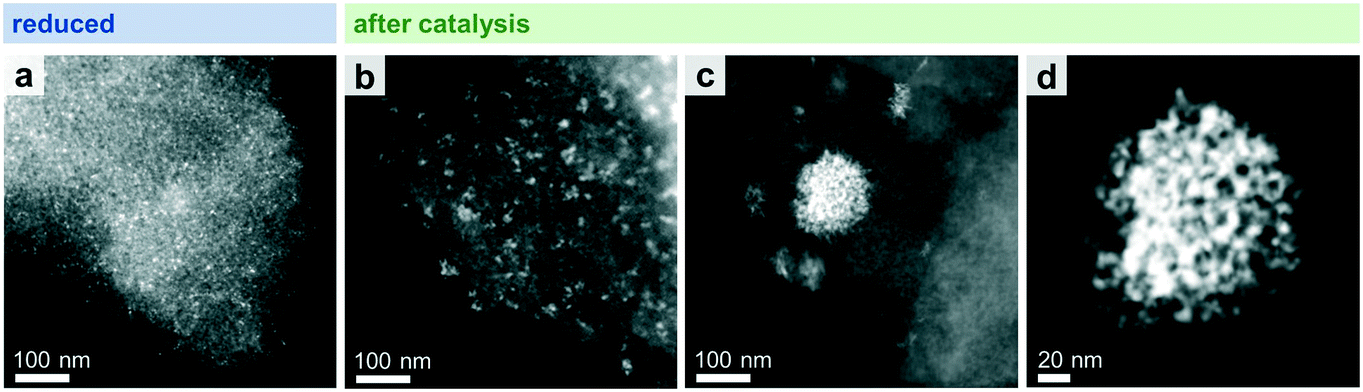 | ||
| Fig. 3 HAADF-STEM images of Rh/SiO2 after (a) in situ reduction (265 °C, 5% H2/N2) and (b–d) catalysis (long-term catalytic study; 54 bar, max. 260 °C; 530 h on stream). (b) Representative domain with smaller agglomerates (<10 particles) and (c) minority domain with larger agglomerates. (d) High-resolution STEM of one single agglomerate from image (c). | ||
While this is the case for most of the domains investigated, some areas are also found containing large agglomerates with sizes of 20–200 nm (Fig. 3c). High-resolution STEM could resolve their individual nanoparticles (Fig. 3d).
In the case of RhMn/SiO2, the STEM-EDX mapping of the freshly reduced sample visualizes that Rh nanoparticles are surrounded by a MnO phase (Fig. 4a). Likewise, the formation of RhMn nanoalloy structures can be excluded, as no overlap between the Rh and Mn EDX signals were found (Fig. 4a). In addition, Mn is probably in an amorphous, oxidized phase as no crystalline particles were observed.
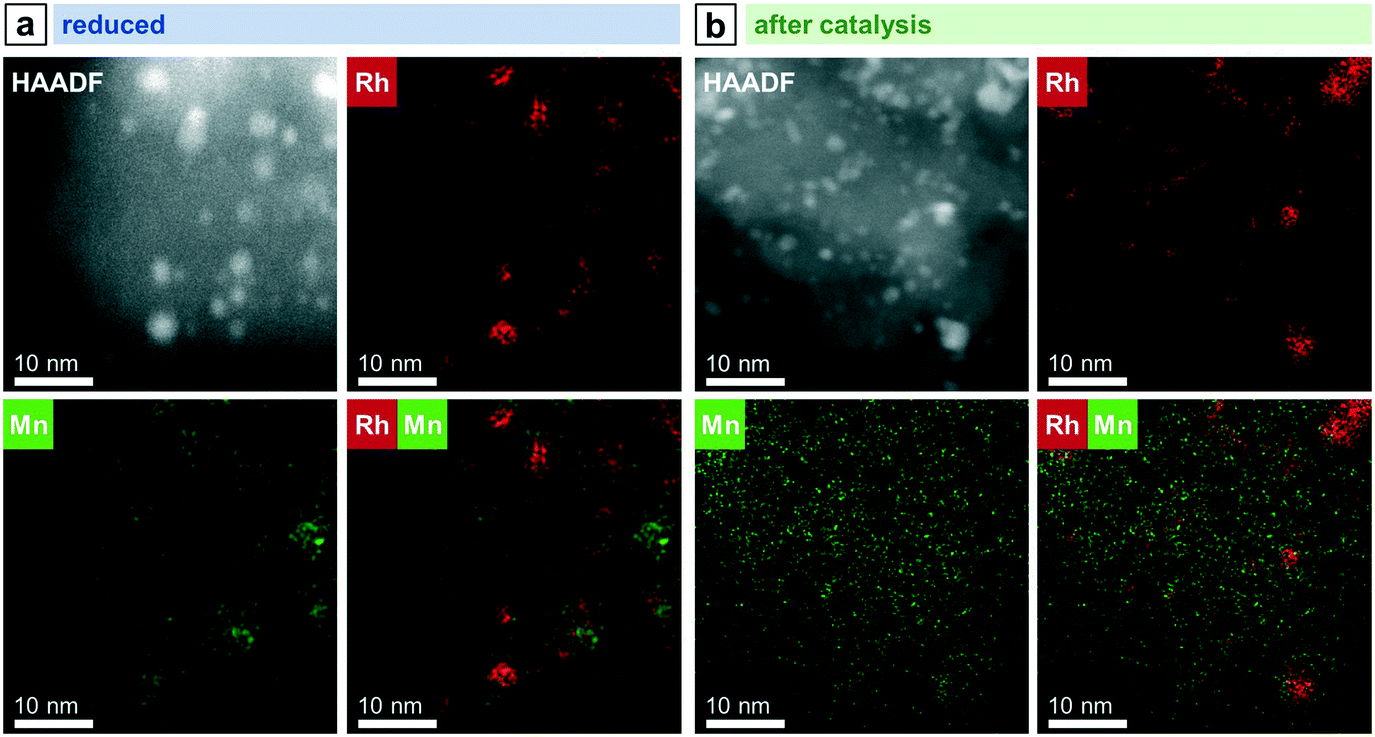 | ||
| Fig. 4 STEM-EDX mappings of RhMn/SiO2 after (a) reduction and (b) catalysis (long-term catalytic study; 54 bar, max. 260 °C; 530 h on stream). Rh L and Mn K EDX signals were used. For additional single-element maps and corresponding EDX spectra see Fig. S5 and S6.† | ||
In contrast to the unpromoted Rh/SiO2 catalyst, the Rh nanoparticles on RhMn/SiO2 stay well-dispersed also after catalysis, and agglomeration of individual particles to the same extent was not observed (Fig. 5a). In addition, corresponding STEM-EDX mapping of RhMn/SiO2 after catalysis reveals a higher dispersion of Mn (Fig. 4b), which is in accordance with previously mentioned XPS results and an X-ray absorption spectroscopy (XAS) study demonstrating a higher Rh dispersion on a Mn-promoted Rh catalyst after exposure to a CO/He atmosphere at elevated temperatures.40 It is consequently assumed that the Rh nanoparticles are anchored through the well-dispersed MnO phase, reducing their mobility over the support. Thus, the amorphous MnO phase prevents mainly sintering and agglomeration of Rh particles.
