
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShort-channel mesoporous SBA-15 silica modified by aluminum grafting as a support for CoRu Fischer–Tropsch synthesis catalysts†
Sophia
Mohammadnasabomran
a,
Carlos
Márquez-Álvarez
 b,
Joaquín
Pérez-Pariente
*b and
Agustín
Martínez
*c
b,
Joaquín
Pérez-Pariente
*b and
Agustín
Martínez
*c
aSchool of Chemistry, College of Science, University of Tehran, Tehran, Iran
bInstituto de Catálisis y Petroleoquímica, ICP-CSIC, C/Marie Curie 2, 28049, Madrid, Spain. E-mail: jperez@icp.csic.es
cInstituto de Tecnología Química, Universitat Politècnica de València – Consejo Superior de Investigaciones Científicas (UPV – CSIC), Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: amart@itq.upv.es
First published on 29th April 2021
Abstract
Highly ordered short-channel mesoporous silica SBA-15 with large pores (11.2 nm) has been synthesized by using tetramethyl orthosilicate (TMOS) as a silica source, the amphiphilic block copolymer Pluronic PE-10400 as a structure-directing agent, and 1,3,5-triisopropylbenzene as a swelling agent. The Al-SBA-15 support was prepared by multiple cycles of grafting of Al sec-butoxide dissolved in toluene on the calcined SBA-15 material, aiming at covering the channel surface with a subnanometric layer of Al–O chemical species. Both the pristine and the Al-containing materials have been used as supports for preparing 20 wt% Co catalysts promoted with 1 wt% Ru and then evaluated for Fischer–Tropsch synthesis (FTS). The samples have been characterized by ICP-OES, elemental analysis, N2 physisorption, XRD, electron microscopy, FTIR-pyridine, H2-TPR, H2 chemisorption, and XPS. The ordered mesopore architecture of the starting support was preserved during the grafting treatment, but the pore size was reduced from 11.2 to 10.0 nm, while the pore volume and surface area also decreased. No segregated alumina phase was detected. It was found that the Al-grafting remarkably improves the Co dispersion and favors a more homogeneous distribution of the metal species along the SBA-15 particles. Under conventional FTS conditions (220 °C, 2.0 MPa, H2/CO = 2), the Al-grafted CoRu/Al-SBA-15 catalyst exhibited a lower TOF for the exposed Co0 sites resulting in a lower CO conversion and cobalt-time-yield (CTY) with respect to the all-silica CoRu/SBA-15 counterpart. However, CoRu/Al-SBA-15 displayed, at constant CO conversion (∼40%), an enhanced selectivity to liquid hydrocarbons (C5–C20) to the detriment of waxes (C21+). While the lower activity of CoRu/Al-SBA-15 was mainly ascribed to a greater abundance of very small (<6 nm) Co0 nanoparticles with a lower TOF, the shift in selectivity towards lighter hydrocarbons was related, in addition to the contribution of sub-6 nm Co0 particles, to an enhanced CO diffusion limitation and to the cracking of long-chain FTS products on the Brønsted acid sites present in this catalyst.
1. Introduction
Ordered mesoporous SiO2, Al2O3, and TiO2 materials have received great attention in catalysis owing to their large surface area and porosity as well as to their relatively homogeneous and, within certain limits, tuneable pore size. In fact, cobalt-based catalysts supported on these mesoporous materials have shown good catalytic performance in the Fischer–Tropsch synthesis (FTS) reaction.1–3 The activity and C5+ selectivity in FTS over supported Co catalysts are known to strongly depend on the dispersion (i.e. particle size) of active Co0 species and diffusion phenomena that are closely related to the textural and chemical properties of the supports.4,5 Therefore, finely tuning the support properties is a plausible, albeit challenging, strategy to design Co-based FTS catalysts with improved catalytic performance. One of the main aspects of FTS catalysts is the interaction between the metal and the support. Hence, strengthening the interaction between Co species and the metal oxide support enhances the metal dispersion but decreases the amount of metallic cobalt surface sites by promoting the formation of hard-to-reduce mixed cobalt–support compounds (e.g. cobalt aluminates in Al2O3-supported catalysts).6 Silica-based materials exhibit a weaker interaction with the supported metal phases in comparison with Al2O3 and TiO2,7 which favours the reducibility of Co oxides albeit at the cost of achieving poorer metal dispersions.The ordered mesoporous SBA-15 silica is considered the most interesting silica-based mesostructure because of its large specific surface area and pore volume, uniformly-sized pores in the range of 4–30 nm, and thick framework walls. Due to their favourable textural properties, SBA-15-supported Co catalysts generally exhibit an enhanced Co dispersion and FTS activity with respect to equivalent catalysts supported on conventional (non-ordered) SiO2 materials.8 Moreover, in comparison with MCM-41 silicas displaying narrower mesopores (typically sizing 2–4 nm), Co/SBA-15 catalysts provide enhanced transport rates for reactants and products during FTS, favourably affecting the activity and selectivity to desired long-chain (C5+) hydrocarbons.8–10 However, as commonly occurs for silica-based supports, the weak metal–support interaction tends to promote undesirable sintering of metal particles during the thermal activation treatments (e.g., calcination–reduction) and FTS reaction that decreases the amount of active Co0 sites available for reaction with a detrimental effect on the overall catalyst performance.
These drawbacks of silica supports can be overcome by alumina (γ-Al2O3), which shows an intermediate Co–support interaction strength, thus exhibiting a relatively high capacity for dispersing the cobalt phases as compared to silica. On the other hand, the cobalt reducibility of alumina-supported catalysts is generally lower than those based on silica and they often require the addition of small amounts of noble metals like Ru or Pt in order to enhance their reducibility. In addition, alumina-based FTS catalysts display good mechanical properties and a high resistance to attrition.5,11–16 However, even mesoporous alumina prepared in the presence of surfactants shows a broad pore size distribution, which makes it difficult to gain precise control over the uniformity of metal particle size and hence over the FTS performance.17 Therefore, it would be desirable to combine in one single support the benefits of the well-defined porous architecture of silica-based ordered SBA-15 materials, and those arising from the interaction between cobalt and alumina supports. This concept has been explored by grafting Al precursors on the surface of SBA-15 silicas resulting in highly accessible Al species, in contrast to the incorporation of Al by direct synthesis that typically leads to a large fraction of Al being located at inaccessible positions deep within the SBA-15 walls.18 The grafting approach was already described earlier to improve the performance of a series of catalysts based on ordered mesoporous silicas (OMS) for processes other than FTS, aiming at combining the architectural features of the silica support with the surface chemical functionality of alumina, as in the case of MCM-41,19 which was later extended to SBA-15.20–22 This strategy was later applied to FTS catalysts. In this way, Hao et al.23 reported the grafting, in supercritical CO2, of aluminum isopropoxide on SBA-15 with a pore size of 4.7 nm, which resulted in segregated alumina outside the surface channel and the consequent location of most of the impregnated Co outside the mesopores. The incorporation of Al in SBA-15 silicas, even in low concentrations, not only improved the cobalt dispersion but also promoted the formation of middle range hydrocarbons (C8–C18) with increased amounts of branched paraffins and less waxes (C21+) due to secondary reactions occurring on the Al-related Brønsted acid sites.24 It is interesting to notice that in most prior studies, the starting silica SBA-15 material used for alumina coating exhibited a relatively small pore, in the range of 5–7 nm, and the particles show a characteristic fiber-like morphology having a length of several microns with the mesoporous channels running all along the fibers. However, for FTS, larger pore diameters as well as shorter channels that could reduce diffusion limitations during the reaction are much more desirable from the viewpoint of both activity and selectivity to long-chain (C5+) hydrocarbons.18,25
In this work, we propose a feasible approach to the synthesis of a support with a large pore size (∼11 nm), well-defined porosity, and short diffusion path by sequential grafting of all-silica short-channel SBA-15 with a suitable aluminum precursor under appropriate conditions. The application of the grafted aluminum-containing SBA-15 material as a support for preparing Ru-promoted Co-based FTS catalysts and the investigation of the correlation between support properties and catalytic performance is another aim of the present study.
