
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a H-Sulfo-POSS catalyst and application in the acetalization of glycerol with 2-butanone to yield a biofuel additive†
Viktor
Söderholm
a,
Jesús
Esteban
 b and
Dieter
Vogt
*a
b and
Dieter
Vogt
*a
aLaboratory of Industrial Chemistry, Department of Biochemical and Chemical Engineering, Technical University of Dortmund, Emil-Figge-Str. 66, D-44227 Dortmund, Germany. E-mail: dieter.vogt@tu-dortmund.de
bDepartment of Chemical Engineering and Analytical Science, The University of Manchester, Manchester M13 9PL, UK
First published on 25th May 2021
Abstract
Nanosized polyhedral oligomeric silsesquioxanes (POSS) have been employed as molecular weight enlarged structures for different purposes owing to their ease of synthesis and modification. In this work, the commercially available compound octaphenyl-POSS has been modified with chlorosulfonic acid to obtain multifunctional acidic materials (H-Sulfo-POSS) with different degrees of functionality in the structure. The synthesized materials have been characterized by ion exchange capacity, degree of sulfonation, 1H-NMR, ATR-IR spectroscopy and SEM. These H-Sulfo-POSS nanocomposites were then applied to the acetalization of glycerol with 2-butanone to yield the corresponding ketal, which is a product of interest as an oxygenate fuel additive. The catalytic activity in dynamic experiments of the H-Sulfo-POSS (2601 h−1) material with the highest degree of sulfonation was even higher than that shown by the molecular catalyst para-toluene sulfonic acid (PTSA) (2256 h−1) and better than Amberlyst 36 (213 h−1); however, upon recycling by centrifugation of the catalyst, the activity of this material was not very stable. On the other hand, the material with the lowest degree of sulfonation showed not only higher activity than Amberlyst 36, but also very stable activity after reutilization throughout 10 runs.
1. Introduction
Silsesquioxanes are hybrid inorganic–organic nanostructured materials that are gaining increasing attention as advanced polymer composites. They originate from the hydrolysis and condensation of silanes with alkoxy groups and can acquire different structures like ladder, cage and partial cages. Polyhedral oligomeric silsesquioxanes (POSS) have become very attractive arrangements of silsesquioxanes that originate from different monomers giving rise to structures of the general formula (RSiO1.5)n, in which R can be a range of organic substituents that lead to styryl-POSS, methacrylate-POSS, norbornyl-POSS, vinyl-POSS, epoxy-POSS or siloxane-POSS, among others.1,2Fig. 1 presents the general structure of a cubic POSS material. | ||
| Fig. 1 The general structure of a cubic polyhedral oligomeric silsesquioxane (POSS). Organic R groups can be unreactive or reactive, with the X substituent being different or the same as R. | ||
Owing to their environmental neutrality, chemical and thermal inertness, high heat resistance features and ability to improve mechanical properties of polymers, they have been widely applied. Many studies have studied POSS or POSS-containing hybrid polymers as fire retardants, high performance materials in nanocomposite membranes or in the synthesis of advanced materials for biomedicine and tissue engineering, electronics or photo- and electro-luminescence devices.2–4
POSS have also seen applications in catalysis owing to their characteristic arrangements, rigid structure and commercial availability, which make them great candidates for molecular weight enlargement (MWE), with the corresponding good recyclability features. For example, a POSS-supported N-heterocyclic carbenes/imidazolium salts on Pd nanoparticles have proven effective and recyclable catalysts for Suzuki–Miyaura cross-coupling reactions with TOFs as high as 1670 h−1.5 Another case is the use of hybrid composites made from octa-aminopropyl POSS hydrochloride salt and ionic liquids onto which Cu or Ag nanoparticles are immobilized. Such catalysts are active and show good recyclability in “click” reactions and in the reduction of 4-nitrophenol.6 Finally, the catalytic activity of Ru complexes, molecular weight enlarged by POSS moieties, in metathesis reactions proved comparable to that of commercially available second generation Grubbs–Hoveyda catalysts. Recycling experiments proved that the retention of the catalyst was high when separating the product with ceramic membranes.7
As a consequence of the development of the biodiesel industry, over the years, glycerol (Gly) has become widely available and there has been an opportunity to use it as a building block for many products of interest.8,9 Among them, glycerol acetals appear as interesting additives in (bio)fuels owing to their oxygenate nature, which have rendered them useful as enhancers of the octane number and cold flow properties as well as successfully reducing particulate emissions and gum formation.10,11
The production of glycerol acetals stems from the acid-catalyzed reaction of glycerol with aldehydes or ketones in a reaction that also generates a molecule of water as a by-product. The reaction will only take place in the presence of an acid catalyst.10,11 The acetal products obtained in this reaction can contain 5-membered rings (dioxolanes) or 6-membered rings (dioxanes), the former being usually kinetically favoured. Fig. 2 presents a scheme of the reaction.
