
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructured hydroxyapatite composites as efficient solid base catalysts for condensation reactions†
Tharun
Jose
a,
Jamal
Ftouni
b and
Pieter C. A.
Bruijnincx
 *a
*a
aOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl
bOmya International AG, Froschackerstrasse 6, CH-4622 Egerkingen, Switzerland
First published on 12th April 2021
Abstract
Herein, we report the use of structured hydroxyapatite composite (SHCs) as highly efficient and recyclable solid base catalysts for various condensation reactions. Catalyst performance as function of catalyst loading, reaction time and reaction temperature were studied in the solventless self-aldol condensation reaction of butyraldehyde to 2-ethylhexenal under mild reaction conditions. SHC catalysts were found to outperform benchmark solid base catalysts such as MgO, TiO2, calcium carbonate and hydroxyapatites. Characterization of the synthesized SHC catalysts by a range of surface analysis, spectroscopic and electron microscopy techniques, showed that a moderate acid/base ratio and high BET surface area to be key to their high efficiency. Furthermore, recycling experiments showed the catalyst to be stable over multiple runs. Moreover, the most active SHC catalyst was investigated in other prototypical condensation reactions such as the Knoevenagel condensation, Claisen–Schmidt condensation and Henry reaction, again showing excellent performance. These results highlight the versatility of these SHC materials and their potential for industrial employment as solid base catalysts.
Introduction
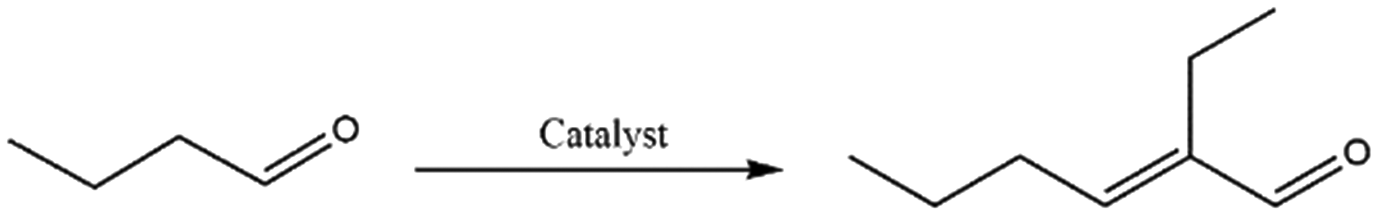
Catalytic condensation reactions are an important tool to upgrade platform chemicals by C–C bond formation to give higher value products used for applications ranging from transportation fuels to fine or specialty chemicals.1,2 Some of the prototypical condensation reactions include the aldol, Knoevenagel, Henry and Claisen–Schmidt condensations, among others. For instance, butyraldehyde's self-aldol condensation product 2-ethylhexenal is used as an intermediate for the synthesis of insecticides, plasticizers and other fine chemicals.3 Its derivative 2-ethylhexanoic acid is, for example, used in cosmetics, pharmaceuticals, paints and coatings.4 Alternatively, the reduction of 2-ethylhexenal yields 2-ethylhexanol, which finds application as or is used as precursor to plasticizers, wetting agents, solvents, fragrances and lubricants5 and is produced at a volume of 2.5 mton per year.6 Knoevenagel and Henry reactions in turn yield, for example, valuable chalcones and nitroarenes used in the production of pharmaceuticals, rubber, plastics, flavonoids and dyes.7–9 Recently, condensation reactions have become even more significant as a tool to upgrade bio-derived feedstock, offering the opportunity to produce longer carbon chain products with high atom economy and with limited generation of (harmless) by-products.10 In particular, aldol condensation reactions are being heavily studied in the context of bio-oil upgrading, as they achieve oxygen removal from the aldehyde fraction of bio-oils, which can make up to 20 wt% of this feed, at no carbon loss. Similarly, such condensation reactions are often central to the upgrading of oxygenated biobased platform molecules.10,11Liquid phase aldol condensation reactions are typically base-catalyzed, either by soluble bases or by solid materials such as alkali-metal containing zeolites,12 alkaline-earth metal oxides,13 and hydrotalcites.14–16 Solid base use can indeed provide considerable advantages in terms of sustainability when it comes to waste generation, catalyst recovery, solvent use, etc.17,18 Challenges in solid base-catalyzed conversions, on the other hand, include deactivation of the catalyst by poisoning, as well as catalyst instability issues, e.g. by leaching of the material into solution.19 To address these challenges, the development of new solid base catalysts that show improved performance and are, preferably, made from readily obtainable materials, is therefore a subject of considerable interest.
In this context, hydroxyapatites (HAPs), naturally occurring minerals with the general formula Ca10(PO4)6(OH)2, have shown to be excellent catalysts for Knoevenagel, Claisen–Schmidt, Henry and aldol condensation reactions.11,20–28 HAP is the most stable calcium phosphate salt and the principal inorganic constituent of human bones and teeth.29,30 HAPs receive considerable attention as their ability to form solid solutions, to accept many anionic and cationic substituents, and to tune its acid/base properties makes them highly versatile as heterogeneous catalysts for liquid-phase condensation reactions.20
HAPs are highly non-stoichiometric solids, with the Ca/P ratio commonly being used as an index of non-stoichiometry (the stoichiometric ratio being 1.67) as well as of their acid–base properties.30,31 It is well known that HAPs are predominantly acidic when Ca/P ∼1.50, show both acidic and basic characteristics when the Ca/P ratio is between 1.50 and 1.67 and are predominantly basic when the ratio is ≥1.67. Carbonate-rich apatites are more basic and typically synthesized by co-precipitation of sodium carbonates, or by means of addition of atmospheric CO2 as CO32− anions, which in turn compensate the charge of excess calcium cations in the apatite solids. The use of calcium carbonate (CC) as a source of calcium for carbonate-rich HAP synthesis has advantages over the normally employed sources, such as calcium nitrate, phosphate, chloride or acetate.11,20,31–33 The drawbacks of these non-CC calcium salts are their relatively high costs and the waste originating from associated counterions and laborious washing steps. Indeed, CC-derived, carbonate-rich HAP materials thus constitute a class of natural and abundant materials that can be exploited for their enhanced basicity, given that their Ca/P ratio is larger than 1.67.34–36 In addition to the catalyst precursors used, the choice of synthesis route, also to a large extent, determines catalyst properties as well as the ease of material synthesis scale-up. For example, HAP synthesis is often carried out via sol–gel procedures, making its production in industrial amounts challenging. Alternatively, a method has been disclosed to synthesize CC-based, carbonate-rich HAP minerals,37 by etching CC and reprecipitating carbonated HAP materials as structured hydroxyapatite composites (SHCs) by externally supplying CO2 in the form of carbonic acid, in the presence of phosphate ions. This procedure allows considerable variation of the CC content in the final material. This synthesis approach thus offers advantages over alternative routes and the acid/base properties of the materials were anticipated to be promising for condensation catalysis. We report the use of these SHC materials as solid base catalysts, first exploring their performance in the self-aldol condensation of butyraldehyde and later expanding the scope to other condensation reactions. Detailed characterization data is presented, as well as the development of a regeneration protocol to allow SHC reuse.
