
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous screening of multiple substrates with an unspecific peroxygenase enabled modified alkane and alkene oxyfunctionalisations†
Anja
Knorrscheidt
a,
Jordi
Soler
 b,
Nicole
Hünecke
a,
Pascal
Püllmann
a,
Marc
Garcia-Borràs
*b and
Martin J.
Weissenborn
*ac
b,
Nicole
Hünecke
a,
Pascal
Püllmann
a,
Marc
Garcia-Borràs
*b and
Martin J.
Weissenborn
*ac
aBioorganic Chemistry, Leibniz Institute of Plant Biochemistry, Weinberg 3, 06120 Halle (Saale), Germany. E-mail: martin.weissenborn@ipb-halle.de
bInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, Carrer Maria Aurèlia Capmany 69, Girona 17003, Catalonia, Spain
cInstitute of Chemistry, Martin Luther University Halle-Wittenberg, Kurt-Mothes-Str. 2, 06120 Halle (Saale), Germany
First published on 17th May 2021
Abstract
A high throughput GC-MS approach was developed, permitting the simultaneous analysis of up to three substrates and six products quantitatively from one reaction mixture. This screening approach was applied to site-saturation libraries of the novel unspecific peroxygenase MthUPO. Using this setup enabled substantial insights from a small mutant library. Enzyme variants were identified exhibiting selective alkene epoxidation and substantially shifted regioselectivities to 2- and 1-octanol formations. Computational modelling rationalised the observed selectivity changes.
Introduction
Fungal unspecific peroxygenases (UPOs) were discovered in 20041–6 and can hydroxylate an extensive substrate scope.7,8 Contrary to the phylogenetically related chloroperoxidases,9 UPOs can hydroxylate non-activated aliphatic sp3-carbons. Epoxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds is also observed for UPOs. The mechanism performs via the activation of hydrogen peroxide by the iron-heme centre. Upon releasing a water molecule, the oxyferryl Fe(IV)O porphyrin cation radical compound I (Cpd I) is formed. This reactive species can perform oxyfunctionalisations (Scheme 1).3
C double bonds is also observed for UPOs. The mechanism performs via the activation of hydrogen peroxide by the iron-heme centre. Upon releasing a water molecule, the oxyferryl Fe(IV)O porphyrin cation radical compound I (Cpd I) is formed. This reactive species can perform oxyfunctionalisations (Scheme 1).3
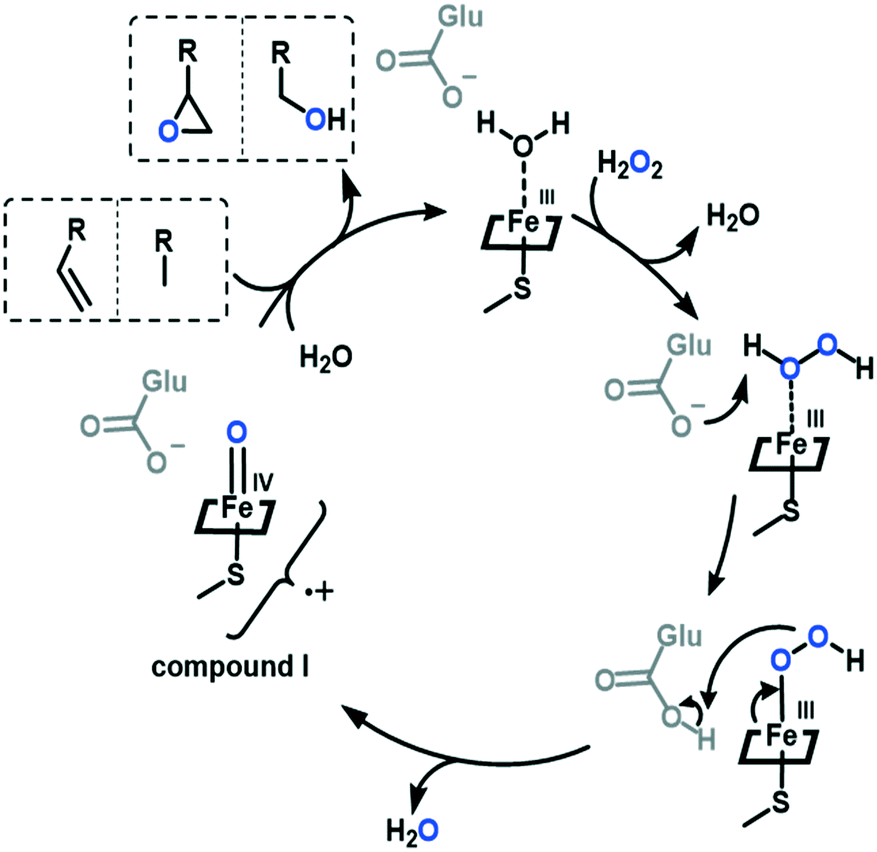 | ||
| Scheme 1 Proposed mechanism for UPOs catalysing alkane and alkene oxyfunctionalisations. | ||
Using this enzyme mechanism for the selective hydroxylation of non-activated carbons is of utmost interest. Thus far, the performed studies on UPOs towards aliphatic hydroxylation reactions were mainly limited to wildtype UPO enzymes and a yeast expression UPO variant.2,4–6,10–12 Although previously demonstrated UPO activities are impressive, their hampered regio- and chemoselectivities represent a limitation for their synthetic and industrial application. This limitation was recently addressed for the first time by combining computational modelling and mutagenesis and led to MroUPO (from Marasmius rotula) and CviUPO (from Collariella virescens) variants with improved chemoselectivities.13,14 Also, recent work in our laboratory addressed this issue by creating chemodivergent variants of MthUPO from Myceliophthora thermophila for benzylic or aromatic oxyfunctionalisations.30
To harness the impressive activities of UPOs for chemo- or regioselective transformations, protein engineering approaches and directed evolution techniques are of particular interest. Directed evolution consists of random or semi-rational mutagenesis to create enzyme libraries assessed for improved enzyme abilities by high throughput analysis. Beneficial mutations are selected and subjected to further rounds of mutagenesis and analysis. This method is extremely successful and has led to the engineering of many selective enzymes.15–17