 | ||
| Fig. 2 Overall scheme of the acetalization of glycerol with a ketone or aldehyde. | ||
Different catalysts have been used for this purpose. PTSA has been utilized for the acetalization of glycerol with formaldehyde,12 acetone13 or benzaldehyde.14 As for heterogeneous catalysts, sulfonic acid-based ion exchange resins like the classic Amberlyst 15 have been used for the reaction of glycerol with aldehydes of different chain length from butanal to decanal,15 Amberlyst 35 and 36 for the production of solketal with acetone16,17 or reacting glycerol with formaldehyde or benzaldehyde.18 Other solid materials include micro or mesoporous materials like zeolites, such as USY or ZSM for reaction with butanal19 or BEA with benzaldehyde,18 or montmorillonite K-10.18
Finally, a polystyrene sulfonate-octavinyl POSS polymeric catalyst (POSS-x-SO3H) has been successful used in the acetalization of glycerol with benzaldehyde, reaching conversions over 92% with good overall selectivity to the acetal products.20 Moreover, the selectivity to the dioxolane product with respect to the dioxane was 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20,20 higher than those obtained with Amberlyst 36 (61:39), zeolite BEA (59:41) or montmorillone K-10 (48:52).18 The reaction with butanone is also possible, but has been somehow understudied considering the similarity of the product with solketal. For this reaction, focus of our present study, there are reports using the following catalysts: H3PW12P40,21 SnCl2,22 AlF3·H2O,23 and Re/SiO2.24
:20,20 higher than those obtained with Amberlyst 36 (61:39), zeolite BEA (59:41) or montmorillone K-10 (48:52).18 The reaction with butanone is also possible, but has been somehow understudied considering the similarity of the product with solketal. For this reaction, focus of our present study, there are reports using the following catalysts: H3PW12P40,21 SnCl2,22 AlF3·H2O,23 and Re/SiO2.24
Given the use of acidic catalysts for this reaction and others involving renewable substrates to further value-added chemicals, materials modified with sulfonic acid functionalities appear as an interesting alternative. Another example of reaction requiring acidic media to take place is the dehydration of sugars like glucose or fructose to obtain 5-hydroxymethylfurfural and levulinic acid,25 which has been reported using a sulfonated hyperbranched poly(arylene oxindole) as catalyst.26 In this sense, H-Sulfo-POSS poses a multifunctional nanomaterial that could be of interest for catalysis. Therefore, this work focuses on the synthesis of this material, its characterization and performance in the acetalization of glycerol, also analyzing its stability upon several cycles of recycling.
2. Experimental
2.1. Materials
For the preparation of the H-Sulfo-POSS catalysts, the following chemicals were used: OP-POSS (Hybrid Plastics, 99%), chlorosulfonic acid (Acros Organics, 97%), dichloromethane (VWR, 99.5%) and chloroform (VWR, 99.8%). The catalytic runs for the synthesis of the glycerol acetal required the use of glycerol (Henkel, 99%), butanone (Aldrich, 99%), ethanol (ITW Reagents, 99%), p-toluenesulfonic acid (Sigma Aldrich, 99%) Amberlyst 36 (Sigma Aldrich).2.2. Synthesis of H-Sulfo-POSS
1H-NMR (Sulfo-POSS-1 sodium salt, 400 MHz, D2O) δ = 7.97 (s, 1H), 7.77 (d, J = 7.3 Hz, 1H), 7.73 (dd, J = 8.0, 1.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H) ppm.
1H-NMR (Na-Sulfo-POSS-2, 400 MHz, D2O) δ = 8.08 (s, 1H), 7.87 (m, 1H), 7.83 (dd, J = 8.1, 1.3 Hz, 1H), 7.58 (m, 1H).
13C-NMR (Na-Sulfo-POSS-2, 101 MHz, D2O) δ = 142.17, 141.59, 137.02, 136.94, 131.59, 130.32, 128.98, 128.41, 126.63, 125.31 ppm.
1H-NMR (Na-Sulfo-POSS-3, 400 MHz, D2O) δ = 8.09 (s, 1H), 7.88 (dd, J = 7.5, 1.3 Hz, 1H), 7.83 (dd, J = 8.0, 1.4 Hz, 1H), 7.60 (m, 1H) ppm.