 | ||
| Fig. 5 Representative overview STEM images of (a) RhMn/SiO2, (b) RhFe/SiO2, and (c) RhMnFe/SiO2 after reduction (265 °C, 5% H2/N2) and after catalysis (long-term catalytic study; 54 bar, max. 260 °C; 530 h on stream). RhFe/SiO2: like Rh/SiO2 (Fig. 3), agglomerates of individual Rh particles can be seen beside relatively small nanoparticles after catalysis. | ||
Complementary STEM overview imaging (Fig. 5b) and STEM-EDX investigations on the reduced RhFe/SiO2 catalyst suggest the presence of well-dispersed Rh nanoparticles and a finely distributed Fe phase before catalytic studies (Fig. S7†). Furthermore, no domains with local enrichments of Rh and Fe in the same areas are found. RhFe nanoalloy formation does thereby not occur during in situ reduction. However, after long-term catalytic study, overlapping Rh and Fe signals in the superimposed STEM-EDX mapping indicate the formation of RhFe nanoalloy structures (Fig. 6a). Corresponding difference maps of Rh L and Fe K of two independent domains visualize the distribution of Fe which is not in the vicinity to Rh (Fig. S9; for all maps and EDX spectra see Fig. S8 and S10†). As no agglomeration of Fe has been observed, it is consequently assumed that the “free” Fe is still finely distributed over the silica support and most likely in an oxidized state.
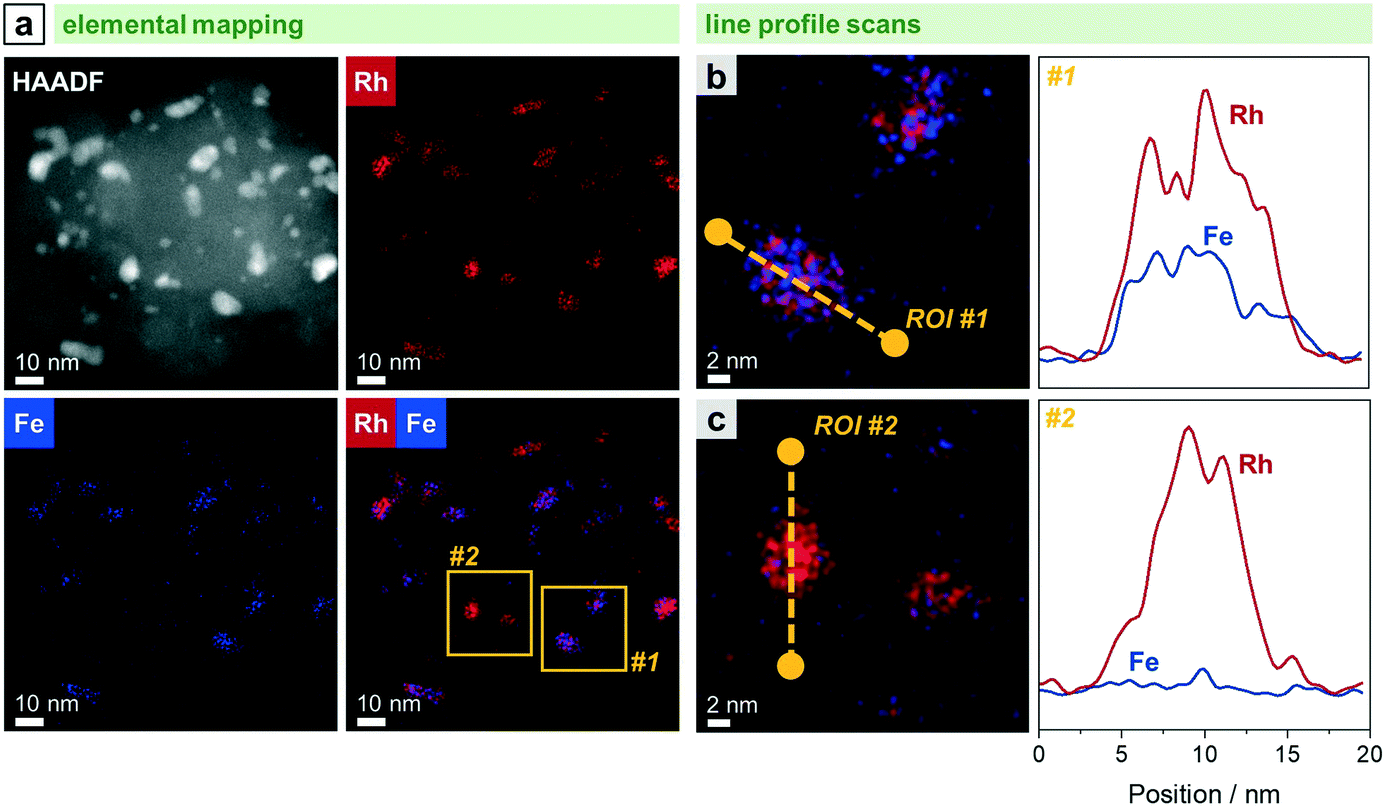 | ||
| Fig. 6 STEM-EDX analyses of RhFe/SiO2 after catalysis (long-term catalytic study; 54 bar, max. 260 °C; 530 h on stream): (a) HAADF micrograph and STEM maps of Fe K and Rh L with domains investigated with line profile scans highlighted in yellow. Elemental line profile scans and corresponding line profiles of Fe K and Rh L for a representative (b) RhFe alloy particle within area #1 and (c) metallic Rh particle within area #2. For additional single-element maps and corresponding EDX spectrum, see Fig. S8.† | ||
The in situ transformation of pure Rh nanoparticles to RhFe nanoalloys during StE reaction is in accordance with our recent study about similar RhFeOx/SiO2 catalysts36 and other reported RhFe nanoalloy structures.20,23,39
Representative elemental line profile scans further proved the alloying of Rh with Fe (Fig. 6b and S11†). Owing to the shape of the corresponding line profile intensities, RhFe bulk alloy formation is rather likely than surface alloy structures. Compositional analyses based on EDX area-selective investigations of several RhFe alloy particles indicate the formation of nanoalloys with an average molar Fe:Rh ratio of 0.34 (Fig. S12 and Table S2†). However, no exact composition could be determined due to different degrees