
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic elucidation of monoalkyltin(IV)-catalyzed esterification†
Lukas A.
Wolzak
ab,
Joen J.
Hermans
 c,
Folkert
de Vries
d,
Keimpe J.
van den Berg
e,
Joost N. H.
Reek
*b,
Moniek
Tromp
*ad and
Ties J.
Korstanje
*a
c,
Folkert
de Vries
d,
Keimpe J.
van den Berg
e,
Joost N. H.
Reek
*b,
Moniek
Tromp
*ad and
Ties J.
Korstanje
*a
aSustainable Materials Characterization, van 't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: t.j.korstanje@uva.nl; moniek.tromp@rug.nl
bBio-inspired, Homogeneous and Supramolecular Catalysis, van ‘t Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl
cMolecular Photonics, van 't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
dFaculty of Science and Engineering, Materials Chemistry – Zernike Institute for Advanced Materials, University of Groningen Nijenborgh 4, 9747 AG Groningen, The Netherlands
eAkzo Nobel Car Refinishes BV, Rijksstraatweg 31, 2171 AJ Sassenheim, The Netherlands
First published on 17th February 2021
Abstract
Monoalkyltin(IV) complexes are well-known catalysts for esterification reactions and polyester formation, yet the mode of operation of these Lewis acidic complexes is still unknown. Here, we report on mechanistic studies of n-butylstannoic acid in stoichiometric and catalytic reactions, analyzed by NMR, IR and MS techniques. While the chemistry of n-butyltin(IV) carboxylates is dominated by formation of multinuclear tin assemblies, we found that under catalytically relevant conditions only monomeric n-BuSn(OAc)3 and dimeric (n-BuSnOAc2OEt)2 are present. Density functional theory (DFT) calculations provide support for a mononuclear mechanism, where n-BuSn(OAc)3 and dimeric (n-BuSnOAc2OEt)2 are regarded as off-cycle species, and suggest that carbon–oxygen bond breaking is the rate-determining step.
Introduction
The synthesis of esters from alcohols and carboxylic acids is an important reaction in organic synthesis of small molecules and polymers.1,2 Although strong Brønsted acids are efficient esterification catalysts, often the milder Lewis acids are preferred since they provide fewer unwanted side products.3 Therefore over the past decades, a variety of Lewis acidic organotin derivatives have been developed and these catalysts are frequently used in industry.4,5 The inherent Lewis acidity of tin(IV) complexes in combination with facile ligand exchange are considered the origin of their catalytic performance in the esterification reaction.6 Especially organometallic mono- and dialkyltin(IV) complexes display good catalytic performance and stability in many transesterification, esterification and polyesterification reactions.7 The presence of the alkyl tails, generally n-butyl or n-octyl, appears to be pivotal for the catalytic behavior of these tin-based catalysts, since strong Lewis acids such as SnCl4 are not considered good esterification catalysts. The dialkyltin(IV) compounds form distinct ladder-like structures, distannoxanes, which can incorporate an alcohol and activate the carbonyl function of the carboxylic acid.8–11 Although still under debate, the catalytic activity of these distannoxanes is often attributed to the unique cooperative behavior of the tin atoms in the distannoxanes.12–14 For applications of distannoxanes as catalysts in transesterification reactions see the work of Otera and co-workers.15–17Monoalkyltin(IV) catalysts have received far less attention than the distannoxanes. Nevertheless the chemistry of n-butyltin(IV) complexes with carboxylic acids has been well studied by X-ray crystallography (Scheme 1).18,19 The complex with the highest carboxylate to tin ratio, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, is n-butyltin tricarboxylate and is synthesized from n-butyltin trichloride and the appropriate silver carboxylate (Scheme 2).20 This hydrolytically unstable complex decomposes in the presence of water to undefined polymeric material or to a ladder-shaped complex, [(n-BuSn(O)OOCR)2-n-BuSn(OOCR)3]2, with a tin to carboxylate stoichiometry of 3:5.21 Further hydrolysis results in the formation of a drum-shaped cluster with a tin to carboxylate stoichiometry of 1:1. This [n-BuSn(O)OOCR]6 cluster can also be obtained from the reaction of polymeric n-butylstannoic acid with a carboxylic acid in the appropriate ratio.22,23 Although all of these multinuclear tin complexes have been synthesized and spectroscopically analyzed, their relevance under catalytic conditions remains largely unexplored. It is well possible that under catalytic conditions these tin clusters disintegrate, in contrast to the distannoxanes, and esterification happens via a mononuclear mechanism. Here, we report on our mechanistic investigation of mono-n-butyltin(IV)-catalyzed esterification. This class of catalysts was studied under catalytically relevant conditions with a variety of spectroscopic techniques complemented with DFT calculations.