2. Experimental
2.1. Materials
Aluminum sec-butoxide Al(O-sec-Bu)3 as an alumina precursor, tetramethyl orthosilicate (TMOS) as a silica precursor, Pluronic PE-10400 as a structure directing agent, and 1,3,5-triisopropylbenzene as a swelling agent were purchased from Sigma-Aldrich (Oakville, Canada). Cobalt nitrate hexahydrate (99.9%) and ruthenium(III) nitrosyl nitrate were purchased from Alfa Aesar (Tewksbury, United States) and used as the main active metal and promoter precursors, respectively. Toluene as a solvent was purchased from Fisher Scientific (United States).2.2. Support synthesis
Short-channel SBA-15 silica samples were prepared using Pluronic PE-10400, a symmetric triblock copolymer comprising poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), according to the procedure described in ref. 26–28. The use of this surfactant instead of the conventional Pluronic P123, under suitable synthesis conditions, allows the formation of thin plate-like hexagonal particles with less than one micron thickness, where the mesoporous channels run perpendicular to the base plane of the particle.For this purpose, 4.5 g of PE-10400 and 0.048 g of NH4F were dissolved in 195 mL of 1.1 M aqueous HCl solution at room temperature in a closed Pyrex container under stirring. After complete dissolution, 6.6 mL of TMOS was added to the solution. As the aim of this work is to prepare large-pore SBA-15 materials, the above procedure was slightly modified in a second preparation by adding to the TMOS solution 0.024 mL of 1,3,5-triisopropylbenzene as a swelling agent. In both cases, the gel obtained was left under stirring for 24 h and 20 °C, and then was heated at 80 °C in the closed container for 1 day. After filtering and washing with 300 mL of ethanol, the solid was dried at room temperature. The surfactant removal was done by extraction with ethanol for 4 h at 80 °C. Finally, the samples were calcined for 6 h at 550 °C at 1 °C min−1 heating rate. The final samples were labeled SP (without an expander) and LPS (with an expander).
2.3. Grafting procedure
The grafting procedure was derived from that reported in ref. 19, but substantial modifications were introduced to gain a better control over the nature of the aluminum coating of the surface. The purpose of the procedure is to form a first layer of mono-anchored Al species on the silica surface, by using a Si–OH/Al alkoxide precursor ratio of one, forming Si–O–Al(OR)2 species. The subsequent hydrolysis of these species produces Al–OH groups that would be able to react with a second dose of Al alkoxide, to form Al–O–Al linkages, and the same procedures could be repeated several times. We have used just three cycles in order to decrease the possibility of forming an undesirable, physically separated, compact alumina phase. Al grafting for preparing Al-SBA-15 was performed as follows. First, 3 g of calcined SBA-15 with expanded pores (LPS) was placed in a three-neck round-bottom flask and kept under vacuum overnight at 100 °C in order to remove any residual water adsorbed on the sample. The required amount of aluminum precursor was estimated by calculating the amount of silanol groups of the LPS sample by TGA analysis (weight loss at T > 560 °C). In this way, 1.2 g of aluminum-tri-sec-butoxide was dissolved in 25 mL of toluene and injected onto the flask containing the dried LPS sample at 50 °C and kept for 12 h at this temperature under stirring. After that, the solvent was removed under vacuum and N2 bubbled through water at room temperature was flowed into the sample flask for 12 h at 100 °C, in order to hydrolyze the alkoxide groups still remaining attached to aluminum. Therefore, we avoid the use of triethylamine and of a hydrolyzing water/ethanol solution as reported in ref. 19. After heating the treated sample at 100 °C overnight under vacuum, the same procedure was repeated two more times to achieve a nominal Al/Si atomic ratio of 0.29. After the third and last injection of the Al precursor, the dried sample was calcined step by step (2 h at 100 °C, 2 h at 200 °C, and 3 h at 300 °C) at 1 °C min−1 heating rate in the presence of air flow (0.1 L min−1). The final sample was labeled ALPS, and its Al/Si atomic ratio was 0.26 as determined by chemical analysis (ICP-OES).2.4. Preparation of catalysts
The required amounts of cobalt nitrate and ruthenium(III) nitrosyl nitrate were dissolved in water and the obtained solution introduced via incipient wetness impregnation to the support pores to achieve nominal metal loadings of 20 wt% Co and 1 wt% Ru. The supports were dried before impregnation at 150 °C overnight. The impregnated samples were dried in an oven at 200 °C for 12 h and then calcined at 300 °C for 3 h with 1 °C min−1 heating rate under flowing air (0.5 L gcat−1 min−1). The final samples were labeled as CRALPS (CoRu/Al-SBA-15) and CRLPS (CoRu/SBA-15).2.5. Characterization methods
Thermogravimetric analysis curves (TG/DTG) were recorded using Perkin Elmer TGA7 equipment from 30 °C to 900 °C with a heating rate of 20 °C min−1 under air flow.Nitrogen adsorption–desorption isotherms were acquired at −196 °C with a Micromeritics ASAP 2420 sorptometer. Prior to analysis, the samples were degassed at 200 °C for 16 h. Total surface areas were calculated by the BET method. The pore size distribution, micropore volume, and mesopore surface area and volume were determined from the adsorption data using the methods of non-local density functional theory (NLDFT) with the kernel function for oxide materials with cylindrical pores.29,30
Chemical analysis of the bare ALPS support and CoRu catalysts was performed by ICP-OES using a Varian 715-ES spectrometer. Prior to the analyses, the samples (ca. 20 mg) were dissolved in a mixed acid solution of HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HF:HCl with a 1:1:3 volume ratio.
:HF:HCl with a 1:1:3 volume ratio.
Scanning electron microscopy (SEM) has been performed using a Hitachi S-4100 field-emission (FE) gun microscope equipped with a BSE-AUTRATA detector using gold-coated powder specimens.
The morphology of the calcined supports and catalyst particles was studied by transmission electron microscopy (TEM) in a Tecnai G2 field emission gun device with EDAX microanalysis working at 200 kV. Samples for TEM studies were prepared by ultrasonic dispersion in ethanol for 1 min. After that, the suspensions were dropped onto a carbon coated copper grid. The TEM-derived Co3O4 particle size distributions for the calcined CRLPS and CRALPS samples were determined by counting about 200 particles in several images taken at different positions on the grid, using the ImageJ software. The Al-containing CRALPS catalyst was further characterized in the same microscope by high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM). Before observation, the calcined sample was reduced in pure H2 for 10 h at 400 °C and subsequently passivated at room temperature under a diluted oxygen flow (0.5 vol% O2 in N2) in order to prevent spontaneous re-oxidation of the bulk metal nanoparticles upon contact with ambient air. The Co0 particle size histogram was generated from the HAADF-STEM images upon measuring about 200 particles. Surface average Co3O4 and Co0 particle sizes (D[3,2]) were calculated from the directly measured particle sizes as D[3,2] = ∑ni·di3/∑ni·di2, where ni is the number of particles with diameter di.
The X-ray diffraction patterns of the calcined catalysts were acquired with a SEIFERT XRD 3000P diffractometer using monochromatized Cu-Kα radiation. Using the Scherrer equation, the average size of the Co3O4 crystallites in the calcined catalysts was estimated from the line broadening of the Co3O4 peak at 2θ of 36.88° using a K shape factor of 0.89.
Characterization of the structure of hexagonally ordered SBA-15 mesoporous materials was carried out by small-angle X-ray scattering. The position of Bragg diffraction peaks provides important information about the type of ordering and unit cell parameters in the SBA-15 mesoporous materials.
Fourier-Transform infrared (FTIR) spectra of adsorbed pyridine were recorded in the transmission mode, in the mid-IR region at 4 cm−1 optical resolution, using a Thermo Nicolet Nexus 670 FTIR spectrometer equipped with an MCT criodetector. Samples were pressed into self-supporting wafers of 13 mm diameter and a thickness of ca. 10 mg cm−2, placed inside an all-glass closed cell provided with CaF2 windows, and degassed at 400 °C for 12 h. Subsequently, the samples were cooled down to 150 °C, saturated with pyridine at 8 Torr for 5 min, and weakly adsorbed pyridine was removed under vacuum at 150 °C for 1 h. The spectra of pretreated and pyridine-adsorbing samples were normalized with respect to the integral intensity of the silica overtones located within the 2090–1730 cm−1 range. Spectra of adsorbed pyridine were obtained by subtracting the normalized spectrum of the pretreated sample from the normalized spectrum of the corresponding pyridine-adsorbing sample. The concentrations of Brønsted and Lewis acid sites were calculated from the integral intensities of the IR bands at ca. 1545 and 1450 cm−1, respectively, using the corresponding molar extinction coefficient reported by Emeis.31
Hydrogen temperature-programmed reduction (H2-TPR) of the calcined catalysts was performed using a Micromeritics Autochem 2910 apparatus. The catalyst samples were first purged in a flow of argon at room temperature. Then, TPR of 30 mg of each sample was performed using 10 vol% hydrogen in an argon gas mixture and the temperature increased up to 900 °C at a heating rate of 10 °C min−1. For determination of the extent of cobalt reduction, 100 mg of sample was reduced under a flow of pure H2 under the same reduction conditions applied in-reactor prior to catalysis (400 °C, 10 h, heating rate of 1 °C min−1). After switching the gas from H2 to Ar and flushing the sample at 400 °C for 30 min, the Ar flow was replaced with a flow of 10 vol% H2 in Ar and the temperature increased from 400 °C to 900 °C at a heating rate of 10 °C min−1. The degree of cobalt reduction (DOR) was calculated from the amount of H2 consumed in this last step assuming that it corresponds to the reduction of CoO (remaining after the previous reduction at 400 °C) to Co0.