of alloying. In this manner, Rh-rich alloy nanoparticles and even pure Rh nanoparticles were found (Fig. 6c). The elemental line profile scan on a representative Rh particle clearly shows the absence of Fe (Fig. 6c). In comparison to the bulk composition obtained from ICP-OES (Fe:Rh = 0.40), about 15% of the Fe is not found in RhFe alloy structures and probably present as oxidic species. However, these findings are in contrast with the previously mentioned Fe 2p XPS results indicating Fe mainly in an oxidized state and no indications from metallic Fe states (Fig. S2†). The formation of a passivation layer during sample handling might explain the apparently high amounts of Fe oxides detected by XPS due to its surface sensitivity. Furthermore, the main reflection in the corresponding X-ray diffractogram is slightly shifted to 2θ = 41.1° compared to the (111) reflection of metallic Rh at 40.9° of the unpromoted Rh/SiO2 (Fig. S13†). This shift and the asymmetric tailing toward higher 2θ values might indicate an underlying contribution of Rh-rich bimetallic RhFe phases which should appear between metallic Rh and RhFe (42.7; C25-1408). However, no reliable information about the exact composition of the bimetallic RhFe phases could be obtained from XRD data due to the relatively broad reflections from the small crystallites.
Combining both elements as modifiers in RhMnFe/SiO2 catalyst also results in a combination of both features observed in the bimetallic catalysts. After the H2 treatment, the Rh nanoparticles are well-dispersed over the support (Fig. 5c), and no correlation of Rh, Mn, and Fe EDX signals was observed in the corresponding STEM-EDX mappings (Fig. 7a). Therefore, it is assumed that Rh is in an unalloyed state surrounded by well-dispersed oxidic Mn and Fe species.
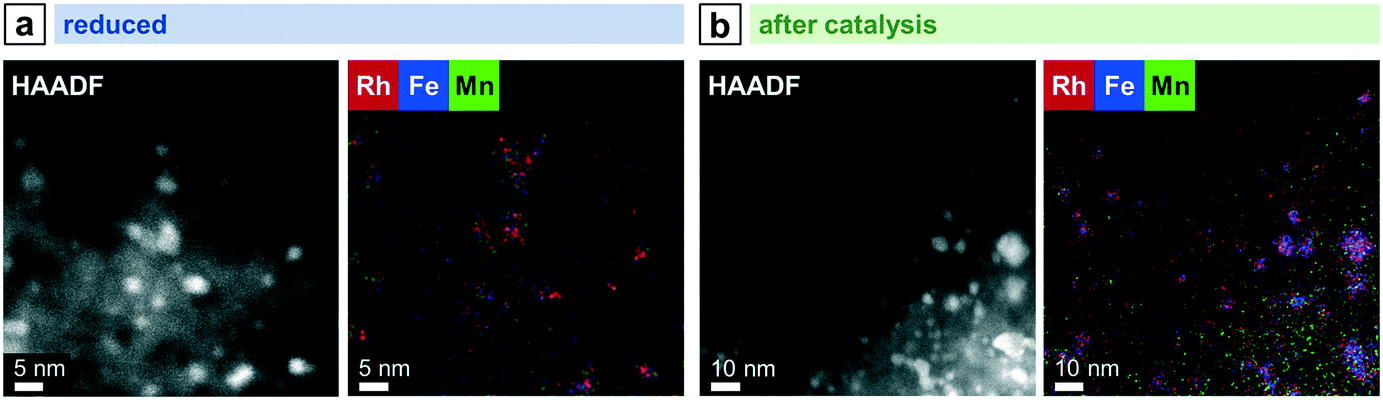 | ||
| Fig. 7 STEM-EDX mappings of RhMnFe/SiO2 after (a) reduction (265 °C, 5% H2/N2) and (b) catalysis (long-term catalytic study; 54 bar, max. 260 °C; 530 h on stream). Rh L, Mn K, and Fe K EDX signals were used. For additional single-element maps and corresponding EDX spectra see Fig. S14 and S15.† | ||
After catalysis, the nanoparticles remain relatively small, as indicated by the overview STEM image (Fig. 5c). The superimposed mapping of Rh and Mn visualizes that MnO is in vicinity to nanoparticles that mainly appear as alloyed RhFe nanostructures probably formed under high-pressure syngas conditions (Fig. 7b). Furthermore, MnO leads to the anchoring of the RhFe alloy nanoparticles and reduces their mobility on the support, similar to the single-promoted RhMn/SiO2 catalyst.