13C-NMR (Na-Sulfo-POSS-3, 101 MHz, D2O) δ = 141.65, 137.00, 136.37, 131.60, 130.33, 128.98, 128.47, 126.79, 125.31 ppm.
2.3. Characterization of H-Sulfo-POSS
2.4. Acetalization of glycerol with 2-butanone and recycling tests
H-Sulfo-POSS-1 was reapplied for recycling experiments, but this time three tubes were run in parallel. In cycle 1, 5 and 10, the reaction was sampled over time. Because sampling removes catalyst from the reaction mixture it will have to be discarded from future cycles. Therefore, the whole experiment started with three reaction vessels. The first vessel was used up for sample over time experiment in cycle 1, the second and third vessels were used up in sampling over time experiments in cycle 5 and 10 respectively. Fig. 3 presents a summary of the process.
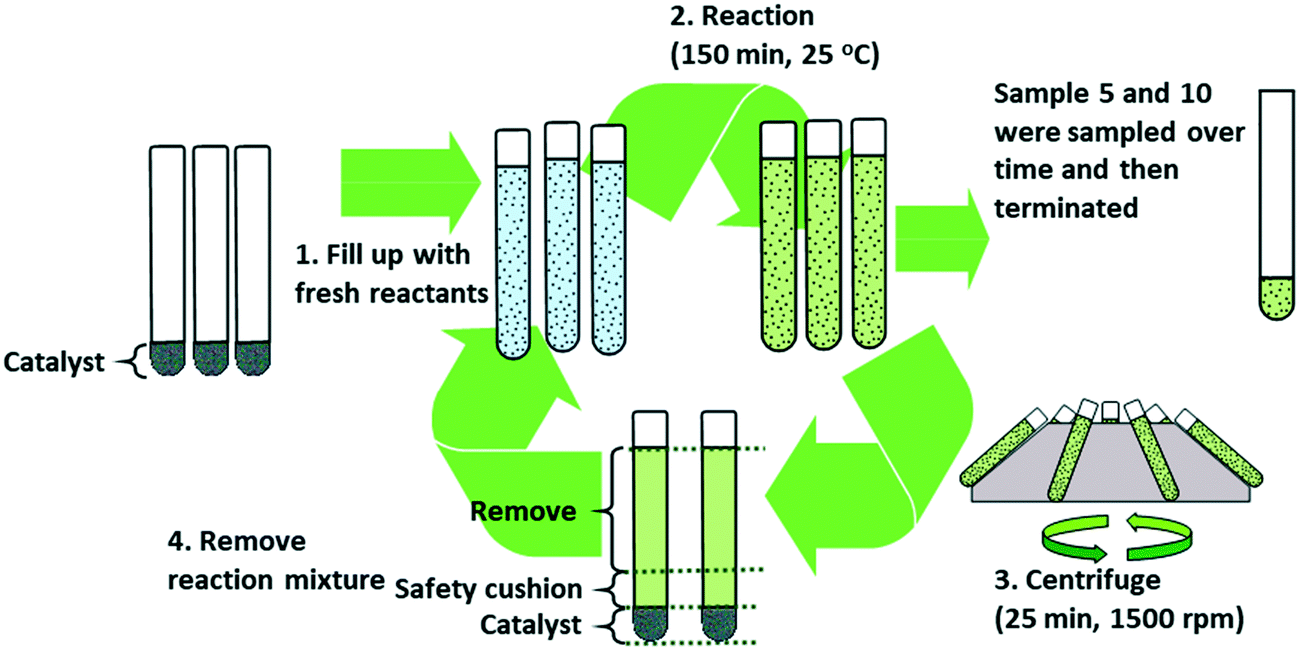 | ||
| Fig. 3 Operation and catalyst recycling strategy by centrifugation-aided sedimentation after each reaction. | ||
3. Results and discussion
3.1. Synthesis and characterization of H-Sulfo-POSS
H-Sulfo-POSS was synthesized by direct chlorosulfonation of octaphenyl-POSS by adding chlorosulfonic acid in excess at 0 °C, then leaving to react over night at room temperature and finally adding water for hydrolysis. The synthetic protocol that was used was based on that of Kafi et al.28 The overall reaction is presented in Fig. 4 below. | ||
| Fig. 4 Synthesis of H-Sulfo-POSS by chlorosulfonation of octaphenyl-POSS and subsequent hydrolysis. | ||
The synthesis was performed three times where small changes in parameters were made between them, and which is presented in Table 1 below. The first two experiments resulted in overnight premature precipitation of (product) material, while the third experiment stayed fully dissolved until water was being added the next day. Each of the three different products have been labelled a specific name (H-Sulfo-POSS-1, -2 or -3).