Cobalt dispersions were determined by H2 chemisorption at 150 °C on Micromeritics ASAP 2010C equipment, by estimating the total gas uptakes in the H2 adsorption isotherms extrapolated to zero pressure. Before measurements, the calcined catalysts (0.25 g) were reduced in situ under flowing pure H2 at 400 °C for 10 h and then degassed at 1.3 Pa. Mean Co0 particle sizes (d) were estimated from the total amount of chemisorbed H2, Co content (from ICP-OES), and degree of cobalt reduction (DOR) assuming a H/Co = 1 atomic ratio stoichiometry and a hemispherical particle geometry with a surface atomic density of 14.6 atoms per nm2, according to eqn (1):
| d (nm) = 96/D | (1) |
X-ray photoelectron spectroscopy (XPS) measurements were performed on SPECS equipment with a UHV system (pressure in the range of 10−10 mbar) equipped with a PHOIBOS 150 9MCD energy analyzer, using a non-monochromatic Al X-ray source (200 W power and 12 kV accelerating voltage). High-resolution XP spectra were recorded with an analyzer pass energy of 20 eV. For analyses, powdered samples were pressed into self-supporting wafers and deposited on the sample holder with double-sided adhesive conductive carbon tape. In order to correct for charging effects, the binding energy (BE) values for the analyzed elements were referenced to the main O 1s emission line assigned to silica (BE set to 532.9 eV (ref. 32–34)). Data analysis was performed using CasaXPS software (v2.3.16). Deconvolution of experimental peaks into components was done using a non-linear, least squares fitting algorithm after Shirley baseline subtraction. XPS peaks were fitted using a 70% Gaussian–30% Lorentzian mixed function. Surface atomic ratios were calculated from the peak area ratios normalized by the corresponding relative sensitivity factors provided by CasaXPS software.
2.6. Catalytic testing
The catalytic performance for FTS of the Ru-promoted Co catalysts supported on SBA-15 silica and grafted Al-SBA-15 materials was evaluated in a tubular down flow fixed bed reactor made up of stainless steel, charged with 0.5 g of catalyst in the calcined form. Prior to the reaction, the catalyst was reduced in situ under a flow of hydrogen (200 mL gcat−1 min−1) at atmospheric pressure and 400 °C for 10 h. After catalyst reduction and cooling down to 100 °C under H2 flow, the reactant gas mixture (CO, H2 and Ar with a 3:6:1 volume ratio) was fed to the reactor, and the temperature and reaction pressure increased up to 220 °C and 2.0 MPa, respectively. In total, the FTS experiments lasted about 16 h according to the following protocol that proved useful to derive relevant structure–performance correlations for Co-based FTS catalysts as reported in our previous studies.25,35,36 Thus, during the first 8 h of reaction (period 1), the space velocity (GHSV) was maintained constant at 7.0 Lsyngas gcat−1 h−1 in order to compare the activity under equal conditions. Then, during the last 8 h (period 2), the GHSV was adjusted for each catalyst to fairly compare the product selectivity at a constant CO conversion of ca. 40%. The reported activity (period 1) corresponded to the averaged data in the time-on-stream (TOS) range of 5–8 h where a nearly constant CO conversion was attained after an initial deactivation, while a stable CO conversion was observed for the whole period 2 (Fig. S1 of the ESI†).
The products were continuously removed from the reactor and passed through two consecutive traps maintained, respectively, at 150 °C and 100 °C (both at the reaction pressure of 2.0 MPa) to retain water and heavier hydrocarbons. CO, CO2 and uncondensed hydrocarbons were regularly monitored online at time intervals of ca. 1 h in a GC (Varian 3800). After each reaction period, the heavier hydrocarbons were collected, weighted, and analyzed offline in the same GC. The overall product distributions were obtained by merging the online and offline GC analyses. The carbon mass balances for the reported experiments were 100 ± 2%. The CO conversion, cobalt-time-yield (CTY, molCO gCo−1 h−1), and carbon-based selectivity of hydrocarbon products were determined using the formulas presented in our previous work.17
3. Results and discussion
3.1. Selection of the SBA-15 silica support
The small-angle X-ray diffraction patterns of the two SBA-15 samples (SP and LPS) are characteristic of materials consisting of a well-ordered two-dimensional hexagonal array of tubular mesopores (Fig. 1). A considerable low-angle shift of the reflections for the sample synthesized in the presence of triisopropylbenzene can be observed, which indicates an enlargement of the unit cell parameter a0 from 119 Å for the sample obtained in the absence of the additive (SP) to 136 Å when the additive is present (sample LPS). This observation is consistent with earlier reports on the effect of this aromatic compound as a pore expander in conventional SBA-15 materials.37 | ||
| Fig. 1 Small-angle X-ray diffraction patterns of calcined SBA-15 materials. | ||
The pore enlargement anticipated by the expansion of the unit cell size is confirmed by adsorption/desorption of nitrogen. The N2 physisorption isotherm of sample LPS (Fig. 2, left) corresponds to type IVa according to the IUPAC classification,38 while the hysteresis loop (H1) is characteristic of the capillary condensation that takes place in the mesopores. The adsorption branch shows a very steep increase at 0.7–0.8 relative pressure, evidencing a very uniform channel diameter, consistent with the narrow pore size distribution displayed in Fig. 2 (right).
 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms (left) and pore size distributions (right) of samples SP (a), LPS (b), ALPS (c), CRLPS (d) and CRALPS (e). Full symbols correspond to the adsorption branch and open symbols to the desorption branch. For clarity, the isotherms have been upshifted by 40 (a), 30 (b), 20 (c), and 10 mmol g−1 (d). | ||
The isotherm of the sample synthesized in the absence of triisopropylbenzene is qualitatively like that of LPS. It can be noticed that the use of triisopropylbenzene produced an increase of the pore diameter by 1.1 nm, from 10.1 to 11.2 nm (Table 1). This is a considerable enlargement that brought the pore dimension to values not only larger than those of previous studies on Al-grafting of SBA-15, but places also the pore size in the suitable range for the successful achievement of the two aims of the present study. First, coating of the pore surface with a thin layer of Al–O species avoided pore plugging and the formation of unwanted physically separated alumina; second, the pore size of the resulting Al-containing material would still be sufficiently large so as to accommodate inside the pores the cobalt nanoparticles whose confinement inside the SBA-15 channels would preclude them from excessive growth.
| Sample | BET surface area (m2 g−1) | Micropore volume (cm3 g−1) | Mesopore diametera (nm) | Mesopore surface area (m2 g−1) | Mesopore volume (cm3 g−1) |
|---|---|---|---|---|---|
| a Pore size corresponding to the maximum of the pore size distribution. | |||||
| SP | 942 | 0.04 | 10.1 | 740 | 0.95 |
| LPS | 583 | 0.04 | 11.2 | 386 | 0.72 |
| ALPS | 318 | 0.04 | 10.0 | 172 | 0.35 |
| CRLPS | 342 | 0.03 | 10.7 | 201 | 0.40 |
| CRALPS | 233 | 0.03 | 10.0 | 125 | 0.25 |
While the pore size of the LPS sample would be quite suitable for our purpose, it should also at the same time possess the appropriate morphology leading to short channels. Fig. 3 shows the scanning electron microscopy images of SP and LPS samples. Plate-like hexagonal structures of 500–600 nm in diameter and 200–300 nm thickness are clearly evident for SP, although small particles with a round-shape morphology are also observed. For the large-pore material LPS, the hexagonal plates are somewhat less abundant, and round-shape particles predominate, which are nevertheless smaller than 500 nm in diameter. The tubular channels in this structure are aligned perpendicular to the basal plane of the particles,26–28 and hence their average length would be in the range of 300–500 nm. Therefore, it can be concluded that sample LPS presents the required pore size and channel length to be used as a starting material for Al-grafting.
