
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-sufficient asymmetric reduction of β-ketoesters catalysed by a novel and robust thermophilic alcohol dehydrogenase co-immobilised with NADH†
Alejandro H. Orrego‡
 abc,
Daniel Andrés-Sanz‡c,
Susana Velasco-Lozanoc,
Mercedes Sanchez-Costab,
José Berenguerb,
José M. Guisan*a,
Javier Rocha-Martin*a and
Fernando López-Gallego*cd
abc,
Daniel Andrés-Sanz‡c,
Susana Velasco-Lozanoc,
Mercedes Sanchez-Costab,
José Berenguerb,
José M. Guisan*a,
Javier Rocha-Martin*a and
Fernando López-Gallego*cd
aDepartment of Biocatalysis, Institute of Catalysis and Petrochemistry (ICP), CSIC, Campus UAM, Cantoblanco, 28049 Madrid, Spain. E-mail: jmguisan@icp.csic.es; javirocha@icp.csic.es
bDepartment of Molecular Biology, Universidad Autónoma de Madrid, Center for Molecular Biology Severo-Ochoa (UAM-CSIC), Nicolás Cabrera 1, 28049 Madrid, Spain
cHeterogeneous Biocatalysis Laboratory, Center for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 182, Donostia San Sebastián, Spain. E-mail: flopez@cicbiomagune.es
dIKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, 48013 Bilbao, Spain
First published on 12th March 2021
Abstract
β-Hydroxyesters are essential building blocks utilised by the pharmaceutical and food industries in the synthesis of functional products. Beyond the conventional production methods based on chemical catalysis or whole-cell synthesis, the asymmetric reduction of β-ketoesters with cell-free enzymes is gaining relevance. To this end, a novel thermophilic (S)-3-hydroxybutyryl-CoA dehydrogenase from Thermus thermophilus HB27 (Tt27-HBDH) has been expressed, purified and biochemically characterised, determining its substrate specificity towards β-ketoesters and its dependence on NADH as a cofactor. The immobilization of Tt27-HBDH on agarose macroporous beads and its subsequent coating with polyethyleneimine has been found the best strategy to increase the stability and workability of the heterogeneous biocatalyst. Furthermore, we have embedded NADH in the cationic layer attached to the porous surface of the carrier. Since Tt27-HBDH catalyses cofactor recycling through 2-propanol oxidation, we achieve a self-sufficient heterogeneous biocatalyst where NADH is available for the immobilised enzymes but its lixiviation to the reaction bulk is avoided. Taking advantage of the autofluorescence of NADH, we demonstrate the activity of the enzyme towards the immobilised cofactor through single-particle analysis. Finally, we tested the operational stability in the asymmetric reduction of β-ketoesters in batch, succeeding in the reuse of both the enzyme and the co-immobilised cofactor up to 10 reaction cycles.
1. Introduction
Today, the biocatalytic asymmetric reduction of β-ketoesters is gaining relevance among chemistry routes to access enantiomerically pure β-hydroxyesters.1–3 The biosynthesis of this family of compounds requires mild reaction conditions (temperature and pressure) and avoids the use of toxic substrates or metallic catalysts.4,5 β-Hydroxyesters are building blocks for food and pharmaceutical industries to be used in the synthesis of functional products.6 For example, methyl 3-hydroxybutyrate has proven to be effective against Alzheimer's disease,7 memory loss8 or osteoporosis.9 Likewise, ethyl (R)-3-hydroxybutyrate is employed in the synthesis of β-lactamases inhibitors,10,11 while its S-isomer is an important intermediate for the synthesis of carbapenems and insect pheromones.12,13 Among all the industrially exploited β-ketoesters, ethyl (S)-4-chloro-3-hydroxybutanoate has one of the largest markets since it is an essential building block for manufacturing cholesterol-lowering drugs.1,14,15Applied biocatalysis mainly exploits recombinant whole-cells expressing mesophilic alcohol dehydrogenases to accomplish the asymmetric reduction of β-ketoesters.16 Alternatively, the use of cell-free enzymes may increase both the enantiopurity of the final product and the scope of the substrate.17,18 Nevertheless, the industrial implementation of cell-free processes demands more robust enzymes. Here, genome mining has succeeded in discovering new enzymes from thermophilic environments in the last decades. For example, the genomes from several thermophiles have been mined to find more stable transaminases, alcohol reductases and ketoreductases.19–23
Together with the discovery of more robust biocatalysts, enzyme immobilization emerges as a complementary strategy to enhance the biocatalyst stability as well as to increase their workability. The immobilised enzymes can be easily separated from the reactants once the reaction is completed and reused for several reaction cycles.24,25 However, recovering an active and stable enzyme upon the immobilization protocol is not trivial, so one needs to screen different immobilization techniques to find the one that maximises both functional features in the final heterogeneous biocatalyst. Moreover, our groups have recently reported that controlling the immobilization chemistry we can also co-immobilise small coenzymes or cofactors with their enzyme partner to create self-sufficient immobilised enzymes, which become independent of the exogenous supply of those molecules. This strategy has allowed co-immobilizing alcohol dehydrogenases and ketoreductases with NAD(P)H,26,27 transaminases with PLP28 and glycosyltransferases with UDP-glucose,29 to operate the corresponding biotransformations using both batch and flow-reactors, without the exogenous addition of those cofactors.
In this work, we report the cloning, expression and characterization of the (S)-3-hydroxybutyryl-CoA dehydrogenase (Tt27-HBDH, EC 1.1.1.157) from the thermophilic organism Thermus thermophilus HB27. This enzyme belongs to the large family of NAD(P)H dependent dehydrogenases (EC 1.1.1.X) that catalyse the asymmetric reduction of acetoacetyl-CoA and its reverse oxidative reaction with exquisite enantioselectivity.30–32 Having this enzyme in hand, we screened different immobilization and post-immobilization protocols to further stabilise both tertiary and quaternary enzyme structures under high temperatures, low pH and high concentration of co-solvents like 2-propanol. Furthermore, the most stable immobilization protocol was also exploited to co-immobilise both Tt27-HBDH and NADH, resulting in a self-sufficient heterogeneous biocatalyst where enzyme and cofactor interplay within the porous environment of the carrier. The immobilised enzyme catalysed both the main reduction reaction and the cofactor recycling one, using 2-propanol as ultimate electron donor. Finally, this self-sufficient heterogeneous biocatalyst was exploited for the asymmetric reduction of β-ketoesters in batch, and both the enzyme and the co-immobilised cofactor were reused for up to 10 reaction cycles.
2. Materials and methods
2.1. Materials
Nicotinamide adenine dinucleotides (NAD+, NADH, NADP+ and NADPH) were purchased from Carbosynth (Berkshire, UK). Ethyl acetoacetate (EAA), ethyl 3-hydroxybutyrate, ethyl 2-chloroacetoacetate and ethyl 4-chloroacetoacetate were purchased from Alfa Aesar (Karlsruhe, Germany). Alcohols, ketones and aldehydes were supplied by Sigma-Aldrich (St. Louis, IL). Polyethyleneimine (PEI) (MW. 25–60 kDa) and polyallylamine (PAA) (MW. 17 kDa) were purchased from Sigma-Aldrich (St. Louis, IL, USA). Plain 6BCL agarose was purchased from Agarose Beads Technologies (Madrid, Spain). Protein concentrations were determined using BCA Protein Assay Kit from Pierce (Rockford, IL, USA). Enzymatic assays were carried out on a V-730 spectrophotometer from JASCO Analitica Spain S.L. (Madrid, Spain). Buffers, mediums and other reagents were obtained from Sigma-Aldrich Co. (St. Louis, IL, USA). Single particle experiments were carried out on a Cytation 5 cell image multi-mode reader from Biotek Instruments Inc. (Winooski, VT, USA). The gene from formate dehydrogenase (FDH) from Candida boidinii were synthetized and cloned in pET28b by GenScript (Piscataway, USA).2.2. Structure homology modelling
Structure prediction was performed using protein model portal (https://swissmodel.expasy.org/).33 For Tt27-HBDH, we used the HBDH from Clostridium acetobutylicum (PDB: 6ACQ) as structure template. The electrostatic potential and surface exposure of each residue was calculated by using the Bluues server.34 Figures were prepared using the PyMOL Molecular Graphics System (version 1.7.4.5, Schrodinger LLC).2.3. Bacterial strains and growth conditions
T. thermophilus HB27 was used as the source of DNA. Escherichia coli BL21 (DE3) was used for the expression of the protein. The thermophile was grown at 70 °C in TB (Thermus broth) under stirring (170 rpm) and E. coli was grown at 37 °C in modified lysogeny broth (LB) medium. Either Ampicillin (100 μg mL−1) or Kanamycin (30 μg × mL−1) was added to the cultures when required for the selection of the plasmids.2.4. Cloning and overexpression of TTC0898 gene
Amplification of (S)-3-hydroxybutyryl-CoA dehydrogenase was carried out using the following oligonucleotides; forward: TTCCATATG![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/i_char_0047_0332.gif) ATGGAGGTCAAGCGGATCGG and reverse TTCGAATTCTCAGCCCACCTTGTTCCCC. These primers include the sequence of restriction enzymes, NdeI and EcoRI (underlined) respectively, for subsequent cloning actions. Purified T. thermophilus DNA was used as template. PCR product was run in a 0.8% agarose gel and, a main band was obtained at the expected size of 873 bp.
ATGGAGGTCAAGCGGATCGG and reverse TTCGAATTCTCAGCCCACCTTGTTCCCC. These primers include the sequence of restriction enzymes, NdeI and EcoRI (underlined) respectively, for subsequent cloning actions. Purified T. thermophilus DNA was used as template. PCR product was run in a 0.8% agarose gel and, a main band was obtained at the expected size of 873 bp.
For the heterologous expression of the enzyme, the purified PCR product was digested with NdeI and EcoRI restriction enzymes and cloned into the pET22b (+) plasmid, which had been previously digested with the same enzymes, under the control of the phage T7 protein 10 promoter. The ligation was performed at 37 °C for 4 h and transformed in E. coli DH5α cells by heat shock. The resulting pET22-TTC898 derived plasmid was purified by conventional methods.