Two technologies are required to perform protein engineering for regio-, and chemoselective hydroxylation reactions using UPOs: a microtiter plate-based enzyme production setup and a high throughput assay, which is able to differentiate between various functional isomers and regioisomers.
The development of heterologous UPO expression systems in the yeast organisms Saccharomyces cerevisiae and Pichia pastoris enabled the UPO production in sufficient amounts using a microtiter plate setup.18–20,29 The remaining challenge was the development of a suitable and versatile assay system.
In the last two decades, substantial progress was made towards the design of smaller but smarter enzyme libraries in the protein engineering field. This is mostly due to significant time and money constraints associated with large mutant libraries' assessment. Advances were achieved by focussing the mutagenesis on specific regions, like the active site or the substrate entrance channel.21,22 Another approach is to limit the available amino acid alphabet to solely include functionally relevant residues.23,24 These extremely successful developments vastly increased the applicability of directed evolution.
Further potential to increase the “smartness” of mutant libraries is by assessing the maximum information of the created variants. Fasan and co-workers approached this by generating a focussed P450 library and screened it with several substrates consecutively. This “fingerprinting method” generated substantially more insights and knowledge about the mutated positions and discovered specific enzyme variant/substrate pairs.25
We hypothesised that screening an enzyme library with the simultaneous assessment of several substrates and products would provide a new dimension to the obtainable insights. The gained knowledge would be [(n − 1) + (m − 1)]-fold increased relative to the assessment of one substrate/product pair, where n is the number of substrates and m the number of quantified products. A tool to permit screenings of this kind would be our recently reported, substrate-independent high throughput analysis with an off-the-shelf GC-MS (gas chromatography-mass spectrometry).26 This method enabled the quantification of an internal standard, the product and the byproduct simultaneously based on the MS separation within the mass analyser (quadrupole) with sample injection intervals of 33 s.
Further expanding this method could enable the assessment of product distributions for different functional isomers with different masses like alkene epoxidation vs allylic hydroxylation, and to differentiate regioisomeric products with the same mass, permitting protein engineering for regioselectivity.
The possible challenge of the simultaneous screening of multiple substrates is the potential inhibitory effect creating false negative results. However, this effect would be comparable to the natural evolution where a vast number of similar substrates were available resulting in highly specialised and specific biocatalysts.
In the present work, we demonstrate the screening of 900 transformants with 3 substrates, 6 products, and one internal standard simultaneously (Fig. 1A). The optimised GC-MS technique proved to be able to differentiate between regioisomers and functional isomers for the substrates octane, cyclohexane and cyclohexene. By only screening a small focused library, three UPO variants with significantly modified regio- and chemoselectivity were identified.
