
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterodinuclear catalysts Zn(II)/M and Mg(II)/M, where M = Na(I), Ca(II) or Cd(II), for phthalic anhydride/cyclohexene oxide ring opening copolymerisation†
Arron C.
Deacy
,
Christopher B.
Durr
,
Ryan W. F.
Kerr
 and
Charlotte K.
Williams
*
and
Charlotte K.
Williams
*
Chemistry Research Laboratory, Department of Chemistry, 12 Mansfield Rd, Oxford OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
First published on 15th March 2021
Abstract
Anhydride and epoxide ring opening copolymerisation is a useful and controlled route to aliphatic, semi-aromatic and functionalised polyesters. Building upon our prior investigations of synergic Zn(II)/Mg(II) and Mg(II)/Co(II) polymerisation catalysts, here, a series of heterodinuclear catalysts for phthalic anhydride/cyclohexene oxide ROCOP are reported. The complexes feature either Zn(II)/M or Mg(II)/M combinations, where M = Na(I), Ca(II) or Cd(II), and are coordinated by an ‘open’ dinucleating ortho-vanillin derived Schiff-base ligand. The ligand features two binding ‘pockets’: 1) a coordination site comprising diphenolate and dimine moieties and 2) a site featuring diphenolate and diether groups. Heterodinuclear complex syntheses are high yielding due in part to the different coordination chemistries of the binding ‘pockets’. All new complexes are fully characterised including by multi-nuclear NMR spectroscopy and X-ray diffraction experiments; several solid state structures are Lewis base adducts with the crystallisation solvents (THF or acetonitrile) and provide insights into the structures of catalytic cycle intermediates. The catalysts show good performances and quantitative formation of ester linkages. They show equivalent activity to the widely investigated [(salen)Cr(III)Cl]/PPNCl systems (TOF = 67 h−1, 0.1 mol%, 100 °C, 17.6 kg mol−1) but lower activity than previously reported Zn(II)/Mg(II) catalysts or than tethered metal-salen catalysts. All the complexes are less active than the Zn(II)/Zn(II) homodinuclear complex proving that catalytic synergy is not provided only by heterodinuclear complexes with different functions; these findings underscore the need for continued exploration of the factors underpinning synergic heterometallic combinations.
Introduction
Polyesters are produced on a >60 Mt per year scale annually and used in sectors including packaging, clothing, household goods, electronics and construction.1–4 Sector growth is driven, in part, by sustainability concerns associated with hydrocarbon polymers. The ester linkage is more easily cleaved than C–C bonds, facilitating mechanical or chemical recycling and, in some cases, allowing for complete biodegradation.1–3,5–7 Today most polyesters are prepared by step-growth methods, but these reactions suffer from low rates and poor control and require high energy input both for reaction conditions (high temperature/gas flows) and rigorous monomer purifications.1,3,8 Cyclic ester ring opening polymerisation (ROP) is fast and well-controlled for aliphatic polyesters; it's used in the production of 1–2 Mt per annum of polylactide from sugar-derived lactide.2 Cyclic ester ROP is driven by relief of ring strain and is unsuitable for substituted lactones, most gamma-butyrolactones and larger cyclic esters which show low thermodynamic polymerisability and low thermal stability.9,10 Using it to make rigid, semi-aromatic or functionalised polyesters requires esoteric monomer syntheses, many showing low yields and multiple steps, and reactions requiring optimisation to overcome ring strain on a case-by-case basis.9,11–14The ring-opening copolymerisation (ROCOP) of cyclic anhydrides and epoxides is a useful alternative to lactone ROP.5,15 It is well controlled, applies monomers (epoxides/anhydrides) already made and used at scale by the polymer industry and produces high yields of functionalised, rigid and/or semi-aromatic polyesters.2 Today, these monomers are petrochemicals but viable bio-based routes have been proposed using corn stover to produce PA and triglyceride by products to make CHO.16–18 The alternating semi-aromatic polyester produced shows a high glass transition temperature (Tg = 133–146 °C) and was recently exploited in high elasticity ABA-triblock polyester thermoplastic elastomers (A: poly(PA-alt-CHO), B: poly(ε-decalactone)).19–23 The elastomers showed excellent recoverability, resilience and wider operating temperature ranges than analogous styrenic thermoplastic elastomers (SBS or SIS), with the added benefit of being fully acid degradable after use.23
The ROCOP catalyst is important since it controls rate, selectivity for ester linkages and polyester molar mass. Most catalysts are tested using phthalic anhydride (PA)/cyclohexene oxide (CHO) ROCOP and hence this is a desirable benchmark reaction for new catalyst development (Fig. 1).5,15,24–28 Amongst the leading PA/CHO ROCOP catalysts are bicomponent metal catalyst + ionic co-catalyst systems (PPNCl), commonly featuring Al(III), Co(III) or Cr(III) coordinated by salen, salophen, triphenolate or porphyrin ligands.25,29–32 For example, one of the more active systems is Cr(III)-salen/PPNCl, which showed a turn-over-frequency (TOF) of approx. 60 h−1 (kobs = 12.0 × 10−5 s−1, 100 °C, [Cr]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PPNCl]0:[PA]0:[CHO]0 = 1:1:200:200. [Cr]0 = 4.05 mM, toluene) (Fig. S1I†).24 Several adaptations have been reported to improve activity, notably by the groups of Lee and Coates, in which the nucleophilic co-catalyst is appended to the phenol or diamine backbone moieties, respectively (Fig. S1II and III†). For example, Lee and co-workers reported a modified Co(III)-salen catalyst showing a TOF of 1900 h−1 ([Co]0:[PA]0:[PO]0 = 1:7500:100000, 80 °C) and Coates and co-workers reported a modified Al(III)-salen catalyst showing a TOF of >100 h−1 ([Al]0:[PA]0:[PO]0 = 1:100:800, 60 °C).33,34 Nonetheless such tethered co-catalyst systems require lengthy and sometimes difficult syntheses.
:[PPNCl]0:[PA]0:[CHO]0 = 1:1:200:200. [Cr]0 = 4.05 mM, toluene) (Fig. S1I†).24 Several adaptations have been reported to improve activity, notably by the groups of Lee and Coates, in which the nucleophilic co-catalyst is appended to the phenol or diamine backbone moieties, respectively (Fig. S1II and III†). For example, Lee and co-workers reported a modified Co(III)-salen catalyst showing a TOF of 1900 h−1 ([Co]0:[PA]0:[PO]0 = 1:7500:100000, 80 °C) and Coates and co-workers reported a modified Al(III)-salen catalyst showing a TOF of >100 h−1 ([Al]0:[PA]0:[PO]0 = 1:100:800, 60 °C).33,34 Nonetheless such tethered co-catalyst systems require lengthy and sometimes difficult syntheses.