:1, is n-butyltin tricarboxylate and is synthesized from n-butyltin trichloride and the appropriate silver carboxylate (Scheme 2).20 This hydrolytically unstable complex decomposes in the presence of water to undefined polymeric material or to a ladder-shaped complex, [(n-BuSn(O)OOCR)2-n-BuSn(OOCR)3]2, with a tin to carboxylate stoichiometry of 3:5.21 Further hydrolysis results in the formation of a drum-shaped cluster with a tin to carboxylate stoichiometry of 1:1. This [n-BuSn(O)OOCR]6 cluster can also be obtained from the reaction of polymeric n-butylstannoic acid with a carboxylic acid in the appropriate ratio.22,23 Although all of these multinuclear tin complexes have been synthesized and spectroscopically analyzed, their relevance under catalytic conditions remains largely unexplored. It is well possible that under catalytic conditions these tin clusters disintegrate, in contrast to the distannoxanes, and esterification happens via a mononuclear mechanism. Here, we report on our mechanistic investigation of mono-n-butyltin(IV)-catalyzed esterification. This class of catalysts was studied under catalytically relevant conditions with a variety of spectroscopic techniques complemented with DFT calculations.
 | ||
| Scheme 1 Stoichiometric reactions of monoalkyltin complexes with carboxylic acids compared to their behavior under catalytic esterification conditions. | ||
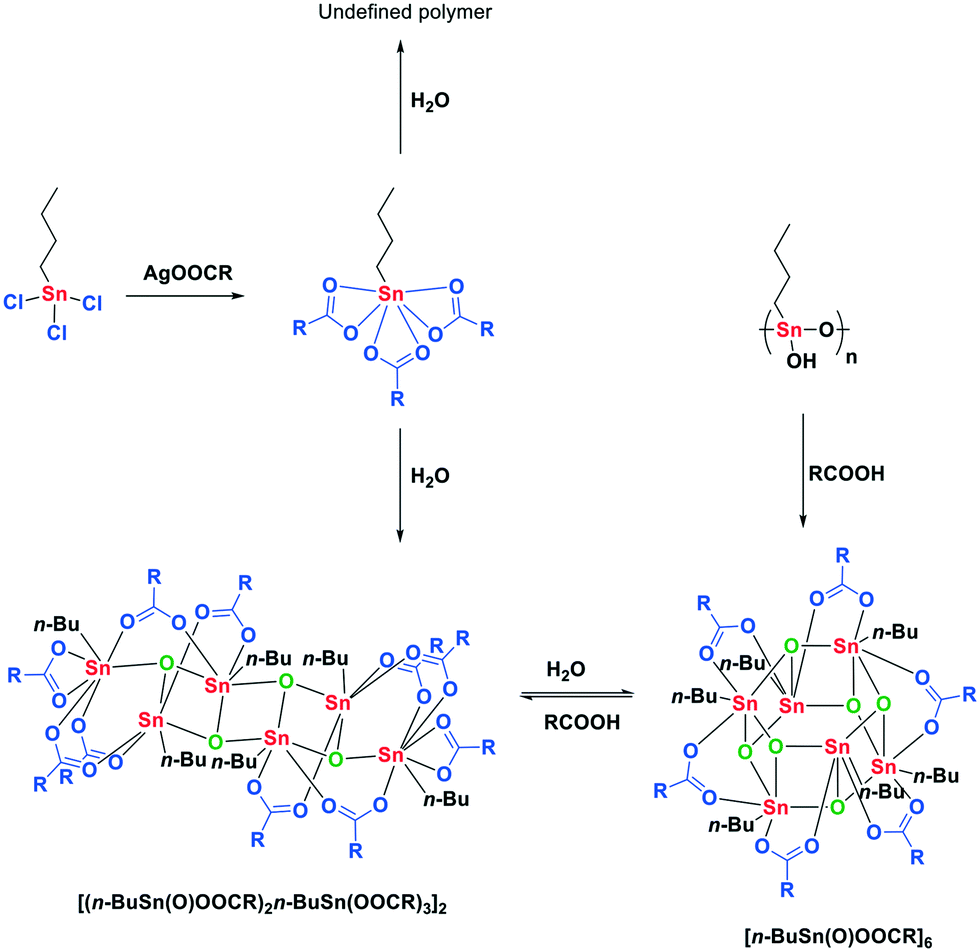 | ||
| Scheme 2 Reaction of monoalkyltin complexes with carboxylic acids. | ||
Results and discussion
We started our investigation by establishing the catalytic activity of various tin(IV) compounds. Also n-butylstannoic acid and acetic and benzoic acid derivatives thereof were examined in a model esterification reaction between benzoic acid and heptanol (in a 1:10 ratio) in the presence of 1 mol% catalyst (Table 1). An excess of alcohol was used in order to exclude the need of dehydrating agents or azeotropic distillation.