 | ||
| Fig. 1 A) UPO catalysed one pot conversion of octane, cyclohexane and cyclohexene (1 mM H2O2, 5 mM of each substrate, 5% (v/v) acetone, 100 mM KPi pH 7, 1 h reaction time at 25 °C using 100 μl of the microtiter plate cultivated S. cerevisiae supernatant containing the corresponding UPO variant). B) Regiospecific mass fragmentation during the ionisation and C) MISER-GC-MS chromatogram of an injected microtiter plate detecting quantitative product amounts of the regio- and chemospecifically formed products. | ||
Results and discussion
We started our technology development using our previous![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) ultiple
ultiple ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) njections in a
njections in a ![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) ingle
ingle ![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) xperimental
xperimental ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) un (MISER) GC-MS method.26 As this method already provided the simultaneous assessment of three analytes, we further challenged the system by employing it to determine the cyclohexene epoxidation/allylic hydroxylation as well as the cyclohexane hydroxylation. Since the MISER GC-MS technique relies on analyte quantification via MS analysis, it is vital that all possible products exhibit different masses. As epoxides tend to derivatise in the GC-liner, we increased the robustness of the setup by performing an acidic workup. This opens the epoxides and forms the stable halohydrins.
un (MISER) GC-MS method.26 As this method already provided the simultaneous assessment of three analytes, we further challenged the system by employing it to determine the cyclohexene epoxidation/allylic hydroxylation as well as the cyclohexane hydroxylation. Since the MISER GC-MS technique relies on analyte quantification via MS analysis, it is vital that all possible products exhibit different masses. As epoxides tend to derivatise in the GC-liner, we increased the robustness of the setup by performing an acidic workup. This opens the epoxides and forms the stable halohydrins.
Since non-activated C–H bonds represent the most challenging compounds for selective oxyfunctionalisations, we considered the bioconversion of octane as a model substrate. Its hydroxylation led to three different regioisomers: 2-, 3- and 4-octanol—all having the identical mass. Additionally, the formed alcohols tend to fragment by electron impact ionisation (Fig. S2†). The resulting small fragments cannot be quantified by MS in complex biological matrices. To allow the simultaneous analysis and quantification of the different octanol regioisomers the enzymatic products were derivatised by N-trimethylsilyl-imidazole (TSIM) to generate the respective silylethers. We were pleased to observe that the increased masses led to regiospecific fragmentation during electron impact ionisation (Fig. 1B). The specific fragmentation permitted the detection, differentiation and quantification of the generated products within the MISER-GC-MS setup (Fig. 1 and S1†).
To compare the MS response and to rule out ion suppression effects of the three octanols and the oxyfunctionalised cyclohexane and cyclohexene products (including the internal standard), the MS response of the seven compounds was analysed individually and as compound mixture in the MISER-GC-MS approach. The single and multiple analyte measurements revealed a linear response thereby verifying the feasibility of the envisioned simultaneous quantification setup (Fig. S3†).
With the ability of the simultaneous analysis of seven compounds in hand, we expanded the workflow to a microtiter plate setup including the following steps: i) microtiter plate cultivation of S. cerevisiae expressing the respective UPO variant, ii) bioconversion of the substrates octane, cyclohexane and cyclohexene by the secreted UPO variant, iii) acidic ring-opening of the epoxide leading to halohydrins, iv) extraction with n-hexane, v) derivatisation of the alcohols with the silylation agent TSIM, vi) washing with water to dispose the emerged imidazole and vii) injection of the 96-well plate samples into the isothermal MISER-GC-MS run (Fig. 1C). The entire MISER-GC-MS workflow was applied for 96 biological replicates yielding a standard deviation of <12% (Table S3†).
To select a UPO with the feasibility to convert unactivated or less activated substrates, we investigated the bioconversions of octane, cyclohexane and cyclohexene with a panel of heterologously expressed UPOs (Fig. 2B). UPO secreting S. cerevisiae transformants were cultivated in 96-well microtiter plates and the corresponding supernatant assessed for their biotransformation. The UPOs from Galerina marginata (GmaUPO) and Marasmius wettsteinii (MweUPO) showed no activity, whereas slight activity was detected for the UPOs from Marasmius rotula (MroUPO) and Chaetomium globosum (CglUPO). However, for these four UPOs the secretion levels were by far the lowest20 and are hence not suitable for the desired workflow in microtiter plates. The engineered yeast-secretion variant AaeUPO* (also referred to as PaDa-I or rAaeUPO in the literature) from Agrocybe aegerita18 demonstrated a pronounced epoxide formation of cyclohexene, allylic hydroxylation, 2- and 3-octanol formation. The highest activities were displayed by the recently discovered MthUPO from Myceliophthora thermophila and TteUPO from Thielavia terrestris. Whereas MthUPO showed substantial amounts of 2-, 3- and 4-octanol as well as the epoxide, TteUPO revealed already a selectivity towards 3- and 2-octanol and decreased activities on the epoxide formation. Cyclohexanol formations were detected only in small amounts for all tested UPOs. As MthUPO exhibited the lower regioselectivity but slightly increased activity for octane oxidation, we selected this UPO for our protein engineering endeavour.