For the overexpression of the enzyme, pET22-TTC898 was transformed in BL21 (DE3) E. coli cells. Colonies were picked and grown in LB containing ampicillin. When an optical density (OD600) of 0.5 was reached, 1 mM IPTG (isopropyl-β-D-thiogalactopyranoside) was added to induce the expression of T7 RNA polymerase, and consequently the expression of Tt27-HBDH. The induced cultures were grown for 3 h at 37 °C and then harvested by centrifugation.
2.5. Purification of Tt27-HBDH
The cells overexpressing Tt27-HBDH were resuspended in 50 mM phosphate buffer pH 7 and broken by sonication. The cell debris was discarded by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 20 min). The soluble crude protein extract was incubated at 70 °C for 45 min to denaturalise the host proteins. The pellet of denatured proteins was discarded after centrifugation (10000 × g for 30 min). The activity and protein concentration were determined in both supernatant and suspension.
000 × g for 20 min). The soluble crude protein extract was incubated at 70 °C for 45 min to denaturalise the host proteins. The pellet of denatured proteins was discarded after centrifugation (10000 × g for 30 min). The activity and protein concentration were determined in both supernatant and suspension.
2.6. Expression and purification of CbFDH
Formate dehydrogenase from Candida boidinii (FDH) was overexpressed in E. coli BL21 (DE3) as described in bibliography.35 In summary, cells transformed with the plasmid pET28-CbFDH were grown at 21 °C until OD600 of 0.6 and induced with 1 mM IPTG. The culture was incubated at 21 °C for 18 h. Finally, the cells were harvested by centrifugation.2.7. Enzyme assay
The redox activity of Tt27-HBDH was spectrophotometrically measured by monitoring the absorbance at 340 nm, corresponding to NADH. The general enzymatic assay consisted on the reduction of a pre-heated solution of 0.2 M of EAA and 0.25 mM NADH at pH 7. Afterward, 100–50 μg of the enzyme was added in order to trigger the reaction. The substrate specificity assay for the reductive reactions was done with 0.25 mM NADH and 10 mM of the corresponding ketone or aldehyde at 65 °C and pH 7. The oxidative reactions were performed with 2.5 mM NAD+ and 25 mM of the corresponding alcohol at 65 °C and pH 7. In all cases, the solutions were pre-heated at 65 °C and 100 μg of Tt27-HBDH were added to trigger the reaction. When indicated, different temperatures and pHs were used. One Tt27-HBDH unit (U) was defined as the amount of enzyme required to either oxidise or reduce 1 μmol of NADH or NAD+, respectively, per minute under given conditions and considering an ε = 6.22 mM−1 cm−1 for NADH at 340 nm.2.8. Kinetic parameters
Kinetic parameters were calculated by measuring initial activity as previously described.36 Oxidative steady-state kinetics were determined using different concentrations of ethyl (S)-3-hydroxybutyrate (0.01–2 M) and NAD+ (0.01–5 mM). Reductive steady-state kinetics were determined using different concentrations of EAA (0.001–1 M), ethyl 2-chloroacetoacetate (1–250 mM) and NADH (0.005–0.5 mM). Activity assays were performed as described above. The activities for each substrate concentration were assayed by triplicate, resulting in a mean value for each substrate concentration. All mean activities were plotted and adjusted to a Michaelis–Menten model.2.9. Molecular weight determination by size exclusion chromatography (SEC)
The molecular mass of the Tt27-HBDH was determined by SEC using a Superdex 200 10/300 GL column equilibrated with 25 mM Tris-HCl buffer pH 7 at a flow rate of 0.5 mL min−1. Column and detector temperature were set at 4 °C. SEC was performed with an ÄKTA purifier system (GE Healthcare, Munich, Germany). A sample of pure soluble Tt27-HBDH (1 mg mL−1) in 25 mM Tris-HCl buffer pH 7 was filtered with a polytetrafluoroethylene membrane of thickness 0.45 μm and 100 μL of the filtered sample was injected in the size exclusion column. The following molecular mass standards were used for calibration: blue dextran (2000 kDa), ferritin (440 kDa), alcohol dehydrogenase from Bacillus stearothermophilus (147 kDa), bovine serum albumin (63 kDa), ovalbumin (43 kDa) and green fluorescent protein from Aequorea victoria (28 kDa).2.10. Immobilization of the enzyme
10 mL of enzyme solution at the indicated pH and temperature was mixed with 1 g of different supports and incubated under gently stirring. At different times, samples of the supernatants and suspensions were withdrawn, and their enzymatic activity was measured. Unless indicated, the experiments were performed offering less than 3 U × g−1 of carrier in order to avoid diffusion problems that could affect the apparent enzyme stability.:10 (w:v). After 2 h, the preparation was vacuum filtered and washed with 10 mM sodium phosphate buffer pH 7.2.11. Coimmobilization of FDH on AG–G@Tt27-HBDH
Starting from the biocatalyst previously prepared with Tt27-HBDH immobilised on AG–G and coated with PEI, the coimmobilization of FDH was conducted by adding 10 volumes of a FDH solution (0.0663 mg mL−1, 0.1 U mL−1) in sodium phosphate buffered solution 10 mM pH 7. After incubating 1 h at 4 °C with orbital shaking, the suspension was filtered and the beads were incubated with 10 volumes of 1,4-butanediol diglycidyl ether (30 mM at pH 7) for 1 h at 4 °C. Finally, the suspension was filtered and washed 3 times with 10 volumes of sodium phosphate buffered solution 10 mM pH 7 and stored at 4 °C.2.12. Coimmobilization of NADH on AG–G@Tt27-HBDH biocatalyst coated with PEI
Once the Tt27-HBDH was immobilised on AG–G and coated with PEI, the resulting matrix was equilibrated with an excess of sodium phosphate buffer 25 mM pH 7. Then, the heterogeneous biocatalyst was incubated with a freshly prepared solution of 1 mM NADH in 10 mM Tris-HCl pH 7 for 1 h at 25 °C. Finally, the heterogeneous biocatalyst with the immobilised cofactor was washed with 2 volumes of 10 mM Tris-HCl pH 7 buffer. The supernatants of the immobilization and the washing step were analysed by UV-vis to quantify the amount of cofactor that was immobilised and then released during the washing steps.2.13. Inactivation of different Tt27-HBDH preparations
The different biocatalysts were incubated at different pH, temperatures or in the presence of 20% 2-propanol. Samples from the incubated suspension were withdrawn at different times and their enzymatic activity measured. The half-life and the stabilization factor were calculated as previously described.402.14. Synthesis of β-hydroxyesters and operational stability of biocatalyst
100 mg of solid biocatalyst (11–33 mgprotein × gcarrier−1) were placed in a micro chromatographic column (Biospin TM, BIO-RAD). The reaction was triggered by adding 1 mL of reaction mixture composed of either 10 or 200 mM of ethyl acetoacetate, 5% (v/v) of isopropyl alcohol in a 10 mM Tris-HCl buffer at pH 8.0, as well as 1 mM of NADH when exogenous cofactor was added. Reactions were incubated either for 5 h at 60 °C or 24 h at 25 °C under gently rotational agitation and then stopped by vacuum filtration. The heterogeneous biocatalysts were washed with 3 volumes of the same buffer once the reaction was completed and then it was re-used for the next cycles. Samples were withdrawn from the reaction bulk at different times but never removing more than 5% of the total reaction volume. Finally, these samples were analysed by gas chromatography. The cofactor total turnover number (TTN) per cycle was calculated as: ethyl 3-hydroxybutyrate (mol) detected after batch reaction/NADH (mol) added in reaction mix or co-immobilised with the enzyme. The enzyme total turnover number was calculated as: ethyl 3-hydroxybutyrate (mol) detected after batch reaction/immobilised enzyme (mol) added to the reaction. The accumulated TTN values mean the sum of all TTN values after each reaction cycle.2.15. Gas chromatography (GC) analysis
Liquid–liquid extraction of organic compounds from the samples was done by the addition of 300 μL of dichloromethane, discarding the aqueous phase. 30–50 mg of anhydrous MgSO4 were added to dry samples before GC analysis. As internal standard, eicosane 2 mM was employed. Gas chromatography analyses were carried out in an Agilent 8890 System gas chromatograph using a column of (5%-phenyl)-methylpolysiloxane (Agilent, J&W HP-5 30 m × 0.32 mm × 25 μm), helium as a carrier gas, and equipped with a flame ionization detector (FID). The temperature of the injector and FID detector were 280 °C and 300 °C, respectively. The separation of compounds was carried out by two sequential temperature ramps: the initial temperature (60 °C) was maintained for 2 min and progressively increased up to 160 °C at a rate of 10 °C min−1. Then, the column temperature was increased 20 °C min−1 for 4 min until 250 °C and maintained for 4 min. Retention times were 5.2 min for ethyl acetoacetate and 5.08 min for ethyl 3-hydroxybutyrate.2.16. Determination of EAA, S-EHB and R-EHB by chiral HPLC
Aqueous reaction samples were analysed by HPLC (Waters 2690) with Lux® 5 μm cellulose-1 (250 mm × 4.6 mm) column, equipped with a PDA detector at 210 nm. Analytes were eluted with a normal mobile phase composed by n-hexane/2-propanol following the gradient 98:2 from 0–4 min, 85–15 from 4–14 min and 98:2 from 14–25 min. Retention times were 6.24 min for EAA, 9.75 min for R-EHB and 11.86 min for S-EHB.
2.17. Determination of chlorinated derivatives by chiral GC
Aqueous reaction samples were liquid–liquid extracted with 1:1 ethyl acetate and dried with anhydrous magnesium sulphate. Then, 2 μL of organic samples were analysed by chiral gas chromatography (Hewlett Packard 5890 Series II), with a chiral column Alpha DEX™ 120 (30 m × 0.25 mm × 0.25 μm film thickness), with an FID detector at 250 °C, injector at 250 °C at isothermal conditions at 115 °C. Retention times: 4-Cl-EAA 8.98 min, 4-Cl-S-EHB 12.63 min, 4-Cl-R-EHB 12.86 min.