 | ||
| Fig. 3 SEM images of samples SP (a and b) and LPS (c and d). | ||
3.2. Characterization of LPS and Al-grafted ALPS supports
Grafting of Al on the calcined LPS sample was carried out as described in the Experimental section, by performing three sequential treatments of the sample with aluminum-tri-sec-butoxide. As evidenced by X-ray diffraction, the nice hexagonal arrangement of the starting LPS remains unaffected by the grafting procedure (Fig. 1). On the contrary, changes in the N2 adsorption properties are clearly observed (Fig. 2). Aluminum grafting produces a noticeable decrease of the mesopore volume and area, and, above all, a decrease of the pore diameter by 1.2 nm, from 11.2 nm for LPS to 10.0 nm for ALPS (Table 1). Moreover, the shape of the hysteresis loop evidences a remarkable homogeneity of the pore size, as also evidenced by its narrow pore size distribution (Fig. 2). Therefore, it can be concluded that the Al-grafting produces a rather uniform coating of the pores with a layer of Al–O species of nearly 0.6 nm thickness. This value would be consistent with the presence of a 3-oxygen layer thick alumina film,39 as expected for the 3-cycle Al-grafting procedure used here. The first layer would correspond to Al attached to Si atoms of the silanol groups originally present in the material, forming Si–O–Al bonds. It is also interesting to notice that the micropores commonly present inside the mesopore walls of SBA-15 have not been affected by the grafting (Table 1), which strongly suggests that Al-species are not present there. It is possible that the bulky aluminum-tri-sec-butoxide cannot access the narrow micropores, leaving them free from Al coating. The representative TEM images in Fig. 4 reveal the ordered honeycomb arrangement of the pores in the hexagonal-shape particles for both LPS and ALPS supports. | ||
| Fig. 4 Representative TEM images of supports LPS (a) and ALPS (b). | ||
XPS provided additional insight into the nature and distribution of the several chemical species present at the sample surface. The Si 2p, Al 2p, and O 1 s XPS spectra of sample ALPS are displayed in Fig. 5. The corresponding fitted XPS parameters and the surface atomic ratios are presented in Table 2.
 | ||
| Fig. 5 Fitted Si 2p, Al 2p, O 1s, and Co 2p core-level spectra of samples ALPS (bottom), CRALPS (middle), and CRLPS (top). Peak intensities of all core-levels have been normalized for each sample by referencing to the corresponding Si 2p peak area. | ||
| ALPS | CRALPS | CRLPS | |||
|---|---|---|---|---|---|
| Core level | BE (eV) | BE (eV) | Area (%) | BE (eV) | Area (%) |
| Si 2p | 103.5 | 103.5 | 103.5 | ||
| Al 2p | 75.3 | 75.3 | |||
| O 1s | 532.9 | 532.9 | 92.3 | 532.9 | 93.4 |
| 530.7 | 7.7 | 530.4 | 6.6 | ||
| Co 2p3/2 (Co3+, Co3O4) | 780.2 | 7.8 | 780.0 | 30.0 | |
| Co 2p3/2 (Co2+, Co3O4) | 781.1 | 3.9 | 781.5 | 15.0 | |
| Co 2p3/2 (Co2+, CoAl2O4) | 781.9 | 32.6 | |||
| Co 2p3/2 satellite | 786.4 | 21.5 | 785.2 | 20.8 | |
| Co 2p1/2 (Co3+, Co3O4) | 795.5 | 4.1 | 795.3 | 15.6 | |
| Co 2p1/2 (Co2+, Co3O4) | 796.7 | 2.0 | 796.9 | 7.8 | |
| Co 2p1/2 (Co2+, CoAl2O4) | 797.4 | 16.9 | |||
| Co 2p1/2 satellite | 804.5 | 11.2 | 804.1 | 10.8 | |
| Atomic ratio | ALPS | CRALPS | CRLPS | ||
| Al/Si | 0.673 | 0.444 | — | ||
| Co/(Si + Al) | — | 0.125 | 0.036 | ||
The Si 2p spectrum shows a dominant signal at 103.5 eV, which is characteristic of SiO2-based materials.34 In addition, a contribution of very small intensity is observed at 101 eV. This BE value is too low to be attributed to Si atoms eventually bonding to Al atoms in Si–O–Al configuration.22,40 Indeed, this signal could correspond to the Auger Al KLL line, which overlaps with the Si 2p line excited by Al Kα radiation.41
The O 1s spectrum shows a single peak at BE of 532.9 eV, characteristic of SiO2 materials. Finally, the Al 2p spectrum is dominated by a single signal that appears at BE of 75.3 eV, attributed to Al present in an aluminosilicate phase.42 It is interesting to notice that the BE of aluminum in segregated (bulk) alumina is 74.5 eV,42,43 but no signal at that BE is observed in the spectrum, which excludes its presence in the material. The very thin Al coating, formed by just 3 Al monolayers, would make the whole spectrum dominated by the nature of the Al atoms directly linked to the framework Si atoms. It can be noticed that the Al/Si atomic ratio determined by XPS (Table 2) is higher than the bulk ratio (0.673 vs. 0.26). This high Al/Si ratio should be attributed to the small analysis depth of XPS, typically less than 5 nm, which allows detection of the whole thin alumina layer covering the silica support but only part of the silica material lying underneath, in the outermost region of the sample particles. Indeed, the results of N2 physisorption show a notable homogeneity of the thickness of the Al-coating layers of the silica channels, which points to a uniform Al/Si ratio all along the particles.
The acidity of the grafted Al-SBA-15 support (ALPS) was studied by FTIR of adsorbed pyridine. As observed in the normalized FTIR-pyridine spectrum shown in Fig. 6, the ALPS support exhibits an intense band at ca. 1455 cm−1, attributed to Al3+-related Lewis acid sites (LAS), in addition to a much weaker band at ca. 1545 cm−1 associated with Brønsted acid sites (BAS). The presence of BAS indirectly evidences the existence of tetrahedral Al species in Si–OH–Al groups on the surface of the Al-grafted support.44 The concentrations of BAS and LAS amounted to 32 and 234 μmol gsup−1, respectively.
 | ||
| Fig. 6 FTIR-pyridine spectra of the bare ALPS carrier (a) and the CRALPS catalyst (b) recorded at a pyridine desorption temperature of 150 °C. The spectra were normalized to the silica overtones (2090–1730 cm−1 range). | ||
3.3. Characterization of CoRu catalysts
As described in the Experimental section, Co and Ru were deposited on the pristine all-silica LPS material (CRLPS catalyst) and the Al-containing sample (CRALPS catalyst). The Co content determined by ICP-OES is similar for both calcined catalysts and amounted to 17.1 and 16.5 wt% for CRLPS and CRALPS, respectively, close to the nominal value (20 wt%). By contrast, the Ru contents are remarkably lower (0.3 wt% for CRLPS and 0.2 wt% for CRALPS) than the targeted 1 wt%. Such a significant loss of Ru is commonly observed in Ru-promoted Co catalysts and is attributed to the formation, catalyzed by Co3O4, of volatile suboxide RuOx species during the air-calcination treatment.45The X-ray diffraction results (Fig. 7) reveal, for both catalysts, the presence of reflections corresponding to the different crystallographic planes of Co3O4 as the only crystalline cobalt phase detected after calcination. In comparison with CRALPS, the Al-free CRLPS catalyst exhibits sharper peaks that suggest larger cobalt oxide crystallites in the latter. Indeed, the Co3O4 average crystal size estimated from peak broadening is 15 nm for CRLPS, whereas it is reduced to 11 nm for the Al-grafted CRALPS sample (Table 3). It is also interesting to notice that, while the average Co3O4 crystallite size in the Al-sample is quite close to the average pore size (10 nm), it largely exceeds the pore diameter in the all-silica catalyst (11.2 nm). Therefore, the presence of the Al-layer in the SBA-15 carrier favors a higher dispersion of the cobalt oxide particles, which can be attributed to the enhanced interaction of the metal precursor species with the support surface relative to the all-silica material, in agreement with previous observations.24,46
 | ||
| Fig. 7 Wide-angle X-ray diffraction patterns of supported CoRu catalysts. | ||
| Catalyst | d(Co3O4) | H2-TPR | H2 chemisorption | STEM | |||
|---|---|---|---|---|---|---|---|
| XRD (nm) | TEMc (nm) | DORa (%) | H2 uptake (μmol g−1) | D(Co0)b (%) | d(Co0)H (nm) | d(Co0)Tc (nm) | |
| a Determined by H2-TPR on pre-reduced (400 °C, 10 h) samples. b Co0 dispersion derived from H2 chemisorption after correction by DOR. c Surface average cobalt particle diameter (D[3,2]). | |||||||
| CRLPS | 15 | 10.1 | 96 | 156 | 11.2 | 8.6 | — |
| CRALPS | 11 | 5.9 | 87 | 192 | 15.8 | 6.1 | 6.2 |
The N2 adsorption/desorption isotherms and pore size distributions of the two CoRu catalysts are shown in Fig. 2, and the derived textural parameters are included in Table 1. It has been estimated that the mean pore size is reduced by 0.5 nm due to deposition of Co and Ru on both supports. However, the shape of the hysteresis cycle suggests larger heterogeneity in the pore diameters of the channels after Co/Ru impregnation. For the Al-grafted sample, the impregnation with Co/Ru produces a more noticeable modification of the hysteresis loop and hence of the fine structure of the pores compared with that in the case of the Al-free support. The shift of the desorption branch of the isotherm toward lower relative pressures evidences a narrowing of the free diameter of the channels, which can be reasonably attributed to the presence of cobalt (and Ru) oxide phases located inside the channels. As observed in Table 1, both catalysts display a lower BET surface area and mesopore volume than their corresponding supports, which is due in part to the dilution effect of the metal oxides present in the samples. Interestingly, while the relative decrease in mesopore volume for the Al-containing CRALPS catalyst (29%) slightly exceeds that expected from the simple dilution effect (ca. 23%), it is much higher for CRLPS (ca. 44%). This fact suggests that a considerable fraction of the inner volume of the CRLPS sample should be blocked by cobalt oxide particles, probably located at the pore mouths of the unidirectional channels of SBA-15. The stronger metal–support interaction would prevent to a large extent this phenomenon in the Al-grafted sample.