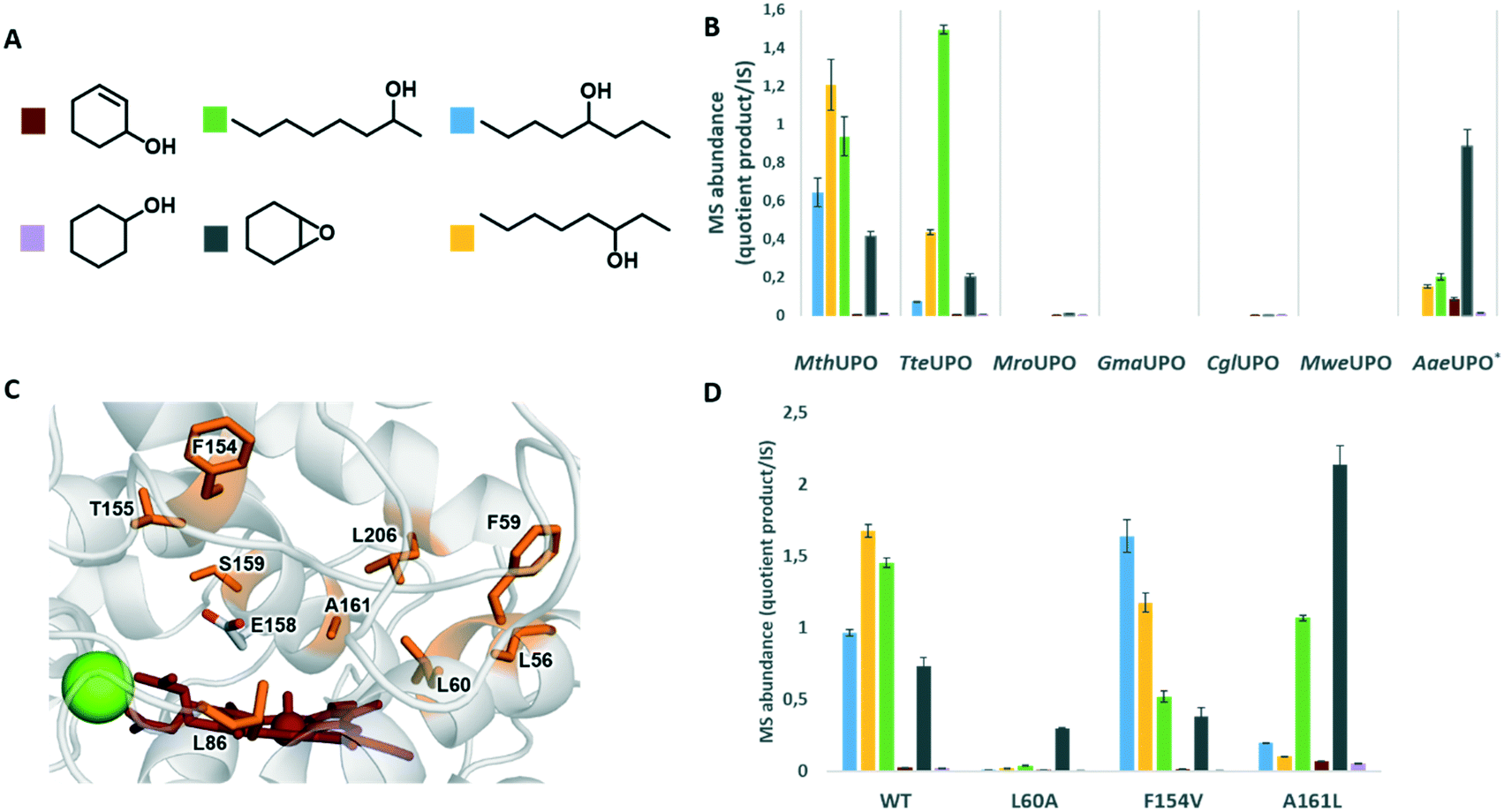 | ||
| Fig. 2 Multiple substrate/product screening of UPOs cultivated in microtiter plate format A) colour legend for the different products, B) comparison of product distribution of several UPOs and C) active site of MthUPO with sites modified by saturation-mutagenesis (highlighted in orange), Mg2+ ion (green sphere) and haem species (red). D) MthUPO variants with shifted chemo- and regioselectivity. UPO catalysed one pot conversion of octane, cyclohexane and cyclohexene (1 mM H2O2, 5 mM of each substrate, 5% (v/v) acetone, 100 mM KPi pH 7, 1 h reaction time at 25 °C using 100 μl of the microtiter plate cultivated S. cerevisiae supernatant containing the corresponding UPO variant). | ||
We started our chemo- and regioselectivity fingerprint studies by investigating the active site using a homology model (Fig. 2C). We aimed to gain the maximum knowledge from a small library within one mutagenesis round and independently saturated nine active site residues with all canonical amino acids: L56, F59, L60, L86, F154, T155, S159, A161 and L206. The mutagenesis was performed using the Golden Mutagenesis cloning technique and its web tool for primer design.27 The mutant library was transformed into S. cerevisiae, assessing around 900 transformants.