|
|
|||
|---|---|---|---|
| Entrya | Complex | Conv. [%] benzoic acid | Yield [%] heptyl benzoate |
| a All reactions were performed with benzoic acid (5 mmol), heptanol (50 mmol), and catalyst (1 mol%, 0.05 mmol), at 150 °C for 6 h. Yield and conversion were determined by GC analysis with pentadecane as internal standard. | |||
| 1 | No cat. | 10 | 6 |
| 2 | Sn(OAc)4 | 23 | 23 |
| 3 | SnCl4·5H2O | 31 | 31 |
| 4 | n-BuSnCl3 | 52 | 50 |
| 5 | (n-BuSnOOH)n (1) | 82 | 82 |
| 6 | [n-BuSn(O)OAc]6 (2) | 87 | 87 |
| 7 | n-BuSn(OAc)3 (3) | 63 | 63 |
| 8 | n-BuSn(OBz)3 (4) | 89 | 87 |
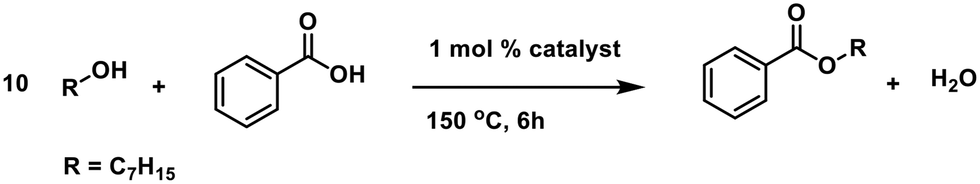
There is a clear difference in catalytic activity between the tin(IV) compounds and the n-butyltin(IV) substituted derivatives, with the latter being more active. The polymeric n-butylstannoic acid (1), the drum-shaped [n-BuSn(O)OAc]6 (2) and monomeric n-BuSn(OBz)3 (4) display the highest activity (Table 1, entries 5, 6 and 8). These three compounds have comparable activity, suggesting that catalytic activity is not related to unique properties of the n-butylstannoic acid polymer. Counter-intuitively, n-BuSn(OAc)3 (3) shows inferior performance, which we ascribe to the slow displacement of the tin-bound acetates for benzoate groups. In addition, an order of 0.74 in catalyst (Fig. S2†) was found for 1 (vide infra for interpretation).
In order to study the behavior of n-butyltin(IV) complexes under catalytically relevant conditions we investigated solutions of 1, 2 and 3 by NMR spectroscopy. With three NMR-active nuclei with spin ½, 119Sn being the most sensitive, Sn NMR provides a valuable tool for examining the coordination environment of tin complexes in solution.24
Various organotin complexes retain their geometry in anhydrous organic solvents, as demonstrated by 119Sn NMR experiments.18,20 In anhydrous CDCl3 a singlet in the 119Sn NMR spectrum at −480 ppm is observed for cluster 2 and from 1H NMR a 1:1 ratio of the acetate groups and the n-butyl tails can be deduced.22,25 For monomer 3 in anhydrous CDCl3 a signal at −532 ppm is present, which is in the expected range of a seven-coordinate tin complex. The 1H NMR spectrum displays the expected 3:1 ratio of the acetate groups and the n-butyl tail. For polymeric 1 no 1H NMR spectra could be obtained due to its insoluble nature in common organic solvents.
After establishing the NMR shifts in CDCl3 we switched to acetic acid-d4 as solvent and measured the tin precursors at various temperatures (Table 2). At 298 K complex 3 shows a singlet at −528 ppm. This minor upfield shift compared to 3 in CDCl3 indicates that the seven-coordinate geometry of n-BuSn(OAc)3 is retained in acetic acid (AcOH). Upon increasing the temperature to 363 K a downfield shift to −518 ppm, Δδ 10.5 ppm, is observed which is caused by a decreased temperature-induced shielding (Table 2, column 2).26 Upon dissolving 2 in AcOD-d4 at 298 K a singlet at −528 ppm, corresponding to monomer 3, and a peak at −553 ppm is observed (Table 2, column 3). Upon heating to 363 K we again only observe a singlet at −517 ppm indicative for complex 3. In addition, this process turned out to be reversible upon lowering the temperature to 298 K. For 1 three peaks at −529, −554 and −592 ppm are present at 298 K (Table 2, column 4). Likewise, heating to 363 K resulted in a single signal at −517 ppm. In addition, in 1H and 13C NMR we observed a similar trend with identical spectra for complexes 1, 2 and 3 (Fig. S41–S47†) at elevated temperatures.
| Temp. (K) | 3 n-BuSn(OAc)3 | 2 [n-BuSn(O)OAc]6 | 1 (n-BuSnOOH)n |
|---|---|---|---|
| a 119Sn chemical shifts are given in ppm relative to Sn(CH3)4. All samples are 0.4 M in Sn and measured in AcOD-d4. | |||
| 298 | −528 | −528 | −529 |
| −553 | −554 | ||
| −592 | |||
| 333 | −523 | −523 | −524 |
| −553 | −554 | ||
| 363 | −518 | −517 | −517 |
These results indicate that these precursors in acetic acid at catalytically relevant temperatures (≥363 K) rapidly form a mononuclear tin tricarboxylate in situ (Fig. 1). To obtain more insight into the origin of the two high-field signals around −554 and −591 ppm, which appear upon solvation of 1 and 2 in AcOD-d4 at 298 K, we treated complex 3 with various equivalents of D2O (Fig. 2). The addition of D2O resulted in identical 119Sn chemical shifts, which reveals that the formation of these species is dependent on the concentration of water, rather than Sn atoms.