| H-Sulfo-POSS | Solvent | V (solvent)/m(OP-POSS) | Eq. of HSO3Cl (per pH) | Est. Sulf. Deg. | Yield |
|---|---|---|---|---|---|
| [−] | [−] | [ml g−1] | [−] | [−] | [%] |
| 1 | CH2Cl2 | 9 | 3.9 | 0.195 | 30 |
| 2 | CHCl3 | 20 | 7.2 | 0.285 | 62 |
| 3 | CHCl3 | 10 | 8.7 | 0.660 | 81 |
The absence of any acidic contamination material in the collected crude H-Sulfo-POSS product had to be safeguarded as they were to be investigated for their acidic catalytic properties. The crude products were therefore first neutralized with NaOH (Na2CO3 was used in the first attempt) and then re-acidified by stirring in concentrated HCl and finally left on high vacuum. During the chlorosulfonation stage, up to eight phenyl groups of each octaphenyl-POSS substrate could have been sulfonated. The collected products therefore contained a varying degree of sulfonation (ratio between sulfonic groups to total number of phenyl groups). The overall degree of sulfonation for each product batch was estimated based on the H-Sulfo-POSS ion-exchange-capacity (IEC) of a sample from that batch. During all three synthetic attempts an initially clear, homogeneous and slightly yellow reaction mixture resulted after all chlorosulfonic acid had been added. Extensive premature precipitation of a white solid occurred over night in the first two experiments. Interestingly these two initial products showed significantly lower IEC than the third, which stayed in solution also until the next day. Some have reported that the degree of sulfonation can be controlled by the excess of chlorosulfonic acid used, where 6–11 equivalents of chlorosulfonic acid to phenyl groups would give about 40% complete sulfonation.29 In contrast, others have reported significantly higher degree of sulfonation when using the same equivalents of chlorosulfonic acid.30 And yet again, others have reported as high as 94% complete sulfonation when only using 1.5 eq. of chlorosulfonic acid to phenyl groups.27 Hence, there is large ambiguity about what the important factors are that determine the degree of sulfonation of octaphenyl-POSS. But in all protocols the temperature has always been kept close to 0 °C while adding the chlorosulfonic acid dropwise. A previous study on sulfonation of phenyl-silanes observed large amounts of Si–C(phenyl) bond cleavage.31 In this work the lower degree of sulfonation of the first two products is simply attributed to the premature precipitation from the reaction mixture. This effectively made their still unreacted phenyl groups inaccessible to the chlorosulfonic acid. The risk of premature precipitation from the reaction mixture seems to increase when more solvent (chloroform or DCM) is used per amount of OP-POSS.
The product that was collected from experiment 3 had an overall degree of sulfonation of 66% after having reacted at room temperature for 12 h. Hence, the rate of the sulfonation reaction itself at room temperature was not very vigorous at all. To reach higher degrees of sulfonation it is certainly important to maintain full solubility through the first stage (hence use a large excess of chlorosulfonic acid), but longer reaction times and slightly increased reaction temperatures are suggested to be tested. All three H-Sulfo-POSS products were obtained as very fine grey/white powders. They were practically insoluble in any of the following solvents (water, ethanol, DMSO, THF, DCM, chloroform). And so, their characterization by NMR had to be done on their associated Na-salts by reacting the H-Sulfo-POSS materials with NaOH.29 Which improved solubility in water for H-Sulfo-POSS-1 to 2 g L−1, and for H-Sulfo-POSS-2 and -3 to approximately 5 g L−1. The yields of the products were not great for any of the experiments, especially bad for H-Sulfo-POSS-1 (30%). The reason for this is mainly due to underestimating the water solubilities of the Na-derivatives during their washing with water. Albeit those steps were necessary to take as to guarantee no remainder of chlorosulfonic, H2SO4 or HCl being present in the final products.
| H-Sulfo-POSS | IEC [meq g−1] | Sulfonation degree of reacidified H-Sulfo-POSS materials | |
|---|---|---|---|
| [−] | Crude | Reacidified | |
| 1 | — | 1.34 | 0.195 |
| 2 | 2.99 | 1.89 | 0.285 |
| 3 | 4.03 | 3.62 | 0.660 |
As can be seen, the crude products had significantly greater IEC than after they had been neutralized and re-acidified again. The reason for this is that contaminating H2SO4, HCl or chlorosulfonic acid remaining in the crude products would by then have been neutralized and removed. But it is also possible that the re-acidification process was not complete and that there existed unprotonated sulfonic acid groups after the re-acidification step. This could be a possibility as when the relatively highly water-soluble Na-Sulfo-POSS is protonated, its solubility decreases and starts precipitating and trapping still yet unprotonated sulfonic groups inside precipitated particles.