Results and discussion
Five SHC materials were used in this study, varying in phase composition and surface area. The mineral compositions of the as-synthesized SHC materials were determined by Rietveld refinement of the powder X-ray diffraction (XRD) patterns (Fig. 2 and Table S1, ESI†). The materials consist mainly of CC and HAP components, with SHC-5 containing an additional amount of octa-calcium phosphate. The CC and HAP percentages in SHC-3, SHC-4 and SHC-5 range from 11–15 and 79–85%, respectively. SHC-1 and SHC-2 are more CC rich at 50% and 79%, respectively. Thermal gravimetric analysis (TGA-DSC) of the SHC samples (Fig. S1†) showed them to be thermally stable up to 600 °C. The SHC solids all have a similar morphology. The SEM images (Fig. 1) showed that the surface of the SHCs is characterized by lamellar HAP surrounded by agglomerated fine CC particulates. It was previously shown by FIB-SEM analysis that the structure of the particles consists of a CC core surrounded by a HAP shell.38 | ||
| Fig. 1 SEM images of the SHC solid samples. | ||
Table 1 displays the bulk (from ICP-OES) and surface (from XPS) compositions of all SHC catalysts and the Ca/P ratio, which, as noted above, is indicative of the acid/base properties of the materials.20,31 The data shows that all the catalysts have a Ca/P ratio higher than 1.67, suggesting them to be predominantly basic (vide infra). Activation is mandatory for most of the solid base catalysts prior to reaction, as basic sites may be blocked by CO2 and H2O, resulting in poor activity.19,39 Hence, all catalysts were subjected to thermal activation at 200 °C for 4 h. The nitrogen physisorption measurements in Table 2 show that the thermal activation procedure did not affect the surface areas or pore volumes. The samples showed a considerable difference in surface area, ranging from 21.0 to 156.2 m2 g−1. For comparison, the BET surface area and total pore volume of commercially available solids such as MgO, TiO2, CaCO3 and HAPs are also included, high (HAP-H) and low (HAP-L) surface area examples for the latter. The nitrogen adsorption/desorption isotherm plots for the SHC catalysts are presented in the ESI† (Fig. S2). The thermal treatment also did not alter the phase composition of the materials of any of the samples, as evidenced by the XRD data displayed in Fig. 2. Moreover, the characteristic, sharp (104) diffraction peak of CC at 34° is decreasing as HAP content increases from SHC-1 to SHC-5; on the contrary an increase in the characteristic, strong (211) reflection of HAP at 37° is seen.
| Catalyst | ICP-OES | XPS | ||||
|---|---|---|---|---|---|---|
| Ca (at%) | P (at%) | Ca/P (at. ratio) | Ca (at%) | P (at%) | Ca/P (at. ratio) | |
| SHC-1 | 87.9 | 12.1 | 7.3 | 14.2 | 6.7 | 2.1 |
| SHC-2 | 79.7 | 20.3 | 3.9 | 13.0 | 6.7 | 1.9 |
| SHC-3 | 83.3 | 16.7 | 4.9 | 14.4 | 7.8 | 1.9 |
| SHC-4 | 79.7 | 20.3 | 3.9 | 14.1 | 6.7 | 2.1 |
| SHC-5 | 70.2 | 29.9 | 2.3 | 14.5 | 8.1 | 1.8 |
| Entry | Catalyst | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | ||
|---|---|---|---|---|---|
| Beforea | Afterb | Beforea | Afterb | ||
| a Before thermal activation. b After thermal activation at 200 °C for 4 h. c Publicly available information on commercial samples; n.d.-not determined. | |||||
| 1 | SHC-1 | 21.0 | 19.3 | 0.03 | 0.13 |
| 2 | SHC-2 | 56.7 | 58.1 | 0.10 | 0.41 |
| 3 | SHC-3 | 156.2 | 160.3 | 0.30 | 0.87 |
| 4 | SHC-4 | 86.7 | 85.5 | 0.19 | 0.18 |
| 5 | SHC-5 | 105.5 | 106.7 | 0.67 | 0.65 |
| 6 | MgO | n.d. | 239.5 | n.d. | 0.24 |
| 7 | TiO2 | n.d. | 7.4 | n.d. | 0.03 |
| 8 | CaCO3 | n.d. | 6.6 | n.d. | 0.05 |
| 9 | HAP-Lc | 9.4 | n.d. | n.d. | n.d. |
| 10 | HAP-Hc | 100 | n.d. | n.d. | n.d. |
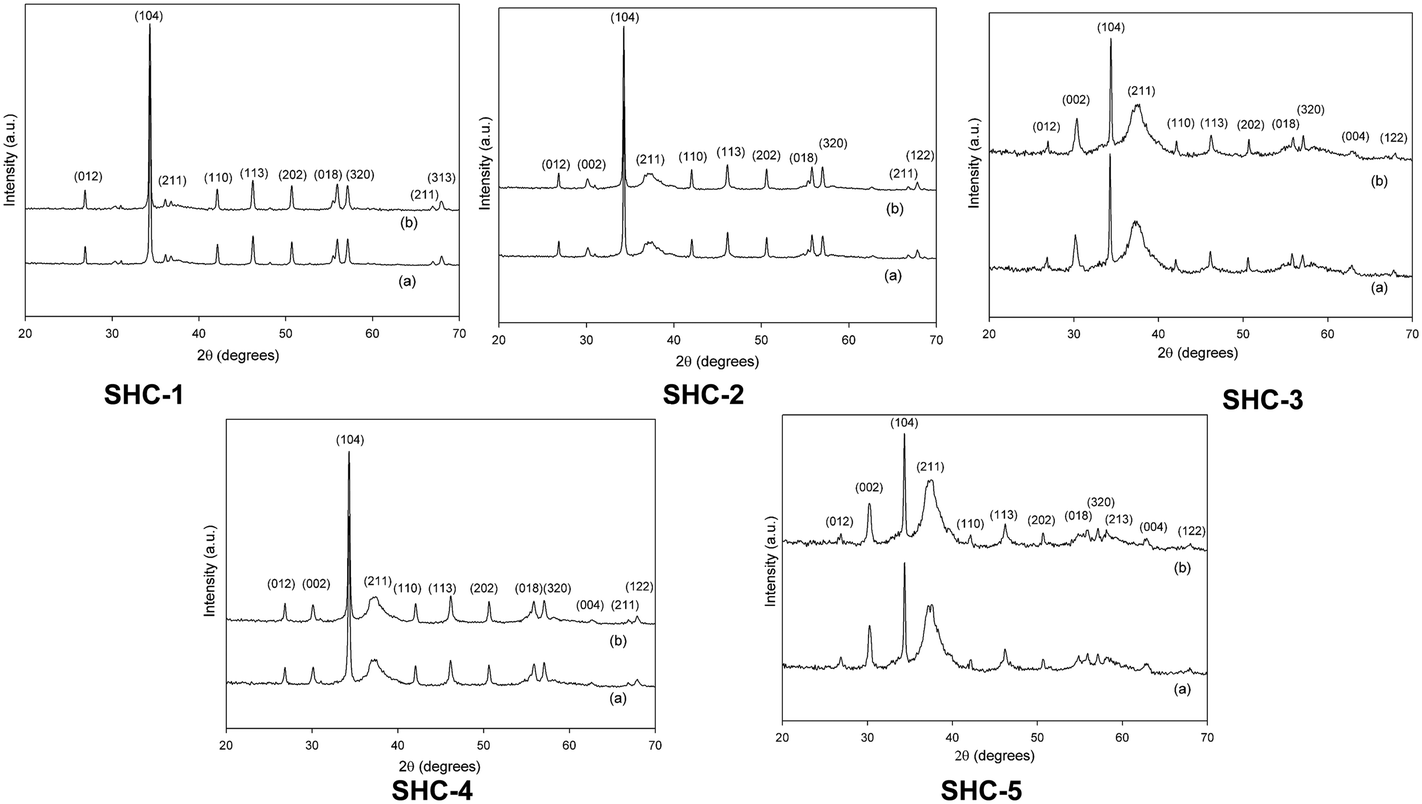 | ||
| Fig. 2 XRD structures of SHC catalysts. (a) Before thermal activation and (b) after thermal activation at 200 °C for 4 h. | ||