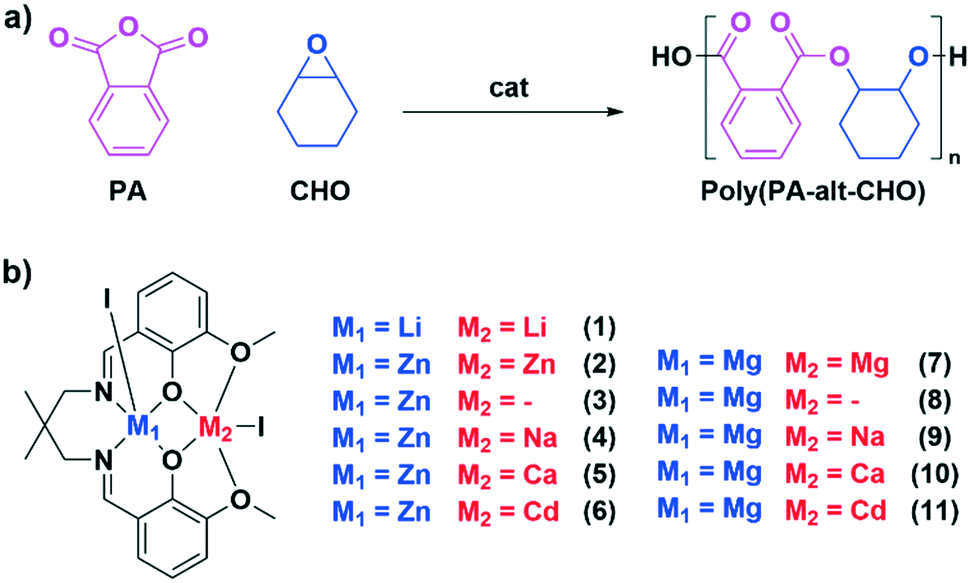 | ||
| Fig. 1 a) Illustrates the ring opening copolymerisation of phthalic anhydride (PA) with cyclohexene oxide to form poly(PA-alt-CHO). b) Displays the catalysts 1–11 targeted in this investigation. | ||
An alternative strategy is to use dinuclear catalysts which operate without co-catalysts and are known to retain activity in ROCOP catalysis at low catalyst loading.17,27 Furthermore, metals from groups 1, 2 and 12 are attractive targets due to their lack of colour, low toxicity and low cost.35–38 Dinuclear catalysts based on both Zn(II) and Mg(II) are well known in the ROCOP of CO2/epoxide,39–41 but are much less explored for the ROCOP of anhydride/epoxide (Fig. S1IV and V†).17 In 2014, we reported di-zinc and di-magnesium catalysts showing good activity, TOF = 97 h−1 (Mg2), 24 h−1 (Zn2) for PA/CHO ROCOP ([Cat]0:[PA]0:[CHO]0 = 1:100:800, [Cat]0 = 12 mM, 100 °C).17,42 Subsequently, other dinuclear catalysts showed impressive PA/CHO ROCOP activities, e.g. [(salen)AlCl]2 (TOF = 750 h−1, [Al2]0:[PPNCl]0:[PA]0:[CHO]0 = 1:100:800, [Al2]0 = 10 mM, 100 °C); [(trisphenolate)Fe]2 (TOF = 1180 h−1, 100 °C, [Fe2]0:[PPNCl]0:[PA]0:[CHO]0 = 1:1:500:2500, [Fe2]0 = 4 mM, 100 °C) although PPNCl was required to observe high activity in the latter case (Fig. S1VI and VII†).43,44
A further advance has been to exploit synergic heterodinuclear catalysts where the mixed metal complex shows significantly better performance than either homodinuclear analogue or mixtures and has been applied in the ROCOP of CO2/epoxide and more recently to lactide ROP.36,37,40,41,45,46 For example, L′ZnMgBr2 catalyst showed 40 fold higher activity in PA/CHO ROCOP compared with an equimolar mixture of L′Zn2Br2:L′Mg2Br2 (TOF = 188 h−1, [Cat]0:[PA]0:[CHO]0, [Cat]0 = 12.5 mM, 100 °C) (L′ = macrocyclic diphenolate tetra(amine) ligand, Fig. S1IV†).40 Catalytic synergy was also observed in the mechanistically similar ROCOP of CO2 with epoxides, with metal combinations of Mg(II)Zn(II), LMg(II)Co(II) and Co(III)K(I) being most active.35–37,41
Here, we investigate heterodinuclear catalysts using metals selected from groups 1, 2, 12. The work builds upon an earlier report of a di-Zn(II) catalyst, coordinated by a dinucleating Schiff base ligand derived from ortho-vanillin, which showed an excellent activity of 198 h−1 in CHO/PA ROCOP ([Zn2]0:[PA]0:[CHO]0 = 1:100:800, [Zn2]0 = 12.5 mM, 100 °C, Fig. S1V†).47 This work targets heterodinuclear Zn(II) or Mg(II) complexes, combined with Na(I) or Ca(II) because the metals show higher ionic character and oxophilicity relative to Zn(II).48 It is hypothesised that more oxophilic metal centres might coordinate the epoxide more strongly as well as labilise the second metal carbonate bond to insertion chemistry. As part of the systematic investigation into M(II)M′(II) combinations, Cd(II) is selected as it shows comparable ionic radii to both Na(I) and Ca(II) but very much lower oxophilicity.48
Results and discussion
To compare heterodinuclear complexes with the high performance di-zinc catalysts it is important to systematically vary only the metal centres and thus the same pro-ligand (H2L) was used throughout. It was successfully synthesised following the previously reported procedure, by adding 2,2-dimethylpropanediamine to ortho-vanillin, in methanol, and stirring the solution for 4 hours (Scheme 1).47 Following solvent removal, the target ligand precipitated and, after washing, was isolated in 93% yield. Successful ligand synthesis was confirmed by 1H NMR spectroscopy by the disappearance of the aldehyde resonances (10.19 ppm) and the formation of imine resonances (8.28 ppm) as well as methylene and methyl resonances from the 2,2-dimethyl propylene backbone (3.45 and 1.04 ppm).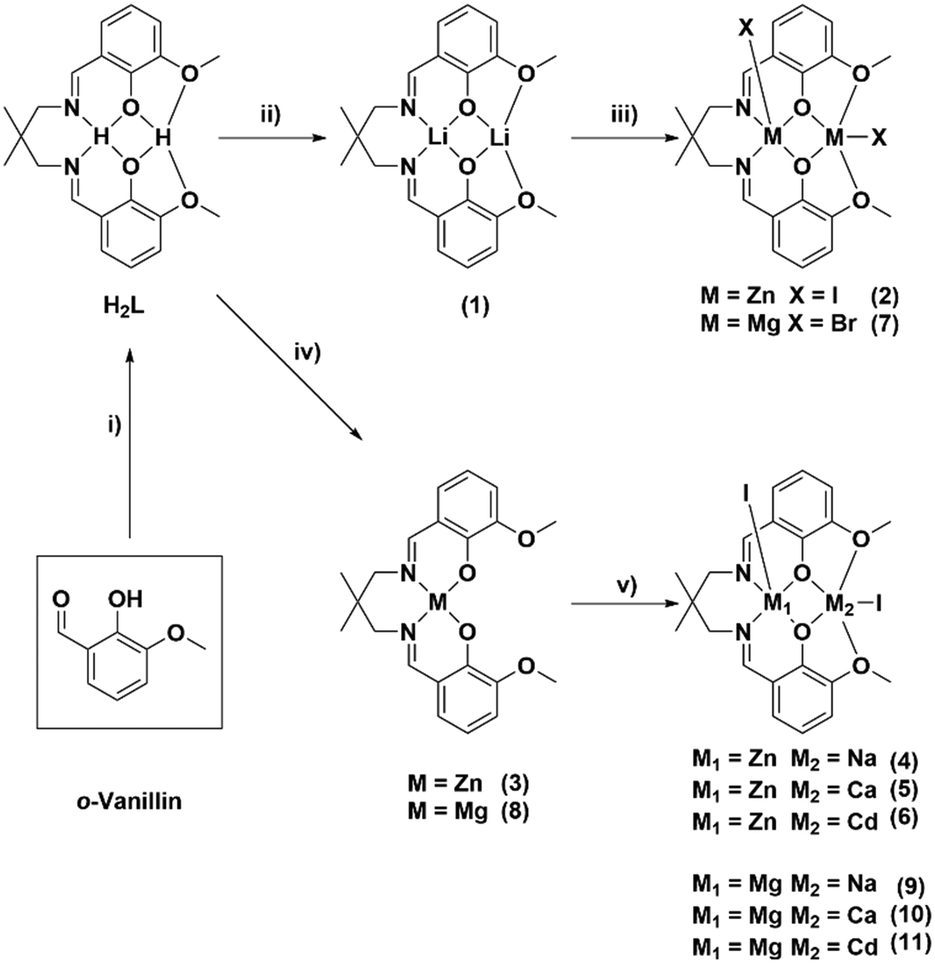 | ||
| Scheme 1 The syntheses of mono-, homo- and heterodinuclear complexes 1–11. i) 2,2-Dimethylpropanediamine, MeOH, 4 h, 25 °C, 93%. ii) 2 equiv. Li(N(Si(CH3)3)2), THF, 4 h, 25 °C, 90%. iii) 2 equiv. ZnI2 or MgBr2, THF, 16 h, 25 °C, >80%. iv) ZnEt2 or Mg(N(Si(CH3)3)2)2, THF, 25 °C, >70%. v) MI2 (M = Na(I), Ca(II) or Cd(II), THF, 25 °C, >70%. | ||