The acid properties of the CRALPS sample were assessed by FTIR-pyridine. The FTIR spectrum in the pyridine region is presented in Fig. 6 along with that of the ALPS support previously discussed. After incorporation of Co and Ru to the ALPS support and following air calcination, the intensity of the Al3+-related band at ca. 1455 cm−1 decreases slightly and a new component appears at 1451 cm−1 ascribed to pyridine coordinated to Co2+/Co3+ cations of the spinel oxide Co3O4 displaying Lewis acidity. The same features are evidenced by looking at the pyridine bands at around 1622 and 1611 cm−1 of LAS associated with Al3+ and Co2+/3+ cations, respectively (Fig. 6). Similar IR-pyridine LAS bands at 1452 and 1612 cm−1 were also observed for pyridine interacting with Co2+ cations in ion-exchanged positions of Co-zeolites.47,48 The lower frequency of the band of pyridine coordinated to cobalt cations indicates a weaker strength of the associated LAS in comparison with those related to aluminum cations. The total concentration of LAS in the CRALPS catalyst, normalized by the mass of the support, is 244 μmol gsup−1, slightly above that of the bare support. Concomitantly, the intensity of the BAS band is somewhat decreased in the CRALPS catalyst with respect to the support, resulting in a density of BAS of 24 μmol gsup−1. Since these values are normalized by the mass of the support, the lower amount of BAS in the catalyst is likely due to the partial coverage of the support surface by the loaded metal (Co + Ru) oxide phases. In conclusion, the above FTIR-pyridine data clearly reveal the presence of BAS on the surface of the CRALPS catalyst, which can be associated with tetrahedrally coordinated Al3+ species in bridged Si–OH–Al groups located probably at the interface between the grafted alumina layer and the silica SBA-15 carrier.
The Co 2p core-level XP spectrum of the calcined catalyst supported on bare silica (CRLPS) shows two asymmetric signals that can be fitted with two split components with a 2:1 area ratio (Fig. 5 and Table 2), which can be assigned to the Co 2p3/2 and 2p1/2 peaks of Co3+ (780.0 and 795.3 eV) and Co2+ (781.5 and 796.9 eV) in Co3O4.49–57 This is in agreement with XRD analysis that identified this spinel oxide as the only cobalt crystalline phase in the calcined material. Furthermore, the presence of the Co3O4 phase would also account for the weak shoulder found at BE around 530.4 eV in the O 1s spectrum, in addition to the main peak at BE of 532.9 eV assigned to the silica phase. In the case of sample CRALPS, the slight shift of both Co 2p peak maxima towards higher BE, besides the higher intensity of the Co 2p shake-up satellites, indicates that this catalyst contains a higher proportion of paramagnetic Co2+ species compared to CRLPS. Indeed, the Co 2p spectrum can be fitted with a split component at 781.9 and 797.4 eV, which can be ascribed to Co 2p3/2 and 2p1/2 peaks of cobalt aluminate,51–60 in addition to the peaks assigned to the Co3O4 phase. The relative peak areas (Table 2) indicate a notably higher proportion of Co in the aluminate phase with respect to the oxide. The fact that Co3O4 is the only phase detected by XRD indicates the amorphous nature of the cobalt aluminate phase formed in sample CRALPS, as commonly observed for Co/Al2O3 FTS catalysts.61
Moreover, despite both catalysts having the same cobalt content, the XPS spectrum of the catalyst prepared with the all-silica support shows much weaker Co 2p components than the catalyst supported on the Al-grafted material. Indeed, the calculated surface Co/Si atomic ratio of sample CRLPS is three times lower than the Co/(Si + Al) ratio of sample CRALPS (Table 2). This result indicates, in agreement with XRD data, that CRLPS contains larger cobalt oxide crystals than CRALPS. Given the dimension of these crystals, sizing in average 15 nm for CRLPS and 11 nm for CRALPS according to XRD (Table 3), the XPS technique can only probe a fraction (the outermost layers) of the cobalt oxide crystals. As the surface to volume ratio of the crystallites decreases with the increase of crystal dimensions, the fraction of cobalt that can be detected by XPS would be smaller for CRLPS. Indeed, the low amount of Co determined by XPS for sample CRLPS in comparison to CRALPS suggests that, on the external surface of catalysts particles, the increase of Co3O4 crystal size in CRLPS with respect to CRALPS is even larger than that found by XRD for the bulk.
H2-TPR experiments have been carried out to investigate the reducibility of the catalysts. The results, plotted in Fig. 8, show two reduction peaks. The low temperature peak (T1) is typically assigned to the reduction of Co3O4 to CoO. The second peak (T2) is assigned to the reduction of small CoO crystals to Co0 species. After Al grafting, T1 shifted to lower temperature, from 200 to 175 °C. On the contrary, T2 increased by 20 °C suggesting that there are smaller CoO crystals.12,61,62 The shoulder at ca. 265 °C in the profile of sample CRLPS may be related to the reduction of larger CoO particles displaying a weaker interaction with the support, probably those located outside the mesopores. Additionally, the Al-SBA-15-supported sample (CRALPS) exhibits a broad reduction feature at temperatures of 500–700 °C that can be attributed to the reduction of Co2+ strongly interacting with the Al-SBA-15 surface, probably with surface Al species (e.g. Co–O–Al) forming a cobalt aluminate-like phase, as was also suggested beforehand based on the analysis of the Co 2p XPS signal.
 | ||
| Fig. 8 H2-TPR profiles of the SBA-15-supported CoRu catalysts. | ||
The degree of cobalt reduction (DOR) was determined by H2-TPR of the catalysts pre-reduced in pure H2 at 400 °C for 10 h, and the results are displayed in Table 3. Although both catalysts showed high DOR values due to the presence of Ru as a cobalt reduction promoter, the all-silica CRLPS catalyst presents a higher DOR (96%) than the Al-containing CRALPS sample (87%). The lower DOR of CRALPS can be related to the presence of a fraction of Co as hardly reducible cobalt aluminate, as discussed previously.
The morphology and particle size distribution of the supported Co3O4 species in the calcined catalysts were characterized by bright-field TEM. It shows that the ordered mesopore architecture of the ALPS (Al-SBA-15) support is preserved and that CRALPS displays smaller Co3O4 particles in comparison with CRLPS (Fig. 9). Indeed, the histograms in Fig. 9 (right panels) show that CRALPS displays a narrower Co3O4 particle size distribution with a surface average particle size (D[3,2]) of 5.9 nm, while CRLPS shows a wider size distribution with a larger D[3,2] of 10.1 nm (Table 3). Considering the pore diameters (10–11 nm, Table 1) and the TEM-derived Co3O4 particle size distributions, it seems evident that a significant fraction of the particles in CRLPS should be located outside the mesochannels while most of them should reside within the mesopores in CRALPS.
