
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of nickel(0) with organochlorides, organobromides, and organoiodides: mechanisms and structure/reactivity relationships
Megan E.
Greaves
 ab,
Elliot L. B.
Johnson Humphrey
a and
David J.
Nelson
*a
ab,
Elliot L. B.
Johnson Humphrey
a and
David J.
Nelson
*a
aWestCHEM Department of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, Scotland, UK. E-mail: david.nelson@strath.ac.uk
bChemical Development, Pharmaceutical Technology and Development, Operations, AstraZeneca, Macclesfield SK10 2NA, UK
First published on 8th April 2021
Abstract
The reactions of nickel(0) complexes with organohalides have been reviewed. The review is divided according to the class of ligand that is present on nickel: phosphine, N-heterocyclic carbene, or bidentate nitrogen ligand. The preferred mechanism of reaction is often determined by a delicate balance of ligand and substrate structure, and relatively small changes can lead to large differences in behaviour. This will have an impact on the progress of catalytic reactions that use organohalide substrates, and may influence ligand selection and/or the scope and limitations of the reaction.
Introduction
Nickel catalysis in organic synthesis
The application of nickel catalysis in organic synthesis has been the subject of widespread investigation during the past ten to fifteen years, and is currently one of the most exciting frontiers in catalysis.1 Many of the advances in nickel catalysis, such as the development of photoredox/cross-coupling reactions2,3 and reductive cross-electrophile coupling4 make use of the fact that nickel will readily access odd-numbered oxidation states.5However, the rather different properties of nickel compared to palladium (for example) lead to additional mechanistic complexity that requires considerable effort and resource to understand. This review therefore focuses on both classic and newer studies that aim to understand reaction mechanisms and structure/reactivity relationships in the reactions of nickel(0) with organohalides.
Reaction mechanisms
Before embarking on a detailed discussion of the literature, it is worthwhile to consider the mechanisms that might be in operation.6 These can be divided into approximately five categories (Scheme 1), although the divisions between some mechanisms may be blurred, and there may be further nuance in the exact details. Nevertheless, we can broadly consider reactions to proceed via one of the following processes.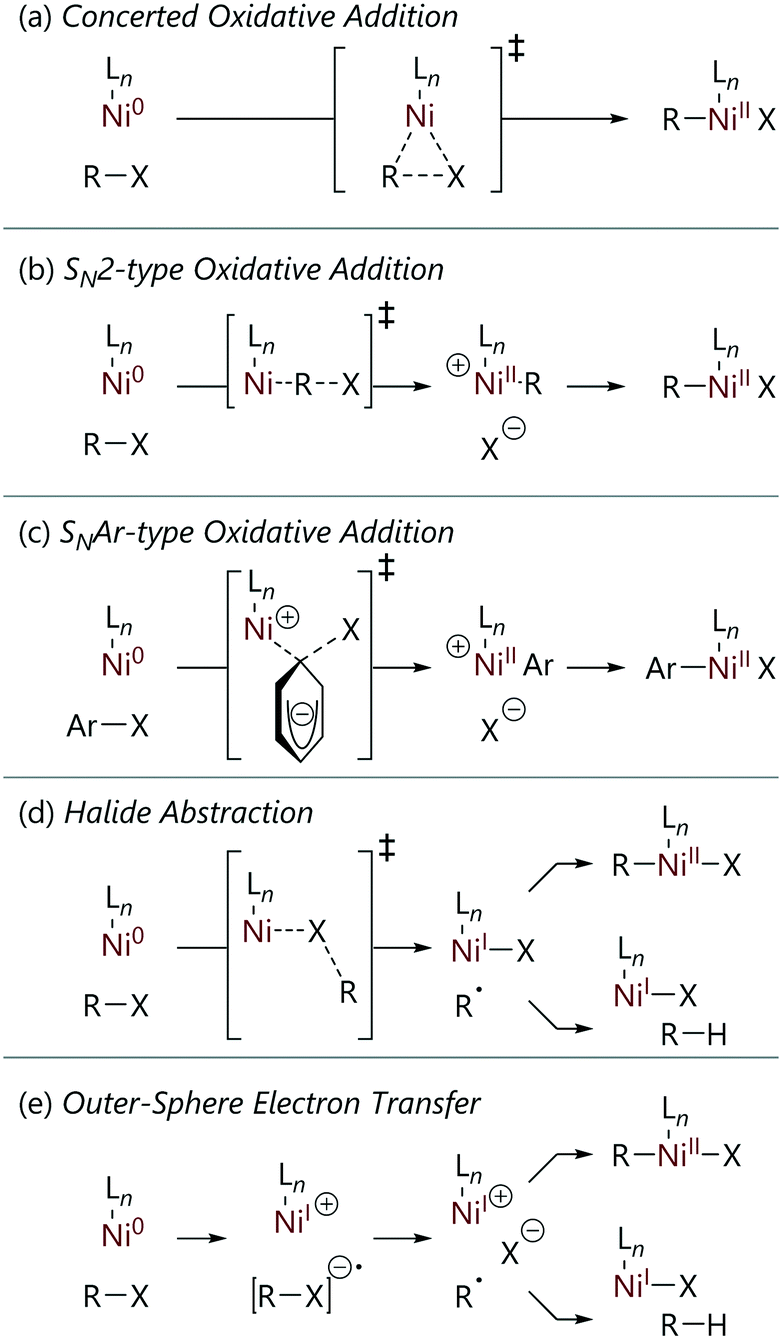 | ||
| Scheme 1 Mechanisms for the reactions of nickel(0) complexes with organohalide substrates. | ||
Tools for mechanistic studies
The tools that can be used to study these reaction mechanisms are rather varied, and will depend on factors including the presence or absence of NMR active nuclides and whether the complexes of interest are paramagnetic or diamagnetic.Some tools that can be used to study and characterise these reaction pathways include:
Scope of the review
This review considers the published literature to date on the topic of the reactions of organochloride, organobromide, and organoiodide compounds with nickel(0) complexes. The review will not consider the reactions of organofluorides, alcohol derivatives, trimethylammonium salts, diazonium salts, or any of the other alternative electrophiles for nickel-catalysed cross-coupling reactions.15,16 The review is divided first by ligand class, and then by substrate type, with the narrative typically in approximately chronological order.Nickel(0) complexes with phosphorus ligands
Reactions with aryl halides
The reactions of aryl halides with phosphine-ligated nickel(0) complexes have received attention as far back as the 1970s. Some aspects of this topic were recently reviewed by Pérez-Garcí and Moret.17In 1970, Fahey reported the products of the reactions of [Ni(η2-C2H4)(PR3)2] (R = Et or Ph) with bromopentafluorobenzene, chlorotrifluoroethylene, 1,2,4-trichlorobenzene, 1,2-dichlorobenzene, and 1-bromo-2-chlorobenzene;18 these were obtained in 6–19% yield and formulated as the trans-[Ni(X)(R′)(PR3)2] complexes based on infrared analysis which revealed a strong band at ca. 420 cm−1 that is proposed to correspond to an asymmetric Ni–P stretching vibration. The regioselectivity of the oxidative addition reaction of 1,2,4-trichlorobenzene was confirmed by quenching the resulting complex with acid; this formed a mixture of 1,2-dichlorobenzene (7%), 1,3-dichlorobenzene (6%), and 1,4-dichlorobenzene (87%). The selectivity for the 2-position was taken as possible evidence of an SNAr-type oxidative addition transition state by comparison of the regiochemistry of the corresponding reaction between 1,2,4-trichlorobenzene and methoxide. However, the relative rate of C–Br versus C–Cl activation (as determined from the outcomes of the reactions of 1-bromo-2-chlorobenzene) was ca. 0.003, much lower than would be expected for an SNAr reaction (ca. 0.1–1) but similar to that observed for the SN2 reactions of alkyl halides and cobaloximes (ca. 0.0015–0.023).
In 1975, Foa and Cassar investigated the rates of oxidative addition of [Ni(PPh3)4] with various para-substituted aryl halides (Scheme 2); the majority of kinetic experiments were conducted with aryl chlorides.19 [Ni(PPh3)3] forms spontaneously when solid [Ni(PPh3)4] is dissolved in solution and so this is the active species.20 The oxidative addition reactions were found to be first order in aryl halide and inhibited by triphenylphosphine when excess ligand was added to kinetic experiments.
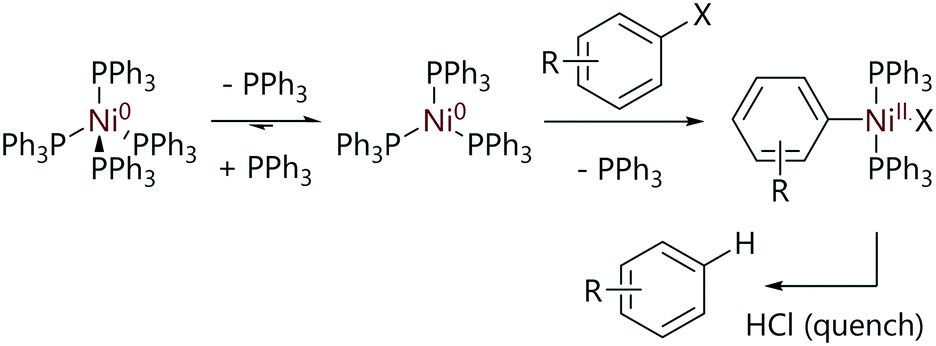 | ||
| Scheme 2 Reactions studied by Foa and Cassar.19 | ||
Relative rates of oxidative addition for differently-substituted aryl halides were measured using competition reactions; in these experiments, equimolar quantities of two aryl halides were mixed and [Ni(PPh3)4] (10 mol%) was added. The reaction mixture was quenched with a saturated HCl/Et2O solution and the proportions of the hydrocarbons formed were quantitatively determined by gas chromatography. These data were used to determine relative rates of reaction, which were plotted using a Hammett treatment (Fig. 1). A good correlation was observed for electron-withdrawing substituents, and a large value of ρ was obtained (>8). A dramatic change of gradient to ca. 0.25 was observed for substituents where σ < 0.23. The relative rates of reaction for aryl chlorides and aryl bromides with the same aryl substitution pattern were often very different depending on the aryl fragment; for haloarenes with electron withdrawing substituents the reaction was approximately one hundred times faster with an aryl bromide compared to a chloride, but this difference was decreased to two to three times when an electron donating substituent was present. The authors proposed that the evidence suggested a concerted three-centre oxidative addition process, although the sudden change in slope of the Hammett plot could be indicative of different reaction mechanisms depending on how electron-rich the substrate is.
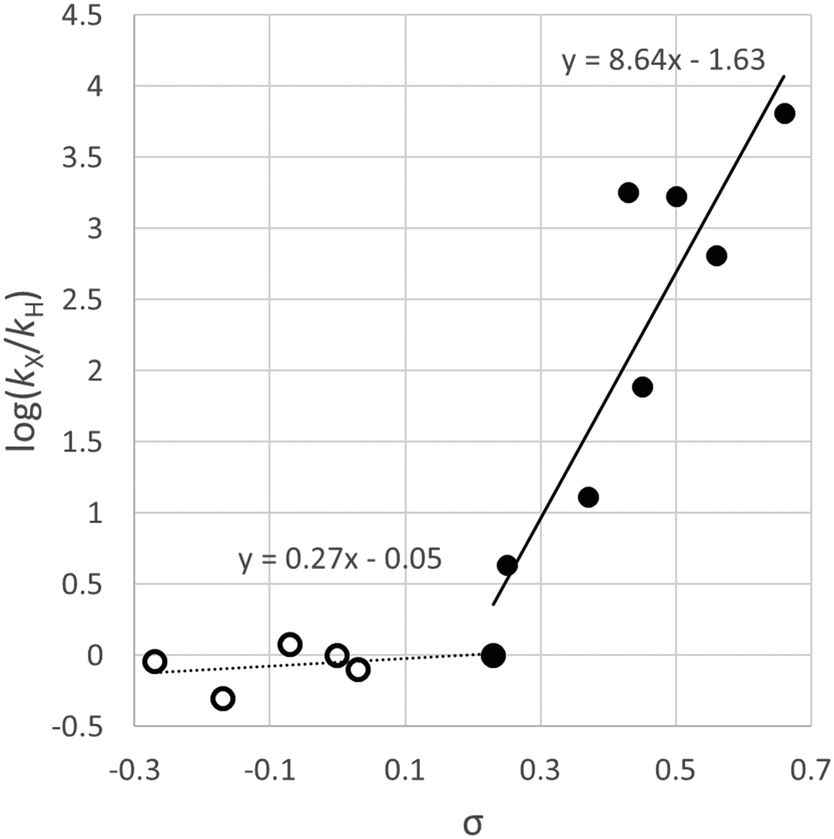 | ||
| Fig. 1 Hammett plot constructed from data reported by Foa and Cassar for the reactions of [Ni(PPh3)4] with substituted aryl chlorides indicating a relatively flat regime (ρ = 0.27) for electron-rich substrates and a steep slope (ρ = 8.6) for electron-poor substrates.19 | ||
In what is widely regarded as the seminal study of oxidative addition to nickel(0), the mechanism of the reaction of [Ni(PEt3)4] with aryl halides was extensively probed in 1979 by Tsou and Kochi.21 The reactions of [Ni(PEt3)4] with various para-substituted aryl halides yielded both [Ni(Ar)X(PEt3)2] and [NiX(PEt3)3] complexes as products, in ratios that depended on the structure of the aryl halide and the reaction solvent (Scheme 3(a)). Notably, a (slow) further reaction of [NiX(PEt3)3] with another molecule of aryl halide was observed, to form [NiX2(PEt3)2] plus an aryl radical that likely abstracts hydrogen from the solvent (Scheme 1). The formation of [Ni(Ar)X(PEt3)2] was monitored via IR spectroscopy and the formation of [NiX(PEt3)3] was monitored via EPR spectroscopy. The formation of [NiX(PEt3)3] via comproportionation between [Ni(PEt3)4] and [Ni(Ar)X(PEt3)2] was ruled out using control experiments.