Gratifyingly, the applied high throughput screening with multiple substrates resulted in the emergence of three variants with significantly modified product distribution: L60A, F154V and A161L (Fig. 2D). Considering the aliphatic and hydrophobic nature of the screened substrates, it is not surprising that all positions were substituted with amino acids harbouring aliphatic side chains. Position F154 and A161 are located within the α-7-helix located above the peroxo-iron complex (Fig. 2C). Whereas A161 is located deeper in the active site, F154 is directly placed in the substrate entrance channel. L60 is found in the active site near the iron centre in a strategic position that could be important to modulate the substrate's approach to the Cpd I species in a reactive binding pose.
A clear shift in octanol regioisomer formation is observed within the multiple substrate screening for variants F154V and A161L. The wild type displayed the highest activity towards 3-octanol formation, followed by 2-octanol and then 4-octanol (Fig. 2D). Variant F154V modified that pattern and trended towards the 4-octanol formation. The most significant shift in regioselectivity for the octanol formation was discovered for variant A161L. According to the high throughput analysis, this variant substantially diminished the 4- and 3-octanol formation while preserving the 2-octanol regioisomer formation.
Besides the regioselectivity shifts for the octane hydroxylation, the bioconversions of cyclohexene and cyclohexane were simultaneously analysed. The primary screening data revealed substantially improved cyclohexene epoxidation by variant A161L while maintaining the high chemoselectivity with low allylic hydroxylations.
Additionally, we were very pleased to discover a selective variant for the epoxidation of cyclohexene over all the competing oxidation reactions. Variant L60A almost abolished the octane transformation but kept a significant cyclohexene oxide formation.
The screened library did not result in any significantly improved variant for cyclohexane hydroxylation or for a shifted cyclohexene oxidation pattern (from epoxidation towards allylic hydroxylation). It might be that additional positions or a combination of mutations (double or triple mutants) are required to access these activities. However, activation barriers for C–H abstraction by Cpd I estimated from density functional theory (DFT) model calculations (Fig. S12–S14†) already indicated that cyclohexane is intrinsically less reactive than cyclohexene epoxidation and octane C–H activation, respectively. DFT calculations also indicated that allylic C–H activation in cyclohexene is energetically less favourable than epoxidation (Fig. S13†).
With the successful screening of 900 transformants, we set our focus on verifying the observed selectivity shifts using purified MthUPO variants and standard single substrate GC-MS analysis.
A syringe pump system was utilised for gradual hydrogen peroxide supply in a two-liquid-phase system with octane as the organic phase and the corresponding purified MthUPO variant identified from the MISER-GC-MS screening. Variant F154V demonstrated 1.8-fold improved turnover numbers (TONs = mol product per mol enzyme) for formation of the 4-octanol (1a) regioisomer relative to the wildtype. Also, a 1.5 and 4-fold decreased formation of 3- (1b) and 2-octanol (1c), respectively, were observed (Fig. 3A and Table S4†), confirming the regioselectivity trend obtained by the GC-MS screening.
 | ||
| Fig. 3 A) Enzymatically catalysed conversion of octane and comparison of the by MISER-GC-MS identified MthUPO variants, B) cyclohexene and 1-methylcyclohexene bioconversion of MthUPO wildtype and A161L to the corresponding epoxide and C) styrene and styrene derivative bioconversion MthUPO wildtype and A161L (standard deviation <8%, reaction conditions for the syringe pump setup are in the ESI†). | ||
Variant A161L also proved the results from the primary screening. A 10-fold decreased formation of 4-octanol (1a) and no 3-octanol (1b) product was detected. However, the 2-octanol (1c) formation was even slightly increased. Most astonishingly, the single GC-MS analysis of variant A161L revealed the formation of 1-octanol (1d)—the least reactive position for C–H activation by Cpd I, as characterised by DFT calculations (Fig. S14†). 38% of the total octane hydroxylation product corresponded to 1-octanol (1d). The wild type or variant F154V display no activity for this transformation (Fig. S4†).