The di-lithium complex (1) was synthesised by pro-ligand reaction with two equivalents of Li(N(Si(CH3)3)2) and was isolated in 90% yield (Scheme 1). It was analysed by NMR spectroscopy (Fig. S2–S4†) and X-ray diffraction (Fig. S5 and Table S1†). Its 1H NMR spectrum showed the complete disappearance of the phenol resonance (13.40 ppm, d8-THF, 298 K) accompanied by lower chemical shift values for the imine and methylene resonances (7.90 and 3.16 ppm, respectively); the NMR data are indicative of lithium coordination within the diphenolate–diimine ligand binding pocket. The small shift in the methoxide resonances (3.76–3.66 ppm) indicates only weak lithium interaction with the diphenolate–diether binding cavity.
The di-Zn(II) complex (2) was synthesised by addition of two equivalents of zinc iodide to complex (1), in THF, at room temperature, and was isolated in 75% yield and analysed by NMR spectroscopy (Fig. S6 and S7†) and X-ray diffraction (Fig. S8 and Table S2†). Its 1H NMR spectrum (d8-THF, 298 K) displays the characteristic diastereotopic splitting of the methylene and methyl proton resonances (4.63/2.95 ppm and 1.03/0.67 ppm, respectively); these specific changes confirm zinc coordination in the diphenolate–diimine pocket. Further confirmation of the di-Zn(II) complex formation came from the deliberate preparation of a mono-Zn complex (3). This was achieved by the reaction of H2L with an equivalent of diethyl zinc and was characterised by NMR spectroscopy (Fig. S9 and S10†) and X-ray diffraction (Fig. S11 and Table S3†). Its 1H NMR spectrum (CDCl3, 298 K) shows very broad methylene resonances (3.41 ppm) characteristic of rapid exchange processes, on the NMR timescale, either arising from monomer–dimer equilibria or from exchange between the different binding pockets. Adding a metal iodide to complex (3), in THF at 25 °C, resulted in the formation of the desired heterodinuclear complexes LZn(II)M, M = Na(I), Ca(II) or Cd(II) (4–6), respectively. All these products were isolated in good yields (>78%) as pale-yellow powders. NMR spectroscopy was useful (Fig. S12–S17†) but could not unambiguously establish heterodinuclear complex formation since both the homodinuclear (2) and heterodinuclear (4–6) complexes show the same number and multiplicity of ligand resonances due to their having the same ligand symmetries (Fig. S18†). The NMR spectrum does, however, display only one set of ligand resonances and thus contamination/formation of homodinuclear analogues, such as di-zinc (2) can be ruled out. Clear evidence for heterodinuclear complex formation was obtained in all cases through single crystal X-ray diffraction studies (vide infra, Fig. S19–S21, Tables S4–S6†) and purity confirmed by elemental analysis.
The di-Mg(II) complex (7) was synthesised by adding two equivalents of magnesium dibromide to complex (1), in THF at 25 °C (Scheme 1). It was isolated as a white powder in high yields (>80%) and characterised by NMR spectroscopy (Fig. S22 and S23†) and X-ray diffraction methods (Fig. S24, Table S7†), with purity confirmed by elemental analysis. In this case, bromide was used as the co-ligand since the analogous iodide precursor is highly insoluble in most organic solvents. In contrast to the di-zinc complex (2), the di-Mg(II) complex (7) fails to show any diastereotopic splitting of the methylene and methyl resonances. This difference is attributed to the contrasting coordination chemistries of the two metals: Zn(II) adopts a penta-coordinate geometry, whereas Mg(II) adopts an octahedral geometry, with the C2-axis rendering the methylene/methyl proton magnetically equivalent. Mono-Mg(II) complex (8) was synthesised by reaction of H2L with Mg(N(Si(CH3)3)2)2, at room temperature, in THF. Its 1H NMR spectrum is very complex and cannot be easily characterised, probably due to the formation of aggregates as observed in the solid state (vide infra, Fig. S25†).49 The target heterodinuclear Mg(II)M complexes, M = Na(I), Ca(II) and Cd(II), (9–11) were synthesised by adding the appropriate metal iodide to complex (8), in THF at 25 °C. All complexes were isolated as off-white powders and were characterised by NMR spectroscopy (Fig. S26–S30†) and X-ray diffraction methods (Fig. S31 and S32, Tables S8 and S9†) with purity confirmed by elemental analysis.