The catalysts were first tested for the self-aldol condensation reaction using butyraldehyde as the benchmark substrate, under various experimental conditions. Initial screening conditions consisted of 130 °C for a reaction time of 22 h under solvent-free conditions at 3 mol% catalyst loading, conditions typically described in literature (see Table S2† for an overview of literature results).40–46 The results, presented in Table 3, show that no conversion was obtained in the absence of a catalyst after 2 or 6 h and a 6% yield of the desired product at 8% conversion after 22 h (entry 1). Entries 2 to 6 are commercially available solid base catalysts such as MgO, TiO2, CaCO3 and HAP, and are included for benchmarking. Among the benchmark solids, HAP-H showed the best catalytic activity up to 6 h, whereas for 22 h MgO and HAP-H showed similar performance, both having higher surface area compared to the rest of the commercial solids. Compared to these conventional solid base catalysts, SHC-3, SHC-4 and SHC-5 (entries 9–11) exhibited excellent catalytic activity. While most SHC catalysts reached full conversion after 22 h, the results after 6 h showed the activity to increase in the order SHC-1 < SHC-2 < SHC-4 < SHC-5 < SHC-3. Overall, under the selected reaction conditions, the desired product (2-ethylhexenal) is obtained in good yield and excellent selectivity throughout. As SHCs contain both CC and HAP phases, a physical mixture of CC and HAP-L catalysts (entry 12), in the same proportion as SHC-3, was also tested; this combination proved to be the least active, underperforming even in comparison with the individual components (entry 4 and 5). A comparison of the best performing SHC material (SHC-3) and HAP-H at the same wt% of added catalyst (Table S3†) again showed the former to perform better. The results thus show that the high HAP content materials (SHC-3, SHC-4 and SHC-5) gave the highest yield.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | |||||
| 2 | 6 | 22 | |||||
| X (%) | Y (%) | X (%) | Y (%) | X (%) | Y (%) | ||
| Reaction conditions: temperature = 130 °C; catalyst loading = 3 mol%; X = conversion, Y = yield; HAP-L = hydroxyapatite with lower surface area (9.4 m2 g−1); HAP-H = hydroxyapatite with higher surface area (100 m2 g−1); entry 12, catalyst loading – CaCO3 = 0.5 mol%, HAP-L = 2.5 mol%; n.d. = not determined; conversion and yield were determined by 1H NMR (CDCl3) using mesitylene as an internal standard. | |||||||
| 1 | — | 0 | 0 | 0 | 0 | 8 | 6 |
| 2 | MgO | 0 | 0 | 30 | 29 | 71 | 70 |
| 3 | TiO2 | 0 | 0 | 9 | 9 | 50 | 47 |
| 4 | CaCO3 | 2 | 2 | 40 | 39 | 60 | 59 |
| 5 | HAP-L | 7 | 7 | 41 | 39 | 62 | 60 |
| 6 | HAP-H | 10 | 10 | 53 | 53 | 70 | 68 |
| 7 | SHC-1 | 0 | 0 | 28 | 28 | 50 | 48 |
| 8 | SHC-2 | 9 | 9 | 29 | 29 | 57 | 55 |
| 9 | SHC-3 | 73 | 72 | 100 | 99 | n.d. | n.d. |
| 10 | SHC-4 | 18 | 18 | 70 | 69 | 88 | 85 |
| 11 | SHC-5 | 58 | 57 | 91 | 89 | 100 | 98 |
| 12 | CaCO3 + HAP-L | 3 | 3 | 10 | 10 | 43 | 43 |
The reaction parameters (reaction time, catalyst loading and reaction temperature) were optimized using the most active catalyst (SHC-3) under solvent-free conditions. As the selectivity for the 2-ethylhexenal product was found to be high throughout (>97%), butyraldehyde conversion will be used to describe catalyst performance. Variation of reaction time showed that after 4 h butyraldehyde conversion is almost complete, showing only a very small gain after that (Fig. S3†). Catalyst loading was varied from 0.5 to 3 mol%, resulting in a butyraldehyde conversion increase from 11 to 96% (Fig. S4†). Generally, catalyst loadings of around 10 mol% or higher are described for the self-aldol condensation reactions (Table S2†).40,45–48 Typically, base catalysts are sensitive to atmospheric CO2 and moisture, and deactivation of basic sites occurs to a greater extent compared to acid catalysts.19,39 Moreover, the condensation reaction generates water that can further tamper the basic active sites and require higher catalyst loadings to obtain good catalytic activity.1 The SHC catalysts, however, already showed excellent results at low loading of 3 mol%. The dependence of butyraldehyde conversion on the reaction temperature was also investigated (Fig. S5†). Expectedly, at lower reaction temperatures, the aldol addition products dominate, while at temperatures above 100 °C, typically only the unsaturated condensation products are detected. At temperatures above 200 °C, further condensation to the trimer products could also be observed.28 With the dimer 2-ethylhexenal being the desired product, we screened by varying the temperature range from 80 to 130 °C. Excellent activity with a selectivity of >99% for the dimer product was obtained at 130 °C.