2.18. In operando single-particle studies
5 μL of 1:10 (w/v) suspension of the Tt27-HBDH immobilised on AG–G and further coated with PEI was placed into the wells of transparent 96-well plates and incubated with a 0.1 mM NADH for 5 min. Then, the reaction was automatically triggered by dispensing a mix composed by ethyl acetoacetate 10 mM in Tris-HCl buffer pH 8 up to a final volume of 200 μl. NADH fluorescence intensity was recorded every 8 s using a Cytation 5 cell image multi-mode reader (Biotek, USA). The sample was observed with a 4× Olympus plan fluorite objective with a numerical aperture of 0.13 and a working distance of 17 mm. During the time-lapse microscopy experiment, the brightfield channel was also recorded to detect any change in the bead positions that might create an artefact for further analysis. Images were taken and recorded with a Blackfly BFLY-U3-2356M (FLIR systems, Oregon, USA) with a resolution of 1920 × 1200 and an IMX249 (Sony, Tokyo, Japan) sensor.
3. Results and discussion
3.1. Expression and purification of recombinant Tt27-HBDH
The gene ttc0898 from Thermus thermophilus HB27 encodes a protein annotated as a 3-hydroxybutyryl-CoA dehydrogenase (EC:1.1.1.157). This enzyme, here named as Tt27-HBDH, is 54–50% identical to the previously described bacterial (S)-3-hydroxybutyryl-CoA dehydrogenases from Clostridium butyricum,30,41Clostridium acetobutylicum,31 and Ralstonia eutropha,42 and 45% identical to the human 3-hydroxyacyl-CoA dehydrogenase.43 The multi-sequence alignment (MSA) of Tt27-HBDH revealed two conserved motifs; the catalytic triad (S119, H140 and N190) and the nucleotide-binding motif formed by both GXGXXG sequence and the polar residues D33, D92, K97 and N117 (Fig. S1†). These motifs were fully conserved in the family of NADH-dependent 3-hydroxyacyl CoA dehydrogenases (Pfam: 3HCDH_N and 3HCDH) as supported by the amino acid conservation plots (Fig. S2†). To better understand the function/structure relationships for this enzyme, we created a structure model based on the X-ray structure of the 3-hydroxybutyryl-CoA dehydrogenase from Clostridium acetobutylicum (PDB: 6ACQ; 54% homology).31 This model confirmed the roles of the conserved residues according to those predicted by the conservation plots (Fig. 1).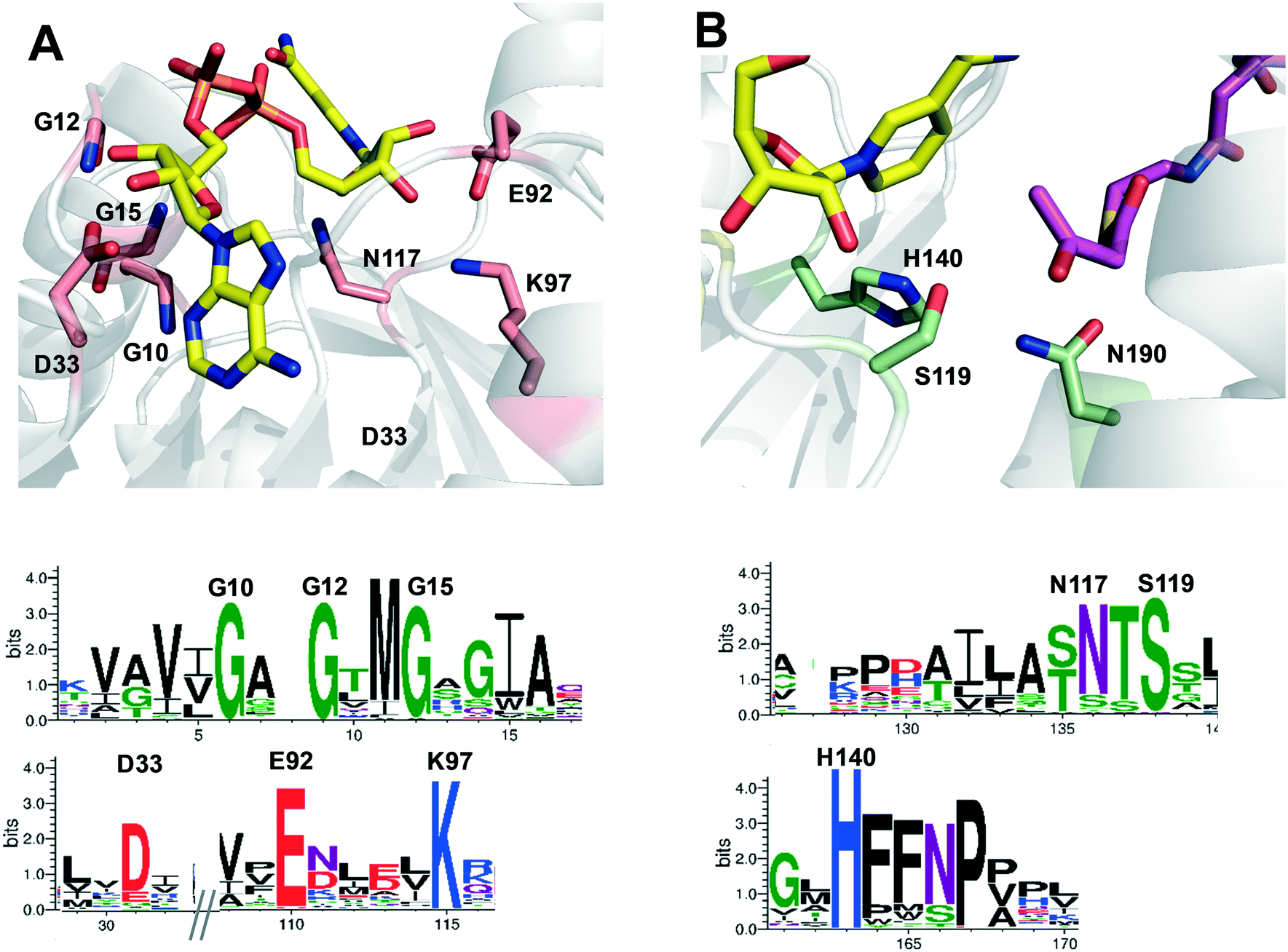 | ||
| Fig. 1 NADH binding cavity (A) and active site (B) from the 3D-model of Tt27-HBDH. Top images highlight the residues involved in the cofactor binding (pink) and the catalytic triad (green) according to the conserved residues identified in the conservation plots (bottom panels). The overlapped NADH and 3-hydroxybutyryl-CoA are coloured in yellow and violet, respectively. | ||
To demonstrate the function of Tt27-HBDH predicted by the MSA, we cloned and expressed the gene in E. coli as heterologous host to ultimately purify and characterise it. The purification protocol consisted in a thermal shock that precipitated the vast majority of the proteins from the lysate but Tt27-HBDH, which remained soluble with a high purity due to its thermophilic nature (Fig. S3†). The apparent electrophoretic mobility of Tt27-HBDH was 32 kDa in agreement with its expected mass according to the primary sequence translated from the ttc0898 gene (31.78 kDa) and similar to the monomeric mass found for other HBDH enzymes.31,41,42 Furthermore, size-exclusion chromatography (SEC) of the pure Tt27-HBDH revealed that this enzyme is a hexamer with an estimated molecular weight of 190 kDa (Fig. S4†). This oligomerization state was also state reported for the homologous enzyme from C. acetobutylicum,31 but different to the C. butyricum (2-mer)30 or R. eutropha (2-mer)42 homologs.