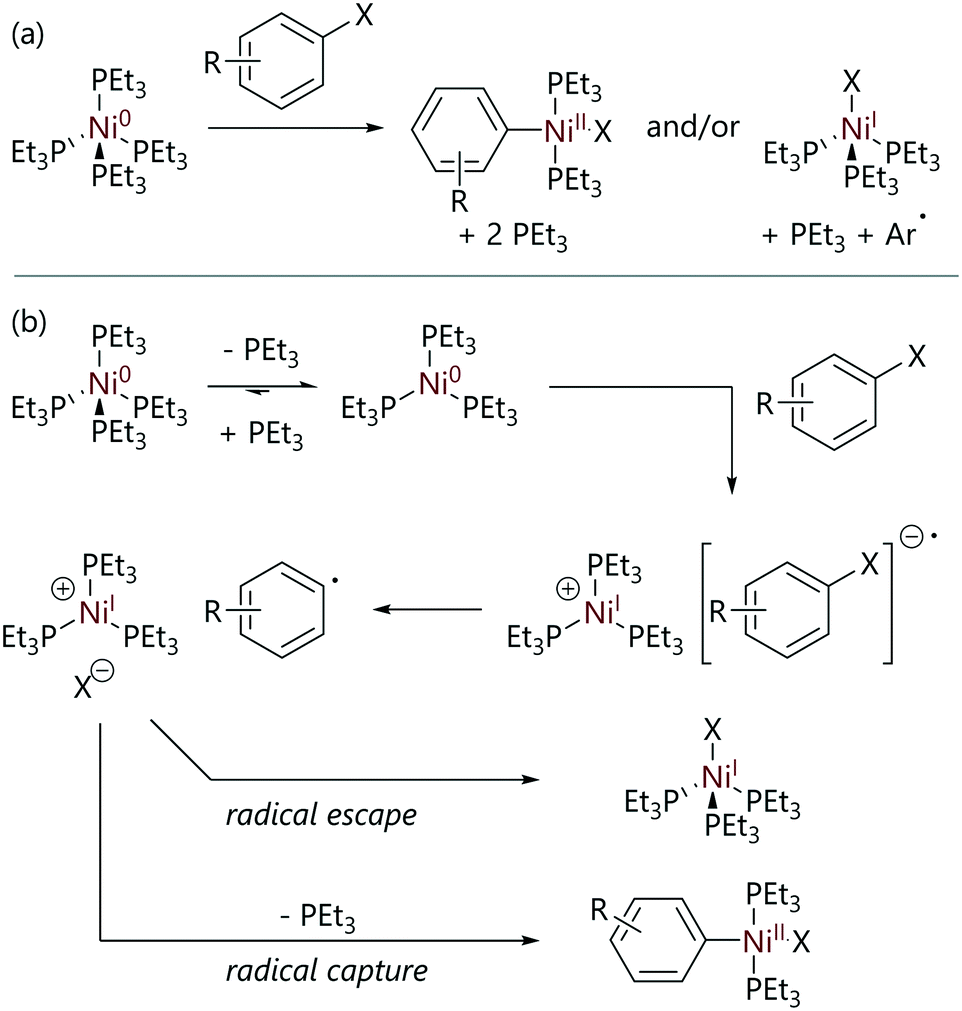 | ||
| Scheme 3 (a) The outcomes of the reactions of [Ni(PEt3)4] with aryl halides (X = Cl, Br, or I) to form [Ni(Ar)X(PEt3)2] and/or [NiX(PEt3)3].21 (b) The reaction mechanism proposed initially. | ||
It was found that a number of factors affect the ratio of [Ni(Ar)X(PEt3)2] to [NiX(PEt3)3] formed during each reaction, including: the identity of the halide; the substitution pattern of the aryl substituent; the reaction solvent; and the phosphine ligand. The quantity of the paramagnetic species [NiX(PEt3)3] was a function of the identity of X (I ≫ Br > Cl); an increase in the amount of nickel(I) formed was also observed in more polar solvents (hexane < toluene < THF). There was no correlation between the product ratio and the identity of the para-substituents on the aryl substrates used, although positively charged substituents increased the amount of [NiX(PEt3)3], whilst negatively charged substituents decreased nickel(I) formation. Although the identity of the ligand attached to nickel would influence the reactivity of nickel(0) initially, the speciation of the nickel(0) species (i.e. [Ni(PEt3)4], [Ni(PEt3)3], or [Ni(PEt3)2]) had no effect on product distribution but the reaction rate was decreased if additional triethylphosphine was added.
In solution, [Ni(PEt3)4] readily dissociates triethylphosphine to form [Ni(PEt3)3] (Keq = 1.2 × 10−2 mol L−1 in benzene at 25 °C).20 After this pre-equilibrium is established, the proposed mechanism for this reaction involves the collapse of a charged ion pair in the rate-limiting transition state (Scheme 3(b)). This proposal was supported by previous studies that showed that single-electron transfer from nickel to an electron-acceptor was feasible; [Ni(PEt3)4] rapidly reduced tetracyanoethylene (amongst other electron-acceptors) to generate radical anions that were detected by EPR spectroscopy.22 The cage collapse of this charged ion pair and the ratios of products formed ([Ni(Ar)X(PEt3)2] or [NiX(PEt3)3]) was proposed to depend on the stability and lifetime of the ion pair, which in turn depends on reaction variables such as the halide identity, the aryl para-substituent, the reaction solvent, and the ligands attached to nickel.
To support this mechanism, cyclic voltammetry was used to study the reactions of [Ni(PEt3)3] with various para-substituted aryl chlorides, bromides and iodides. A Hammett plot with a positive slope, indicating a build-up of negative charge in the transition state, was consistent with the formation of a radical anion. A linear relationship was found between the rate constants for oxidative addition of [Ni(PEt3)3] to aryl halides and the reduction of aryl halides, suggesting a similar transition state.
Funes-Ardoiz et al. later explored this system using DFT calculations of a [Ni(PMe3)4] model system, with some calculations on the corresponding PMe2Ph, PMePh2, and PPh3 systems. Chloro-, bromo-, and iodobenzene were used as model substrates. This study concluded that the nickel-containing products – [Ni(Ar)X(PMe3)2] and [NiX(PMe3)3] – arise from two competing pathways: an SNAr-type oxidative addition, without an Meisenheimer intermediate, to [Ni(PMe3)3] leads to the formation of [Ni(Ar)X(PMe3)2] and a halide abstraction mechanism to the formation of [NiX(PMe3)3] (Fig. 2).23
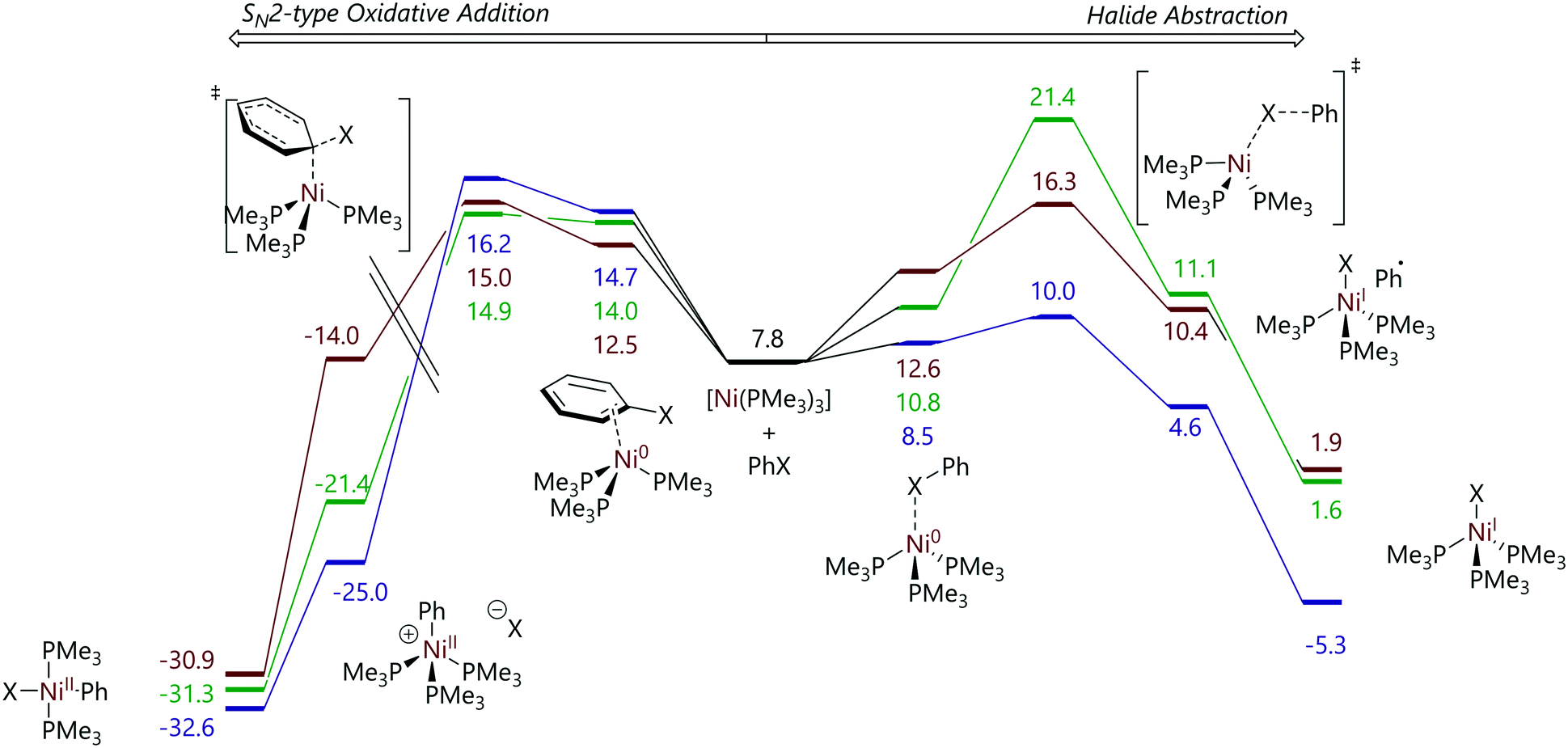 | ||
| Fig. 2 Profiles for the reactions of chloro- (green), bromo- (brown), and iodobenzene (purple) with [Ni(PMe3)3] from DFT calculations. Energies are free energies in kcal mol−1 and are quoted relative to [Ni(PMe3)4]. Left: SNAr-type oxidative addition for which the barriers are relatively insensitive to halide identity. Right: Halide abstraction for which the barriers are strongly dependent on halide identity.23 Note that the energy scale on the left-hand side is discontinuous. | ||
Both mechanisms were shown to proceed through [Ni(PMe3)3] as opposed to [Ni(PMe3)4], [Ni(PMe3)2], or [Ni(PMe3)]; the latter two species were found to be rather high in energy. The identity of halide that was used had little influence on the energetics of the SNAr mechanism; however, a broad range of energies were obtained for the halide abstraction pathway for aryl chlorides, bromide and iodides, consistent with the rather different C–Cl, C–Br, and C–I bond strengths. The reaction selectivity was found to depend on the relative energies of the transition states of the SNAr type mechanism and the halide abstraction mechanism. The SNAr reaction was favoured for chlorobenzene, similar in energy to halide abstraction for bromobenzene, and much higher in energy than halide abstraction for iodobenzene. Microkinetic modelling using DFT-derived energies allowed the experimentally-observed ratio of products to be reproduced to a reasonable degree of accuracy.
Experimental evidence for these two competing mechanisms was provided by Manzoor et al. who studied the reactions of [Ni(PPh3)4] with halobenzene substrates using NMR spectroscopy.24 Typical stoichiometric experiments involved exposing [Ni(PPh3)4] to the substrate at low temperatures in the NMR spectrometer, followed by gradually increasing the temperature in stages and observing the sample composition. The major product of the reactions of [Ni(PPh3)4] with chlorobenzene or bromobenzene was found to be (diamagnetic) trans-[Ni(Ph)X(PPh3)2], which was characterised by 1H NMR spectroscopy (Scheme 4(a)). However, when iodobenzene was used as the substrate the major product was paramagnetic [NiI(PPh3)3], consistent with previous observations by Kochi whilst studying reactions of [Ni(PEt3)4].21
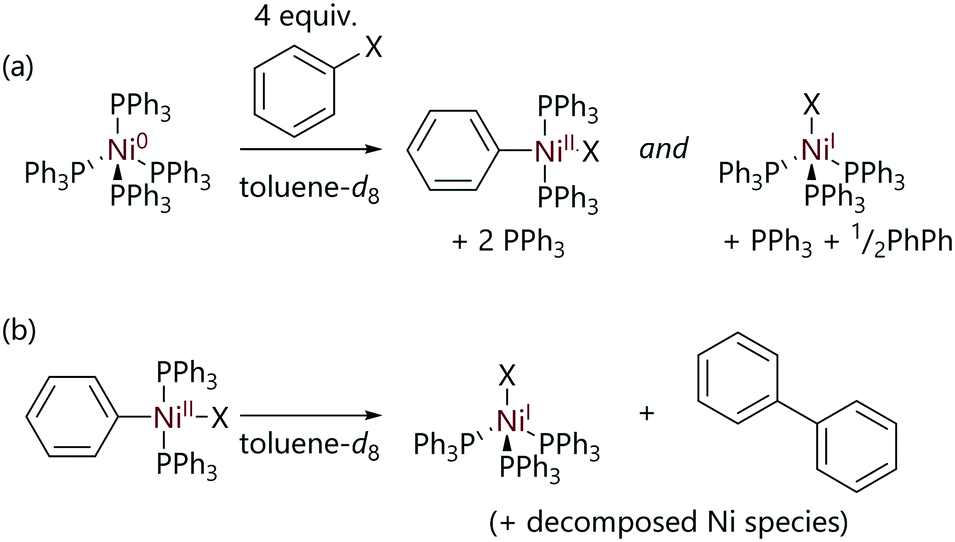 | ||
| Scheme 4 (a) The reactions of [Ni(PPh3)4] with halobenzene substrates to form trans-[Ni(Ph)X(PPh3)2] and [NiX(PPh3)3] complexes. (b) The decomposition of trans-[Ni(Ph)X(PPh3)2] to form [NiX(PPh3)3] and biphenyl.24 | ||
After the oxidative addition of chlorobenzene to nickel(0) was complete, the slow decomposition of trans-[Ni(Ph)(X)(PPh3)2] to [NiX(PPh3)3] and biphenyl was observed at 298 K (Scheme 4(b)).24 However, when isolated crystals of trans-[Ni(Ph)(X)(PPh3)2] were dissolved in toluene-d8, full decomposition of the sample was observed to occur within minutes, suggesting that the presence of PPh3 inhibits the decay of the nickel(II) complex. This is in contrast to observations made by Kochi, where the analogous complex with the smaller, more electron-rich triethylphosphine ligand (i.e. [Ni(Ar)X(PEt3)2]) was found to be stable in solution at room temperature indefinitely.21 This observation has been attributed to the better donor properties of triethylphosphine compared to triphenylphosphine meaning that triethylphosphine does not readily dissociate under the same conditions.
The mechanism of the decomposition of trans-[Ni(Ph)X(PPh3)2] to [NiX(PPh3)3] and biphenyl was probed by conducting DFT calculations (Fig. 3).24 The dissociation of triphenylphosphine from trans-[NiCl(Ph)(PPh3)2] was proposed to be followed by a reaction with a second molecule of trans-[NiCl(Ph)(PPh3)2] to form a chloride-bridge dinuclear intermediate. This dinuclear species could then lead to [NiCl2(PPh3)] and [Ni(Ph)2(PPh3)2], with reductive elimination of biphenyl from the latter species followed by comproportionation to form two molecules of [NiCl(PPh3)2] which then coordinate a third triphenylphosphine ligand. Alternatively, binuclear reductive elimination might form biphenyl and two nickel(I) species directly. These pathways were both found to be feasible thermodynamically, but some transition states were not located, and are indeed rather challenging to locate.