 | ||
| Fig. 1 Formation of n-butyltin triacetate upon dissolution of multimetallic n-butyltin complexes in carboxylic acid at catalytically relevant temperatures. | ||
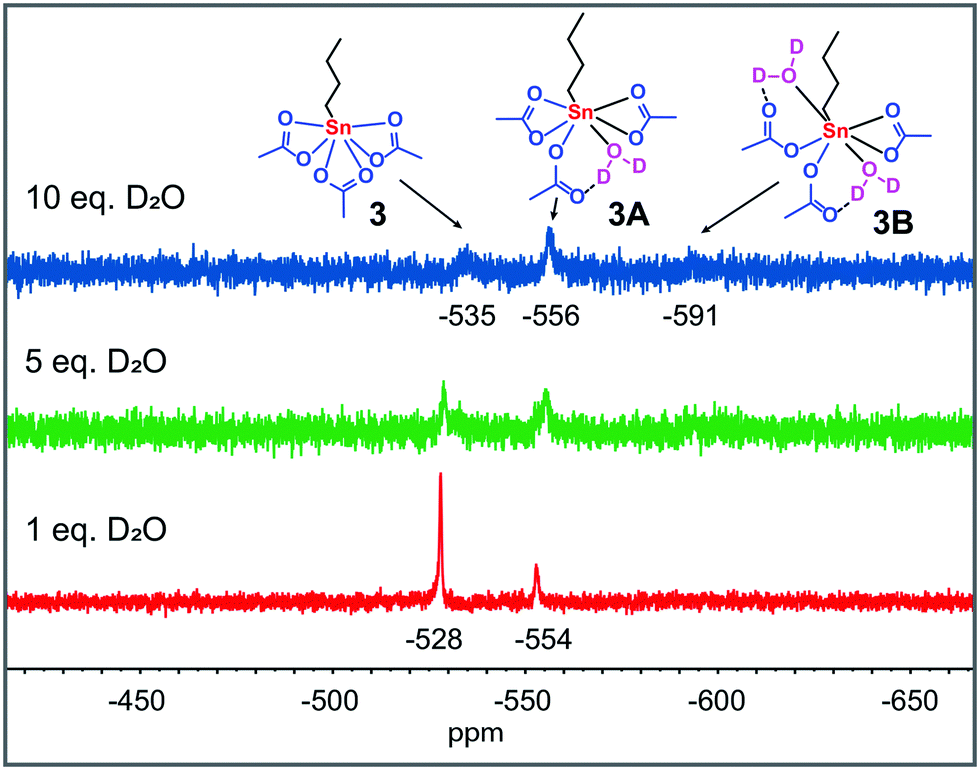 | ||
| Fig. 2 119Sn{H} NMR spectra (186 MHz, 298 K) of the reaction of 3 (0.4 M) with various equivalents of D2O in AcOD-d4 at 298 K. 119Sn chemical shifts are given in ppm relative to Sn(CH3)4. | ||
Upon dissolution in AcOD-d4, the oxo- and hydroxo-moieties in 2 and 1 become protonated and subsequently water is formed in situ. Therefore, we propose that these two shifts emerge from 3 with either one (−554–−556 ppm) or two (−591–−592 ppm) D2O molecules coordinated. Similar chemical shifts are observed for the addition of D2O to 3 dissolved in a 1:1 mixture of AcOH and EtOH (Fig. S49†).
More structural information on the formed tin complexes was accessed via119Sn NMR DFT calculations on a B3LYP/TZVPP all electron level of theory.27,28 Evaluation of the 119Sn NMR chemical shift of various conformers of 3 with one (Fig. S5†) and two water (Fig. S6†) molecules coordinated to the central tin atom resulted in two structures with a good match between experimental and calculated 119Sn NMR chemical shift (Fig. 3).
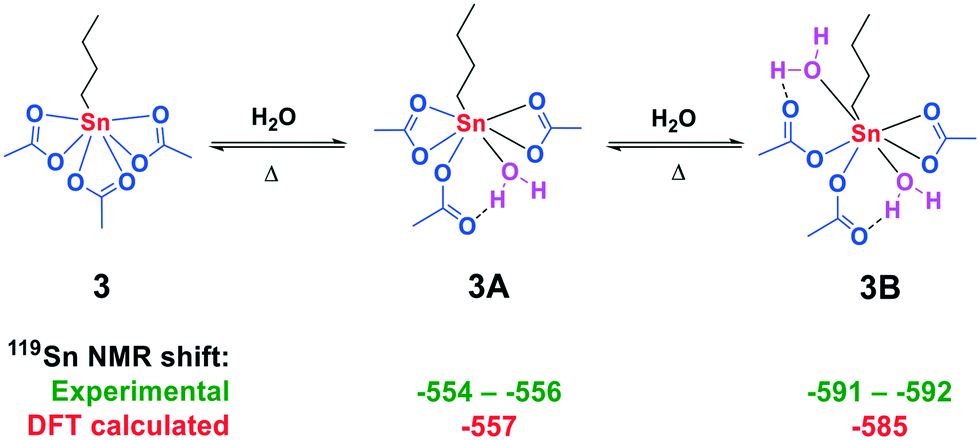 | ||
| Fig. 3 Formation of adducts 3A and 3B from n-BuSn(OAc)3 with H2O in AcOH. Experimental (green) and calculated (red) 119Sn chemical shifts (in ppm). | ||
For the monohydrate complex 3A, one water molecule is bound trans to the n-butyl group and one of the acetate groups has changed coordination mode from bidentate to monodentate in order to accommodate a hydrogen bond between the carbonyl oxygen and the proton of water. For structure 3B a second water molecule is bound to the central tin core which results in two of the acetate groups adopting a monodentate coordination mode. The coordination of water to structure 3A and 3B proved to be reversible upon increasing the temperature, resulting in the sole formation of 3 (Fig. S50 and S51†).