Solid state structure: X-ray crystallography
Single crystals, suitable for X-ray diffraction experiments, were obtained for all the new complexes by pentane diffusion into a saturated solution of the target complex in THF at −40 °C, unless stated otherwise (Fig. 2 and 3). The solid-state structures were useful to inform about both the speciation and nuclearity of the complexes. Complex (1) is a dimer (Fig. S5, Table S1†) with each Li(I) atom adopting a penta-coordinate, square based pyramidal geometry (τ = 0.06). The aggregation of two di-lithium complexes forms a cubane structure (Li4O4), a motif which is very common in lithium coordination chemistry.50–52 The dimeric structure was found to be retained in the solution phase through 2D DOSY NMR analysis (THF, 298 K, Fig. S33†) giving a diffusion coefficient of 8.91 × 10−10 m2 s−1 and hence a hydrodynamic radius of 5.1 Å which is in-line with the radius calculated from the crystallographic molecular structure of 5.3 Å (half-dimer radius = 4.5 Å). Adding zinc iodide to complex (1) allowed isolation of a LZn(II)Li(I) (A) complex, which was characterised using single crystal X-ray diffraction (Fig. S34, Table S10†). Both Zn(II) and Li(I) centres adopt penta-coordinate, square based pyramidal geometries (Zn(II) τ = 0.14, Li(I) τ = 0.01). To balance charge, the Zn(II) ion is also coordinated to an iodide co-ligand, whereas the Li(I) ion coordinates an additional THF molecule from the crystallisation solvent. The isolation of this intermediate suggests that the formation of the di-Zn(II) complex (1) occurs by stepwise metatheses reactions, with zinc first being coordinated by the diphenolate–diimine binding cavity. Adding a second equivalent of zinc iodide to (A) resulted in the isolation of di-Zn(II) complex (2) where the Zn(II) (Zn1) coordinated by the diphenolate–diimine pocket remains penta-coordinate (τ = 0.13) and the second Zn(II) (Zn2) is coordinated only by the diphenolates, without any coordination from the two ether groups, in a tetrahedral geometry. The coordination sphere for Zn2 is completed by an iodide co-ligand and a THF molecule (crystallisation solvent). The facility for THF coordination indicates Zn2 is the more Lewis acidic of the two Zn(II) centres, consistent with its coordination geometry and the presence of two imine donors (electron donating groups) coordinating to Zn1. The site of THF coordination is interesting as it provides insight into the likely metal at which epoxide coordination should occur during PA/CHO ROCOP catalysis.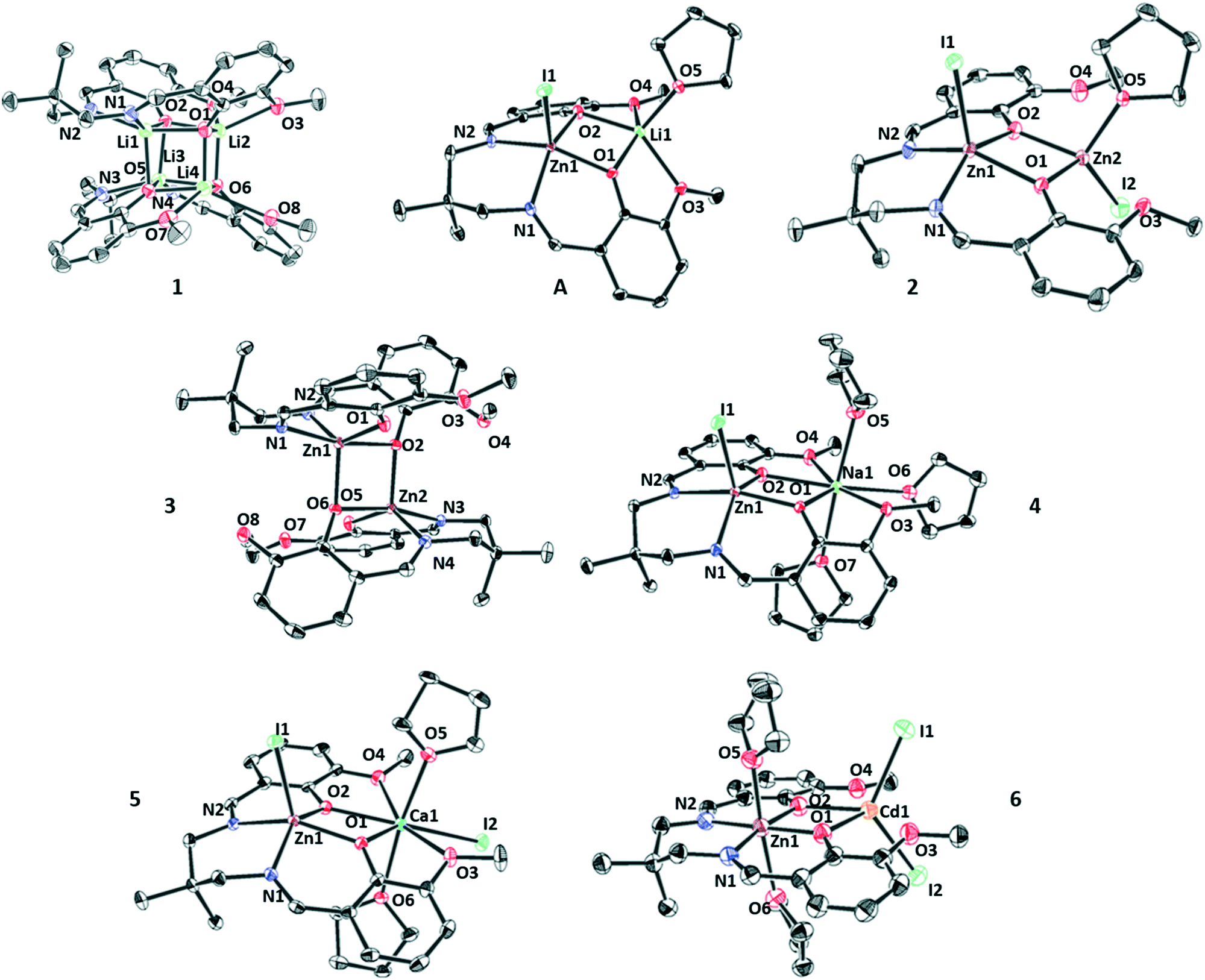 | ||
| Fig. 2 ORTEP representations of the molecular structures of complexes 1–6. These structures are obtained by single crystal X-ray diffraction experiments. Complex disorder issues and H-atoms are omitted, for clarity, and the thermal ellipsoids are represented at 40% probability. | ||
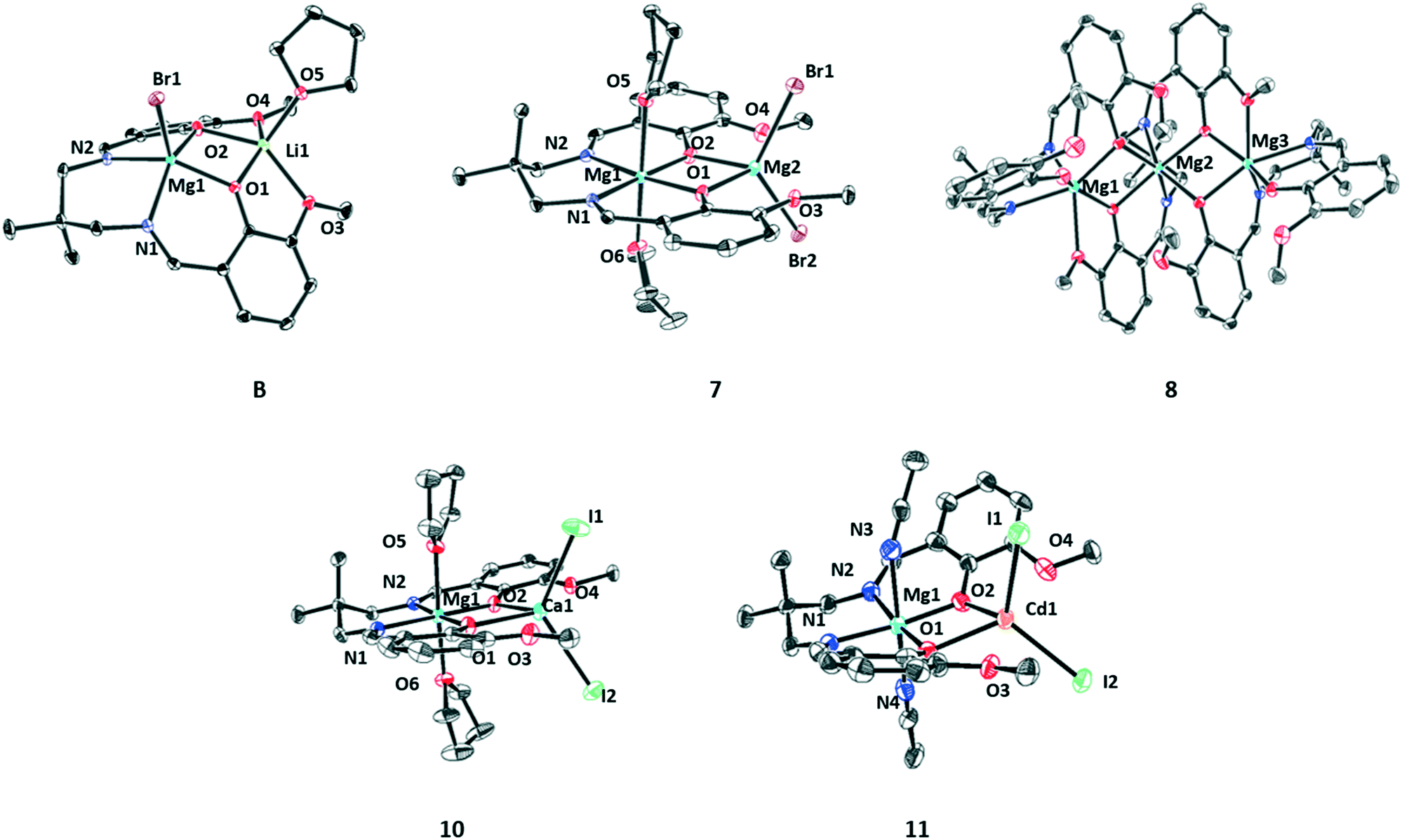 | ||
| Fig. 3 ORTEP representation of the molecular structures of complexes 7–11. These structures are obtained by single crystal X-ray diffraction experiments. Complex disorder issues and H-atoms are omitted, for clarity, and the thermal ellipsoids are represented at 40% probability. | ||
A single crystal of the mono-Zn(II) complex (3) was isolated by pentane diffusion into a saturated solution of the complex in chloroform at 25 °C. Its solid-state structure is dimeric with the Zn–O coordination resulting in a half cubane structure (Zn2O2). Both zinc centres adopt identical distorted penta-coordinate geometries (τ = 0.53). Both (4) and (5) form the desired heterodinuclear structures (monomeric) in the solid state. In both complexes, the Zn(II) centres adopt penta-coordinate, square pyramidal geometries (4τ = 0.26, 5τ = 0.09) and are coordinated by the diphenolate–diimine moieties and an iodide co-ligand. Both alkali(ne) earth metals adopt heptacoordinate, pentagonal bipyramidal geometries. For Na(I), the coordination chemistry involves the diphenolate–diether groups from the ligand and three THF molecules. In the case of Ca(II), it is the ligand diphenolate–diether pocket, two trans-axial THF molecules and an iodide co-ligand to balance the extra charge (in comparison to (4)).