Thus far, UPO's only reported ability to perform terminal hydroxylations on linear alkanes was reported for MroUPO, where the terminal alcohol was identified as an intermediate that was further oxidised to the corresponding carboxylic acid.12
In the case of MthUPO A161L, the variant exhibited no octanol overoxidation as no aldehyde or carboxylic acid were detected (Fig. S4†). This specificity further renders variant A161L highly significant.
To gain insights into the switch in the hydroxylation pattern observed due to the A161L mutation, we performed molecular dynamics (MD) simulations as previously shown for UPOs.28,30 MD simulations with octane substrate bound in wild type MthUPO showed that the preferential binding conformation of octane corresponds to a buried mode that allows octane to maximise the hydrophobic interactions with multiple residues in the active site (L56, L60, F63, F154 and L206). Within this conformation, C4/C3 and C2 are accessible to Cpd I for the homolytic hydrogen abstraction from the C–H bond (Fig. 4 and S15 and S16†).
 | ||
| Fig. 4 Catalytically relevant binding modes as characterised from MD simulations with A) octane bound in wild type (WT) MthUPO; B) octane bound in A161L variant and C) cyclohexene bound in A161L variant. Substrates, haem cofactor and important active site and catalytic residues are shown in sticks format. The three mutated positions are highlighted in orange, substrates are shown in purple, and structural Mg2+ ion as a green sphere. | ||
The inclusion of the bulkier A161L mutation in this inner active site position prevents the octane from binding deeper in the active site. It explores an additional conformation in which it partially occupies the entrance channel (Fig. 4 and S18†). From this new binding mode, in which octane is flanked by F59, F63, F159 and L206 residues, only the terminal positions C1/C2 can effectively approach the Cpd I catalytic species for hydroxylation.
We then focused on analysing the determined activities for cyclohexene. Variant A161L resulted in slightly improved TONs of 4380 compared to the wild type for the formation of the epoxide 2 in a two-liquid-phase system (Fig. 3B). MD simulations with cyclohexene bound in wild type and A161L variant indicated that the preferential and more buried binding pose explored by cyclohexene in the wild type is displaced towards occupying a binding position near the entrance channel in A161L variant (Fig. S19 and S20†). In this position, cyclohexene interacts with F154 and F63 aromatic side chains (Fig. 4C and S22†), similarly to what is observed for octane (Fig. 4B). We hypothesised that by utilising a prochiral cyclohexene, this improved positioning would lead to substantially improved stereoselectivities. By using 1-methyl-1-cyclohexene, the enantioselectvitiy was indeed significantly improved in the epoxide 3 formation from the wildtype (17% ee) to variant A161L (75% ee).
Variant L60A, which displayed the selective epoxidation of cyclohexene in presence of octane, showed decreased conversions relative to the wildtype in the single substrate setup with 1160 TONs (Table S5†). MD simulations performed with cyclohexene and octane independently bound in L60A variant indicate that this mutation induces an enlargement of the active site region near the Cpd I active species. Cyclohexene bound in this wider active site can explore catalytically competent poses concerning Cpd I active species (Fig. S21†). However, octane can barely explore reactive conformations towards the Cpd I due to the less tight binding and its higher flexibility in this enlarged active site (Fig. S17†). Together with the intrinsic energetically less favourable octane hydroxylation than competing cyclohexene epoxidation (see DFT calculations Fig. S13 and S14†), the latter is proposed to be responsible for the observed substrate selective oxidations.
Based on these results, we were highly interested to employ variant A161L in the epoxidation of styrene derivatives (Fig. 3C). The expected styrene oxide (4) formation was improved 4.5-fold relative to the wildtype to 5030 TONs. Besides this significantly improved activity, variant A161L also exhibited a shift in stereoselectivity. Whereas the wildtype exhibited the formation of the epoxide 4 in the R-conformation with 32% ee, variant A161L produced excess of the S-enantiomer with a 28% ee. Comparing variant A161L to the wildtype when using α-methyl-styrene, resulted in similar observation. A161L showed an excess in the formation of the epoxide 5 and an excess of the S-enantiomer (29% ee), whereas the wild type exhibited selectivities for the R-enantiomer formation (77% ee, (Fig. S7†).