The effect of water on 3 in AcOH was further studied by ATR-FTIR spectroscopy via the addition of various equivalents of H2O to 3 in AcOH. Vibrations at 1588 (νCOO), 660 (νSn–O) and 568 cm−1 (νSn–O) appeared to be correlated to the H2O concentration (Fig. S52 and S53†). Similar experiments using H218O resulted in a small shift in one of the Sn–O vibrational modes from 568 to 554 cm−1 (Fig. 4). This isotope effect further underlines the interaction between 3 and the oxygen atom from the water molecule(s). Furthermore, experiments with D2O instead of H2O resulted only in a minor shift in the COO vibrational mode from 1588 to 1575 cm−1 (Fig. S54†).
 | ||
| Fig. 4 ATR-FTIR spectra of 3 in AcOH with 2, 5 and 10 equivalents of H216O (dashed lines) and H218O (solid lines). | ||
Knowing its behavior in AcOH in both the presence and absence of water, we turned our attention to the 119Sn NMR spectrum of 3 in anhydrous EtOH at 298 K. Under these conditions two signals at −532 and −541 ppm were observed, revealing the partial conversion of 3 into another seven-coordinate tin complex (Fig. S55†). The structure of this complex was further elucidated with ATR-FTIR which gave a strong vibration at 1020 cm−1, indicative of a C–O stretching vibration (Fig. S58†).29 In addition, a vibration at 1711 cm−1 revealed the presence of uncoordinated AcOH. Only minor isotope effects were observed upon changing EtOH to EtOD-d6 (Fig. S59†). These findings indicate the exchange of one of the acetate groups for an ethoxy group. Subsequently, an aliquot of this mixture was studied with LIFDI-MS which resulted in the detection of the fragments [SnOAc3]+ and [n-BuSnOAc2OEt–n-BuSnOAcOEt]+ (Fig. S60 and S61†). The in situ formation of the dimer (n-BuSnOAc2OEt)2 (5) is in agreement with a seven-coordinate environment around the tin core as indicated by 119Sn NMR. In addition, the dimer 5 was independently synthesized via the reaction of n-BuSn(OAc)3 and n-BuSnOEt3, and has a 119Sn NMR chemical shift of −544 ppm in CDCl3.30 The bridging mode of the ethoxy groups in 5 becomes apparent from the distinct quaternary carbon signal at 182.61 ppm observed for the acetate groups in the 13C NMR spectra at 233 K, indicating that all acetate groups are in a terminal position (Fig. S33†). Discrimination between the different conformers is possible upon comparison of the experimental and DFT-calculated 119Sn NMR chemical shift together with the calculated free energy differences (Fig. 5).
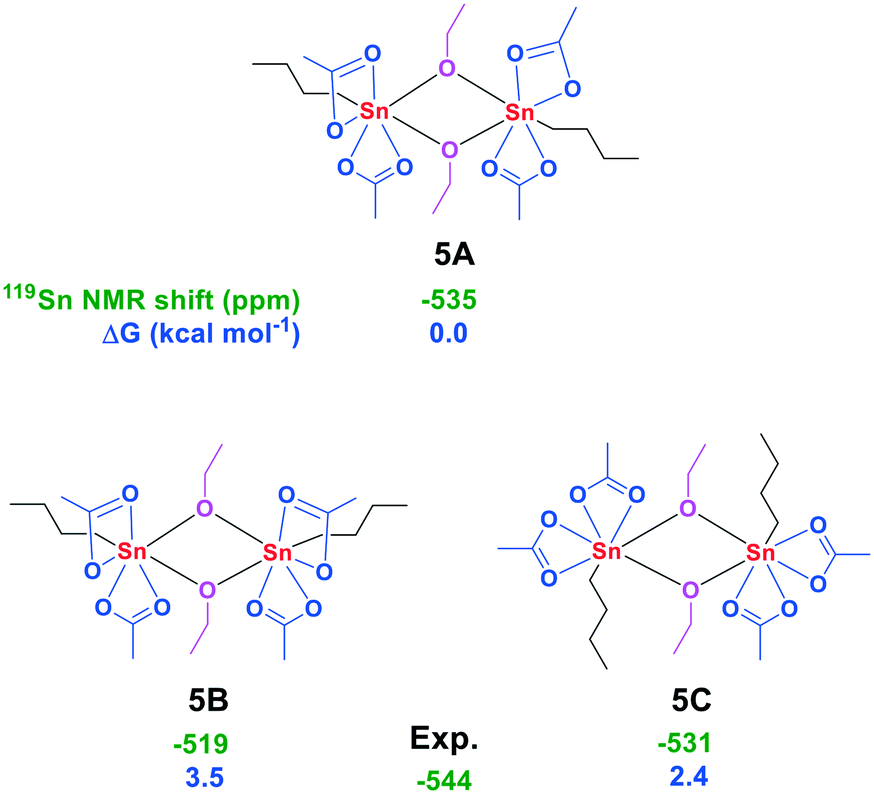 | ||
| Fig. 5 Conformers of 5 with their 119Sn NMR chemical shift (green, in ppm) and calculated free energies (blue, in kcal mol−1 relative to n-BuSn(OAc)3). | ||
These data together point to the formation of the isomer with both n-butyl groups occupying the trans-position with respect to the bridging ethoxy groups and transoid with respect to the other n-butyl group (complex 5A). Solvation of 5A in AcOH resulted in a 119Sn NMR chemical shift at −529 ppm, which indicates complete conversion to 3 (Fig. S62†). We thus propose the partial formation of the dimer 5A when 3 is dissolved in EtOH, while addition of AcOH results in the backwards reaction to 3.