As presented in Table 1, XPS data showed that all the SHC catalysts have a Ca/P ratio higher than 1.67 and would act as base catalysts, but this ratio did not correlate with catalytic activity (Table 3). The BET surface area of the SHC catalysts (Table 2) did correlate with catalytic activity (SHC-3 > SHC-5 > SHC-4 > SHC-2 > SHC-1), i.e. the catalytic activity increases along with the catalysts BET surface area. Similarly, the pore volume also followed the similar trend as of BET surface area, except for SHC-4. SHC-4 (0.18 cm3 g−1) showed a lower pore volume compared to SHC-2 (0.42 cm3 g−1), whereas BET surface area, as well as surface Ca/P ratio (from XPS) for SHC-4, was found to be higher compared to the SHC-2 catalyst. In addition, SHC-4 contains more acidic and basic sites than SHC-2, as discussed in the next section. In general, the crystallinity, BET surface area and catalytic activity all negatively correlate with an increasing carbonate content for the SHC materials, in agreement with literature.31
The acid–base properties of the SHC solid catalysts and the commercially available HAPs were studied with NH3 and CO2 TPD (Table 4). Detailed distribution of acidic/basic sites is presented in the Table S4.† The SHC catalysts that showed good catalytic activity (SHC-3, SHC-4 and SHC-5), exhibited a good share of both basic and acidic sites. For SHC-2 and SHC-5, the number of acidic sites was found to be slightly higher compared to the number of basic sites, even though the Ca/P ratios of these catalysts were higher than 1.67. Similar results have been previously reported for the HAP catalysts, as shown in Table 4, entries 8–11.28,31Fig. 3 shows the CO2 desorption profiles, indicative of the nature of the basic sites, for the most active catalysts (SHC-3, SHC-4 and SHC-5) and Fig. S6† for SHC-1 and SHC-2. It is known that a combination of moderate acidic and basic sites is beneficial for catalytic activity for aldol condensation reactions.11,28 The results here are in line with this, with e.g. the least active SHC-1 material possessing only weak basic sites (Fig. S6†). The most active catalysts (SHC-3 and SHC-5) mainly have basic sites of moderate strength (temperature of desorption between 150–400 °C) and fewer weaker ones (<150 °C). SHC-4, in contrast, has more weak basic sites than moderate ones. In line with this, the probability for basic site accommodation increases with pore volume,19 and SHC-4 is at the lower end of the pore volume range with 0.18 cm3 g−1. As noted above, some acidity is also important for the self-aldol condensation reaction, e.g., to aid the dehydration step. Acidity measurements with NH3-TPD were comparable to the CO2-TPD results, with SHC-3 giving the highest number of acidic sites. The acidity of all the SHC catalysts was found to be in the moderate range (150–400 °C), as shown in Fig. 4.
| Entry | Catalyst | ICP-OES | CO2-TPD | NH3-TPD | Ref. |
|---|---|---|---|---|---|
| Ca/P atomic ratio | Total number of basic sites (mmol g−1) | Total number of acidic sites (mmol g−1) | |||
| Hap = stoichiometric hydroxyapatite; Hap-CO3 = carbonated hydroxyapatite; HapE-NaCO3 = sodium-containing carbonate-rich hydroxyapatite; Ca-HA = hydroxyapatite; ref. = references. | |||||
| 1 | SHC-1 | 7.13 | 0.05 | 0.03 | This work |
| 2 | SHC-2 | 3.93 | 0.08 | 0.09 | |
| 3 | SHC-3 | 4.95 | 0.33 | 0.19 | |
| 4 | SHC-4 | 3.88 | 0.15 | 0.13 | |
| 5 | SHC-5 | 2.34 | 0.08 | 0.12 | |
| 6 | HAP-L | 1.85 | 0.06 | 0.02 | |
| 7 | HAP-H | 1.89 | 0.13 | 0.11 | |
| 8 | Hap | 1.69 | 0.10 | 0.76 | 31 |
| 9 | Hap-CO3 | 1.70 | 0.13 | 0.65 | 31 |
| 10 | HapE-NaCO3 | 2.39 | 0.10 | 0.14 | 31 |
| 11 | Ca-HA | 1.67 | 0.02 | 0.09 | 28 |
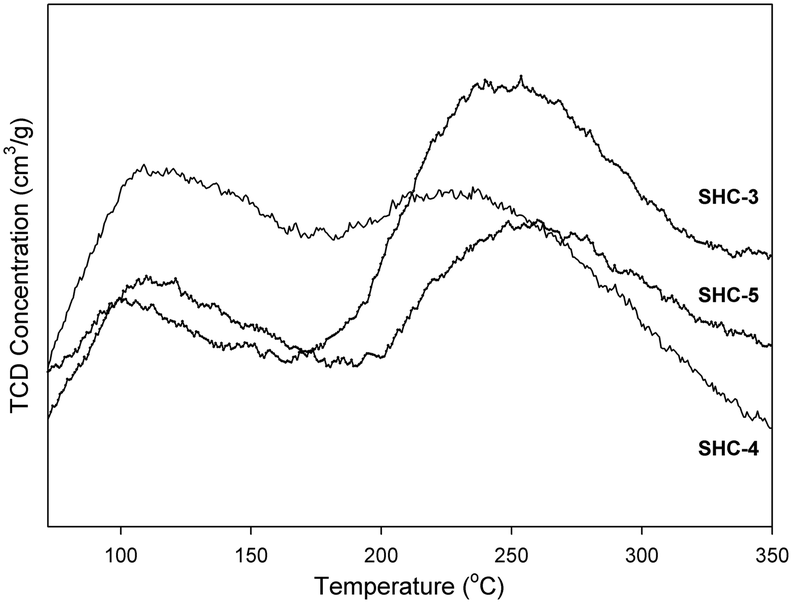 | ||
| Fig. 3 CO2-TPD curves for the SHC solids. Weak basic sites ≤150 °C, moderate basic sites = 150–400 °C. | ||
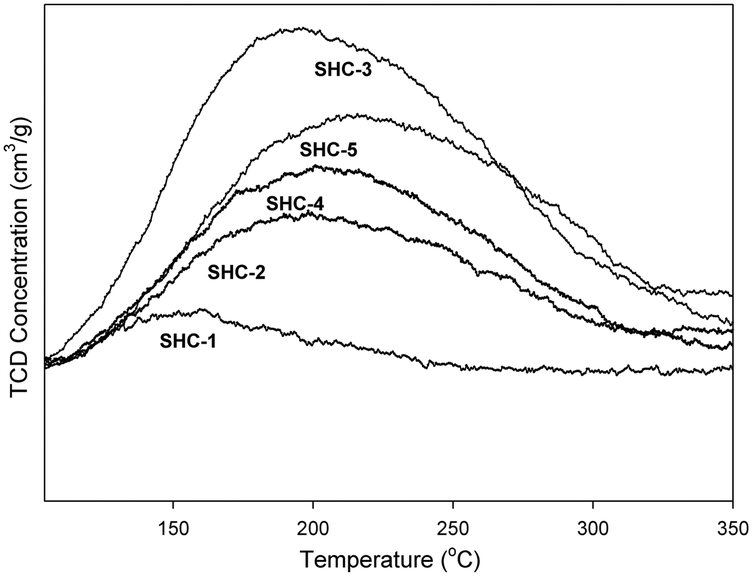 | ||
| Fig. 4 NH3-TPD curves for the SHC solids. Moderate acidic sites = 150–400 °C. | ||
That striking the right balance in strength and ratio of basic and acid sites is more important than only strong basicity, is illustrated by the fact that while MgO has both more and stronger basic sites than SHC-3, it is less active in the condensation reaction (Table 3 and Fig. S7†). Commercial hydroxyapatite (HAP-H), on the other hand, has a comparable, but basic sites are distributed as a border line between weaker and moderate region than the active SHC catalysts (Fig. S8† and Table 4), resulting in a lower activity (compare to SHC-3, SHC-4 and SHC-5) (Table 3). For HAP-L, all basic sites are of weak strength (Fig. S8†). In summary, the presence of moderate acidic and basic sites is, as anticipated, vital to impart good catalytic activity in this aldol condensation reaction.