Lastly, we assessed cis-methyl-styrene for its epoxidation. We were surprised to note that as products for the WT and the variant A161L the allylic hydroxylation and overoxidation to the aldehyde was observed instead of the expected epoxidation. This activity contrasts with the well-characterised AaeUPO with its high selectivity for cis-methyl-styrene epoxidation.4
Conclusions
In the present work, we have performed protein engineering targeting the recently characterised MthUPO to address its limitations in terms of low chemo-, and regioselectivity towards aliphatic carbon hydroxylation and CC bond epoxidation, respectively. Since no versatile assay was available to screen for these selectivities, we developed a high throughput screening allowing the quantification of seven analytes simultaneously. We used an off-the-shelf GC-MS without further modifications rendering this method feasible and accessible for many academic laboratories. As the product distinction and quantification is based on mass spectrometric abundance, this allowed the distinction of products with different masses like allylic hydroxylation vs epoxidation and substrate preference directly from the reaction product mixtures. We further expanded the system to allow the simultaneous quantification of regioisomers with identical masses by derivatisation allowing their quantification by regiospecific fragmentations during ionisation. This multiple substrate screening approach allows gaining the maximal information of an enzyme library thereby significantly decreasing the required screening amount and efforts. A unique mass (fragment) is indispensable for the MISER-GC-MS methodology, which required in this work an additional laboratory work-up to access several small products in parallel. Seven wild type UPOs were screened towards their activity and selectivity for octane, cyclohexane and cyclohexene oxidation simultaneously. The novel MthUPO was selected and conducted to mutagenesis at nine selected positions resulting in a focused enzyme library of 900 transformants. Screening this relatively small library resulted in identifying three enzyme variants with divergent chemo- and regioselectivities. One variant (F154V) exhibited pronounced activity towards the 4-octanol formation, and L60A almost exclusively performed cyclohexene epoxidations. Most strikingly was the discovery of A161L variant, that revealed 38% regioselectivity towards the terminal hydroxylation of octane—an activity otherwise only once observed with UPOs.12 Computational modelling based on DFT calculations and substrate-bound MD simulations was performed to rationalise the observed reactivity patterns. MD simulations showed that reshaping the active site due to specific mutations was responsible for modulating the preferential substrate binding poses, controlling the final chemo- and regioselectivities observed.
The presented data showed utterly new insights into UPO activities. The employed method permits vastly increased insights into small enzyme libraries that will be extremely useful for coming efforts in UPO and general enzyme engineering and laboratory evolution.
Author contributions
A. K. and M. J. W. designed the project and the experiments for the method development of multiple substrate MISER-GC-MS. P. P established the original S. cerevisiae expression system for UPOs. A. K. performed the mutagenesis and A. K. and N. H. performed the S. cerevisiae cultivations. The multiple substrate bioconversion, the analysis by MISER-GC-MS and the hit verification was done by A. K.; and J. S. and M. G. B designed the computational modelling. J. S. performed the DFT calculations and MD simulations under M. G. B. supervision, and both analysed the computational data. M. J. W., M. G. B. and A. K. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. W, A. K., and N. H. thank the Bundesministerium für Bildung und Forschung (“Biotechnologie 2020+ Strukturvorhaben: Leibniz Research Cluster”, 031A360B) for generous funding. P. P. thanks the Landesgraduiertenförderung Sachsen-Anhalt for a PhD scholarship. The authors thank Eugen Schell for fruitful discussions. M. G. B. thanks the Generalitat de Catalunya AGAUR for a Beatriu de Pinós H2020 MSCA-Cofund 2018-BP-00204 project, the Spanish MICINN (Ministerio de Ciencia e Innovación) for PID2019-111300GA-I00 project, and J. S. thanks the Spanish MIU (Ministerio de Universidades) for a predoctoral FPU fellowship FPU18/02380. The computer resources at MinoTauro and the Barcelona Supercomputing Center BSC-RES are acknowledged (RES-QSB-2019-3-262 0009 and RES-QSB-2020-2-0016).References
- R. Ullrich, J. Nuske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 Search PubMed.
- S. Bormann, A. Gomez Baraibar, Y. Ni, D. Holtmann and F. Hollmann, Catal. Sci. Technol., 2015, 5, 2038–2052 Search PubMed.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 Search PubMed.
- E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Angew. Chem., Int. Ed., 2011, 50, 10716–10719 Search PubMed.
- Y. Ni, E. Fernandez-Fueyo, A. Gomez Baraibar, R. Ullrich, M. Hofrichter, H. Yanase, M. Alcalde, W. J. van Berkel and F. Hollmann, Angew. Chem., Int. Ed., 2016, 55, 798–801 Search PubMed.
- J. Dong, E. Fernandez-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 Search PubMed.
- M. Hofrichter, H. Kellner, R. Herzog, A. Karich, C. Liers, K. Scheibner, V. W. Kimani and R. Ullrich, in Grand Challenges in Fungal Biotechnology, ed. H. Nevalainen, Springer International Publishing, Cham, 2020, pp. 369–403 Search PubMed.