With the knowledge of the behavior of the components under various conditions in hand, we turned our attention to following the catalyst under catalytically relevant conditions. The catalytic esterification of acetic acid and ethanol at 363 K, in the presence of molecular sieves to mimic the azeotropic distillation conditions commonly applied, was monitored over time (18.7% yield ethyl acetate after 1 h, Fig. S66†). During catalysis, aliquots of the catalytic reaction mixture were examined by 119Sn NMR spectroscopy. At 363 K a single 119Sn chemical shift is observed which moves upfield from −525 to −539 ppm over a time period of 60 minutes (Fig. S64†), which we attribute to a shift in equilibrium between monomer 3 and dimer 5A. 119Sn NMR measurements of the same samples at 298 K revealed two broad signals with a chemical shift of −535 and −555 ppm after 5 minutes reaction time (Fig. 6). These two signals can be assigned to 3 and monohydrated complex 3A respectively. After 30 minutes reaction time three broad signals at −545, −553 and −590 ppm were observed. The two upfield signals originate from n-BuSn(OAc)3 with either one or two water molecules coordinated (structures 3A and 3B), while the signal at −545 ppm is consistent with the dimer 5A. After 60 minutes reaction time two new unidentified signals at −550 and −553 are present but the main contributions are still from structures 3A, 3B and 5A.
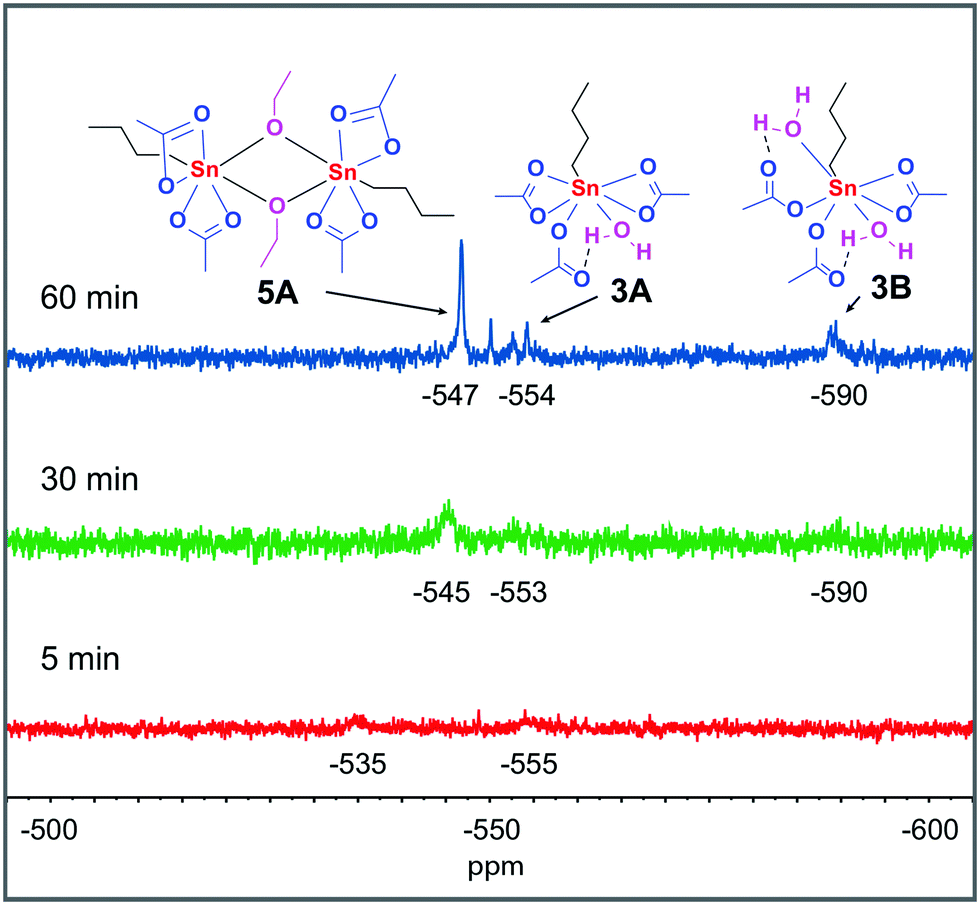 | ||
| Fig. 6 Time dependent 119Sn{H} NMR spectra (186 MHz, 298 K) of complex 3 in a 1:1 mixture of AcOH and EtOH. All samples are 0.4 M in Sn and measured unlocked. | ||
The adducts of 3 with H2O (structure 3A and 3B) are only observed at 298 K, at elevated temperatures water is expected to be expelled from these complexes and monomeric 3 is formed, vide supra.