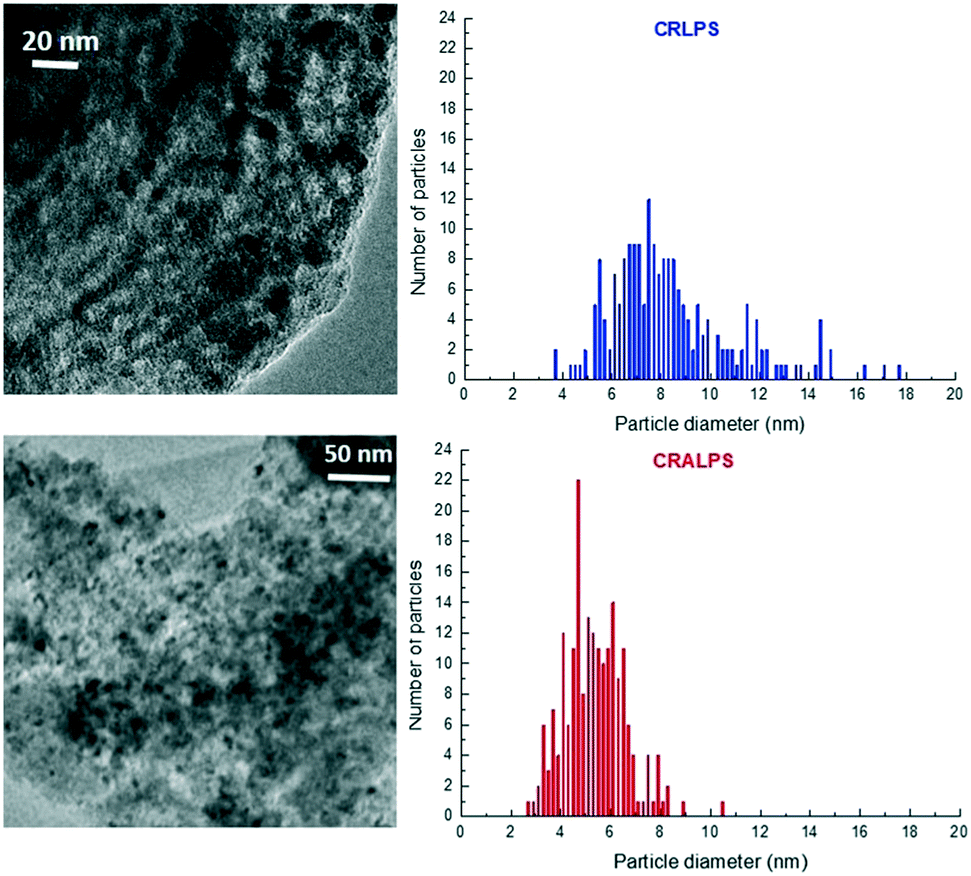 | ||
| Fig. 9 Representative bright-field TEM images (left) and corresponding Co3O4 particle size distributions (right) for calcined CRLPS (top) and CRALPS (bottom) catalysts. | ||
The cobalt dispersion in the catalysts after reduction in H2 at 400 °C was determined by H2 chemisorption. The results (Table 3) clearly reveal a higher H2 uptake for the Al-grafted CRALPS catalyst (192 μmol g−1) than for CRLPS (156 μmol g−1), which translates into a higher Co0 dispersion in the former (15.8 vs. 11.2%). The mean Co0 particle sizes estimated from the obtained metal dispersions after correction by DOR amounted to 8.6 nm for CRLPS and 6.1 nm for CRALPS (Table 3).
The CRALPS catalyst was additionally characterized by HAADF-STEM after reduction in H2 at 400 °C and surface passivation. As inferred from the representative STEM image shown in Fig. 10 (inset), Co0 nanoparticles appear to be uniformly distributed mainly within the mesoporous channels of the Al-SBA-15 carrier. The STEM-derived distribution of cobalt particle sizes (Fig. 10a) reveals a narrow size distribution in the 4–7 nm range, with a surface average Co0 particle diameter of 6.2 nm, virtually matching the mean particle size of 6.1 nm obtained by H2 chemisorption (Table 3). Moreover, STEM-EDX mapping confirms the presence of Co homogeneously distributed throughout the pore network of the grafted Al-SBA-15 carrier (Fig. 10b).
 | ||
| Fig. 10 a) Cobalt particle size distribution and representative HAADF-STEM image (inset), and b) STEM-EDAX mapping of the reduced and passivated CRALPS catalyst. | ||
3.4. Catalytic performance for FTS of CoRu/(Al-)SBA-15 catalysts
The FTS activities, expressed as CO conversion, cobalt-time-yield (CTY), and turnover frequency, (TOF) of catalysts CRLPS and CRALPS are compared in Table 4 for the experiments at a constant GHSV of 7.0 Lsyngas gcat−1 h−1 (period 1). As shown in Table 4, the Al-free CRLPS catalyst exhibits about 1.7-fold higher FTS activity in terms of both CO conversion and CTY than the CRALPS catalyst based on the Al-grafted SBA-15 support. Moreover, the intrinsic activity per active Co0 site or TOF, based on H2 chemisorption data, is also about two-fold higher for the catalyst supported on the all-silica SBA-15 material (CRLPS). At this point, we should note that the contribution of metallic Ru species (known to be active for FTS) to the overall activity of our catalysts was estimated to be, at maximum, 9.5% and 5.2% for CRLPS and CRALPS, respectively, as detailed in the ESI.† These values are in good agreement with that reported in an earlier work by Iglesia and co-workers, who estimated a contribution of Ru species of less than 10% to the total activity of a CoRu/TiO2 catalyst loaded with 11.6 wt% Co and 0.14 wt% Ru.3 Therefore, the observed differences in activity between CRLPS and CRALPS catalysts can be mainly attributed to changes in the properties of active Co0 sites, as will be discussed later.On the other hand, the product selectivity of CRLPS and CRALPS catalysts at a constant CO conversion of ca. 40% (period 2) is presented in Table 5. It can be seen there that both catalysts display a very low selectivity to CO2 (≤0.3%), as anticipated from the known low activity of cobalt-based FTS catalysts for the competing water-gas shift reaction. The distribution of hydrocarbons, however, is clearly different, with CRALPS exhibiting a higher selectivity to light (C1 and C2–C4) and liquid (C5–C20) hydrocarbons than CRLPS. Remarkably, the selectivity to long chain hydrocarbons (C21+), typically referred to as FT waxes, is much higher for CRLPS (14.0%) than for CRALPS (1.9%). This shift in the hydrocarbon distribution upon incorporation of Al in the SBA-15 silica is clearly evidenced in the corresponding ASF distribution plots shown in Fig. S2,† where the concentration of hydrocarbons for CRALPS starts to decline drastically above C20.
| Catalyst | CO2 selectivity (% C) | Hydrocarbon distribution (% C) | ||||
|---|---|---|---|---|---|---|
| C1 | C2–C4 | C5–C20 | C21+ | C5+ | ||
| CRLPS | 0.2 | 11.4 | 10.2 | 64.4 | 14.0 | 78.4 |
| CRALPS | 0.3 | 15.0 | 14.5 | 68.6 | 1.9 | 70.5 |
The activity of Co-based FTS catalysts is known to be mainly dictated by the size of the supported Co0 nanoparticles through the previously reported TOF–particle size dependence, by which a drastic decline in the TOF of surface Co0 sites occurs for nanoparticles sizing below ca. 6 nm.63,64 In the present study, the calcined Al-grafted CRALPS catalyst was shown by XRD and TEM to exhibit a greater cobalt dispersion resulting in smaller Co3O4 particles in comparison with CRLPS (Table 3). According to H2 chemisorption measurements, the CRALPS catalyst also shows a higher Co dispersion than CRLPS after reduction in H2 at 400 °C, with average Co0 particle sizes of 6.1 and 8.6 nm, respectively (Table 3). Moreover, the Co0 particle size distribution for as-reduced CRALPS derived from HAADF-STEM analysis revealed that a large fraction of the nanoparticles (∼60%) in this catalyst displays sizes below 6 nm (Fig. 9), for which a lower TOF can be expected. Although the as-reduced CRLPS was not characterized by HAADF-STEM, both the mean size and size distribution of Co3O4 nanoparticles in the calcined sample measured by TEM (Fig. 8) and the average Co0 particle size determined by H2 chemisorption (Table 3) indicate a much lower relative amount of sub-6 nm Co0 nanoparticles in this catalyst with respect to CRALPS. Therefore, the lower TOF measured for CRALPS can be accounted for by its larger proportion of intrinsically less active small Co0 nanoparticles, resulting in a lower CO conversion and CTY for this catalyst with respect to CRLPS (Table 4). In this regard, by taking advantage of the ability of the Al-grafted ALPS support to efficiently disperse the Co nanoparticles, a suitable strategy to improve the activity of CRALPS could be to increase the Co loading until an average Co0 particle size of around 8 nm is achieved. In doing so, a significant enhancement of the FTS activity would be expected as the result of an increase of both the average TOF and the total amount of exposed Co0 sites with near-optimum TOF.
The dependence of the FTS product selectivity on catalyst properties is less straightforward than the activity due to the influence, besides other factors, of diffusion-related phenomena occurring within the catalyst pores which are filled with liquid hydrocarbons under industrially relevant reaction conditions.65,66 Iglesia and co-workers modelled the intrapellet diffusion resistance during FTS through the χ parameter, which relates to several measurable catalyst properties according to the following equation (eqn (2)):65
| χ (m) = (R02·ε·θCo)/rp | (2) |
According to Iglesia's diffusion model,65 the selectivity to long chain (C5+) hydrocarbons first increases with increasing χ up to values of around 160 × 1016 m due to an enhanced limitation for the diffusion of α-olefins favouring their re-adsorption on Co0 sites and participation in C–C chain growth processes. Above this χ value, i.e. under stronger diffusion limitations, the intra-pore diffusion of CO starts to be restricted causing a local increase in the H2/CO ratio around the active Co0 sites located within the pores. Consequently, the hydrogenation of CxHy intermediates is favoured causing a decline in the C5+ selectivity in favour of lighter hydrocarbons. In our case, the value of the χ parameter amounted to 162 × 1016 m for CRLPS and to 246 × 1016 m for CRALPS (Table 6). The higher χ value of CRALPS is mostly due to its lower mean pore radius (rp) and higher density of Co sites per unit area (θCo) determined by its higher Co dispersion and lower BET surface area in comparison with CRLPS. Moreover, it has to be considered that the diffusion restrictions in the 1D mesochannels of SBA-15 would be enhanced with respect to conventional 3D porous carriers for which the correlation between the χ parameter and the C5+ selectivity established by Iglesia was derived. Therefore, the calculated χ values for our CRLPS and CRALPS catalysts suggest that, in this case, the C5+ selectivity is determined by the intra-pore diffusion of CO, as also concluded in earlier studies for Co supported on SBA-15 materials.18 This is supported by the almost identical olefin/paraffin ratios for selected hydrocarbons (C2–C4) obtained for the two catalysts (Table 6), since a decrease in this ratio would be expected at increasing values of χ in the regime where the C5+ selectivity is governed by the re-adsorption of α-olefins.65 Therefore, according to the calculated χ values, the Al-grafted CRALPS catalyst shows higher resistance to the diffusion of CO than CRLPS, which may explain at least in part the lower selectivity to C5+, particularly to waxes (C21+), and higher selectivity to lighter hydrocarbons (C1–C4) in the former (Table 5).