The assignment of the spectra was not trivial as each sample contained a mixture of Sulfo-POSS molecules with a variety of different degrees of sulfonation. The singlet at 8.04 represents the proton at the ortho-position to both POSS and the sulfonic group.28,29 The peaks slightly downfield were not possible to be individually identified due to peak overlap of protons associated with both sulfonated and non-sulfonated phenyl groups.
 | ||
| Fig. 5 ATR-FTIR spectra of octaphenyl-POSS (OP-POSS) and the three H-Sulfo-POSS catalysts. | ||
The IR spectrum of the starting material (OP-POSS) shows some distinguishable bands that were identified as belonging to aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H stretch vibrations (around 3025 cm−1), Si–C-bending vibrations (at 1425 cm−1) and Si–O–Si stretching vibrations (at 1086 and 750–800 cm−1). After the aromatic electrophilic sulfonation a few very distinguishable bands associated with the –SO3H group (1050, 1120 and 1170 cm−1) appeared.32 Also, the broad band at 3370 cm−1 can be attributed to O–H stretching absorption bands, indicating strong H-bonding by its broad shape. The band at around 1650 cm−1 is attributed to the meta-substitution of the phenyl rings of octaphenyl-POSS.29 The spectra have not been scaled and can only be interpreted qualitatively and not quantitatively.
C and C–H stretch vibrations (around 3025 cm−1), Si–C-bending vibrations (at 1425 cm−1) and Si–O–Si stretching vibrations (at 1086 and 750–800 cm−1). After the aromatic electrophilic sulfonation a few very distinguishable bands associated with the –SO3H group (1050, 1120 and 1170 cm−1) appeared.32 Also, the broad band at 3370 cm−1 can be attributed to O–H stretching absorption bands, indicating strong H-bonding by its broad shape. The band at around 1650 cm−1 is attributed to the meta-substitution of the phenyl rings of octaphenyl-POSS.29 The spectra have not been scaled and can only be interpreted qualitatively and not quantitatively.
 | ||
| Fig. 6 Field emission scanning electron microscopy (FE-SEM) pictures of the prepared catalysts. (a) H-Sulfo-POSS-1; (b) H-Sulfo-POSS-2 and (c) H-Sulfo-POSS-3. | ||
The H-Sulfo-POSS-1 compound forms particles of varying sizes from as small as 9 μm up to approximately 250 μm, where the larger particles look like a very compact and consistent assembly of smaller particles. At the maximum zoom on the particles, the surface looks highly uneven and rough, showing a high degree of porosity. A similar scenario is observed with the H-Sulfo-POSS-2 material, in which the roughness appears to give high access to the active catalytic sites. The H-Sulfo-POSS-3 material shows significant amounts of very fine particles scattered at the 2 μm and 800 nm zoom.
3.2. Catalytic tests of H-Sulfo-POSS in the acetalization of glycerol with butanone
 | ||
| Fig. 7 The equilibrium yield in the ketalisation of glycerol with butanone at different molar ratios. Conditions: 16 h, 25 °C, ethanol (0.8 ml), glycerol (0.5 g, 5.4 mmol), butanone (according to ratio), catalyst: H-Sulfo-POSS-1 (1 mol%). | ||
As may be observed, the molar excess of 2-butanone significantly affects the equilibrium, as observed with other aldehydes and ketones in other acetalization reactions.10 For the future experimental protocol it was decided that a glycerol/butanone ratio of 1:6 was suitable where a maximum yield of 82.3% at equilibrium is to be expected. Ethanol was identified as a suitable solvent for the otherwise immiscible glycerol and ketones34 to become fully miscible. This is important to avoid mass transfer limitations owing to the presence of two liquid phases during the first moments of the reaction. Also, using the additional solvent ethanol has shown not to affect the equilibrium conversion of the reaction33 since it does not compete for the acetalization reaction. This would require the presence of two –OH groups (such as in the case of glycerol or glycols); thus, having only one –OH, ethanol would only form a hemiacetal, which are very unstable species and are indeed not observed.35
 | ||
| Fig. 8 Reaction profiles for the ketalisation of glycerol with butanone using PTSA, Amberlyst-36, H-Sulfo-POSS-1, -2 and -3 as catalysts. Conditions: 25 °C. Ethanol (1.7 g), glycerol (1 g, 10.8 mmol), butanone (4.7 g, 65.2 mmol, 6 equiv. per glycerol), catalyst loading of 0.1 mol% (molH+/molgly). | ||
The higher degree of sulfonation gave a more active catalyst as can be seen from the relative order in activity between H-Sulfo-POSS-1, -2 and -3. This may be a consequence of an enhanced interaction between the substrates and catalyst due to a higher presence of SO3− moieties that initiate the reaction mechanism with the hydroxyl functions of glycerol.35 H-Sulfo-POSS-3 presents similar or even slightly better activity than PTSA. Amberlyst-36 indeed presents a lower activity, undoubtedly caused by mass transfer limitations into its porous structure.