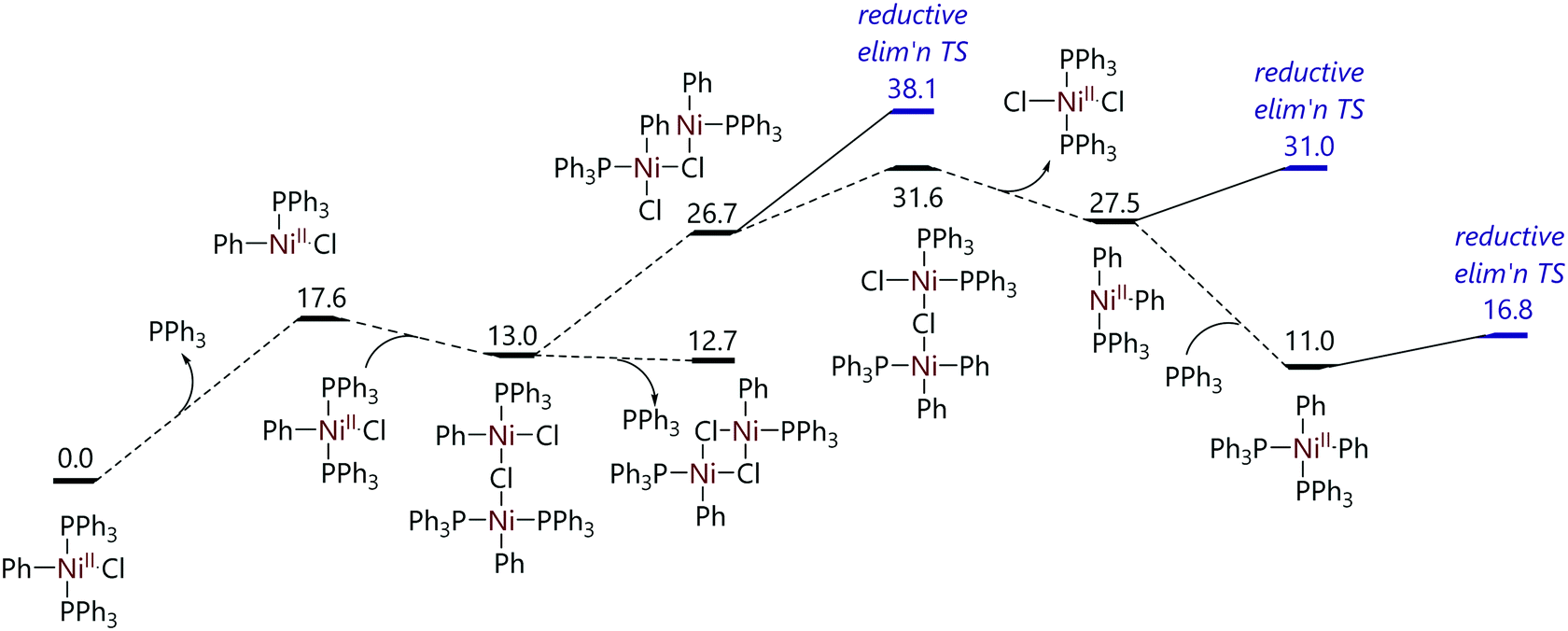 | ||
| Fig. 3 Proposed pathway for the decomposition of trans-[NiCl(Ph)(PPh3)2] to [NiCl(PPh3)3].24 | ||
The reactions of [Ni(PCy3)2] with organohalides have been less well-studied, perhaps due to the need to prepare this complex from the reduction of the corresponding dihalide with reducing metals.25 The oxidative addition of chloroarenes to [Ni(PCy3)2] is reported to lead to [Ni(Ar)Cl(PCy3)2] complexes, which decompose to form biphenyl and [NiCl(PCy3)2].26 The corresponding reactions with alkyl halides lead to nickel(I) and nickel(II) products and mixtures of alkanes and alkenes.
The oxidative addition reactions of nickel(0) complexes with bidentate ligands can often be complicated by catalyst speciation. Many such ligands lead to very stable four-coordinate [Ni(L)2] complexes that can be poorly reactive in catalysis.27 As part of a comprehensive study of the trifluoromethylthiolation of aryl chlorides, Yin et al. screened several bidentate phosphines including XantPhos, (±)-BINAP, dppf, dcpf, dppm, dppe, and dppp (dppf = 1,1′-bis(diphenylphosphino)ferrocene; dcpf = 1,1′-bis(dicyclohexylphosphino)ferrocene; dppm = bis(diphenylphosphino)methane; dppe = 1,2-bis(diphenylphosphino)ethane; dppp = 1,3-bis(diphenylphosphino)propane). The latter three ligands and (±)-BINAP were ineffective, while XantPhos and the ferrocene derivatives gave moderate conversions; dppf was taken forward in the optimisation. Experimental studies established that [Ni(COD)2]/dppf reacts with aryl chlorides to form [NiCl(dppf)] as the ultimate product, while [Ni(COD)2]/dppe leads to poorly reactive [Ni(dppe)2]. Computational studies suggested a three centre concerted oxidative addition mechanism and confirmed the significant energy barrier to replacing a dppe ligand in [Ni(dppe)2] with chlorobenzene (33.1 kcal mol−1), explaining the poor reactivity of dppe–nickel species in the catalytic reaction. In addition, isolated [Ni(COD)(dppf)] performed better in catalysis than the same complex formed in situ by [Ni(COD)2]/dppf, suggesting an inhibitory role for COD.
Amatore and Jutand have generated coordinatively-unsaturated [Ni(dppe)] in situ from [NiCl2(dppe)] using electrochemical methods;28 this decomposes to half an equivalent of [Ni(dppe)2] in the absence of organohalide but in the presence of bromobenzene it forms [NiBr(Ph)(dppe)]. The rate constant for oxidative addition is ca. 105 L mol−1 s−1, consistent with the highly reactive nature of this bent, coordinatively-unsaturated species.
A study by Guard et al. in the same year noted the formation of [NiCl(dppf)] in reactions catalysed by dppf–nickel, regardless of the oxidation state of the (pre-)catalyst used or additional ligands present on that (pre-)catalyst.29 The role of nickel(I) in catalysis is still a subject of active research and is beyond the scope of the present review. The comproportionation mechanism for dppf–nickel systems has been studied using DFT calculations.30 It has been proposed that this comproportionation can be minimised by switching to bulkier 1,1-bis(dicyclohexylphosphino)ferrocene ligands.31
Nicolas et al. examined the oxidative addition of chloroarenes to [Ni(η2-C6H5Me)(dcpp)], in the context of nickel-catalysed Negishi cross-coupling reactions, using experimental and computational methodology (dcpp = 1,3-bis(dicyclohexylphosphino)propane).32 [NiCl(Ar)(dcpp)] complexes were observed experimentally. Oxidative addition via a concerted transition state was found to have a rather low activation energy (ΔG‡ = 12.9 kcal mol−1), with either transmetalation or the exchange of biaryl for chloroarene found to be the most energetically-demanding step. Lavoie et al. characterised a similar transition state for a [Ni(PAd-DalPhos)] species by DFT, and noted that it was similar in energy to oxidative addition to an analogous nickel(I) amide complex (PAd-DalPhos = 8-(2-(di-o-tolylphosphaneyl)phenyl)-1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphaadamantane).33
Bajo et al. studied the reactions of model nickel complex [Ni(COD)(dppf)] with a bidentate phosphine ligand with aryl halides (and other aryl electrophiles).34 It was previously known that these reactions lead to [NiX(dppf)] complexes as the final nickel-containing species. The oxidative addition reactions of various aryl electrophiles to [Ni(COD)(dppf)]35 were monitored via31P NMR spectroscopy (Scheme 5). The reactions were found to be first order with respect to [Ni(COD)(dppf)] and aryl halide, and were inhibited by added COD. On the basis of the kinetic data, an initial reversible displacement of COD with aryl halide was proposed, followed by an irreversible oxidative addition step. The nickel(II) oxidative addition product – i.e. [Ni(Ar)X(dppf)] – typically underwent rapid comproportionation with [Ni(COD)(dppf)] to form [NiX(dppf)] which was the only nickel-containing product observed; these species were characterised using EPR spectroscopy and single crystal X-ray diffraction analysis. Notably, comproportionation was sufficiently fast that no oxidative addition products were observed in the 31P NMR spectra at ambient temperatures. The oxidative addition product was observed as two doublets in the 31P NMR spectra at lower temperatures and when more sterically-hindered ortho-substituted aryl halides were employed as substrates.
 | ||
| Scheme 5 The oxidative addition of aryl halides and other aryl electrophiles to [Ni(COD)(dppf)].34 | ||
It was also noted that naphthyl substrates are much more reactive than aryl substrates in oxidative addition to [Ni(COD)(dppf)] (by ca. six- to eight-fold).34 This is consistent with the observation that studies that involve very challenging substrates such as aryl ethers tend to demonstrate higher yields with naphthyl and other extended aromatic systems.
Three mechanisms previously proposed for oxidative addition of aryl halides to nickel(0) and palladium(0) complexes were considered. Radical reactions were ruled out as the addition of TEMPO had no impact on the rates of oxidative addition; however, subsequent studies have shown that TEMPO is not always a reliable radical probe in this system as it can react directly with nickel(0).36 The activation parameters obtained were similar to those for the oxidative addition of aryl halides to [Pd(PPh3)4].37 Notably, the Hammett plot obtained by Bajo et al. showed that for electron-rich substrates ρ = 1.2 whereas electron-poor substrates appeared to show an opposing trend, consistent with a change in rate-determining step. The shallow gradients obtained from the Hammett plots ruled out an SNAr-type mechanism7 and a small ΔS‡ of 0(3) cal K−1 mol−1 led to a three-centre transition state being proposed (see Scheme 1).
Following on from this study, Cooper et al. have investigated how common functional groups can interact with nickel(0) complexes and influence the rate and selectivity of oxidative addition (Scheme 6);38 model nickel complexes with dppf ligands were utilised, and were compared to the analogous palladium complexes in Suzuki–Miyaura cross-coupling reactions.39 Aldehyde- and ketone-substituted aryl chlorides underwent unexpectedly rapid oxidative addition to [Ni(COD)(dppf)]. The kinetic data obtained implied that the previously described three-centred concerted mechanism was in operation;34 however, the initial ligand exchange (aryl halide for COD) was thought to be favoured when the aryl halide bears an aldehyde or ketone, shifting the initial equilibrium towards the [Ni(η2-ArX)(dppf)] complex. Aldehydes and ketones are known to be excellent ligands for low valent nickel complexes,40–43 with nickel complexes that bear a bidentate phosphine ligand (typically dtbpe) giving rise to some interesting electronic effects that have been examined in detail by Kennepohl and Love (dtbpe = 1,2-di(tert-butylphosphino)ethane).44,45 Amides and esters did not benefit from this coordination effect. In the reaction between [Ni(COD)(dppf)] and 4-chlorobenzaldehyde, an intermediate was observed that presented two doublets in the 31P NMR spectrum; these were attributed to the η2(CO)-complex.
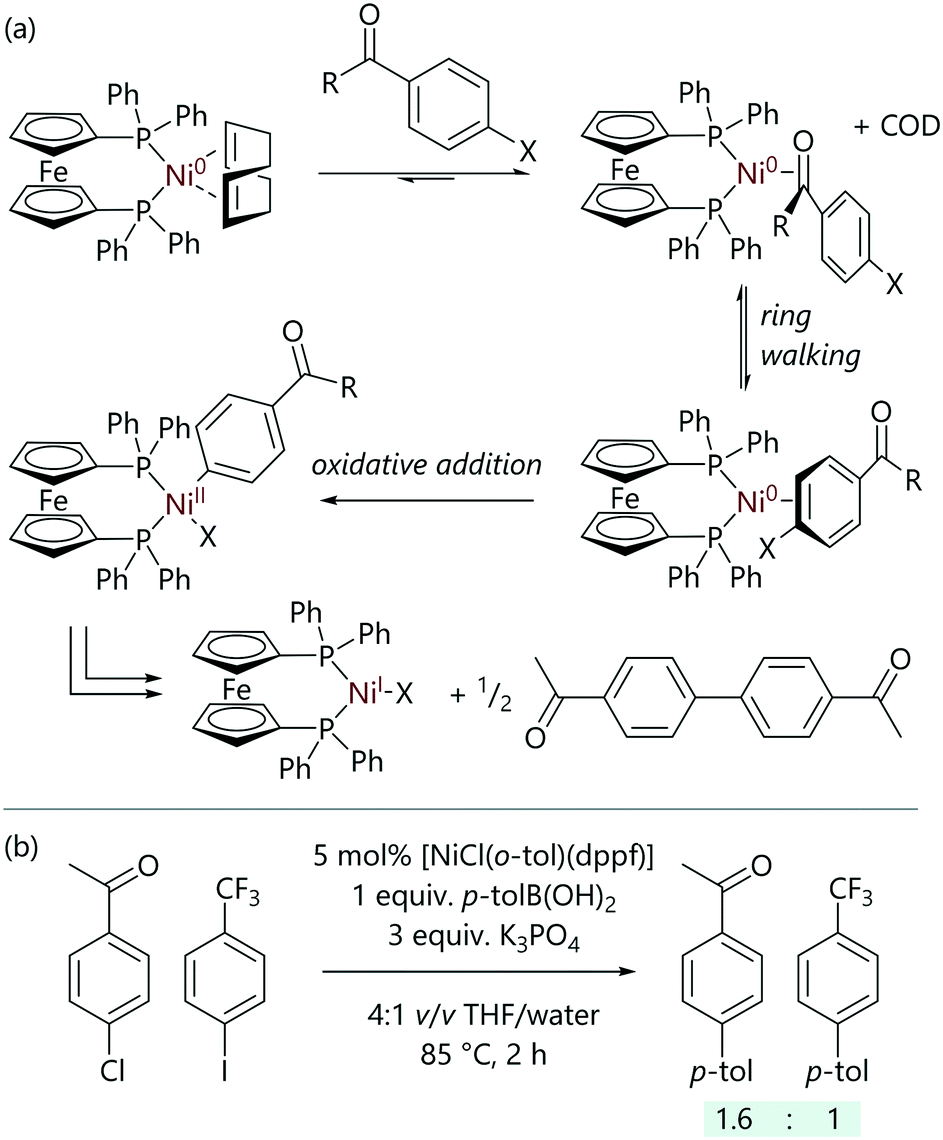 | ||
| Scheme 6 (a) Ring-walking processes in the oxidative addition of aryl halides with aldehyde and ketone functional groups to [Ni(COD)(dppf)]. (b) The effect of aldehyde and ketone coordination on selectivity in Suzuki–Miyaura cross-coupling reactions.38 | ||
This exceptional reactivity of aromatic halides bearing aldehyde and ketone groups was then exploited to achieve selective Suzuki–Miyaura cross-coupling reactions;38 in inter- and intramolecular competition experiments, selective cross-coupling was achieved at the site that was in conjugation with the aldehyde or ketone. This selectivity was such that the normal order of reactivity of aryl halides (I > Br > Cl) could be overturned, and was attributed to a ‘ring-walking’ process where the nickel moves from the initial site of coordination across a π-system to the aryl halide.46–51 This proposal was supported by DFT calculations.
However, aldehydes and ketones (without halide functional groups) can act as inhibitors in these reactions, which prevents a potential drawback to the use of nickel catalysis.38 Palladium has been shown not to interact strongly enough to induce these selectivity and inhibition effects observed with nickel.39
In light of the much wider scope of electrophiles that are reactive with nickel in cross-coupling catalysis,15,52 there can be selectivity challenge in nickel catalysis if the substrate is highly functionalised. Two recent studies have explored these selectivity challenges in some detail.
Entz and Russell et al. examined the case of C–Cl versus C–OTs activation in nickel-catalysed Suzuki–Miyaura cross-coupling reactions using a combination of experiment and theory.53 Bajo et al. had previously shown that the oxidative addition of p-F3CC6H4Cl to [Ni(COD)(dppf)] was ca. three times faster than the corresponding reaction with p-F3CC6H4OTs.34 Computational studies of the reaction of 4-chlorophenyl tosylate were conducted initially using model species [Ni(PMe3)2], [Ni(PPh3)2], and [Ni(PCy3)2] and ΔΔG‡ (i.e. ΔG‡OTs − ΔG‡Cl) was evaluated in each case (Scheme 7(a)).53 It was noted that the trimethylphosphine complex ought to prefer C–OTs oxidative addition (ΔΔG‡ = −3.0) while the triphenylphosphine complex should prefer C–Cl activation (ΔΔG‡ = +2.0) and the tricyclohexylphosphine complex should be less selective (ΔΔG‡ = −1.2). The structures of the (concerted) oxidative addition transition states revealed favourable interactions between the oxygen of the sulfonate and the nickel centre in the oxidative addition of the tosylate; these were more pronounced for trimethylphosphine due to its smaller steric impact.