Selectivity at iso-conversion and reactivity of Rh/Mn/Fe/SiO2 catalysts
To investigate the influence of the described structural models of Mn and/or Fe-promoted Rh/SiO2 catalysts on their reactivity in the conversion of syngas, all four catalysts are compared at process-relevant reaction conditions (Fig. 8). As the addition of Fe and Mn to Rh/SiO2 leads to a significant increase in CO conversion, a preliminary catalytic study was conducted to identify the catalyst amounts necessary for iso-conversions after equilibration (>120 h on stream). Based on this study, the amounts of catalysts were varied to yield approx. 5% CO conversion. Only 33% (RhMnFe), 30% (RhMn), and 20% (RhFe) of promoted catalysts were required to match the conversion level of pure Rh/SiO2 catalyst. This procedure was necessary as the product selectivities of Rh-based catalysts are highly dependent on conversion during StE reaction. | ||
| Fig. 8 Comparison of (a) product selectivities at iso-conversion and (b) product formation rates among the four catalysts investigated. Below each column, CO conversion (a) and total CO consumption rate (b) are given. Reaction conditions: 54 bar, 260 °C, H2:CO:N2:Ar = 60:20:10:10, 41.7 mL min−1 total flow per reactor, GHSVs = 2500–12500 h−1, 170–190 h on stream. Catalyst amounts were varied to yield approx. 5% CO conversion: V(Rh) = 1.0 mL, V(RhMn) = 0.2 mL, V(RhFe) = 0.3 mL, V(RhMnFe) = 0.33 mL. | ||
Rh/SiO2 and RhMn/SiO2 catalysts show a similar overall product spectrum at iso-conversions (Fig. 8a). The main differences are significantly increased C2+ oxygenate and suppressed methane selectivities. An improvement in the C2+ oxygenate selectivity by the addition of Mn is well-known for Rh/SiO2 catalysts, as reported in previous studies.28,30,40 However, a clear explanation is still missing, and the role of Mn as a modifier or promoter for Rh-based catalysts is still elusive, to the best of our knowledge. By considering the aforementioned structural model of RhMn/SiO2, we propose that the MnO phase stabilizes the relatively small Rh nanoparticles and mainly prevents agglomeration. As the product spectrum does not change essentially, we assume that MnO has mainly an effect on the structural properties. This assumption is in accordance with an FTIR study indicating that Mn does not change Rh′s electronic properties.41 Owing to the stability of Rh nanoparticles and the almost retained Rh dispersion, the RhMn/SiO2 catalyst demonstrated the highest observed CO consumption rate among the catalysts tested (Fig. 8b).
In contrast, the conversion of syngas on RhFe/SiO2 yields methanol as main product with a selectivity of 50% (Fig. 8a). Therefore, it is apparent that the addition of Fe leads to a tremendous loss of C–C coupling ability and thus to an overall C2+ oxygenate selectivity below 20%. We reasoned that this strongly altered product spectrum is most likely caused by the close interaction of Rh and Fe. Therefore, the proposed alloying with Fe might have a vital impact on the electronic properties of Rh/SiO2 catalysts. Therefore, the changed electronic structure leads to fast hydrogenation of initially formed CHxO* (x = 1–3) surface fragments, which, in turn, are the main reaction intermediates for the formation of C2+ oxygenates.
Once again, the combination of Mn and Fe as modifiers on RhMnFe/SiO2 catalysts also leads to combined effects on their structural and electronic properties. This finding is also reflected in the catalyst reactivity (Fig. 8a). Compared to the monometallic Rh/SiO2 catalyst, RhMnFe/SiO2 shows an increased C2+ oxygenate selectivity in general and the highest ethanol selectivity among the catalysts investigated. Furthermore, the selectivity towards methanol is increased, accompanied by a reduced selectivity towards C2+ alkanes and alkenes.
