
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Propylene synthesis via isomerization–metathesis of 1-hexene and FCC olefins†
Gyula
Novodárszki
 a,
Blanka
Szabó
a,
Róbert
Auer
b,
Katalin
Tóth
b,
László
Leveles
b,
Róbert
Barthos
a,
Gábor
Turczel
a,
Zoltán
Pászti
a,
József
Valyon
a,
Magdolna R.
Mihályi
*a and
Róbert
Tuba
*a
a,
Blanka
Szabó
a,
Róbert
Auer
b,
Katalin
Tóth
b,
László
Leveles
b,
Róbert
Barthos
a,
Gábor
Turczel
a,
Zoltán
Pászti
a,
József
Valyon
a,
Magdolna R.
Mihályi
*a and
Róbert
Tuba
*a
aInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Magyar tudósok körútja 2, P.O. Box 286, 1519 Budapest, Hungary. E-mail: tuba.robert@ttk.hu
bMOL, Hungarian Oil and Gas Public Limited Company, Október huszonharmadika u. 18, 1117 Budapest, Hungary
First published on 19th July 2021
Abstract
Conversion of 1-hexene or olefins obtained by fluid catalytic cracking (FCC) to propylene via isomerization–metathesis (ISOMET) was investigated using ethylene as a cross-coupling agent. Zeolite H-beta (HBEA) was applied as an isomerization catalyst. The olefin metathesis (OM) catalysts were about 12 wt% molybdena, supported on zeolite beta (MoO3/HBEA), and γ-alumina (MoO3/Al2O3). HBEA-supported catalyst with a lower molybdena content (6 wt%) was also investigated. The catalysts were characterized by X-ray diffractometry (XRD), H2-temperature-programmed reduction (H2-TPR), and Fourier transform infrared (FT-IR), visible Raman, in situ ultraviolet-visible (UV/VIS) and XPS spectroscopy. It was shown that HBEA is a highly active and robust catalyst of double-bond isomerization. Applying a physical mixture of HBEA and 12MoO3/Al2O3 catalyst at 150 °C and 3 bar ethylene pressure, 60% conversion of 1-hexene to propylene was attained. Interestingly, quantitative conversion to propylene was achieved after reactivation of the deactivated catalyst in an argon atmosphere at 550 °C. It was found that the pre-treatment of the catalyst with olefins such as ethylene before inert gas activation resulted in significant catalyst activity improvement. This suggests that the adsorbed olefins may play a key role in the formation of active metal centers during the catalyst reactivation process. The catalyst mixture also had good performance in the conversion of FCC olefins to propylene. The MoO3/HBEA catalysts have rendered reasonable activity; however, the catalysts showed a significantly shorter lifetime than the alumina-containing catalyst mixture.
Introduction
Olefin metathesis (OM) is a powerful and versatile method of organic chemistry. During OM, carbon atoms of two C=C double bonds are reorganized to new double-bond-containing molecules. These reactions are very selective and require mild reaction conditions. The atom economy of the synthetic procedures is often 100%, i.e., all the starting materials are incorporated into the products.Nature is abundant in bio-based materials containing the olefin bond. Low-value olefins can also be found in vast amounts in petrochemical by-products. These chemicals often appear as an underutilized feedstock.
With the advent of alternative fuels and electric cars, the worldwide demand for mineral oil-based fossil fuels will certainly decrease. However, the need for high-value innovative materials, including advanced polymers, steadily increases.1–3 Unfortunately, at present, these demands cannot be met by using renewable feedstocks only. Therefore, there is still a constant need for the development of high atom-economic chemical procedures for the efficient and environmentally benign conversion of petrochemicals to high-value materials.
Propylene is an emerging bulk chemical, the key monomer of polypropylene plastic and other commodity chemicals. It is mainly obtained as a by-product of ethylene production in steam cracker units.4,5 Possibilities to control the ratio of ethylene to propylene in the cracked product mixture are limited. As a result, cracking units are currently producing excess ethylene, while the increasing demand for propylene remains uncovered.6–9 Propylene can also be synthesized on a large scale by propane dehydrogenation as well as Fischer–Tropsch synthesis and methanol-to-olefin conversion.3,10–12 Propylene can also be produced selectively via OM under moderate reaction conditions. The most general example is the ethenolysis of 2-butene giving two propylene molecules.1,4,5,7,9,11–14
There are several industrial examples for OM of olefinic hydrocarbon over heterogeneous catalysts such as the Shell Higher Olefin Process (SHOP) for detergent production,15 the Philips Triolefin Process for synthesis of 2-butene16 and the Olefin Conversion Technology used by ABB Lummus Global for propylene production.17 The most widely used catalyst systems include Mo (ref. 11, 14 and 18–22) W (ref. 3, 4, 6, 8, 9, 12, 18 and 23–25) or Re (ref. 2, 26 and 27) oxides. Mo- and W-based catalysts are widely used in the petrochemical industry. Some new OM reactions, catalysed by metal oxide, have been reviewed recently.10,28
A special case of OM is isomerization–metathesis (ISOMET). During ISOMET, the olefinic bonds migrate along the hydrocarbon chain (isomerization), which is followed by a cross-metathesis reaction with another olefin, a cross-coupling agent. A special case of OM, when the cross-coupling agent is ethylene, is called ethenolysis.10,29 In the case of linear mono-olefins, the complete ISOMET using ethylene as a cross-coupling agent theoretically ends up with propylene as the only end product (Scheme 1).
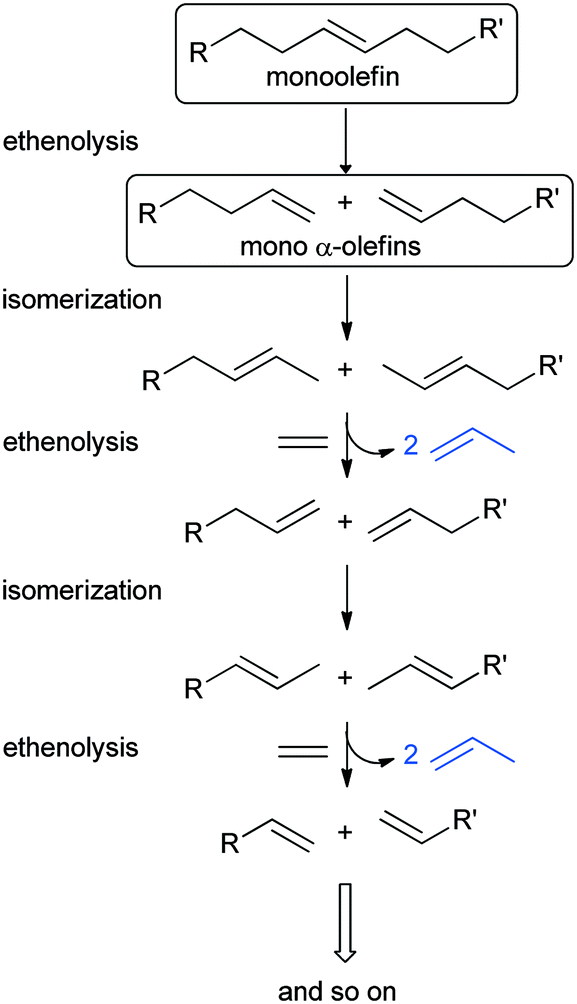 | ||
| Scheme 1 The conversion of C >5 olefins into propylene via ISOMET with ethylene. | ||
The ISOMET of C4 and C5 olefins producing propylene is one of the most widely investigated areas. Interestingly, industrially applied systems are mainly based on W (5–10% on silica) or Re (5–10% on alumina) catalyst systems.4,11,30–32 The ISOMET process of higher olefins (C4–C9) and the mixtures thereof has also been patended.33 Re2O7, WO3 and MoO3 were used as OM catalysts, whereas RuO2, MgO and K2CO3 were applied as isomerization catalysts. Another patent application describes a process for converting an olefin feed containing butenes, diolefins and polyolefins to propylene with ethylene using molybdenum as a metathesis catalyst and MgO, K2CO3, and K2O as isomerization catalysts.34 In both cases, the use of a fluid catalytic cracking (FCC) mixture is mentioned as a source of olefin feed, but only after appropriate refining.35,36
The FCC and FCC light fractions are abundant in C >5 olefin components. Furthermore, not only petrochemical streams but also materials derived from renewable feedstocks may contain C >5 olefins. For example, the ethenolysis of oleic acid produces 1-decene,29,37 while the ethenolysis of linoleic acid gives 1-hexene. Both vegetable oils are highly abundant in nature.38 Light olefins (propylene, butenes, butadiene) are also available from biomass-originated bioethanol5,39 and biobutanol40 and from biomass pyrolysis.6 Both micro- and mesoporous silicates, aluminosilicates were shown to be effective as supports for molybdena in the OM reaction.20–22
In the present study, zeolite beta was chosen as the isomerization catalyst for the ISOMET of 1-hexene model compound and FCC fractions. Beta is a large-pore zeolite with a three-dimensional structure of 12-membered ring channels.41 Owing to its large pore size, strong acid sites and high chemical and thermal stability, it is used as a catalyst in the petrochemical industry and fine chemistry. It is also utilized as an adsorbent.42 Zeolite beta has already been applied as a support itself43,44 or mixed with Al2O3 in the OM of 2-butene with ethylene to propylene.45 However, it has not been used in the ISOMET of long-chain olefins yet, where the role of zeolite is the double-bond isomerization of the reactant and intermediate alpha-olefin products. The dispersed MoOx, the active component of the oxide-supported catalysts, was shown to be present as isolated or oligomeric surface species as well as crystalline particles on a high-surface-area oxide support.28
Papers reporting isomerization metathesis using C5+ alkenes as raw materials for propylene synthesis are rare.46,47 In particular, there is no information in the literature about the synthesis of propylene from C5+ alkenes via ISOMET using HBEA as isomerization and MoOx/Al2O3 as olefin metathesis catalysts. This paper describes a zeolite-supported molybdena (MoO3/HBEA) catalyst system having isomerization (HBEA) as well as metathesis (MoO3) activity. The mixed bed of alumina-supported molybdena and zeolite beta (MoO3/Al2O3 + HBEA) ISOMET catalyst is also reported. MoO3 loading of about 12 wt% was applied on both supports. Based on the work of Li and co-workers, for HBEA, a catalyst containing lower MoO3 loading (6 wt%) was also investigated.48 The catalysts were characterized and investigated in the ISOMET of 1-hexene model compound with ethylene. Conversion of FCC having high olefin content to propylene was also studied. It has been found that the initial catalytic activity can be significantly improved by simple high-temperature heat treatment of the deactivated ISOMET catalyst in an inert gas.