Catalyst recycling or reusability is evidently important from economic and environmental points of view.49,50 Deactivation may even be more of a concern for solid base catalysts compared to solid acid catalysts particularly in liquid phase reactions at elevated temperatures with polar substrates.39 To validate the potential for reuse of the SHC solid catalysts, recycling experiments were performed with SHC-3. After each run, the catalyst was recuperated, washed thoroughly with solvent, dried, weighed and subsequently applied in an ensuing run. As a slight amount of material is inevitably lost during such a protocol, the substrate amount was adjusted to keep the catalyst loading at 3 mol% in each reuse (Table 5). In general, no physical change to the catalyst, nor dissolution of the material into solution were noticed. Entries 1 and 2 show the first recycle experiments with SHC-3, highlighting a drastic drop-in catalytic activity in the second run, giving only an 11% yield after 4 h. Extending the reaction time to 22 h led to an increase in yield to around 55%, a level that was maintained also upon subsequent recycling. These results thus demonstrate the catalyst to still be active under these recycling conditions, but much less so than the fresh catalyst. XRD analysis (Fig. S9†), for entry 1, showed that the structure remained intact after the first recycle. Similarly, SEM images (Fig. S10†) did not show any visual change in SHC-3's morphology. Encouragingly, ICP analysis of the reaction mixture of entry 1 (Table S5†), after separating the catalyst by filtration, showed no leaching of Ca or P (entry 3, Table S5†); elemental analysis however, did show an increase in carbon content (entry 2, Table S5†), suggesting carbonaceous material deposition to cause the drop in catalytic activity by blockage of active sites.39,51–53 Changing the catalyst washing solvent from diethyl ether to more polar methanol resulted in an improved catalytic activity upon reuse (entry 2, Table 5), but did not yet lead to full restoration of activity. As carbon deposition may likely also be a function of total basicity/acidity, this was expected to be less of an issue for some of the other SHC materials, thus providing a payoff between (initial) activity and stability. Indeed, upon switching to SHC-4 (entry 3, Table 5), we noted that activity could be close to fully maintained upon recycling up to four times. Again, as for SHC-4 XRD analysis showed that crystallinity was preserved (Fig. S11†). That SHC-3 showed a more dramatic loss in activity than SHC-4 correlates with the larger loss of BET surface area and basic sites (CO2-TPD) of the former (Table S6†), in line with deactivation by carbon deposition.
| Entry | Catalyst | Washing solvent | Run no. | Yield (%) | |
|---|---|---|---|---|---|
| 4 h | 22 h | ||||
| Reaction conditions: aldehyde = butyraldehyde; temperature = 130 °C; catalyst loading = 3 mol%; results from NMR; IS = mesitylene; MeOH = methanol; n.d. = not determined. | |||||
| 1 | SHC-3 | Ether | 1 (fresh) | 88 | n.d. |
| 2 | 11 | 55 | |||
| 3 | 7 | 58 | |||
| 4 | 13 | 57 | |||
| 2 | MeOH | 1 (fresh) | 89 | n.d. | |
| 2 | 21 | ||||
| 3 | SHC-4 | Ether | 1 (fresh) | 42 | n.d. |
| 2 | 36 | 74 | |||
| 3 | 35 | 73 | |||
| 4 | 35 | 73 | |||
| 4 | SHC-4 | MeOH | 1 (fresh) | 42 | n.d. |
| 2 | 33 | ||||
| 3 | 39 | ||||
| 4 | 39 | ||||
| 5 | SHC-3 | MeOH | 1 (fresh) | 90 | |
| 2 | 25 | ||||
| 3 | 64 | ||||
| 4 | 66 | ||||
Further improvement of the catalyst regeneration protocol involved reversal of active site blockage by CO2 or H2O.19,39,52 Indeed, thermal reactivation after washing further improved recyclability. Table 5, entries 4 and 5, illustrates the recycling experiments for both SHC-4 (entry 4) and SHC-3 (entry 5) where after each cycle the catalyst has been subjected to a thermal reactivation and washing using methanol as the solvent. For both catalysts (entries 4 and 5), after the first fresh run, the catalyst was subjected to a thermal activation at 200 °C for 4 h, whereas for runs 3 and 4 the thermal activation was performed at 200 °C overnight. For both catalysts, a small drop-in activity was still seen in the 2nd run; whereas an improvement in the catalytic performance was observed in runs 3 and 4. Analysis of the spent and regenerated materials by FT-IR (Fig. S12†) again points at the formation of carbonaceous materials as primary cause for catalyst deactivation. For both SHC-3 and SHC-4, IR spectra of the catalysts recovered after first run showed more carbonaceous materials, as indicated by bands seen between 3000–2800 cm−1, which can be attributed to aldehyde C–H and/or C–H alkyl stretching vibrations; additionally, the spectra of the SHC-3 catalyst also showed a broad OH peak around 3400 cm−1.54,55 As previously stated, prior to the 2nd run, the recovered catalysts were subjected to thermal activation at 200 °C for only 4 h, whereas for runs 3 and 4, the thermal activation was done overnight. The resulting improvement seen in activity for runs 3 and 4, coincided with the disappearance of the peaks around 3000 and 3400 cm−1, after the 4th run. The results thus show that the catalyst activity can be close to completely regained by proper thermal reactivation and selection of catalyst washing solvent. Overall, SHC-4 catalyst showed better recyclability, even though its catalytic activity was on the lower end, compared to the SHC-3 catalyst. The difference in pore volume (0.87 vs. 0.18 cm3 g−1 for SHC-3 and SHC-4, respectively), while initially unfavorable for activity, could then have the upside of allowing less coke to be accommodated.