Long-term and thermal stability of Rh/Mn/Fe/SiO2 catalysts
Two independent long-term catalytic studies were performed to investigate the influence of Mn and Fe on the stability of Rh/SiO2 catalysts during the conversion of syngas. With over 22 days (530 h) for the long-term catalytic study and over 14 days (350 h) on stream for the high temperature investigation, these catalytic experiments are the longest tests of Rh-based catalysts in the conversion of syngas to oxygenates reported in literature, to the best of our knowledge. These long periods of time on stream (TOS) are necessary to investigate Rh catalysts regarding their stability and performance in CO hydrogenation, as 120 h on stream are required to obtain stable catalytic behavior (see formation phase in Fig. 9).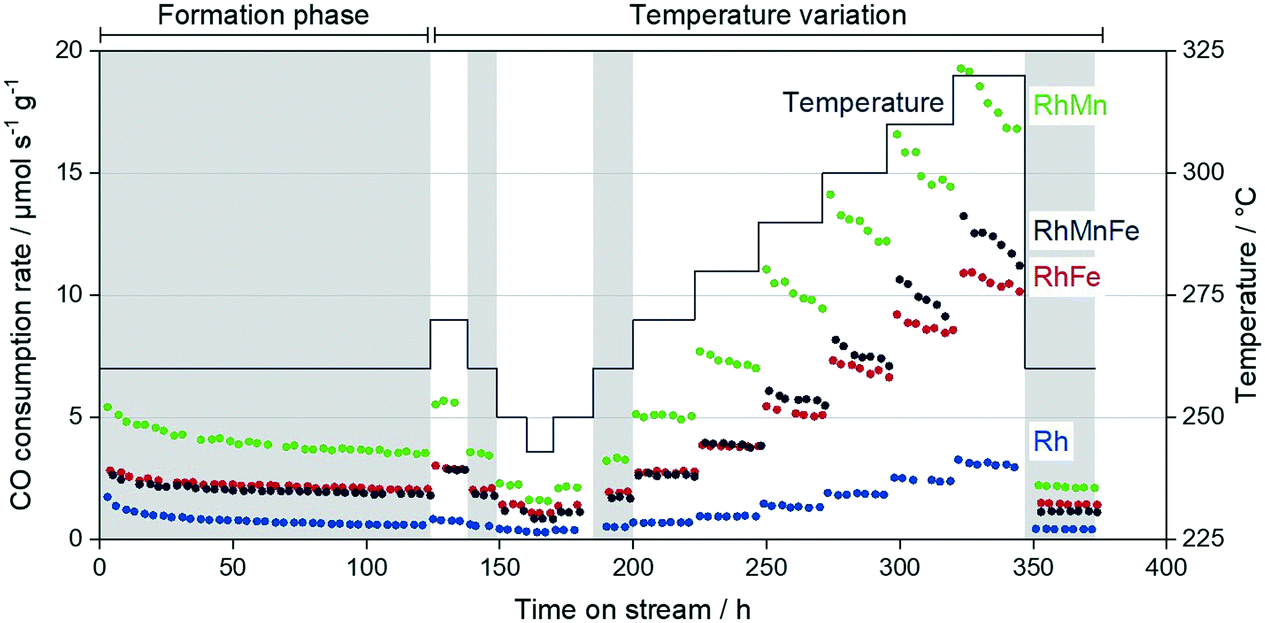 | ||
| Fig. 9 Overall CO consumption rate of Rh/SiO2, RhMn/SiO2, RhFe/SiO2, and RhMnFe/SiO2 over TOS during high temperature stability investigation. Reaction conditions: 54 bar, 243–320 °C, H2:CO:N2:Ar = 60:20:10:10, 41.7 mL min−1 total flow per reactor, GHSVs = 2500–12500 h−1. Catalyst amounts were varied to yield approx. 5% CO conversion: V(Rh) = 1.0 mL, V(RhMn) = 0.2 mL, V(RhFe) = 0.3 mL, V(RhMnFe) = 0.33 mL. Reference conditions are highlighted in grey. | ||
During this initial formation phase, all catalysts lose activity in terms of the overall CO consumption rate. This deactivation follows a similar trend as observed for particle growth after long-term catalytic investigation (Table 2). It is consequently assumed that sintering contributes to the deactivation of Rh-based catalysts under CO hydrogenation conditions, which has also been proposed for similar Rh-based catalysts.28 Besides migration of particles followed by coalescence, the migration of atoms (Ostwald ripening) has been described as the most important sinter mechanism.42 In this manner, a CO-assisted mechanism of sintering through Ostwald ripening has been proposed for supported cobalt catalysts under similar reaction conditions. The in situ formation of cobalt subcarbonyl species, Co(CO)x (x = 1–3), may increase the mobility of cobalt atoms over the hydroxylated support surface.43 Moreover, a theoretical study has been reported suggesting a similar sinter mechanism for Rh-based catalysts.44 As the formation of Rh(CO)2 dicarbonyl species under CO-containing atmosphere has been postulated,38 it is consequently proposed that sintering through migration of Rh(CO)x (x = 1, 2) carbonyl species is also relevant for supported Rh catalysts under high-pressure syngas conditions. However, further experimental and theoretical investigations are required for a clear statement.
Besides the described loss in activity, selectivities also gradually change during the initial formation phase. These changes are mainly caused by the increased mobility of Mn and Fe on the supports, yielding probably the formation of Rh–MnO interfacial sites and/or RhFe nanoalloy structures. In our high temperature investigation, we observed critical temperatures above 280–290 °C, which cause an accelerated loss in activity within each iso-thermal step (Fig. 9; TOS > 250 h). Interestingly, this deactivation is more pronounced for the Mn-containing catalysts and has a minor impact on Rh/SiO2 and RhFe/SiO2 catalysts.
To illustrate these differences in deactivation, linear regression analyses were performed for each individual temperature step, and the resulting activity loss rates were compared among the four catalysts investigated (Table S3; Fig. S16†). The Mn-containing catalysts show significant activity loss already above temperatures of 280 °C, whereas Rh/SiO2 and RhFe/SiO2 are still stable in terms of their CO consumption rates at these temperatures. The fastest deactivation represented in the lowest activity loss rate value of −0.13 μmol s−1 gcat−1 hTOS−1 was determined for the RhMn/SiO2 catalyst. With this value, the Mn-promoted catalyst deactivates three times faster than the single-promoted RhFe/SiO2 catalyst. Similar trends can be seen for the multi-promoted RhMnFe/SiO2 with the second-lowest activity loss rate of −0.08 μmol s−1 gcat−1 hTOS−1 (Table S3†).