Results and discussion
The molybdena content of the catalysts, determined by ICP-OES, is shown in Table 1. The ISOMET activity of 13MoO3/HBEA and the physical mixture of 12MoO3/Al2O3 and HBEA was higher than that of 6MoO3/HBEA (vide infra). The effect of the amount of MoO3 loading on HBEA zeolite (70%)/Al2O3(30%) has been investigated by Li et al. using the above-mentioned solid support mixture; a 6–8% molybdena loading was found to be optimal.48 The characterization of 13MoO3/HBEA and 12MoO3/Al2O3 is presented below, whereas the properties of 6MoO3/HBEA are included in the ESI.†| Catalyst | MoO3a (wt%) | SSAb (m2 g−1) | H2 uptakec (mmol g−1) | H/Moc |
|---|---|---|---|---|
| a Determined by ICP-OES. b Specific surface area (SSA) determined by the Brunauer–Emmett–Teller method. c Calculated from the TPR curve measured up to 800 °C. | ||||
| Al3O3 | — | 192 | — | — |
| 12MoO3/Al2O3 | 11.7 | 184 | 2.38 | 5.9 |
| HBEA (Si/Al = 12) | — | 480 | — | — |
| 13MoO3/HBEA | 13.5 | 235 | 2.61 | 5.6 |
| 6MoO3/HBEA | 6.1 | 476 | 1.17 | 5.6 |
Catalyst characterization
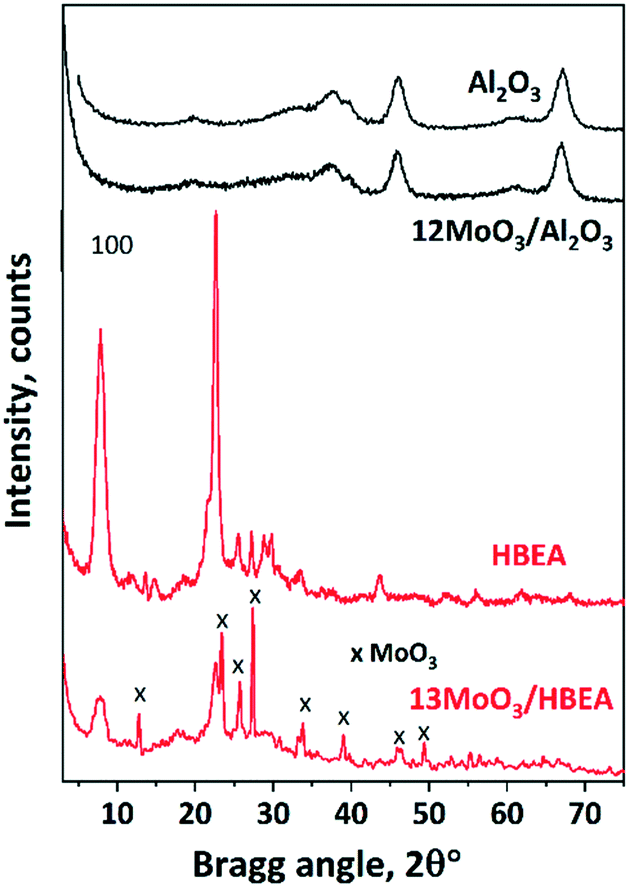 | ||
| Fig. 1 XRD patterns of supports and catalysts calcined at 500 °C. | ||
The monolayer coverage of the used γ-Al2O3 having a specific surface area of 192 m2 g−1 corresponds to about 19.2 wt% MoO3 content.49 The absence of the MoO3 XRD reflections suggests that MoO3 crystallites are well dispersed on the alumina surface; thus they are X-ray amorphous species. The obtained results confirmed that alumina-supported MoO3 catalysts of a high dispersion can be obtained below monolayer coverage or even at MoO3 contents higher than that corresponding to the monolayer coverage.50,51 Upon Mo loading (13.5 wt% MoO3) the characteristic reflections of HBEA at 2θ = 7.8° and 22.6° significantly decreased, as was also shown by others.43,52
In addition, intense reflections typical of orthorhombic MoO3 (JCPDS 35-609) appeared in the XRD pattern of 13MoO3/HBEA at 2θ = 27.4°, 23.4° and 25.7°. The average MoO3 particle size, calculated from the Scherrer equation using the 2θ = 27.4° reflection, was about 90 nm. At lower MoO3 loading (6MoO3/HBEA), no crystalline MoO3 phase was detected by XRD and the diffraction lines of the zeolite support were more intense (Fig. S1†). No crystalline Al2(MoO4)3 phase was detected in these samples. Formation of this phase was found in samples treated at high temperature (680 °C).43 In our case, however, the MoO3 content was not high and the decomposition temperature of the Mo precursor was lower, 500 °C, so the formation of the aluminium molybdate phase is unlikely in any of the supports.
As expected, the specific surface area of the molybdena-loaded catalyst was smaller than those of the corresponding supports. The difference in the surface areas depends on the pore size of the support (Table 1). For γ-Al2O3, which is mesoporous, the specific surface area decreased only slightly (from 192 to 184 m2 g−1). The specific surface area of the microporous zeolite catalyst with 12 wt% MoO3 loading, however, was only half of that of the zeolite support (235 and 480 m2 g−1). At 6 wt% MoO3 content the SSA of HBEA did not decrease (476 m2 g−1).
Temperature-programmed reduction by hydrogen (H2-TPR)
The reducibility of the molybdenum species in 12MoO3/Al2O3 and 13MoO3/HBEA was investigated by temperature-programmed H2 reduction (Fig. 2). Two reduction peaks were observed on the H2-TPR curve of the 12MoO3/Al2O3 catalyst. The low-temperature peak in the range of 300–600 °C with a maximum of 450 °C represents the reduction of multilayered and octahedral Mo(VI) to Mo(IV).50,53 The tetrahedral Mo (IV) species has stronger interaction with the Al2O3 support, leading to a reduction temperature in the range of 600–800 °C. In this higher temperature region the reduction of Mo(IV) to Mo(0) takes place.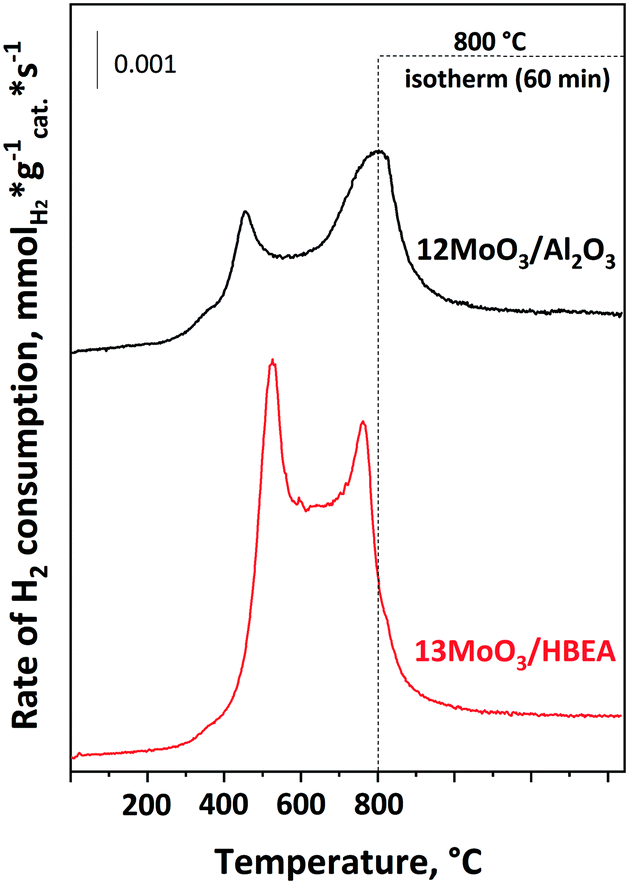 | ||
| Fig. 2 H2-TPR curves of the 12MoO3/Al2O3 and 13MoO3/HBEA catalysts. The catalysts were treated in O2 flowing at a rate of 30 ml min−1 for 1 h at 500 °C, cooled to 25 °C and then heated at a rate of 10 °C min−1 up to 800 °C for 1 h in a flow of a 9% H2/N2 mixture. | ||
For the 13MoO3/HBEA catalyst, two reduction peaks were observed in the temperature range of 300–650 °C and 650–800 °C. The peak at lower temperature is assigned to reduction of Mo(VI) to Mo(IV) as in the case of the 12MoO3/Al2O3 catalyst. The second peak represents the complete reduction of Mo(IV) to Mo(0).54 A similar TPR curve was observed for 6MoO3/HBEA (Fig. S2†). Table 2 shows that the hydrogen uptake, expressed in the H/Mo ratio, in the Al2O3-supported sample was 5.9. This amount of hydrogen consumption is near to the amount needed for the total reduction of MoO3 to Mo(0) (6). Molybdena has been shown to react with the zeolitic protons during oxidative decomposition of the heptamolybdate precursor.55 At 500 °C, MoOx oligomers migrate into the channels and react with the Brønsted acid sites of the zeolite to form ditetrahedral Mo species, i.e. (Mo2O5)2+ cations. These cationic Mo species can only be reduced at temperatures above 800 °C.54,56Table 1 shows that the H/Mo ratio was 5.6 for both MoO3/HBEA catalysts, suggesting that some Mo may occupy cationic positions and cannot be reduced under our TPR conditions. In line with the above results, the loss of Brønsted acid sites was also observed for these samples (vide infra). Before the catalytic experiments, the calcined catalysts were activated in situ in a flow of Ar at 550 °C. Thermal autoreduction of Mo(VI) cannot be detected by H2-TPR.
| Catalyst | TOS (h) | Propylene yield (%) | |
|---|---|---|---|
| Activateda | Reactivatedb | ||
| a Activation: the ex situ calcined catalyst was in situ pre-treated in a flow of Ar (50 ml min−1) at 550 °C for 2 h. The activated catalyst was cooled to the target temperature in a flow of Ar and the ISOMET reaction was performed. b Reactivation: after 3 h TOS, the reactant feed was stopped and the total pressure was reduced to atmospheric pressure. The catalyst was purged with a flow of Ar (50 ml min−1) for 30 min at the reaction temperature to remove olefins, then heated to 550 °C at a ramp rate of 10 °C min−1 and maintained at this temperature for 2 h. The reactivated catalyst thus obtained was cooled to the target temperature in a flow of Ar and the ISOMET reaction was performed again. | |||
| 75 | 0.5 | 10 | 20 |
| 3 | 6 | 16 | |
| 100 | 0.5 | 15 | 59 |
| 3 | 12 | 19 | |
| 125 | 0.5 | 29 | 86 |
| 3 | 5 | 17 | |
| 150 | 0.5 | 19 | 82 |
| 3 | 1 | 10 | |
FT-IR spectroscopy of adsorbed pyridine
For the ISOMET reaction, the catalyst must contain Brønsted acid sites that promote double-bond isomerization of terminal olefins. Pyridine adsorption, followed by FT-IR spectroscopy, was used to characterize the acidity of the supported molybdena catalyst (Fig. 3). Brønsted and Lewis acid sites of the catalysts can be distinguished by the characteristic 19b/8a ring vibrations of pyridinium ions and pyridine bound to Lewis acid sites.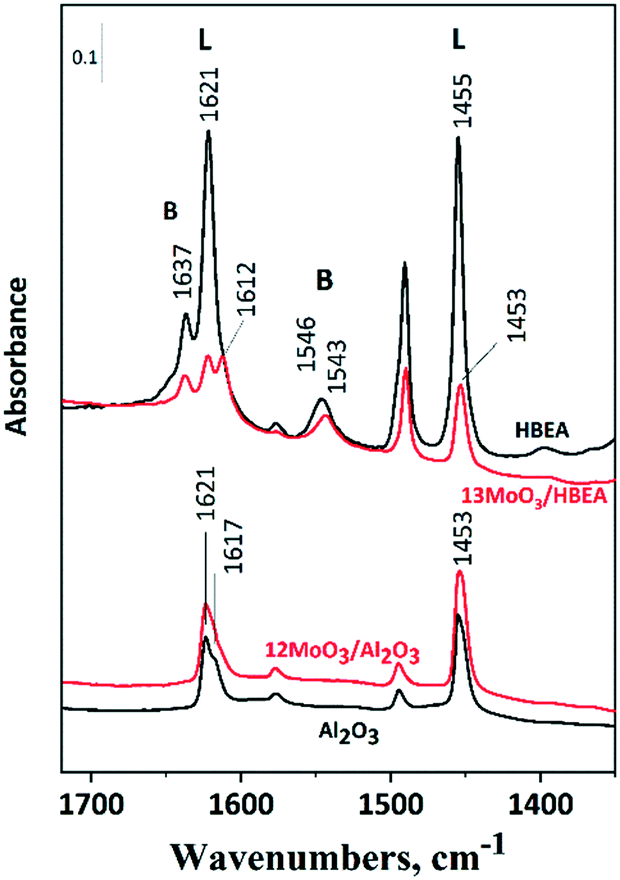 | ||
| Fig. 3 FT-IR spectra of pyridine. The samples were activated in high vacuum at 550 °C for 1 h. Pre-treated samples were allowed to come in contact with Py vapour at 5 mbar pressure at 200 °C for 30 min, then evacuated for 30 min. After evacuation, the spectra were recorded at room temperature. Labels L and B indicate the characteristic bands for Py bonded to Lewis and Brønsted acid sites, respectively. | ||
These vibrations of the two species appear in different regions of the spectrum, i.e., at 1545/1637 cm−1 and around 1455/1620 cm−1, respectively. Fig. 3 shows that the HBEA support has both Brønsted and Lewis acid sites. In zeolites, Brønsted acidity is due to protons compensating for the negative charge on the zeolite framework generated by tetrahedral framework aluminum. Extra-framework aluminum represents Lewis acid sites. A high number of defect sites caused by crystallographic faults is typical for the structure of zeolite beta.57 Thus, trigonal framework aluminum near the silanol nests shows Lewis acidity. Upon Mo loading, the number of both Brønsted and Lewis acid sites decreased significantly. The FT-IR results suggest that molybdenum reacts with zeolitic protons and occupies cationic positions as ditetrahedral Mo ions (Mo2O5)2+, as proposed by Ding et al.58 The amount of Brønsted acid sites in the zeolite sample was 0.71 mmol g−1, measured by the ammonium ion exchange capacity. Comparing the band intensities of the pyridinium ions around 1545 cm−1, the number of Brønsted acid sites decreased by 36%, indicating that 0.12 mmol g−1 Mo is in the cationic position, which is about 15% of the total Mo content. In addition, surface MoOx species can be bound in the zeolite silanol nests both in the micropores and on the outer surface of the zeolite crystallites. These species cause a significant decrease in the number of Lewis acid sites.