To complement our experimental findings under catalytically relevant conditions, we have further examined the reaction mechanism for the n-BuSn(OAc)3-catalyzed esterification by DFT calculations at the BP86-D3/def2-TZVP//M06-2X-D3/def2-TZVP/def2-QZVPP level of theory (Scheme 3).31 The observed off-cycle resting states, monomer 3 and dimer 5A, are equal in free energy, and both can lead to the formation of intermediate D (ΔG = 4.5 kcal mol−1) which is the starting points of the catalytic cycle. The tin-bound ethoxy group can subsequently accommodate another ethanol molecule via a hydrogen bond (structure E). Next, nucleophilic attack (ΔG‡ = 18.0 kcal mol−1) results in intermediate F, where the hydrogen atom of the ethanol is accepted by the Lewis basic ethoxy group. A rotation (ΔG‡ = 17.3 kcal mol−1), which requires the breakage of a hydrogen bond, pre-organizes intermediate G for the carbon–oxygen bond breaking step. The actual carbon–oxygen bond breaking has the highest barrier with ΔG‡ = 20.0 kcal mol−1 and can be regarded as the rate determining step. The collapse of the tetrahedral intermediate G as turnover-limiting step is consistent with previously reported metal-catalyzed esterification and amidation reactions.32,33 From structure H water is expelled, which was bound via two hydrogen bonds, and finally the ester is replaced by a new acetic acid molecule. Overall the reaction is slightly exergonic with ΔΔG = −1.9 kcal mol−1.
 | ||
| Scheme 3 Proposed catalytic cycle for the esterification of ethanol and acetic acid catalyzed by 3. Hydrogen bonds are indicated with black dashed lines and free energy values are given in kcal mol−1 relative to 3. | ||
The experimentally observed off-cycle resting states 3 and 5A can both form the amphoteric mixed alkoxide/carboxylate (structure D) which is the starting point of the monomeric catalytic cycle. Furthermore, a monomeric catalytic cycle, with two off-cycle resting states, one monomeric and the other dimeric, being in equilibrium with each other and equal in energy, is in agreement with the experimentally determined catalyst order of 0.74, vide supra.
The in situ formation of this amphoteric catalyst is essential since it can act as a Lewis acid to activate the carboxylic acid, and as a Brønsted base to deprotonate the alcohol during nucleophilic attack. The importance of having an amphoteric catalyst in esterification reactions is not only restricted to tin-based catalysts as we have recently demonstrated for titanium-based esterification catalysts.34 Furthermore, the function of the n-butyl tail on the tin catalyst is to enforce a seven-coordinate tin center which remains during the whole catalytic cycle as established by DFT calculations.
Conclusions
We have demonstrated here that n-butyl substituted tin(IV) complexes have a unique coordination chemistry which results in a monomeric tin catalyst under catalytic conditions. Although the commonly applied esterification catalyst n-butyl stannoic acid is a polymer in the solid state, it transforms to monomeric 3 and dimeric 5A as off-cycle resting states under catalytically relevant conditions, as proven by NMR, ATR-FTIR and MS measurements. DFT calculations lend support to a monomeric mechanism where structures 3 and 5A are regarded as off-cycle species. In this mechanism the carbon–oxygen bond breaking step is the rate determining step. Furthermore, water formed during the esterification reaction has limited effect on the active catalyst, since the water adducts 3A and 3B where only observed at 298 K. These findings shed new light on the role of the n-butyl tail and the nuclearity of the class of mono-n-butyl tin esterification catalysts.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is part of the Advanced Research Center for Chemical Building Blocks, ARC CBBC, which is co-founded and co-financed by the Netherlands Organization for Scientific Research (NWO, contract 736.000.000) and the Netherlands Ministry of Economic Affairs and Climate. In addition, the authors thank NWO for funding VENI grants 722.016.012 (to T. J. K.) and 016.Veni.192.05 (to J. J. H.). The authors thank Dr. Andreas Ehlers and Ed Zuidinga for NMR spectroscopy and mass spectrometry support.Notes and references
- J. Otera and J. Nishikido, Esterification. Methods, Reactions, and Applications, Wiley-VCH Verlag GmbH & Co., Weinheim, 2nd edn, 2010 Search PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University press, Oxford, 2nd edn, 2012 Search PubMed.