The finding that THF preferentially coordinates to the alkali(ne) earth metals, as opposed to zinc likely arises from both the higher oxophilicity values (Ca(II) = 0.3, Zn(II) = 0.2) and higher Lewis acidity values (Na(I) = 21.1, Ca(II) = 19.5 and Zn(II) = 10.9, hardscale).48 The intermetallic distances for Zn(II)/Na(I) and Zn(II)/Ca(II) are very similar at 3.56(6) Å and 3.57(9) Å, respectively. Complex (6) also shows the expected heterodinuclear structure (monomeric) where the Zn(II) centre adopts a hexa-coordinate, octahedral geometry, coordinated by the diphenolate–diimine ligand binding pocket and two THF molecules (crystallisation solvent). The Cd(II) centre adopts a tetrahedral geometry, coordinated by the di-phenolate moieties of the ligand and two iodide co-ligands. In this case, the neutral Lewis base, THF, molecules are both coordinated to Zn(II), this is rationalised by its smaller ionic radius and, therefore, a higher Lewis acidity.
Adding an equivalent of magnesium bromide to complex (1) resulted in the isolation of crystals of LMg(II)Li (B) which is isostructural with complex (A) (Fig. S35, Table S11†). Both the metals, Mg(II) and Li(I) adopt penta-coordinate square pyramidal geometries (Mg(II) τ = 0.07, Li(I) τ = 0.06), with the Mg(II) being coordinated by the ligand diphenolate–diimine moieties and the bromide, whilst the Li(I) is coordinated by the ligand diphenolate–diether moieties and a THF molecule (crystallisation solvent). The addition of a second equivalent of magnesium bromide to (B) allowed the isolation of di-Mg(II) complex (7). In its structure the Mg(II) coordinated by the diphenolate–diimine binding pocket adopts a hexacoordinate, octahedral geometry with two additional THF molecules. The other Mg(II) adopts a tetrahedral geometry and is coordinated by the diphenolate–diether moieties of the ligand and two bromide co-ligands. The intermetallic separations for complexes (7) and (B) are 3.11(7) Å and 3.00(1) Å, respectively.
A single crystal of complex (8) was obtained via the slow evaporation of a THF solution at 25 °C. It has an aggregated trimeric structure (L3Mg3) where each Mg(II) centre adopts a hexa-coordinate octahedral geometry. The structure of heterodinuclear complex (10) is consistent with that of complex (7) whereby Mg(II) adopts a hexa-coordinate, octahedral geometry and is coordinated by the diphenolate–diimine pocket of the ligand and two THF molecules, whilst the Ca(II) adopts a tetrahedral geometry and is coordinated by the phenolate ligand moieties and two iodide co-ligands. Single crystals of complex (11) were isolated via vapour diffusion of pentane into a saturated acetonitrile solution at −40 °C. It shows the expected heterodinuclear complex formation, again with Mg(II) being coordinated by the diphenolate–diimine ligand binding pocket and with the octahedral geometry being achieved by the coordination of two acetonitrile molecules. The Cd(II) adopts a tetrahedral geometry and is coordinated by the ligand diphenolates and two iodide co-ligands.
Ring-opening copolymerisation catalysis
The series of dinuclear complexes, both homodinuclear complexes (1), (2) and (7) and heterodinuclear complexes (4–6) and (9–11), were each tested as catalysts for phthalic anhydride and cyclohexene oxide ring opening copolymerisation (Table 1). Each polymerisation was conducted at 100 °C, using 0.1 mol% catalyst loading, neat CHO (9.9 M) and at a relative loading of [cat]0:[PA]0:[CHO]0 = 1:100:1000. Polymerisations were monitored using in situ ATR-IR spectroscopy which allowed correlation of the relative intensity of a characteristic carbonyl stretch for phthalic anhydride (1825 cm−1) against time to be used to assess polymerisation rates. All the polymerisations were monitored until complete anhydride conversion (>99%) whereupon an aliquot was removed for NMR spectroscopic and GPC analysis. All the catalysts show excellent selectivity for polyester formation (>99%) without any detectable ether linkages, by 1H NMR spectroscopy. Further, all reactions are well controlled, with polyester molar mass values correlating well with expected values (Mn(theoretical) = 24.6 kg mol−1) and narrow dispersity (Đ = 1.03–1.30). All polymerisations showed bimodal molar mass distributions characterised by the higher mass peak being approximately twice that of the lower. Such bimodality is very common in epoxide ROCOP catalysis and arises from reaction between the epoxide solvent and adventitious water resulting in the formation of 1,2-cyclohexanediol.53–57 The diol functions as a chain transfer agent, i.e. it undergoes rapid exchange with the catalyst–polymer chain, and since it is a telechelic initiator, results in molar mass values which are twice those of chains initiated from iodide/bromide groups of the catalyst.54
| Entry | Catalyst | Time (h) | Conv.b (%) | Poly(ester) linkagesc (%) | TOFd (h−1) | k obs (×10−6 s−1) | M n [Đ]f (kg mol−1) |
|---|---|---|---|---|---|---|---|
|
a Reaction conditions = [Cat]0:[PA]0:[CHO]0 = 1:100:1000. [Cat]0 = 10 mM, 100 °C.