In the spectrum of 12MoO3/Al2O3 the intensities of the bands characteristic of Lewis acid sites slightly increase at 1453 and 1621 cm−1 compared to the bare support (Fig. 3). Highly polymerized MoOx species were suggested to be responsible for the additional Lewis acid sites at this Mo content.59 An approximately 10% decrease in Brønsted sites was observed on 6MoO3/HBEA relative to the HBEA support (Fig. S3†).
Raman spectroscopy
The structure of supported molybdenum oxide was investigated by Raman spectroscopy under ambient conditions in the hydrated state. The Raman spectra of 12MoO3/Al2O3 and 13MoO3/HBEA are presented in Fig. 4. In the spectrum of 12MoO3/Al2O3, the bands observed at 953, 910 and 355 cm−1 are assigned to the symmetric stretching, asymmetric stretching, and bending vibrations of the terminal Mo=O bond of octahedral MoO6 species in hydrated heptamolybdate, respectively.60 In addition, the bands at 566 and 222 cm−1 are due to the Mo–O–Mo symmetric stretching and deformation vibrations of these molybdena species, respectively. These results confirm that at 12 wt% molybdena loading octahedral species dominate on alumina. It should be mentioned that tetrahedral MoO4 units are also present as the Raman band at 910 cm−1 is typical of the symmetrical stretching vibrations of the terminal Mo=O in tetrahedral molybdena species.61 This observation was also confirmed by UV-vis spectroscopy. The intense and narrow bands in the spectrum of 13MoO3/HBEA are due to crystalline MoO3,62 indicating that this catalyst contains the MoO3 phase, which is also supported by the XRD data (Fig. 1). At 6 wt% molybdena loading, the intensity of the 970 cm−1 band increased compared to that of the zeolite support (Fig. S4†). This indicates that the catalyst contains tetrahedral MoOx species.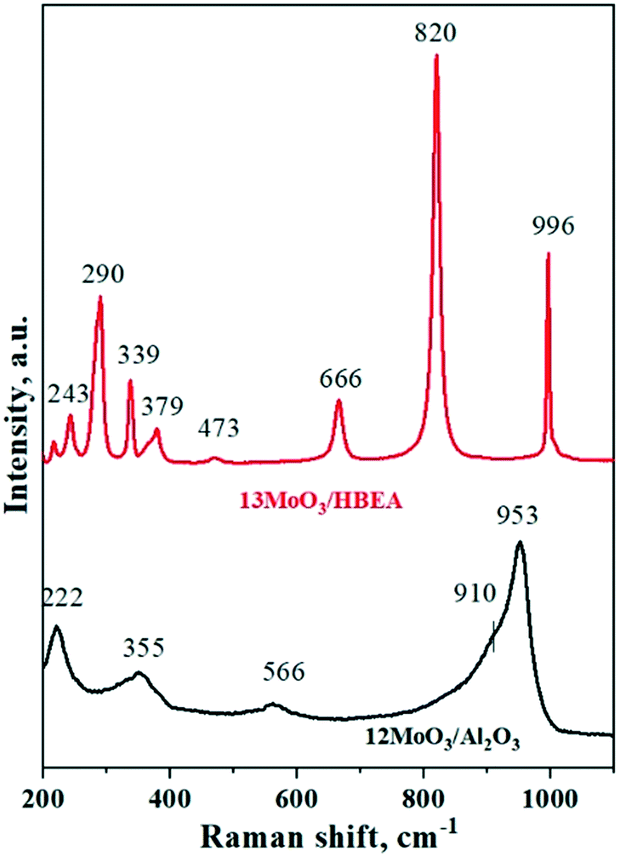 | ||
| Fig. 4 Raman spectra of 12MoO3/Al2O3 and 13MoO3/HBEA under ambient conditions. | ||
In situ UV-vis spectroscopy
In situ UV-vis experiments were performed to determine the type of MoOx species in the calcined 12MoO3/Al2O3 and 13MoO3/HBEA catalysts and to follow changes in the degree of polymerization of MoOx during in situ activation in an inert atmosphere (Ar) at temperatures increased up to 550 °C (Fig. 5). Both calcined catalysts show a strong absorption band below 330 nm. The bands at 250 and around 290 nm are due to the ligand–metal charge transfers (LMCTs) from oxygen to Mo(VI) in highly dispersed and small linear-chained tetrahedral MoOx, respectively. LMCT in polymerized octahedral MoOx gives rise to a band at around 330 nm.59,63–65 For 12MoO3/Al2O3, the intensity of the absorption band at 250 nm becomes weaker above 200 °C, while the band at 330 nm gains strength. This result suggests that during thermal treatment in inert atmosphere tetrahedral MoOx is transformed to polymerized octahedral MoOx species over alumina-supported molybdena. Comparing the relative intensity of the bands below 300 nm and at 330 nm in the spectra of 13MoO3/HBEA, similar but smaller changes were observed, indicating that a smaller portion of tetrahedral MoOx species is transformed to octahedral MoOx species on the zeolite-supported molybdena. MoO3 located in microporous channels is less capable of forming longer MoOx chains. They can be formed only on the outer surface of the zeolite crystals, but in small amounts only because the outer surface is only approx. 4–5% of the specific surface area of the zeolite. In the spectrum of the calcined 13MoO3/HBEA the presence of a weak absorption band at around 400 nm is due to the crystalline MoO3 phase, which was also confirmed by XRD. Crystalline MoO3 was shown to be catalytically inactive.66 In the spectrum of 6MoO3/HBEA, at higher temperatures, those tetrahedral MoOx species that are surrounded by other Mo atoms cannot be detected (Fig. S5†). This result suggests that during heat treatment in Ar, MoOx species migrate into the microporous channels and occupy cationic positions in the zeolite.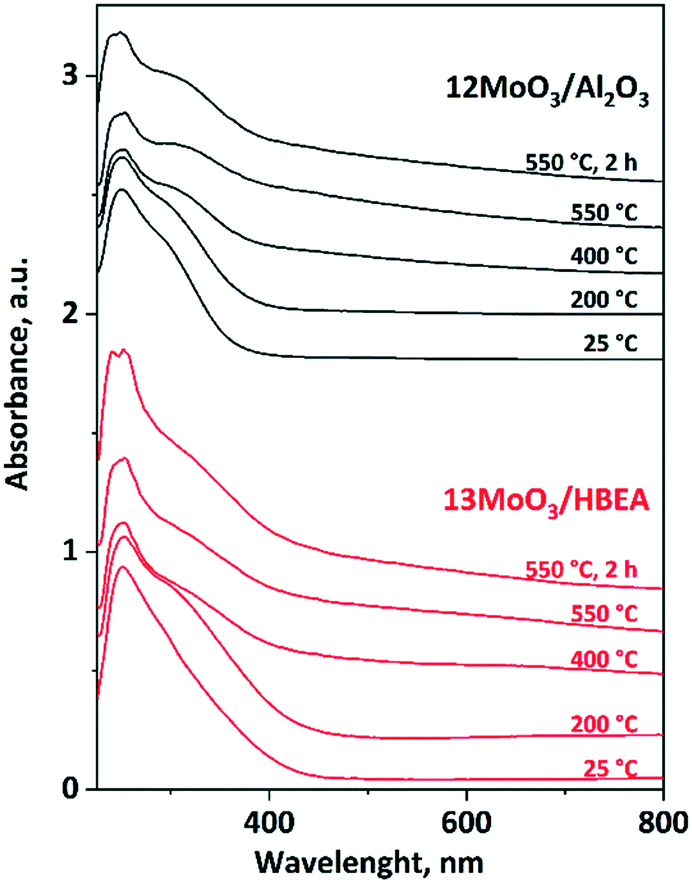 | ||
| Fig. 5 In situ UV-vis spectra of the 12MoO3/Al2O3 and 13MoO3/HBEA catalysts. The catalysts were heated at a rate of 10 °C min−1 up to 550 °C for 2 h in an Ar flow. | ||
XPS measurements
Quantitative evaluation of the XPS data indicated that the Mo oxide content of the MoO3/Al2O3 catalyst was around 12 wt%, in good agreement with the nominal composition. Spectral features of Al (Al 2p binding energy of 74.7 eV, Al KLL kinetic energy of 1386.8 eV and the Auger parameter defined as their sum at 1461.5 eV) as well as the main O 1s peak around 531.4 eV all pointed to an oxidized/hydroxylated environment for the Al(III) cations,67,68 in agreement with literature data on similar catalysts.69 No significant changes in the chemical environment of Al were observed during the treatments. In addition, no sign of dissolution of Mo into the alumina was detected. The Mo 3d5/2–3/2 spin orbit doublet of well-dispersed MoO3 on alumina has a rather broad line shape with a 3d5/2 binding energy of around 233 eV,69–71 which is somewhat higher than the value characteristic for bulk MoO3 (232.5 eV).72 While the shift is attributed to the strong interaction of the oxidized Mo species with the alumina support,70 the broadening can be the result of a charging effect70 but may also indicate the existence of a range of slightly different environments for the adsorbed Mo(VI) ions.73 As shown in Fig. 6a, these features are well reproduced in the Mo 3d spectrum of the calcined 12MoO3/Al2O3 sample exposed to air. The spectrum can be well modelled by a single peak pair with the 3d5/2 component at 233.2 eV, indicating the exclusive presence of Mo(VI) ions.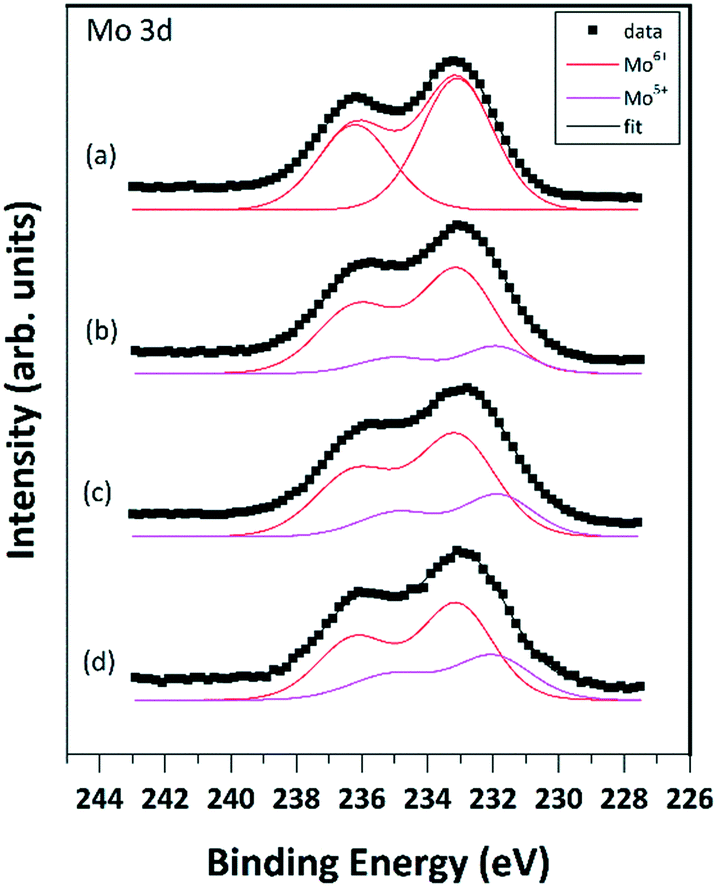 | ||
| Fig. 6 Mo 3d spectra of the calcined 12MoO3/Al2O3 sample exposed to air (a), after activation in Ar at 550 °C for 1 h (b), subsequent C2H4 exposure at 100 °C for 1 h (c) and after reactivation in Ar at 550 °C for 1 h (d). | ||
Treatment in Ar at 550 °C resulted in a marginal shift of the maximum of the spectrum towards lower binding energies and further apparent broadening (Fig. 6b). Spectral modelling revealed that these changes can be interpreted as the result of the appearance of a new Mo 3d doublet with its 3d5/2 peak at around 231.7–231.9 eV.