Additionally, as seen in Table 3, the TOF of our synthesized catalysts is much higher than an industrial reference ion exchange resin like Amberlyst 36 and also a molecular reference catalyst like PTSA. There is not an abundance of studies on the acetalization reaction between glycerol and butanone, and none that shows any reaction profiles or TOF. In contrast to other works in literature with catalysts like the heteropolyacid H3PW12O40 (ref. 21) and with rhenium supported on silica,24 the activity in terms of TOF appears to be higher. This is remarkable, considering that the reaction conditions employed in the other cases were more favourable. In one case the molar excess of butanone to glycerol is as high as 20,24 which makes the contact between butanone and glycerol more favourable as the dispersed droplets of the latter are significantly smaller (i.e., there is a larger interfacial area) than when using lower molar excesses.36 In the other case, the molar excess of butanone is also higher, but more relevantly for catalytic activity, the temperature is 328 K compared to our experiments at room temperature (298 K).24 However, it should be noted that in the mentioned references21,24 the yield of the acetal product was measured at high conversions of glycerol, close to the thermodynamic equilibrium of the reaction,24 which is far too high to measure meaningful TOFs.
| Catalyst | T (K) | Molar excess ketone: butanone | Glycerol conversiona (%) | TOF (h−1) | Ref. |
|---|---|---|---|---|---|
| a Glycerol conversion at which the TOF was calculated. b H3PW12O40 = heteropolyacids. c Calculated from the data in the reference. | |||||
| H-Sulfo-POSS-1 | 298 | 6 | 26.5 | 1587 | This work |
| H-Sulfo-POSS-2 | 298 | 6 | 30.7 | 1844 | This work |
| H-Sulfo-POSS-3 | 298 | 6 | 43.4 | 2601 | This work |
| Amberlyst 36 | 298 | 6 | 3.6 | 213 | This work |
| PTSA | 298 | 6 | 37.6 | 2256 | This work |
| Re/SiO2 | 328 | 10 | 92.6 | 115 | 24 |
| H3PW12O40b (1 mol%) | 298 | 20 | 74 | 70.7c | 21 |
 | ||
| Fig. 9 Yield per recycle run for the H-Sulfo-POSS catalysts using the centrifugation approach. Conditions: 25 °C, 150 min, ethanol (1.7 g), glycerol (1 g, 10.8 mmol), butanone (4.7 g, 65.2 mmol, 6 equiv. per glycerol), H-Sulfo-POSS catalysts loaded at 0.1% (molH+/molgly). | ||
The H-Sulfo-POSS-3 catalyst initially gave the highest yield of the ketal as expected from the TOF values shown in Table 3. However, with each cycle the yield of the ketal was rapidly decreasing. H-Sulfo-POSS-2 showed an intermediate high initial yield of ketal with a significant but limited drop in the yield for the first few cycles. The H-Sulfo-POSS-1 catalyst showed an intermediate initial yield of the ketal of about 50%; however, this yield remained almost unchanged throughout 10 cycles. Obviously, H-Sulfo-POSS-3, which shows the highest solubility of the three H-Sulfo-POSS catalysts, will be washed out relatively easily. This is expected in H-Sulfo-POSS materials whose sulfonation degree is higher than 50% (ref. 29) as is the case of H-Sulfo-POSS-3, which exhibits 66% as indicated in Table 2. In case of a soluble catalyst like H-Sulfo-POSS-3 separation by membrane filtration with an appropriate pore size could be an alternative procedure to be assessed.