The applicability of the SHC materials was finally tested in other condensation reactions, using SHC-3 in prototypical Knoevenagel, Claisen–Schmidt and Henry reactions. The reaction conditions were optimized similarly to the self-aldol condensation reactions and the results are displayed in Table 6. Entry 1 shows the results of the Knoevenagel condensation reaction between benzaldehyde and malononitrile to produce 2-benzilidenemalononitrile, a versatile chemical building block. Compared to the self-aldol condensation reaction, the Knoevenagel reaction worked well at lower reaction temperatures too. SHC-3 also proved to be active, albeit more moderately, in chalcone synthesis through the Claisen–Schmidt condensation reaction of benzaldehyde and acetophenone (entry 2). Finally, the Henry reaction between benzaldehyde and nitromethane gave β-nitrostyrene as the main product. The Henry reaction is well-known for the reversible nature of the nitroaldol addition product and after 2 h of reaction, both the nitroaldol product (2-nitro-1-phenylethanol) and the condensation product (β-nitrostyrene) were detected with yields of 33% and 67%, respectively. With dehydration being irreversible, the selectivity to the latter further increased with reaction time to 92% and >99% after 6 h and 22 h, respectively, with conversions of 29 and 69%.
| Entry | Reactions | Product | T (°C) | Time (h) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 6 | 22 | 44 | ||||||||
| X (%) | Y (%) | X (%) | Y (%) | X (%) | Y (%) | X (%) | Y (%) | ||||
Reaction conditions: catalyst loading = 3 mol%; catalyst: SHC-3; T = temperature; X = conversion, Y = yield; entry 1, substrate 1 = benzaldehyde, substrate 2 = malononitrile; entry 2, substrate 1 = benzaldehyde, substrate 2 = acetophenone; entry 3, substrate 1 = benzaldehyde, substrate 2 = nitromethane; substrate ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2; n.d. = not determined; conversion and yield were determined by 1H NMR (CDCl3) using mesitylene as an internal standard. :2; n.d. = not determined; conversion and yield were determined by 1H NMR (CDCl3) using mesitylene as an internal standard.
|
|||||||||||
| 1 | Knoevenagel condensation |
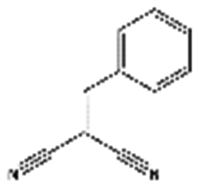
|
70 | 40 | 38 | 87 | 83 | 100 | 95 | n.d. | |
| 130 | 67 | 67 | 93 | 92 | 100 | 96 | |||||
| 2 | Claisen–Schmidt condensation |
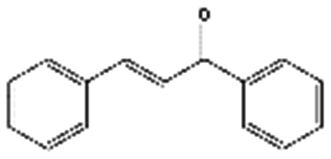
|
130 | 19 | 17 | 39 | 35 | 75 | 74 | 98 | 97 |
| 3 | Henry reaction |
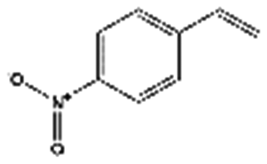
|
130 | 14 | 9 | 29 | 27 | 69 | 68 | 96 | 95 |
Conclusions
SHC materials, consisting of a combination of CC and HAP phase and made from readily available minerals, were found to be excellent solid base catalysts for the self-aldol condensation of butyraldehyde. A high HAP content proved beneficial for catalysis with the materials containing ca. 15% CC and 85% HAP, respectively, performing best. As evident from the control experiments, careful synthesis and close interaction between the HAP and CC phases seems key to provide the best catalytic activity for SHC catalysts, as the physical mixtures of HAP and CC or individual components proved much less effective. Investigation of various reaction parameters indicated that the butyraldehyde conversion reached its maximum when the reaction was carried out with 3 mol% of catalyst loading, at 130 °C for 4 h. Catalytic activity increased with BET surface area, as well as with mesoporosity. Striking a balance in the amount, nature and ratio of acidic and basic sites is crucial for catalytic performance in the self-aldol condensation reaction; all active SHC catalysts combine a higher number of moderate acidic and basic sites with some weak acidic and basic sites. Upon proper regeneration by washing and thermal reactivation, the SHC catalysts proved to be recyclable. Furthermore, the SHC catalysts also performed well in other condensation reactions such as the Knoevenagel condensation, Claisen–Schmidt condensation and Henry reactions.In general, compared to widely employed solid base catalysts such as MgO and HAP, SHC catalysts performed better in the self-aldol condensation reaction of butyraldehyde. The SHC materials are industrially available, porous, and stable structures made from naturally abundant and non-toxic precursors and have now been shown to perform well as solid base catalysts for condensation reactions. As such, these highly active solid base catalysts, of which the acid–base and morphological properties can be carefully tuned, provide a promising new addition to the existing portfolio of industrially employed solid base catalysts.
Experimental
Unless otherwise noted, all commercial reagents were used as received without further purification. Butyraldehyde (≥99.5%), malononitrile (≥99%), nitromethane (≥99%), mesitylene (98%), HAP-L nanopowder (<200 nm, particle size, ≥97%), HAP-H (5 μm, particle size) and TiO2 nanopowder (<100 nm particle size, 99.5%) were purchased from Sigma-Aldrich. Benzaldehyde (98+%), acetophenone (98%), CC (98+%) and MgO (98%) were obtained from Acros organics.SHCs were obtained from Omya International. The core–shell SHC materials consist of a CC core and a shell a lamellar HAP surrounded by agglomerated fine CC particulates. The samples are produced by etching CC particles and reprecipitating modified surface structure with in situ or externally supplied CO2 in the form of carbonic acid. To facilitate the in situ production of CO2, acids such as phosphoric acid can be used, resulting in a mixed mineral morphology. Variations in the etching process produce a range of morphologies with recrystallized surfaces, consisting of incorporated hydroxyapatite (HAP) in the case of phosphoric acid.
XRD patterns were recorded using a Bruker D2 Phaser powder X-ray diffractometer using a Co radiation source, Co Kα = 1.789 Å. Measurements were carried out between 10–70° 2θ using a scan speed of 0.5 s per step. TGA was conducted using Mettler Toledo TGA/DSC 3+. The samples were heated from 25 up to 600 °C with a ramp of 25 °C and a 10 min hold at 105 °C and 500 °C, with an air flow of 80 ml min−1. XPS experiments were carried out in a Kratos AXIS Ultra DLD spectrometer using a monochromatic Al Kα radiation (hυ = 1486.6 eV) operating at 225 W (15 mA, 15 kV). Instrument base pressure was 5 × 10−10 Torr. SEM images were recorded on two different instruments, (a) for general characterization: a Sigma VP field emission scanning electron microscope (Carl Zeiss AG, Germany) and a variable pressure secondary electron detector (VPSE) and/or secondary electron detector (SE) with a chamber pressure of about 50 Pa, (b) for recycle experiments: the surface images were taken with a ThermoScientific phenom Pro X microscope operated at 10 kV acceleration voltage. The Ca and P contents of the SHC solids were analysed via ICP-OES technique using a Perkin Elmer Avio 500 device. The textural properties of the SHC solids were analysed by recording an N2 physisorption isotherms with a Micromeritics Tristar 3000 setup. The samples were outgassed prior to performing the measurement overnight at 300 °C under a N2 flow. Surface areas were determined using the Brunauer–Emmett–Teller (BET) theory. The carbon content of the SHC catalysts was obtained from EA and it was carried out using a Fisons NA1500 NCS analyser. Infrared spectra were recorded using a Perkin Elmer Spectrum Two FT-IR spectrometer.