According to its binding energy, this new component was attributed to Mo(V) species.72 The abundance of the Mo(V) species clearly increased upon ethylene exposure (Fig. 6c) and some further increase was observed after the subsequent treatment in Ar at 550 °C (Fig. 6d). Although the combination of the Mo(VI) and Mo(V) states adequately modeled the observed line shapes, the broad peaks can overlap the weak Mo(IV) signal, so their presence, especially after the ethylene exposure and the subsequent re-activation, cannot be completely ruled out.
1-Hexene isomerization over HBEA catalyst
Preliminary experiments were carried out to investigate the isomerization of 1-hexene model compound using the HBEA catalyst at 75 °C and atmospheric pressure in an inert Ar gas atmosphere. At nearly 90% 1-hexene conversion, the yields of cis- and trans-2-hexene and cis- and trans-3-hexene were 70% and 20%, respectively. (Fig. 7). This result indicates that double bond shift and cis–trans rearrangement are the main reactions in the conversion of 1-hexene over zeolite beta, consistent with earlier results on large- and medium-pore zeolites, i.e., H-Y and H-ZSM-5. Side reactions such as polymerization are negligible.74–76 It was demonstrated that under these conditions, HBEA is highly robust. No catalyst deactivation was observed during a time on stream (TOS) of 10 h.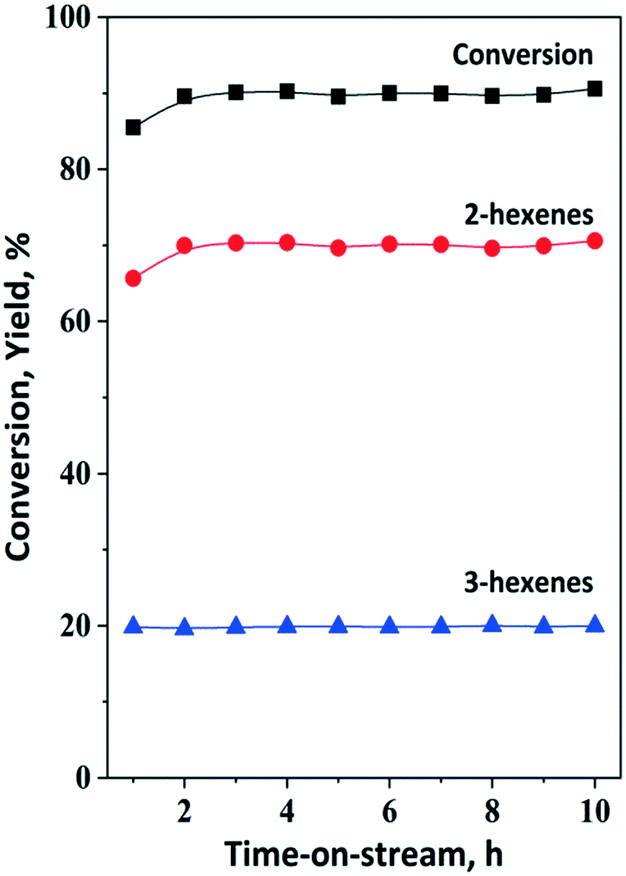 | ||
| Fig. 7 Isomerization of 30% 1-hexene (1-H) solution using the HBEA catalyst at 75 °C and 3 gcatg1−H−1 h space time under 3 bar ethylene pressure. The calcined catalyst was pretreated in situ in a flow of Ar (50 ml min−1) at 450 °C for 2 h at atmospheric pressure. | ||
The ISOMET of 1-hexene with ethylene was performed at 3 bar total pressure of ethylene and at an ethylene/1-hexene molar ratio of 10. In order to make sure that the isomerization is not affected by ISOMET conditions the isomerization reaction was performed in the presence of 3 bar ethylene (optimal ethylene pressure for OM, Fig. S7†) as well. The results showed that neither the catalytic activity nor the product selectivity was affected. Similar conversion and reaction product distribution to that in the absence of ethylene were observed. The influence of reaction temperature on the isomerization conversion and product distribution has been investigated at 75 (optimal reaction temperature for OM), 100, 125 and 150 °C and 3 gcatg1−H−1 h space time under 3 bar ethylene pressure. It could be concluded that even at 75 °C neither the conversion of 1-hexene nor the product distribution has been changed significantly.
By using the optimized OM (ethenolysis) condition, the catalyst activity did not decrease within ten hours of the reaction (Fig. 7). The yield of 2-hexenes achieved 66% (55% trans-2-hexene and 11% cis-2-hexene) after 1 hour TOS. HBEA is a highly robust isomerization catalyst showing high activity and stability under optimized metathesis reaction conditions.
The 1-hexene isomerization activity of the MoO3-containing HBEA catalysts was also investigated. The catalysts were pretreated in an O2 flow at 550 °C for 2 h to keep Mo in the Mo(VI) state. It was found that the isomerization activity of HBEA significantly dropped upon impregnating with MoO3. With increasing MoO3 content, the number of Brønsted acid sites decreases (shown by FT-IR spectroscopy), and therefore the isomerization activity also decreases. Actually, at 6 wt% loading 80% 1-hexene isomerization was observed (versus 90% on neat HBEA), while at 12 wt% loading the isomerization activity was around 60% after one hour time on stream.
At 12 wt% molybdena content the isomerization activity was halved after 5 hours time on stream, whereas 6MoO3/HBEA showed good stability in 1-hexene isomerization.
1-Hexene ISOMET using MoO3/HBEA catalysts
Theoretically, (not only) terminal olefins such as 1-hexene can be completely converted to propylene by using ethylene in sequential isomerization and ethenolysis steps (ISOMET) (Scheme 2). Two types of sites are required, Brønsted sites for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double-bond isomerization of 1-hexene and the intermediate alpha-olefins formed (1-pentene, 1-butene) and active MoOx sites for the conversion of isomerized olefins (2- and 3- hexenes, 2-pentenes, 2-butenes) with ethylene to give propylene and lower alpha-olefins (1-pentene, 1-butene).
C double-bond isomerization of 1-hexene and the intermediate alpha-olefins formed (1-pentene, 1-butene) and active MoOx sites for the conversion of isomerized olefins (2- and 3- hexenes, 2-pentenes, 2-butenes) with ethylene to give propylene and lower alpha-olefins (1-pentene, 1-butene).
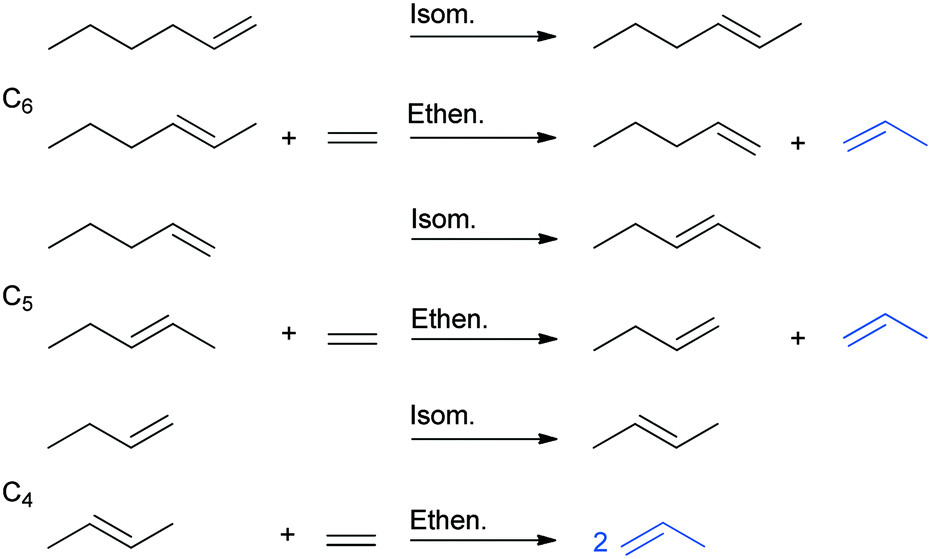 | ||
| Scheme 2 Subsequent isomerization and ethenolysis steps in ISOMET of 1-hexene with ethylene to propylene. (The pathway of the 3-hexene isomer is not shown here). | ||
The pathway of the 3-hexene isomer to propylene is shorter, as its reaction with ethylene gives 1-butene, which is converted to propylene in the isomerization and ethenolysis steps (not shown in Scheme 2).
In the following series of experiments, HBEA was used not only as an isomerization catalyst but also as a support for MoO3. The reactions were carried out under 3 bar ethylene pressure and 3 gcatg1−H−1 h space time in the temperature range of 75–150 °C. The catalyst was pre-treated in inert gas at 550 °C and the activated catalyst thus obtained was tested in the ISOMET reaction for 3 h time on stream (TOS). After activation, the catalyst was regenerated under the same conditions as the pre-treatment. The activity of the reactivated catalyst was also studied in the ISOMET reaction.
On the activated 13MoO3/HBEA catalyst, the propylene yield was low, ranging from 10% to 29% at 0.5 h TOS (Table 2). It was 10% at 75 °C; with increasing reaction temperature it reached its maximum (29%) at 125 °C and then at 150 °C decreased to 19%. Reactivation, i.e., heat treatment in an inert atmosphere at high temperature (550 °C) after the ISOMET reaction, however, resulted in a significant increase in the initial activity of the 13MoO3/HBEA catalyst. At 75 °C, the propylene yield doubled, while at higher temperatures the amount of propylene was 3 and 4 times as high on the reactivated catalyst as on the freshly activated catalyst. Thus, at 125 and 150 °C more than 80% propylene yield can be obtained over reactivated 13MoO3/HBEA at 0.5 h TOS. However, the catalyst is rapidly deactivated under all conditions. The rate of deactivation was higher at higher temperatures, and higher on the reactivated catalyst. During 3 h TOS, at 75 °C the propylene yield decreased from 10% to 6% and from 20% to 16% on the activated and reactivated catalyst, respectively. At 125 °C, the initial activity of the activated and reactivated catalyst decreased from 29% to 5% and from 86% to 17%, respectively, in three hours (Table 2). The 13MoO3/HBEA catalyst, in which the active sites of both the isomerization and the OM reactions are located in a microporous aluminosilicate support, is rapidly deactivated due to the polymerization of olefins.77 In particular, at 150 °C, after three hours TOS the propylene yield dropped significantly from 85% to 10%.