3.2. Substrate specificity of Tt27-HBDH
Tt27-HBDH accepted both NADH and NADPH as redox cofactor, but its enzymatic activity was 6-fold higher with the first one. Using NADH as cofactor, the redox activity was tested with a battery of ketones, aldehydes and alcohols (Table 1). As predicted, we found its highest oxidation and reduction activities towards ethyl 3-hydroxybutyrate and its corresponding β-ketoester (EAA), respectively. This enzyme was also able to reduce halogenated derivatives of EAA. Noteworthy, Tt27-HBDH was fully enantioselective towards ethyl (S)-3-hydroxybutyrate as demonstrated by its null activity towards the R-isomer. This exquisite enantiopreference matches the enantioselectivity found for other HBDHs from the same family,31,41,42 other native thermophilic alcohol dehydrogenase21,44 and some microbial engineered ketoreductases.45,46 In contrast, the Tt27-HBDH enantioselectivity towards β-ketoesters is the opposite to the one found for the (R)-3-hydroxybutyryl-CoA dehydrogenase from Ralstonia eutropha,47 the R-selective ketoreductases from Lactobacillus kefir48 and Neurospora crassa49 and the alcohol dehydrogenase 2 from T. thermophilus.22 The native enzyme showed an extremely narrow substrate specificity according to its low activity towards other ketoesters, β-ketodiesters, aldehydes and ketones (Table 1). Remarkably, Tt27-HBDH was active with 2-propanol, which shows its potential capacity to self-recycle the NADH cofactor in the presence of this alcohol as proven with other ketoreductases.50–52 Despite the limited substrate scope, Tt27-HBDH emerges as a promising enzyme to produce halogenated and non-halogenated enantiopure β-hydroxyesters, which play important roles as building blocks in the food and pharmaceutical industries.6| Substrate | Specific activity (U mg−1) | Relative activity (%) | |
|---|---|---|---|
| Specific activity was determined for the soluble and pure enzyme at 65 °C and pH 7. The substrate concentration was 25 mM for alcohols and 10 mM for ketones and aldehydes. The relative activity of each substrate regarding the measured activity towards ethyl (S)-3-hydroxybutyrate (100%). | |||
| Aldehydes and ketones | Ethyl acetoacetate | 1.92 | 43.3 |
| Cyclohexanone | 0 | 0 | |
| Ethyl 4,4,4-trifluoroacetoacetate | 0.2 | 4.4 | |
| D,L-Glyceraldehyde | 0.06 | 1.26 | |
| Propiophenone | 0 | 0 | |
| Acetophenone | 0 | 0 | |
| Octanal | 0 | 0 | |
| 2-Ketohexanoic acid | 0 | 0 | |
| Ethyl benzoylacetate | 0 | 0 | |
| Phenylglyoxylic acid | 0 | 0 | |
| Acetone | 0 | 0 | |
| 2,2,2-Trifluoroacetophenone | 0 | 0 | |
| Ethyl levulinate | 0.08 | 1.8 | |
| Dimethyl 1,3-acetonedicarboxylate | 0.42 | 9.4 | |
| Ethyl 3-oxohexanoate | 0 | 0 | |
| Ethyl 4-chloroacetoacetate | 1.05 | 23.7 | |
| Ethyl 2-chloroacetoacetate | 1.91 | 42.9 | |
| Alcohols | Glycerol | 0.003 | 0.07 |
| 2,2,2-Trifluoroethanol | 0 | 0 | |
| Cyclohexanol | 0.03 | 0.7 | |
| 1-Phenylethanol | 0.05 | 1.1 | |
| 1-Phenyl-1-propanol | 0 | 0 | |
| Ethyl 3-hydroxybutyrate | 2.64 | 59.5 | |
| Ethyl (S)-3-hydroxybutyrate | 4.44 | 100 | |
| Ethyl (R)-3-hydroxybutyrate | 0 | 0 | |
| 2-Phenyl-1-propanol | 0 | 0 | |
| Ethanol | 0 | 0 | |
| 2-Propanol | 0.06 | 1.3 | |
| 2-Hydroxypentanoic acid | 0 | 0 | |
3.3. Biochemical characterization
The Michaelis–Menten kinetic parameters of Tt27-HBDH were determined towards the most active substrates in both oxidative and reductive directions (Table 2 and Fig. S5†). While the kcat values fall in the range reported for other HBDHs, the KM values towards both ethyl β-keto- and hydroxy-esters are significantly higher than the corresponding values reported for other members of this family. Since this family of enzymes accepts acetoacetyl-CoA and 3-hydroxybutyryl-CoA as natural substrates,30,41–43,47 Tt27-HBDH seems to bind ethyl β-ketoesters with lower affinity as the substrate lacks the coenzyme A (CoA). As found for other HBDH, the pantothenic moiety binds strongly the substrate through a conserved asparagine duplex (Asn143 and Asn223 in Tt27-HBDH) that establish a hydrogen-bond interaction with the CoA molecule.30,31,41,42,53 This enzyme was 8.7 fold more catalytically effective for the reduction than for the oxidation under neutral pH according to the data shown in Table 2. This trend aligns with the behaviour of other dehydrogenases from the same microorganism.21,22 Remarkably, when the substrate is halogenated at position 2, the reduction reaction was 15 fold more efficient than when non-halogenated β-ketoesters were used.| K M (mM) | k cat (s−1) | k cat/KM (M−1 s−1) | |
|---|---|---|---|
| The steady-state kinetics parameters were calculated at pH 7 and 65 °C (see Materials and methods section). Kinetic constants for NADH and NAD+ were determined with 1 M EAA and 2 M ethyl 3-hydroxybutyrate, respectively. Kinetic constants for EAA and ethyl 2-chloroacetoacetate were determined with NADH 0.25 mM. Kinetic constants for ethyl (S)-3-hydroxybutyrate (S)-EHB) were determined with NAD+ 2.5 mM. | |||
| EAA | 325.5 | 21.1 | 64.9 |
| (S)-EHB | 343 | 7.33 | 21.4 |
| NADH | 0.0334 | 20.3 | 606660 |
| NAD+ | 0.186 | 12.9 | 69400 |
| Ethyl 2-chloroacetoacetate | 144.6 | 143.5 | 990 |
Next, we determined both the optimum pH and temperature for the free Tt27-HBDH through both oxidation and reduction directions (Fig. 2). The optimal pH varies depending on direction of the redox reaction, as the reduction and oxidation maximum activities were found at pH 5 and 9, respectively (Fig. 2A). On the other hand, the optimal temperature for the reaction was 85 °C, with a specific activity of 55 U mg−1 under the assay conditions (Fig. 2B), showing up the thermophilic nature of the protein.
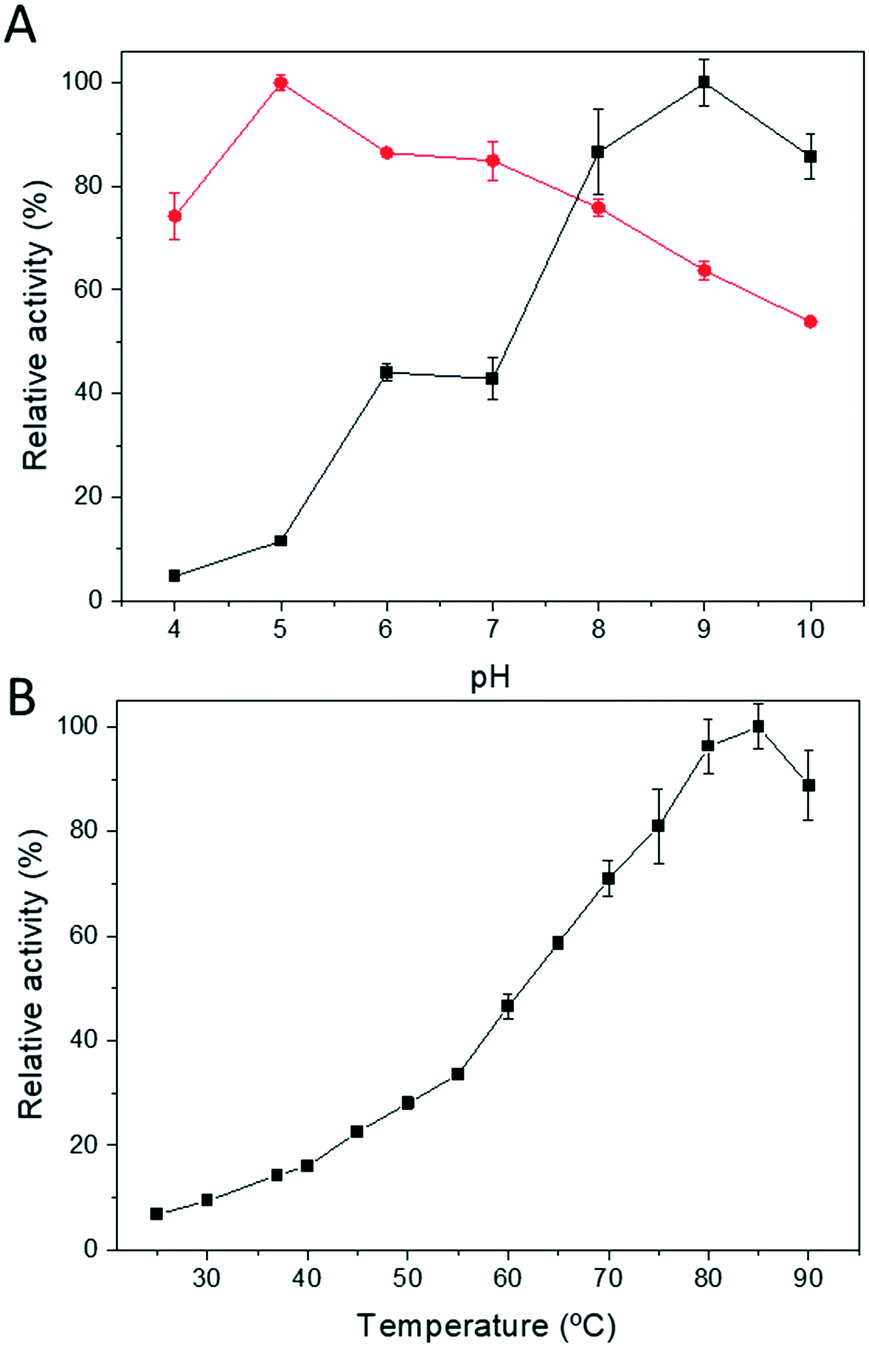 | ||
| Fig. 2 Influence of pH (A) and temperature (B) on the enzymatic activity of soluble Tt27-HBDH. The pH effect on reduction activity towards EAA using NADH (red circles) and oxidation activity towards ethyl 3-hydroxybutyrate using NAD+ (black squares). Temperature effect on the reductive enzymatic activity. For both experiments, the relative activity was calculated assigning 100% to the highest measured activity at one particular temperature and pH. | ||
3.4. Immobilization of Tt27-HBDH
Despite the intrinsic thermostability of Tt27-HBDH, we immobilised this enzyme on different carriers with the aim of re-using the biocatalyst and utilizing it under drastic working conditions (such as extreme pHs and in presence of solvents). We screened different immobilization strategies that include reversible and irreversible immobilization chemistries where the enzymes are differently oriented on the carrier surface. In a first approach, the enzyme was reversibly immobilised on porous agarose microbeads coated with different cationic polymers such as polyethyleneimine (PEI) of 25 kDa (AG-PEI) and polyallylamine (PAA) of 17 kDa (AG–PAA). These functionalised carriers immobilise the enzymes through ionic interactions established with the most acidic enzyme regions.39 In a second approach, Tt27-HBDH was immobilised on porous agarose beads activated with glyoxyl groups (short aliphatic aldehydes) (AG–G); one of the most successful immobilization chemistries reported up to now. The aldehyde groups promote an intense multipoint covalent attachment between the enzyme and the carrier forming reversible Schiff bases that need to be further reduced under mild conditions.37 This immobilization chemistry allows the orientation of the enzyme through the surface regions that expose the highest content of Lys residues.37,38 The number of bonds formed between the enzyme and the carrier was controlled through incubating the immobilization for different times; from 1 to 24 h as described elsewhere.54Table 3 shows the two major immobilization parameters; immobilization yield and recovered activity upon the immobilization, determined for all the heterogeneous biocatalyst herein presented. In all cases, immobilization yields were higher than 94%, but the recovered activities (immobilization effectiveness) varied 85–50% depending on the immobilization chemistry. The high recovered activity upon all immobilization protocols may rely on the thermophile origin of Tt27-HBDH. It seems that the higher intrinsic stability of the thermophile enzymes55,56 allows them to better resist the immobilization conditions as well as the formation of intense attachments with the carrier that may cause negative structural distortions.