Although the deactivation of the Mn-containing catalysts is more pronounced within each temperature step, the percentage changes in CO consumption rate between reference conditions (TOS: 190–199 h and >352 h) are comparable among the promoted catalysts with values ranging from −25.5 to −34.1% (Table 3).
| Catalyst | Step | TOS (h) | CO consumption rate (μmol s−1 gCat−1) | Percentage change (%) | Deactivation factor |
|---|---|---|---|---|---|
| Rh/SiO2 | Formation phase | 3 | 1.74 | −66.6 | 2.99 |
| 121 | 0.58 | ||||
| Temperature variation | 190–197 | 0.52 | −18.8 | 1.23 | |
| 351–372 | 0.42 | ||||
| RhMn/SiO2 | Formation phase | 3 | 5.42 | −34.7 | 1.53 |
| 122 | 3.54 | ||||
| Temperature variation | 190–197 | 3.27 | −34.1 | 1.52 | |
| 352–373 | 2.16 | ||||
| RhFe/SiO2 | Formation phase | 4 | 2.82 | −26.7 | 1.36 |
| 123 | 2.07 | ||||
| Temperature variation | 191–198 | 1.94 | −25.5 | 1.34 | |
| 353–374 | 1.44 | ||||
| RhMnFe/SiO2 | Formation phase | 5 | 2.63 | −31.5 | 1.46 |
| 124 | 1.80 | ||||
| Temperature variation | 192–199 | 1.71 | −33.1 | 1.50 | |
| 353–374 | 1.14 |
We consequently assume that the deactivation during temperature variation does not only result from a growth in particle size. This is also in agreement with our particle size investigations mentioned above, showing that the mean particle sizes of Rh nanoparticles on Mn-containing catalysts are only slightly increased after the catalytic testing (Table 2). For this reason, the deactivation might be caused by a temperature-induced surface restructuring, and it seems to be that this restructuring is reversible when returning to lower temperatures at the reference conditions. At these conditions, all four catalysts show stable CO consumption rates again. A further deactivation of the catalysts has not been observed during additional 20 h on stream at 260 °C. Therefore, it is assumed that temperatures above 280 °C facilitate MnO overlayer formation like catalysts in a strong or reactive metal–support interaction state.45–47 With longer reaction times, more active Rh sites might be blocked by MnO, which, in turn, leads to the strong deactivation behavior during the individual temperature steps above 290 °C (Fig. 9). This partial coverage of Rh active sites by MnO is in agreement with an FTIR study reported by Ojeda et al.34 and with a combined XAS and FTIR investigation by Schwartz and co-workers.41
Conclusions
To gather a fundamental understanding of promoter effects in the complex multi-promoted RhMnFe/SiO2 catalyst, the monometallic Rh/SiO2, and single-promoted catalysts, RhMn/SiO2 and RhFe/SiO2, were systematically investigated in four different states: calcined, reduced, after a long-term catalytic study, and after a high temperature investigation at industrially relevant high-pressure conditions (243–320 °C, 54 bar). The thorough analysis of each catalyst in the different states with integral and local characterization methods led to specific structural models before and after catalytic investigations (Scheme 2). These structural models provide a detailed view on compositions, electronic properties, and morphologies of silica-supported Rh-based catalysts.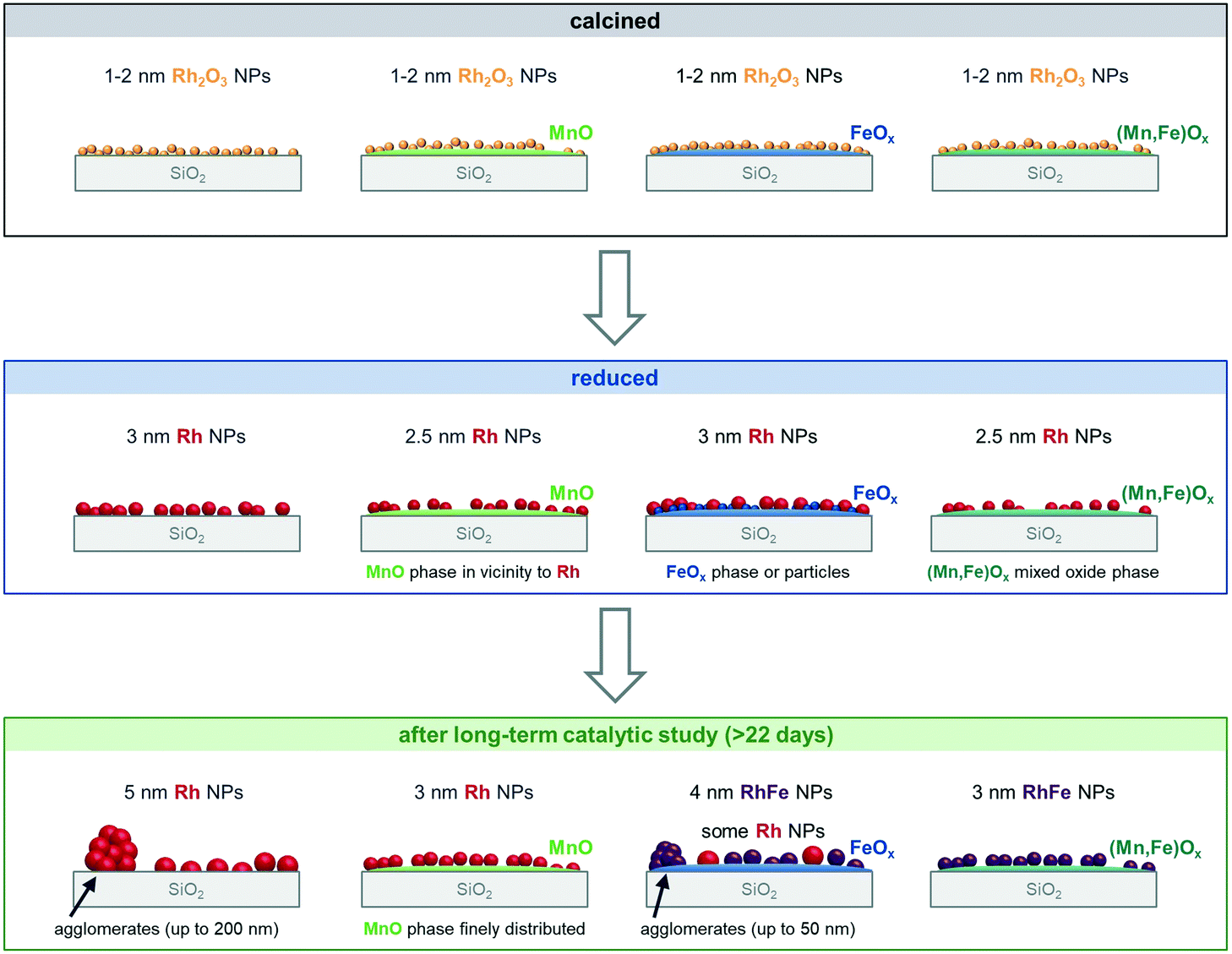 | ||
| Scheme 2 Structural models of Rh/SiO2, RhMn/SiO2, RhFe/SiO2, and RhMnFe/SiO2 after calcination, in situ H2 treatment and long-term catalytic study. Representation of the different morphologies based on combined analysis of XRD, STEM with EDX mapping, selective-area EDX analyses, elemental line profile scans, and XPS. | ||