| Catalyst | Olefin/paraffin ratio | Branched/linear ratio | |||||
|---|---|---|---|---|---|---|---|
| C2 | C3 | C4 | C5 | C6 | C7 | C8 | |
| CRLPS | 0.09 | 1.20 | 0.89 | 0.03 | 0.03 | 0.05 | 0.07 |
| CRALPS | 0.08 | 0.98 | 0.92 | 0.04 | 0.05 | 0.09 | 0.11 |
In addition to diffusion-related issues, the FTS selectivity is also influenced by the size of the supported Co0 nanoparticles. In this respect, besides displaying lower TOFs, small Co0 nanoparticles sizing below ca. 6 nm were also reported to be intrinsically more selective to methane at the expense of C5+ hydrocarbons.63,64 This occurs as C–C chain growth is assumed to require relatively large ensembles of Co0 atoms that are more abundant at the terraces of larger nanoparticles.67,68 Thus, the presence of a significant proportion of sub-6 nm nanoparticles in CRALPS (Fig. 10) could also account for its lower C5+ selectivity (and higher C1 selectivity) with respect to CRLPS.
Finally, the presence of Brønsted acid sites in the CRALPS catalyst (Fig. 6) may promote the cracking of long chain primary FT products (e.g., heavier olefins) producing a shift in the hydrocarbon distribution towards lighter fractions, as previously reported for cobalt catalysts supported on acidic Al-doped SBA-15 silicas.23,24 In order to determine whether the acid catalysis contributed to a significant extent to the product distribution of the Al-grafted CRALPS catalyst, we analysed the branched/linear ratio for individual hydrocarbons in the C5–C8 range (including both olefins and paraffins) for the two studied catalysts (Table 6). The results showed a higher branched/linear ratio, increasing with carbon number, for CRALPS that indicates the occurrence of acid catalysis in this catalyst. Therefore, it seems reasonable to assume that the presence of Brønsted acid sites associated with Al species in Al–OH–Si groups would contribute, via isomerization and cracking of heavier hydrocarbons, to the changes in product selectivity observed for CRALPS.
4. Conclusions
Aluminum was introduced as a uniform layer (ca. 0.6 nm thickness) on the surface of a short-channel mesostructured SBA-15 silica with enlarged pores (11.2 nm pore diameter) by three sequential grafting steps using aluminum-tri-sec-butoxide as a precursor. According to FTIR-pyridine measurements, the grafted Al-SBA-15 sample (Al/Si = 0.26) exhibited, besides Al3+-related Lewis acid sites, Brønsted acid sites associated with tetrahedral Al3+ species in Al–OH–Si groups located at the interface between the Al layer and the silica surface. Both the parent SBA-15 silica and the grafted Al-SBA-15 material were employed as supports for preparing, via incipient wetness impregnation, Ru-promoted Co catalysts with nominal loadings of 20 wt% Co and 1 wt% Ru. As a consequence of the stronger Co–support interaction induced by the presence of surface Al species in the grafted sample, as ascertained by H2-TPR, the CRALPS catalyst displayed a higher cobalt dispersion than that supported on the SBA-15 silica (CRLPS) both after air-calcination at 300 °C (XRD, TEM) and after reduction in H2 at 400 °C (H2 chemisorption, STEM). The mean Co0 particle size in the as-reduced samples, derived from H2 chemisorption measurements, was 8.6 nm for CRLPS and 6.1 nm for CRALPS. Moreover, the Co0 particle size histogram for CRALPS obtained by HAADF-STEM revealed a high proportion (∼60%) of small nanoparticles sizing below 6 nm in this catalyst.When evaluated for the Fischer–Tropsch synthesis (FTS) reaction under industrially relevant conditions (220 °C, 2.0 MPa, H2/CO = 2), the Al-containing CRALPS catalyst displayed a CO conversion and a cobalt-time-yield (CTY) notably lower than CRLPS. The inferior FTS activity of CRALPS was related to the lower intrinsic TOF of the exposed Co0 sites in the small (<6 nm) nanoparticles abundantly present in this catalyst, resulting in a TOF of 3.3 × 10−2 s−1. In comparison, the catalyst supported on the all-silica SBA-15 carrier (CRLPS) comprising larger Co0 particles exhibited a higher TOF of 6.9 × 10−2 s−1. Moreover, the presence of the grafted Al species on the SBA-15 silica surface induced significant changes in the product selectivity of the corresponding CoRu catalyst. Hence, at a constant CO conversion of ca. 40%, the selectivity to waxes (C21+) was remarkably reduced from 14.0% for CRLPS to only 1.9% for CRALPS while the selectivity to liquid hydrocarbons (C5–C20), precursors of synthetic liquid fuels, was increased from ca. 64.4% to 68.6% accompanied by an increase in the formation of branched hydrocarbons. The changes in product selectivity observed for CRALPS were concluded to arise from the contribution of three main factors: the intrinsic lower C5+ selectivity of the abundant sub-6 nm Co0 nanoparticles, the enhanced restriction for the intra-pore diffusion of CO, and the isomerization and cracking of heavier hydrocarbons catalyzed by the Brønsted acid sites existing in the Al-containing catalyst.
Author contributions
Sophia Mohammadnasabomran: resources, investigation, writing – original draft. Carlos Márquez-Álvarez: resources, investigation, writing – reviewing and editing. Joaquín Pérez-Pariente: conceptualization, supervision, writing – reviewing and editing. Agustín Martínez: conceptualization, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M. acknowledges the MINECO of Spain for financial support through the Severo Ochoa (SEV2016-0683) and RTI2018-102161 projects. C. M.-A. and J. P.-P. also acknowledge funding of the Spanish Ministry of Science Innovation and Universities (project PID2019-107968RB-I00, AEI, Spain). S. M. acknowledges the organization of student affairs (Iran) for a sabbatical leave grant. The authors are thankful to the Microscopy Service of the Universitat Politècnica de València for its assistance in microscopy characterization. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- E. Iglesia, Stud. Surf. Sci. Catal., 1997, 107, 153 CrossRef CAS.
- S. Strorsater, B. Totdal, J. C. Walmsley, B. S. Tanem and A. Holmen, J. Catal., 2005, 236, 139 CrossRef.
- E. Iglesia, S. L. Soled, R. A. Fiato and G. H. Via, J. Catal., 1993, 143, 345 CrossRef CAS.
- D. Wei, J. G. Goodwin, R. Oukaci and A. H. Singleton, Appl. Catal., A, 2001, 210, 137 CrossRef CAS.
- A. A. Adesina, Appl. Catal., A, 1996, 138, 345 CrossRef CAS.
- S. L. Soled, E. Iglesia, R. A. Fiato, J. E. Baumgartner, H. Vroman and S. Miseo, Top. Catal., 2003, 26, 101 CrossRef CAS.
- J. Panpranot, J. G. Goodwin Jr. and A. Sayari, Catal. Today, 2002, 77, 269 CrossRef CAS.
- A. Martínez, C. López, F. Márquez and I. Díaz, J. Catal., 2003, 220, 486 CrossRef.
- P. Concepción, C. López, A. Martínez and V. F. Puntes, J. Catal., 2004, 228, 321 CrossRef.
- J. R. A. Sietsma, J. D. Meeldijk, J. P. den Breejen, M. Versluijs-Helder, A. J. van Dillen, P. E. de Jongh and K. P. de Jong, Angew. Chem., Int. Ed., 2007, 46, 4547 CrossRef CAS PubMed.
- D. Wei, J. G. Goodwin, R. Oukaci and A. H. Singleton, Appl. Catal., A, 2001, 210, 137 CrossRef CAS.
- G. Jacobs, T. K. Das, Y. Zhang, J. Li, G. Racoillet and B. H. Davis, Appl. Catal., A, 2002, 233, 263 CrossRef CAS.