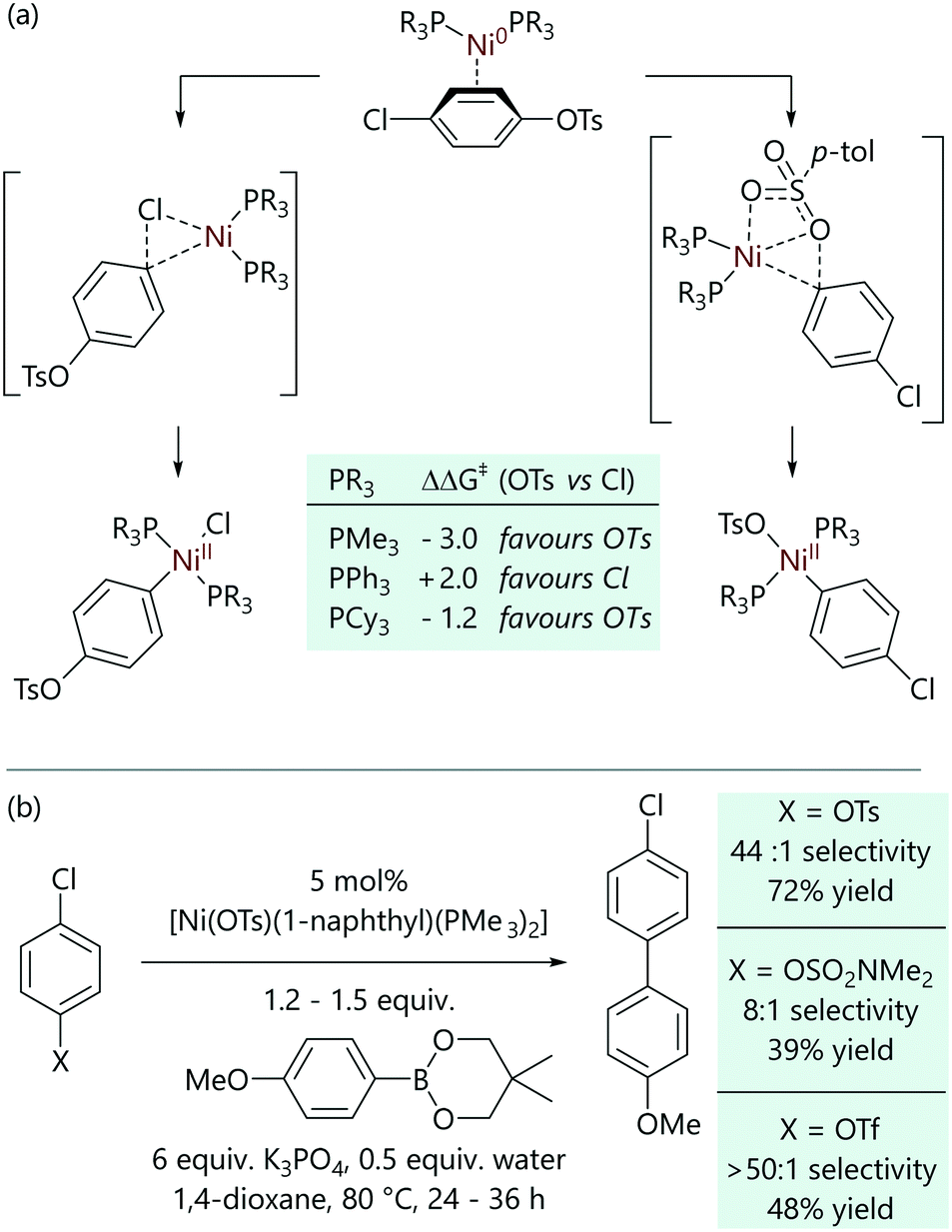 | ||
| Scheme 7 (a) Oxidative addition transition states for the possible reactions of 4-chlorophenyl tosylate with [Ni(PR3)2] complexes. (b) Examples of selective cross-coupling at the C–O bond in the presence of a C–Cl bond, enabled by the use of a trimethylphosphine–nickel pre-catalyst; selectivity was determined by GC analysis, while yields are isolated yields.53 | ||
Experimental studies in which [Ni(COD)2] plus a phosphine ligand were exposed to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1-chloronaphthalene and naphthyl 1-tosylate allowed the corresponding oxidative addition products to be observed by 31P{1H} NMR spectroscopy, and thus for their ratio to be measured.53 Triarylphosphines and the bidentate dcpf ligand were overwhelmingly selective for C–Cl oxidative addition, while trialkylphosphines and arylalkylphosphines produced mixtures. Dimethylphenylphosphine and trimethylphosphine were the only ligands that were selective for C–OTs oxidative addition (in ratios of 1:5.7 and 1:6.3, respectively). This then led to the development of cross-coupling conditions that allowed a range of chloro/tosylate, chloro/triflate, and chloro/sulfamate substrates to be selectively coupled at the C–O site, leaving the aryl chloride intact for further reactions (Scheme 7(b)).
:1 mixture of 1-chloronaphthalene and naphthyl 1-tosylate allowed the corresponding oxidative addition products to be observed by 31P{1H} NMR spectroscopy, and thus for their ratio to be measured.53 Triarylphosphines and the bidentate dcpf ligand were overwhelmingly selective for C–Cl oxidative addition, while trialkylphosphines and arylalkylphosphines produced mixtures. Dimethylphenylphosphine and trimethylphosphine were the only ligands that were selective for C–OTs oxidative addition (in ratios of 1:5.7 and 1:6.3, respectively). This then led to the development of cross-coupling conditions that allowed a range of chloro/tosylate, chloro/triflate, and chloro/sulfamate substrates to be selectively coupled at the C–O site, leaving the aryl chloride intact for further reactions (Scheme 7(b)).
Jacobs and Keaveney exploited the reversibility of C–Cl bond activation and the different reactivity of arylnickel(II) fluorides and arylnickel(II) chlorides to achieve C–F selective Hiyama coupling reactions.54 [Ni(Ar)Cl(PR3)2] complexes will not react directly with silanes, while [Ni(Ar)F(PR3)2] complexes can.55 A Hiyama cross-coupling between 1-fluoronaphthalene and trimethoxyphenylsilane was achieved employing [Ni(COD)2]/PCy3 as the catalytic system (Scheme 8). Notably, under these optimised conditions, no conversion was observed for 1-chloronaphthalene. In a competition experiment with both 1-fluoronaphthalene and 1-chloronaphthalene present, no coupled product was observed.
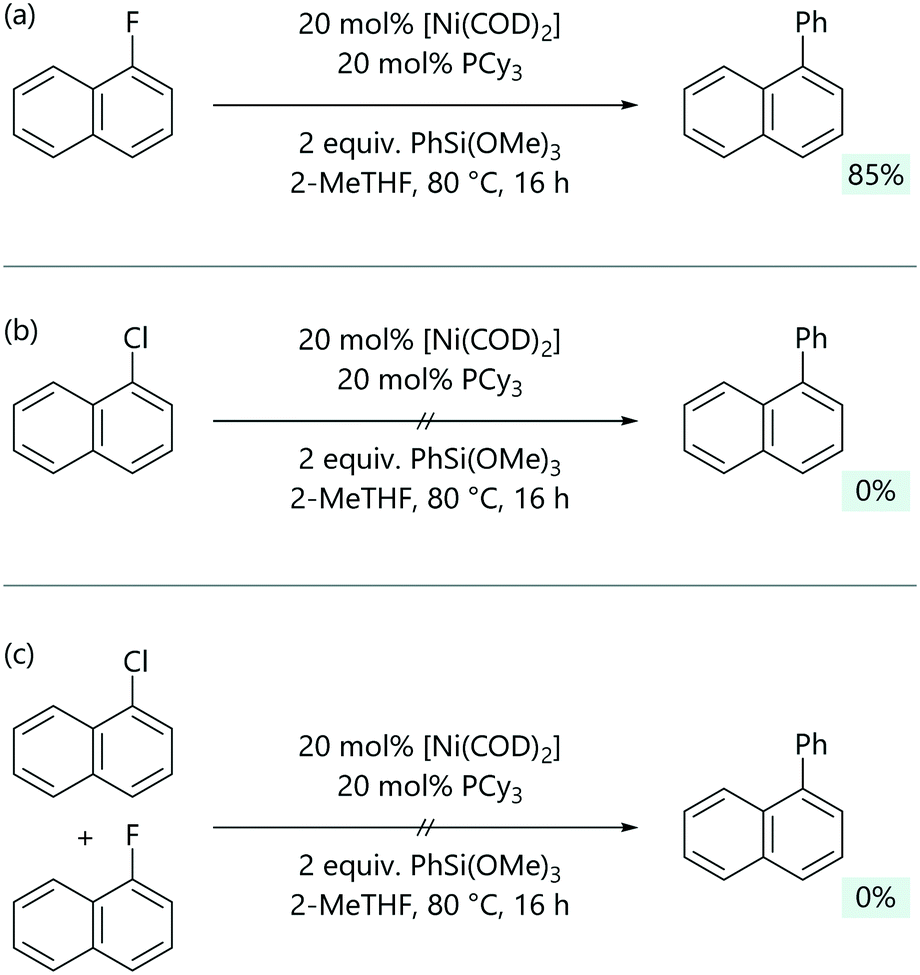 | ||
| Scheme 8 (a) The base-free Hiyama coupling of aryl fluorides. (b) Chloroarenes are unreactive under the same conditions, because the arylnickel(II) chloride cannot directly transmetalate. (c) Chloroarenes poison the otherwise productive reactions of fluoroarenes.54 | ||
DFT calculations were used to gain insight into the mechanism of this reaction.54 Optimisation experiments suggested a [Ni(PCy3)] active species, as an increase in the ligand to nickel ratio led to a decrease in the yield, and so the calculated free energy profile started from [Ni(COD)(2-MeTHF)(PCy3)] (Grel = 0). As expected, the barrier for the concerted oxidative addition of 1-fluoronaphthalene (Grel = 28.4 kcal mol−1) was much higher than that for 1-chloronaphthalene (Grel = 8.0 kcal mol−1). For transmetalation, a low energy transition state could be located for the Ni–F pathway (Grel = 2.1 kcal mol−1), but transmetalation was much more energetically demanding for the Ni–Cl pathway (>50 kcal mol−1). Consistent with the experimental observations from the competition reaction, the irreversible oxidative addition with 1-chloronaphthalene is faster than reaction with 1-fluoronaphthalene and forms the oxidative addition product (Grel = −26.1 kcal mol−1) which ultimately inhibits cross-coupling.
Developing a catalyst system to promote reversible C–Cl oxidative addition would induce the desired selectivity for reaction at the C–F bond over reaction at the C–Cl bond. Whilst the oxidative addition of 1-fluoronapthalene does not proceed if the ligand is too bulky, the reversible oxidative addition of 1-chloronapthalene required a bulky ligand.54 The contradictory results encountered meant that the desired selectivity for C–F over C–Cl oxidative addition is difficult to achieve, however the concept of selectively destabilising the C–Cl oxidative addition product is a promising avenue for future work.
Pérez-García et al. have studied the reactions of nickel(0) complexes with a tris(phosphine) ligand (Scheme 9).56 The treatment of the nickel(0) complex with haloarenes led to the formation of well-defined nickel(II) oxidative addition products, of which one example was structurally characterised by X-ray diffraction analysis.
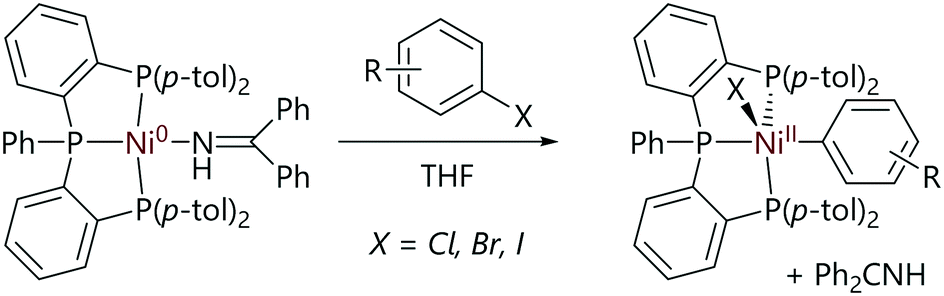 | ||
| Scheme 9 Oxidative addition reactions of a tris(phosphine)nickel(0) complex.56 | ||
Kinetic studies were carried out in the presence of excess imine ligand and aryl bromide revealed kinetic behaviour that was pseudo-first order in the nickel complex, first order in aryl halide, and inverse first order in imine. This is consistent with a pre-equilibrium in which the imine ligand is exchanged for the aryl halide before oxidative addition. A Hammett correlation (using σ− substituent constants) revealed a significant and positive reaction constant (ρ = 2.6), which is consistent with a three-centre oxidative addition transition state, although it is not possible to decouple the electronic effects on the pre-equilibrium from the electronic effects on the transition state energy. An Eyring analysis yielded a negative entropy of activation (ΔS‡ = −18(2) cal K−1 mol−1), suggesting an associative ligand exchange prior to oxidative addition. DFT calculations revealed that a three-centre concerted oxidative addition mechanism was reasonable, with halide abstraction being much less favourable (ΔΔG‡ = +7.2 kcal mol−1).
Reactions with vinyl halides
Relatively few studies into the mechanism of oxidative addition of vinyl halides to nickel have been carried out. This may be due to the difficult synthesis and the ultimate stability of complex vinyl halides, as studies that have been conducted have utilised simple halides.Fahey has studied the oxidative addition of vinyl halides to nickel complexes.18,57 Vinyl bromide and chloroprene reacted rapidly with [Ni(COD)(PEt3)2] to give the corresponding halo-organo-nickel complexes. The product from oxidative addition of the vinyl bromide decomposed at ambient temperatures and therefore was not subjected to full characterisation. However, the nickel product from the oxidative addition of the chloroprene was stable under argon for months and was fully characterised as the trans-allyl nickel complex (Scheme 10).
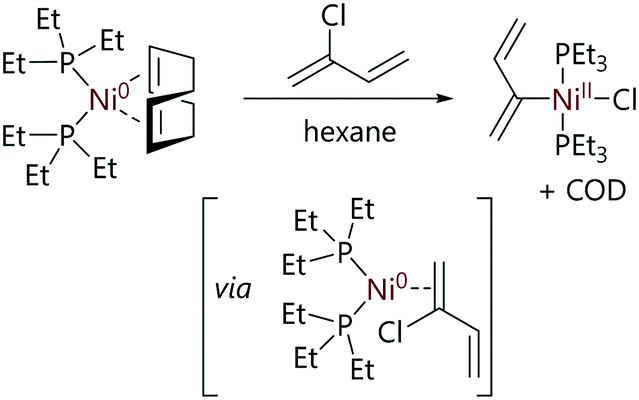 | ||
| Scheme 10 Reaction of [Ni(COD)(PEt3)2] with chloroprene to form a stable allyl product.57 | ||
The proposed mechanism for the reaction of [Ni(COD)(PEt3)2] with vinyl halides suggest a coordination of the π-system to nickel(0) followed by formation of the oxidative addition product (Scheme 10).57 The intermediates in this mechanism were not isolated and therefore the mechanism was speculative but it may be similar to the ring-walking processes known to occur with for nickel complexes (vide supra). Radical processes were ruled out for vinyl substrates; vinyl radicals exhibit very poor stability, but more importantly the stereochemistry was retained when trans-β-bromostyrene was used as a substrate for oxidative addition. While no rate constants were measured, it was noted that vinyl halides reacted rapidly with [Ni(COD)(PEt3)2], while aryl halides required elevated temperatures of 50–60 °C.