The H-Sulfo-POSS-1 catalyst was further investigated in recycling experiments whereby sample over time experiments were performed either at the start (cycle 1), at the 5th cycle or at the 10th cycle. A sample over time experiment is essentially equivalent to a destructive sample. Therefore, the recycling experiment setup started with three reaction vessels running in parallel, when a time-resolved experiment had to be conducted that reaction vessel was terminated from further recycling runs as sampling removes significant portions of catalyst from the reaction mixture. The results from those experiments are presented in Fig. 10.
 | ||
| Fig. 10 Reaction profile of the ketalisation of glycerol using fresh and recycled H-Sulfo-POSS as catalyst and comparison with Amberlyst 36. Conditions: 25 °C, 150 min, ethanol (1.7 g), glycerol (1 g, 10.8 mmol), butanone (4.7 g, 65.2 mmol, 6 equiv. per glycerol), H-Sulfo-POSS-1 catalysts loaded at 0.1% (molH+/molgly). | ||
The reaction profile is very similar in cycle 1, 5 and 10 and in all three instances showing a more active system than that based on the Amberlyst-36 catalyst.
4. Conclusions
Three batches of sulfonated octaphenyl-POSS, H-Sulfo-POSS, were synthesized, each of which with a slightly higher degree of sulfonation. The H-Sulfo-POSS catalysts were characterized by NMR, IR and SEM. The IEC was measured for all H-Sulfo-POSS catalysts to determine their degree of sulfonation. The solubility of the H-Sulfo-POSS was extremely low in all the solvents that were investigated. The Na-Sulfo-POSS on the other hand was slightly soluble in water and other polar solvents, which then allowed them to be analyzed by NMR. The H-Sulfo-POSS compounds were investigated for their catalytic activity in the ketalisation reaction of glycerol with butanone. The catalytic activity of each of the synthesized H-Sulfo-POSS catalysts in this reaction increased with the degree of sulfonation, the most active catalyst, H-Sulfo-POSS-3 showing a TOF as high as 2601 h−1. The activity of H-Sulfo-POSS-2 and -3 was similar to PTSA while H-Sulfo-POSS-1 was somewhat less active albeit still considerably more active than the industrial reference Amberlyst-36. This is explained by the higher ion exchange capacity that these modified nanocomposites exhibit as the charge density is increased with a higher degree of sulfonation. Centrifugation was explored as a way of recycling the H-Sulfo-POSS catalysts. H-Sulfo-POSS-1 showed good recyclability over 10 centrifugation-recycling runs, which is explained by the lower solubility of H-Sulfo-POSS-1 as compared to H-Sulfo-POSS-2 and -3. These sulfonated materials show an outstanding prospects for biomass conversion to value-added products and other biofuels in other reactions starting from glycerol, such as esterifications and etherifications, or sugars, like the dehydration to furans. Further work could lead to modifying the POSS material and then functionalizing them to tune their solubility and activity. In addition, due to their size, there is potential catalyst recycling employing membrane technologies.Author contributions
Viktor Söderholm: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing-original draft, writing-review & editing. Jesús Esteban: conceptualization, data curation, formal analysis, supervision, visualization, writing-original draft; writing-review & editing. Dieter Vogt: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – TRR 63 “Integrierte chemische Prozesse in flüssigen Mehrphasensystemen” (Teilprojekt D3) – 56091768.References
- G. Li, L. Wang, H. Ni and C. U. Pittman, J. Inorg. Organomet. Polym., 2001, 11, 123–154 CrossRef CAS.
- W. Zhang, G. Camino and R. Yang, Prog. Polym. Sci., 2017, 67, 77–125 CrossRef CAS.
- X. Chen and L. F. Dumée, Adv. Eng. Mater., 2019, 21, 1800667 CrossRef.
- W. Zhang and A. H. E. Müller, Prog. Polym. Sci., 2013, 38, 1121–1162 CrossRef CAS.
- S. Mohapatra, T. Chaiprasert, R. Sodkhomkhum, R. Kunthom, S. Hanprasit, P. Sangtrirutnugul and V. Ervithayasuporn, ChemistrySelect, 2016, 1, 5353–5357 CrossRef CAS.
- A. Akbari, A. Naderahmadian and B. Eftekhari-Sis, Polyhedron, 2019, 171, 228–236 CrossRef CAS.
- A. Falk, J. M. Dreimann and D. Vogt, ACS Sustainable Chem. Eng., 2018, 6, 7221–7226 CrossRef CAS.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- C. H. C. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. M. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- A. Cornejo, I. Barrio, M. Campoy, J. Lazaro and B. Navarrete, Renewable Sustainable Energy Rev., 2017, 79, 1400–1413 CrossRef.