b Expressed as a percentage of PA vs. the theoretical maximum (100%) determined by comparison of the relative integrals of the 1H NMR (CDCl3) spectrum of resonances assigned to polyester (4.80–5.26 ppm) against the sum of integrals for PA (7.97 ppm), polyester and ether (3.22–3.64 ppm).
c Expressed as a percentage of ester linkage vs. the theoretical maximum (100%) determined by comparison of the relative integrals of the 1H NMR (CDCl3) spectrum of resonances due to polyester against ether.
d Turn-over-frequency (TOF) = TON/time (h).
e
k
obs (μs−1) determined from the gradient of the linear fit to [PA] vs. time (s) data.
f Determined by GPC (gel-permeation chromatography) in THF using narrow Mn polystyrene standards with dispersity (Đ) given in brackets, two values for Mn are reported due to bimodal molar mass distributions.
g [cat]0:[PA]0:[CHO]0 = 1:100:800, [cat]0 = 12.5 mM, 100 °C.40
h [Cr]0:[PPN3]0:[PA]0:[CHO]0 = 1:1:200:200, [Cr]0 = 4 mM in Tol, 100 °C.24
i [Al2]0:[PA]0:[CHO]0 = 1:2:250:1000, [Al2] = 10 mM, 50 °C.44
j [Fe2]0:[PPNCl]0:[PA]0:[CHO]0 = 1:1:500:2500, [Fe2]0 = 4 mM, 100 °C.43
n.d. = not determined. |
|||||||
| 1 | Zn(II)/Zn(II) (2) | 0.6 | >99 | >99 | 149 ± 7 | 500 ± 25 | 8.5 [1.05] |
| 18.0 [1.03] | |||||||
| 2 | Zn(II)/Na(I) (4) | 2.8 | >99 | >99 | 29 ± 2 | 130 ± 7 | 7.4 [1.14] |
| 18.5 [1.08] | |||||||
| 3 | Zn(II)/Ca(II) (5) | 1.4 | >99 | >99 | 62 ± 3 | 220 ± 11 | 6.0 [1.08] |
| 19.1 [1.12] | |||||||
| 4 | Zn(II)/Cd(II) (6) | 1.3 | >99 | >99 | 67 ± 3 | 240 ± 12 | 5.6 [1.23] |
| 17.0 [1.08] | |||||||
| 5 | Mg(II)/Mg(II) (7) | 2.8 | >99 | >99 | 35 ± 2 | 140 ± 7 | 8.7 [1.11] |
| 17.0 [1.08] | |||||||
| 6 | Mg(II)/Na(I) (9) | 4.0 | >99 | >99 | 10 ± 1 | 40 ± 2 | 9.8 [1.08] |
| 21.5 [1.13] | |||||||
| 7 | Mg(II)/Ca(II) (10) | 3.2 | >99 | >99 | 25 ± 2 | 100 ± 5 | 5.3 [1.11] |
| 16.8 [1.18] | |||||||
| 8 | Mg(II)/Cd(II) (11) | 1.6 | >99 | >99 | 64 ± 3 | 220 ± 10 | 9.2 [1.14] |
| 24.0 [1.07] | |||||||
| 9g | L′Mg(II)/Zn(II) | 0.5 | 94 | >99 | 188 | n.d. | 5.0 [1.09] |
| 10.9 [1.04] | |||||||
| 10h | (Salen)Cr(III)Cl/PPNCl | 3.3 | >99 | >99 | 61 | 120 | 17.0 [1.12] |
| 11i | [(Salen)Al(III)]2/PPNCl | 0.3 | 90 | >99 | 750 | n.d. | 9.8 [1.14] |
| 12j | [(Trisphenolate)Fe(III)]2/PPNCl | 0.3 | 58 | >99 | 1160 | n.d. | 7.3 [1.24] |
Before comparing catalyst activity values, it is instructive to examine the typical kinetic features of the PA/CHO ROCOP. Analysis of the anhydride concentration–time data shows a linear fit indicative of a zero order rate dependency, as has commonly been observed for other catalysts in this field.58–62
The data is consistent with phthalic anhydride insertion being pre-rate limiting (fast) with the rate determining step likely involving metal carboxylate attack on (second) metal coordinated epoxide. Accordingly, a chain-shuttling mechanism is proposed and illustrated for the most active di-zinc catalyst (2) (Fig. 4).39,41,63 The mechanism is underpinned by the solid state data which showed a THF molecule coordinated at Zn2 (also coordinated to the diphenolate groups of the ligand). Thus, in the proposed mechanism, the epoxide is coordinated at Zn2 and initiation occurs by attack of the Zn1-iodide to ring-open the epoxide and generate a Zn2-alkoxide intermediate. This intermediate undergoes rapid reaction with phthalic anhydride to generate a Zn1-carboxylate intermediate. The coordination of another epoxide at Zn2 allows the cycle to renew (propagation). Concurrently, it is proposed the second iodide group migrates between Zn1 and Zn2 in an equal and opposite manner to the growing polymer chain. Such chain-shuttling mechanisms were previously proposed for other di- and multi-nuclear catalysts for CO2/epoxide ROCOP.37,63–66
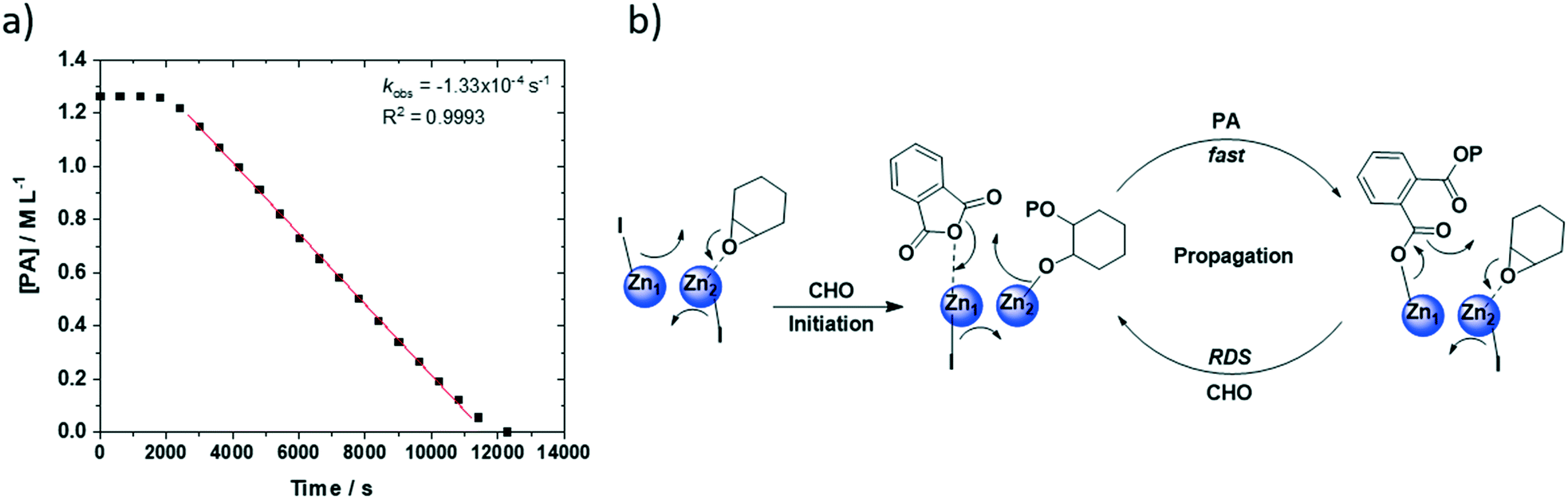 | ||
| Fig. 4 a) Linear fit plot of [PA] vs. time (s) for PA/CHO ROCOP using catalyst (4). Data points are determined using in situ ATR-IR spectroscopy, by measuring the change in absorption intensity for PA (1825 cm−1) and are calibrated to the correct concentration using aliquots analysed by 1H NMR spectroscopy. b) The proposed chain-shuttling PA/CHO ROCOP mechanism where the rate limiting step is attributed to metal carboxylate attack on a (second) metal coordinated epoxide molecule. | ||