Reactions with alkyl halides
Stille and Cowell have studied the reactions of benzyl halides with [Ni(PPh3)4] as part of efforts to develop methodology for the formation of esters by alkoxycarbonylation.58 They noted poor stereochemical control of substrates where the benzylic centre had point stereochemistry, which is suggestive of (planar) radical intermediates; in addition, compounds with hydrogen atoms at the adjacent position were affected by β-hydride elimination.Zhang et al. utilised a nickel(0) complex with tetradentate P2S2 ligand ((o-(Ph2P)C6H4CH2SCH2)2) for a reactivity study with allyl and alkyl halides (in the presence of NaBPh4).59 Oxidative addition to allyl halides led to cationic allylnickel(II) species. The reactions with alkyl halides produced [Ni–R]+ complexes, which underwent β-hydride elimination to form nickel(II) hydride complexes.
Kehoe et al. used variable temperature NMR spectroscopy to elucidate the mechanism of oxidative addition of various alkyl halides to [Ni(PPh3)4].60 The 31P NMR spectrum of [Ni(PPh3)4] at room temperature contains a single broad resonance (δP = 24 ppm). When cooled to 200 K, the spectrum contains two resonances (δP = 8, 25 ppm) which are attributed to free PPh3 and [Ni(PPh3)3], respectively, confirming that the dominant species in the reaction mixture is [Ni(PPh3)3]; Keq for this dissociation is >106 mol L−1.20 The reaction of [Ni(PPh3)3] with iodoalkanes yielded alkanes and alkenes as the major products, plus a nickel hydride as the minor product (Scheme 11). This product ratio suggested oxidative addition was occurring followed by a subsequent β-H elimination event. The observation that the rate decreases in the order t-BuX > s-BuX > n-BuX (X = Cl, Br, I) is consistent with a radical mechanism. Experimental evidence, plus evidence from previous work published by Tsou and Kochi,21 led to halide abstraction being proposed as the mechanism of reaction, which is followed by a subsequent β-H elimination step.
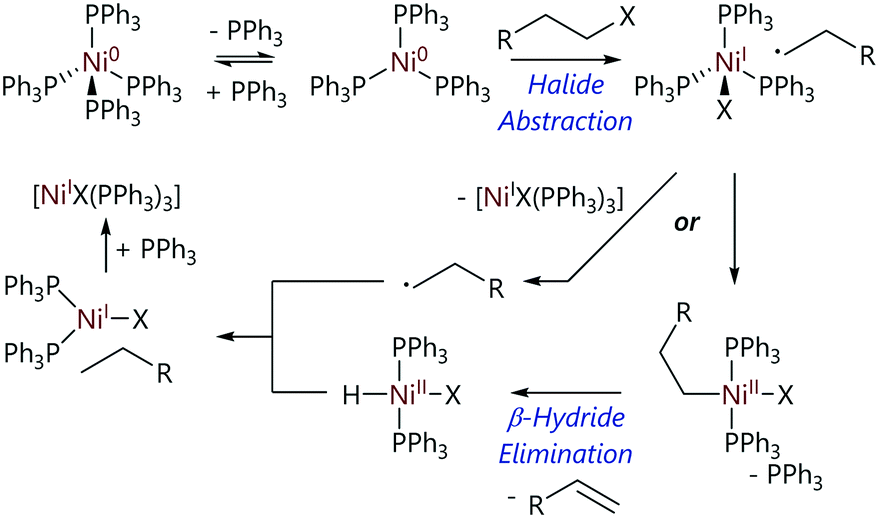 | ||
| Scheme 11 Proposed mechanism for the reactions of alkyl halides with [Ni(PPh3)4].60 | ||
The low concentrations of the nickel hydride complex found in the reaction mixtures prompted further investigation to determine how this was being consumed.60 It was established that alkyl radicals have the ability to escape the cage and do not necessarily all recombine to form the formal (NiII) oxidative addition product. These radicals have three preferred reactions: self-reaction, disproportionation (to form alkane plus alkene), or abstraction of a proton from either the solvent or nickel hydride complex. Equal amounts of alkene and alkane products, consistent with disproportionation, were observed; however, a lack of longer chain alkanes that would be consistent with combination reactions ruled out self-combination. There was also no evidence of any deuterated products, ruling out hydrogen atom abstraction from solvent molecules (reactions were carried out in toluene-d8). The conclusion was that the alkyl radicals escaping from the cage were reacting preferentially with the nickel hydride, and this was supported using DFT calculations, providing an explanation for the low concentrations of nickel hydrides observed.
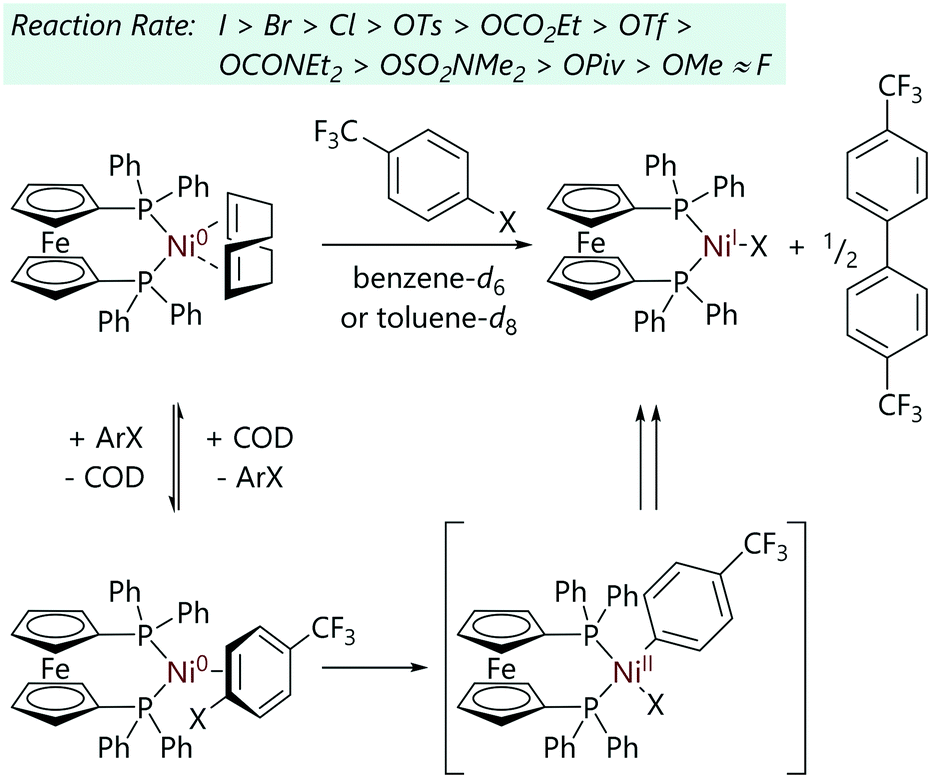
Greaves et al. explored the mechanism of the reaction of [Ni(COD)(dppf)] with (2-haloethyl)benzene substrates.36 In contrast to the analogous reactions with aryl halides, these reactions proceeded via [Ni(dppf)2], a species which exists in equilibrium (Keq = 6.8) with [Ni(COD)(dppf)] and excess dppf. The reactions of these alkyl halides with [Ni(COD)(dppf)], in the presence of dppf, were monitored by 31P NMR spectroscopy. Reactions were first order with respect to [Ni(COD)(dppf)], dppf and alkyl halide, and showed an inverse first order dependence on COD concentration. A product study revealed that [NiX(dppf)] was the final nickel-containing product, and this was detected by EPR spectroscopy. The evolution of dihydrogen was confirmed by 1H NMR spectroscopy and the presence of styrene was confirmed by GC-MS analysis. A reaction mechanism that explains the observed products was proposed (Scheme 12). An initial reversible displacement of COD with dppf yielded the active species [Ni(dppf)2], which then undergoes halide abstraction and radical recombination to form [Ni(CH2CH2Ph)X(dppf)] (not observed by 31P NMR spectroscopy). A β-H elimination event followed by dissociation of styrene yields [Ni(H)(X)(dppf)] which undergoes comproportionation to form [NiX(dppf)] and dihydrogen.
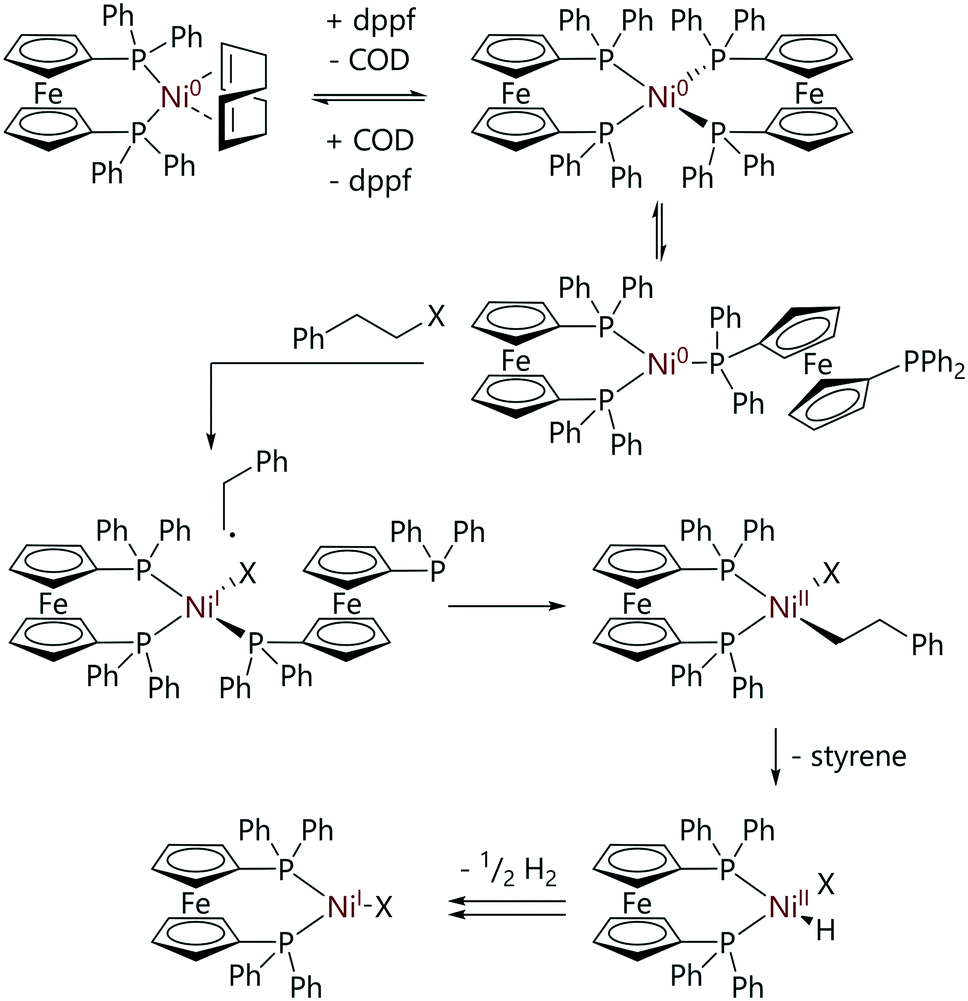 | ||
| Scheme 12 Proposed mechanism for the reactions of alkyl halides with [Ni(COD)(dppf)].36 | ||
The involvement of radicals in the reaction was confirmed as reaction rates dramatically increased when tert-butyl bromide or (2-bromopropyl)benzene were employed as substrates; these reactions were too fast to monitor by NMR spectroscopy.36 There were two mechanistic possibilities: outer sphere electron transfer and halide abstraction. Outer sphere electron transfer was ruled out as a mechanistic possibility, as [Ni(dppe)2], which should be capable of outer sphere electron transfer, did not undergo reaction with the (2-haloethyl)benzene substrates. DFT calculations confirmed a halide abstraction mechanism was most likely for these reactions, operating via a [Ni(dppf)2] active species.
Nickel(0) complexes with chelating nitrogen ligands
The reactions of nickel(0) complexes with bipyridine ligands (or similar) are arguably far less well understood than for other ligand classes. There is an emerging picture that suggests that, because these ligands are redox non-innocent, nickel(I)61 plays a more significant role in the corresponding catalytic reactions.62,63 Vicic has studied the terpyridine systems in some detail, revealing that many [NiX(terpy)] complexes – which on inspection appear to be nickel(I) – are in fact better described as nickel(II) with a radical on the ligand (terpy = 2,2′:6′,2′′-terpyridine);64,65 a full and detailed discussion of that work is somewhat beyond the scope of this review – which focusses on nickel(0) – but interested readers are directed towards the relevant manuscripts.The reported reactivity of this class of nickel(0) complex with aryl halides typically leads to the invocation of odd-numbered oxidation states, although some two-electron (oxidative addition) reactivity has also been noted. For example, Shields and Doyle reported the synthesis of diamagnetic square planar complexes [NiCl(o-tol)(dtbpy)] and [NiCl(p-tol)(dtbpy)] from the reaction of [Ni(COD)2], dtbpy, and the corresponding chloroarene in THF (dtbpy = 4,4-di(tert-butyl)bipyridine);66 these structures were supported by 1H and 13C{1H} NMR data and elemental analysis.
Weix used the observed difference in reactivity between aryl and alkyl halides to achieve the selective sp2–sp3 reductive cross-electrophile coupling reactions of aryl halides with alkyl halides;4,67 these reactions employ a NiII pre-catalyst, and use an external reductant to form the active Ni0 species in situ. In most cases, the ligands are substituted bipyridine or phenanthroline ligands. The oxidative addition of the aryl halide is selectively achieved (versus the reaction of the alkyl halide) due to a difference in rate. This arylnickel(II) intermediate was found to stoichiometrically react with alkyl halides to re-form the initial pre-catalyst, without the requirement for an external reductant. Radical-clock experiments carried out using (cyclopropyl)methyl bromide as the alkyl halide confirmed the presence of radical intermediates, as only the ring-opened product was observed.68 Increased amounts of ring-opened product were observed when the nickel concentration was increased, suggesting radical formation and reaction were occurring at different points of the catalytic cycle. Finally, from an analysis of the products of quenched reaction mixtures, the oxidative addition product was found to be the resting state of the catalyst explaining the selectivity of the reaction. These observations result in the proposed catalytic cycle shown in Scheme 13.