- M. Hofrichter, H. Kellner, M. J. Pecyna and R. Ullrich, Adv. Exp. Med. Biol., 2015, 851, 341–368 Search PubMed.
- A. Zaks and D. R. Dodds, J. Am. Chem. Soc., 1995, 117, 10419–10424 Search PubMed.
- S. Peter, M. Kinne, X. Wang, R. Ullrich, G. Kayser, J. T. Groves and M. Hofrichter, FEBS J., 2011, 278, 3667–3675 Search PubMed.
- E. D. Babot, J. C. del Rio, L. Kalum, A. T. Martinez and A. Gutierrez, Biotechnol. Bioeng., 2013, 110, 2323–2332 Search PubMed.
- A. Olmedo, C. Aranda, J. C. Del Rio, J. Kiebist, K. Scheibner, A. T. Martinez and A. Gutierrez, Angew. Chem., Int. Ed., 2016, 55, 12248–12251 Search PubMed.
- J. Carro, A. González-Benjumea, E. Fernández-Fueyo, C. Aranda, V. Guallar, A. Gutiérrez and A. T. Martínez, ACS Catal., 2019, 9, 6234–6242 Search PubMed.
- A. González-Benjumea, J. Carro, C. Renau-Mínguez, D. Linde, E. Fernández-Fueyo, A. Gutiérrez and A. T. Martínez, Catal. Sci. Technol., 2020, 10, 717–725 Search PubMed.
- F. H. Arnold, Nat. Biotechnol., 1998, 16, 617–618 Search PubMed.
- W. P. C. Stemmer, Nature, 1994, 370, 389–391 Search PubMed.
- Z. Sun, Q. Liu, G. Qu, Y. Feng and M. T. Reetz, Chem. Rev., 2019, 119, 1626–1665 Search PubMed.
- P. Molina-Espeja, E. Garcia-Ruiz, D. Gonzalez-Perez, R. Ullrich, M. Hofrichter and M. Alcalde, Appl. Environ. Microbiol., 2014, 80, 3496–3507 Search PubMed.
- P. Molina-Espeja, S. Ma, D. M. Mate, R. Ludwig and M. Alcalde, Enzyme Microb. Technol., 2015, 73–74, 29–33 Search PubMed.
- P. Püllmann, A. Knorrscheidt, J. Münch, P. R. Palme, W. Hoehenwarter, S. Marillonnet, M. Alcalde, B. Westermann and M. J. Weissenborn, bioRxiv, 2020, DOI:10.1101/2020.07.22.216432.
- M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891–903 Search PubMed.
- G. Li, P. Yao, R. Gong, J. Li, P. Liu, R. Lonsdale, Q. Wu, J. Lin, D. Zhu and M. T. Reetz, Chem. Sci., 2017, 8, 4093–4099 Search PubMed.
- G. Qu, R. Lonsdale, P. Yao, G. Li, B. Liu, M. T. Reetz and Z. Sun, ChemBioChem, 2018, 19, 239–246 Search PubMed.
- Z. Sun, P. T. Salas, E. Siirola, R. Lonsdale and M. T. Reetz, Bioresour. Bioprocess., 2016, 3, 44 Search PubMed.
- K. Zhang, S. El Damaty and R. Fasan, J. Am. Chem. Soc., 2011, 133, 3242–3245 Search PubMed.
- A. Knorrscheidt, P. Püllmann, E. Schell, D. Homann, E. Freier and M. J. Weissenborn, ChemCatChem, 2020, 12, 4788–4795 Search PubMed.
- P. Püllmann, C. Ulpinnis, S. Marillonnet, R. Gruetzner, S. Neumann and M. J. Weissenborn, Sci. Rep., 2019, 9, 10932 Search PubMed.
- M. Municoy, A. Gonzalez-Benjumea, J. Carro, C. Aranda, D. Linde, C. Renau-Minguez, R. Ullrich, M. Hofrichter, V. Guallar, A. Gutierrez and A. T. Martinez, ACS Catal., 2020, 10, 13584–13595 Search PubMed.
- P. Püllmann and M. J. Weissenborn, bioRxiv, 2020, DOI:10.1101/2020.12.23.424034.
- A. Knorrscheidt, J. Soler, N. Hünecke, P. Püllmann, M. Garcia-Borras and M. J. Weissenborn, ChemRxiv, 2020, DOI:10.26434/chemrxiv.13265618.v1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy02457k |
| This journal is © The Royal Society of Chemistry 2021 |