In terms of catalyst activity values, some clear trends can be observed from the series of new compounds. Complexes without any initiating groups, i.e. (1), (3) and (8) are totally inactive giving confidence that catalysis occurs by a coordination–insertion mechanism. The di-Zn(II) complex (2) shows the highest activity with a TOF of 149 h−1 and a pseudo-zero order rate coefficient of kobs = 500 × 10−6 s−1. The values were closely related to those previously reported for the di-Zn(II) acetate complex (TOF = 198 h−1), although under slightly lower catalyst concentration (10 mM versus 12.5 mM), which was expected due to the propagating species being identical.47 Di-Zn(II) (2) is four times more active than di-Mg(II) complex (7), under identical conditions. For both complexes, the solid-state structures show coordinated THF molecules and this suggests that epoxide coordination is likely to be effective, especially since reactions are conducted in neat epoxide. Thus, the different activity values are attributed to the enhanced nucleophilicity of the zinc-carboxylate counterpart. Also relevant is the different coordination chemistry of (2) vs. (7): di-zinc complex (2) features iodide co-ligand coordinated at each of the two zincs whereas di-Mg(II) complex (7) features both bromides at the Mg(II) ion coordinated only by phenolate groups from the ligand. The halide ligands are the expected sites for coordination of the propagating chain and kinetic analyses suggest an anionic carboxylate resting state. According to the chain shuttling mechanism, it is tentatively proposed that the zinc carboxylate nucleophilicity is increased by the presence of imine donors and that the magnesium-carboxylate nucleophilicity is reduced by their absence and the lack of coordination of the ether groups. Consistent with this notion, is the finding that the reactivity trend (di-Zn(II) > di-Mg(II)) is exactly opposite to that observed for macrocyclic di-Zn(II) and di-Mg(II) catalysts, where the latter is around four times more active than the former for the same reaction.17 In the macrocyclic di-Mg(II) complex, both Mg(II) centres are coordinated by (electron donating) amine groups consistent with the notion that such donors are important to enhance metal-carboxylate nucleophilicity and hence accelerate rates.
The di-Zn(II) complex (2) represents a significant increase in both activity (149 h−1vs. 3 h−1) and selectivity (>99% vs. 86%) compared to a similar di-Zn(II) complex featuring benzotriazole donors coordinating the second Zn(II) centre, albeit under slightly differing conditions ([Cat]0:[PA]0:[CHO]0 = 1:100:100 in toluene (5 mL), 110 °C, 24 h). The two additional ether donors appear important in regulating epoxide coordination, at the other Zn(II) centre, and in preventing the formation of ether linkages.27
The two complexes with Cd(II) coordination, (6) and (11) show equivalent activity values (TOF = 65 h−1, kobs = 230 × 10−6 s−1). The molecular structures are isostructural and feature the neutral donors (THF) coordinated at Zn(II) or Mg(II) and both halides coordinate at Cd(II). The solid-state structures suggest that during catalysis the epoxide should be coordinated at Zn(II) or Mg(II) and be attacked by a common Cd(II)-carboxylate intermediate. The closely analogous rates are fully consistent with the chain shuttling mechanism and supports the hypothesis that the rate determining step is metal carboxylate attack on coordinated epoxide.
The other zinc heterodinuclear complexes (4) and (5) show Zn(II) coordinated by diphenolate–diimine binding pocket. Following the proposal that the rate determining step involves Zn(II)-carboxylate nucleophilic attack on (second) metal coordinated epoxide. The heterodinuclear magnesium complex (10) features a structure analogous to (7) and accordingly the magnesium(II) centre should coordinate the epoxide which is attacked by the Ca(II)-carboxylate nucleophile. All the heterodinuclear complexes are less active than di-Zn(II) complex (2) and show the same activity trend∼: Zn(II) ≫ Cd(II) > Mg(II) > Ca(II) > Na(I).