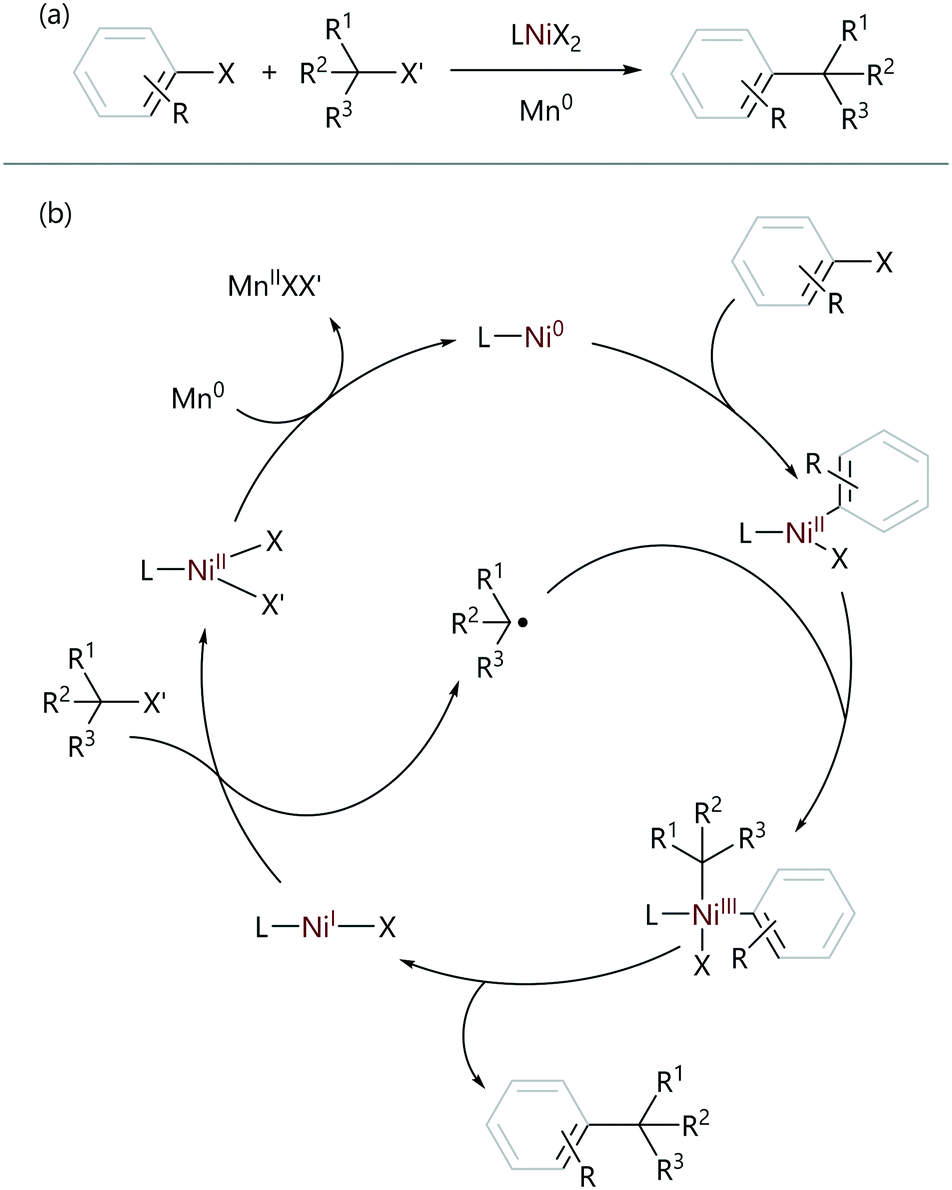 | ||
| Scheme 13 (a) Reductive cross-electrophile cross-coupling reactions. (b) The proposed mechanism for the reductive cross-electrophile coupling reactions of sp2- and sp3-organohalides.4,67 | ||
Kalvet et al. and Jover have both examined the nickel-catalysed trifluoromethylthiolation of aryl halides using DFT calculations.62,63 Jover has conducted a DFT study of the alternative full cycles for the trifluoromethylthiolation reaction of iodobenzene catalysed by 4,4′-di(methoxy)-2,2′-bipyridine (dmbpy) ligated nickel(0) (Scheme 14).63 Two pathways from [Ni(dmbpy)2] were considered. The first was oxidative addition via a three-centre transition state, followed by transmetalation and reductive elimination (i.e. a Ni0/NiII cycle). The second was halide abstraction on the triplet surface to form a nickel(I) species that then mediates a NiI/NiIII cycle proceeding via transmetalation, oxidative addition, and reductive elimination. For the Ni0/NiII cycle, the highest energy structure was the oxidative addition transition state at 26.3 kcal mol−1vs. [Ni(dmbpy)2]. However, the formation of [NiI(dmbpy)] from [Ni(dmbpy)2] is facile; this occurs via a minimum energy crossing point from the singlet surface to the triplet surface (Grel = 3.0 kcal mol−1), halide abstraction (Grel = 3.6 kcal mol−1), and then ligand dissociation from [NiI(dmbpy)2] (Grel = 8.3 kcal mol−1).
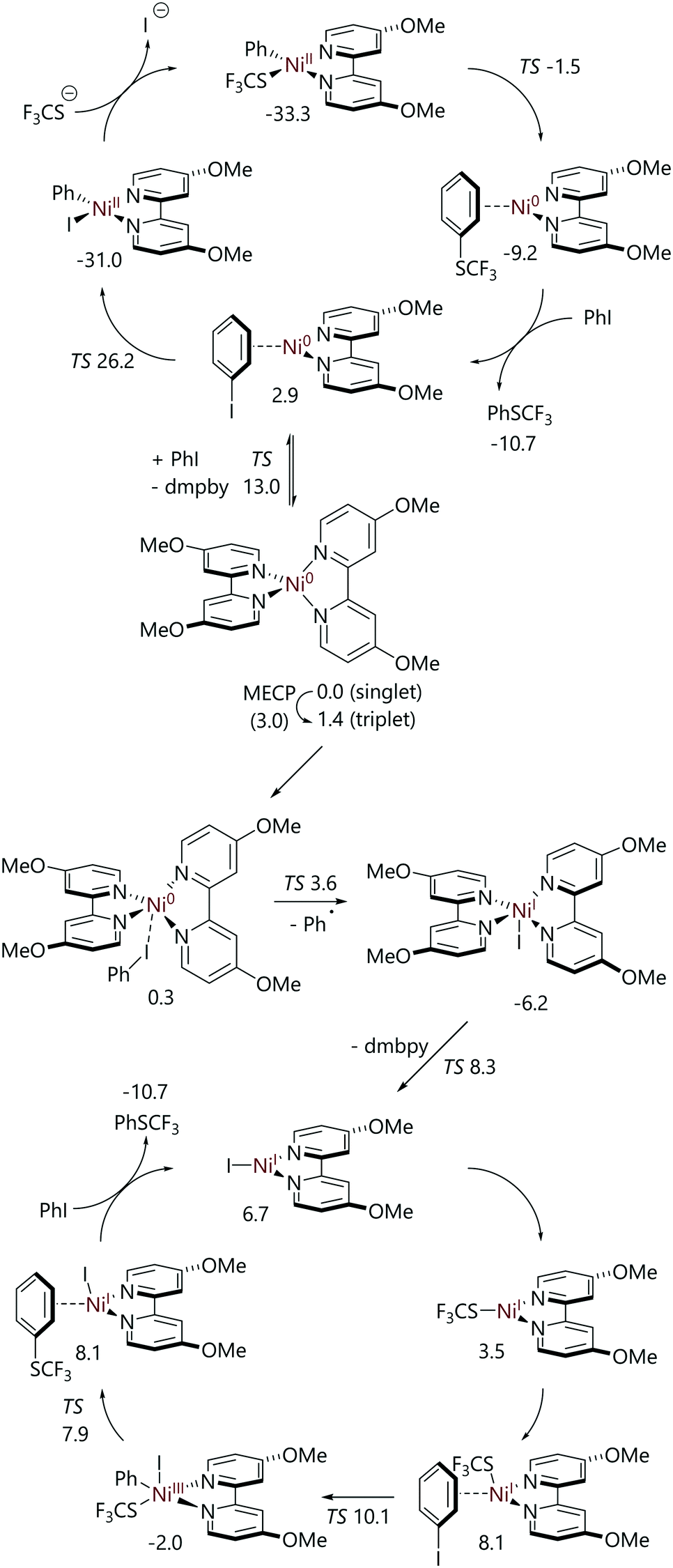 | ||
| Scheme 14 Catalytic cycles for iodobenzene trifluoromethylthiolation catalysed by [Ni(dmbpy)2], as determined from DFT calculations.63 | ||
While [NiI(dmbpy)] is 6.7 kcal mol−1 higher in energy than [Ni(dmbpy)2] plus iodobenzene, the NiI/NiIII catalytic cycle that it can mediate features very low barriers, with the slowest step being oxidative addition to [Ni(SCF3)(dmbpy)] (Grel = 10.1 kcal mol−1).
Mohadjer Beromi et al. have studied the reactions of aryl halides with bipyridine-nickel(0) and phenanthroline-nickel(0) complexes that were formed in situ (Scheme 15).9 In the case of the 4,4′-di(tert-butyl)-2,2′-bipyridine (dtbbpy) complex, the reaction of [Ni(COD)(dtbbpy)] with excess chlorobenzene led to [Ni(μ-Cl)(dtbbpy)]2 which was characterised by methods including NMR spectroscopy and X-ray crystallography;9 its magnetic moment was found to be 2.605μB (Evans's method). This dimer is EPR silent, and so cannot be detected or characterised using EPR spectroscopy. It can either be described as two ferromagnetically-coupled nickel(I) centres or as two nickel(II) centres with radical anion ligands; XPS data are consistent with a +2 oxidation state at nickel. This dimer could be converted to [NiCl2(dtbbpy)] using trityl chloride, which formed Gomberg's dimer as the byproduct. Alternatively, reactions with 2,4,6-trialkylphenylmagnesium bromide generated [Ni(Ar)2(dtbbpy)] complexes.
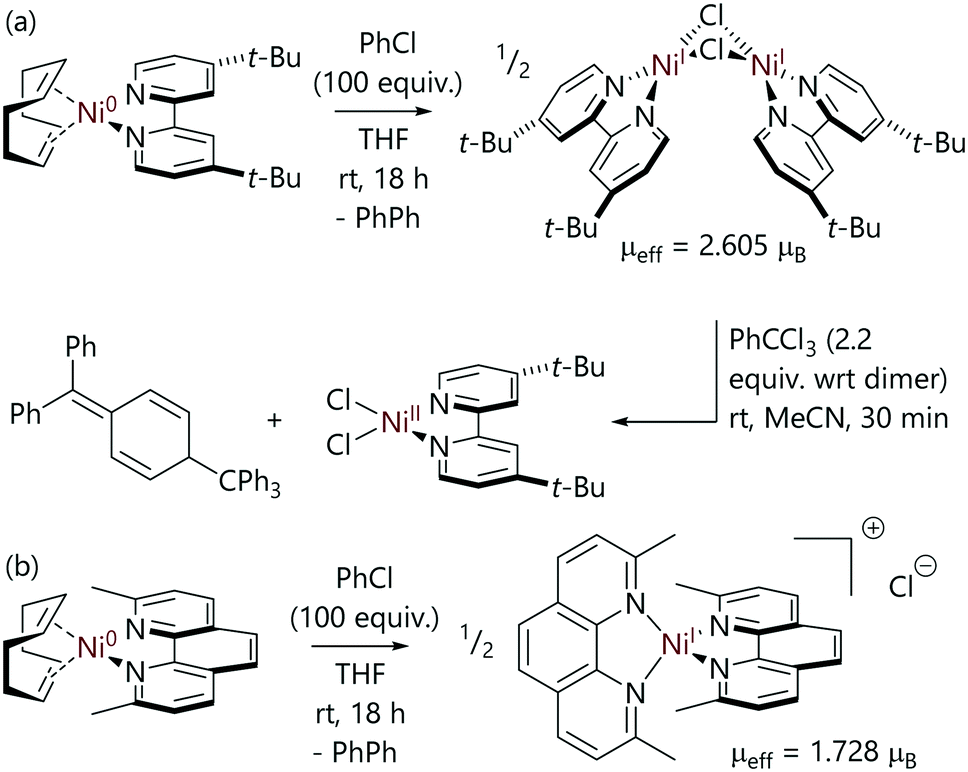 | ||
| Scheme 15 Reactions between [Ni(COD)(L)] complexes and chlorobenzene, where L is (a) a bidentate bipyridine or (b) a 2,9-dimethylphenanthroline ligand.9 | ||
In contrast, the reactions of neocuproine (2,9-dimethyl-1,10-phenanthroline, neoc) complex [Ni(COD)(neoc)]69 with excess chlorobenzene resulted in the formation of monomeric nickel(I) complex [Ni(neoc)]2Cl, which has an outer sphere chloride counterion (μeff = 1.728μB).9 This species was characterised by techniques including EPR spectroscopy. The outcomes of these reactions are therefore quite sensitive to ligand structure.
Nickel(0) complexes with N-heterocyclic carbene ligands
There is significant interest in the use of N-heterocyclic carbenes as ligands for nickel catalysis, especially for the activation of stronger bonds such as those in aryl ethers. The synthesis of NHC-bearing nickel catalysts70 and their applications in a variety of catalytic reactions71,72 have recently been reviewed. Nickel(0) complexes with one or two NHC ligands have been shown to react with organohalides via one or two electron steps.There are relatively few well-defined nickel(0) complexes with a single NHC ligand, and these are largely limited to [Ni(NHC)(η2,η2-1,6-hexadiene)],73 [Ni(NHC)(η6-arene)],74 and [Ni(NHC)(η2-olefin)2]75–77 complexes. [Ni(IPr)(η2,η2-1,6-hexadiene)] was found to react with allyl chlorides to form well-defined [Ni(allyl)Cl(IPr)] complexes (IPr = 1,3-bis(2,6-di(iso-propyl)phenyl)imidazol-2-ylidene).73
De Aguirre et al. have modelled each step of a tandem photocatalysis/cross-coupling reaction in which 2-iodoacetamide and 1-octene are converted to the corresponding indoline (Fig. 4).78 The catalyst formed in situ (from [Ni(COD)2] plus IPr) was proposed to be [Ni(IPr)(η2-1-octene)]; halide abstraction to form [Ni(I)(IPr)(η2-1-octene)] plus an aryl radical was found to be significantly more energetically favourable than oxidative addition (ΔΔG‡ = 13.7 kcal mol−1). The remainder of the work led the authors to conclude that a Ni0/NiI/NiII/NiIII mechanism was operative for this reaction.