This catalyst contains 90 nm-size MoO3 crystals, which block the micropores; therefore we have halved the MoO3 content of the catalyst. No crystalline MoO3 phase was detected by XRD in 6MoO3/HBEA and only the low-intensity Raman bands indicate the presence of a small amount of MoOx species; however, the catalyst showed lower activity in 1-hexene ISOMET (Table S1†).
At 75 °C the activity of the two catalysts was about the same, but as the temperature increased, the propylene yield on 6MoO3/HBEA hardly changed. The number of active sites is presumably decreasing during the reaction as carbonaceous deposits may gradually block the micropores; thus the reactant molecules cannot access the active sites located inside the pores. Not only coking but also the low oxidation state of Mo is a reason for the decreased activity (vide infra).
On MoO3/HBEA catalysts, the Brønsted acid sites of HBEA zeolite support protonate the 2-hexene to form the carbenium ion intermediate of double-bond isomerization.77–79 Zhang et al.11 reported that both the number of Brønsted acid sites and the high dispersion of MoO3 species have an important role in determining the OM activity.
Brønsted acid sites not only catalyse double-bond isomerization of alpha-olefins but are also involved in the OM reactions, i.e., participate in the generation of active metal carbene species, which are the active site of the OM reactions. Previous work suggested that Brønsted acid OH groups interact with adjacent metal oxides, for example, MoOx, WOx, or ReOx, for the generation of OM active carbene species.80,81 Recent results showed that using the MoOx/SBA-15 catalyst, surface Brønsted acidic OHs coordinated to Mo(VI) protonate propylene and isopropoxide species are formed upon propylene adsorption.82 Such species are further oxidized by the lattice oxygen of MoOx to gas-phase acetone, yielding reduced Mo(IV). This species reacts with gas-phase propylene to form the OM active Mo(VI) alkylidene species. When Brønsted acid sites and MoOx species were located on the microporous support, rapid deactivation occurred during the ISOMET reaction.
Complementary characterization techniques revealed that in the 13MoO3/HBEA sample Mo exists in highly dispersed tetrahedral and polymerized octahedral MoOx species, which are active in the OM reaction. However, a fraction of Mo is present as catalytically inactive crystalline MoO3 phase. Tentatively it is presumed that the lower activity of 6MoO3/HBEA is most probably due to the formation of hardly accessible MoOx species located in the microporous channels.
In order to achieve a more active and stable catalyst for the ISOMET of 1-hexene with ethylene, molybdena was loaded onto a gamma-alumina support (12MoO3/Al2O3) because at 12 wt% loading only highly dispersed MoOx is formed. Further experiments were performed with a mixed bed of 12MoO3/Al2O3 (50%) and zeolite HBEA (50%) catalysts.
1-Hexene ISOMET using 12MoO3/Al2O3 and HBEA catalyst mixture
The activity and stability of the physical mixture of 12MoO3/Al2O3 and HBEA was studied in the ISOMET reaction of 1-hexene at 75 °C, ethylene pressure of 3 bar and 6 and 12 gcatg1−H−1 h space time (Fig. 8A and A′, B and B′) and compared with MoO3/HBEA. In these experiments, we compared the properties of catalysts with the same molybdena content but different weights, i.e., a mixture of 1 g of 12MoO3/Al2O3 + 1 g of HBEA as well as 1 g of 13MoO3/HBEA. Over the freshly activated catalyst, the propylene yield was 33% after 0.5 h TOS. However, after reactivation of the catalyst the propylene yield more than doubled and reached a level of nearly 80%.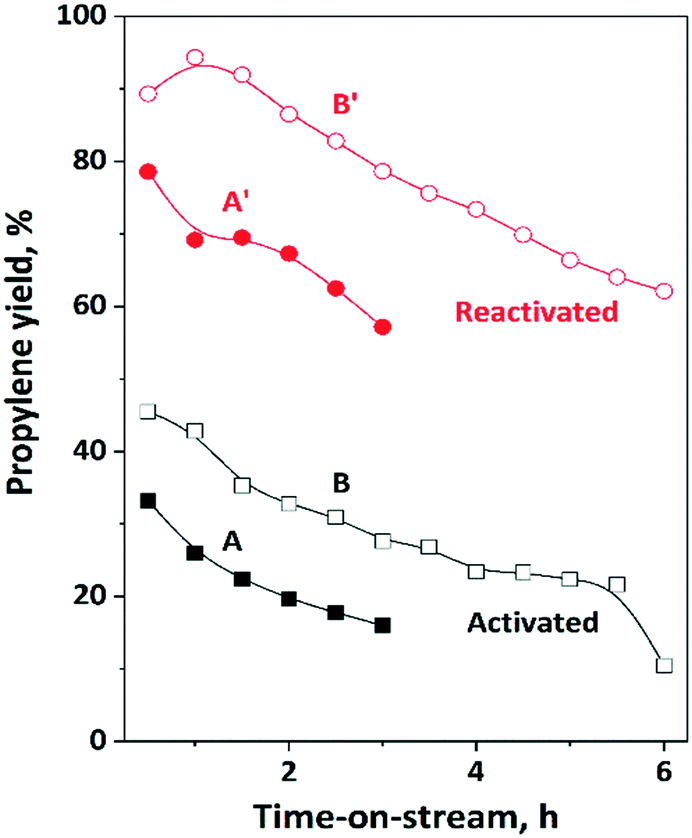 | ||
| Fig. 8 Propylene yield in ISOMET of 1-hexene (1-H) with ethylene over activated and reactivated catalyst mixture of HBEA and MoO3/Al2O3 as a function of time on stream (TOS) at space time of 6 (A, A′) and 12 (B, B′) gcatg1−H−1 h, at 75 °C, 3 bar ethylene pressure, and ethylene/1-hexene ratio of 10. Activation: the mixture of ex situ calcined catalyst was in situ pre-treated in a flow of Ar (50 ml min−1) at 550 °C for 2 h. Reactivation: after 3 h TOS in the ISOMET reaction, the catalyst was purged with Ar flow (50 ml min−1) at 550 °C for 2 h (see Table 2 for detailed conditions). After activation and reactivation the catalyst was cooled to the reaction temperature in a flow of Ar (50 ml min−1) and the ISOMET reaction was performed again. | ||
Repeated reactivation gave similar high propylene yield (Fig. S9†). Not only the initial activity of the catalyst improved significantly but the lifetime of the catalyst also increased. (Fig. 8A and A′).
The propylene yield on the activated catalyst mixture decreased from 33% to 18% in 3 h TOS. Deactivation of the reactivated catalyst is slower; the propylene yield dropped from 79% to only 57% in 3 h. Significantly lower initial activity and faster deactivation were observed for 13MoO3/HBEA (Table 2). Over the activated and reactivated 13MoO3/HBEA catalyst in three hours TOS the propylene yield decreased from 10% to 6% and 20% to 16%, respectively. The higher activity of the 12MoO3/Al2O3 catalyst compared to 13MoO3/HBEA is due to the higher concentration of active MoOx species on alumina than on zeolite (microporous aluminosilicate), as confirmed by XRD and Raman and UV-vis spectroscopy. Higher space time resulted in a significantly higher propylene yield. The catalyst reactivation has shown significantly higher (>90%) propylene yield and longer catalyst lifetime (Fig. 8B and B′).
Further experiments were carried out to investigate the influence of the reaction temperature on the overall propylene yield (Fig. 9). At 125 °C, the propylene yield was around 50%. The catalyst activity did not change as a function of TOS. After reactivation of the catalyst, a propylene yield of more than 90% can be obtained. Some catalyst deactivation was observed; however, after 3 h TOS still 80% propylene yield could be measured.
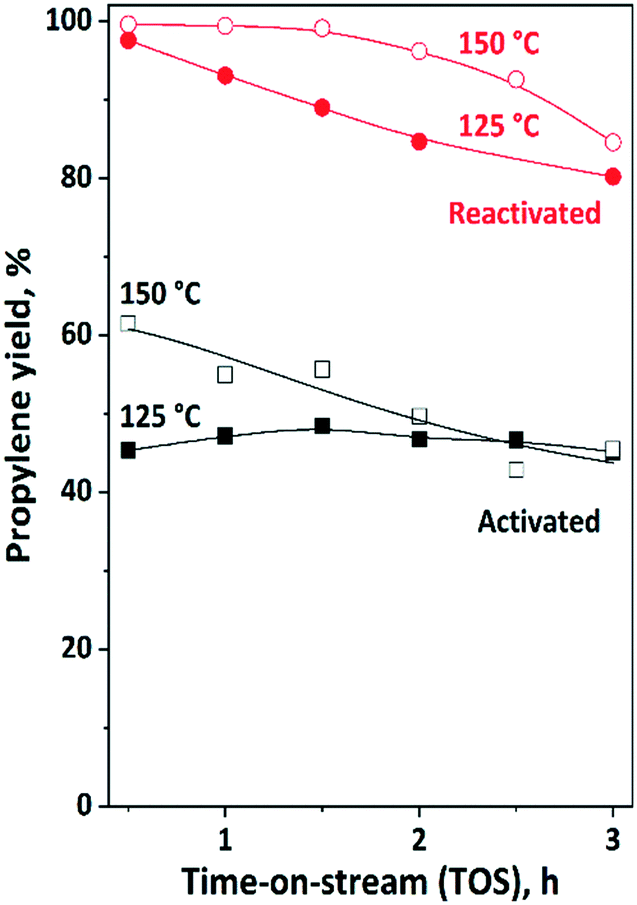 | ||
| Fig. 9 Propylene yield in ISOMET of 1-hexene (1-H) with ethylene over activated and reactivated catalyst mixture of HBEA and 12MoO3/Al2O3 as a function of time on stream (TOS) at 125 °C and 150 °C, 6 gcatg1−H−1 h space time, 3 bar ethylene pressure, and ethylene/1-hexene ratio of 10. Activation: the mixture of ex situ calcined catalyst was in situ pre-treated in a flow of Ar (50 ml min−1) at 550 °C for 2 h. Reactivation: after 3 h TOS in the ISOMET reaction the catalyst was purged with an Ar flow (50 ml min−1) at 550 °C for 2 h (see Table 2 for detailed conditions). After activation and reactivation, the catalyst was cooled to the reaction temperature in a flow of Ar (50 ml min−1) and the ISOMET reaction was performed under the conditions described above. | ||
By increasing the reaction temperature up to 150 °C, even higher yield and longer catalyst lifetime was observed. After the catalyst reactivation approximately 100% propylene yield was found, which was maintained up to two hours. From the third hour of the reaction the propylene yield slightly decreased; however, it was still high (>80%). As was observed after the first run and catalyst reactivation, the catalyst performance was always significantly higher.
Activation and reactivation of 12MoO3/Al2O3 and HBEA catalyst mixture
Conventional pre-treatment of the heterogeneous OM catalysts includes high-temperature calcination and inert gas purging. It has also been shown that olefin pre-treatment at low or high temperatures can improve the initial activity. Amakawa et al.82 reported that heat treatment in inert gas at 550 °C for 2 hours after the room-temperature olefin adsorption doubled the catalytic activity of MoO3/SBA-15. Surface isopropoxide species were supposed to form and activate surface Mo(VI) sites by reduction to Mo(IV) and formation of C3 oxygenate (acetone).This reduced Mo species react with propylene and give active Mo(VI)–alkylidene species. Two orders of magnitude increase in activity was observed over silica-supported MoO3 and WO3 when propylene adsorption was performed at high temperature, 550 °C and 700 °C, respectively.83 The high-temperature activation was explained by a pseudo-Wittig mechanism.