Temperature programmed desorption of ammonia (NH3-TPD) measurements were performed using a Micromeritics ASAP2920 apparatus. 0.1 g of sample was dried in situ under an He flow with a temperature ramp of 5 °C min−1 up to 400 °C. Subsequently, the sample was cooled to 100 °C; at this point, NH3 pulses of 25.3 cm3 min−1 were applied. The sample was then heated to 600 °C with a ramp of 5 °C min−1 to induce desorption of NH3. A procedure similar to the one described for NH3-TPD was employed for temperature programmed desorption of CO2 (CO2-TPD), the main difference being the lower temperature (50 vs. 100 °C) at which CO2 pulses are fed to the sample. For calculating the number of acidic or basic sites, it was assumed that only one molecule of NH3 or CO2 can adsorb on a single site. Detailed procedure in ESI† (E1).
The aldol condensation of butyraldehyde was performed in a batch reaction system, under vigorous magnetic stirring and a nitrogen atmosphere. Prior to the reaction, all the SHC solids were thermally activated at 200 °C for 4 h. In a typical experiment, a 50 mL two-necked flask connected to a reflux condenser was filled with 55.6 mmol of butyraldehyde, 3 mol% SHC (mol% = catalyst loading (mmol)/butyraldehyde (mmol) × 100) of catalyst (SHC solids) and mesitylene as the internal standard. After passing the N2 through the headspace of the reaction system and setting a constant, vigorous stirring rate, the reaction mixture was heated up to the required temperature (80–130 °C). The progress of the reaction was monitored by taking samples from the reaction media at different intervals of time (2–22 h). For the recycling experiments, the used catalyst was filtered, washed thoroughly with a solvent (ether or methanol), dried at 200 °C, weighed and then applied in a subsequent run. The conversion and yield were determined by 1H NMR (CDCl3). The 1H spectra (400 MHz) and 13C spectra (100 MHz) were recorded on an Agilent MRF400 or a Varian AS400 spectrometer at 25 °C. The chemical shifts are reported in the standard δ notation of parts per million, referenced to residual peak of the solvent, as determined relative to TMS (δ = 0 ppm).
All the other condensation reactions were carried out similarly to the aldol condensation reaction. For the Knoevenagel condensation reaction, the substrates employed were benzaldehyde and malononitrile, similarly for the Claisen–Schmidt condensation reaction, benzaldehyde and acetophenone were used and the substrates, benzaldehyde and nitromethane for the Henry reaction. In all cases the substrate ratio is maintained 1 (benzaldehyde):2 and the catalyst loading of 3 mol% was used, and temperatures of 130 °C was employed except for Knoevenagel condensation, where a lower reaction temperature (70 °C) was also used.
Aldol condensation reaction, 2-ethyl-2-hexenal: 1H NMR (CDCl3, 400 MHz) δ = 0.98 (m, 6H), 1.56 (m, 2H), 2.36 (m, 4H), 6.43 (t, J = 7.5 Hz 1H), 9.36 (s, 1H). 13C NMR (100 MHz, CDCl3); δ = 13.47, 14.02, 17.40, 22.10, 30.82, 145.53, 154.77, 195.26.
Knoevenagel condensation reaction, 2-benzylidene-malononitrile: 1H NMR (400 MHz, CDCl3) δ (ppm): 7.55 (t, J = 7.6 Hz, 2H), 7.64 (t, J = 7.4 Hz, 1H), 7.78 (s, 1H), 7.91 (d, J = 7.3 Hz, 2H), 13C NMR (100 MHz, CDCl3); δ = 82.86, 112.48, 113.51, 129.60, 130.70, 134.54, 159.88.
Claisen–Schmidt condensation reaction, benzylideneacetophenone (chalcone): 1H NMR (CDCl3, 100 MHz) δ = 7.40–7.67 (m, 9H), 7.82 (d, J = 15.7 Hz, 1 H), 8.03 (dd, J = 8.4, 1.4 Hz, 2 H) ppm. 13C NMR (400 MHz, CDCl3); δ = 122.26, 128.44, 128.59, 128.65, 128.77, 130.65, 132.91, 134.58, 137.26, 145.03, 198.24.
Henry reaction, 2-nitrovinylbenzene (β-nitrostyrene): 1H NMR (CDCl3, 400 MHz) δ 7.41–7.51 (m, 3H), 7.54–7.57 (m, 2H), 7.59 (d, J = 13.7 Hz, 1H), 8.01 (d, J = 13.7 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3); δ = 129.10, 129.39, 130.08, 130.15, 132.11, 137.69, 139.03.
Conflicts of interest
T. J., J. F. and P. C. A. B. are listed as inventors on a patent application filed by Omya International related to the results described in the manuscript.Acknowledgements
The authors thank Dr. Craig DePorter, Dr. Daniel E. Gerard, Dr. Nicole Russ and Sarah Gysin, from Omya International, for their support. Omya International is kindly acknowledged for financial support.Notes and references
- S. Fakirov, Prog. Polym. Sci., 2019, 89, 1–18 CrossRef CAS.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247–268 CAS.
- S. K. Sharma and R. V. Jasra, Ind. Eng. Chem. Res., 2011, 50, 2815–2821 CrossRef CAS.
- R. Raju and K. Prasad, Tetrahedron, 2012, 68, 1341–1349 CrossRef CAS.
- R. D. Ashford, Ashford's Dictionary of industrial chemicals, Wavelength, Saltash, 2011 Search PubMed.
- G. J. Kelly, F. King and M. Kett, Green Chem., 2002, 4, 392–399 RSC.
- A. Miller, W. Keener, M. Watwood and F. Roberto, Appl. Microbiol. Biotechnol., 2002, 58, 183–188 CrossRef CAS PubMed.
- E. Rohrmann, R. G. Jones and H. A. Shonle, J. Am. Chem. Soc., 1944, 66, 1856–1857 CrossRef CAS.
- K. Ramawat and J. M. Mérillon, Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes, 2013 Search PubMed.
- L. Wu, T. Moteki, A. A. Gokhale, D. W. Flaherty and F. D. Toste, Chem, 2016, 1, 32–58 CrossRef CAS PubMed.
- E. G. Rodrigues, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Green Chem., 2014, 16, 4870–4874 RSC.
- M. J. Climent, A. Corma, H. Garcia and J. Primo, J. Catal., 1991, 130, 138–146 CrossRef CAS.
- A. M. Frey, J. Yang, C. Feche, N. Essayem, D. R. Stellwagen, F. Figueras, K. P. de Jong and J. H. Bitter, J. Catal., 2013, 305, 1–6 CrossRef CAS.