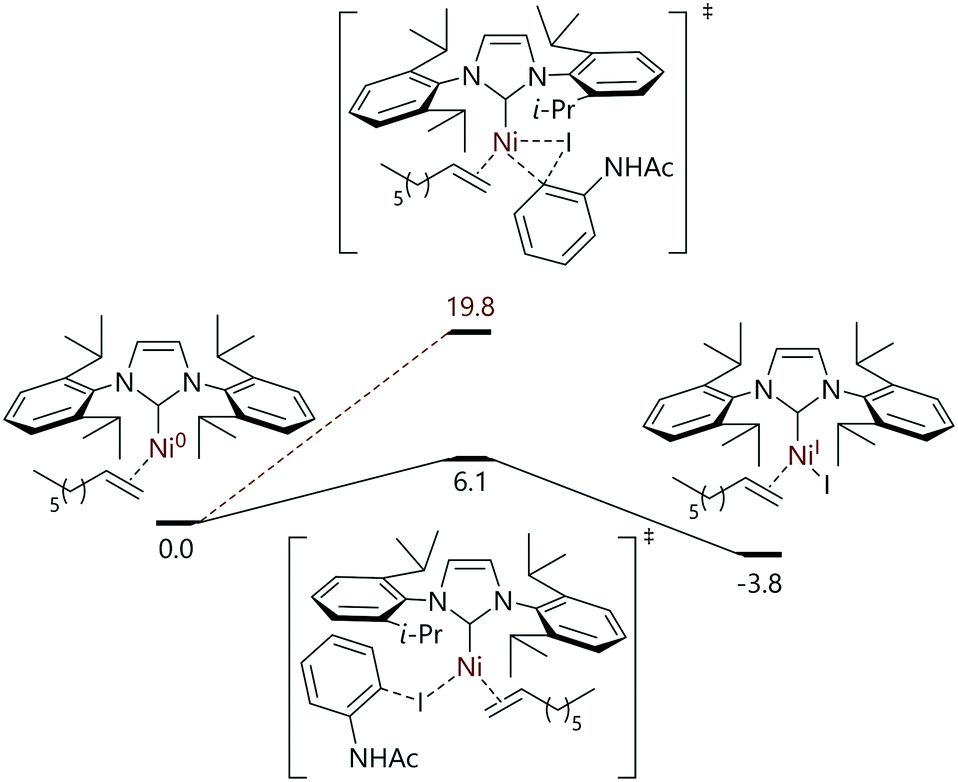 | ||
| Fig. 4 Halide abstraction as the first step in a tandem photocatalysis/cross-coupling reaction mechanism in which four oxidation states of nickel are invoked.78 | ||
Nicasio and Maseras have mapped out the Buchwald–Hartwig cross-coupling reactions of heteroaryl halides catalysed by [Ni(IPr)(η2-styrene)2] (Fig. 5).79 The reactions of 2-chloropyridine with [Ni(IPr)(η2-styrene)2] or [Ni(η2-C6H5Me)(IPr)] led to a 2:3 mixture of two nickel(II) complexes: a nickel monomer bound to the nitrogen and C2 of pyridine, and another in which two 2-pyridyl units bridge two nickel centres. Both structures were characterised using a number of techniques, including single crystal X-ray diffraction analysis. The use of 2-chloro-6-tert-butylpyridine prevented the formation of a 2-pyridyl-bridged species, and allowed the isolation of the monomeric complex. DFT analysis of this reaction pathway suggested that the reaction proceeded via the dissociation of (only) one styrene ligand, followed by coordination of 2-chloropyridine via the nitrogen atom. Rearrangement to an η2-complex and oxidative addition (via an SN2-type transition state) delivers the experimentally-observed [Ni(IPr)(κ2-N,C-2-pyridyl)] complex; a halide abstraction transition state was not located computationally, but it was noted that the halide abstraction step would produce products that were higher in energy by ca. 20 kcal mol−1 (Grel = 8.2–12.7 kcal mol−1).
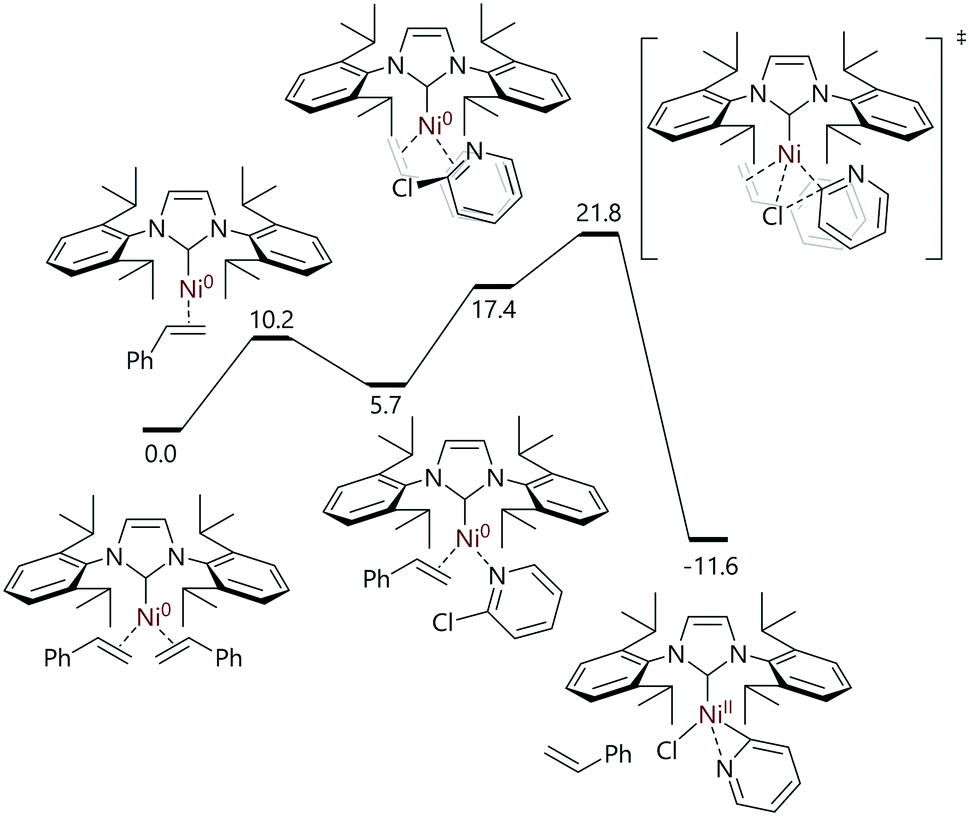 | ||
| Fig. 5 Oxidative addition as the first step in the [Ni(IPr)(η2-styrene)2]-catalysed amination of (hetero)aryl halides.79 | ||
In contrast, the reaction of 3-chloropyridine with [Ni(η2-C6H5Me)(IPr)] produced a trimeric [NiCl(IPr)(μ-κ1-C:κ1-N-3-pyridyl)]3 complex (in 82% isolated yield). Unlike the 2-pyridyl complexes, this species was catalytically inactive; it was noted that the reactions of 3-chloropyridine catalysed by [Ni(IPr)(η2-styrene)2] also failed to produce the desired product.
The mechanistic landscape for the reactions of [Ni(NHC)2] complexes with organohalides is determined by the structure of the NHC ligand, with small and large NHC ligands often leading to quite different reactivity for the corresponding nickel(0) complex. The reactions between organohalides and [Ni(IMeMe)2] complexes lead to well-defined trans-[Ni(R)(X)(IMeMe)2] complexes (Scheme 16(a)); however, [Ni(Me)(I)(IMeMe)2] decomposes rapidly once formed (IMeMe = 1,3,4,5-tetramethylimidazol-2-ylidene).80
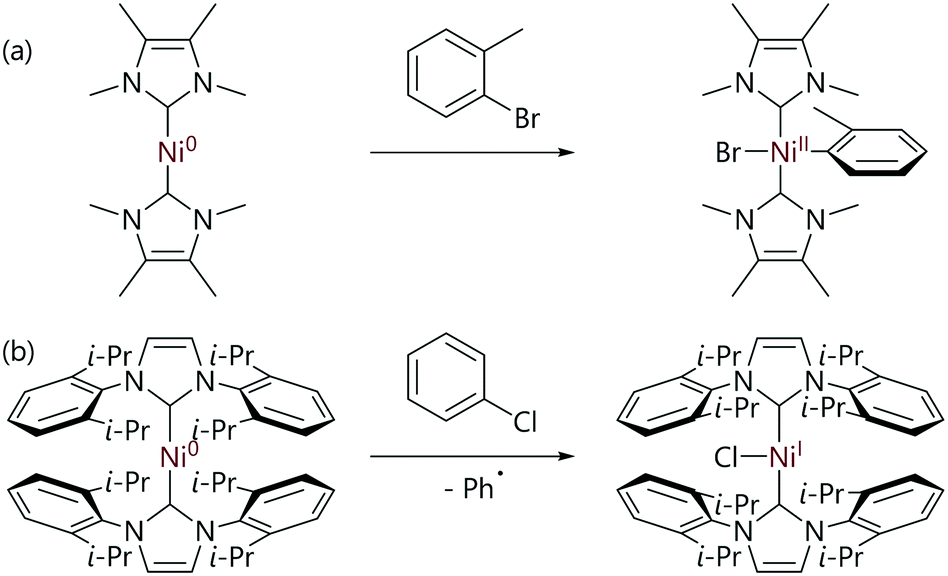 | ||
| Scheme 16 (a) An example of the oxidative addition of aryl halides to a [Ni(NHC)2] complex with a small NHC ligand.80 (b) An example of the reaction of an aryl halide with an [Ni(NHC)2] complex with a relatively large NHC ligand.81 | ||
Similar, well-behaved two electron behaviour is observed in reactions between [{Ni(IiPr)2}2(μ-η2:η2-COD)] and aryl halides, which form [Ni(Ar)(X)(IiPr)2] (IiPr = 1,3-di(iso-propyl)imidazol-2-ylidene).83,84 Reactions with aroyl halides led to the corresponding acylnickel(II) halide species.85
In contrast, the reactions of aryl halides with [Ni(NHC)2] complexes with larger NHC ligands such as IMes and IPr lead to nickel(I) complexes of the form [NiX(NHC)2] (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) (Scheme 16(b)).81,86 [NiCl(IPr)2] exists in equilibrium with [Ni(μ-Cl)(NHC)]2 species (often referred to as ‘Sigman's dimers’)87 plus free NHC,81 which can be used to prepare [NiCl(NHC)(PR3)] complexes by reaction with the corresponding phosphine ligand.88,89
Nelson and Maseras investigated this difference in reactivity as a function of NHC size using DFT calculations.82 The mechanistic proposal that was developed suggested that the difference in reactivity was due to the accessibility of the η2-complex that precedes oxidative addition. In the case of smaller NHC ligands, the linear C–Ni–C arrangement can bend relatively easily to accommodate an η2-aryl halide ligand, while for larger NHC ligands such a change in geometry is prohibitively expensive energetically (Fig. 6).
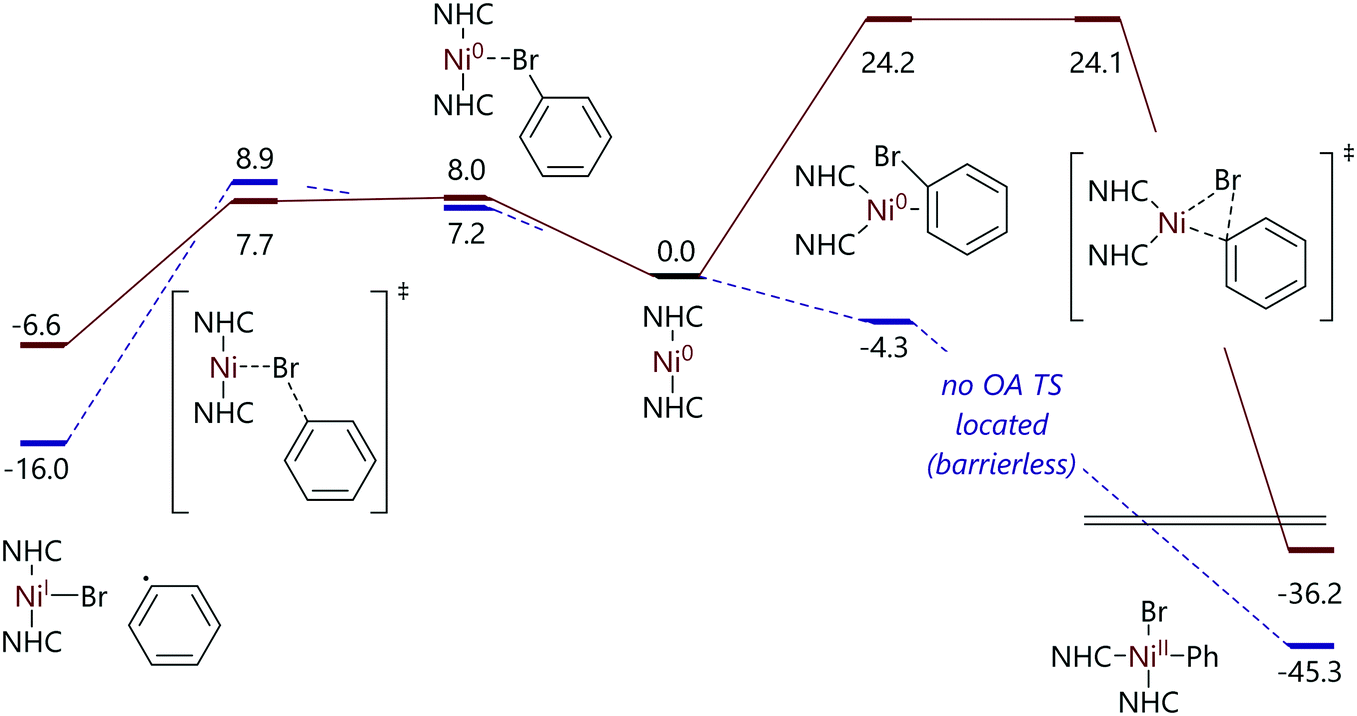 | ||
| Fig. 6 Free energy profiles for the reactions of bromobenzene with [Ni(IMeMe)2] and [Ni(IMes)2].82 | ||
Summary and outlook
The reactions of nickel(0) complexes with organohalides proceed via a range of different mechanisms, and can lead to nickel(I) and/or nickel(II) products. It is apparent that the precise outcomes and mechanisms are a function of both substrate and ligand structure, as well as the coordination number of the nickel(0) complex.The reactions of nickel(0) complexes with monodentate phosphines with aryl halides can proceed via halide abstraction or oxidative addition, while complexes with bidentate phosphines appear to proceed via two-electron chemistry. However, the reactions of alkyl halides clearly involve one electron steps such as halide abstraction, but in some cases this is followed by the recombination of the radical with the nickel(I) complex.
Bipyridine-type ligands have long been known to be redox non-innocent, and this is reflected in the reactions of the corresponding nickel(0) complexes. The evidence so far suggests an important role in catalysis for nickel(I) complexes with these ligands, especially in the developing field of tandem photocatalysis/cross-coupling.90
The third class of complex that has been examined in this review is NHC-nickel(0) complexes. The reactions of [Ni(NHC)(L)n] complexes can proceed via one or two-electron processes, but the factors that determine the pathway of choice in a given reaction remain relatively poorly understood. The situation for [Ni(NHC)2] complexes is rather more well established, with computational evidence suggesting that the size of the NHC ligands in such complexes influences how favourable oxidative addition is versus halide abstraction, with the latter requiring far less perturbation of the [Ni(NHC)2] complex from a linear coordination geometry.
There remain a number of practical and conceptual challenges in understanding the reactions of nickel(0) with organochlorides, organobromides, and organoiodides, and some of these are described here:
• Much of our understanding is still based on stoichiometric experiments, which can tell us what is feasible but tells us relatively little about rates; the collection of robust kinetic data for these reactions can be key to understanding the mechanistic features and, importantly, for distinguishing between possible pathways.
• Gathering data, and especially gathering kinetic data, for these systems requires a model complex that is both sufficiently stable to obtain in pure form yet sufficiently reactive to be representative of a process occurring during a catalytic reaction. We have conducted a number of (as yet unpublished) preliminary studies where we have struggled to either isolate the pure nickel(0) complex, where the necessary ancillary ligands make the complex poorly reactive; or where the model complex is insufficiently soluble for a robust kinetic study.