Activation in H2 or Ar: the ex situ calcined catalyst was in situ pre-treated in 5% H2/Ar or Ar flow (50 ml min−1) at 550 °C for 2 h. Reactivation in Ar: after 3 h TOS in 1-hexene ISOMET the catalyst was purged in a flow of Ar (50 ml min−1) for 0.5 h at 75 °C to desorb olefins, then heated to 550 °C at a ramp rate of 10 °C min−1 and maintained at this temperature for 2 h. Reactivation in O2: after 3 h TOS in 1-hexene ISOMET the catalyst was purged in a flow of O2 (50 ml min−1) for 0.5 h at 75 °C to desorb olefins, heated to 550 °C at a ramp rate of 10 °C min−1 and maintained at this temperature for 2 h, and then purged with Ar (50 ml min−1) for 0.5 h and cooled to the reaction temperature. Activation in ethylene: after Ar-activation ethylene was fed into the reactor for 1 h at 75 °C then changed to Ar flow for 0.5 h and heated to 550 °C at a ramp rate of 10 °C min−1 and maintained at this temperature for 2 h.
Others state that the active species is Mo(V).84 Thus, there is still some debate in the scientific community as to which catalyst oxidation state results in high catalytic activity. The surface Mo(VI) oxide species are known to be catalytically inactive; Mo(V) and Mo(IV) are shown to be active. However, lowering the oxidation state results in catalytically inactive species again; therefore the appropriate activation is crucial for the high catalyst activity.
Fig. 10 shows the effect of different activation and reactivation on propylene yield in 1-hexene ISOMET over the 12MoO3/Al2O3–HBEA catalyst mixture. No propylene was formed over the 12MoO3/Al2O3 and HBEA mixture oxidized in situ at 550 °C, confirming that surface Mo(VI) oxide species are not active in the OM reaction. The experiment was stopped after 0.5 h TOS without waiting for the catalyst to be activated in situ during the reaction, as proved by Amakawa et al.62
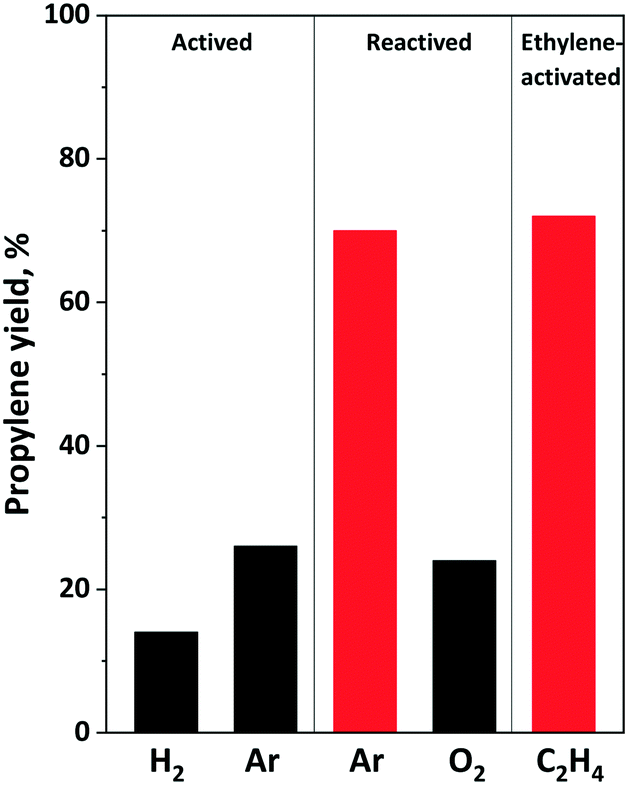 | ||
| Fig. 10 Propylene yield over activated and reactivated 12MoO3/Al2O3–HBEA catalyst mixture in ISOMET of 1-hexene at 75 °C for 1 h TOS, 6 gcatg1−H−1 h space time under 3 bar ethylene pressure and ethylene/1-hexene ratio of 10. | ||
The catalyst pretreated in a 5% H2/Ar mixture at 550 °C for 2 hours has the lowest activity (Fig. 10 and S8†). At 75 °C and 3 bar ethylene pressure the propylene yield was about 15%. Higher propylene yield (∼28%) was obtained over Ar-activated catalyst (Fig. 8 and 10). However, after performing the ISOMET reaction for 3 h TOS and treatment again in inert gas flow at 550 °C for 2 h, improved activity was observed. The propylene yield increased to 80%. When the catalyst was reactivated in an O2 flow at 550 °C for 1 h followed by purging with an Ar flow for 2 h no catalyst activity improvement was observed. It remained the same as that of the Ar-activated catalyst. Fig. 10 shows that improved ISOMET activity was also observed when only the cross-coupling agent ethylene was fed into the reactor at 75 °C (actually, self-metathesis proceeds) and the catalyst was heat-treated in Ar at 550 °C (ethylene-activated catalyst). These results suggest that after 1-hexene ISOMET or ethylene OM, during the Ar reactivation the remaining olefins may also participate in the generation of the catalytically active molybdenum species.83 With calcination of the catalyst in O2 flow at 550 °C Mo–carbene species are decomposed and Mo is oxidized to Mo(VI).
The XPS results show that during inert gas treatment at 550 °C some of the Mo(VI) are reduced to Mo(V) species. The ethylene-activated catalyst contains more Mo(V) species. The high catalyst performance can also be explained by the partial reduction of Mo initiated by the hydrocarbon residues remaining on the catalyst surface.
Propylene synthesis via ISOMET of FCC fractions using 12MoO3/Al2O3 and HBEA catalyst mixture
Our further aim was to develop an OM-based chemical process for a mixture produced by FCC cracking (further named as crude FCC and FCC light). The analysis of FCC light and FCC fractions revealed that the olefin content is about 37 wt% FCC light and 22 wt% crude FCC and approximately 50% of the olefins are 2-olefins (Fig. S10†). The main olefin components of the FCC light fraction are C5–C7 olefins; however, some C4 and C8 components could also be detected. Experiments have been carried out to synthetize propylene from the crude FCC fraction. Theoretically, regardless of the nature of the linear mono olefins (either terminal or internal) all olefins can be converted to propylene via ISOMET using ethylene (Scheme 1). In terms of the weight of olefins, approximately 100 tons of C4–C8 olefin mixture can be converted to 200 tons of propylene by using 100 tons ethylene.The ISOMET of FCC light and FCC fractions was carried out without any pretreatment at 75 °C and 3 bar ethylene pressure. The FCC light fraction contained mainly C5–C7 olefin components. Due to the relatively high olefin content (35–37%) and purity, high propylene yield (70%) and long catalyst lifetime were obtained (Fig. 11). Following the reactivation significantly higher (90%) propylene yield and longer catalyst lifetime were observed. After 8 hours TOS the catalyst remained still active and the propylene yield reached 50%. Compared to the 12MoO3/Al2O3–HBEA catalyst mixture, significantly lower propylene yield could be obtained using the 12MoO3/HBEA catalyst (Fig. S11†).
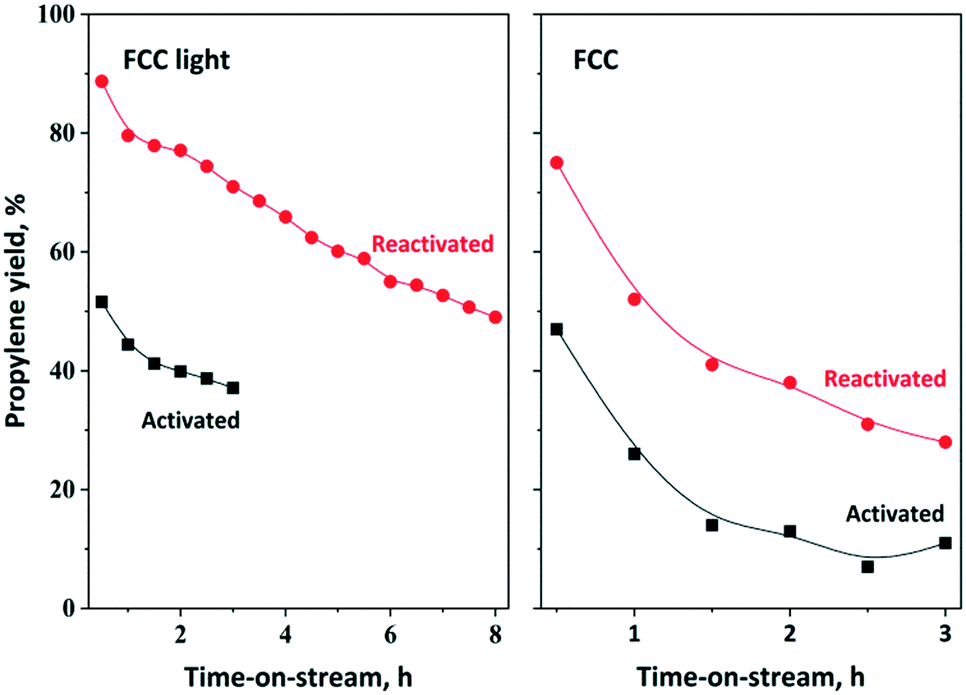 | ||
| Fig. 11 Propylene yield in ISOMET of FCC light and FCC over activated and reactivated HBEA and 12MoO3/Al2O3 catalyst mixture as a function of time on stream at 75 °C and 6 gcatgolefin−1 h space time under 3 bar ethylene pressure. Activation: the mixture of ex situ calcined catalyst was in situ pre-treated in a flow of Ar (50 ml min−1) at 550 °C for 2 h. Reactivation: after 3 h TOS in ISOMET reaction the catalyst was purged with Ar flow (50 ml min−1) at 550 °C for 2 h (see Table 2 for detailed conditions). After activation and reactivation, the catalyst was cooled to the reaction temperature in a flow of Ar (50 ml min−1) and the ISOMET reaction was performed under the conditions described above. | ||
The ISOMET of FCC showed that after 0.5 h TOS slightly higher than 45% propylene yield was obtained, which declined steadily in the next two hours (Fig. 11). The catalyst reusability was also investigated. It was found that experiments carried out with reactivated catalyst resulted in significantly higher (75%) propylene yield and longer catalyst lifetime.
Experimental
Catalyst preparation
The MoO3/Al2O3 olefin MET catalyst was prepared by incipient wetness impregnation of the alumina support. Commercial γ–Al2O3 (Ketjen, CK 300) was first calcined at 500 °C for 8 h (at a heating rate of 10 °C min−1) and then impregnated with ammonium heptamolybdate solution. The sample was air-dried at 120 °C for 6 h and then slowly heated (2 °C min−1) to 500 °C and kept at this temperature for 12 h. The MoO3 content of the air-calcined catalyst was 11.7 wt%, so the catalyst is designated as 12MoO3/Al2O3.MoO3/HBEA ISOMET catalysts were prepared by wet impregnation of H-Beta (HBEA; Si/Al = 12; SÜD-Chemie AG, München) with ammonium heptamolybdate solution. Before impregnation, zeolite HBEA was calcined at 500 °C for 8 h. After impregnation, the solvent was evaporated by keeping the preparation at 120 °C for 6 h. The samples were then heated to 500 °C in air and kept at this temperature for 12 h. The air-calcined samples are designated as xMoO3/HBEA. The MoO3 contents of the catalysts were 13.5 and 6.1 wt%.