- J. J. Li, Fischer-Speier esterification, in Name Reactions, Springer, Berlin, Heidelberg, 2002 Search PubMed.
- A. G. Davies, J. Chem. Res., 2010, 34, 181–190 CrossRef CAS.
- S. Kenneth, WO2017/091437A1, 2013, p. 21.
- M. R. Meneghetti and S. M. P. Meneghetti, Catal. Sci. Technol., 2015, 5, 765–771 RSC.
- M. A. Hossain, M. A. Mohamed Iqbal, N. M. Julkapli, P. San Kong, J. J. Ching and H. V. Lee, RSC Adv., 2018, 8, 5559–5577 RSC.
- J. Otera, N. Dan-oh and H. Nozaki, Tetrahedron, 1993, 49, 3065–3074 CrossRef CAS.
- J. Otera, N. Dan-oh and H. Nozaki, J. Org. Chem., 1991, 56, 5307–5311 CrossRef CAS.
- T. Yano, K. Nakashima, J. Otera and R. Okawara, Organometallics, 1985, 4, 1501–1503 CrossRef CAS.
- J. Bonetti, C. Gondard, R. Petiaud and A. Michel, J. Organomet. Chem., 1994, 481, 7–11 CrossRef CAS.
- J. Otera, N. Dan-oh and H. Nozaki, J. Org. Chem., 1991, 56, 5307–5311 CrossRef CAS.
- E. Crawford, T. Lohr, E. M. Leitao, S. Kwok and J. S. McIndoe, Dalton Trans., 2009, 9110–9112 RSC.
- B. Jousseaume, C. Laporte, M. C. Rascle and T. Toupance, Chem. Commun., 2003, 1428–1429 RSC.
- J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- J. Otera, Acc. Chem. Res., 2004, 37, 288–296 CrossRef CAS.
- D. L. An, Z. Peng, A. Orita, A. Kurita, S. Man-E, K. Ohkubo, X. Li, S. Fukuzumi and J. Otera, Chem. – Eur. J., 2006, 12, 1642–1647 CrossRef CAS.
- V. Chandrasekhar, C. G. Schmid, S. D. Burton, J. M. Holmes, R. O. Day and R. R. Holmes, Inorg. Chem., 1987, 26, 1050–1056 CrossRef CAS.
- V. Chandrasekhar, S. Nagendran and V. Baskar, Coord. Chem. Rev., 2002, 235, 1–52 CrossRef CAS.
- H. H. Anderson, Inorg. Chem., 1964, 3, 912–914 CrossRef CAS.
- R. R. Holmes, C. G. Schmid, V. Chandrasekhar, R. O. Day and J. M. Holmes, J. Am. Chem. Soc., 1987, 109, 1408–1414 CrossRef CAS.
- V. B. Mokal, V. K. Jain and R. T. Tiekink, J. Organomet. Chem., 1991, 407, 173–180 CrossRef CAS.
- M. C. Sharps, D. A. Marsh, L. N. Zakharov, J. E. Hutchison and D. W. Johnson, Cryst. Res. Technol., 2017, 52, 1–7 CrossRef.
- J. C. Martins, M. Biesemans and R. Willem, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 271–322 CrossRef CAS.
- G. Prabusankar and R. Murugavel, Organometallics, 2004, 23, 5644–5647 CrossRef CAS.
- J. D. Kennedy, J. Chem. Soc., Perkin Trans. 2, 1977, 2, 242–248 RSC.
- G. Casella, F. Ferrante and G. Saielli, Inorg. Chem., 2008, 47, 4796–4807 CrossRef CAS.
- P. Avalle, R. K. Harris and R. D. Fischer, Phys. Chem. Chem. Phys., 2002, 4, 3558–3561 RSC.
- C. T. Lynch, K. S. Mazdiyasni, J. S. Smith and W. J. Crawford, Anal. Chem., 1964, 36, 2332–2337 CrossRef CAS.
- D. P. Gaur, G. Srivastava and R. C. Mehrotra, J. Org. Chem., 1973, 63, 221–231 CrossRef CAS.
- C. Müller, D. M. Andrada, I. A. Bischoff, M. Zimmer, V. Huch, N. Steinbrück and A. Schäfer, Organometallics, 2019, 38, 1052–1061 CrossRef.
- P. Villo, O. Dalla-Santa, Z. Szabó and H. Lundberg, J. Org. Chem., 2020, 85, 6959–6969 CrossRef CAS.
- H. Lundberg, F. Tinnis, J. Zhang, A. G. Algarra, F. Himo and H. Adolfsson, J. Am. Chem. Soc., 2017, 139, 2286–2295 CrossRef CAS.
- L. A. Wolzak, J. I. van der Vlugt, K. J. van den Berg, J. N. H. Reek, M. Tromp and T. J. Korstanje, ChemCatChem, 2020, 12, 5229–5235 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2049109. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cy00184a |
| This journal is © The Royal Society of Chemistry 2021 |