• On a related note, we would urge caution in interpreting ligand comparisons that rely on the formation of an active ligand-nickel complex in situ. The combination of a ligand and [Ni(COD)2] does not guarantee the formation of a single well-defined species.91
• The role of nickel(I) in catalysis is somewhat behind the scope of this review, although recent work in the area is producing convincing evidence that nickel(I) complexes can mediate some reactions.62,63,91–93 However, the involvement of nickel(I) versus nickel(0) can be difficult to infer, and the mechanism(s) by which a nickel(I) active species might form in a given reaction are often unclear.
• Identifying the products of reactions can be challenging; these could be paramagnetic nickel(I) complexes or paramagnetic or diamagnetic nickel(II) complexes, for example. While nickel(0) and square planar nickel(II) complexes lend themselves to analysis using techniques such as NMR spectroscopy, nickel(I) species are often best studied using EPR spectroscopy, and tetrahedral nickel(II) species are challenging to characterise by either of these methods. This is further complicated by the potential onwards reactions of nickel(I) or nickel(II) complexes.
• Some substrate classes are less well-studied and are consequently less well understood. The reactions of aryl halides have been explored relatively widely, while alkyl halides are of significant recent interest for us and other researchers. The reactions of vinyl halides with nickel(0) seem to be poorly explored, despite the potential for interesting coordination effects.38,39,47
• The reactions of nickel(0) complexes with phosphine ligands form the bulk of the literature in this area, with fewer studies of systems with NHC, bipyridine-type, or mixed (C, N/C, P/N, P) systems. This is likely linked to the challenges in studying these systems using spectroscopic tools, while 31P NMR spectroscopy is a very convenient tool for monitoring phosphine systems. The redox non-innocent94 nature of polypyridyl ligand scaffolds provides further challenges; the dimeric, paramagnetic, EPR-silent(!) nickel(I) complex isolated by Hazari's team9 serves as an example of a system that must have led to significant confusion before a crystal structure was obtained.
Despite these many challenges, this area remains an area of active investigation for us, providing a number of opportunities for academic studies that can lead to useful and impactful applications in catalysis.
The mechanistic complexity of nickel catalysis can in some cases be viewed as a somewhat negative aspect of this field; however, the diversity of reaction pathways that are available will undoubtedly support and underpin the development of new catalytic methodology.
Author contributions
MEG: investigation, writing – original draft; ELBJH: investigation, writing – original draft; DJN: investigation, supervision, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MEG thanks AstraZeneca and the EPSRC for an Industrial CASE Studentship (EP/R512114/1). ELBJH thanks the University of Strathclyde for a Research Excellence Studentship. DJN thanks the University of Strathclyde for a Chancellor's Fellowship (2014–2018). We are grateful to all co-workers, collaborators, and funders – past and present – for their contributions to our research programme in nickel catalysis.Notes and references
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- J. A. Labinger, Organometallics, 2015, 34, 4784–4795 CrossRef CAS.
- M. Portnoy and D. Milstein, Organometallics, 1993, 12, 1665–1673 CrossRef CAS.
- M. M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC.
- M. Mohadjer Beromi, G. W. Brudvig, N. Hazari, H. M. C. Lant and B. Q. Mercado, Angew. Chem., Int. Ed., 2019, 58, 6094–6098 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, ACS Catal., 2019, 9, 6803–6813 CrossRef CAS.
- P. Morgante and R. Peverati, Int. J. Quantum Chem., 2020, 120, e26332 CrossRef CAS.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- N. Fey, B. M. Ridgway, J. Jover, C. L. McMullin and J. N. Harvey, Dalton Trans., 2011, 40, 11184–11191 RSC.
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2010, 111, 1346–1416 CrossRef PubMed.
- A. Arévalo and J. J. García, Eur. J. Inorg. Chem., 2010, 2010, 4063–4074 CrossRef.
- P. M. Pérez-García and M.-E. Moret, Chimia, 2020, 74, 495–498 CrossRef PubMed.
- D. R. Fahey, J. Am. Chem. Soc., 1970, 92, 402–404 CrossRef CAS.
- M. Foa and L. Cassar, J. Chem. Soc., Dalton Trans., 1975, 2572–2576 RSC.
- C. A. Tolman, W. C. Seidel and L. W. Gosser, J. Am. Chem. Soc., 1974, 96, 53–60 CrossRef CAS.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS.
- I. H. Elson, D. G. Morrell and J. K. Kochi, J. Organomet. Chem., 1975, 84, C7–C10 CrossRef CAS.
- I. Funes-Ardoiz, D. J. Nelson and F. Maseras, Chem. – Eur. J., 2017, 23, 16728–16733 CrossRef CAS PubMed.
- A. Manzoor, P. Wienefeld, M. C. Baird and P. H. M. Budzelaar, Organometallics, 2017, 36, 3508–3519 CrossRef CAS.
- G. Favero, A. Morvillo and A. Turco, Gazz. Chim. Ital., 1979, 109, 27–28 CAS.
- A. Morvillo and A. Turco, J. Organomet. Chem., 1981, 208, 103–113 CrossRef CAS.
- A. L. Clevenger, R. M. Stolley, N. D. Staudaher, N. Al, A. L. Rheingold, R. T. Vanderlinden and J. Louie, Organometallics, 2018, 37, 3259–3268 CrossRef CAS.
- C. Amatore and A. Jutand, Organometallics, 1988, 7, 2203–2214 CrossRef CAS.
- L. M. Guard, M. Mohadjer Beromi, G. W. Brudvig, N. Hazari and D. J. Vinyard, Angew. Chem., Int. Ed., 2015, 54, 13352–13356 CrossRef CAS PubMed.
- M. Mohadjer Beromi, A. Nova, D. Balcells, A. M. Brasacchio, G. W. Brudvig, L. M. Guard, N. Hazari and D. J. Vinyard, J. Am. Chem. Soc., 2017, 139, 922–936 CrossRef CAS PubMed.
- E. L. Barth, R. M. Davis, M. Mohadjer Beromi, A. G. Walden, D. Balcells, G. W. Brudvig, A. H. Dardir, N. Hazari, H. M. C. Lant, B. Q. Mercado and I. L. Peczak, Organometallics, 2019, 38, 3377–3387 CrossRef CAS PubMed.
- E. Nicolas, A. Ohleier, F. D'Accriscio, A.-F. Pécharman, M. Demange, P. Ribagnac, J. Ballester, C. Gosmini and N. Mézailles, Chem. – Eur. J., 2015, 21, 7690–7694 CrossRef CAS PubMed.
- C. M. Lavoie, R. McDonald, E. R. Johnson and M. Stradiotto, Adv. Synth. Catal., 2017, 359, 2972–2980 CrossRef CAS.
- S. Bajo, G. Laidlaw, A. R. Kennedy, S. Sproules and D. J. Nelson, Organometallics, 2017, 36, 1662–1672 CrossRef CAS.
- G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164–4172 CrossRef CAS PubMed.
- M. E. Greaves, T. O. Ronson, G. C. Lloyd-Jones, F. Maseras, S. Sproules and D. J. Nelson, ACS Catal., 2020, 10, 10717–10725 CrossRef CAS PubMed.
- C. Amatore and F. Pfluger, Organometallics, 1990, 9, 2276–2282 CrossRef CAS.
- A. K. Cooper, D. K. Leonard, S. Bajo, P. M. Burton and D. J. Nelson, Chem. Sci., 2020, 11, 1905–1911 RSC.
- A. K. Cooper, P. M. Burton and D. J. Nelson, Synthesis, 2020, 52, 565–573 CrossRef CAS.
- T. T. Tsou, J. C. Huffman and J. K. Kochi, Inorg. Chem., 1979, 18, 2311–2317 CrossRef CAS.
- R. Countryman and B. R. Penfold, J. Cryst. Mol. Struct., 1972, 2, 281–290 CrossRef CAS.
- A. N. Desnoyer, E. G. Bowes, B. O. Patrick and J. A. Love, J. Am. Chem. Soc., 2015, 137, 12748–12751 CrossRef CAS PubMed.
- D. J. Mindiola, R. Waterman, D. M. Jenkins and G. L. Hillhouse, Inorg. Chim. Acta, 2003, 345, 299–308 CrossRef CAS.
- A. N. Desnoyer, W. He, S. Behyan, W. Chiu, J. A. Love and P. Kennepohl, Chem. – Eur. J., 2019, 25, 5259–5268 CrossRef CAS PubMed.
- W. He and P. Kennepohl, Faraday Discuss., 2019, 220, 133–143 RSC.
- D. Strawser, A. Karton, O. V. Zenkina, M. A. Iron, L. J. W. Shimon, J. M. L. Martin and M. E. van der Boom, J. Am. Chem. Soc., 2005, 127, 9322–9323 CrossRef CAS PubMed.
- O. V. Zenkina, A. Karton, D. Freeman, L. J. W. Shimon, J. M. L. Martin and M. E. van der Boom, Inorg. Chem., 2008, 47, 5114–5121 CrossRef CAS PubMed.
- O. V. Zenkina, O. Gidron, L. J. W. Shimon, M. A. Iron and M. E. van der Boom, Chem. – Eur. J., 2015, 21, 16113–16125 CrossRef CAS PubMed.
- M. Orbach, S. Shankar, O. V. Zenkina, P. Milko, Y. Diskin-Posner and M. E. van der Boom, Organometallics, 2015, 34, 1098–1106 CrossRef CAS.
- N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15258–15259 CrossRef CAS PubMed.
- J. A. Bilbrey, A. N. Bootsma, M. A. Bartlett, J. Locklin, S. E. Wheeler and W. D. Allen, J. Chem. Theory Comput., 2017, 13, 1706–1711 CrossRef CAS PubMed.
- J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC.
- E. D. Entz, J. E. A. Russell, L. V. Hooker and S. R. Neufeldt, J. Am. Chem. Soc., 2020, 142, 15454–15463 CrossRef CAS PubMed.
- E. Jacobs and S. T. Keaveney, ChemCatChem, 2021, 13, 637–645 CrossRef CAS.
- C. A. Malapit, J. R. Bour, C. E. Brigham and M. S. Sanford, Nature, 2018, 563, 100–104 CrossRef CAS PubMed.
- P. M. Pérez-García, A. Darù, A. R. Scheerder, M. Lutz, J. N. Harvey and M.-E. Moret, Organometallics, 2020, 39, 1139–1144 CrossRef PubMed.
- D. R. Fahey and J. E. Mahan, J. Am. Chem. Soc., 1977, 99, 2501–2508 CrossRef CAS.
- J. K. Stille and A. B. Cowell, J. Organomet. Chem., 1977, 124, 253–261 CrossRef CAS.
- A. Zhang, C. Wang, X. Lai, X. Zhai, M. Pang, C.-H. Tung and W. Wang, Dalton Trans., 2018, 47, 15757–15764 RSC.
- R. Kehoe, M. Mahadevan, A. Manzoor, G. McMurray, P. Wienefeld, M. C. Baird and P. H. M. Budzelaar, Organometallics, 2018, 37, 2450–2467 CrossRef CAS.
- C. Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347–5399 RSC.
- I. Kalvet, Q. Guo, G. J. Tizzard and F. Schoenebeck, ACS Catal., 2017, 7, 2126–2132 CrossRef CAS PubMed.
- J. Jover, Catal. Sci. Technol., 2019, 9, 5962–5970 RSC.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175–13183 CrossRef CAS PubMed.
- J. T. Ciszewski, D. Y. Mikhaylov, K. V. Holin, M. K. Kadirov, Y. H. Budnikova, O. Sinyashin and D. A. Vicic, Inorg. Chem., 2011, 50, 8630–8635 CrossRef CAS PubMed.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192–16197 CrossRef CAS PubMed.
- M. Newcomb, in Encyclopedia of Radicals in Chemistry, Biology and Materials, 2012, DOI:10.1002/9781119953678.rad007.
- The nickel(0) species may be [Ni(COD)(neoc)], [Ni(neoc)]2, or a mixture of the two, but the ligand was added as 1 molar equivalent with respect to nickelz.
- A. P. Prakasham and P. Ghosh, Inorg. Chim. Acta, 2014, 431, 61–100 CrossRef.
- V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef CAS.
- M. Henrion, V. Ritleng and M. J. Chetcuti, ACS Catal., 2015, 5, 1283–1302 CrossRef CAS.
- J. Wu, J. W. Faller, N. Hazari and T. J. Schmeier, Organometallics, 2012, 31, 806–809 CrossRef CAS.
- Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi and S. Ogoshi, Organometallics, 2014, 33, 1276–1282 CrossRef CAS.
- M. J. Iglesias, J. F. Blandez, M. R. Fructos, A. Prieto, E. Álvarez, T. R. Belderrain and M. C. Nicasio, Organometallics, 2012, 31, 6312–6316 CrossRef CAS.
- A. J. Nett, S. Cañellas, Y. Higuchi, M. T. Robo, J. M. Kochkodan, M. T. Haynes, J. W. Kampf and J. Montgomery, ACS Catal., 2018, 8, 6606–6611 CrossRef CAS PubMed.
- S. Felten, S. F. Marshall, A. J. Groom, R. T. Vanderlinden, R. M. Stolley and J. Louie, Organometallics, 2018, 37, 3687–3697 CrossRef CAS.
- A. de Aguirre, I. Funes-Ardoiz and F. Maseras, Angew Chem., Int. Ed., 2019, 58, 3898–3902 CrossRef CAS PubMed.
- S. G. Rull, I. Funes-Ardoiz, C. Maya, F. Maseras, M. R. Fructos, T. R. Belderrain and M. C. Nicasio, ACS Catal., 2018, 8, 3733–3742 CrossRef CAS.
- D. S. McGuinness, K. J. Cavell, B. W. Skelton and A. H. White, Organometallics, 1999, 18, 1596–1605 CrossRef CAS.
- S. Miyazaki, Y. Koga, T. Matsumoto and K. Matsubara, Chem. Commun., 2010, 46, 1932–1934 RSC.
- D. J. Nelson and F. Maseras, Chem. Commun., 2018, 54, 10646–10649 RSC.
- T. Zell, M. Feierabend, B. Halfter and U. Radius, J. Organomet. Chem., 2011, 696, 1380–1387 CrossRef CAS.
- T. Zell, P. Fischer, D. Schmidt and U. Radius, Organometallics, 2012, 31, 5065–5073 CrossRef CAS.
- T. Zell and U. Radius, Z. Anorg. Allg. Chem., 2013, 639, 334–339 CrossRef CAS.
- K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546–2552 CrossRef CAS PubMed.
- B. R. Dible, M. S. Sigman and A. M. Arif, Inorg. Chem., 2005, 44, 3774–3776 CrossRef CAS PubMed.
- S. Nagao, T. Matsumoto, Y. Koga and K. Matsubara, Chem. Lett., 2011, 40, 1036–1038 CrossRef CAS.
- K. Matsubara, Y. Fukahori, T. Inatomi, S. Tazaki, Y. Yamada, Y. Koga, S. Kanegawa and T. Nakamura, Organometallics, 2016, 35, 3281–3287 CrossRef CAS.
- J. Twilton, P. Zhang, M. H. Shaw, R. W. Evans and D. W. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997–2009 CrossRef CAS PubMed.
- T. Inatomi, Y. Fukahori, Y. Yamada, R. Ishikawa, S. Kanegawa, Y. Koga and K. Matsubara, Catal. Sci. Technol., 2019, 9, 1784–1793 RSC.
- R. J. Somerville, C. Odena, M. F. Obst, N. Hazari, K. H. Hopmann and R. Martin, J. Am. Chem. Soc., 2020, 142, 10936–10941 CrossRef CAS PubMed.
- Perhaps we should refer to these systems as 'redox guilty'.
| This journal is © The Royal Society of Chemistry 2021 |