Catalyst characterization
X-ray powder diffraction patterns of the catalysts were recorded by a Philips PW 1810/3710 diffractometer equipped with a graphite monochromator. CuKα radiation (λ = 0.1541 nm) was used. The X-ray tube was set at 40 kV and 35 mA current. The scan step size was 0.02 degrees 2-theta, whereas the scan time was five seconds in each step.The specific surface area (SSA) of the catalysts was calculated from the N2 adsorption isotherms using the Brunauer–Emmett–Teller (BET) method. The adsorption isotherms were measured at −196 °C using a Thermo Scientific Surfer automated gas sorption instrument. Before measurement, samples were evacuated at 250 °C for 2 h in high vacuum (∼10−6 mbar).
The reducibility of the catalysts was studied using temperature-programmed H2-reduction (H2-TPR). About 100 mg of sample was treated in a 30 ml min−1 O2 flow at 500 °C for 1 h in a quartz reactor tube (6 mm ID). To obtain the TPR curve the samples were cooled to 25 °C, exposed to a 30 ml min−1 flow of a 9.0 vol% H2/N2 mixture and then heated up to 800 °C at a rate of 10 °C min−1. The reactor effluent was passed through a trap cooled by liquid nitrogen (−196 °C) to remove water from the gas flow. The rate of H2 uptake was recorded using a thermal conductivity detector (TCD).
The acidity of the catalysts was characterized using a Nicolet Impact Type 400 spectrometer applying the self-supported wafer technique and using an in situ cell. The acid site concentrations were determined using transmission Fourier-transform infrared (FT-IR) spectra of the adsorbed pyridine (Py) on Brønsted- and/or Lewis acid sites of the catalysts. IR spectra of the sample were recorded at room temperature, averaging 32 scans at a resolution of 2 cm−1. Spectra were normalized to a wafer thickness of 5 mg cm−2. Difference spectra of adsorbed Py were generated by subtracting the spectrum of the wafer before pyridine adsorption from the spectrum of the wafer loaded by pyridine.
The in situ UV/vis spectra were collected using a Thermo Scientific Evolution 300 UV/vis spectrophotometer equipped with a Praying Mantis diffuse reflectance accessory and a high temperature and pressure reaction chamber. The catalysts were diluted 50–50 wt% with BaSO4.
Raman analyses were carried out at the Research and Industrial Relations Center of the Faculty of Science, Eötvös Loránd University of Budapest, using a HORIBA JobinYvon LabRAM HR 800 Raman microspectrometer. A frequency doubled Nd-YAG green laser with a 532![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm excitation wavelength was employed, displaying 10–20mW on the sample surface. An OLYMPUS ×50 (N.A. = 0.6) objective was used to focus the laser. For Raman analyses, a 100μm confocal hole, 600 grooves per mm optical grating, and 20–120 s cumulated exposition time were used. The spectral resolution of measurements was 3.0 cm−1.
nm excitation wavelength was employed, displaying 10–20mW on the sample surface. An OLYMPUS ×50 (N.A. = 0.6) objective was used to focus the laser. For Raman analyses, a 100μm confocal hole, 600 grooves per mm optical grating, and 20–120 s cumulated exposition time were used. The spectral resolution of measurements was 3.0 cm−1.
The XPS measurements using self-supported wafers were performed by using an Omicron EA 125 electron spectrometer in the fixed analyser transmission mode; photoelectrons were excited by non-monochromatized MgKα (1253.6 eV) radiation. Detailed spectra were recorded with a pass energy of 30 eV, providing a resolution of around 1 eV. Spectra were collected in the as-received state of the catalysts, after 2 h annealing in 300 mbar Ar at 550 °C (simulated activation), after 3 h exposure to 100 mbar C2H4 at around 100 °C (simulated reaction) and after repeated activation at 550 °C. The treatments were performed in a preparation chamber attached to the electron spectrometer so no exposure to the atmosphere was needed during sample transfer. Spectra were processed with the CasaXPS package [N. Fairley, “CasaXPS: Spectrum Processing Software for XPS, AES and SIMS,” Cheshire, 2006, http://www.casaxps.com], while quantitative evaluation of the data was performed with the XPSMultiQuant package [M. Mohai, “XPS MultiQuant: Multimodel XPS Quantification Software,” Surf. Interface Anal. 36 (2004) 828–832, M. Mohai, “XPS MultiQuant: Multi-model X-ray photoelectron spectroscopy quantification program,” 2011, http://aki.ttk.hu/XMQpages/XMQhome.php]. As the samples were insulating, charge compensation was performed by setting the binding energy of the main C 1s peak (arising from adventitious hydrocarbon) of the as-received catalysts to 285.0 eV.
Catalytic test
The catalytic experiments were carried out in a downstream, fixed-bed, stainless-steel tube reactor (12 mm ID). The reactor was loaded with 2 g of catalyst grains prepared by compression of the sample powder, crushing and sieving (0.315–0.63 mm) and the flow rate of the reactant liquid mixture (1,5–6 ml h−1) was varied. The reactor temperature was controlled using an Omron E5CN controller. The liquid mixture (1-hexene in hexane solution (30%), FCC; FCC light) was fed into the reactor by a high-pressure micro pump (Teledyne Isco, Model 100DM). The gas was introduced by a mass flow controller (Aalborg GFC17). The total pressure and the reaction temperature were varied in the range of 1–50 bar and 25–150 °C, respectively. Using water cooling, the reactor effluent was separated to liquid and gas products. The liquid product mixtures were analysed by GC-MS (Shimadzu QP-2010) using a 60 m ZB-WAX PLUS capillary column. The gaseous reactor effluent was analysed for detection of propylene and light hydrocarbons using an on-line gas chromatograph (Varian 3300) with a flame ionization detector (FID) and applying a 30 m Supelco (alumina/chloride) capillary column.The propylene yield as outcome parameter was determined; in some cases complete mass balance including the quantitative distribution of the intermediate species was calculated.
Catalyst activation/reactivation
Activation and reactivation of the catalysts were performed in an inert gas (Ar) flow at 550 °C for 2 h.Activation: the ex situ calcined catalysts were in situ pre-treated in a flow of Ar (50 ml min−1) at atmospheric pressure and 550 °C for 2 h. The activated catalyst was cooled to the target temperature in a flow of Ar and the ISOMET reaction was performed.
Reactivation: after 3 h TOS, the reactant feed was stopped and the catalyst was purged with a flow of Ar (50 ml min−1) for 30 min at reaction temperature and atmospheric pressure. Then it was heated to 550 °C at a ramp rate of 10 °C min−1 and maintained at this temperature for 2 h. The reactivated catalyst thus obtained was cooled to the target temperature in a flow of Ar and the ISOMET reaction was performed again.
After the ISOMET reaction, the catalyst was purged with an Ar flow at 75 °C for 0.5 h and the effluent was analysed by GC-MS. It was found that the reactant and the intermediate olefins (ethylene, propylene, butenes, pentenes and hexenes) completely desorbed from the catalyst surface within 30 min. With increasing temperature longer olefins/paraffins (>C6) were detected by GC-MS. Above 300 °C there were no further organic species observed.
General procedure for isomerization with HBEA
The isomerization catalyst (HBEA) was pre-treated in situ in Ar (50 ml min−1) flow in the reactor at atmospheric pressure and 450 °C for 2 h. After catalyst pre-treatment the reactor was cooled to the target temperature under an Ar flow. The isomerization of 1-hexene was investigated at the temperature range of 75–150 °C, 6 gcatg1−hexene−1 h space time in Ar flow at atmospheric pressure.Conclusions
Heterogeneous molybdena catalysts were prepared using alumina and zeolite beta (HBEA) as support. The catalysts and their mixtures were characterized and studied in the ISOMET of 1-hexene and FCC olefins. It was shown that the Brønsted sites of HBEA catalyze the double-bond isomerization, while the MoOx species supported on alumina or HBEA are active in the ethenolysis of olefins to give propylene as the final product from both 1-hexene and FCC olefins. Over a mixture of 12MoO3/Al2O3 and HBEA catalysts, quantitative conversion of 1-hexene to propylene could be achieved at 150 °C, 3 bar ethylene pressure and 6 gcatg1−hexene−1 h space time. The catalyst mixture also had good performance in the ethenolysis of FCC olefins. Relative to the 12MoO3/Al2O3–HBEA system, the 13MoO3/HBEA catalyst rendered lower activity and significantly shorter catalyst lifetime presumably due to the lower amount of highly dispersed MoOx sites formed on the zeolite.The deactivated ISOMET catalyst system could be reactivated by treatment in an argon flow at 550 °C for 2 hours. The reactivated catalysts showed higher initial activity and stability than the fresh catalysts pre-treated in the same way. It has been demonstrated that treatment of the catalyst with olefins such as ethylene before inert gas activation resulted in significant catalyst activity improvement. It indicates that the adsorbed olefins may play a key role in the formation of active metal centers during the catalyst reactivation process. Activating the catalyst either in a strongly reducing or in an oxidizing environment, however, led to the loss of activity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the project No. VEKOP-2.3.2-16-2017-00013 financed by the European Union and the State of Hungary, co-financed by the European Regional Development Fund. The financial support of the National Research, Development and Innovation (NRDI) Office (2019-2.1.13-TÉT_IN-2020-00043) is also greatly acknowledged. Further thanks are due to COST Action CA17128 (LignoCOST) “Establishment of a Pan-European Network on the Sustainable Valorization of Lignin” for initiating this work. The authors also thank Dr. László E. Aradi for the Raman experiments and the anonymous reviewers for the fruitful suggestions.References
- R. Beucher, R. D. Andrei, C. Cammarano, A. Galarneau, F. Fajula and V. Hulea, ACS Catal., 2018, 8, 3636–3640 CrossRef CAS.
- S. Vorakitkanvasin, W. Phongsawat, K. Suriye, P. Praserthdam and J. Panpranot, RSC Adv., 2017, 7, 38659–38665 RSC.
- G. Zuo, Y. Xu, J. Zheng, F. Jiang and X. Liu, RSC Adv., 2018, 8, 8372–8384 RSC.
- G. Chen, M. Dong, J. Li, Z. Wu, G. Wang, Z. Qin, J. Wang and W. Fan, Catal. Sci. Technol., 2016, 6, 5515–5525 RSC.
- R. D. Andrei, M. I. Popa, F. Fajula, C. Cammarano, A. Al Khudhair, K. Bouchmella, P. H. Mutin and V. Hulea, ACS Catal., 2015, 5, 2774–2777 CrossRef CAS.
- W. Jiang, X. Mo, S. Feng, F. Xu, G. Zhou, H. Zhou, C. Xu and B. Chen, Mol. Catal., 2017, 442, 49–56 CrossRef CAS.
- W. Jiang, R. Huang, P. Li, S. Feng, G. Zhou, C. Yu, H. Zhou, C. Xu and Q. Xu, Appl. Catal., A, 2016, 517, 227–235 CrossRef CAS.
- S. Lwin and I. E. Wachs, ACS Catal., 2017, 7, 573–580 CrossRef CAS.
- H. Liu, S. Huang, L. Zhang, S. Liu, W. Xin and L. Xu, Catal. Commun., 2009, 10, 544–548 CrossRef CAS.
- V. Hulea, Catal. Sci. Technol., 2019, 9, 4466–4477 RSC.
- D. Zhang, X. Li, S. Liu, X. Zhu, F. Chen and L. Xu, Chem. – Asian J., 2015, 10, 1647–1659 CrossRef CAS PubMed.
- K. Gayapan, S. Sripinun, J. Panpranot, P. Praserthdam and S. Assabumrungrat, RSC Adv., 2018, 8, 11693–11704 RSC.
- M. Stoyanova, U. Bentrup, H. Atia, E. V. Kondratenko, D. Linke and U. Rodemerck, Catal. Sci. Technol., 2019, 9, 3137–3148 RSC.