- W. Bing, L. Zheng, S. He, D. Rao, M. Xu, L. Zheng, B. Wang, Y. Wang and M. Wei, ACS Catal., 2018, 8, 656–664 CrossRef CAS.
- O. Kikhtyanin, D. Kadlec, R. Velvarská and D. Kubička, ChemCatChem, 2018, 10, 1464–1475 CrossRef CAS.
- W. Bing, H. Wang, L. Zheng, D. Rao, Y. Yang, L. Zheng, B. Wang, Y. Wang and M. Wei, Green Chem., 2018, 20, 3071–3080 RSC.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- Y. Ono, J. Catal., 2003, 216, 406–415 CrossRef CAS.
- L.-B. Sun, X.-Q. Liu and H.-C. Zhou, Chem. Soc. Rev., 2015, 44, 5092–5147 RSC.
- A. Fihri, C. Len, R. S. Varma and A. Solhy, Coord. Chem. Rev., 2017, 347, 48–76 CrossRef CAS.
- A. Smahi, A. Solhy, H. El Badaoui, A. Amoukal, A. Tikad, M. Maizi and S. D. Sebti, Appl. Catal., A, 2003, 250, 151–159 CrossRef CAS.
- S. D. Sebti, R. Tahir, R. Nazih, A. Saber and S. D. Boulaajaj, Appl. Catal., A, 2002, 228, 155–159 CrossRef CAS.
- N. Elazarifi, A. Ezzamarty, J. Leglise, L.-C. D. Ménorval and C. Moreau, Appl. Catal., A, 2004, 267, 235–240 CrossRef CAS.
- S. Sebti, A. Solhy, R. Tahir and A. Smahi, Appl. Catal., A, 2002, 235, 273–281 CrossRef CAS.
- A. Solhy, R. Tahir, S. Sebti, R. Skouta, M. Bousmina, M. Zahouily and M. Larzek, Appl. Catal., A, 2010, 374, 189–193 CrossRef CAS.
- N. Neelakandeswari, G. Sangami, P. Emayavaramban, R. Karvembu, N. Dharmaraj and H. Y. Kim, Tetrahedron Lett., 2012, 53, 2980–2984 CrossRef CAS.
- Z. D. Young, S. Hanspal and R. J. Davis, ACS Catal., 2016, 6, 3193–3202 CrossRef CAS.
- A. M. Hernández-Giménez, J. Ruiz-Martínez, B. Puértolas, J. Pérez-Ramírez, P. C. A. Bruijnincx and B. M. Weckhuysen, Top. Catal., 2017, 60, 1522–1536 CrossRef.
- T. J. Webster, E. A. Massa-Schlueter, J. L. Smith and E. B. Slamovich, Biomaterials, 2004, 25, 2111–2121 CrossRef CAS PubMed.
- S. Kannan, J. H. G. Rocha and J. M. F. Ferreira, J. Mater. Chem., 2006, 16, 286–291 RSC.
- L. Silvester, J.-F. Lamonier, R.-N. Vannier, C. Lamonier, M. Capron, A.-S. Mamede, F. Pourpoint, A. Gervasini and F. Dumeignil, J. Mater. Chem. A, 2014, 2, 11073–11090 RSC.
- T. Hara, S. Kanai, K. Mori, T. Mizugaki, K. Ebitani, K. Jitsukawa and K. Kaneda, J. Org. Chem., 2006, 71, 7455–7462 CrossRef CAS PubMed.
- D. A. Nowicki, J. M. S. Skakle and I. R. Gibson, J. Mater. Chem. A, 2018, 6, 5367–5377 RSC.
- D. Pham Minh, N. D. Tran, A. Nzihou and P. Sharrock, Ind. Eng. Chem. Res., 2013, 52, 1439–1447 CrossRef CAS.
- C. Verwilghen, M. Chkir, S. Rio, A. Nzihou, P. Sharrock and G. Depelsenaire, Mater. Sci. Eng., C, 2009, 29, 771–773 CrossRef CAS.
- D. M. Roy and S. K. Linnehan, Nature, 1974, 247, 220–222 CrossRef CAS PubMed.
- J. Ftouni, T. Jose and P. C. A. Bruijnincx, CH Pat., 20199667.5-1109, 2020 Search PubMed.
- M. Farzan, R. Roth, G. Québatte, J. Schoelkopf, J. Huwyler and M. Puchkov, Pharmaceutics, 2019, 11, 32 CrossRef CAS PubMed.
- I. Sádaba, M. López Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- X. Zhang, H. An, H. Zhang, X. Zhao and Y. Wang, Ind. Eng. Chem. Res., 2014, 53, 16707–16714 CrossRef CAS.
- H. A. Patel, S. K. Sharma and R. V. Jasra, J. Mol. Catal. A: Chem., 2008, 286, 31–40 CrossRef CAS.
- T. Jose, N. Sudheesh and R. S. Shukla, J. Mol. Catal. A: Chem., 2010, 333, 158–166 CrossRef CAS.
- D. Sun, S. Moriya, Y. Yamada and S. Sato, Appl. Catal., A, 2016, 524, 8–16 CrossRef CAS.
- D. Liang, G. Li, Y. Liu, J. Wu and X. Zhang, Catal. Commun., 2016, 81, 33–36 CrossRef CAS.
- X. Xu, H. Meng, Y. Lu and C. Li, RSC Adv., 2018, 8, 30610–30615 RSC.
- Z. Wang, P. Fongarland, G. Lu and N. Essayem, J. Catal., 2014, 318, 108–118 CrossRef CAS.
- Y. Watanabe, K. Sawada and M. Hayashi, Green Chem., 2010, 12, 384–386 RSC.
- A. Corma, V. Fornés, R. M. Martín-Aranda and F. Rey, J. Catal., 1992, 134, 58–65 CrossRef.
- M. Marafi and A. Stanislaus, Resour., Conserv. Recycl., 2008, 52, 859–873 CrossRef.
- J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC.
- M. Guisnet and P. Magnoux, Appl. Catal., A, 2001, 212, 83–96 CrossRef CAS.
- C. Sievers, Y. Noda, L. Qi, E. M. Albuquerque, R. M. Rioux and S. L. Scott, ACS Catal., 2016, 6, 8286–8307 CrossRef CAS.
- O. Kikhtyanin, V. Kelbichová, D. Vitvarová, M. Kubů and D. Kubička, Catal. Today, 2014, 227, 154–162 CrossRef CAS.
- J. Shangguan, C.-H. Li, M.-Q. Miao and Z. Yang, New Carbon Mater., 2008, 23, 37–43 CrossRef.
- S. Zeng, S. Liu, Y. Qi, L. Cui, Q. Dai and C. Bai, RSC Adv., 2018, 8, 11462–11468 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00102g |
| This journal is © The Royal Society of Chemistry 2021 |