Compared more broadly to other catalysts in this field, the di-Zn(II) complex (2) showed equivalent activity to a heterodinuclear Zn(II)/Mg(II) catalyst, which itself showed a 40-fold increase in activity compared to the di-zinc/di-magnesium analogues. In contrast, in this work all the heterodinuclear combinations underperformed compared to the di-Zn(II) complex. In fact, a similar observation was made for another series of heterodinuclear catalysis for carbon dioxide/CHO ROCOP where a macrocyclic ancillary ligand was coordinated with Zn(II)M, where M = Li(I), Na(I), K(I), Ca(II), Al(III), Ga(III) and In(III); all combinations were less active than the di-Zn(II) catalyst.35,38 Thus, the Zn(II)Mg(II) combination is somewhat special amongst the set of metals in providing heterodinuclear synergy. To target this desirable combination, attempts were made to prepare Zn(II)Mg(II) complexes from the precursors (3) and (8). Unfortunately, isolation of a pure heterodinuclear complex was unsuccessful and the 1H NMR spectrum of the products showed signals attributed to both Zn(II)Mg(II) and Mg(II)Zn(II) combinations (i.e. where both Zn(II) and Mg(II) were coordinated by each binding pocket). The formation of both complexes was attributed to the low selectivity of the phenolate–ether binding pocket and the tendency for mono-Zn(II)/Mg(II) complexes to undergo rapid metal exchange processes which prevent stabilisation of a particular metal into a single binding pocket. Overall, the di-Zn(II) (2) shows equivalent activity to well-known (salen)Cr(III)Cl/PPNCl catalyst systems (kobs = 120 × 10−6 s−1) but is at least an order of magnitude slower than the best PA/CHO ROCOP catalysts featuring di-Al(III) or di-Fe(III) metal centres.43,44
Conclusions
The synthesis and polymerisation catalytic activity of a series of heterodinuclear complexes, coordinated by a dinucleating Schiff-base ortho-vanillin ligand, were reported. The ligand features diphenolate–diimine and diphenolate–diether binding pockets. Depending on the metal selections, different coordination chemistries resulted and their formation was rationalised, in part, by the tendency to coordinate the less oxophilic metal in the diphenolate–diimine site and the more oxophilic metal in the other site. The complexes were single-component catalysts for the ring-opening copolymerisation of phthalic anhydride with cyclohexene oxide and showed good activities, high selectivity for ester linkage formation and reasonable polymerisation control. None of the heterodinuclear complexes showed catalytic synergy. This finding shows that simply preparing heterodinuclear complexes with different metals for epoxide coordination/nucleophile location is not sufficient to confer synergic behaviour. Given the synergy observed for other Zn(II)Mg(II) catalysts, coordinated by a symmetrical macrocycle ancillary ligand, explorations of other ‘open’ Schiff base ligands featuring more electron donating substituents are warranted. The preparation of Co(II)Mg(II) complexes is also recommended since this combination was recently shown to be synergic in carbon dioxide/cyclohexene oxide ring opening copolymerisation.36 Detailed kinetic investigations revealed the rate limiting step involves Co(II) carbonate attack on a Mg(II) coordinated epoxide, indicating that a future target set of complexes should feature Co(II) coordinated in the diphenolate–diimine pocket and Mg(II) in the diphenolate(–diether) pocket.36 Very recently, Co(III)/M(I) catalysts, coordinated by an asymmetrical macrocyclic ligand showed excellent activity in carbon dioxide/propene oxide ROCOP.37 Investigations of other heterodinuclear Co(III)/M(I) catalysts, coordinated by the same ortho-vanillin Schiff-base ligand described here, will be reported in due course.Author contributions
CKW and ACD designed the catalysts; rationalized the characterization and copolymerization results and wrote the manuscript. ACD carried out all experimental work. X-ray crystallographic data were collected and analysed by CBD (complexes 1–6, 8–10) and RWFK (complexes 7 and 11), respectively.Conflicts of interest
CKW is a director of Econic Technologies.Acknowledgements
The EPSRC (EP/S018603/1, EP/K014668/1) and Econic Technologies (CASE award to ACD) are acknowledged for research funding.References
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef PubMed.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef.
- F. Stempfle, P. Ortmann and S. Mecking, Chem. Rev., 2016, 116, 4597–4641 CrossRef PubMed.
- A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef PubMed.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858–13861 CrossRef PubMed.
- H. Nakajima, K. Loos, S. Ishizu and Y. Kimura, Macromol. Rapid Commun., 2018, 39, 1700865 CrossRef PubMed.
- T. Liu, T. L. Simmons, D. A. Bohnsack, M. E. Mackay, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 6040–6047 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- S. Giarola, C. Romain, C. K. Williams, J. P. Hallett and N. Shah, Chem. Eng. Res. Des., 2016, 107, 181–194 CrossRef CAS.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
- X. Yu, J. Jia, S. Xu, K. U. Lao, M. J. Sanford, R. K. Ramakrishnan, S. I. Nazarenko, T. R. Hoye, G. W. Coates and R. A. DiStasio, Nat. Commun., 2018, 9, 2880 CrossRef PubMed.
- H. Li, H. Luo, J. Zhao and G. Zhang, Macromolecules, 2018, 51, 2247–2257 CrossRef CAS.
- G. Si, L. Zhang, B. Han, Z. Duan, B. Li, J. Dong, X. Li and B. Liu, Polym. Chem., 2015, 6, 6372–6377 RSC.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- H.-Y. Ji, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 641–648 RSC.
- C.-Y. Yu, H.-J. Chuang and B.-T. Ko, Catal. Sci. Technol., 2016, 6, 1779–1791 RSC.
- L.-F. Hu, C.-J. Zhang, H.-L. Wu, J.-L. Yang, B. Liu, H.-Y. Duan and X.-H. Zhang, Macromolecules, 2018, 51, 3126–3134 CrossRef CAS.
- K. Bester, A. Bukowska, B. Myśliwiec, K. Hus, D. Tomczyk, P. Urbaniak and W. Bukowski, Polym. Chem., 2018, 9, 2147–2156 RSC.
- H. K. Ryu, D. Y. Bae, H. Lim, E. Lee and K.-s. Son, Polym. Chem., 2020, 11, 3756–3761 RSC.
- C. Martín, A. Pizzolante, E. C. Escudero-Adán and A. W. Kleij, Eur. J. Inorg. Chem., 2018, 2018, 1921–1927 CrossRef.
- M. Hatazawa, R. Takahashi, J. Deng, H. Houjou and K. Nozaki, Macromolecules, 2017, 50, 7895–7900 CrossRef CAS.
- J. Y. Jeon, S. C. Eo, J. K. Varghese and B. Y. Lee, Beilstein J. Org. Chem., 2014, 10, 1787–1795 CrossRef PubMed.
- B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed.
- A. C. Deacy, C. B. Durr, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2018, 57, 15575–15583 CrossRef CAS PubMed.
- A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS PubMed.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- A. C. Deacy, C. B. Durr and C. K. Williams, Dalton Trans., 2020, 49, 223–231 RSC.
- P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- C. Romain, J. A. Garden, G. Trott, A. Buchard, A. J. P. White and C. K. Williams, Chem. – Eur. J., 2017, 23, 7367–7376 CrossRef CAS PubMed.
- Z. Shi, Q. Jiang, Z. Song, Z. Wang and C. Gao, Polym. Chem., 2018, 9, 4733–4743 RSC.
- J. Li, Y. Liu, W.-M. Ren and X.-B. Lu, J. Am. Chem. Soc., 2016, 138, 11493–11496 CrossRef CAS PubMed.
- W. Gruszka, A. Lykkeberg, G. S. Nichol, M. P. Shaver, A. Buchard and J. A. Garden, Chem. Sci., 2020, 11, 11785–11790 RSC.
- A. J. Gaston, Z. Greindl, C. A. Morrison and J. A. Garden, Inorg. Chem., 2021, 60, 2294 CrossRef CAS PubMed.
- A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS PubMed.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2011, 50, 9242–9243 CrossRef CAS PubMed.
- L. M. Jackman and B. D. Smith, J. Am. Chem. Soc., 1988, 110, 3829–3835 CrossRef CAS.
- D. J. MacDougall, J. J. Morris, B. C. Noll and K. W. Henderson, Chem. Commun., 2005, 456–458 RSC.
- X. Zhao, T. Wu, S.-T. Zheng, L. Wang, X. Bu and P. Feng, Chem. Commun., 2011, 47, 5536–5538 RSC.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S.-i. Han, H. Yun, H. Lee and Y.-W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS PubMed.
- G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- Z.-Q. Wan, J. M. Longo, L.-X. Liang, H.-Y. Chen, G.-J. Hou, S. Yang, W.-P. Zhang, G. W. Coates and X.-B. Lu, J. Am. Chem. Soc., 2019, 141, 14780–14787 CrossRef CAS PubMed.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- N. J. Van Zee, M. J. Sanford and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 2755–2761 CrossRef CAS PubMed.
- T. Stößer, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 2668 CrossRef PubMed.
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef PubMed.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- H. Nagae, R. Aoki, S.-n. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS.
- S. Kissling, P. T. Altenbuchner, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, U. B. Seemann and B. Rieger, Chem. – Eur. J., 2015, 21, 8148–8157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2057256–2057267. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cy00238d |
| This journal is © The Royal Society of Chemistry 2021 |