| 1. Carrier | 2. Immobilization time (h) | 3. Immobilization yielda (%) | 4. Recovered activityb (%) | 5. Half-life (h)/stabilization factor (SF) | |
|---|---|---|---|---|---|
| 80 °C, pH 7c,d | 70 °C, pH 4.5c,d | ||||
| The preparations were done as described in Materials and methods section. a Immobilization yield (%) = 100 × (1 − (activity in the supernatant upon the immobilization/offered activity)). b Recovered activity (%) = (measured activity upon the immobilisation)/(offered activity × (immobilisation yield/100)) × 100. c Half-life was calculated according to the Materials and methods section. d Stabilization factor is the ratio between the biocatalyst half-life and the soluble Tt27-HBDH half-life (12 h at 80 °C and pH 7, and 0.5 h at 70 °C and pH 4.5, using a concentration of 20 μg mL−1). All the data are the mean values of three separate experiments where the error value was never higher than 5%. | |||||
| AG–G | 1 | 97.4 | 57.2 | 71 h (5.9) | 195 h (390) |
| 3 | 98 | 53.7 | 43 h (3.6) | 238 h (476) | |
| 6 | 99 | 51 | 56 h (4.7) | 149 h (298) | |
| 24 | 100 | 50 | 32 h (2.7) | 90 h (184) | |
| AG–PEI | 2 | 94 | 85 | 4.2 h (0.35) | 41 h (82) |
| AG–PAA | 2 | 97 | 85 | 12.6 h (1.05) | 11 h (22) |
3.5. Stability of immobilised Tt27-HBDH preparations
Next, we incubated the different immobilised preparations at 80 °C/pH 7 and at 70 °C/pH 4.5 to assess their stability (Table 3). Based on such thermal stability studies, we selected the Tt27-HBDH immobilised on AG–G for 3 h as the optimal heterogeneous biocatalyst for further studies. This heterogeneous biocatalyst was 476 times more stable than its soluble counterpart under high temperature and acidic pH, suggesting a rigidification of the enzyme tertiary structure as observed for other multimeric enzymes immobilized on AG–G.22,57,58 Unfortunately, this immobilization protocol failed to stabilize the hexameric structure of Tt27-HBDH as some subunits were still released after incubating the immobilised enzyme under denaturing conditions (Fig. S6†).When we inspected the enzyme 3D model and analysed its exposed surface area and electrostatics through the Bluues server (Fig. 3A),34 we found a cluster of Lys (K4, K52, K56, K58, K81) highly accessible ε-NH2 (>70 Å2) with local pKa below 11. These Lys fulfil the criteria to efficiently interact with the aldehydes at the surface of AG–G. Moreover, such Lys cluster surrounds the N-terminus (the most reactive amine group in the enzyme surface) forming two symmetrical clusters of reactive primary amines that are displayed in the same interaction plane and prone to react with the carrier (Fig. 3B). If the immobilization took place through that region, a maximum of three subunit would be irreversibly attached to the carrier, in accordance with the SDS-PAGE results (Fig. S6†). In the light of both experimental data and the structure-based information, we suggest that the above-mentioned Lys-rich region likely drives the immobilization of Tt27-HBDH on AG–G.
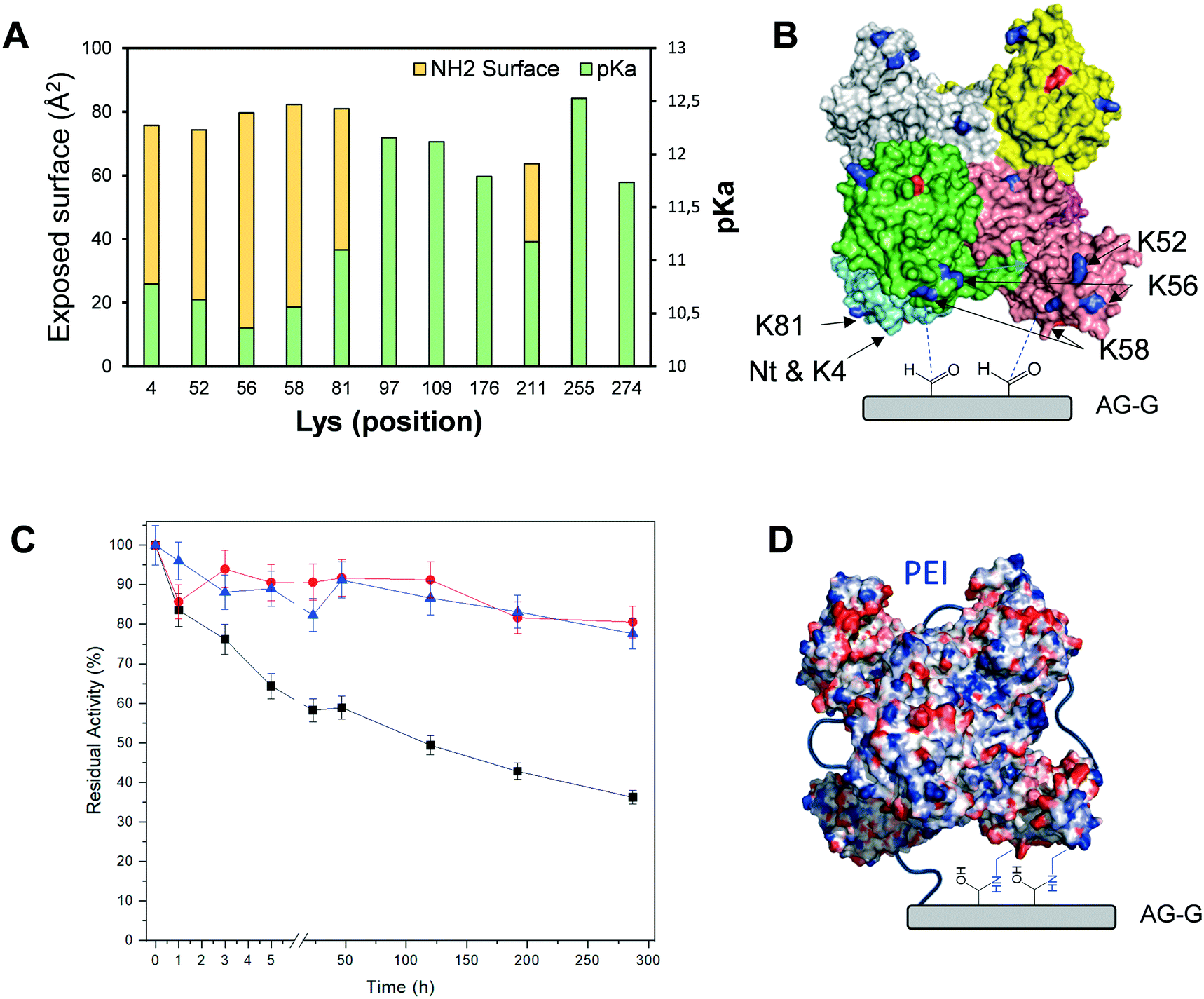 | ||
| Fig. 3 Exposed surface and pKa of all Lys residues from the primary sequence of Tt27-HBDH estimated with Bluues server (A). Surface of Tt27-HBDH hexamer 3D-model, highlighting the N-terminus (red) and the most exposed Lys (blue) with pKa below 11. The different colours represent the six subunits forming the enzyme quaternary structure according to the 3D-model (B). Inactivation courses of Tt27-HBDH immobilised on AG–G for 3 h (black squares) and further coated with PEI of 25 kDa (red circles) and 60 kDa (blue triangles). The inactivation conditions were 60 °C, pH 4,5 and 20% 2-propanol (C). Electrostatic surface of Tt27-HBDH hexamer 3D-model using Bluues server (D). All protein images were created with Pymol version 1.7.4.5. | ||
To avoid the subunit dissociation during the thermal inactivation and to ultimately increase the thermal stability, we coated the immobilised enzyme with PEI. Our aim was to establish an intramolecular ionic cross-liking between the acid residues of the two subunits and the positive charges of PEI, as well as intermolecular irreversible bonds between the primary amine groups of PEI and the aldehydes of the carrier. We tested two different sizes of PEI (25 and 60 kDa) to make different coated heterogeneous biocatalysts. As result, the post-immobilization polymeric coating, regardless of the PEI size, greatly stabilises the immobilised enzymes under highly denaturing conditions (60 °C, pH 4.5 and 20% of 2-propanol) at the expense of reducing the expressed activity of the biocatalyst by 40%. These inactivation conditions were extremely severe for the free enzyme since it was fully inactivated after only 15 min. Under these denaturing conditions, the coated heterogeneous biocatalysts retained more than 80% of their initial activity after 300 h of incubation, unlike the non-coated enzyme immobilised on AG–G for 3 h, which lost 65% of its activity after the same incubation time (Fig. 3C). According to the excellent stabilization achieved by the PEI cross-linking upon the enzyme immobilization, we suggest that medium-size cationic polymers are able to cross-link the six subunits of Tt27-HBDH through the negative charges displayed in the enzyme surface (Fig. 3D), consequently stabilizing its quaternary structure. Moreover, the hydrophilic microenvironment created by the PEI coating may prevent the enzyme inactivation under the presence of high co-solvent concentration such as 2-propanol. The rather fast inactivation of the soluble enzyme made impossible to accurately assess to what extent both the immobilization and the subsequent post-immobilization coating contribute to the enzyme stabilization. However, assuming that the half-life time of the immobilised and coated enzyme is more than 300 h and the one of the free enzyme is less than 15 min under 60 °C, pH 4.5 and 20% 2-propanol, we may approximate that the stabilization factor should fall in the range of the thousands.
We selected 60 °C, pH 4.5 and 20% of 2-propanol as inactivation conditions because they are highly relevant for the application of Tt27-HBDH in the asymmetric reductions of β-ketoester and their chlorinated derivatives. Low pHs are desired to prevent the spontaneous decomposition of substrate at pH above 7,18,59 high temperatures increase the enzyme activity and high concentration of 2-propanol can play a dual role as solvent to increase the substrate solubility, and as co-substrate of Tt27-HBDH to self-recycle the redox cofactor during the asymmetric reduction. Having an immobilised biocatalyst with such outstanding stability under these conditions, we decided to further use it for the asymmetric reduction of β-ketoesters.