Comparing the specific nanostructures before and after long-term catalytic studies (>22 days on stream) allowed us to ascribe individual reactivities to intrinsic promoter effects of Mn and Fe on silica-supported Rh catalysts. The distinct interactions of Rh with Mn and/or Fe develops during a relatively long time on stream of about 120 h under reaction conditions. Fe serves as an electronic modifier on Rh/SiO2 probably through the in situ formation of RhFe nanoalloy structures under the influence of high-pressure syngas conditions at elevated temperatures. Although the detailed STEM-EDX analysis suggested RhFe nanoalloy formation, no indications could be determined from XPS. For this reason, additional investigations might be required to clarify the Rh and Fe speciation. Nevertheless, the presumable change in Rh′s electronic properties might reason the alteration of the overall product spectrum resulted in less C–C coupling abilities and fast hydrogenation of CHO* surface fragment to methanol. On the contrary, MnO does not substantially change the intrinsic product spectrum of Rh/SiO2 and serves more likely as a structural modifier. In both Mn-containing catalysts, RhMn/SiO2 and RhMnFe/SiO2, respective Rh and/or RhFe nanoparticles are anchored to the silica-support by a MnO phase in their vicinity. With this structural feature, overall CO consumption rates and C2 oxygenate selectivity are significantly enhanced. MnO might be present as an overlayer, which probably blocks active Rh adsorption sites and gets more pronounced at temperatures above 260 °C. As this effect is of reversible nature, it might result from strong metal–promoter interaction similar to catalysts in reactive metal–support interaction states.
To conclude, the individual nanostructures represented as simplified structural models provide atomistic insights into the role of Mn and Fe in single- and multi-promoted Rh/SiO2 catalysts. The influence of both promoters on the deactivation and thermal stability could be clarified through long-term catalytic studies at process-relevant reaction conditions with more than 22 days on stream time. We foresee that these structural models will serve as a basis for improved catalyst design strategies and more sophisticated computational modeling efforts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted in the framework of the BasCat collaboration between BASF SE, TU Berlin, Fritz Haber Institute (FHI) of the Max Planck Society and was partially funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat. We would like to thank Dr. Frank Girgsdies for XRD, Dr. Shuang Li and Dr. Johannes Schmidt for XPS measurements. Dr. Michael Geske for technological advice, Felix Schuster and Martin Konrad for their assistance in laboratory work, Stephen Lohr and Jan Meißner for technical assistance. We would like to gratefully acknowledge Dr. Walid Hetaba and Dr. Thomas Lunkenbein for the opportunity to be part of the ChemiTEM project at the FHI.References
- V. Subramani and S. K. Gangwal, Energy Fuels, 2008, 22, 814–839 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514 RSC.
- P. C. Ellgen and M. Bhasin, US Pat., 4096164, 1976 Search PubMed.
- M. Ao, G. H. Pham, J. Sunarso, M. O. Tade and S. Liu, ACS Catal., 2018, 8, 7025–7050 CrossRef CAS.
- J. Hu, Y. Wang, C. Cao, D. C. Elliott, D. J. Stevens and J. F. White, Catal. Today, 2007, 120, 90–95 CrossRef CAS.
- S. Chen, A. M. Abdel-Mageed, D. Li, J. Bansmann, S. Cisneros, J. Biskupek, W. Huang and R. J. Behm, Angew. Chem., Int. Ed., 2019, 58, 10732–10736 CrossRef CAS PubMed.
- A. Rezvani, A. M. Abdel-Mageed, T. Ishida, T. Murayama, M. Parlinska-Wojtan and R. J. Behm, ACS Catal., 2020, 10, 3580–3594 CrossRef CAS.
- A. M. Abdel-Mageed, A. Klyushin, A. Knop-Gericke, R. Schlögl and R. J. Behm, J. Phys. Chem. Lett., 2019, 10, 3645–3653 CrossRef CAS PubMed.
- Y. H. Zhao, K. Sun, X. Ma, J. Liu, D. Sun, H. Y. Su and W. X. Li, Angew. Chem., Int. Ed., 2011, 50, 5335–5338 CrossRef CAS PubMed.
- R. Zhang, M. Peng and B. Wang, Catal. Sci. Technol., 2017, 7, 1073–1085 RSC.
- W. Mao, J. Su, Z. Zhang, X. C. Xu, D. Fu, W. Dai, J. Xu, X. Zhou and Y. F. Han, Chem. Eng. Sci., 2015, 135, 301–311 CrossRef CAS.
- A. S. Lisitsyn, S. A. Stevenson and H. Knözinger, J. Mol. Catal., 1990, 63, 201–211 CrossRef CAS.
- S. Wang, W. Guo, H. Wang, L. Zhu and K. Qiu, Catal. Lett., 2014, 144, 1305–1312 CrossRef CAS.
- G. Chen, X. Zhang, C.-Y. Guo and G. Yuan, C. R. Chim., 2010, 13, 1384–1390 CrossRef CAS.