- A. R. Trifoi, P. S. Agachi and T. Pap, Renewable Sustainable Energy Rev., 2016, 62, 804–814 CrossRef CAS.
- V. R. Ruiz, A. Velty, L. L. Santos, A. Leyva-Pérez, M. J. Sabater, S. Iborra and A. Corma, J. Catal., 2010, 271, 351–357 CrossRef CAS.
- N. Suriyaprapadilok and B. Kitiyanan, Energy Procedia, 2011, 9, 63–69 CrossRef CAS.
- R. R. Pawar, S. V. Jadhav and H. C. Bajaj, Chem. Eng. J., 2014, 235, 61–66 CrossRef CAS.
- P. H. R. Silva, V. L. C. Gonçalves and C. J. A. Mota, Bioresour. Technol., 2010, 101, 6225–6229 CrossRef CAS.
- J. Esteban, F. Garcia-Ochoa and M. Ladero, Green Process. Synth., 2017, 6, 79–89 CAS.
- J. Esteban, M. Ladero and F. Garcia-Ochoa, Chem. Eng. J., 2015, 269, 194–202 CrossRef CAS.
- J. Deutsch, A. Martin and H. Lieske, J. Catal., 2007, 245, 428–435 CrossRef CAS.
- H. Serafim, I. M. Fonseca, A. M. Ramos, J. Vital and J. E. Castanheiro, Chem. Eng. J., 2011, 178, 291–296 CrossRef CAS.
- Y. Leng, J. Zhao, P. Jiang and D. Lu, Catal. Sci. Technol., 2016, 6, 875–881 RSC.
- M. J. da Silva, A. A. Julio and F. C. S. Dorigetto, RSC Adv., 2015, 5, 44499–44506 RSC.
- M. J. da Silva, M. de Oliveira Guimaraes and A. A. Julio, Catal. Lett., 2015, 145, 769–776 CrossRef.
- S. Guidi, M. Noe, P. Riello, A. Perosa and M. Selva, Molecules, 2016, 21, 657 CrossRef PubMed.
- M. Kapkowski, W. Ambrożkiewicz, T. Siudyga, R. Sitko, J. Szade, J. Klimontko, K. Balin, J. Lelątko and J. Polanski, Appl. Catal., B, 2017, 202, 335–345 CrossRef CAS.
- J. Esteban, A. J. Vorholt and W. Leitner, Green Chem., 2020, 22, 2097–2128 RSC.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 RSC.
- D. Aili, T. Allward, S. M. Alfaro, C. Hartmann-Thompson, T. Steenberg, H. A. Hjuler, Q. Li, J. O. Jensen and E. J. Stark, Electrochim. Acta, 2014, 140, 182–190 CrossRef CAS.
- A. Kafi, Q. Li, T. Chaffraix, J. Khoo, T. Gengenbach and K. J.-C. Magniez, Polymer, 2018, 137, 97–106 CrossRef CAS.
- S. Subianto, M. K. Mistry, N. R. Choudhury, N. K. Dutta and R. Knott, ACS Appl. Mater. Interfaces, 2009, 1, 1173–1182 CrossRef CAS.
- C. Hartmann-Thompson, A. Merrington, P. I. Carver, D. L. Keeley, J. L. Rousseau, D. Hucul, K. J. Bruza, L. S. Thomas, S. E. Keinath, R. M. Nowak, D. M. Katona and P. R. Santurri, J. Appl. Polym. Sci., 2008, 110, 958–974 CrossRef CAS.
- E. Carlier, A. Revillon, A. Guyot and P. Baumgartner, React. Polym., 1993, 21, 15–25 CrossRef CAS.
- B. Decker, C. Hartmann-Thompson, P. I. Carver, S. E. Keinath and P. R. Santurri, Chem. Mater., 2010, 22, 942–948 CrossRef CAS.
- M. R. Nanda, Z. Yuan, W. Qin, H. S. Ghaziaskar, M.-A. Poirier and C. C. Xu, Fuel, 2014, 117, 470–477 CrossRef CAS.
- J. Esteban, A. J. Vorholt, A. Behr, M. Ladero and F. Garcia-Ochoa, J. Chem. Eng. Data, 2014, 59, 2850–2855 CrossRef CAS.
- V. Calvino-Casilda, K. Stawicka, M. Trejda, M. Ziolek and M. A. Bañares, J. Phys. Chem. C, 2014, 118, 10780–10791 CrossRef CAS.
- H. Murasiewicz and J. Esteban, Ind. Eng. Chem. Res., 2019, 58, 6933–6947 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00344e |
| This journal is © The Royal Society of Chemistry 2021 |