3.6. Application of Tt27-HBDH biocatalyst in asymmetric reduction
We exploited the Tt27-HBDH immobilised on AG-G and further coated with PEI for the asymmetric reduction of EAA as model β-ketoester. To assure the in situ recycling of NADH, we evaluated two different regeneration systems (Table 4); 1) a self-recycling approach exploiting the side oxidative activity of Tt27-HBDH towards 2-propanol and 2) the orthogonal cofactor recycling using formate dehydrogenase (FDH) and formic acid as partner enzyme and electron donor, respectively. Table 4 shows the conversion of EAA into ethyl 3-hydroxybutyrate using different soluble and immobilised biocatalysts. Tt27-HBDH immobilised on AG–G and coated with PEI (AG–G@Tt27-HBDH(PEI)) yielded 98% substrate conversion using 2-propanol as electron donor in 24 h. However, when FDH was ionically absorbed and further covalently cross-linked on the PEI layer over the immobilised Tt27-HBDH (AG–G@Tt27-HBDH(PEI)/FDH) using diglycidyl ether as cross-linking reagent, the heterogeneous biocatalyst was only able to reach the 77% yield under the same reaction conditions (Table 4). The conversion of EAA into ethyl 3-hydroxybutyrate was confirmed by GC and NMR (Fig. S7 and S8†). In the light of these results, the NADH regeneration seems to be more efficient when AG–G@Tt27-HBDH(PEI) self-recycles the cofactor than when using the orthogonal system with the two co-immobilised enzymes. On the contrary, the soluble enzymes reached 100% yields regardless of the recycling system. The low performance of the Tt27-HBDH/FDH co-immobilised system is likely due to the low activity recovered by FDH upon the immobilization; its specific activity was reduced to 14% in the co-immobilised preparation (Table S1†). Both soluble and immobilised enzymes yielded the S-isomer of ethyl 3-hydroxybutyrate with an ee (%) > 99% according to chiral GC analysis (Fig. S9†), demonstrating that Tt27-HBDH enantioselectivity was preserved upon the immobilization protocol.|
|
|||
|---|---|---|---|
| Biocatalyst | Cofactor | Conversion (%) | ee (S) (%) |
| Conversions were determined after a 24 h reaction at 25 °C. (1) Monoenzymatic system is composed of immobilised Tt27-HBDH on glyoxyl-agarose beads treated with polyethyleneimine. (2) Bienzymatic system is composed of co-immobilised CbFDH on the monoenzymatic system. All reaction mixtures consisted in 10 mM EAA 5% (v/v) 2-propanol in Tris-HCl buffer 10 mM pH 8 at 25 °C and those ones for soluble enzyme and NADH-soluble also contains NADH 1 mM. The conversion was calculated after 24 h of reaction. e.e = ((R – S)/(R + S)) × 100. All the data are the mean values of three separate experiments where the error value was never higher than 5%. | |||
| Tt27-HBDH | Soluble | 100 ± 0.0 | 99 |
| Tt27-HBDH + FDH | Soluble | 100 ± 0.0 | 99 |
| AG–G@Tt27-HBDH (PEI) | Soluble | 97.6 ± 0.3 | 99 |
| AG–G@Tt27-HBDH (PEI)/FDH | Soluble | 76.9 ± 6.7 | 99 |
| AG–G@Tt27-HBDH (PEI-NADH) | Immobilised | 99.3 ± 0.1 | 99 |
AG–G@Tt27-HBDH(PEI) was also tested for the asymmetric reduction of ethyl 4-chloroacetoacetate, giving rise to 6.7% yield although the substrate conversion reached 44%. The unexpected difference between the production of the 3-hydroacyl derivative and the conversion of the β-ketoester may rely on the lability of the chlorinated substrates under aqueous reaction conditions. Unlike EAA, the chromatographic areas corresponding to its chlorinated counterpart dramatically decayed in the absence of any enzyme (Fig. S10†), supporting the fact that the conversion observed for this substrate is the result of the spontaneous chemical hydrolysis of ethyl 4-chloroacetoacetate and its subsequent decarboxylation, as previously described.60,61 Despite the spontaneous degradation of the substrate, we assessed the enantiopurity of the formed products through chiral GC, obtaining the R-isomer with an ee > 99% (Fig. S11†). The opposite CIP of the chlorinated derivatives explains that Tt27-HBDH yields the R-isomer instead the S-isomer obtained for the ethyl 3-hydroxybutyrate.
3.7. Fabrication and kinetic characterization of self-sufficient heterogeneous biocatalysts of Tt27-HBDH
In the last years, our groups have been able to co-immobilise different alcohol dehydrogenases and ketoreductases with their corresponding redox cofactors.26,27,62 As result, we obtain self-sufficient heterogeneous biocatalysts, which require no exogenous supply of NAD(P)H. Harnessing the PEI layer needed to stabilise the quaternary structure of Tt27-HBDH, we co-immobilised the NADH with the enzyme through ionic interactions (AG–G@Tt27-HBDH(PEI-NADH)). The cofactor was effectively bound to the agarose microbeads in less than 15 min (Fig. S12†). We managed to immobilise 5.6 μmol of NADH per gram of carrier containing 11.04 mg of enzyme after two washes that removed all the cofactors molecules weakly bound to the carrier (Table S2†). This load is similar to the ones obtained for the same architecture we previously reported, where a different alcohol dehydrogenase from T. thermophilus HB27 and a formate dehydrogenase from Candida boidinii and NAD+ were co-immobilised.26 As we reported elsewhere,26,27 the cofactor establishes an association/dissociation equilibrium between its negatively charged phosphate groups and the positive amine groups of the PEI layer. To demonstrate that immobilised Tt27-HBDH effectively utilises the co-immobilised NADH, we performed single-particle studies monitoring the autofluorescence of the NADH through time-lapse fluorescence microscopy (Fig. 4 and S12†). The relative fluorescence intensity of NADH rapidly decayed when EAA was dispensed to the reaction well. After 20 min, the fluorescence intensity within the beads was less than 10% of the initial signal, indicating that all molecules of NADH were oxidised to NAD+ as consequence of the asymmetric reduction of the β-ketoesters (Fig. 4A and B). Control experiments demonstrate that NADH remains inside agarose microbeads for up to 50 min, demonstrating that the cofactor molecules diffuse inside the porous beads to reach the enzyme activity sites but do not abandon them (Fig. S12A†). Even the addition of 2-propanol caused negligible loss of the relative fluorescence within the beads (Fig. S12B†). Therefore, the immobilised NADH was only consumed by the co-immobilised Tt27-HBDH when EAA is added to the solution.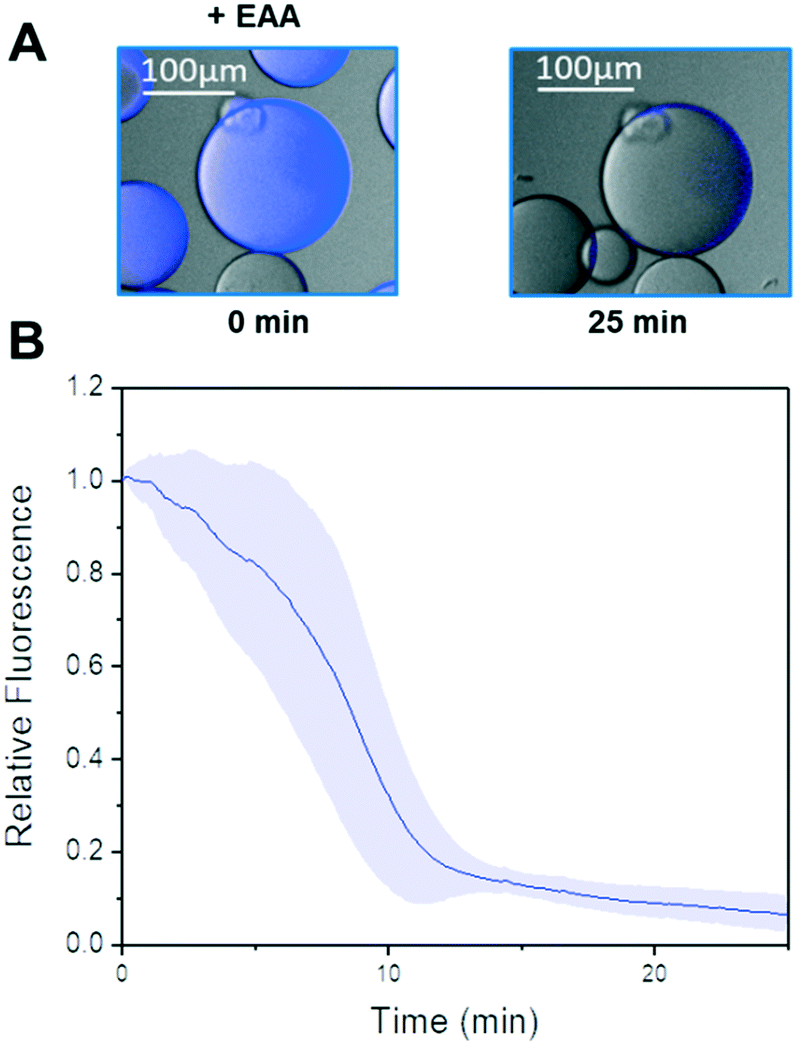 | ||
| Fig. 4 In operando kinetics analysis based on single particle experiments. Overlay of bright-field and fluorescence images from agarose microbeads co-immobilizing Tt27-HBDH and NADH incubated with 10 mM EAA and 5% 2-propanol (v/v) in 10 mM Tris-HCl pH 8.0 buffer (A). Time courses for the consumption of NADH catalysed by different beads (n = 5) of AG–G@Tt27-HBDH(PEI) in presence of EAA (B). The solid line and the shadow represent the mean value and the standard deviation of the relative fluorescence at each time point, considering the value of 1 to the relative fluorescence units of each bead at time 0. | ||