- N. Yang, J. S. Yoo, J. Schumann, P. Bothra, J. A. Singh, E. Valle, F. Abild-Pedersen, J. K. Nørskov and S. F. Bent, ACS Catal., 2017, 7, 5746–5757 CrossRef CAS.
- Y. Wang, Z. Song, D. Ma, H. Luo, D. Liang and X. Bao, J. Mol. Catal. A: Chem., 1999, 149, 51–61 CrossRef CAS.
- Y. Wang, H. Luo, D. Liang and X. Bao, J. Catal., 2000, 196, 46–55 CrossRef CAS.
- J. Yu, J. Yu, Z. Shi, Q. Guo, X. Xiao, H. Mao and D. Mao, Catal. Sci. Technol., 2019, 9, 3675–3685 RSC.
- T. Hartman, R. G. Geitenbeek, G. T. Whiting and B. M. Weckhuysen, Nat. Catal., 2019, 2, 986–996 CrossRef CAS.
- M. Haider, M. Gogate and R. Davis, J. Catal., 2009, 261, 9–16 CrossRef CAS.
- R. M. Palomino, J. W. Magee, J. Llorca, S. D. Senanayake and M. G. White, J. Catal., 2015, 329, 87–94 CrossRef CAS.
- P. Carrillo, R. Shi, K. Teeluck, S. D. Senanayake and M. G. White, ACS Catal., 2018, 8, 7279–7286 CrossRef CAS.
- J. Wang, Q. Zhang and Y. Wang, Catal. Today, 2011, 171, 257–265 CrossRef CAS.
- W. Liu, S. Wang, T. Sun and S. Wang, Catal. Lett., 2015, 145, 1741–1749 CrossRef CAS.
- P. Carrillo, R. Shi, S. D. Senanayake and M. G. White, Appl. Catal., A, 2020, 608, 117845 CrossRef CAS.
- J. Gao, X. Mo and J. G. Goodwin Jr., J. Catal., 2009, 268, 142–149 CrossRef CAS.
- X. Huang, D. Teschner, M. Dimitrakopoulou, A. Fedorov, B. Frank, R. Kraehnert, F. Rosowski, H. Kaiser, S. Schunk, C. Kuretschka, R. Schlögl, M.-G. Willinger and A. Trunschke, Angew. Chem., Int. Ed., 2019, 58, 8709–8713 CrossRef CAS PubMed.
- M. Dimitrakopoulou, X. Huang, J. Kröhnert, D. Teschner, S. Praetz, C. Schlesiger, W. Malzer, C. Janke, E. Schwab, F. Rosowski, H. Kaiser, S. Schunk, R. Schlögl and A. Trunschke, Faraday Discuss., 2018, 208, 207–225 RSC.
- P. Preikschas, J. Bauer, X. Huang, S. Yao, R. Naumann d'Alnoncourt, R. Kraehnert, A. Trunschke, F. Rosowski and M. Driess, ChemCatChem, 2019, 11, 885–892 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- G. C. Smith, J. Electron Spectrosc. Relat. Phenom., 2005, 148, 21–28 CrossRef CAS.
- Y. Liu, F. Göeltl, I. Ro, M. R. Ball, C. Sener, I. B. Aragão, D. Zanchet, G. W. Huber, M. Mavrikakis and J. A. Dumesic, ACS Catal., 2017, 7, 4550–4563 CrossRef CAS.
- M. Ojeda, M. L. Granados, S. Rojas, P. Terreros, F. J. Garcia-Garcia and J. L. G. Fierro, Appl. Catal., A, 2004, 261, 47–55 CrossRef CAS.
- K. O'Connell and J. R. Regalbuto, Catal. Lett., 2015, 145, 777–783 CrossRef.
- P. Preikschas, M. Plodinec, J. Bauer, R. Kraehnert, R. Naumann d'Alnoncourt, R. Schlögl, M. Driess and F. Rosowski, ACS Catal., 2021, 11, 4047–4060 CrossRef CAS.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- A. Suzuki, Y. Inada, A. Yamaguchi, T. Chihara, M. Yuasa, M. Nomura and Y. Iwasawa, Angew. Chem., Int. Ed., 2003, 42, 4795–4799 CrossRef CAS PubMed.
- N. Yang, A. J. Medford, X. Liu, F. Studt, T. Bligaard, S. F. Bent and J. K. Nørskov, J. Am. Chem. Soc., 2016, 138, 3705–3714 CrossRef CAS PubMed.
- H. Bao, X. Sun, Z. Jiang, Y. Huang and J. Wang, Chin. J. Catal., 2014, 35, 1418–1427 CrossRef CAS.
- V. Schwartz, A. Campos, A. Egbebi, J. J. Spivey and S. H. Overbury, ACS Catal., 2011, 1, 1298–1306 CrossRef CAS.
- E. D. Goodman, A. C. Johnston-Peck, E. M. Dietze, C. J. Wrasman, A. S. Hoffman, F. Abild-Pedersen, S. R. Bare, P. N. Plessow and M. Cargnello, Nat. Catal., 2019, 2, 748–755 CrossRef CAS PubMed.
- M. Claeys, M. E. Dry, E. van Steen, P. J. van Berge, S. Booyens, R. Crous, P. van Helden, J. Labuschagne, D. J. Moodley and A. M. Saib, ACS Catal., 2015, 5, 841–852 CrossRef CAS.
- R. Ouyang, J.-X. Liu and W.-X. Li, J. Am. Chem. Soc., 2013, 135, 1760–1771 CrossRef CAS PubMed.
- S. Penner and M. Armbrüster, ChemCatChem, 2015, 7, 374–392 CrossRef CAS.
- E. Braunschweig, J. Catal., 1989, 118, 227–237 CrossRef CAS.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00421b |
| This journal is © The Royal Society of Chemistry 2021 |