- W. Ma, G. Jacobs, R. A. Keogh, D. B. Bukur and B. H. Davis, Appl. Catal., A, 2012, 437–438, 1 CrossRef CAS.
- T. K. Das, G. Jacobs, P. M. Patterson, W. A. Conner, J. Li and B. H. Davis, Fuel, 2003, 82, 805 CrossRef CAS.
- K. Shimur, T. Miyazaw, T. Hanaok and S. Hirat, Appl. Catal., A, 2015, 494, 1 CrossRef.
- S. Mohammadnasabomran, A. Tavasoli and Y. Zamani, React. Kinet., Mech. Catal., 2019, 128, 217 CrossRef CAS.
- S. Mohammadnasabomran, A. Tavasoli, Y. Zaman, C. Márquez-Álvarez, J. Pérez-Pariente and A. Martínez, Fuel Process. Technol., 2020, 205, 106433 CrossRef CAS.
- M. Lualdi, G. Di Carlo, S. Lögdberg, S. Järås, M. Boutonnet, V. La Parola, L. F. Liotta, G. M. Ingo and A. M. Venezia, Appl. Catal., A, 2012, 443–444, 76 CrossRef CAS.
- M. V. Landau, E. Dafa, M. L. Kaliya, T. Sen and M. Herskowitz, Microporous Mesoporous Mater., 2001, 49, 65 CrossRef CAS.
- P. Madhusudhan Rao, A. Wolfson, S. Kababya, S. Vega and M. V. Landau, J. Catal., 2005, 232, 210 CrossRef.
- M. Baca, E. la Rochefoucauld, E. Ambroise, J.-M. Krafft, R. Hajjar, P. P. Man, X. Carrier and J. Blanchard, Microporous Mesoporous Mater., 2008, 110, 232 CrossRef CAS.
- C. M. A. Parlett, L. J. Durndell, A. Machado, G. Cibin, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, Catal. Today, 2014, 229, 46 CrossRef CAS.
- Q. Q. Hao, Y. H. Zhao, H. H. Yang, Z. T. Liu and Z. W. Liu, Energy Fuels, 2012, 26, 6567 CrossRef CAS.
- Y. Cai, X. Xu, H. Wang, L. Wang, L. Chen, R. Li, J. Ding, H. Wan and G. Guan, Ind. Eng. Chem. Res., 2018, 57, 3844 CrossRef CAS.
- G. Prieto, A. Martínez, R. Murciano and M. A. Arribas, Appl. Catal., A, 2009, 367, 146 CrossRef CAS.
- V. Gascón, I. Díaz, C. Márquez-Álvarez and R. M. Blanco, Molecules, 2014, 19, 7057 CrossRef PubMed.
- P. Linton and V. Alfredsson, Chem. Mater., 2008, 20, 2878 CrossRef CAS.
- P. Linton, A. R. Rennie, M. Zackrisson and V. Alfredsson, Langmuir, 2009, 25, 4685 CrossRef CAS PubMed.
- M. L. Ojeda, J. M. Esparza, A. Campero, S. Cordero, I. Kornhauser and F. Rojas, Phys. Chem. Chem. Phys., 2003, 5, 1859 RSC.
- P. I. Ravikovitch, S. C. Ó. Domhnaill, A. V. Neimark, F. Schüth and K. K. Unger, Langmuir, 1995, 11, 4765 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, ed. J. Chastain, Perkin-Elmer Corporation, Physical Electronics Division, Minnesota, 1995 Search PubMed.
- J. Agúndez, C. Ares, C. Márquez-Álvarez and J. Pérez-Pariente, Mol. Catal., 2020, 488, 110922 CrossRef.
- B. Herreros, H. He, T. L. Barr and J. Klinowski, J. Phys. Chem., 1994, 98, 1302 CrossRef CAS.
- G. Prieto, M. I. S. De Mello, P. Concepción, R. Murciano, S. B. C. Pergher and A. Martínez, ACS Catal., 2015, 5, 3323 CrossRef CAS.
- F. Bertella, C. W. Lopes, A. C. Foucher, G. Agostini, P. Concepción, E. A. Stach and A. Martínez, ACS Catal., 2020, 10, 6042 CrossRef CAS.
- L. Cao, T. Man and M. Kruk, Chem. Mater., 2009, 21, 1144 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CAS.
- V. Maurice, I. Bennour, S. Zanna, L. H. Klein and P. Marcus, J. Phys. Chem. C, 2010, 114, 7132 CrossRef CAS.
- A. K. Bhattacharya, D. R. Pyke, R. Reynolds, G. S. Walker and C. R. Werrett, J. Mater. Sci. Lett., 1997, 16, 1 CrossRef.
- H. Seyama, M. Soma and A. Tanaka, Chem. Geol., 1996, 129, 209 CrossRef CAS.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST Standard Reference Database 20, web version 4.1, 2012 Search PubMed.
- E. A. Mel'gunova, A. N. Shmakov, Y. V. Larichev and M. S. Mel'gunov, Kinet. Catal., 2009, 50, 456 CrossRef.
- S. Chen, J. Li, Y. Zhang, Y. Zhao and J. Hong, Catal. Sci. Technol., 2013, 3, 1063 RSC.
- J. Hong, E. Marceau, A. Y. Khodakov, L. Gaberová, A. Griboval-Constant, J.-S. Girardon, C. L. Fontaine and V. Briois, ACS Catal., 2015, 5, 1273 CrossRef CAS.
- J. M. Cho, C. I. Ahn, C. Pang and J. W. Bae, Catal. Sci. Technol., 2015, 5, 3525 RSC.
- L. B. Pierella, C. Saux, S. C. Caglieri, H. R. Bertorello and P. G. Bercoff, Appl. Catal., A, 2008, 347, 55 CrossRef CAS.
- A. García-Trenco and A. Martínez, Appl. Catal., A, 2012, 411–412, 170 CrossRef.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS.
- M. M. Natile and A. Glisenti, Chem. Mater., 2002, 14, 3090 CrossRef CAS.
- A. T. Najafabadi, A. A. Khodadadi, M. J. Parnian and Y. Mortazavi, Appl. Catal., A, 2016, 511, 31 CrossRef.
- B. A. Fouetio Kengne, A. M. Alayat, G. Luo, A. G. McDonald, J. Brown, H. Smotherman and D. N. McIlroy, Appl. Surf. Sci., 2015, 359, 508 CrossRef.
- Z. Li, J. Wu, J. Yu, D. Han, L. Wu and J. Li, J. Mol. Catal. A: Chem., 2016, 424, 384 CrossRef CAS.
- O. A. Kungurova, A. A. Khassin, S. V. Cherepanova, A. A. Saraev, V. V. Kaichev, N. V. Shtertser, G. K. Chermashentseva, E. Yu. Gerasimov, E. A. Paukshtis, O. V. Vodyankina, T. P. Minyukova and G. Abou-Jaoudé, Appl. Catal., A, 2017, 539, 48 CrossRef CAS.
- Z. Li, L. Wu, D. Han and J. Wu, Fuel, 2018, 220, 257 CrossRef CAS.
- E. Østbye Pedersen, I.-H. Svenum and E. A. Blekkan, J. Catal., 2018, 361, 23 CrossRef.
- L. Macheli, A. Roy, E. Carleschi, B. P. Doyle and E. van Steen, Catal. Today, 2020, 343, 176 CrossRef CAS.
- A. M. Hilmen, D. Schanke, K. F. Hanssen and A. Holmen, Appl. Catal., A, 1999, 186, 169 CrossRef CAS.
- G. Jacobs, J. A. Chaney, P. M. Patterson, T. K. Das, J. C. Maillot and B. H. Davis, J. Synchrotron Radiat., 2004, 11, 414 CrossRef CAS PubMed.
- D. J. Moodley, A. M. Saib, J. van de Loosdrecht, C. A. Welker-Nieuwoudt, B. H. Sigwebela and J. W. Niemantsverdriet, Catal. Today, 2011, 171, 192 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692 CrossRef CAS PubMed.
- M. D. Shannon, C. M. Lok and J. L. Casci, J. Catal., 2007, 249, 41 CrossRef CAS.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holwijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956 CrossRef CAS PubMed.
- G. Prieto, A. Martínez, P. Concepción and R. Moreno-Tost, J. Catal., 2009, 266, 129 CrossRef CAS.
- E. Iglesia, Appl. Catal., A, 1997, 161, 59 CrossRef CAS.
- E. Iglesia, S. C. Reyes and R. J. Madon, J. Catal., 1991, 129, 238 CrossRef CAS.
- C. J. Weststrate, P. van Helden and J. W. Niemantsverdriet, Catal. Today, 2016, 275, 100 CrossRef CAS.
- A. Banerjee, A. P. van Bavel, H. P. C. E. Kuipers and M. Saeys, ACS Catal., 2017, 7, 5289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy02418j |
| This journal is © The Royal Society of Chemistry 2021 |