3.8. Re-use and operational stability of the self-sufficient heterogeneous biocatalyst in the asymmetric reduction of ethyl acetoacetate
Once demonstrated the functionality of the immobilised Tt27-HBDH towards the NADH absorbed to the PEI layer of the heterogeneous biocatalyst, we performed the asymmetric reduction of EAA without supplying exogenous cofactor in batch. Fig. 5A shows that the co-immobilization of the enzyme and the cofactor increases both the reaction rate and the product yield after 24 h, when compared to the immobilised enzyme using exogenous cofactor. Expectedly, using both soluble enzyme and cofactor, we observed a higher synthetic rate likely due to the 70% activity reduction suffered upon the enzyme immobilization and the PEI coating (Fig. 5A). However, the soluble system is hard to reuse after one cycle; on the contrary, the heterogeneous biocatalysts herein utilised are easily separated from the reaction products through vacuum filtration and reused for consecutive discontinuous reaction cycles. Immobilised Tt27-HBDH using both co-immobilised and soluble cofactor were re-used for up to 10 cycles maintaining more than 60% product conversion after each 24 h cycle (Fig. 5B). Remarkably, the co-immobilization of both enzyme and cofactor on the same carrier enabled to reach a TTN of 145 and 5223 for the NADH and Tt27-HBDH after the 10th reaction cycle, respectively. The same immobilised enzyme but using the soluble NADH reached similar enzyme TTN but limited the cofactor TTN to a maximum of 10, since the turnovers of NADH could not be accumulated cycle after cycle. The discontinuous use of the self-sufficient heterogeneous biocatalyst was able to produce 25.2 mg of ethyl (S)-3-hydroxybutyrate per mg of NADH with a productivity that ranges 0.066–0.027 g × L−1 × h−1 from cycle 1 to cycle 10.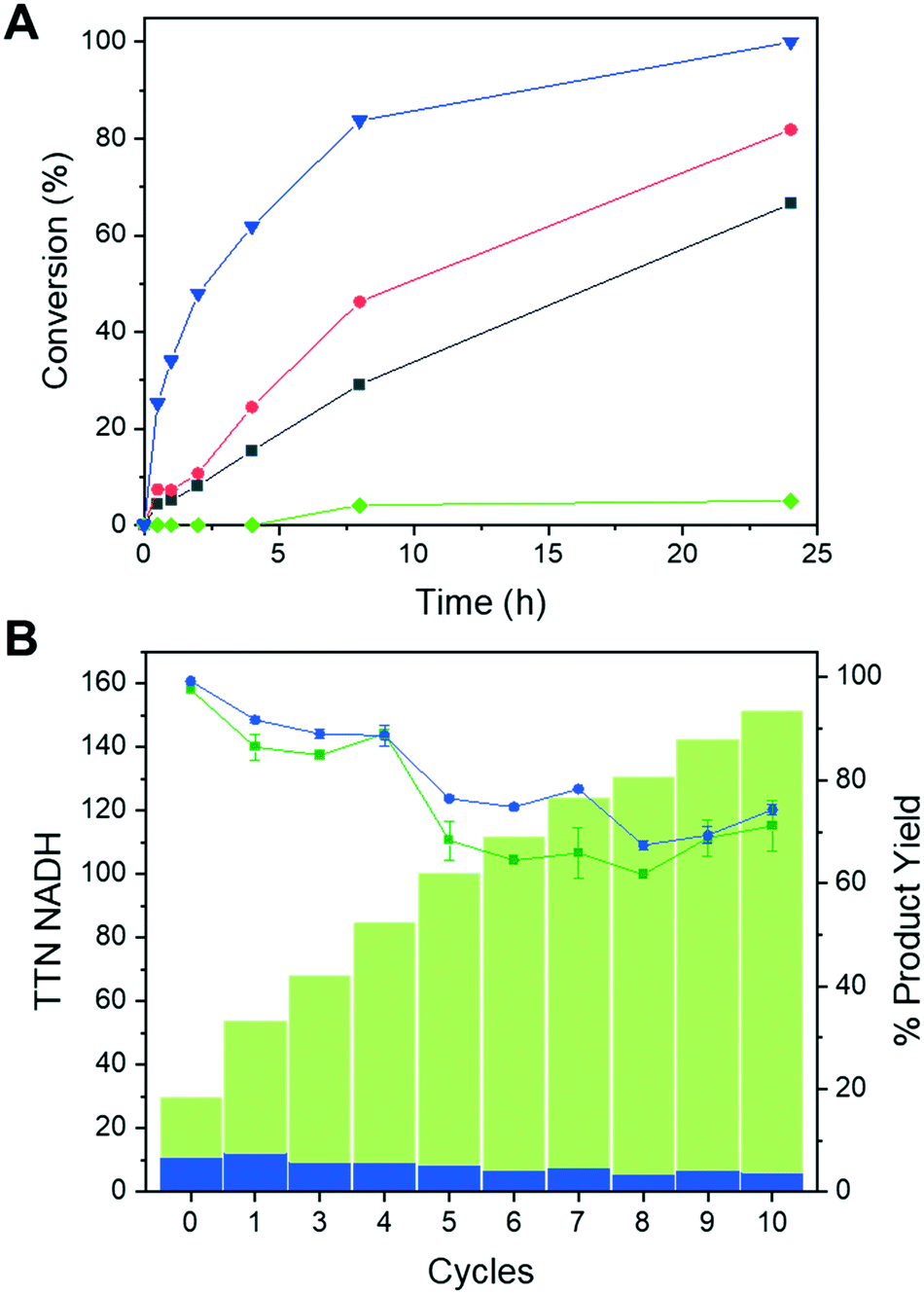 | ||
| Fig. 5 Reaction course and TTN of ethyl acetoacetate asymmetric reduction by AG–G@Tt27-HBDH(PEI) with soluble or co-immobilised NADH. (A) 10% load of Tt27-HBDH immobilised biocatalyst with (red circles) or without (grey squares) co-immobilised NADH as well as soluble Tt27-HBDH (1.1 mg mL−1) (blue triangles) were incubated with 10 mM EAA, 5% 2-propanol (0.66 M), 1 mM NADH in 10 mM Tris-HCl at pH 8.0, for 24 h at 25 °C with orbital agitation and a total reaction volume of 5 mL. AG–G carrier without enzyme (green diamonds) incubated under the same conditions was used as control. (B) Product yield and total turnover number during 10 cycles of 24 h batch reactions at 25 °C. Chromatographic yield (%) of the asymmetric reduction of EAA catalysed by AG–G@Tt27-HBDH (PEI) with (green squares) or without (dark blue circles) co-immobilised NADH in consecutive batch reactions. Reactions were carried out as in panel A. Green bars represent the cofactor accumulated TTN after each consecutive batch reaction cycle. Blue bars represent the single-reaction cofactor TTN using soluble cofactor. | ||
To further intensify the process, we increased the protein load in AG–G@Tt27-HBDH (PEI) up to 32.3 mg per gram with a similar load of NADH (4.84 μmolNADH × g−1carrier). At 200 mM EAA, this self-sufficient heterogeneous biocatalyst converted 89% of the substrate into the corresponding β-hydroxyester after 24 h of reaction at 25 °C (Fig. S13A†). When the temperature was raised up to 60 °C, the conversion ramped up to 99% after only 5 h (Fig. S13A†) as expected from the optimum temperature of this enzyme. Finally, after 5 cycles of 4 h reaction at 60 °C, the conversion remained higher than 90% (Fig. S13B†), obtaining a remarkable accumulated cofactor TTN of 1040, an enzyme accumulated TTN of 5503 producing a total of 123.64 mg of ethyl (S)-3-hydroxybutyrate.
Conclusions
We mined the genome of Thermus thermophilus HB27 to clone and heterologously express the gene ttc0898 that encodes a 3-hydroxybutyryl-CoA dehydrogenase (EC:1.1.1.157), named as Tt27-HBDH. This novel enzyme was purified and biochemically characterised to prove its thermostability, its substrate specificity towards β-ketoesters and its dependence on NADH. Remarkably, the enzyme was able to oxidise 2-propanol; a side-reaction that combined with the asymmetric reduction of β-ketoesters enables the enzyme cofactor self-recycling. We further optimised the immobilization of Tt27-HBDH through screening a set of immobilization chemistries and conditions. As result, we fabricate a self-sufficient heterogeneous biocatalyst where the enzyme is irreversibly attached to agarose-based macroporous beads functionalised with aldehyde groups and further coated with a PEI layer that allows the reversible adsorption of NADH through ionic interactions. In this heterogeneous biocatalyst, both enzyme and cofactor are confined within the same porous space but the reversible binding of NADH lets it travel between the active sites of the irreversibly bound enzymes. The co-immobilization of both Tt27-HBDH and its cofactor generates a self-sufficient heterogeneous biocatalyst highly stable under high temperatures and acid pH that efficiently performs the asymmetric reduction of EAA to yield enantiomerically pure (S)-ethyl 3-hydroxybutyrate without exogenous supply of NADH and using 2-propanol as ultimate hydride donor. Finally, we also proved the high operational stability of this self-sufficient immobilised enzyme in a discontinuous batch process where the solid biocatalyst was filtered after each operation cycle and re-used for the next one. This work contributes to expand the increasing number of examples where cofactor is part of the biocatalyst, extending the operational use of both. We envision this biocatalyst architecture as a sustainable, efficient and selective solution for the manufacturing pharmaceutical and polymer building blocks.Author contribution
DAS and AHO did the experimental work related to the immobilization and the biotransformation and participated in the discussion of the results. MSC and JB performed and supervised the cloning and expression of the protein. JRM, JMG and FLG conceptualize the work and analyse the results. JRM and FLG wrote the manuscript and all authors participated in the editing and revision processes. Finally, FLG, JMG and JB raised the funds to carry out this investigation.Conflicts of interest
There are no conflicts of interests.Acknowledgements
FLG, JRM and JMG thank the funding of European Union (EU) project HOTZYMES (grant agreement no. 829162) under H2020-FETOPEN program. FLG and DAS thank the received funding from the European Research Council (ERC-Co-2018 #818089). Funding from IKERBASQUE to FLG is also acknowledged. This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720. JB thanks the Institutional Grant from the Foundation Ramón Areces. We also acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- Q. Ye, P. Ouyang and H. Ying, Appl. Microbiol. Biotechnol., 2011, 89, 513–522 CrossRef CAS PubMed.
- Y. H. Cui, P. Wei, F. Peng, M. H. Zong and W. Y. Lou, RSC Adv., 2018, 8, 9970–9978 RSC.