- X. Li, X. Zhu, D. Zhang, F. Chen, P. Zeng, S. Liu, S. Xie and L. Xu, J. Energy Chem., 2013, 22, 145–150 CrossRef CAS.
- J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- R. L. Banks, D. S. Banasiak, P. S. Hudson and J. K. Norell, Chem. Eng. Prog., 1979, 75, 73 Search PubMed.
- G. Parkinson, Chem. Eng., 2001, 108, 27–35 CAS.
- T. Otroshchenko, O. Reinsdorf, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2019, 9, 5660–5667 RSC.
- O. V. Klimov, O. S. Alekseev and A. N. Startsev, React. Kinet. Catal. Lett., 1995, 56, 143–150 CrossRef CAS.
- M. A. Ibrahim, M. N. Akhtar, J. Čejka, E. Montanari, H. Balcar, M. Kubů and S. S. Al-Khattaf, J. Ind. Eng. Chem., 2017, 53, 119–126 CrossRef CAS.
- H. Balcar, D. Mishra, E. Marceau, X. Carrier, N. Žilková and Z. Bastl, Appl. Catal., A, 2009, 359, 129–135 CrossRef CAS.
- R. D. Andrei, M. I. Popa, C. Cammarano and V. Hulea, New J. Chem., 2016, 40, 4146–4152 RSC.
- N. Poovarawan, K. Suriye, J. Panpranot, W. Limsangkass, A. Guntida, F. J. C. S. Aires and P. Praserthdam, Catal. Today, 2018, 309, 43–50 CrossRef CAS.
- S. Maksasithorn, P. Praserthdam, K. Suriye and D. P. Debecker, Microporous Mesoporous Mater., 2015, 213, 125–133 CrossRef CAS.
- D. P. Debecker, M. Stoyanova, U. Rodemerck, F. Colbeau-Justinc, C. Boissère, A. Chaumonnot, A. Bonduelle and C. Sanchez, Appl. Catal., A, 2014, 470, 458–466 CrossRef CAS.
- M. Selva, S. Guidi, A. Perosa, M. Signoretto, P. Licence and T. Maschmeyer, Green Chem., 2012, 14, 2727–2737 RSC.
- L. Sang, S. L. Chen, G. Yuan, M. Zheng, J. You, A. Chen, R. Li and L. Chen, J. Nat. Gas Chem., 2012, 21, 105–108 CrossRef CAS.
- S. Lwin and I. E. Wachs, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- M. Sibeijn and J. C. Mol, J. Mol. Catal., 1992, 76, 345–358 CrossRef CAS.
- P. Schwab, R. Schulz, M. Schulz, B. Breitscheidel and G. Meyer, US6166279, 2000.
- N. P. Christopher, E. Mazoyer, M. Taoufik, J. M. Basset, P. T. Barger and J. E. Rekoske, US8704029, 2014.
- S. Choi and B. Ramachandran, US9611193, 2017.
- M. Röper and J. Stephan, US2004/0242944A1, 2004.
- V. A. Dang, L. Zhang, J. T. Ruszkay, E. Morales and S. T. Coleman, WO2017/079509Al, 2017.
- C. Zhang, X. Liu, T. Liu, Z. Jiang and C. Li, Appl. Catal., A, 2019, 575, 187–197 CrossRef CAS.
- Y. Zhang, H. Wang, L. Zhao, J. Gao and C. Xu, Sep. Purif. Technol., 2019, 228, 115757 CrossRef CAS.
- C. P. Park, M. M. Van Wingerden, S. Y. Han, D. P. Kim and R. H. Grubbs, Org. Lett., 2011, 13, 2398–2401 CrossRef CAS PubMed.
- P. D. Nieres, J. Zelin, A. F. Trasarti and C. R. Apesteguía, Catal. Sci. Technol., 2016, 6, 6561–6568 RSC.
- B. Szabó, G. Novodárszki, Z. May, J. Valyon, J. Hancsók and R. Barthos, Mol. Catal., 2020, 491, 110984 CrossRef.
- S. Jeong, H. Kim, J. H. Bae, D. H. Kim, C. H. F. Peden, Y. K. Park and J. K. Jeon, Catal. Today, 2012, 185, 191–197 CrossRef CAS.
- J. B. Higgins, R. B. LaPierre, J. L. Schlenker, A. C. Rohrman, J. D. Wood, G. T. Kerr and W. J. Rohrbaugh, Zeolites, 1988, 8, 446–452 CrossRef CAS.
- B. Yilmaz and U. Müller, Top. Catal., 2009, 52, 888–895 CrossRef CAS.
- X. Li, W. Zhang, S. Liu, X. Han, L. Xu and X. Bao, J. Mol. Catal. A: Chem., 2006, 250, 94–99 CrossRef CAS.
- J. Guan, G. Yang, D. Zhou, W. Zhang, X. Liu, X. Han and X. Bao, Catal. Commun., 2008, 9, 2213–2216 CrossRef CAS.
- S. Liu, S. Huang, W. Xin, J. Bai, S. Xie and L. Xu, Catal. Today, 2004, 93–95, 471–476 CrossRef CAS.
- E. A. Buluchevskii, L. F. Saifullina, A. V. Lavrenov, T. R. Karpova and N. A. Glazov, Russ. J. Appl. Chem., 2017, 90, 1893–1899 CrossRef CAS.
- M. Röper, G. P. Schindler and J. Stephan, EP1379489A1, 2002.
- X. Li, W. Zhang, S. Liu, L. Xu, X. Han and X. Bao, J. Phys. Chem. C, 2008, 112, 5955–5960 CrossRef CAS.
- E. Hillerová, H. Morishige, K. Inamura and M. Zdražil, Appl. Catal., A, 1997, 156, 1–17 CrossRef.
- B. Wang, G. Ding, Y. Shang, J. Lv, H. Wang, E. Wang, Z. Li, X. Ma, S. Qin and Q. Sun, Appl. Catal., A, 2012, 431–432, 144–150 CrossRef CAS.
- I. E. Wachs, Catal. Today, 1996, 27, 437–455 CrossRef CAS.
- E. Mannei, F. Ayari, M. Mhamdi, M. Almohalla, A. Guerrero Ruiz, G. Delahay and A. Ghorbel, Microporous Mesoporous Mater., 2016, 219, 77–86 CrossRef CAS.
- S. Rajagopal, H. J. Marini, J. A. Marzari and R. Miranda, J. Catal., 1994, 147, 417–428 CrossRef CAS.
- Y. Song, C. Sun, W. Shen and L. Lin, Appl. Catal., A, 2007, 317, 266–274 CrossRef CAS.
- R. W. Borry, Y. H. Kim, A. Huffsmith, J. A. Reimer and E. Iglesia, J. Phys. Chem. B, 1999, 103, 5787–5796 CrossRef CAS.
- A. Martínez, E. Peris, M. Derewinski and A. Burkat-Dulak, Catal. Today, 2011, 169, 75–84 CrossRef.
- P. A. Wright, W. Zhou, J. Pérez-Pariente and M. Arranz, J. Am. Chem. Soc., 2005, 127, 494–495 CrossRef CAS PubMed.
- W. Ding, G. D. Meitzner and E. Iglesia, J. Catal., 2002, 206, 14–22 CrossRef CAS.
- T. Hahn, U. Bentrup, M. Armbrüster, E. V. Kondratenko and D. Linke, ChemCatChem, 2014, 6, 1664–1672 CrossRef CAS.
- H. Knözinger and H. Jezlorowski, J. Phys. Chem., 1978, 82, 2002–2005 CrossRef.
- H. Hu and I. E. Wachs, J. Phys. Chem., 1995, 99, 10897–10910 CrossRef CAS.
- G. Mestl and H. Knozinger, Langmuir, 1998, 7463, 3964–3966 CrossRef.
- C. C. Williams, J. G. Ekerdt, J. M. Jehng, F. D. Hardcastle, A. M. Turek and I. E. Wachs, J. Phys. Chem., 1991, 95, 8781–8791 CrossRef CAS.
- C. C. Williams, J. G. Ekerdt, J. M. Jehng, F. D. Hardcastle and I. E. Wachs, J. Phys. Chem., 1991, 95, 8791–8797 CrossRef CAS.
- E. L. Lee and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 14410–14425 CrossRef CAS.
- D. P. Debecker, B. Schimmoeller, M. Stoyanova, C. Poleunis, P. Bertrand, U. Rodemerck and E. M. Gaigneaux, J. Catal., 2011, 277, 154–163 CrossRef CAS.
- J. T. Kloprogge, L. V. Duong, B. J. Wood and R. L. Frost, J. Colloid Interface Sci., 2006, 296, 572–576 CrossRef CAS PubMed.
- M. Harju, S. Areva, J. B. Rosenholm and T. Mäntylä, Appl. Surf. Sci., 2008, 254, 5981–5989 CrossRef CAS.
- B. M. Reddy, B. Chowdhury, E. P. Reddy and A. Fernández, Appl. Catal., A, 2001, 213, 279–288 CrossRef CAS.
- Y. V. Plyuto, I. V. Babich, I. V. Plyuto, A. D. Van Langeveld and J. A. Moulijn, Appl. Surf. Sci., 1997, 119, 11–18 CrossRef CAS.
- P. Novotný, S. Yusuf, F. Li and H. H. Lamb, J. Chem. Phys., 2020, 152, 044713 CrossRef PubMed.
- J. Baltrusaitis, B. Mendoza-Sanchez, V. Fernandez, R. Veenstra, N. Dukstiene, A. Roberts and N. Fairley, Appl. Surf. Sci., 2015, 326, 151–161 CrossRef CAS.
- W. Grünert, A. Y. Stakheev, W. Mörke, R. Feldhaus, K. Anders, E. S. Shpiro and K. M. Minachev, J. Catal., 1992, 135, 269–286 CrossRef.
- B. W. Wojciechowski and A.-N. Ko, Int. J. Chem. Kinet., 1983, 15, 1249–1274 CrossRef.
- J. Abbot, A. Corma and B. W. Wojciechowski, J. Catal., 1985, 92, 398–408 CrossRef CAS.
- M. Pérez-Luna, A. Cosultchi, J. A. Toledo-Antonio and L. Díaz-Garcia, Catal. Lett., 2009, 128, 290–296 CrossRef.
- X. Li, W. Zhang, X. Li, S. Liu, H. Huang, X. Han, L. Xu and X. Bao, J. Phys. Chem. C, 2009, 113, 8228–8233 CrossRef CAS.
- X. Li, A. Zheng, J. Guan, X. Han, W. Zhang and X. Bao, Catal. Lett., 2010, 138, 116–123 CrossRef CAS.
- X. Li, J. Guan, D. Zhou, G. Li, X. Han, W. Zhang and X. Bao, J. Mol. Struct.: THEOCHEM, 2009, 913, 167–172 CrossRef CAS.
- X. Xiaoding, C. Boelhouwer, D. Vonk, J. I. Benecke and J. C. Mol, J. Mol. Catal., 1986, 36, 47–66 CrossRef.
- A. Andreini, X. Xiaoding and J. C. Mol, Appl. Catal., 1986, 27, 31–40 CrossRef CAS.
- K. Amakawa, S. Wrabetz, J. Kröhnert, G. Tzolova-Müller, R. Schlögl and A. Trunschke, J. Am. Chem. Soc., 2012, 134, 11462–11473 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, T. L. Drake, W. Wu, Y. Lin, E. Weitz, L. D. Marks and P. C. Stair, ACS Catal., 2016, 6, 5740–5746 CrossRef CAS.
- B. Zhang, N. Liu, Q. Lin and D. Jin, J. Mol. Catal., 1991, 65, 15–28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00269d |
| This journal is © The Royal Society of Chemistry 2021 |