- G. W. Zheng and J. H. Xu, Curr. Opin. Biotechnol., 2011, 22, 784–792 CrossRef CAS PubMed.
- R. Wohlgemuth, Curr. Opin. Microbiol., 2010, 13, 283–292 CrossRef CAS PubMed.
- S. M. Thomas, R. DiCosimo and V. Nagarajan, Trends Biotechnol., 2002, 20, 238–242 CrossRef CAS PubMed.
- D. Gamenara and P. Domínguez de María, Biotechnol. Adv., 2009, 27, 278–285 CrossRef CAS PubMed.
- J. Zhang, Q. Cao, S. Li, X. Lu, Y. Zhao, J.-S. Guan, J.-C. Chen, Q. Wu and G.-Q. Chen, Biomaterials, 2013, 34, 7552–7562 CrossRef CAS PubMed.
- X.-H. Zou, H.-M. Li, S. Wang, M. Leski, Y.-C. Yao, X.-D. Yang, Q.-J. Huang and G.-Q. Chen, Biomaterials, 2009, 30, 1532–1541 CrossRef CAS PubMed.
- X.-Q. Xiao, Y. Zhao and G.-Q. Chen, Biomaterials, 2007, 28, 3608–3616 CrossRef CAS PubMed.
- J.-Y. He, L.-M. Zhou, P. Wang and L. Zu, Process Biochem., 2009, 44, 316–321 CrossRef CAS.
- B. Wipf, E. Kupfer, R. Bertazzi and H. G. W. Leuenberger, Helv. Chim. Acta, 1983, 66, 485–488 CrossRef CAS.
- K. Mori, Tetrahedron, 1989, 45, 3233–3298 CrossRef CAS.
- N. Oguni and Y. Ohkawa, J. Chem. Soc., Chem. Commun., 1988, 1376–1377 RSC.
- M. Ošlaj, J. Cluzeau, D. Orkić, G. Kopitar, P. Mrak and Z. Časar, PLoS One, 2013, 8, e62250 CrossRef PubMed.
- R. N. Patel, Biomolecules, 2013, 3, 741–777 CrossRef PubMed.
- Z.-Q. Liu, J.-J. Ye, Z.-Y. Shen, H.-B. Hong, J.-B. Yan, Y. Lin, Z.-X. Chen, Y.-G. Zheng and Y.-C. Shen, Appl. Microbiol. Biotechnol., 2015, 99, 2119–2129 CrossRef CAS PubMed.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC.
- S. Shimizu, M. Kataoka, M. Katoh, T. Morikawa, T. Miyoshi and H. Yamada, Appl. Environ. Microbiol., 1990, 56, 2374–2377 CrossRef CAS PubMed.
- M. H. Sung, K. Tanizawa, H. Tanaka, S. Kuramitsu, H. Kagamiyama and K. Soda, J. Bacteriol., 1990, 172, 1345–1351 CrossRef CAS PubMed.
- S. Mathew, K. Deepankumar, G. Shin, E. Y. Hong, B.-G. Kim, T. Chung and H. Yun, RSC Adv., 2016, 6, 69257–69260 RSC.
- A. Pennacchio, B. Pucci, F. Secundo, F. la Cara, M. Rossi and C. A. Raia, Appl. Environ. Microbiol., 2008, 74, 3949–3958 CrossRef CAS PubMed.
- J. Rocha-Martín, D. Vega, J. M. Bolivar, A. Hidalgo, J. Berenguer, J. M. Guisán and F. López-Gallego, Bioresour. Technol., 2012, 103, 343–350 CrossRef PubMed.
- E. E. Ferrandi, A. Previdi, I. Bassanini, S. Riva, X. Peng and D. Monti, Appl. Microbiol. Biotechnol., 2017, 101, 4963–4979 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- R. N. Patel, Bioorg. Med. Chem., 2018, 26, 1252–1274 CrossRef CAS PubMed.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, E. San Sebastian, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. López-Gallego, Chem. – Eur. J., 2017, 23, 16843–16852 CrossRef PubMed.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- L. Trobo-Maseda, A. H. Orrego, J. M. Guisan and J. Rocha-Martin, Int. J. Biol. Macromol., 2020, 157, 510–521 CrossRef CAS PubMed.
- E. J. Kim, J. Kim, J. W. Ahn, Y. J. Kim, J. H. Chang and K. J. Kim, J. Microbiol. Biotechnol., 2014, 24, 1636–1643 CrossRef CAS PubMed.
- M. Takenoya, S. Taguchi and S. Yajima, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2018, 74, 733–740 CrossRef CAS PubMed.
- G. W. Haywood, A. J. Anderson, L. Chu and E. A. Dawes, FEMS Microbiol. Lett., 1988, 52, 259–264 CrossRef CAS.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. G. Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- I. Walsh, G. Minervini, A. Corazza, G. Esposito, S. C. E. Tosatto and F. Fogolari, Bioinformatics, 2012, 28, 2189–2190 CrossRef CAS PubMed.
- E. S. da Silva, V. Gomez-Vallejo, Z. Baz, J. Llop and F. Lopez-Gallego, Chem. – Eur. J., 2016, 22, 13619–13626 CrossRef CAS PubMed.
- W. W. Cleland, Adv. Enzymol. Relat. Areas Mol. Biol., 1977, 45, 273–387 CAS.
- J. M. Guisán, Enzyme Microb. Technol., 1988, 10, 375–382 CrossRef.
- G. Fernández-Lorente, F. Lopez-Gallego, J. M. Bolivar, J. Rocha-Martin, S. Moreno-Perez and J. M. Guisán, Curr. Org. Chem., 2015, 19, 1–13 CrossRef.
- R. Torres, C. Mateo, M. Fuentes, J. M. Palomo, C. Ortiz, R. Fernández-Lafuente, J. M. Guisan, A. Tam and M. Daminati, Biotechnol. Prog., 2002, 18, 1221–1226 CrossRef CAS PubMed.
- O. Romero, J. M. Guisán, A. Illanes and L. Wilson, J. Mol. Catal. B: Enzym., 2012, 74, 224–229 CrossRef CAS.
- E.-J. Kim and K.-J. Kim, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2014, 70, 485–488 CrossRef CAS PubMed.
- J. Kim, J. H. Chang and K. J. Kim, Biochem. Biophys. Res. Commun., 2014, 448, 163–168 CrossRef CAS.
- J. J. Barycki, L. K. O'Brien, J. M. Bratt, R. Zhang, R. Sanishvili, A. W. Strauss and L. J. Banaszak, Biochemistry, 1999, 38, 5786–5798 CrossRef CAS PubMed.
- J. Rocha-Martin, D. E. Vega, Z. Cabrera, J. M. Bolivar, R. Fernandez-Lafuente, J. Berenguer and J. M. Guisan, Process Biochem., 2009, 44, 1004–1012 CrossRef CAS.
- Y. Ni, C.-X. Li, H.-M. Ma, J. Zhang and J.-H. Xu, Appl. Microbiol. Biotechnol., 2011, 89, 1111–1118 CrossRef CAS PubMed.
- G.-W. Zheng, Y.-Y. Liu, Q. Chen, L. Huang, H.-L. Yu, W.-Y. Lou, C.-X. Li, Y.-P. Bai, A.-T. Li and J.-H. Xu, ACS Catal., 2017, 7, 7174–7181 CrossRef CAS.
- J. Kim, J. H. Chang, E. J. Kim and K. J. Kim, Biochem. Biophys. Res. Commun., 2014, 443, 783–788 CrossRef CAS PubMed.
- A. Weckbecker and W. Hummel, Biocatal. Biotransform., 2006, 24, 380–389 CrossRef CAS.
- N. Richter and W. Hummel, Enzyme Microb. Technol., 2011, 48, 472–479 CrossRef CAS PubMed.
- Z. Yang, H. Fu, W. Ye, Y. Xie, Q. Liu, H. Wang and D. Wei, Catal. Sci. Technol., 2020, 10, 70–78 RSC.
- K. Inoue, Y. Makino and N. Itoh, Appl. Environ. Microbiol., 2005, 71, 3633–3641 CrossRef CAS PubMed.
- M. Amidjojo and D. Weuster-Botz, Tetrahedron: Asymmetry, 2005, 16, 899–901 CrossRef CAS.
- R. C. Taylor, A. K. Brown, A. Singh, A. Bhatt and G. S. Besra, Microbiology, 2010, 156, 1975–1982 CrossRef CAS PubMed.
- J. Pedroche, M. del Mar Yust, C. Mateo, R. Fernández-Lafuente, J. Girón-Calle, M. Alaiz, J. Vioque, J. M. Guisán and F. Millán, Enzyme Microb. Technol., 2007, 40, 1160–1166 CrossRef CAS.
- A. Barzegar, A. A. Moosavi-Movahedi, J. Z. Pedersen and M. Miroliaei, Enzyme Microb. Technol., 2009, 45, 73–79 CrossRef CAS.
- V. Carbone, A. Hara and O. El-Kabbani, Cell. Mol. Life Sci., 2008, 65, 1464–1474 CrossRef CAS PubMed.
- A. H. Orrego, L. Trobo-Maseda, J. Rocha-Martin and J. M. Guisan, Enzyme Microb. Technol., 2017, 105, 51–58 CrossRef CAS PubMed.
- A. H. Orrego, F. López-Gallego, A. Espaillat, F. Cava, J. M. Guisan and J. Rocha-Martin, ChemCatChem, 2018, 10, 3002–3011 CrossRef CAS.
- J. Pan, G.-W. Zheng, Q. Ye and J.-H. Xu, Org. Process Res. Dev., 2014, 18, 739–743 CrossRef CAS.
- I. Chin-Joe, P. M. Nelisse, A. J. J. Straathof, J. A. Jongejan, J. T. Pronk and J. J. Heijnen, Biotechnol. Bioeng., 2000, 69, 370–376 CrossRef CAS PubMed.
- J.-Y. Houng and J.-S. Liau, Biotechnol. Lett., 2003, 25, 17–21 CrossRef CAS PubMed.
- E. Muñoz-Morales, S. Velasco-Lozano, A. I. Benítez-Mateos, M. J. Marín, P. Ramos-Cabrer and F. López-Gallego, Catalysts, 2019, 9, 896 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00268f |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |