
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterolytic cleavage of dihydrogen (HCD) in metal nanoparticle catalysis†
Israel
Cano
 *a,
Luis M.
Martínez-Prieto
*b and
Piet W. N. M.
van Leeuwen
c
*a,
Luis M.
Martínez-Prieto
*b and
Piet W. N. M.
van Leeuwen
c
aApplied Physics Department, University of Cantabria, 39005 Santander, Spain. E-mail: israel.canorico@unican.es
bInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas (UPV-CSIC), Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: luismiguel.martinez@csic.es
cLaboratoire de Physique et Chimie des Nano-Objects, INSA-Toulouse, 135 Avenue de Rangueil, 31077 Toulouse, France
First published on 21st January 2021
Abstract
The heterolytic cleavage of H2 into H+ and H−, and the subsequent transfer of these hydrogen species to polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X groups, is a useful strategy to obtain high selectivities in catalytic hydrogenation reactions, which are one of the most important chemical processes. In the homogeneous field, this transformation is catalyzed by a wide variety of metal–ligand complexes, leading to the selective reduction of ketones, aldehydes, and imines, to name a few. In many cases, a ligand–metal cooperative mechanism promotes the activation of the hydrogen molecule; that is, a Lewis basic site of the ligand binds the proton, and simultaneously, a coordinated metal atom acts as a Lewis acid and takes the hydride. Similar to homogeneous catalysts based on organometallic complexes, in the field of metal nanoparticle catalysis the use of supports, ligands and additives can promote heterolytic H2 splitting by a cooperative mechanism with the metal, and thus produce the selective reduction of thermodynamically more stable unsaturated groups. This successful approach has allowed very high selectivities to be obtained, with both supported and non-supported metal nanoparticles (MNPs). This Perspective aims to carry out a critical review of recent examples on heterolytic cleavage of dihydrogen mediated by MNPs, making a selection of the most representative works in search for general features and new steps to be followed. As will be shown, promising advances have been made through the use of supported MNPs functionalized by organic ligands, which combine the advantages of homogeneous and heterogeneous catalysts (activity, selectivity, stability and recyclability), and thus represent a new research area with tremendous industrial interest.
X groups, is a useful strategy to obtain high selectivities in catalytic hydrogenation reactions, which are one of the most important chemical processes. In the homogeneous field, this transformation is catalyzed by a wide variety of metal–ligand complexes, leading to the selective reduction of ketones, aldehydes, and imines, to name a few. In many cases, a ligand–metal cooperative mechanism promotes the activation of the hydrogen molecule; that is, a Lewis basic site of the ligand binds the proton, and simultaneously, a coordinated metal atom acts as a Lewis acid and takes the hydride. Similar to homogeneous catalysts based on organometallic complexes, in the field of metal nanoparticle catalysis the use of supports, ligands and additives can promote heterolytic H2 splitting by a cooperative mechanism with the metal, and thus produce the selective reduction of thermodynamically more stable unsaturated groups. This successful approach has allowed very high selectivities to be obtained, with both supported and non-supported metal nanoparticles (MNPs). This Perspective aims to carry out a critical review of recent examples on heterolytic cleavage of dihydrogen mediated by MNPs, making a selection of the most representative works in search for general features and new steps to be followed. As will be shown, promising advances have been made through the use of supported MNPs functionalized by organic ligands, which combine the advantages of homogeneous and heterogeneous catalysts (activity, selectivity, stability and recyclability), and thus represent a new research area with tremendous industrial interest.
 Israel Cano | Israel Cano developed his PhD in the field of homogenous catalysis at the University of Huelva. After a short postdoctoral stay at the University of Basque Country, he worked as a postdoctoral researcher with Prof. van Leeuwen at ICIQ and INSA (2012–2016). Then, he was awarded with a Marie Sklodowska-Curie fellowship to work with Prof. Jairton Dupont at the University of Nottingham (2016–2019). Since 2019, he has been a senior researcher in the group of Dr. Rafael Valiente at the University of Cantabria. His interests lie in the development of new materials with catalytic and optical applications: coordination compounds, ionic liquids, metal nanoparticles, and rare-earth metal based nanoparticles. |
 Luis M. Martínez-Prieto | Luis M. Martínez-Prieto received his PhD degree in Organometallic Chemistry in 2012, working at the IIQ (Seville, Spain) under the supervision of Prof. Cámpora. He then moved with Prof. Chaudret to the LCC (Toulouse, France) for a postdoctoral stay. His research focused on organometallic nanoparticles (MNPs). In 2015, he joined the lab of Prof. van Leeuwen at the LPCNO, exploring the use of MNPs as catalysts. In 2017, he was awarded with a “Juan de la Cierva” fellowship that allowed him to start an independent career working on confined/supported metal catalysts and magnetic catalysis at the ITQ (Valencia, Spain) in the team of Prof. Corma. |
 Piet W. N. M. van Leeuwen | Piet W. N. M. van Leeuwen after completing his PhD in the area of coordination chemistry in Leiden, Piet van Leeuwen started with Shell Research Amsterdam in 1968 and worked on organometallic chemistry and homogeneous catalysis. Since 1978 he has been head of the section “Fundamental aspects of homogeneous catalysis”. In 1990 he founded the homogeneous catalysis group at the University of Amsterdam and moved there full-time in 1994. From 2000 till 2005 he was a part-time professor of industrial homogeneous catalysis in Eindhoven and director of the National-Research-School-Combination-Catalysis. In 2004 he started as a group leader in ICIQ in Tarragona till 2015. He then moved to INSA-Toulouse, where he works in LPCNO. Since 2009 his work has focused on ligand effects in metal nanoparticle catalysis. |
1. Introduction
1.1 Homogeneous systems
Catalytic hydrogenation of unsaturated organic substrates is one of the most abundant chemical transformations. It uses both homogeneous and heterogeneous catalysts, and for the latter it is one of the oldest reactions marking the start of the discipline with the well-known studies of Sabatier. Large-scale applications involving methanation of carbon oxides, ammonia synthesis, and hydrogenation of fats are over a century old. Homogeneous metal complex catalytic systems were discovered only halfway through the 20th century; several simple metal salts showed hydrogenation activity, but copper salts stood out in the early days. The late start of homogeneous metal catalysis and the relative ease of molecular studies in solution led to a strong focus on mechanistic aspects right from the start. Activation of dihydrogen by metal complexes has been a main theme in chemistry ever since the beginning of homogeneous catalysis.1,2 In the copper catalysis mentioned above, heterolytic cleavage of dihydrogen (HCD) dominated the mechanistic scene, i.e. a copper salt and a base would react with H2 producing a hydride at copper together with the conjugate acid of the base (Scheme 1, reaction (1)). We won't go into the details of the valence state of Cu, but as one can see during this reaction the valence of the metal does not change. Further reaction can reduce the metal ion, as investigated by Halpern. Three reactions for hydrogen activation were initially distinguished by Halpern, the second being homolytic cleavage, particularly studied for bimetallic complexes, e.g. Co carbonyls and Co cyanides, in which the dihydrogen molecule splits into two hydrogen “radicals” (Scheme 1, reaction (2)). This “radical” view was certainly stimulated by the organic radical reactions shown by the same complexes. The third reaction by which dihydrogen engages in homogeneous catalysis is oxidative addition to low valent metal complexes, which became popular with the new rhodium hydrogenation catalysts, e.g. the Wilkinson catalyst,3 soon expanded by enantioselective versions, notably by Kagan,4 again with important mechanistic insight contributed by Halpern.5 In an oxidative addition reaction, both hydrogen atoms formally accept an electron from the transition metal and become hydride anions and the metal increases its valence state by two (Scheme 1, reaction (3)). In particular for 16-electron square-planar group 9 and 10 complexes, this has been a popular model in the classroom, as it explains well the changes in the complexes that take place during the reaction. Reaction (2), mechanistically the homolytic pathway, can also be regarded as an oxidative addition to a bimetallic complex, the valence of each metal being raised by one, and this would be in line with the organometallic formalism of hydrides.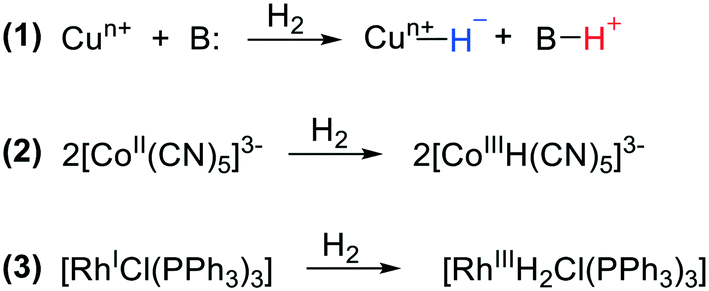 | ||
| Scheme 1 (1) Heterolytic H2 cleavage by a copper salt and a base. (2) Homolytic H2 cleavage by a Co cyanide complex. (3) Homolytic H2 cleavage with an oxidative addition reaction by the Wilkinson catalyst. | ||
The oxidative addition model has dominated the mechanistic work on hydrogenation for decades, perhaps until the late 1980s. However, as was already pointed out by Pauling,6 the charge distribution in a complex does not change as much as the formal description would suggest upon oxidative addition, and the bond between hydrogen and the “electronegative” noble metal should be regarded as covalent and the hydride formed does not have a “hydridic” character in middle and late transition metal hydrides.7 This can be illustrated by many examples of group 9 and 10 metals of which the hydrides are stable in the presence of strong acids, not quite a property of main group or early transition metal (ETM) hydrides. For example, the stable species (diphosphine)PdH+ (abundant in palladium carbonylation chemistry), various metal hydrides used in heterolytic hydrogenation of alkenes, and (triphosphine)PdH+ (for electrochemical reduction of CO2) do not react with acid.8–10 “Oxidative addition” of hydrogen to electropositive main group metals such as elemental Ca and Mg leads to hydride species (because of their reversibility considered as hydrogen storage materials),11,12 but these hydrides are usually not active in alkene hydrogenation and less so as catalysts. Hydrides of electropositive metals such as LiAlH4 can be used for the stoichiometric hydrogenation of CO and CN bonds, but under more forcing conditions they may react with alkenes, as is well known for aluminium, used on a large industrial scale for oligomerization of ethene, although chain transfer is stoichiometric.
Hydrides of immobilized metal complexes were reviewed by Copéret, who noted that the hydrides of group 4–6 early-transition-metals are “hydridic” in character. Numerous reports exist about their application in alkene conversions.13 Their preparation usually involves hydrogenolysis of organometallic precursors and rarely oxidative addition or heterolytic cleavage of H2; in view of their “very hydridic character they react fast with protic solvents, etc.”.
For the synthetic chemist and the electrochemist, the “thermodynamic hydricity” of hydrides is a useful yardstick that can tell about the reactivity of the hydride in important reactions such as reduction of aldehydes and CO2 or water splitting; the hydricity tells us whether a hydride can be transferred from the metal to CO2, or the reverse.14,15 Thermodynamic hydricity, or hydride donating ability, is the free energy required to heterolytically cleave a metal hydride bond (it is a measurable and/or calculable quantity, expressed in kJ mol−1; we will use “hydridic” (character) in the usual loose, qualitative way). For instance, Kubiak et al.15 described very nicely the linear relationship between hydricity and reversible reduction potential of M(diphosphine)2n+ complexes and their corresponding hydrides, and indicated which ones are thermodynamically capable of transferring a hydride to CO2 (overall thermodynamics requires a base to complete the reaction to formate rather than formic acid). In simple catalyst complexes of this type, hydricity parallels catalyst reactivity, as reported by Wiedner, Linehan et al.16 In many practical hydrogenation catalysts of hetero atom–carbon bonds, however, this is not the case, as the catalytic process involves multiple interactions with Lewis acid sites, bases, and protic sites.
HCD on a transition metal with the formal formation of a proton and a hydride anion does not normally produce a hydridic hydride as one can imagine from the reaction already studied by Halpern in which H2 reacts with Ag+ to give “AgH”, Ag being only slightly less electronegative than hydrogen (Pauling values 1.9 (Ag) and 2.1 (H), respectively; Pd in the example above has 2.2). The unstable “AgH” may decompose into Ag and H+ and this way the silver salt is reduced.
After two decades of a dominating position of oxidative addition in mechanistic studies, heterolytic cleavage came to the forefront again in the 1990s with the revival of the “other” Wilkinson catalyst,17 the one based on Ru instead of Rh, via the work of Noyori and Shvo on hydrogenation and transfer hydrogenation but also in relation to the work on hydricity cited above.18–20 The development of the chemistry of dihydrogen complexes contributed to new insights as, for instance, dihydrogen complexes of Ru were found to react with amines giving a ruthenium hydride and an ammonium salt, as was shown in 1990 by Chinn and Heinekey.21 An early example in which a basic ligand functionality fulfils the role of the base was reported by Fryzuk, who described the heterolytic cleavage of H2 across an Ir–amide bond of a P–N–P ligand.22 Later, however, they reported that H2 activation occurred in this particular instance by oxidative addition–reductive transfer rather than as a direct heterolytic cleavage of H2.23 Ever since, a wide variety of ligand involvements in metal-catalysed reactions have been reported and reviewed (pincer catalysis,24–26 outer-sphere hydrogenation catalysis,27 functional ligands,28 models for hydrogenase,29 cooperating ligands,30 ligand-assisted proton transfer,31–34 outer-sphere hydrogen transfer,35etc.). In particular for the heterolytic hydrogenation of CO2, ligand-assisted pathways are of crucial importance.36 HCD in such systems was reviewed by Ito and Ikariya.37
1.2 Heterogeneous systems
In heterogeneous catalysis, the earliest hydrogenation reaction known is that of the platinum catalysed combination of hydrogen and oxygen in Döbereiner's lamp with the use of the catalyst discovered by the Davy nephews,38 commercialized in 1823.39 The French chemist Paul Sabatier is considered the father of the hydrogenation process (Nobel prize 1912, shared with Victor Grignard). In 1897, he discovered that small amounts of nickel catalysed the addition of hydrogen to molecules such as carbon dioxide in what is now known as the Sabatier process. A few years later, Wilhelm Normann disclosed in patents the hydrogenation of liquid oils, the beginning of fat hardening which became a huge industry. Other reactions comprise the hydrogenation of nitrogen (Haber–Bosch process, 1905) and the hydrogenation of coal-derived carbon monoxide (Fischer–Tropsch process, 1922) which converts coal to liquid fuels. Thus, the early heterogeneous hydrogenations have been game changers in the world's energy, agriculture, and food manufacture. The understanding of heterogeneous hydrogenation on a molecular level is usually stated to begin with the publication of Horiuti and Polanyi,40 while taking into account various findings by others such as physi- and chemisorption of substrates and dissociative chemisorption of dihydrogen41 described by Langmuir (Nobel prize 1932). Also in 1934, the Farkas brothers in collaboration with Rideal reported that the transfer of hydrogen to ethene, hitherto described as a transfer of the H2 molecule to ethene on Ni and Pt surfaces, occurs stepwise, H by H (Fig. 1a).42 Based on calculations and kinetics, the preferred mechanism for H/D exchange and hydrogenation of ethene by Horiuti and Polanyi involves the formation of ethylmetal intermediates, taking place in their view at as many as 3 or 4 surface metal atoms without particular placement mentioned of the four metal atoms (Fig. 1b). A discarded mechanism was actually what we would call today a σ-bond metathesis at two adjacent metal atoms (thus a five-membered ring ⋯M–H⋯C–H⋯M′⋯, Fig. 1c), instead of one metal atom as we know from organometallic complexes (four-membered ring, Fig. 1d).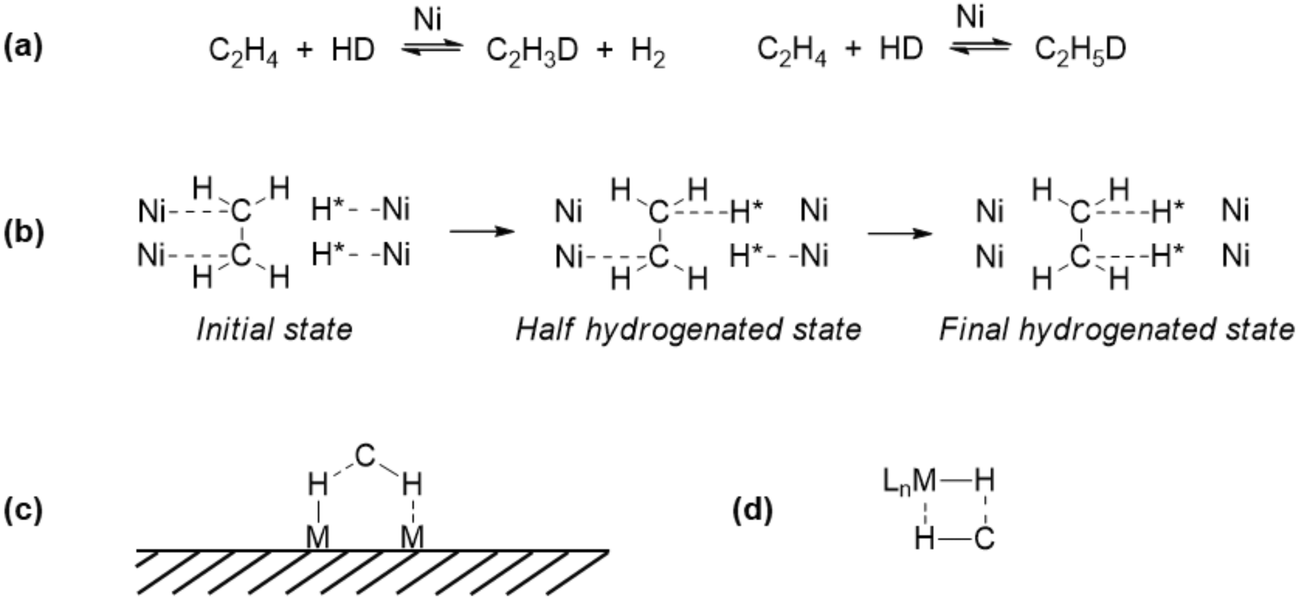 | ||
| Fig. 1 (a) Exchange and addition reactions of ethylene and HD mediated by Ni reported by the Farkas brothers and Rideal, which led them to propose that transfer of H2 to ethylene on Ni and Pt surfaces occurs stepwise. (b) Mechanism for hydrogenation of ethene by Horiuti and Polanyi. (c) Five-membered ring σ-bond metathesis. (d) Four-membered ring σ-bond metathesis. | ||
Polanyi's “insertion” mechanism has stood the test of time with flying colours, as has the dissociative chemisorption of dihydrogen into hydrogen atoms on the metal surface. One of the key contributions of transition-metal surfaces to catalysis is their ability to facilitate the dissociation of the stable H2 molecules into H atoms adsorbed on the surface together with multiple C–H activation/isomerization mechanisms.43,44 As an illustration of the latter, we mention (1) the two adsorption modes of ethene on Pt, η2-Pt and η2-μ2-Pt2, named (CC) and di-σ(CC) in heterogeneous catalysis, (2) ethyl–Pt and vinyl–Pt in several coordination modes, (3) μ2-ethylidene–Pt2 and μ3-ethylidyne–Pt3, etc. (Fig. 2).45 A seminal paper discussing similarities between the reaction of C–H and H–H on surfaces and complexes was published by Saillard and Hoffmann.46 A major difference between solid metal hydrides and metal complex hydrides is the issue of hydridic character, which is completely missing for hydrides on metals. While one might hope that for metal complexes a single parameter that gives an indication of this property can be found, the hydrides on (and below) metal surfaces cannot be treated like this, because the charge, or better its reactivity, is the property of a complicated ensemble of atoms (e.g. metal face, shape, size, coverage, binding mode, mobility of metal atoms and hydrides, other ligands, etc.).47 The huge number of reports about homolytic cleavage on many metals did not lead to simple comparisons of reactivities with those of metal complex hydrides.
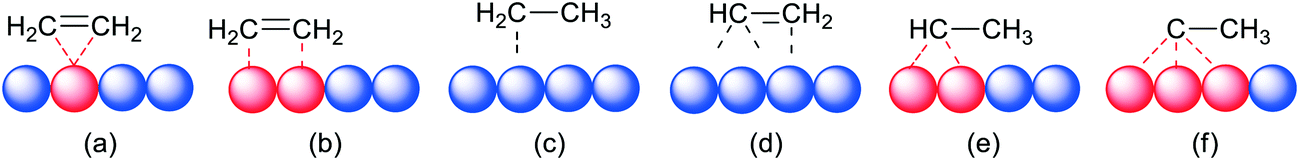 | ||
| Fig. 2 Adsorption modes of ethene on Pt: (a) η2-Pt (CC); (b) η2-μ2-Pt2 (di-σ(CC)); (c) ethyl and (d) vinyl in different coordination modes; (e) μ2-ethylidene–Pt2; (f) μ3-ethylidyne–Pt3. | ||
However, in addition to the overwhelming number of publications using this homolytic paradigm, since the 1960s there has also been ample support for ionic mechanisms, although, to the best of our knowledge, not for purely metallic systems. Two lines of research will be discussed, heterolytic cleavage onto metal oxide catalysts and spill-over from metal catalysts to oxidic supports. HCD on solid oxide catalysts was reported in the 1950s and in hindsight its relation to the homogeneous, heterolytic mechanism has been noted. Trapnell et al. reported that a variety of metal oxides catalysed the exchange of H2 and D2 at mild temperatures.48 One mechanism proposed concerned heterolytic cleavage on non-reducible metal oxides (Fig. 3a, the same as for homogeneous systems, Scheme 1, reaction (1)); another one for reducible oxides, coined “homolytical” or “radicalar”, concerned the transfer of a proton to oxygen and an electron to the metal (Fig. 3b). Chromium oxide gel catalysed the deuteration of 1-hexene giving mostly dideuteriohexane as found by Burwell, while metallic catalysts give quite different patterns characterized by extensive multiple exchange due to reversible reactions and the various bonding modes of alkenes,43 which is indicative of different mechanisms for metal and metal oxide catalysts.49,50 In this early example from 1960, the cleavage of H2 on α-Cr2O3 as a step in the hydrogenation of 1-hexene was named “heterolytic dissociative adsorption at pair sites”, and although the valence state of Cr might have been (II) or (III),51 in Burwell's view it did not change during the hydrogen activation, as is typical of both the heterogeneous and the homogeneous mechanism for HCD.52 Interestingly, the early hydrogenation work involved apolar substrates such as alkenes, and not the typical substrates one would expect for an otherwise heterolytic mechanism.53
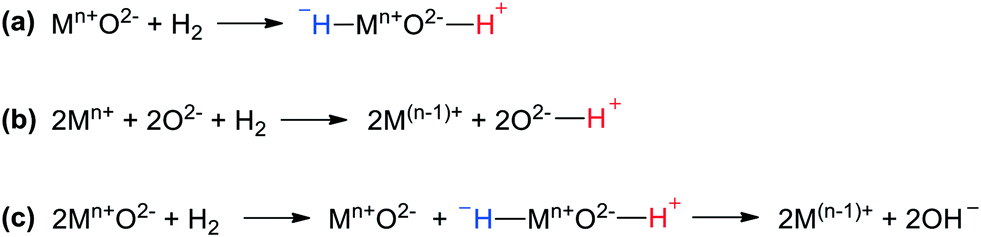 | ||
| Fig. 3 (a) HCD on non-reducible metal oxide catalysts. (b) Homolytic cleavage of H2 on reducible metal oxide catalysts. (c) HCD followed by formation of a homolytic product on reducible metal oxide catalysts. | ||
In an HCD on a metal oxide, a metal hydride and a hydroxide are formed, and the energy gain should largely compensate for the loss of the binding energy of H2 (436 kJ mol−1; upper values for Pt–H 260 kJ mol−1 and O–H 460 kJ mol−1). For most transition metals, the balance will allow the addition of dihydrogen to their metal oxide. Liu reported a combined DFT and experimental study of metal ions on silica and found that the species with the highest heat of metal-hydride formation were the most active ones for propene hydrogenation under mild conditions.54 Late, high-valent transition metals and post-transition metals were much more active than early transition metals, although the formation of the M(H)–O(H) intermediate is not always thermodynamically favorable. Amongst the active metals were also indium and zinc, in accord with the DFT calculations on hydride formation. Clearly, for electropositive main group metals this will mostly not be the case and hydride formation does not occur. For such reactions in bulk materials, thermodynamics prohibit the formation of metal hydrides, e.g. aluminium and zinc. However, for zinc atoms located at certain edges of particles or nanoparticles the balance may be favorable or close to thermodynamically feasible, or as an accessible transition state, sometimes en route to reduction of Zn.55 In a catalytic process, the required energy will add to the overall activation barrier, but catalysis via metal hydrides may still occur.56 For instance, Pydko and Van Santen calculated that ethane will cleave heterolytically on certain Zn ions in a zeolite to give ethyl-“zinc” and hydroxide, and ethene and hydrogen via β-elimination.57 Hydrogen activation on non-reducible oxides has long been under debate, but evidence in favour of it has accumulated. For instance, Copéret and co-workers have recently shown that γ-Al2O3, when treated at appropriately high temperatures, can activate H2 and catalyse the hydrogenation of simple alkenes. The thermal treatment generates defect sites on the alumina surface, which effectively function as frustrated Lewis pairs (FLPs, vide infra).58
Above we mentioned already the addition of dihydrogen to reducible metal oxides, a process that leads to metal reduction by the electrons and formation of hydroxy groups. Like the addition to noble metals, the process is called homolytic, but radicalar has also been used. García-Melchor and López studied as an example of such a process the addition of H2 to CeO2, a reducible oxide, not very active in catalytic hydrogenation.59 The analysis pointed out that dissociative H2 adsorption takes place through a heterolytic pathway followed by the transfer of a hydrogen atom that finally yields the homolytic product (Fig. 3c). From the numerous examples of ETM oxides, we mention the work by Calatayud et al.,60 who studied H2 dissociation on various rutile TiO2 facets by DFT. The topology of the surface had a moderate effect on kinetics and thermodynamics as all four surfaces initially gave heterolytic dissociation to hydride–hydroxyl surface pairs (as in the study by López), which then rearranged by proton and electron transfer to the thermodynamically more favourable reduction product.
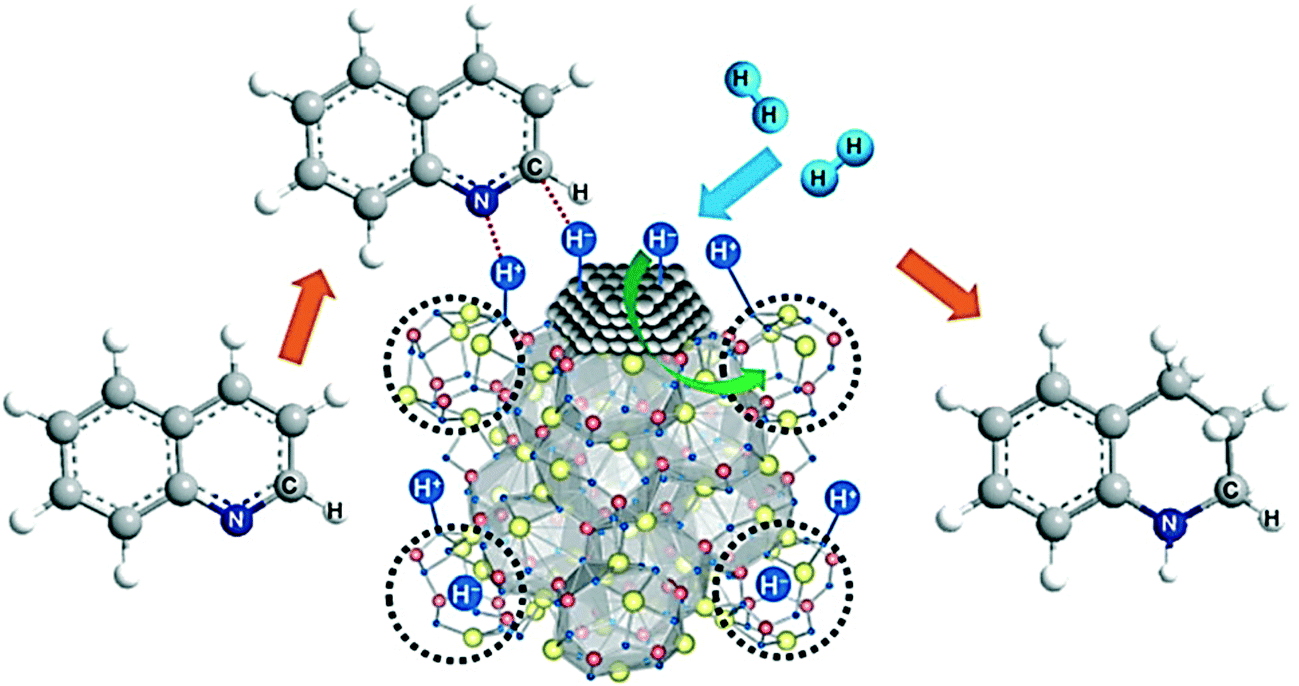
We now turn briefly to the phenomenon called spill-over, which is relevant to nanoparticle catalysed hydrogenations. Prins reviewed spill-over in detail a few years ago.61 The name spill-over was coined by Boudart to explain a phenomenon first described by Khoobiar a few years earlier.62,63 Khoobiar described in 1964 that Pt particles on a yellow WO3 support under a hydrogen atmosphere apparently catalysed the reduction of WO3 to bluish, reduced oxides, while hydrogen alone did not react with WO3 under the same conditions. It was proposed that dihydrogen undergoes dissociative chemisorption on Pt, and that the H-atoms migrate (spill over) to the support and convert to a proton and an electron, see Fig. 4. The proton binds to an oxygen anion and the electron reduces W(VI) to W(V). Both species are mobile in/on the support and further spill-over can occur. Water and alcohols enhance the process and mobility of the protons by providing a more favourable binding of the protons. Huizinga and Prins provided EPR evidence for the reduction of the TiO2 support by H2 dissociated on Pt particles (Fig. 4).64
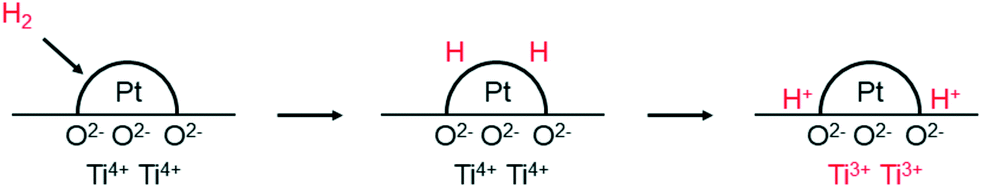 | ||
| Fig. 4 Adsorption of H2 on Pt and spill-over of H atoms to the TiO2 support, producing protons and Ti3+ cations. Adapted with permission from ref. 64. Copyright 1981. American Chemical Society. | ||
Clearly, for this process to occur one needs a reducible support, but n-type semiconductors such as ZnO may also undergo spill-over in such a way, the electrons being stored in the conduction band.61 Many publications report on the participation of spilled-over hydrogen in catalysis on non-reducible oxides (alumina, silica); other scientists remain skeptical and many different explanations for the influence of nearby supports have been brought forward. For spill-over to silica and alumina the debate continues, as it is difficult to exclude other explanations. However, Van Bokhoven et al. have shown that, under highly controlled laboratory experiments in a nanofabricated system, spill-over from Pt particles to iron oxide particles was much more effective on reducible titania than on “non-reducible” alumina.65,66
A further effect of hydrogen spill-over to reducible oxides is the creation of oxygen deficiencies at the surface due to formation of water and reduction of the metal. The open metal sites are now available for coordination to a Lewis-donor substrate, e.g. the aldehyde of an unsaturated enal. Somorjai and Baker have brought this forward as an explanation for the high activity and high selectivity for alcohol formation in the hydrogenation of unsaturated aldehydes by Pt NPs on reducible oxides compared to non-reducible oxides.67,68 Calculations showed that hydrogen atom transfer to the support-coordinated aldehyde is a favoured process, while in the absence of Lewis base–acid interaction it is not (a H atom and an electron are transferred to the organic moiety). It was proposed that the alkoxy/alkoxide formed migrates to the Pt surface to react with a second H atom, although in the general view of heterolytic mechanisms a proton transfer from the support to alkoxide seems a more logical step. Note that the preferred reaction of unmodified Pt and Pd with hydrogen and unsaturated aldehydes is hydrogenation of the CC bond.69 Unlike homogeneous systems, the presence of nearby protons on a support was usually not taken into account in the mechanistic considerations and calculations in the hydrogenation of unsaturated ketones and aldehydes with Pt, Pd, Ru, and Au catalysts.70 As in homogeneous systems the game is open to several possibilities, with initiation by proton transfer, atom transfer, hydride transfer or migration, or simultaneous (outer-sphere) transfer of the hydride and proton.19,71,72 For the present discussion the conclusion that the participation of hydride ions in spill-over systems is the least probable amongst the mechanistic pathways suffices.
The spill-over mechanism received a lot of interest,61,73 but less attention has been paid to the interaction of dihydrogen at the edges of metal particles and support materials. In the last decade, however, mechanistic interpretations involving the bifunctional behaviour of the metal and support, in particular for NPs, have gained importance. At the edges of MNPs on supports one can imagine an HCD reaction similar to that on metal oxides or in metallic complexes with the metal as the hydride acceptor and the oxide as the base (Fig. 5), as was suggested by Fujitani et al. to explain the high activity of small, supported Au NPs in HD exchange reactions; the larger the perimeter interface, the higher the catalytic activity.74 Proof was provided by García et al., who described the HCD by Au NPs on ceria NPs, leading to the concomitant formation of Au–H and positively charged bridged OH groups, besides the generation of water from framework oxygen atoms, as studied by vibration spectroscopy.75 Interestingly, the reaction of the hydrogen loaded catalyst with CO2 and O2 gave formic acid and hydrogen peroxide, respectively. Following this discovery, heterolytic cleavage has slowly re-established its position in the reports on heterogeneous hydrogenation pathways and evidence is also growing.59,76–78
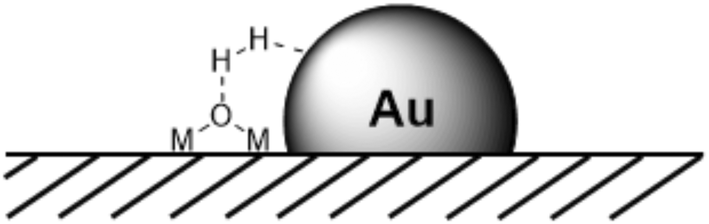 | ||
| Fig. 5 Heterolytic H2 cleavage at the edges of MNPs (for example AuNPs) supported on metal oxide surfaces. | ||
It should be borne in mind that all processes described above for supported metal particles can take place simultaneously: homolytic cleavage on the metal only, spill-over to reducible and non-reducible oxides, heterolytic cleavage at the perimeters (which might also be succeeded by spill-over of hydrogen atoms in either direction), and heterolytic cleavage on the support. The species formed will be related to different catalytic actions and control of the catalyst performance will require control over the formation of all species.
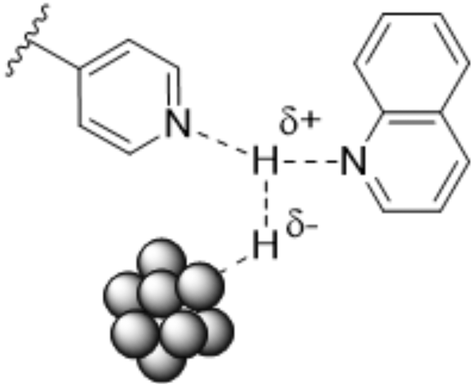
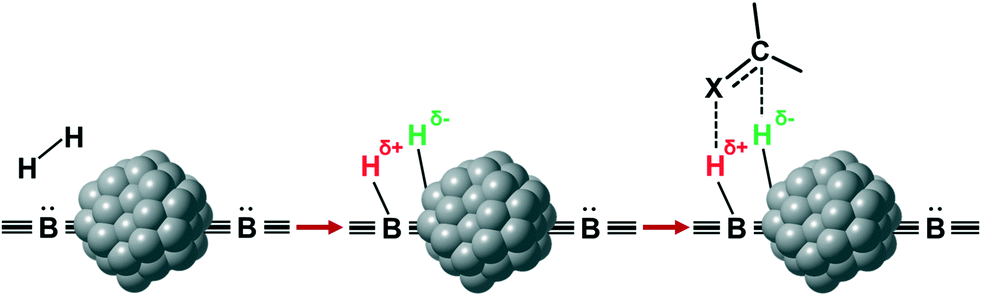
Potentially, a cleaner method to achieve heterolytic dihydrogen activation on MNPs could be the use of ligands on the NPs or stabilizing agents surrounding the NPs. For practical purposes, MNPs in solution have the disadvantage that they are difficult to separate from the reaction mixture after reaction, but nanofiltration or immobilization on passive supports might solve this. Fig. 6a illustrates the basic idea; a nearby base on the metal surface aids the HCD, the same way as in homogeneous systems (Scheme 1, reaction (3)) and on metal oxides (Fig. 3a).79 In the last decade, this technique has found applications at increasing speed and, for instance, Rossi et al. reported highly active Au/SiO2 alkyne semi-hydrogenation catalysts in which piperazine was used as a ligand and base for a highly efficient HCD (vide infra for more details).80 One of us introduced secondary phosphine oxides (SPOs) on MNPs to this end, as it was thought that the P-atom would bind to the metal and the O-atom would be free as a proton acceptor, while a neighbouring metal atom can bind a hydride (Fig. 6b).81 Both for piperazine and SPO, the heterolytic action was supported by DFT calculations; for both heterolytic pathways the activation barrier was considerably lowered compared to the homolytic ones.80,82 When the reaction occurs across unbound M–B (base) sites, the system is called a frustrated Lewis pair, FLP,83 but for bound M–B sites the term FLP is also used, as in the area of FLP chemistry and catalysis.84
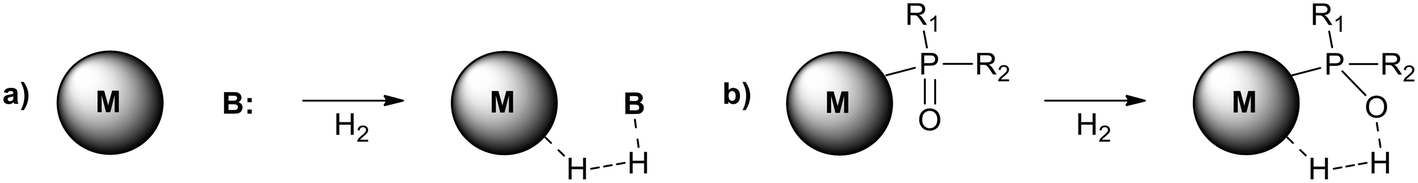 | ||
| Fig. 6 Heterolytic splitting of H2 by (a) MNPs and a nearby base; (b) SPO-ligated MNPs. | ||
1.3 Enantioselective hydrogenation
The best proof for ligand or metal–ligand complex involvement in catalysis is the use of chiral ligands and the induction of chirality in the product. As far as we could trace this back, asymmetric metal catalysis probably started in the area of heterogeneous catalysis when Yoshiharu Izumi et al. used Pd on silk fibres for the hydrogenation of oxime derivatives to produce chiral amino acid precursors with modest ees.85 The authors used several metals and subsequently they switched to metal catalysts, especially RANEY® Ni, modified with chiral hydroxyacids, as reviewed in 1971.86 Cinchona molecules have been widely employed as chiral modifiers and accelerators on metal catalysts since their introduction by Orito et al.87 They obtained 79% ee for the hydrogenation of keto-esters with Pt/C. The scope has since then been extended, the mechanism has been studied in detail, many ligand variations have been added, industrial applications were investigated, etc.88 Notably, the substrates are activated ketones. For the chiral catalysts mentioned here, one usually invokes a hydrogen bond interaction between the ligand and the substrate to enforce a certain orientation of the latter with respect to the catalyst surface. The cinchona molecules contain a tertiary amine that functions as a proton receptor/donor and often it was thought that for the creation of this ammonium donor a protic solvent was needed. However, it was shown that in aprotic solvents and the gas-phase, the proton can also be delivered by the Pt-surface (as a spill-over, one might say),89,90 although Hahn and Baiker described the process consistently as H atom transfer.91 Mechanistically, the chemistry of these modifying chiral ligands on the surface could be closely related to heterolytic cleavage. However, in the vast literature on ligand-induced, heterogeneous, asymmetric hydrogenations, heterolytic cleavage does not seem to play a role in the discussions and we mention this catalysis only here in the introduction.1.4 Frustrated Lewis pairs
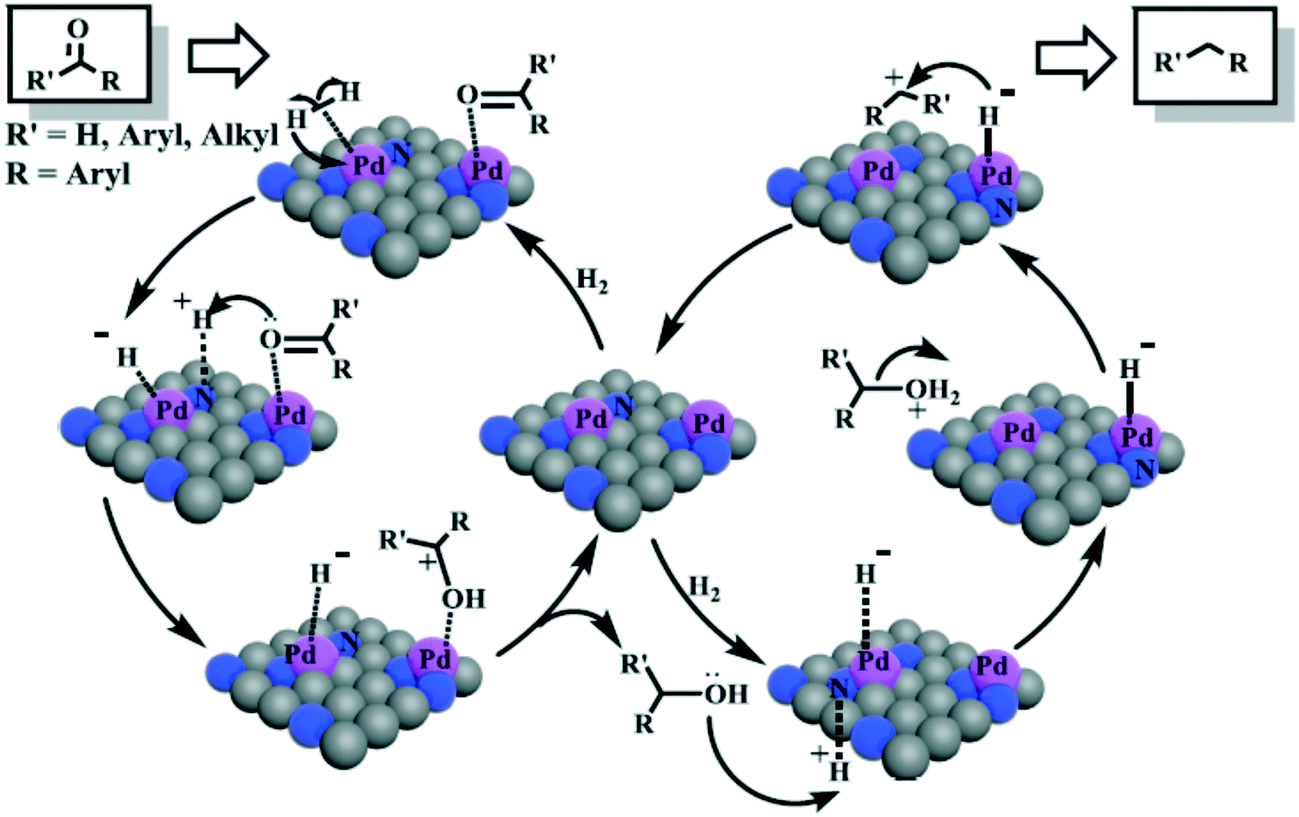
About 15 years ago, Erker and Stephan introduced a new concept and route for the HCD, viz. the use of FLPs.92 Typically, the initial Lewis base and acid contained main group elements such as phosphorus and boron as the central atoms. Even more exciting was the finding that the reagent can be used for the hydrogenation of alkenes.93 The boron Lewis acids used contain fluoro-substituted aryl groups and thus the resulting hydrides are not very hydridic. Transition metal based FLPs (TMFLPs) have also been developed with borane and alumane as the Lewis acids. In the reactions of TMFLPs, the proton is transferred to the transition metal and in several examples the hydride may well possess a hydridic character, but no data on hydricity have been reported.84,94 Apart from the two ligand-modified Au NPs mentioned above that were categorized as FLPs, there are only a few uses of FLPs on the surface of MNPs so far. To facilitate a practical use of FLP hydrogenation, several ways for immobilization have been studied. Solid state FLP catalysts were reviewed by Qu et al.95 The review features FLPs taken from the solution work of which one partner of the pair is immobilized on a solid. This work closely resembles the solution work, and indeed MNPs are not involved. Intramolecular FLPs have been immobilized on the usual range of supports, including polymers. Several of the now FLP classified examples coincide with the metal oxide examples included above, but we want to single out one particular support here, viz. N-doped carbon materials in which the N atoms are assigned the role of the Lewis base and Pd NPs the role of the Lewis acid (see section 3.1).96In this perspective we focus on hydrogenation catalysis by metal nanoparticles that are mechanistically based on heterolytic cleavage of dihydrogen. HCD into H+ and H−, and their subsequent transfer of these hydrogen species to polarized CX groups is a useful stratagem for obtaining high selectivities in hydrogenation catalysis.97 Only reports that explicitly comment on this mechanism will be considered and preferably the mechanistic suggestions will be supported by experiments or theoretical calculations. Firstly, we will discuss MNPs in solution stabilized by ligands and polymers, followed by MNPs on various supports, including a particular section on supported MNPs functionalized with organic ligands. Finally, HCD by “isolated” metal oxide NPs will be examined.
2. Heterolytic cleavage of H2 on MNPs in solution
2.1 MNPs stabilized by polymers
First, we will discuss MNPs synthesized by reaction of metal precursors and reducing agents in the presence of polymers that prohibit the formation of bulk metal by stabilizing the MNPs formed initially. Among the different types of nanoparticle-based systems employed to carry out catalytic transformations that involve a heterolytic cleavage of H2, MNPs stabilized by polymers have shown a limited efficacy. This is probably due to the fact that heterolytic H2 activation requires an intimate contact between the metal surface atoms and the stabiliser to promote the desired cooperative effect. There are only two reports on HCD mediated by MNPs stabilised with polymers. Sánchez-Delgado and co-workers described the synthesis of ruthenium nanoparticles (RuNPs) of 1–2 nm in size immobilised on cross-linked poly(4-vinylpyridine) (PVPy).98 This catalytic system was employed for the hydrogenation of quinoline (substrate/Ru = 84, methanol, 30 bar H2, 120 °C; TOF = 66 h−1), leading to the formation of 1,2,3,4-tetrahydroquinoline with high selectivity. Catalytic experiments by adding an external base or acid and with solvents of different polarities suggested an ionic mechanism through a heterolytic activation of dihydrogen. It was proposed that RuNPs are stabilised in the vicinity of the pyridine groups of the polymer (Fig. 7a), in such a way that H2 is heterolytically cleaved through the cooperation between RuNPs and basic N–pyridine atoms of the polymer (Fig. 7b). Subsequently, an outer sphere concerted transfer of proton and hydride species to the substrate would take place (Scheme 1, reaction (1)), in a way similar to homogeneous ruthenium systems.99 Highly polar solvents provide higher activities, which supports a heterolytic activation of dihydrogen. Similarly, the addition of small amounts of a base such as triethylamine (Et3N) promotes heterolytic H2 splitting and, as a result, a faster hydrogenation is observed (TOF = 120 h−1). On the other hand, an increase of activity was also achieved by the use of acetic acid as an additive (TOF = 180 h−1). The authors suggested that the acid may protonate the N–pyridyl atoms, which would facilitate the interaction with the substrate molecules by hydrogen bonding. However, given that the well-known ability of Ru to activate H2 by itself and that RuNPs prepared by reduction with NaBH4 or H2 usually have a layer of hydrides,100 one might wonder if another hydrogenation mechanism might be operative.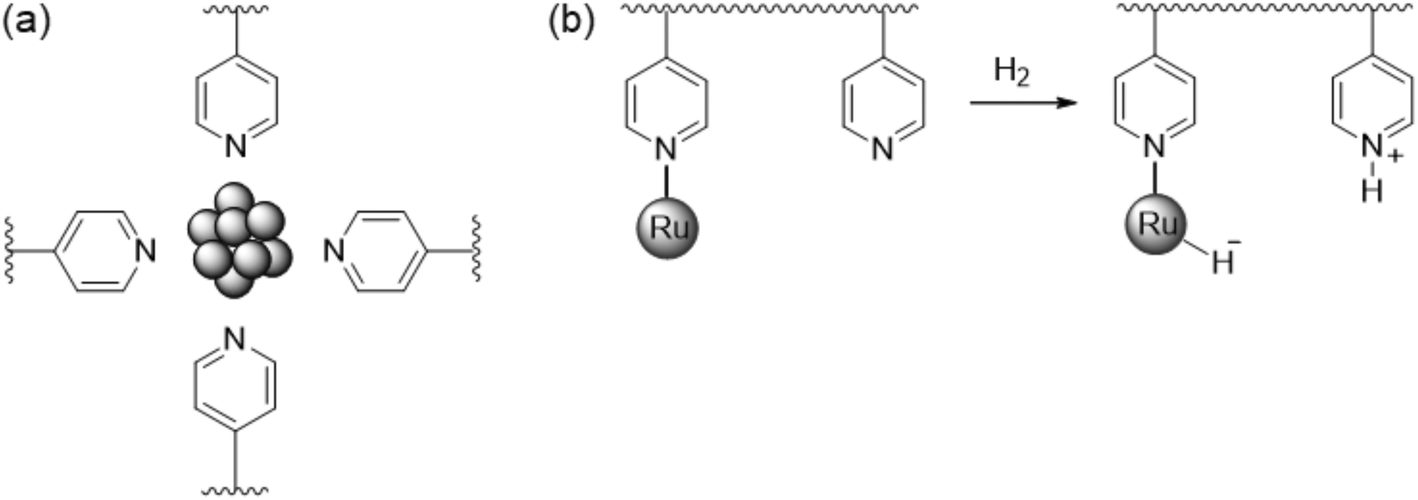 | ||
| Fig. 7 (a) Representative scheme of RuNPs stabilised by poly(4-vinylpiridine). (b) Possible heterolytic H2 cleavage on PVPy-stabilised RuNPs.98 | ||
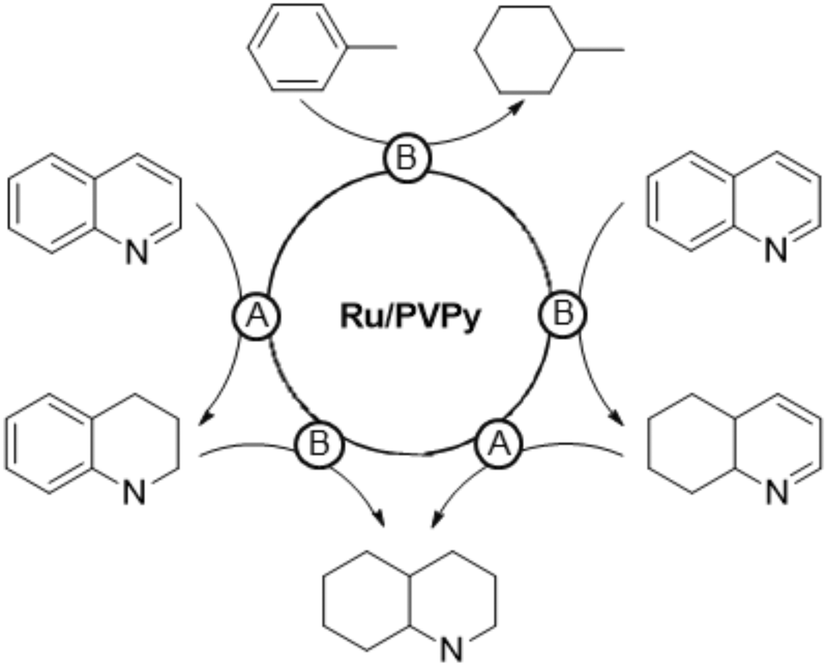
In a following study,101 PVPy-stabilised RuNPs of 3.1 nm were applied as a hydrogenation catalyst of arenes (TOF = 82 h−1 for toluene at 10 bar H2, 120 °C, THF, substrate/Ru = 100) and N-heteroaromatic compounds (TOF = 66 h−1 for quinoline at 30 bar H2, 120 °C, methanol, substrate/Ru = 84). The system is highly active and maximal TOFs of 2000 h−1 and 150 h−1 for toluene and quinoline, respectively, were achieved at 50 bar H2 and 150 °C. Powder XRD and XPS analyses showed that the RuNPs are mainly in the Ru0 state, while a series of experiments provided insights into the catalytic reaction mechanism. Interestingly, two types of active sites that produce two different hydrogenation pathways were identified: one associated with a polar mechanism for CN bond hydrogenation (A), and the other associated with a non-polar mechanism for the common hydrogenation of the aromatic ring (B). An increase in the catalytic activity in the reduction of quinoline was observed with an increase in the solvent polarity, which points to an HCD for the hydrogenation of N-heteroaromatics. This was supported by the results obtained after adding 10 eq. of an external base (Et3N) or acid (HBF4) to the reaction mixture, which led to an enhancement of the activity from 66 h−1 up to 120 h−1 and 180 h−1, respectively. The heterolytic H2 activation would occur through a cooperative effect in which the N atoms of the polymer operate as a Lewis base and neighbouring ruthenium atoms act as a Lewis acid (Fig. 8). In addition, hydrogen bonding between N atoms of the substrate and pyridine groups of the support (or the N atom of Et3N) would favour the protonation of the former. As in the previous case,98 the acid would promote the protonation of N–pyridyl atoms of the polymer or substrate, thus leading to the same hydrogen bonded intermediate (Fig. 8). On the other hand, no effect of solvent polarity or addition of an external base was observed in the hydrogenation of toluene, which suggests a homolytic cleavage of dihydrogen for the reduction of arenes.
 | ||
| Fig. 8 Heterolytic cleavage of H2 mediated by a RuNPs/PVPy system. | ||
Substrate competition experiments with quinoline and toluene supported the existence of these two hydrogenation pathways taking place on different active sites (Scheme 2). Toluene is hydrogenated only after quinoline is fully reduced to decahydroquinoline, which indicates that the heterocyclic ring is hydrogenated at one type of active site (type A), while the carbocyclic ring of quinoline is reduced at another site (type B). Thus, toluene is hydrogenated once reduction of quinoline finishes and type B sites are unblocked. Finally, selective poisoning experiments with thiophene provided further evidence of this dual hydrogenation mechanism. This compound inhibits the hydrogenation of both toluene and the carbocyclic ring of quinoline, suggesting that the thiophene molecule selectively blocks type B active sites.
 | ||
| Scheme 2 Proposed hydrogenation pathways through two different types of active sites.101 | ||
2.2 MNPs stabilized by ligands
Different strategies have been developed to achieve a heterolytic splitting of dihydrogen in MNP catalysis. However, in comparison with supported MNPs, there is little precedent for HCD with homogeneous ligand-stabilized nanoparticles dispersed in solution. A successful approach developed by van Leeuwen and coworkers entails the use of ligands that act both as a stabilising agent for MNPs and a heterolytic activator for H2 through a cooperative effect with a neighbouring metal atom. Secondary phosphine oxides (SPOs) were employed to stabilise nanoparticles with applications as hydrogenation catalysts, in such a way that SPO ligands and surface metal atoms act cooperatively in HCD, which favours the selective reduction of polarized unsaturated groups. Indeed, RuNPs stabilized by SPOs showed an increase in selectivity towards the carbonyl group in the hydrogenation of acetophenone in comparison with a classic tertiary monophosphine (from 26 to 47%). The participation of P–O moieties in H2/D2 exchange reactions supported the heterolytic H2 splitting mediated by surface ruthenium atoms and P–O groups.81 Next, these authors described the synthesis of gold nanoparticles (AuNPs) ligated by a specific SPO, tert-butyl(naphthalen-1-yl)phosphine oxide.102 The SPO-ligated AuNPs were exceptionally selective towards the carbonyl functionality in the hydrogenation of a wide range of substituted aldehydes (Scheme 3). | ||
| Scheme 3 Selective hydrogenation of substituted aldehydes catalysed by SPO-ligated AuNPs (conversion, selectivity). Reaction conditions: AuNPs (0.01 mmol Au), substrate (0.5 mmol), THF/hexane (5 mL), 18 hours, 40–60 °C, 40 bar H2.102 | ||
Several control experiments demonstrated that the SPO ligand plays a crucial role in the catalytic process, thus supporting a hydrogenation mechanism through an HCD in which the oxygen atom of the SPO acts as a Lewis base and binds the proton, H+, whereas a neighbouring gold atom operates as a Lewis acid and takes the hydride, H− (Scheme 4).
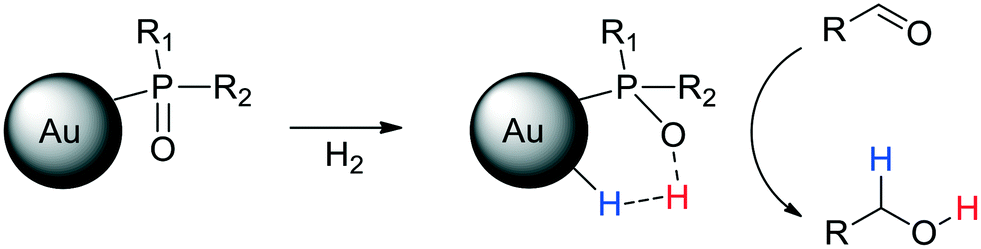 | ||
| Scheme 4 Heterolytic splitting of H2 by SPO-ligated AuNPs and the following hydrogenation of aldehydes. | ||
A subsequent study revealed that the degree of polarization of the PO bond is the key for the HCD, as was proved by cross polarization magic angle spinning (CP-MAS) NMR spectroscopy.103 To this end, a series of AuNPs ligated by different SPOs were synthesized and thoroughly characterized, showing important differences in the morphology and catalytic properties depending on the substituents in the SPO. CP-MAS NMR demonstrated that AuNPs stabilised with aryl SPOs exhibit a strong polarity of the PO bond, thus favouring the HCD. These nanoparticles present Au(I) atoms and SPO anions at the surface and show very high selectivity in aldehyde hydrogenation (Scheme 3). On the other hand, AuNPs ligated by aliphatic phosphine oxides display low polarity in the PO bond, which hampers the heterolytic H2 activation. These alkyl-stabilised AuNPs have both Au(I) and Au(0) atoms at the NP surface, and also contain POH species. Consequently, they display active sites of different nature, leading to lower activity and selectivity in the hydrogenation of substituted aldehydes (Scheme 5).
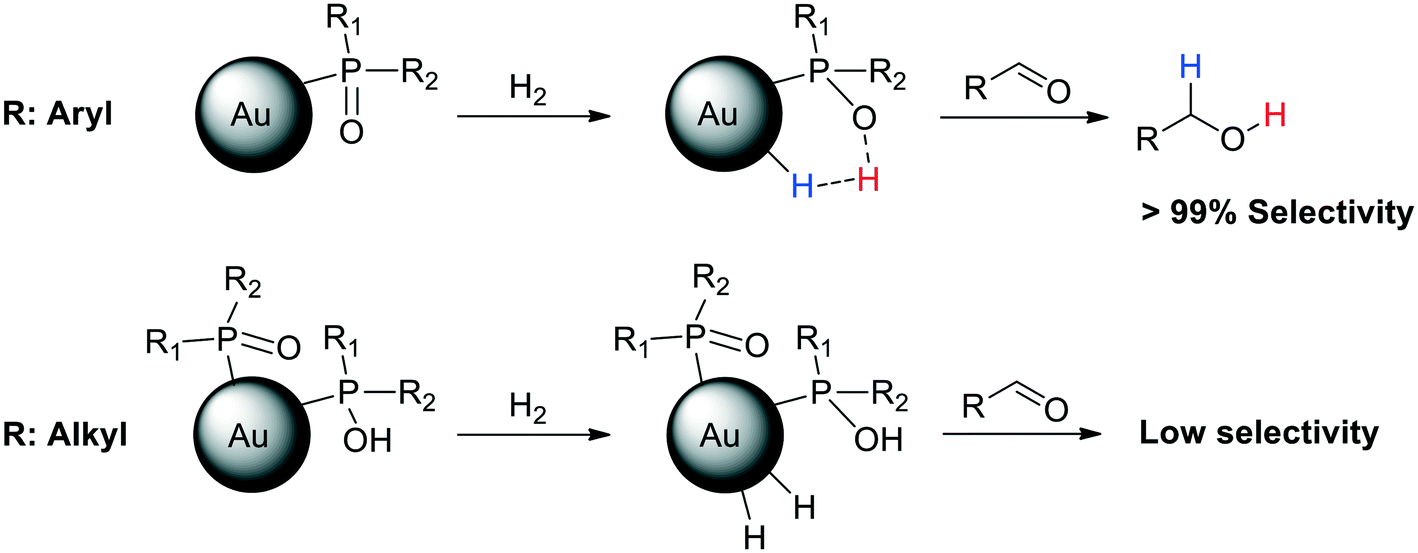 | ||
| Scheme 5 AuNPs ligated by aryl SPOs exhibit a strong polarity of the PO bond, which favours the HCD and thus the selective hydrogenation of aldehydes. AuNPs ligated by alkyl SPOs show a low polarity of the PO bond, which hinders the heterolytic H2 activation. These AuNPs display low selectivity in aldehyde hydrogenation.103 | ||
This heterolytic hydrogenation mechanism by SPO-stabilised AuNPs was further supported through theoretical calculations.82 A cooperative effect was identified at the AuNP–SPO interface by DFT studies, which acts as a frustrated Lewis pair (FLP). This FLP favours the heterolytic H2 splitting and its addition to the CO bond of acrolein by a concerted mechanism, leading to the selective formation of the unsaturated alcohol (Fig. 9). Crucially, the active sites available at the NP surface and the basicity difference between substrate and SPO ligand determine the activity of the AuNPs.
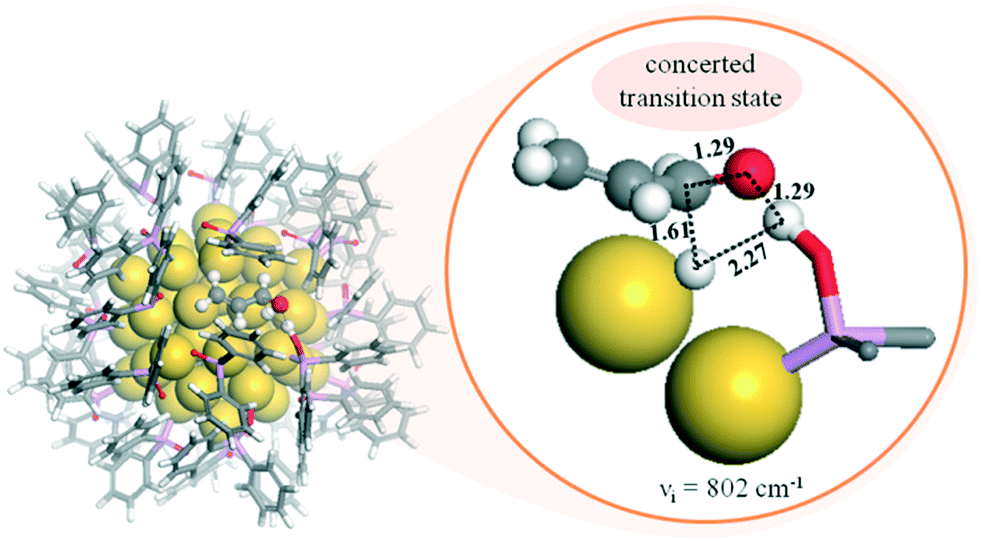 | ||
| Fig. 9 Left: DFT adsorption configuration of 27 Ph2PO (SPO) ligands on a Au55 NP. Right: Structure, distances (Å), and imaginary frequency (cm−1) determined for the transition state corresponding to the concerted mechanism in the selective hydrogenation of the CO bond of acrolein on a SPO-ligated AuNP. The inset displays the formation of the four-membered ring and the distances. Yellow spheres: gold atoms. Stick models: adsorbed Ph2PO ligands. Color codes: C, grey; O, red; H, white; P, pink. Reprinted with permission from ref. 82. Copyright 2017. American Chemical Society. | ||
Following this approach, the same authors proposed a heterolytic H2 activation in the hydrogenation of several unsaturated aldehydes catalysed by SPO-ligated IrNPs.104,105 Indeed, very high chemoselectivities towards the carbonyl group were observed in the hydrogenation of cinnamaldehyde (99%), p-cyanobenzaldehyde (>99%) and 2-octynal (96% selectivity), suggesting a heterolytic splitting of H2 mediated by the oxygen atom of the SPO and a neighbouring iridium atom. Interestingly, the catalytic activity of these NPs was compared with those of an analogous Ir–SPO complex and several supported IrNPs previously described.105 The SPO-stabilised IrNPs showed lower catalytic activity but higher robustness than the complex, providing high selectivities for aldehydes that poison the molecular catalyst, e.g. cyano- and alkyne-substituted aldehydes, whereas the supported IrNPs were generally less selective.
There are other reports describing a HCD carried out by ligand-stabilized MNPs via other mechanisms rather than a ligand–metal cooperation. In this context, very high selectivities were obtained by Jin et al. in the hydrogenation of α,β-unsaturated carbonyl compounds catalyzed by the atomically precise Au25(SR)18 cluster (SR = thiolate).106 The authors suggested that the hydrogenation process occurs by a homolytic mechanism involving the adsorption of H2 at low coordination surface gold atoms and activation of the CO group on electron-rich gold sites located in the core. By contrast, Jiang and coworkers proposed that the H2 activation by Au25(SR)18 clusters takes place through a heterolytic splitting mechanism in which H2 is cleaved by the CO group of the substrate and a surface gold atom.107 DFT studies analysed the possible pathways for the hydrogenation of benzalacetone catalysed by Au25(SR)18, discarding a direct dihydrogen activation on the cluster as that proposed by Jin. Indeed, the transition state corresponding to this path displays a barrier of 2.14 eV, which indicates that this route is very unlikely at the temperature at which the catalytic reaction was performed (0 °C).106 Instead, the theoretical calculations revealed a first coadsorption of both benzalacetone and dihydrogen on the Au25(SR)18 cluster, and a subsequent HCD through the cooperation of a Au atom of the cluster and the carbonyl oxygen atom of benzalacetone (Scheme 6). The study also shows that the process is assisted by one molecule of ethanol (solvent), which forms a hydrogen bond with the OH group of the partially hydrogenated reactant. Then, the hydride species generated at a Au atom is transferred to this intermediate, leading to the selective formation of the unsaturated alcohol product.
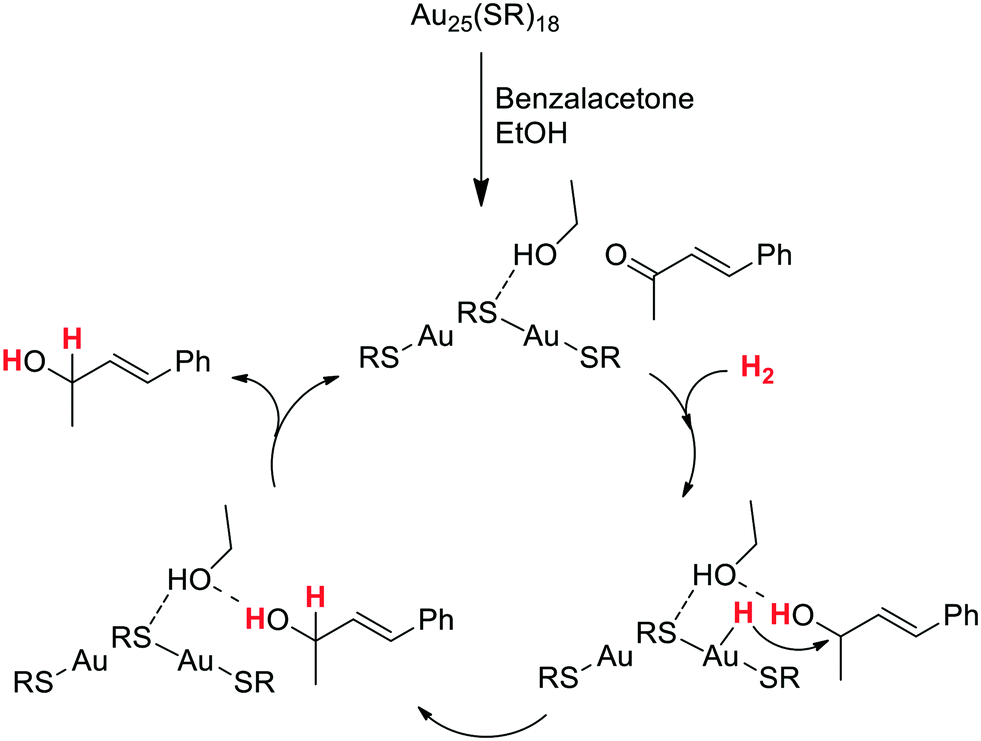 | ||
| Scheme 6 Proposed catalytic cycle for the chemoselective hydrogenation of benzalacetone catalyzed by the Au25(SR)18 cluster. The cluster is represented as RS–Au–SR–Au–SR.107 | ||
DFT calculations are in good agreement with the 100% selectivity observed at 0 °C. Indeed, the energy profile shows an activation energy of 0.90 eV for the ethanol-assisted process, while the barriers in the solvent-free route are 0.99 and 1.12 eV for the hydrogenation of CO and CC bonds, respectively (Fig. 10).
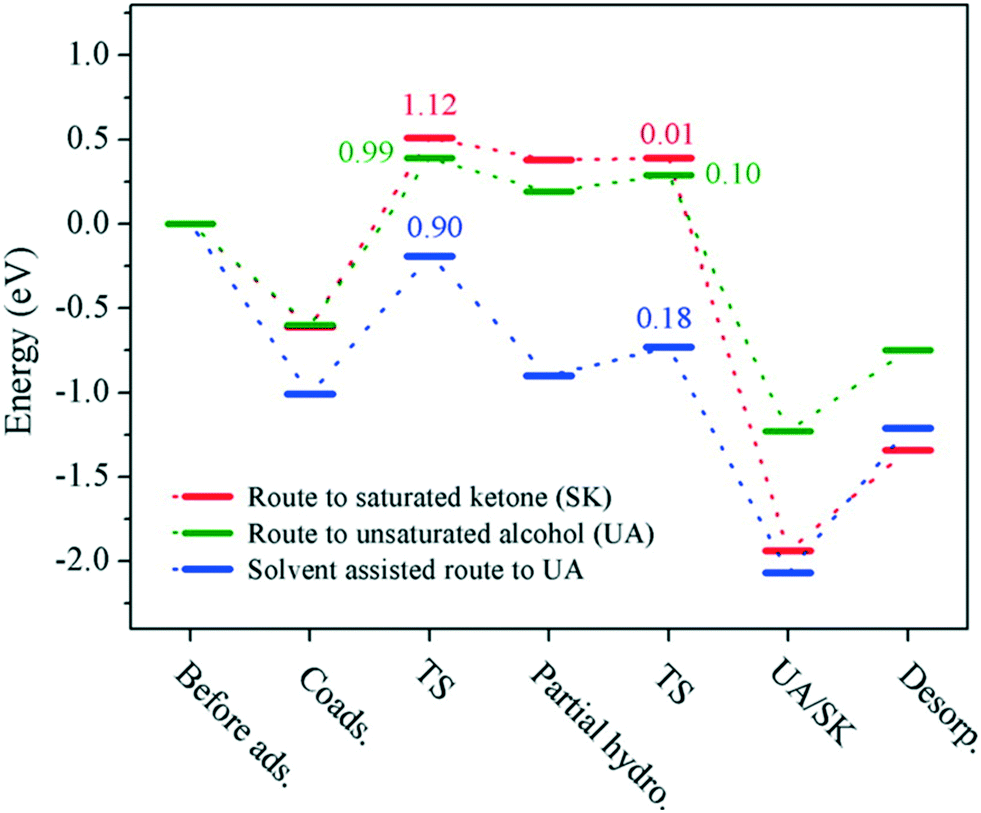 | ||
| Fig. 10 Energy profiles for the chemoselective hydrogenation of benzalacetone to the corresponding saturated ketone (red) and unsaturated alcohol through solvent-free (green) and solvent-assisted (blue) pathways. Reprinted with permission from ref. 107. Copyright 2015. American Chemical Society. | ||
The use of a base as an additive is a very successful strategy to achieve a heterolytic H2 splitting in MNP catalysis. In line with this, Jin and coworkers proposed a heterolytic mechanism in the selective hydrogenation of 4-nitrobenzaldehyde catalysed by gold nanoparticles and nanorods stabilised with citrate and CTABr, respectively (Scheme 7).108 Both Au catalysts are very selective, but the nanorods are more active than the NPs and allow an efficient recycling by centrifugation. 100% selectivity towards 4-nitrobenzyl alcohol was observed at 80 °C and 20 bar H2 in a transformation that requires the additional use of pyridine. In fact, no reaction was observed in the absence of a base. It was suggested that pyridine binds the proton of H2 and forms a PyH+ pyridinium species, whereas the gold catalyst abstracts the corresponding hydride, H−. These PyH+ and H− would also favour the adsorption of the substrate through the aldehyde and nitro groups (–CHO and –NO2).
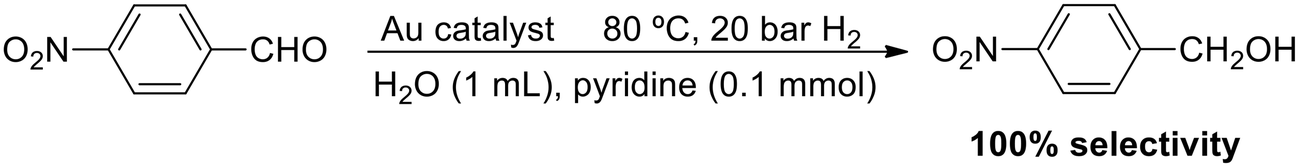 | ||
| Scheme 7 Chemoselective hydrogenation of 4-nitrobenzaldehyde catalysed by AuNPs and Au nanorods. | ||
Previously, Jin described the same process under analogous conditions (H2O as a solvent, 80 °C, 20 bar H2, 0.1 mmol pyridine) catalysed by water-soluble thiolate-stabilized gold clusters ligated by glutathione (H-SG) and captopril (Capt), such as Au15(SG)13, Au18(SG)14, Au25(SG)18, Au25(Capt)18, and Au38(SG)24.109 These atomically precise clusters provided complete selectivity towards the nitrobenzyl alcohol product, for which DFT studies revealed that both –NO2 and –CHO functionalities interact with the S–Au–S surface of the cluster. In parallel, the cluster would activate the H2 molecule with the aid of pyridine. There is no mention of heterolytic H2 splitting but the prerequisite of pyridine points toward a base-assisted heterolytic process.
In a further step, CeO2-supported Au25(SR)18 clusters were employed as a catalyst for aldehyde hydrogenation.110 Interestingly, the presence of a Lewis acid in the reaction medium such as Cu+, Cu2+, Ni2+ and Co2+ enhances the catalytic activity, while the use of a base (NH3 or pyridine) is also required. Once more, 100% selectivity towards the alcohol product was obtained in the reduction of 4-nitrobenzaldehyde, among other substrates. UV-vis spectroscopy, MALDI and ESI-MS showed the generation of new cluster Au25−n(SR)18−n (n = 1–4) species, and DFT calculations elucidated the structure and mechanism for the speciation of Au24(SR)17, which involves the abstraction of a Au–SR fragment from the Au25(SR)18 cluster with the participation of a Lewis acid. As a result, the Au24(SR)17 species may present different open metal sites for adsorption of substrates: a gold atom of the cluster core (Au1), a gold atom on a staple motif (Au2), and the metal ion employed as a Lewis acid (M1). The DFT studies allowed the authors to propose a hydrogenation mechanism for each of these active sites, all of them involving a base-assisted heterolytic H2 cleavage (Fig. 11). In this way, higher conversions are observed with stronger bases such as NH3, which activates the H–H bond and abstracts a H+ to generate NH4+.
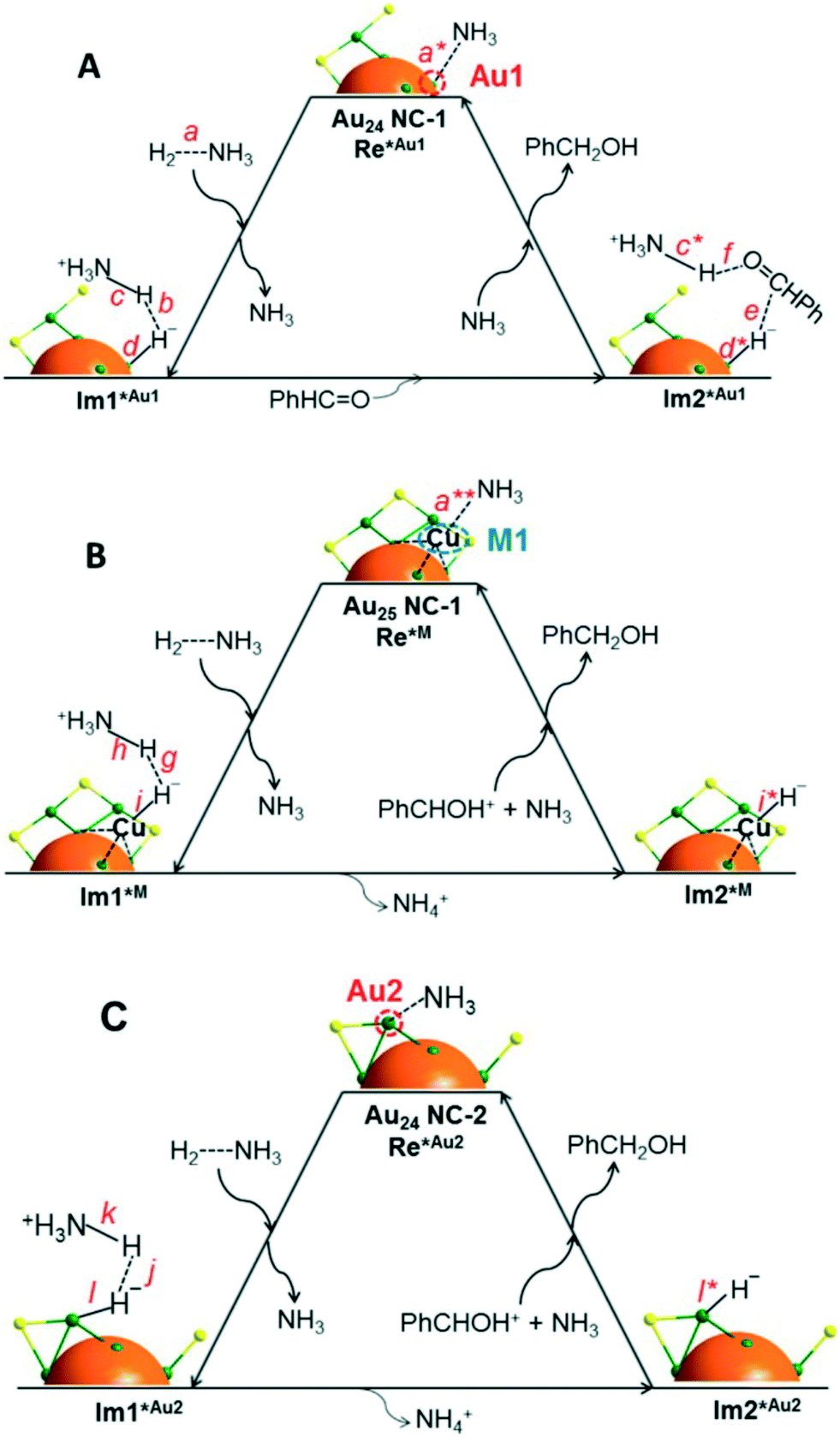 | ||
| Fig. 11 Proposed catalytic mechanism for benzaldehyde hydrogenation in the presence of NH3 on (A) a gold atom of the cluster core (Au1), (B) the metal ion employed as a Lewis acid (M1), and (C) a gold atom on a staple motif (Au2). Colours: Au, green; S, yellow. Reprinted with permission from ref. 110. Copyright 2015. American Chemical Society. | ||
Recently, the same authors described the heterolytic hydrogenation of nitriles mediated by thiolate-stabilised nickel Ni6(SR)12 clusters. The clusters consist of a double crown in which the Ni atoms form a hexagonal ring, while the twelve S atoms are disposed as bridging Ni–S–Ni below and above the ring plane (Fig. 12).111 Consequently, the Ni sites are shielded by these sulphur atoms, which limits the access of substrates. Indeed, very low conversions and complete selectivities towards the primary amine were obtained in the hydrogenation of 4-cyanotetrahydropyran. Interestingly, the use of NH3 dramatically enhances the catalytic activity. DFT calculations indicate that the Ni6(SR)12 cluster is activated through the cleavage and subsequent insertion of H2 into a Ni–S–Ni unit, leading to the formation of a hetero-bridge Ni–H–H–S chain, which undergoes heterolytic H2 cleavage. In this case, NH3 does not act as a heterolytic activator for dihydrogen. Instead, this base would play a crucial role by preventing a recombination reaction that regenerates the cluster structure and leads to the deactivation of the catalyst. The theoretical studies reveal that NH3 is adsorbed on the double crown and partially cleaves the Ni–S–Ni bridge, thus maintaining two Ni sites (Ni–NH3 and Ni–S) open.
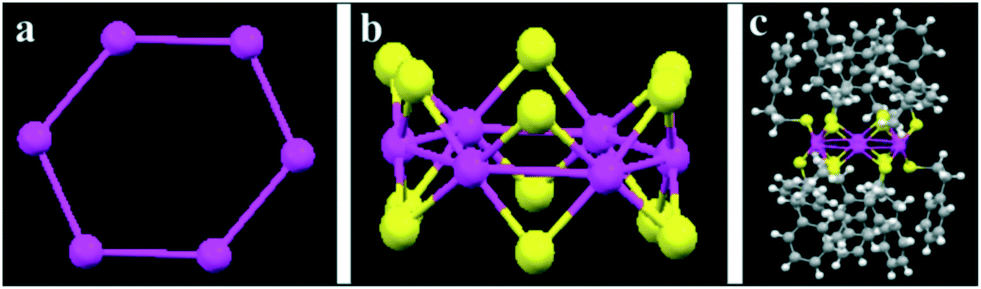 | ||
| Fig. 12 Structure of the Ni6(SR)12 cluster (R: SC2H4Ph): (a) Ni6 structure. (b) Framework of Ni6 plus 12 S atoms. (c) Crystal structure. Colors: Ni, magenta; S, yellow; C, gray; H, white. Reproduced from ref. 111 with permission from The Royal Society of Chemistry. | ||
Finally, the work of Häkkinen and Zheng is worth noting, in which atomically precise copper-hydride nanoclusters (CuNCs) ligated by thiolates were employed as a catalyst for ketone hydrogenation.112 The [Cu25H10(SR)18]3− CuNC contains 10 hydrides that serve as the hydrogen source for the catalytic process. DFT calculations using formaldehyde as a model substrate reveal that this molecule takes a hydride from the cluster to generate a [Cu25H9–OCH3(SR)18]3− intermediate containing an alkoxy group, which then reacts with dihydrogen through two possible catalytic pathways (Fig. 13). In the first route, H2 is heterolytically cleaved in the proximity of the Cu–O bond to form a proton that binds to the alkoxy group and affords the alcohol product, whereas the corresponding hydride species restores the cluster. For the second pathway, it is proposed that the alkoxy intermediate could bind a second hydrogen atom and form the product. Subsequently, the dissociation of H2 leads to the replenishing of these two hydride vacancies and thus the regeneration of the catalyst. Theoretical studies indicate that both routes are kinetically and thermodynamically feasible, and that there is no preference for a particular one. However, additional experimental proofs and the study of the solvent-accessible surface area suggest that the second route is the preferred pathway.
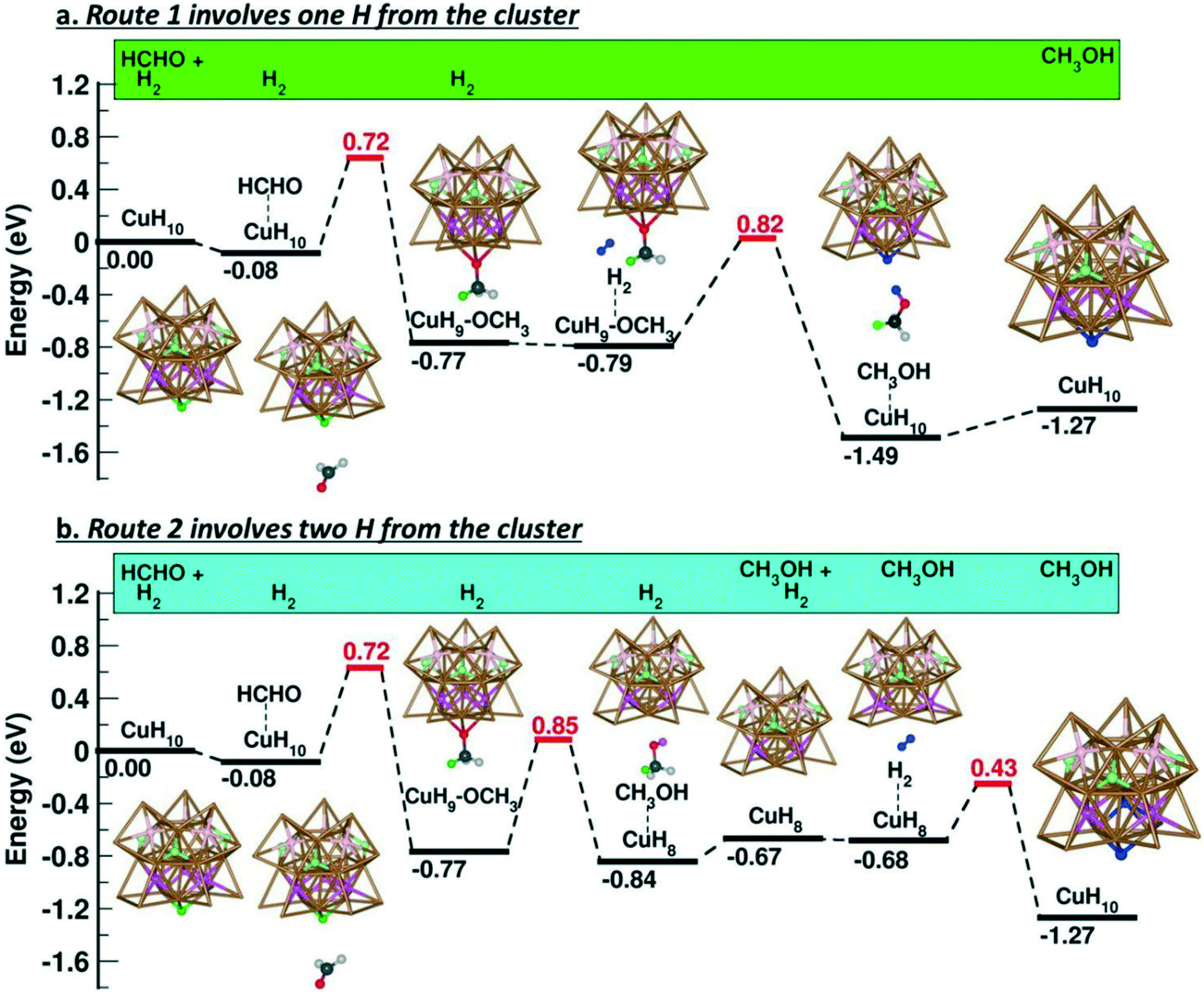 | ||
| Fig. 13 Energy profiles of pathways 1 and 2 for the hydrogenation of formaldehyde catalysed by [Cu25H10(SR)18]3−. Colours: frame of the copper cluster, brown; hydrides, green, pale green, pink and pale pink; C (HCHO), dark grey; H (HCHO), light grey; O (HCHO), red; H (H2), blue. Reprinted with permission from ref. 112. Copyright 2019. American Chemical Society. https://pubs.acs.org/doi/10.1021/acsnano.9b02052. Further permissions related to the material excerpted should be directed to the ACS. | ||
3. Supported MNPs
3.1 Ligand-free MNPs
Supported transition metal catalysts are of great importance for the industrial synthesis of numerous chemicals.113,114 These industrial catalysts are often based on expensive metals well-dispersed on cheap, high-surface area, porous supports. A high dispersion is convenient since most of the existing metals on the surface are accessible for the substrates and ready for catalysis.115,116 In this respect, supported metal nanoparticles (MNPs) have been demonstrated to be a promising type of green catalyst. Their large surface area, together with their higher stability and recyclability compared to MNPs in solution, makes supported MNPs ideal catalysts for industrial applications.117 In fact, supports not only increase the stability and recyclability of MNPs, but they can also be used to modify the MNPs' electronic/steric properties, and therefore alter the catalytic properties.As was mentioned in the Introduction, selective hydrogenation of polar functional groups (CN, CS or CO) with homogeneous catalysts is in several cases due to a metal ligand cooperation, which dissociates H2 in a heterolytic way (forming heteroatom–H+/metal–H− pairs). On the other hand, it has been established that supported-MNPs activate hydrogen in a homolytic way, forming sometimes O–H+ on the support through hydrogen spillover.61 In addition to this, a cooperative activation of H2 can be performed by metal–basic site interfaces. Indeed, well-defined nanostructured materials based on MNPs immobilized on basic supports have been demonstrated to be promising catalysts for selective hydrogenation reactions. Basic functional groups of the support in close proximity to active metal sites promote the heterolytic H2 cleavage and thereby boost the hydrogenation of some substrates, such as heteroaromatics, aldehydes, ketones or carboxylic acids. Here, MNPs and the basic centres of the supports work together in the dissociation of H2 through a heterolytic mechanism (Fig. 5). In addition, the hydrogen transfer occurs through an outer sphere mechanism in which there is no direct interaction between the MNP surface and the substrate (Fig. 14), and prevents poisoning of the catalyst.
 | ||
| Fig. 14 MNP and basic support working in synergy for heterolytic H2 dissociation and the subsequent hydrogenation of R2CX (where X = N, S, O) through an outer sphere mechanism. | ||
The function of supports is generally to avoid MNP aggregation and to facilitate the recyclability of the catalysts. However, the number of reports in which the support plays an active role in the hydrogenation mechanism is growing fast. Indeed, there is a great variety of examples in the literature about heterolytic H2 activation by supported MNPs. Following previous work with polymer-stabilized MNPs (see section 2.1),98,101 Sanchez-Delgado et al. reported one of the first examples in 2012 by using Pd nanoparticles supported on MgO (Pd/MgO) for the hydrogenation of alkenes, quinolines and biodiesel.118 The high activity of Pd/MgO in comparison with commercial Pd/SiO2 and Pd/Al2O3 was explained by an ionic hydrogenation mechanism promoted by the combination of MNPs and the basic sites of the support as shown in Fig. 5. Shortly thereafter, the same research group reported the analogous catalyst containing Ru NPs (Ru/MgO), which is capable of hydrogenating heteroaromatics without catalyst poisoning, normally observed in the presence of nitrogen- and sulfur-containing species.119 This tolerance to poisoning is probably due to the surface ionic hydrogenation pathways shown in Fig. 14. Indeed, after mechanistic studies, they proposed a dual homolytic/heterolytic mechanism, in which two kinds of active sites operate simultaneously: i) the surface oxygen atoms of the support (basic centres) next to metal active sites (Ru atoms) for HCD (site A, Fig. 15); ii) Ru atoms far away from the basic support act as common metallic sites splitting H2 homolytically (site B, Fig. 15). All this explains the high activity and stability of Ru/MgO in the hydrogenation of N- and S-heteroaromatics. This novel mechanism was verified through a competition test between quinolone and toluene, where the hydrogenation of toluene was basically blocked until most of the quinolone was hydrogenated. In addition, an inhibition experiment with thiophene confirmed that after blocking the Ru surface with the heteroarene, the hydrogenation of toluene was totally suppressed, but instead the N-heterocyclic ring of quinolone was fully hydrogenated. A similar dual homolytic/heterolytic hydrogenation mechanism was proposed for Rh nanoparticles immobilized on basic supports, such as MgO, CaO or SrO.120 Here, it was observed that the hydrogenation rate of N-heterocycles increases with the basicity of the support (Rh/MgO < Rh/CaO < Rh/SrO).
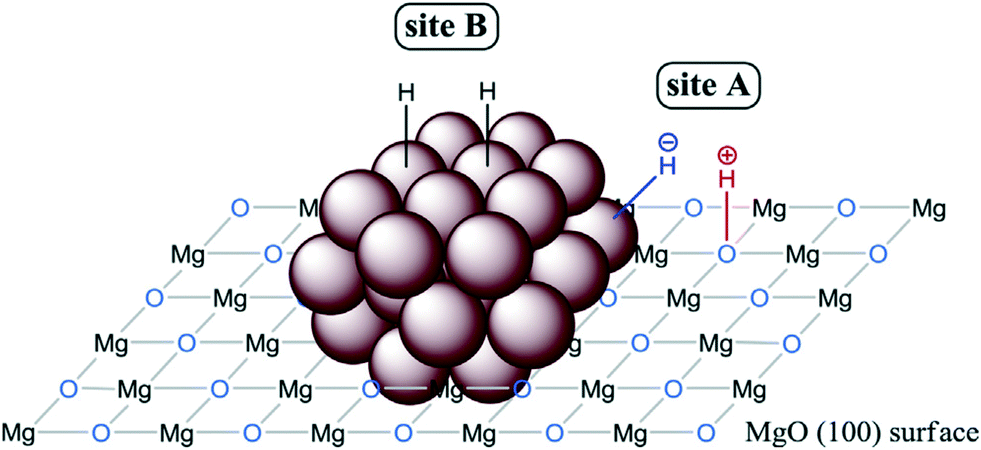 | ||
| Fig. 15 Illustration of the active sites necessary for the dual homolytic/heterolytic hydrogenation mechanism proposed for Ru/MgO. Reprinted with permission from ref. 119. Copyright 2014. Elsevier. | ||
Jagadeesh in collaboration with Beller reported a series of Co and Fe heterogeneous catalysts prepared by pyrolysis of molecular complexes with nitrogen ligands immobilized on activated carbon.121,122 The resulting catalysts, non-noble MNPs supported on N-doped graphene, are able to activate dihydrogen by HCD.123 More specifically, N-doped graphene coated Co- and Fe catalysts show a high activity/selectivity in the hydrogenation of nitroarenes under mild conditions (70 °C and 20 bar). Here, the basicity of the graphene support is essential for the HCD. Zhang et al. recently described another interesting study about HCD in MNPs supported on a N-doped carbon material.96b More specifically, a new catalyst based on Pd NPs supported on nitrogen-doped porous carbon (Pd/NC-BT) gave a high activity in hydrodeoxygenation of a great number of carbonyl compounds, affording the corresponding alkanes. Here, again, the nitrogen atoms promote the HCD in cooperation with the Pd NPs. Fig. 16 shows the proposed mechanism where first the H2 molecule and the carbonyl compound adsorb on the catalyst surface. Then, HCD generates N–H+ and Pd–H− species, which react with the polarized carbonyl group, generating the corresponding alcohol. Finally, the hydrogenolysis of the alcohol group also takes place through the heterolytic cleavage of another H2 molecule. The same group reported a similar system, nitrogen-doped carbon supported Pd NPs, for the selective hydrogenation of quinolone derivatives into the corresponding 1,2,3,4-tetrahydroquinolines.96a Here, in the same way as in the previous case, the nitrogen atoms act as a Lewis base promoting the HCD. Rossi and collaborators proposed an analogous cooperative action between gold and nitrogen atoms for Au NPs encapsulated in nitrogen-doped carbon and supported on TiO2 (Au@N-doped carbon/TiO2).124 Here, as well as in the previous examples, N atoms facilitate the heterolytic H2 cleavage, which also explains the high activity of this catalyst in the semihydrogenation of alkynes to alkenes.
 | ||
| Fig. 16 Proposed mechanism for the hydrodeoxygenation of carbonyl compounds with Pd/NC-BT. Reprinted with permission from ref. 96b. Copyright 2018. Elsevier. | ||
Along the same line, Martínez-Prieto et al. recently proposed the same dual mechanism as Fig. 15 for H2 dissociation in the selective hydrogenation of palmitic acid into the corresponding alcohol (1-hexadecanol) catalysed by Ru NPs supported on reduced-graphene oxide doped with N (Ru/NH2–rGO).125 Here, the basic centres that promote HCD are the amino and pyrrolic groups present in the N-doped graphene (Fig. 17b). To distinguish between sites A and B, they used the hydrogenation of acetophenone as a model reaction (Fig. 17a). They proposed that the ketone groups of acetophenone are hydrogenated by site A, while the aromatic rings are hydrogenated on the Ru surface, site B. Comparing the initial hydrogenation rates of Ru NPs supported on reduced graphene oxide doped (Ru/NH2–rGO) and non-doped with N (Ru/rGO), it was observed that Ru/NH2–rGO hydrogenates the ketone much faster than the phenyl group. On the other hand, for Ru/rGO, which mainly contains site B, the hydrogenation of the ketone is slower than that of the aromatic ring (Fig. 17b and c). Therefore, they concluded that site A in Ru/NH2–rGO enhances the hydrogenation of carbonyl groups in both substrates, acetophenone and palmitic acid.
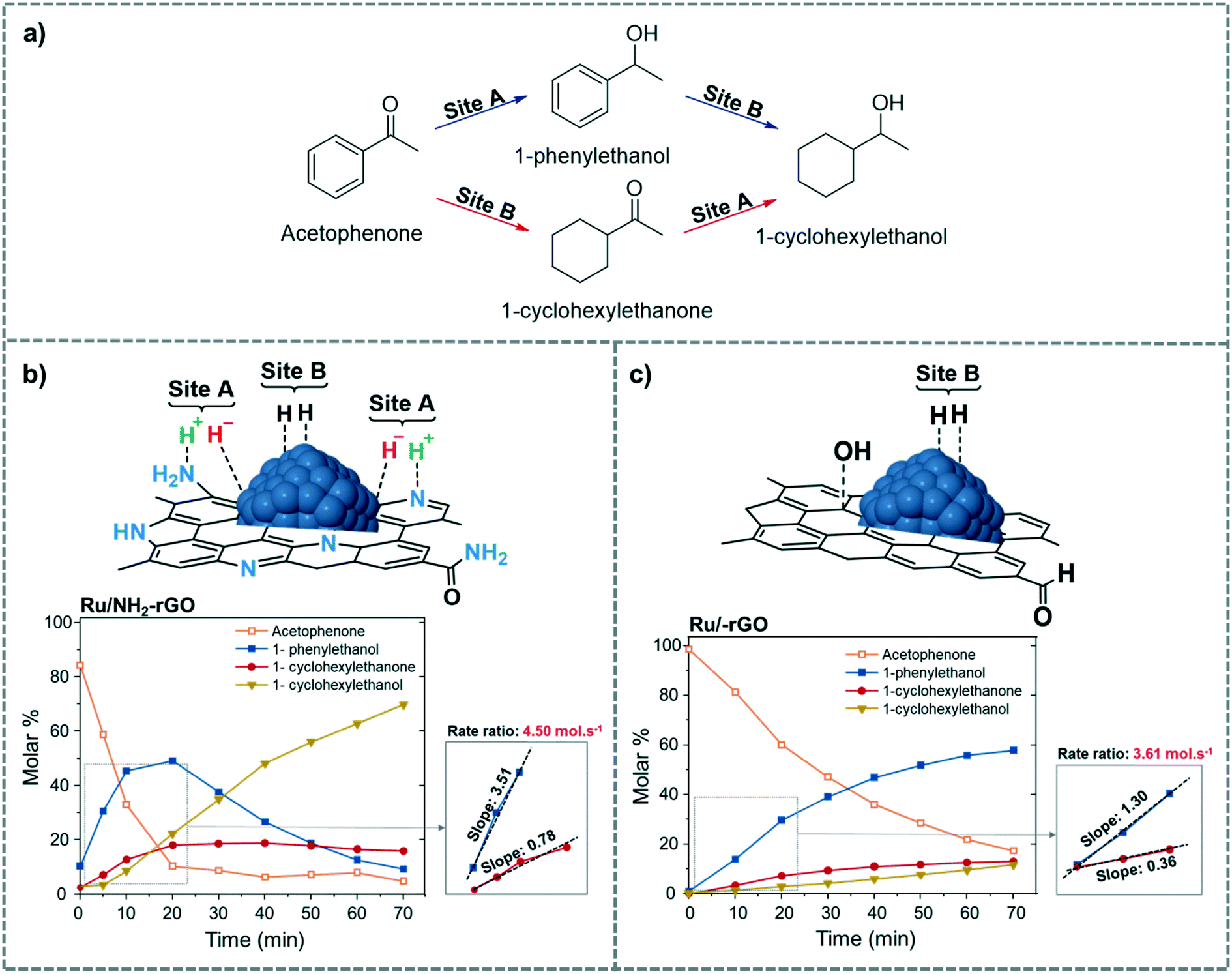 | ||
| Fig. 17 Possible pathways for the hydrogenation of acetophenone to 1-cyclohexylethanol (a). Proposed active sites and time course of the product yield during the hydrogenation of acetophenone using (b) Ru/NH2–rGO and (c) Ru/rGO as catalysts. Adapted with permission from ref. 125. Copyright 2019. Elsevier. | ||
In 2017, Hosono et al. reported a new heterogeneous catalyst based on Ru NPs supported on nanocages of 12CaO·7Al2O3 (Ru/HT-C12A7), which exhibits a high chemo-selectivity in the hydrogenation of heteroarenes.126 Ru/HT-C12A7 hydrogenates N- and O-heteroarenes with high yields in a solvent-free system. The high activity and stability were explained by the high percentage of strong basic sites of HT-C12A7 in close proximity to the Ru atoms, which promotes HCD (Fig. 18). In our opinion, this unique nanocage with many basic sites that facilitate the formation of H+ is a promising material for heterolytic H2 activation by supported MNPs.
 | ||
| Fig. 18 Pathway proposed by Hosono et al. for the selective hydrogenation of heteroarenes over Ru/HT-C12A7. Reprinted with permission from ref. 126. Copyright 2017. Elsevier. | ||
Heterolytic dissociation of H2 on CeO2-supported gold nanoparticles (Au/CeO2) was experimentally detected for the first time in 2010 by García, Corma et al. combining inelastic neutron scattering (INS) and FT-IR spectroscopy. Indeed, after treating Au/CeO2 with H2 at 150 °C, they observed that HCD takes place leading to a hydride coordinated to gold and a proton bonded to a ceria oxygen.75 More specifically, they detected a new band at 400–600 cm−1 by INS corresponding to bridging hydroxyl groups, which disappeared after exposure to CO2 or O2. In addition, by FT-IR they observed the chemisorption of hydrogen on gold through the formation of an Au–H band, which also disappeared after treatment with CO2 or O2. The assignments of these bands were confirmed by isotopic labelling experiments with D2 and 18O2. With these results, they provided experimental evidence about the “direct” heterolytic H2 cleavage over immobilized MNPs, which is in contrast with the hitherto commonly accepted reactivity of supported transition metal catalysts, homolytic cleavage followed by spillover.
HCD on core-gold/shell-ceria nanomaterials (Au@CeO2) was reported by Kaneda et al.127 Here, the great number of interface sites between the gold core (5 nm) and the ceria shell (4 nm thickness) maximizes heterolytic H2 dissociation (Fig. 19a–c), and explains the high selectivity of this material in semi-hydrogenation reactions of alkynes, which possess a higher electrophilicity than alkenes. A couple of experiments were carried out to confirm this ionic H2 cleavage: i) the first order dependence of the reaction rate on H2 pressure in the semihydrogenation of phenylacetylene (Fig. 19d), which points to the participation of the polar hydrogen species in the hydrogenation mechanism; ii) the strong inductive effect on the increment of the reaction rate when using an acetylene with an electron-acceptor group (CF3) (Fig. 19e). The same nanomaterial but supported on hydrotalcite (Au@CeO2/HT) exhibits also a high selectivity in the hydrogenation of unsaturated aldehydes and ketones towards the corresponding unsaturated alcohols.128 A similar selectivity due the above-mentioned H2 heterolytic cleavage was observed by the same group using the analogous silver material (Ag@CeO2) supported on ceria.129
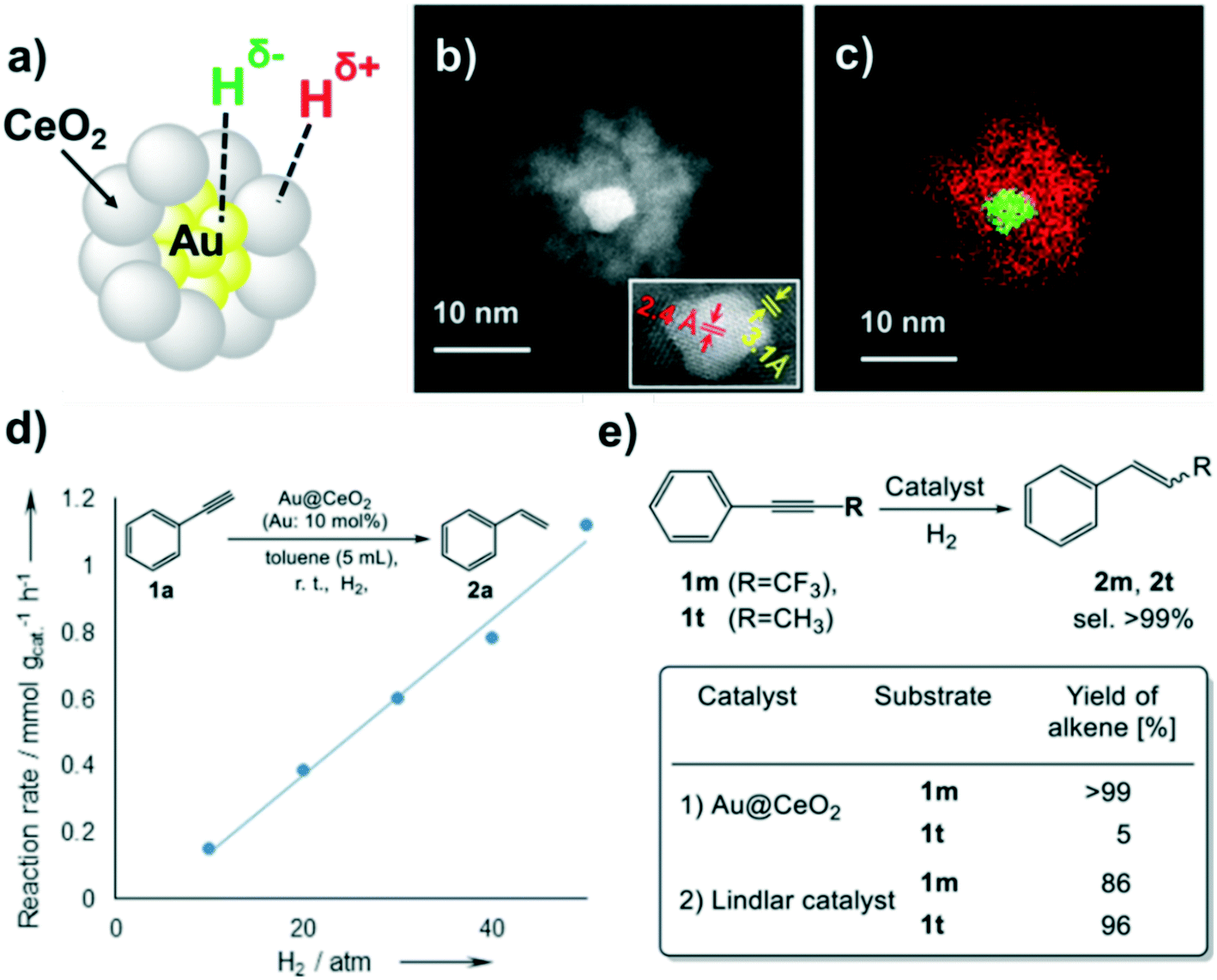 | ||
| Fig. 19 (a) Illustration of Au@CeO2. (b) HAADF-STEM image of Au@CeO2 (inset: HRTEM image showing the lattice spacing). (c) Elemental mapping of Au@CeO2 (Au: green, Ce: red). (d) Reaction rate vs. H2 pressure. (e) Hydrogenation of alkynes using Au@CeO2 or a Lindlar catalyst. Reprinted with permission from ref. 128. Copyright 2016. American Chemical Society. | ||
High levels of chemoselectivity in the hydrogenation of substituted nitroaromatics were also observed by Shimizu et al. by using Au/Al2O3 as a catalyst.130 More specifically, 2.5 nm Au NPs supported on Al2O3 selective hydrogenate –NO2 in the presence of other reducible groups such as vinyl or carbonyl groups. The reason for this high selectivity is the cooperative work between the metal and basic sites at the nanoparticle–support interface, which heterolytically cleave H2. The electrophilic Hδ+ and nucleophilic Hδ− species generated at the metal–support interfaces are transferred to the polar nitro group, forming hydroxylamine, which is subsequently hydrogenated by a similar transfer mechanism to yield the final aniline (Scheme 8).
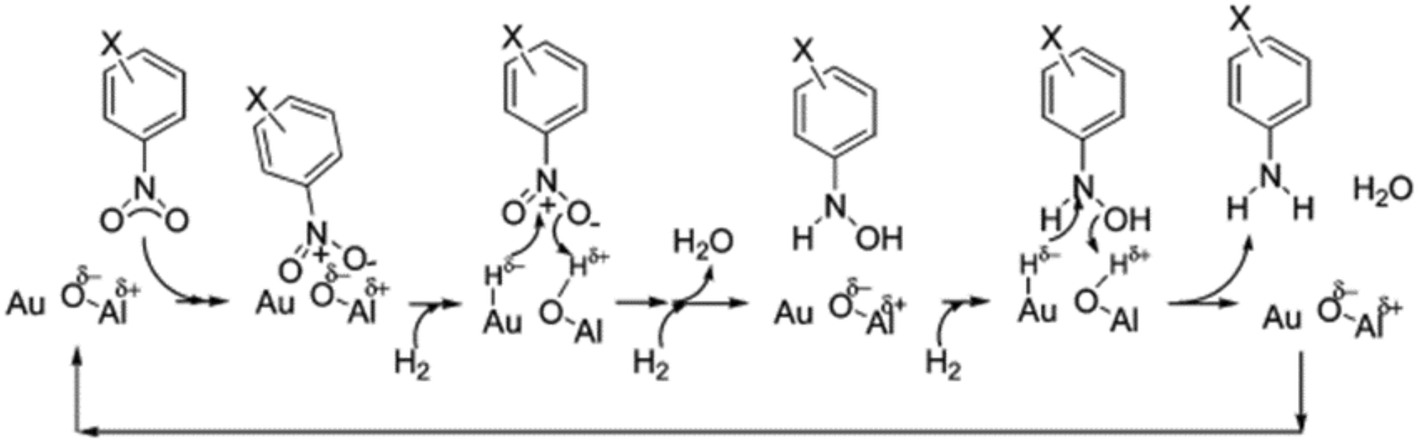 | ||
| Scheme 8 Mechanism proposed by Shimizu et al. for the selective hydrogenation of nitroaromatic compounds catalyzed by Au/Al2O3. Reprinted with permission from ref. 130. Copyright 2009. American Chemical Society. | ||
So far, we have observed that HCD by supported MNPs is highly dependent on the nature of the support. In addition, it can be also influenced by the solution surrounding the MNPs, as has been recently investigated by Dupont et al. More specifically, they observed that hydrogen activation proceeds, as expected, via heterolytic cleavage on a clean surface of Au NPs supported on γ-Al2O3 (Au/γ-Al2O3). However, after encapsulating these supported-Au nanoparticles in ionic liquids (IL-Au/γ-Al2O3), H2 activation goes through a homolytic dissociation.131 As a result, the two catalysts show totally different pathways in the selective hydrogenation of a wide scope of substrates.
Chandler et al.78 provided experimental and theoretical evidence about HCD by supported metal nanoparticles. More specifically, based on DFT calculations with Au nanorods (Au NRs) supported on TiO2, they suggested that Au–H− and O–H+ species are generated. H/D isotopic exchange experiments and kinetic studies supported the proposed mechanism. Apart from offering significant evidence for HCD on metal–support interfaces, this study also showed how water molecules block these interfaces for heterolytic H2 activation. By DFT calculations, Nie et al. investigated the influence of cluster size and metal–support interaction in the adsorption and dissociation of H2 over anatase TiO2 (101) and (001) surfaces.132 The Au atoms that are active in HCD are neutral in charge and located on the edges and corners of the nanoparticle. In particular, HCD occurs through the transition state O2−–H+–H−–Au on both lattices, (101) and (001) (Fig. 20a). Moreover, the existence of oxygen vacancies influences the stability of the adsorbed Au NPs and H2 molecules, but it does not affect significantly the energy barrier for H2 activation. Along the same lines are the studies of Larese et al. about the influence of the support on the hydrogenation of cinnamaldehyde to cinnamyl alcohol by using a series of Au/ZnO catalysts.133 They prepared three different types of Au/ZnO catalysts: Au/rod-tetrapod ZnO, Au/porous ZnO, and Au/ZnO-CP synthesized by co-precipitation. Au/ZnO-CP, which presents the highest dispersion and smallest NP size, showed the best results (100% selectivity to cinnamyl alcohol at 94.9% conversion) (Fig. 20b). Therefore, it can be concluded that the Au–ZnO interaction is a key parameter for achieving a highly selective catalyst for CO hydrogenation.
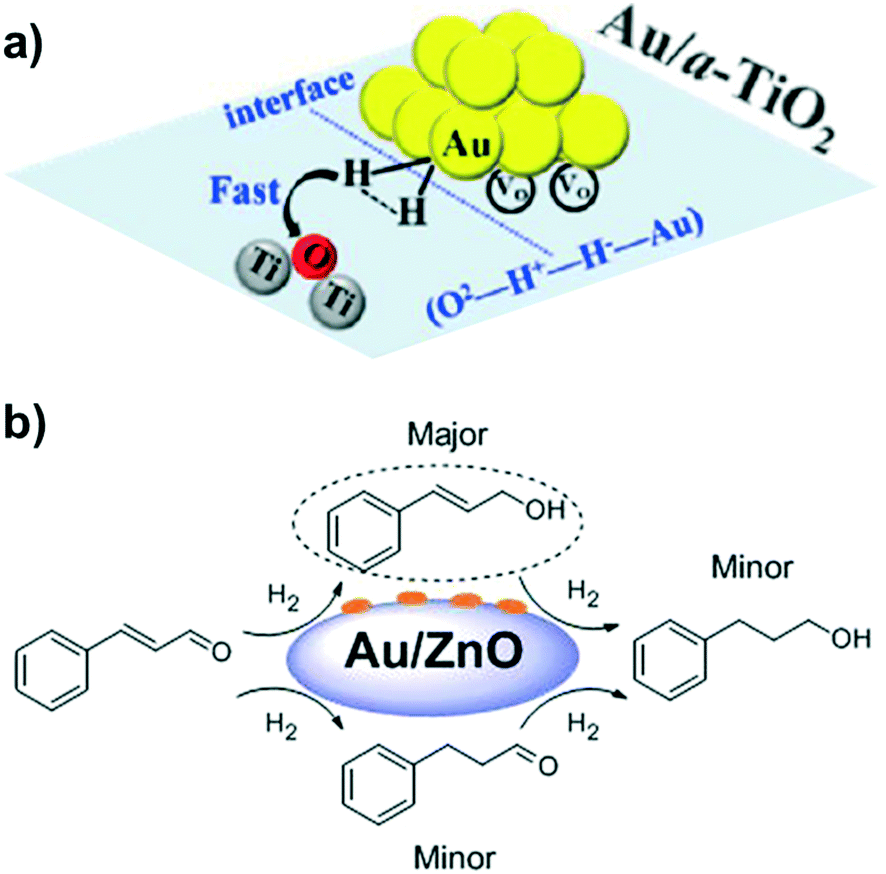 | ||
| Fig. 20 (a) Adsorption and dissociation of H2 at the Au/TiO2 interface. (b) Selective hydrogenation of cinnamaldehyde to the corresponding unsaturated alcohol over ZnO-supported Au NPs. Reprinted with permission from ref. 132 and 133, respectively. Copyrights 2018 and 2015. American Chemical Society. | ||
It is not always the support that provides the basic centres next to metal sites needed for heterolytic activation of H2. In 2013, Tomishige and co-workers reported silica supported Ir NPs partially covered with ReOx (Ir–ReOx/SiO2) as a highly active catalyst for the chemoselective hydrogenation of unsaturated aldehydes into the corresponding alcohols.134 Here, the basic sites are ReOx species that work together with the Ir atoms in HCD. Later, the same group observed a similar selectivity and activity in the hydrogenation of α,β-unsaturated ketones by using an Fe cation modified Ir/MgO catalyst [Ir/MgO+Fe(NO3)3], being one of the best examples of a heterogeneous catalyst that presents high selectivity (up to 90% at full conversion) towards the corresponding α,β-unsaturated alcohol.135 Here, the cation Fe2+ next to the anion O2− forms the actives sites for HCD. (Fig. 21a). Also, Tomishige et al. reported that the presence of Ren+ (Re3+ and Re4+) species on silica-supported RePd bimetallic catalysts (Re–Pd/SiO2) is responsible for HCD in the hydrogenation of stearic acid (Fig. 21b). On the other hand, the Pd(0) atoms facilitate the adsorption of stearic acid at the metal surface, and promote the reduction and dispersion of Ren+ species.136
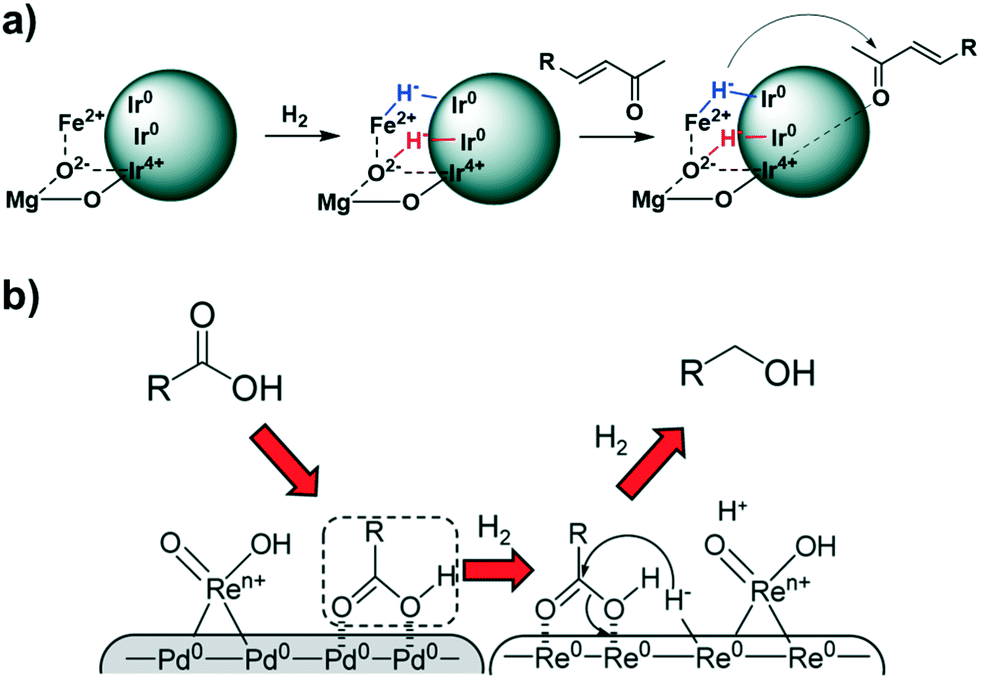 | ||
| Fig. 21 Proposed mechanism for the hydrogenation of carbonyl compounds over (a) Re–Pd/SiO2 and (b) Ir/MgO+Fe(NO3)3. Reprinted with permission from ref. 135 and 136, respectively. Copyrights 2017 and 2015. American Chemical Society. | ||
In addition to metal oxide supports (MgO, CeO2, Al2O3, TiO2 or ZnO), silica and carbon-based supports, other materials such as Fe3O4 or layered double hydroxides (LDHs) have been reported for heterolytic H2 activation by supported MNPs. For example, Pei, Qiao and co-workers reported gold nanoparticles on flower-like magnetite (flower-like Au/Fe3O4) as an efficient catalyst for selective hydrogenation of crotonaldehyde to the corresponding alcohol.137 This catalyst (Fig. 22a), prepared by deposition of Au on hematite and a subsequent reduction, presents a stronger metal–support interaction and a higher activity/selectivity than the comparable catalyst AuNPs supported on Fe3O4, e.g., with flower-like Au/Fe3O4 the selectivity is higher than 76% at full conversion. The proposed mechanism involves HCD on the interface sites Au–O–Fe and the interaction of crotonaldehyde with the hydroxyl groups on magnetite next to gold atoms. The proton of the hydroxyl group attacks the oxygen of the carbonyl, and the H-atom with the hydride character from HCD (Hδ−) is added to the carbon in the α position. Finally, the other hydrogen atom, partially positively charged (Hδ+), replaces the hydrogen of the hydroxyl group, closing the catalytic cycle (Fig. 22b). Due to its magnetic character, this catalyst presents exceptional recovery and recycling properties.
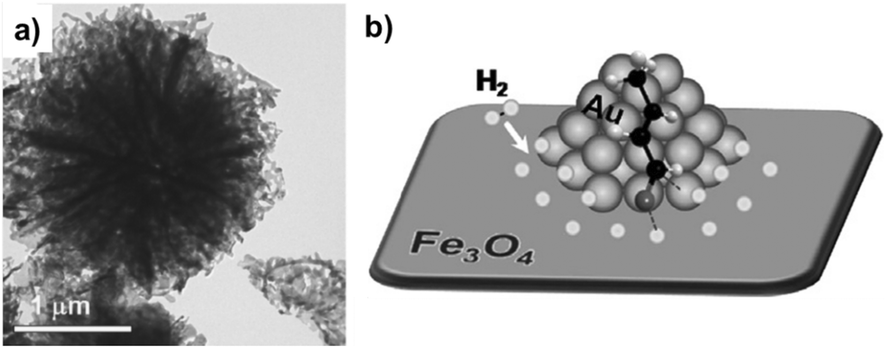 | ||
| Fig. 22 (a) TEM image of flower-like Au/Fe3O4. (b) Illustration of the selective hydrogenation of crotonaldehyde to crotyl alcohol at the interface of flower-like Au/Fe3O4 catalyst. O: gray, H: white, and C: black. Reprinted with permission from ref. 137. Copyright 2011. Elsevier. | ||
In 2011, Kaneda et al. described a cooperative effect between Au NPs and basic sites of layered double hydroxides (LDHs) for HCD. In particular, they selectively deoxygenate epoxides into alkenes (99% selectivity) by using a hydrotalcite-supported gold catalyst (Au/HT).138 The active species, Au–Hδ− and HT–Hδ+ (Scheme 9a), were identified by FT-IR after treatment of the catalyst with molecular hydrogen. The same catalytic system was used for the formation of terminal alkenes through the selective hydrogenolysis of allylic carbonates.139 Here, the proposed mechanism also involves the formation of Au–Hδ− and HT–Hδ+ species at the Au–HT interface. Once the active hydrogen species are formed, the allylic carbonate reacts with HT–Hδ+ to form the Au–allyl intermediate, which is finally attacked by the hydride (Au–Hδ−) to yield the terminal alkene (Scheme 9b).
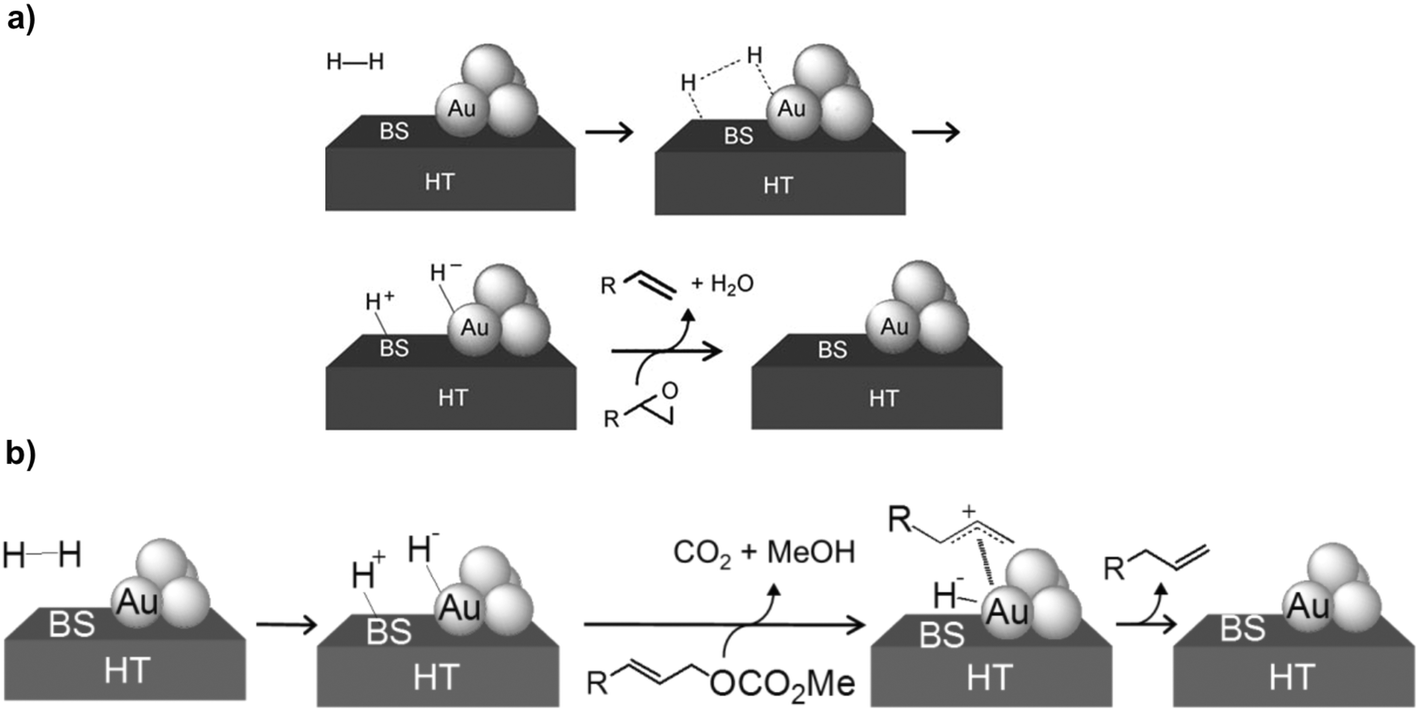 | ||
| Scheme 9 Heterolytic activation of H2 at the interface between gold atoms and basic sites (BS) of HT in (a) the deoxygenation of epoxides and (b) the hydrogenolysis of allylic carbonates. Reprinted with permission from ref. 138 and 139, respectively. Copyright 2011. Wiley. Copyright 2012. Royal Society of Chemistry. | ||
A more recent example concerning HCD at the interface of metal and basic sites of a layered double hydroxide was recently reported by Xiao and coworkers.140 The formation of polar hydrogen species on this Zn–Ti LDH supported PdAu catalyst (Zn–Ti LDHs) is responsible for the selective hydrogenation of phenylacetylene to styrene. The PdAu alloy nanoparticles are placed between the layers of LDHs, and strongly interact with them. This strong interaction facilitates the heterolytic hydrogenation, and makes Zn–Ti LDH an active and selective catalyst in the semihydrogenation of phenylacetylene (90% selectivity at full conversion), suppressing the hydrogenation of styrene after full conversion. In addition, this material was presented as a promising catalyst for industrial applications due to its high stability and effective recyclability.
To finish this section, it is worth mentioning that atomically dispersed catalysts on oxide supports are highly efficient in HCD. Here, the presence of metal–oxygen couples only, instead of metal–metal pairs necessary for homolytic dissociation of H2, makes the heterolytic dissociation the predominant way to cleave hydrogen. A clear example of that is the work of Zheng et al. concerning atomically dispersed palladium atoms on TiO2.141 Through a new photochemical procedure, stable dispersed palladium atoms on ethylene glycolate (EG)-stabilized ultrathin TiO2 nanosheets (Pd1/TiO2) were prepared. The large number of Pd–O interfaces of the system essentially activates dihydrogen in a heterolytic way, which is reflected in an exceptional stability and high catalytic activity in the hydrogenation of carbonyl groups. For example, the atomically dispersed Pd catalyst exhibits a TOF much higher than commercial Pd catalysts in the hydrogenation of aldehydes, without any decrease of the activity during the catalytic process.
3.2 Supported MNPs functionalized with organic ligands
Unifying the advantages of ligand-stabilized and supported MNPs for heterolytic activation of dihydrogen, supported MNPs functionalized with organic ligands are presented as high potential catalysts for selective hydrogenation reactions.142 Theoretically, they possess the stability and recyclability of supported MNPs together with the specific activity of metals influenced by coordinated ligands. At first, one might think that surface ligands would block the active sites of MNPs available for catalysis, but the ligands also modify the NP surface electronically and sterically, thereby changing the catalytic activity and selectivity, in the same way as they do in organometallic chemistry. Additionally, ligands can play an important role in the reaction mechanisms through metal–ligand interactions, like in the heterolytic activation of H2 in ligand-stabilized MNP catalysis discussed above (see section 2.2). In addition to the geometric and electronic effects induced by ligands to the metal surface143–145 and the metal–ligand cooperation,146 there is another possible effect based on substrate–ligand interactions.147,148 The possibility to control the selectivity of a given reaction by ligand–substrate interactions is reminiscent of biological catalysts, where substrates bind to enzymes through non-covalent interactions. To sum up, the functionalization of supported MNPs to improve their catalytic performance is a new strategy with high potential that has been scarcely explored.Since this novel approach is still in its infancy, we have only found one example in the literature about HCD by supported ligand-functionalized MNPs. In 2017, Rossi et al. presented supported-Au NPs functionalized with basic nitrogen compounds as a frustrated Lewis pair able to dissociate dihydrogen heterolytically.80 This HCD lead to a high activity in the selective hydrogenation of alkynes into cis-alkenes. After a screening study of a series of amines with different structures and pKa values, they observed that nitrogen-containing bases with two nitrogen atoms showed a higher activity than those with only one (Fig. 23a). Indeed, piperazine-functionalized Au NPs supported on silica (Au/SiO2) exhibited full conversion and 100% selectivity to styrene in the hydrogenation of phenylacetylene, giving the best result among all the N-containing bases studied. A large amount of the amine is necessary to have a higher activity and to suppress the hydrogenation of styrene to ethylbenzene. For instance, when the substrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amine:Au ratio is 100:1:1, the conversion is very low, only 13% of the alkene is observed. However, when the ratio is 100:100:1, there was a full conversion and a selectivity to styrene of 100%. Therefore, the amount of piperazine not only plays an important role in the activation of Au/SiO2, but also improves the selectivity of the catalyst, blocking the hydrogenation of alkenes into alkanes. Other parameters such as the solvent, temperature and support used also influence the activity, obtaining the best results in EtOH, at 100 °C with piperazine-functionalized Au/SiO2. Employing these conditions, numerous terminal and internal alkynes were selectively hydrogenated to cis-alkenes with excellent yields. Here, as well as in homogeneous catalysis, the ligands coordinated on the metal surface promote the HCD. Au/SiO2 without any amine on the surface is not active in the hydrogenation of alkynes (conversion <1%), but after functionalizing it with piperazine, it is able to activate hydrogen in a heterolytic way and hydrogenate selectively triple bonds. Fig. 23b shows the mechanism proposed by the authors, which starts with the adsorption of piperazine on the gold surface to form the active catalyst. Then, HCD takes place at the Au–N interface. Subsequently, the adsorbed alkyne reacts with the hydride of the pair Au–Hδ−, forming the vinyl intermediate. Finally, this intermediate is attacked by the N–Hδ+ ion, affording the alkene and regenerating the catalyst. To the best of our knowledge, this is one of the few examples of HCD over ligand-functionalized supported metal nanoparticles so far, but this versatile concept can be easily adapted to other catalytic systems with other metals and basic ligands. Thanks to that, we will be able to reach catalytic reactivities characteristic of molecular complexes with tunable heterogeneous systems, having the advantages of both catalysts: (i) homogeneous: activity and selectivity; and (ii) heterogeneous: stability and recyclability.
:amine:Au ratio is 100:1:1, the conversion is very low, only 13% of the alkene is observed. However, when the ratio is 100:100:1, there was a full conversion and a selectivity to styrene of 100%. Therefore, the amount of piperazine not only plays an important role in the activation of Au/SiO2, but also improves the selectivity of the catalyst, blocking the hydrogenation of alkenes into alkanes. Other parameters such as the solvent, temperature and support used also influence the activity, obtaining the best results in EtOH, at 100 °C with piperazine-functionalized Au/SiO2. Employing these conditions, numerous terminal and internal alkynes were selectively hydrogenated to cis-alkenes with excellent yields. Here, as well as in homogeneous catalysis, the ligands coordinated on the metal surface promote the HCD. Au/SiO2 without any amine on the surface is not active in the hydrogenation of alkynes (conversion <1%), but after functionalizing it with piperazine, it is able to activate hydrogen in a heterolytic way and hydrogenate selectively triple bonds. Fig. 23b shows the mechanism proposed by the authors, which starts with the adsorption of piperazine on the gold surface to form the active catalyst. Then, HCD takes place at the Au–N interface. Subsequently, the adsorbed alkyne reacts with the hydride of the pair Au–Hδ−, forming the vinyl intermediate. Finally, this intermediate is attacked by the N–Hδ+ ion, affording the alkene and regenerating the catalyst. To the best of our knowledge, this is one of the few examples of HCD over ligand-functionalized supported metal nanoparticles so far, but this versatile concept can be easily adapted to other catalytic systems with other metals and basic ligands. Thanks to that, we will be able to reach catalytic reactivities characteristic of molecular complexes with tunable heterogeneous systems, having the advantages of both catalysts: (i) homogeneous: activity and selectivity; and (ii) heterogeneous: stability and recyclability.
 | ||
| Fig. 23 (a) Top: Semihydrogenation of alkynes to alkenes using Au/SiO2 functionalized with nitrogen-containing bases. Bottom: Experimental reaction rates vs. computed activation energies for heterolytic H2 activation at N ligand–Au(111) interfaces. Inset: Illustration about heterolytic H2 cleavage over piperazine-functionalized AuNPs supported on SiO2. (b) Proposed mechanism via H2 cleavage. Reprinted with permission from ref. 80. Copyright 2017. American Chemical Society. | ||
4. Metal oxide NPs
The dissociation of H2 on “bulk” metal oxides through a HCD mechanism has a longstanding tradition (section 1.2). This transformation has been proposed to occur on non-reducible oxides with the concomitant formation of MH−/OH+ pairs. On the other hand, a homolytic cleavage that implies the generation of two O–H bonds has been suggested for reducible metal oxides (vide supra). Both mechanisms have long been under discussion, and indeed García-Melchor and López demonstrated that they are not excluding one another and may be interconnected (e.g. as in Fig. 3c).59 A good example of this debate is evidenced in an excellent theoretical study of Pacchioni et al.149 DFT calculations showed that an HCD is energetically preferred on extended surfaces (bulk) of ZrO2, for which Zr–H−/O–H+ pairs are generated without reduction of zirconia. Conversely, a homolytic H2 cleavage involving the formation of two O–H groups and reduction of Zr4+ to Zr3+ is the favoured mechanism for ZrO2 NPs of 1.5 nm in size. In the present section, we focus on the use of “isolated” metal oxide nanoparticles as a tool to achieve a HCD process.In this context, Knözinger and coworkers described a heterolytic hydrogen cleavage on MgO NPs of 5–8 nm in size.150 The MgO NPs, obtained by chemical vapour deposition, were exposed to H2, leading to a HCD and the subsequent formation of Mg–H− and O–H+ species on the NP surface. Under UV irradiation, the formed hydrides serve as the electron source to generate different types of colour centres at the surface and thus characterize electron-trapping sites. IR and EPR studies suggested that the surface MgO sites responsible for the heterolytic cleavage form colour centres because their respective Mg2+ species operate as a trap for electrons from hydrides.
HCD was also achieved by porous CeO2 nanorods that exhibit a high density of surface defects.151 The regulation of surface defects is key for the heterolytic splitting, since it allows the creation of oxygen vacancies. The elimination of one oxygen atom involves the reduction of two surface Ce cations (Ce4+ → Ce3+) and the production of one oxygen vacancy. These vacancies avoid the electronic interaction of Ce species with linked O atoms; that is, the removal of O atoms directly bonded to Ce prevents the generation of a classic Lewis acid–base adduct (Fig. 24).
 | ||
| Fig. 24 Design of FLP in CeO2(110) NPs. Top: Optimized structure of (a) ideal CeO2 NPs; (b) CeO2 NPs with one oxygen vacancy; (c) CeO2 NPs with two adjacent oxygen vacancies. Bottom: Electron-density isosurface of (d) ideal CeO2 NPs; (e) CeO2 NPs with one oxygen vacancy; (f) CeO2 NPs with two oxygen vacancies. Electron-density isosurfaces plotted at 0.01 e bohr−3. Colour bar shows the electrostatic potential scale. Reprinted with permission from ref. 151. Copyright 2017. Springer Nature. | ||
Conversely, the high concentration of surface defects leads to independent acidic and basic sites. Two neighbouring surface Ce3+ cations function as a Lewis acid, while an adjacent but unbonded lattice oxygen operates as a Lewis base, in such a way that the ensemble acts as a FLP that promotes the HCD (Fig. 24). Indeed, DFT calculations showed a low activation energy (0.17 eV) for the heterolytic dissociation of the H–H bond.
The CeO2 nanorods were employed as hydrogenation catalysts for alkenes and alkynes. Very high activities and excellent selectivities were observed in the hydrogenation of a series of substituted alkenes, while a decrease in selectivity was noted for alkynes (20 mg CeO2, 1 mmol substrate, toluene as a solvent, 6–30 bar, 100 °C, 12–24 h).
The HCD is a useful strategy to reduce CO2 and thus obtain high-added value products. For instance, Pérez-Ramírez, López et al. described a heterolytic H2 splitting mechanism in the hydrogenation of CO2 mediated by In2O3 NPs of ca. 8 nm in size.152 These NPs were found to be an efficient catalyst for the synthesis of methanol with high selectivity, for which kinetic experiments showed a lower apparent activation energy in comparison with that of the reverse water-gas shift reaction (CO2 + H2 → CO + H2O). Indeed, a deactivation of the catalyst and therefore an inhibition process by H2O was observed. This was supported by kinetic investigations, in which an apparent reaction order of −0.9 was determined for this molecule. On the other hand, the conversion of CO2 to methanol and thus the selectivity towards the latter is favored at higher H2 pressures, in line with the apparent reaction order with respect to this reactant (0.5).
Experimental and DFT studies demonstrated that the In2O3 (1 1 1) surface termination is the most exposed one and responsible for the process. The calculations showed that this termination is partially reduced by H2 through a homolytic mechanism, leading to the generation of OH groups, the concomitant H2O release and the formation of an oxygen vacancy surrounded by 3 indium atoms. Then, the methanol synthesis process occurs in several steps (Scheme 10): (1) a heterolytic splitting of H2 and CO2 activation takes place at this site surrounded by an In3O5 ensemble. The H2 molecule is activated as a Hδ+–Hδ− species by the binding of Hδ+ to one O atom and Hδ− to one indium atom, in such a way that the electron density of the latter is delocalized in the 3 indium atoms of In3O5. (2) A transfer of one H− to CO2 generates a formate (CHO2−) species, which forms a formic acid (CHOOH) intermediate by a subsequent transfer of H+. (3) Afterwards, a new H2 molecule is heterolytically transferred to this intermediate through the formation of CH2OOH by H− insertion and its following protonation to generate methanediol (CH2(OH)2). (4) A third H2 molecule is heterolytically adsorbed, which implies the dissociation of an OH group from the CH2(OH)2 intermediate to fill the oxygen vacancy and the subsequent formation of formaldehyde (CH2O). (5) Finally, this molecule interacts with the H− arising from this third HCD and evolves to methoxide (CH3O−), which is protonated and forms methanol (CH3OH).
 | ||
| Scheme 10 Hydrogenation of CO2 through a HCD catalyzed by In2O3 NPs to form methanol. | ||
The introduction of OH groups on the surface of In2O3 NPs leads to catalysts with a completely different behavior. The ability of nanostructured hydroxylated indium oxide (In2O3−x(OH)y) to reduce CO2 through a process involving HCD has been studied in depth by Singh, Ozin et al.153 In this case, we are dealing with a transformation that produces CO by the reverse water-gas shift reaction (CO2 + H2 → CO + H2O). The In2O3−x(OH)y surface presents Lewis acid (In) and Lewis base (InOH) sites. These sites are spatially separated due to the presence of oxygen vacancies, and thus may function as a surface FLP with the ability to heterolytically cleave H2 (Scheme 11).154 Then, the hydrogenated FLP adsorbs CO2, which is converted to CO and H2O as a co-product.
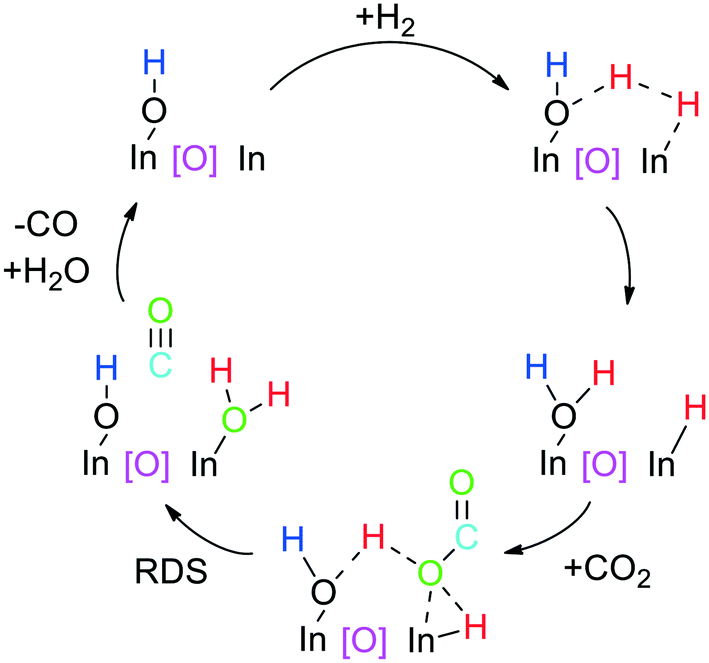 | ||
| Scheme 11 Proposed catalytic cycle for the reverse water-gas shift reaction mediated by nanostructured In2O3−x(OH)y. RDS: rate-determining step. | ||
DFT calculations showed that an increase in temperature hardly affects the process. However, the energy barriers corresponding to the HCD and CO2 adsorption decrease by 0.15 and 0.19 eV at 180 °C, respectively. This enhancement of activity is associated to a larger spatial separation between the Lewis acid (In) and the O of the Lewis base (InOH) of the FLP.155 Importantly, the nanocatalysts based on In2O3−x(OH)y are even more active under light, as photoexcitation reduces the activation barrier by about 20 kJ mol−1.156 Indeed, experimental and theoretical studies demonstrated that the photoactivation promotes the generation of electrons and holes trapped at the Lewis base (electron trapping) and Lewis acid (hole trapping) sites, respectively (Fig. 25). As a result, the Lewis acid and Lewis base active sites located in the vicinity of oxygen vacancies become more acidic and more basic, and thus the surface FLP shows an increased activity.156,157
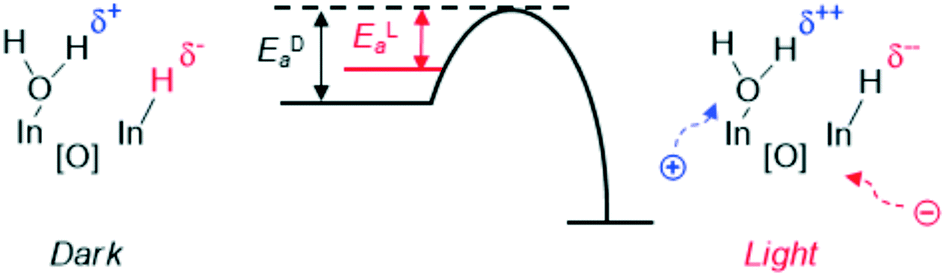 | ||
| Fig. 25 Representation of the difference in the experimental activation energy for the reverse water-gas shift reaction in the dark (black) and in the light (red) mediated by nanostructured In2O3−x(OH)y. Adapted with permission from ref. 153. Copyright 2016. American Chemical Society. | ||
In addition, Ozin and coworkers reported the photocatalytic hydrogenation of CO2 catalyzed by cuprous oxide (Cu2O) nanocubes of ca. 500 nm in size.158 A process involving HCD also led to the reverse water-gas shift reaction. This inexpensive semiconductor shows great potential as a photocatalyst, but exhibits a high tendency towards irreversible disproportionation (Cu2O → Cu + CuO) due to its redox instability, which hinders its use as a photocatalyst. Interestingly, the synthesis provided Cu2O nanocubes in which the surface contains OH groups, oxygen vacancies and Cu sites with different oxidation states (0, I and II). This makes the redox disproportionation reversible, which improves the stability of Cu2O nanocubes and thus allows the CO2 reduction through HCD.
The Cu2O nanocubes exhibit high activity and selectivity in the reverse water-gas shift reaction under very mild conditions (25 °C, intense light of 50 suns of blue, green and red visible light, CO2/H2 = 5, total pressure ca. 4 bar). A CO production of 70.3 mmol gcat−1 h−1 was obtained, which was increased up to 88 mmol gcat−1 h−1 by removal of coproduced water with dehydrated zeolite LTA-3A. In addition, 139.6 mmol gcat−1 h−1 was achieved by a long-term stability experiment (60 h).
The authors suggested that H2 is heterolytically cleaved by an FLP consisting of Cu2O and Cu2(OH)3Cl (Cu(I)2−xCu(II)xO1+x/2−y/2(OH)y). In this FLP, OH acts as a Lewis base and Cu(II) operates as a Lewis acid, leading to a HCD as follows: Cu(II)⋯OH + H2 → Cu(II)H⋯OH2. In addition, the presence of Cu sites with different oxidation states favours electron delocalization (Cu(II)H⋯OH2 ↔ Cu(I)H⋯OH2). Finally, a catalytic cycle was proposed on the basis of in situ DRIFTS studies (Scheme 12), while different control experiments demonstrated that light is a prerequisite for the process.
 | ||
| Scheme 12 Proposed mechanism for photocatalytic hydrogenation of CO2 mediated by Cu2O nanocubes. | ||
An HCD occurs on the surface of Cu2O nanocubes (3 and 4), where electron delocalization between Cu(I) and Cu(II) sites also takes place (4). Then, H2O is removed, thus generating an oxygen vacancy (5). This vacancy facilitates the adsorption of CO2 as a carbonate-like species (1), which eventually converts to CO (2).
5. Conclusions and outlook
For a long time, selective hydrogenation of polar functional groups (CO, CN or CS) has been mostly carried out in the laboratory using homogeneous catalysts, in which H2 is heterolytically activated by a metal ligand cooperation. On the other hand, MNPs and supported metal catalysts normally cleave hydrogen in a homolytic way, showing in general lower selectivities than homogeneous catalysts. However, with the advent of new and controlled protocols for MNP synthesis, and the possibility to functionalize them with organic molecules, the use of MNPs for HCD is gaining attention from the scientific community.
This article collects recent examples of well-defined metal nanocatalysts for the heterolytic activation of H2 by metal–ligand or metal–support cooperation. Here, the catalytic H2 dissociation is promoted by the presence of both surface ligands or support basic sites. Even more exciting is the functionalization of supported MNPs with organic molecules to facilitate the heterolytic H2 cleavage. In this way, we can enhance the selectivity of almost any supported-MNPs. These novel catalytic systems combine the advantages of ligand-stabilized MNPs (selectivity) and supported MNPs (stability and recyclability). In the majority of examples the proof for HCD is absent or only circumstantial, but above we have collected several neat examples that report spectroscopic proof and/or DFT calculations proving the feasibility of HCD. Yet, more joint theoretical–experimental studies about the ligand–metal surface interactions are needed in order to design more efficient and selective catalysts that operate through HCD.
In addition to functionalized supported MNPs, the future directions for research in this area could be oriented to the deposition of single atoms on MNPs as the simplest way to modify their reactivity. A recent example about this was reported by Pérez-Ramírez and López, where chalcogen-modified copper catalysts showed an increased selectivity in the electrochemical reduction of CO2 to formic acid.159 In their study, sulfur-, selenium and tellurium active centers are present on the MNP surface as adatoms, and participate in the hydrogenation mechanism playing a dual role: (i) transferring hydrides to the CO2 molecule and (ii) suppressing the formation of CO. In a similar way, chalcogen-modified MNPs could perform HCD by a cooperative work between chalcogen adatoms and surface metal atoms, just like in homogeneous systems.160
Another interesting prospect in this research area is the use of FLPs as modifying ligands, such as phosphinoboranes or aminophosphines. The main advantage of the functionalization of MNPs with FLPs is to reverse the addition of H2. In a typical HCD reaction (metal–basic centre), the hydride coordinates to the metal and it will not be “hydridic”, but in TMFLP systems the addition is inverted; this means that the transition metal accepts the proton and the Lewis acid (i.e. borane) the hydride, giving rise to a highly “hydridic” hydride which might be of interest for the course of the catalytic reaction.84 Over the last few years, FLPs have been an important source of inspiration in chemistry, and we expect that metal nanoparticle catalysis could also take advantage of this.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Dr Luis M. Martínez-Prieto thanks Instituto de Tecnología Química (ITQ), Consejo Superior de Investigaciones Científicas (CSIC), Universitat Politècnica de València (UPV) for the facilities, and Severo Ochoa excellence programme (SEV-2016-0683), “Juan de la Cierva” programme (IJCI-2016-27966) and Primero Proyectos de Investigación (PAID-06-18; SP20180088) for financial support.Notes and references
- R. G. Dackers and J. Halpern, Kinetics of the homogenous reaction between cupric acetate and molecular hydrogen in aqueous solution, Can. J. Chem., 1954, 32, 969–978 CrossRef.
- J. Halpern, Homogeneous Catalytic Activation of Molecular Hydrogen by Metal Ions and Complexes, J. Phys. Chem., 1959, 63, 398–403 CrossRef CAS.
- F. H. Jardine, J. A. Osborn, G. Wilkinson and J. F. Young, Homogeneous catalytic hydrogenation and hydroformylation of acetylenic compounds, Chem. Ind., 1965, 560 ( J. Chem. Soc. A , 1966 , 1711 ) CAS.
- T. P. Dang and H. B. Kagan, Asymmetric syntheses of hydratropic acid and amino-acids by homogeneous catalytic hydrogenation, J. Chem. Soc. D, 1971, 481 RSC.
- J. Halpern, Homogeneous catalytic hydrogenation: A retrospective account, J. Organomet. Chem., 1980, 200, 133–144 CrossRef CAS.
- L. Pauling, The modern theory of valency, J. Chem. Soc., 1948, 1461–1467 RSC.
- I. Dance, Misconception of reductive elimination of H2, in the context of the mechanism of nitrogenase, Dalton Trans., 2015, 44, 9027–9037 RSC.
- P. W. N. M. van Leeuwen, M. A. Zuideveld, B. H. G. Swennenhuis, Z. Freixa, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz and A. L. Spek, Alcoholysis of Acylpalladium(II) Complexes Relevant to the Alternating Copolymerization of Ethene and Carbon Monoxide and the Alkoxycarbonylation of Alkenes: the Importance of Cis-Coordinating Phosphines, J. Am. Chem. Soc., 2003, 125, 5523–5540 CrossRef CAS.
- R. M. Bullock and J.-S. Song, Ionic Hydrogenations of Hindered Olefins at Low Temperature. Hydride Transfer Reactions of Transition Metal Hydrides, J. Am. Chem. Soc., 1994, 116, 8602–8612 CrossRef CAS.
- D. L. DuBois and D. E. Berning, Hydricity of transition-metal hydrides and its role in CO2 reduction, Appl. Organomet. Chem., 2000, 14, 860–862 CrossRef CAS.
- C. J. Webb, A review of catalyst-enhanced magnesium hydride as a hydrogen storage material, J. Phys. Chem. Solids, 2015, 84, 96–106 CrossRef CAS.
- V. A. Yartys, et al., Magnesium Based Materials for Hydrogen Based Energy Storage: Past, Present and Future, Int. J. Hydrogen Energy, 2019, 44, 7809–7859 CrossRef CAS.
- C. Copéret, D. P. Estes, K. Larmier and K. Searles, Isolated Surface Hydrides: Formation, Structure, and Reactivity, Chem. Rev., 2016, 116, 8463–8505 CrossRef.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Thermodynamic Hydricity of Transition Metal Hydrides, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, Hydricity of Transition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS , and references therein.
- M. S. Jeletic, E. B. Hulley, M. L. Helm, M. T. Mock, A. M. Appel, E. S. Wiedner and J. C. Linehan, Understanding the Relationship Between Kinetics and Thermodynamics in CO2 Hydrogenation Catalysis, ACS Catal., 2017, 7, 6008–6017 CrossRef CAS.
- P. S. Hallman, D. Evans, J. A. Osborn and G. Wilkinson, Selective catalytic homogeneous hydrogenation of terminal olefins using tris(triphenylphosphine)hydridochlororuthenium(II); hydrogen transfer in exchange and isomerisation reactions of olefins, Chem. Commun., 1967, 305–306 RSC.
- R. Noyori, M. Ohta, Y. Hsiao, M. Kitamura, T. Ohta and H. Takaya, Asymmetric synthesis of isoquinoline alkaloids by homogeneous catalysis, J. Am. Chem. Soc., 1986, 108, 7117–7119 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- Y. Blum, D. Czarkie, Y. Rahami and Y. Shvo, (Cyclopentadienone)ruthenium carbonyl complexes - a new class of homogeneous hydrogenation catalysts, Organometallics, 1985, 4, 1459–1461 CrossRef CAS.
- M. S. Chinn and D. M. Heinekey, Dihydrogen complexes of ruthenium. 2. Kinetic and thermodynamic considerations affecting product distribution, J. Am. Chem. Soc., 1990, 112, 5166–5175 CrossRef CAS.
- M. D. Fryzuk and P. A. MacNeil, Amides of rhodium and iridium stabilized as hybrid multidentate ligands, Organometallics, 1983, 2, 682–684 CrossRef CAS.
- M. D. Fryzuk, P. A. MacNeil and S. J. Rettig, Ancillary ligand involvement in the activation of dihydrogen by iridium (III) complexes, Organometallics, 1985, 4, 1145–1147 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Direct synthesis of amides from alcohols and amines with liberation of H2, Science, 2007, 317, 790–792 CrossRef CAS.
- S. Werkmeister, J. Neumann, K. Junge and M. Beller, Pincer-Type Complexes for Catalytic (De)Hydrogenation and Transfer (De)Hydrogenation Reactions: Recent Progress, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef CAS.
- J. R. Khusnutdinova and D. Milstein, Metal–ligand cooperation, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS.
- O. Eisenstein and R. H. Crabtree, Outer sphere hydrogenation catalysis, New J. Chem., 2013, 37, 21–27 RSC.
- B. Askevold, H. W. Roesky and S. Schneider, Learning from the Neighbours: Improving Homogeneous Catalysts with Functional Ligands Motivated by Heterogeneous and Biocatalysis, ChemCatChem, 2012, 4, 307–320 CrossRef CAS.
- F. Gloaguen and T. B. Rauchfuss, Small molecule mimics of hydrogenases: hydrides and redox, Chem. Soc. Rev., 2009, 38, 100–108 RSC.
- H. Gruezmacher, Cooperating Ligands in Catalysis, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef.
- S. Kuwata and T. Ikariya, Unsymmetrical Pincer-Type Ruthenium Complex Containing β-Protic Pyrazole and N-Heterocyclic Carbene Arms: Comparison of Brønsted Acidity of NH Groups in Second Coordination Sphere, Chem. – Eur. J., 2014, 20, 9539–9542 CrossRef.
- N. E. Smith, W. H. Bernskoetter and N. Hazari, The Role of Proton Shuttles in the Reversible Activation of Hydrogen via Metal-Ligand Cooperation, J. Am. Chem. Soc., 2019, 141, 17350–17360 CrossRef CAS.
- D. B. Grotjahn, Bifunctional organometallic catalysts involving proton transfer or hydrogen bonding, Chem. – Eur. J., 2005, 11, 7146–7153 CrossRef CAS.
- P. W. N. M. van Leeuwen, I. Cano and Z. Freixa, Secondary Phosphine Oxides: Bifunctional Ligands in Catalysis, ChemCatChem, 2020, 12, 3982–3994 CrossRef CAS.
- L. V. A. Hale and N. K. Szymczak, Hydrogen Transfer Catalysis beyond the Primary Coordination Sphere, ACS Catal., 2018, 8, 6446–6461 CrossRef CAS.
- N. Onishi, G. Laurenczy, M. Beller and Y. Himeda, Recent progress for reversible homogeneous catalytic hydrogen storage in formic acid and in methanol, Coord. Chem. Rev., 2018, 373, 317–332 CrossRef CAS.
- M. Ito and T. Ikariya, Catalytic hydrogenation of polar organic functionalities based on Ru-mediated heterolytic dihydrogen cleavage, Chem. Commun., 2007, 5134–5142 RSC.
- A. J. B. Robertson, The early history of catalysis, Platinum Met. Rev., 1975, 19, 64–69 CAS.
- R. Hoffmann, Döbereiner's Lighter, Am. Sci., 1998, 86, 326 Search PubMed.
- I. Horiuti and M. Polanyi, Exchange Reactions of Hydrogen on Metallic Catalysts, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC.
- R. Masel, Principles of Adsorption and Reaction on Solid Surfaces, Wiley Interscience, New York, 1996, p. 244 Search PubMed.
- A. Farkas, L. Farkas and E. K. Rideal, Experiments on Heavy Hydrogen IV— The Hydrogenation and Exchange Reaction of Ethylene with Heavy Hydrogen, Proc. R. Soc. London, Ser. A, 1934, 146, 630–639 CAS.
- F. Zaera, Key unanswered questions about the mechanism of olefin hydrogenation catalysis by transition-metal surfaces: a surface-science perspective, Phys. Chem. Chem. Phys., 2013, 15, 11988–12003 RSC.
- Y. Dong and F. Zaera, Kinetics of hydrogen adsorption during catalytic reactions on transition metal surfaces, Catal. Sci. Technol., 2017, 7, 5354–5364 RSC.
- J. Haubrich and K. Wandelt, Adsorption of Unsaturated and Multifunctional Molecules: Bonding and Reactivity, in Surface and Interface Science: Solid-Gas Interfaces I, ed. K. Wandelt, 1st edn, Wiley-VCH Verlag GmbH, 2015, ch. 40, pp. 529–627 Search PubMed.
- J.-Y. Saillard and R. Hoffmann, C–H and H–H activation in transition-metal complexes and on surfaces, J. Am. Chem. Soc., 1984, 106, 2006–2026 CrossRef CAS.
- M. K. Oudenhuijzen, J. A. van Bokhoven, J. T. Miller, D. E. Ramaker and D. C. Koningsberger, Three-Site Model for Hydrogen Adsorption on Supported Platinum Particles: Influence of Support Ionicity and Particle Size on the Hydrogen Coverage, J. Am. Chem. Soc., 2005, 127(5), 1530–1540 CrossRef CAS.
- D. A. Dowden, N. Mackenzie and B. M. W. Trapnell, The catalysis of H2—D2 exchange by oxides, Proc. R. Soc. London, Ser. A, 1956, 237(1209), 245–254 CAS.
- R. L. Burwell, Jr. and A. B. Littlewood, Interaction of deuterium with olefins and alkanes on chromium oxide gel, J. Am. Chem. Soc., 1956, 78, 4170–4171 CrossRef.
- R. L. Burwell, Jr., A. B. Littlewood, M. Cardew, G. Pass and C. T. H. Stoddart, Reactions between Hydrocarbons and Deuterium on Chromium Oxide Gel. I, J. Am. Chem. Soc., 1960, 82, 6272–6280 CrossRef.
- R. L. Burwell, Jr., J. F. Read, K. C. Taylor and G. L. Haller, Adsorptive and Catalytic Properties of Chromia, Z. Phys. Chem., 1969, 64, 18–25 CrossRef.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis, Understanding the Art, Kluwer AP, Dordrecht, The Netherlands, 2004, pp. 48–50 Search PubMed.
- A. L. Dent and R. J. Kokes, Hydrogenation of Ethylene by Zinc Oxide, J. Phys. Chem., 1969, 73, 3772–3790 CrossRef CAS.
- (a) C. Liu, J. Camacho-Bunquin, M. Ferrandon, A. Savara, H. Sohn, D. Yang, D. M. Kaphan, R. R. Langeslay, P. A. Ignacio-de Leon and S. Liu, et al., Development of Activity–Descriptor Relationships for Supported Metal Ion Hydrogenation Catalysts on Silica, Polyhedron, 2018, 152, 73–83 CrossRef CAS; (b) C. Plascencia, L. A. Curtiss and C. Liu, Hydrogen Activation by Silica Supported Metal Ion Catalysts: Catalytic Properties of Metals and Performance of DFT Functionals, J. Phys. Chem. A, 2019, 123, 171–186 CrossRef CAS.
- H. F. Wilson and A. S. Barnard, Thermodynamics of Hydrogen Adsorption and Incorporation at the ZnO(10
![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 0) Surface, J. Phys. Chem. C, 2015, 119, 26560–26565 CrossRef CAS.
0) Surface, J. Phys. Chem. C, 2015, 119, 26560–26565 CrossRef CAS. - H. A. Aleksandrov, G. N. Vayssilov and N. Roesch, Heterolytic dissociation and recombination of H2 over Zn,H-ZSM-5 zeolites—A density functional model study, J. Mol. Catal. A: Chem., 2006, 256, 149–155 CrossRef CAS.
- E. A. Pydko and R. A. van Santen, Activation of Light Alkanes over Zinc Species Stabilized in ZSM-5 Zeolite: A Comprehensive DFT Study, J. Phys. Chem. C, 2007, 111, 2643–2655 CrossRef.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J. M. Basset, Heterolytic Splitting of H2 and CH4 on γ-Alumina as a Structural Probe for Defect Sites, J. Phys. Chem. B, 2006, 110, 23944–23950 CrossRef CAS.
- M. García-Melchor and N. López, Homolytic Products from Heterolytic Paths in H2 Dissociation on Metal Oxides: The Example of CeO2, J. Phys. Chem. C, 2014, 118, 10921–10926 CrossRef.
- B. Wei, F. Tielens and M. Calatayud, Understanding the Role of Rutile TiO2 Surface Orientation on Molecular Hydrogen Activation, Nanomaterials, 2019, 9, 1199 CrossRef CAS.
- R. Prins, Hydrogen Spillover. Facts and Fiction, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS.
- M. Boudart, M. A. Vannice and J. E. Benson, Adlineation, portholes and spillover, Z. Phys. Chem., 1969, 64, 171–177 CrossRef CAS.
- S. Khoobiar, Migration of Hydrogen Atoms on Platinum—Alumina Catalysts from Particle to Neighboring Particles, J. Phys. Chem., 1964, 68, 411–412 CrossRef CAS.
- T. Huizinga and R. Prins, Behavior of Ti3+ Centers In the Low- and High-Temperature Reduction of Pt/TiO2, Studied by ESR, J. Phys. Chem., 1981, 85, 2156–2158 CrossRef CAS.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Catalyst Support Effects on Hydrogen Spillover, Nature, 2017, 541, 68–71 CrossRef CAS.
- C. Spreafico, W. Karim, Y. Ekinci, J. A. van Bokhoven and J. VandeVondele, Hydrogen Adsorption on Nanosized Platinum and Dynamics of Spillover onto Alumina and Titania, J. Phys. Chem. C, 2017, 121, 17862–17872 CrossRef CAS.
- G. Kennedy, G. Melaet, H. L. Han and G. A. Somorjai, et al., In situ spectroscopic investigation into the active sites for crotonaldehyde hydrogenation at the Pt nanoparticle−Co3O4 interface, ACS Catal., 2016, 6, 7140–7147 CrossRef CAS.
- X. Yang, Y. Mueanngern, Q. A. Baker and L. R. Baker, Crotonaldehyde hydrogenation on platinum–titanium oxide and platinum–cerium oxide catalysts: selective C=O bond hydrogen requires platinum sites beyond the oxide–metal interface, Catal. Sci. Technol., 2016, 6, 6824–6835 RSC.
- I. Cano and P. W. N. M. van Leeuwen, Selective Hydrogenation of Aldehydes and Ketones, in Recent Advances in Nanoparticle Catalysis, ed. P. W. N. M. van Leeuwen and C. Claver, Springer Nature, Switzerland, 2020, Molecular Catalysis 1, pp. 345–405 Search PubMed.
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, Mechanistic Insights on the Hydrogenation of α,β-Unsaturated Ketones and Aldehydes to Unsaturated Alcohols over Metal Catalysts, ACS Catal., 2012, 2, 671–683 CrossRef CAS.
- R. M. Bullock, Catalytic Ionic Hydrogenations, Chem. – Eur. J., 2004, 10, 2366–2374 CrossRef CAS.
- J. S. M. Samec, J.-E. Backvall, P. G. Andersson and P. Brandt, Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- References citing ref. 61.
- T. Fujitani, I. Nakamura, T. Akita, M. Okumura and M. Haruta, Hydrogen Dissociation by Gold Clusters, Angew. Chem., Int. Ed., 2009, 48, 9515–9518 CrossRef CAS.
- R. Juárez, S. F. Parker, P. Concepción, A. Corma and H. García, Heterolytic and heterotopic dissociation of hydrogen on ceria-supported gold nanoparticles. Combined inelastic neutron scattering and FT-IR spectroscopic study on the nature and reactivity of surface hydrogen species, Chem. Sci., 2010, 1, 731–738 RSC.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms, Chem. Rev., 2020, 120, 683–733 CrossRef CAS.
- N. C. Nelson and J. Szanyi, Heterolytic Hydrogen Activation: Understanding Support Effects in Water−Gas Shift, Hydrodeoxygenation, and CO Oxidation Catalysis, ACS Catal., 2020, 10, 5663–5671 CrossRef CAS.
- T. Whittaker, K. B. S. Kumar, C. Peterson, M. N. Pollock, L. C. Grabow and B. D. Chandler, H2 Oxidation over Supported Au Nanoparticle Catalysts: Evidence for Heterolytic H2 Activation at the Metal-Support Interface, J. Am. Chem. Soc., 2018, 140(48), 16469–16487 CrossRef CAS.
- M. A. Ortuño and N. López, Reaction mechanisms at the homogeneous–heterogeneous frontier: insights from first principles studies on ligand-decorated metal nanoparticles, Catal. Sci. Technol., 2019, 9, 5173–5185 RSC.
- J. L. Fiorio, N. López and L. M. Rossi, Gold−Ligand-Catalyzed Selective Hydrogenation of Alkynes into cis-Alkenes via H2 Heterolytic Activation by Frustrated Lewis Pairs, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS.
- E. Rafter, T. Gutmann, F. Löw, G. Buntkowsky, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, Secondary phosphineoxides as pre-ligands for nanoparticle stabilization, Catal. Sci. Technol., 2013, 3, 595–599 RSC.
- N. Almora-Barrios, I. Cano, P. W. N. M. van Leeuwen and N. López, Concerted chemoselective hydrogenation of acrolein on secondary phosphine oxide decorated gold nanoparticles, ACS Catal., 2017, 7, 3949–3954 CrossRef CAS.
- D. W. Stephan and G. Erker, Frustrated Lewis Pair Chemistry: Development and Perspectives, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS.
- R. M. Bullock and G. M. Chambers, Frustration across the periodic table: heterolytic cleavage of dihydrogen by metal complexes, Philos. Trans. R. Soc., A, 2017, 375, 20170002 CrossRef.
- S. Akabori, S. Sakurai, Y. Izumi and Y. Fujii, An Asymmetric Catalyst, Nature, 1956, 178, 323–324 CrossRef CAS.
- Y. Izumi, Methods of asymmetric synthesis. Enantioselective catalytic hydrogenation, Angew. Chem., Int. Ed. Engl., 1971, 10, 871–881 CrossRef CAS.
- Y. Orito, S. Imai and S. Niwa, Asymmetric Hydrogenation of Methyl Pyruvate using Pt-C Catalyst modified with Cinchonidine, Nippon Kagaku Kaishi, 1979, 8, 1118–1120 CrossRef ; Chem. Abstr., 1979:592483, CAN 91:192483.
-
(a) F. Meemken and A. Baiker, Recent Progress in Heterogeneous Asymmetric Hydrogenation of CO and CC Bonds on Supported Noble Metal Catalysts, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS;
(b) M. Studer, H.-U. Blaser and C. Exner, Enantioselective Hydrogenation using Heterogeneous Modified Catalysts: An Update, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS;
(c) T. Bürgi and A. Baiker, Heterogeneous Enantioselective Hydrogenation over Cinchona Alkaloid Modified Platinum: Mechanistic Insights into a Complex Reaction, Acc. Chem. Res., 2004, 37, 909–917 CrossRef;
(d) H.-U. Blaser and M. Studer, Cinchona-Modified Platinum Catalysts: From Ligand Acceleration to Technical Processes, Acc. Chem. Res., 2007, 40, 1348–1356 CrossRef CAS;
(e) F. Zaera, Chiral Modification of Solid Surfaces: A Molecular View, J. Phys. Chem. C, 2008, 112, 16196–16203 CrossRef CAS.
- (a) N. Maeda, K. Hungerbuehler and A. Baiker, Asymmetric Hydrogenation on Chirally Modified Pt: Origin of Hydrogen in the N–H–O Interaction between Cinchonidine and Ketone, J. Am. Chem. Soc., 2011, 133, 19567–19569 CrossRef CAS; (b) A. Vargas, G. Santarossa and A. Baiker, Exchange of Hydrogen between a Platinum Surface and a Tertiary Amine: An ab Initio Molecular Dynamics Investigation, J. Phys. Chem. C, 2011, 115, 1969–1977 CrossRef CAS.
- M. von Arx, N. Dummer, D. J. Willock, S. H. Taylor, R. P. K. Wells, P. B. Wells and G. J. Hutchings, Observation of High Enantioselectivity for the Gas Phase Hydrogenation of Methyl Pyruvate using Supported Pt Catalysts Pre-modified with Cinchonidine, Chem. Commun., 2003, 1926–1927 RSC.
- K. R. Hahn and A. Baiker, Comparative Density Functional Theory Study of Cinchonidine and Hydrogen Coadsorption on Platinum Group Metals (Rh, Ir, Pd, and Pt) and Its Implications on Surface Chiral Site Formation, J. Phys. Chem. C, 2020, 124, 18020–18030 CrossRef CAS.
- (a) G. C. Welchand and D. W. Stephan, Facile Heterolytic Cleavage of Dihydrogen by Phosphines and Boranes, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef; (b) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Rapid Intramolecular Heterolytic Dihydrogen Activation by a Four-membered Heterocyclic Phosphane-borane Adduct, Chem. Commun., 2007, 5072–5074 RSC.
- D. W. Stephan, Frustrated Lewis Pairs: From Concept to Catalysis, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS.
- S. R. Flynn and D. F. Wass, Transition metal frustrated Lewis pairs, ACS Catal., 2013, 3, 2574–2581 CrossRef CAS.
- Y. Ma, S. Zhang, C.-R. Chang, Z.-Q. Huang, J. C. Ho and Y. Qu, Semi-solid and solid frustrated Lewis pair catalysts, Chem. Soc. Rev., 2018, 47, 5541–5553 RSC.
- (a) Y. Ren, Y. Wang, X. Li, Z. Zhang and Q. Chi, Selective hydrogenation of quinolines into 1,2,3,4-tetrahydroquinolines over a nitrogen-doped carbon-supported Pd catalyst, New J. Chem., 2018, 42, 16694–16702 RSC; (b) S. Wang, P. Zhou, L. Jiang, Z. Zhang, K. Deng, Y. Zhang, Y. Zhao, J. Li, S. Bottle and H. Zhu, Selective deoxygenation of carbonyl groups at room temperature and atmospheric hydrogen pressure over nitrogen-doped carbon supported Pd catalyst, J. Catal., 2018, 368, 207–216 CrossRef CAS.
- H. Berke, Conceptual approach to the reactivity of dihydrogen, ChemPhysChem, 2010, 11, 1837–1849 CAS.
- R. A. Sánchez-Delgado, N. Machalaba and N. Ng-a-qui, Hydrogenation of quinoline by ruthenium nanoparticles immobilized on poly(4-vinylpyridine), Catal. Commun., 2007, 8, 2115–2118 CrossRef.
- C. A. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, Mechanism of Asymmetric Hydrogenation of Ketones Catalyzed by BINAP/1,2-Diamine–Ruthenium(II) Complexes, J. Am. Chem. Soc., 2003, 125, 13490–13503 CrossRef CAS.
- J. M. Asensio, D. Bouzouita, P. W. N. M. van Leeuwen and B. Chaudret, σ-H–H, σ-C–H, and σ-Si–H Bond Activation Catalyzed by Metal Nanoparticles, Chem. Rev., 2020, 120, 1042–1084 CrossRef CAS.
- M. Fang, N. Machalaba and R. A. Sánchez-Delgado, Hydrogenation of arenes and N-heteroaromatic compounds over ruthenium nanoparticles on poly(4-vinylpyridine): a versatile catalyst operating by a substrate-dependent dual site mechanism, Dalton Trans., 2011, 40, 10621–10632 RSC.
- I. Cano, A. M. Chapman, A. Urakawa and P. W. N. M. van Leeuwen, Air-Stable Gold Nanoparticles Ligated by Secondary Phosphine Oxides for the Chemoselective Hydrogenation of Aldehydes: Crucial Role of the Ligand, J. Am. Chem. Soc., 2014, 136, 2520–2528 CrossRef CAS.
- I. Cano, M. A. Huertos, A. M. Chapman, G. Buntkowsky, T. Gutmann, P. B. Groszewicz and P. W. N. M. van Leeuwen, Air-Stable Gold Nanoparticles Ligated by Secondary Phosphine Oxides as Catalyst for the Chemoselective Hydrogenation of Substituted Aldehydes: a Remarkable Ligand Effect, J. Am. Chem. Soc., 2015, 137, 7718–7727 CrossRef CAS.
- I. Cano, L. M. Martínez-Prieto, P. F. Fazzini, Y. Coppel, B. Chaudret and P. W. N. M. van Leeuwen, Characterization of secondary phosphine oxide ligands on the surface of iridium nanoparticles, Phys. Chem. Chem. Phys., 2017, 19, 21655–21662 RSC.
- I. Cano, L. M. Martínez-Prieto, B. Chaudret and P. W. N. M. van Leeuwen, Iridium versus Iridium: Nanocluster and Monometallic Catalysts Carrying the Same Ligand Behave Differently, Chem. – Eur. J., 2017, 23, 1444–1450 CrossRef CAS.
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Atomically Precise Au25(SR)18 Nanoparticles as Catalysts for the Selective Hydrogenation of α,β-Unsaturated Ketones and Aldehydes, Angew. Chem., Int. Ed., 2010, 49, 1295–1298 CrossRef CAS.
- R. Ouyang and D.-e. Jiang, Understanding Selective Hydrogenation of α,β-Unsaturated Ketones to Unsaturated Alcohols on the Au25(SR)18 Cluster, ACS Catal., 2015, 5, 6624–6629 CrossRef CAS.
- G. Li, C. Zeng and R. Jin, Chemoselective Hydrogenation of Nitrobenzaldehyde to Nitrobenzyl Alcohol with Unsupported Au Nanorod Catalysts in Water, J. Phys. Chem. C, 2015, 119, 11143–11147 CrossRef CAS.
- G. Li, D.-e. Jiang, S. Kumar, Y. Chen and R. Jin, Size Dependence of Atomically Precise Gold Nanoclusters in Chemoselective Hydrogenation and Active Site Structure, ACS Catal., 2014, 4, 2463–2469 CrossRef CAS.
- G. Li, H. Abroshan, Y. Chen, R. Jin and H. J. Kim, Experimental and Mechanistic Understanding of Aldehyde Hydrogenation Using Au25 Nanoclusters with Lewis Acids: Unique Sites for Catalytic Reactions, J. Am. Chem. Soc., 2015, 137, 14295–14304 CrossRef CAS.
- X. Chai, T. Li, M. Chen, R. Jin, W. Ding and Y. Zhu, Suppressing the active site-blocking impact of ligands of Ni6(SR)12 clusters with the assistance of NH3 on catalytic hydrogenation of nitriles, Nanoscale, 2018, 10, 19375–19382 RSC.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala, H. Häkkinen, B. K. Teo and N. Zheng, Atomically Precise, Thiolated Copper–Hydride Nanoclusters as Single-Site Hydrogenation Catalysts for Ketones in Mild Conditions, ACS Nano, 2019, 13, 5975–5986 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Heterogeneous Catalysts for the One-Pot Synthesis of Chemicals and Fine Chemicals, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS.
- A. Corma and H. Garcia, Crossing the borders between homogeneous and heterogeneous catalysis: developing recoverable and reusable catalytic systems, Top. Catal., 2008, 48, 8–31 CrossRef CAS.
- M. Haruta, When Gold Is Not Noble: Catalysis by Nanoparticles, Chem. Rec., 2003, 3, 75–87 CrossRef CAS.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Domínguez, A. Leyva-Pérez and A. Corma, Small gold clusters formed in solution give reaction turnover numbers of 10(7) at room temperature, Science, 2012, 338, 1452–1455 CrossRef CAS.
- (a) D. Astruc, L. Feng and J. R. Aranzaes, Nanoparticles as Recyclable Catalysts: The Frontier between Homogeneous and Heterogeneous Catalysis, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS; (b) A. V. Biradar, A. A. Biradar and T. Asefa, Silica–Dendrimer Core–Shell Microspheres with Encapsulated Ultrasmall Palladium Nanoparticles: Efficient and Easily Recyclable Heterogeneous Nanocatalysts, Langmuir, 2011, 27, 14408–14418 CrossRef CAS; (c) Y.-W. Lee, S.-B. Han, A.-R. Ko, H.-S. Kim and K.-W. Park, Glycerol-mediated synthesis of Pd nanostructures with dominant {111} facets for enhanced electrocatalytic activity, Catal. Commun., 2011, 15, 137–140 CrossRef CAS; (d) N. R. Shiju and V. V. Guilants, Recent developments in catalysis using nanostructured materials, Appl. Catal., A, 2009, 356, 1–17 CrossRef CAS.
- R. Rahi, M. Fang, A. Ahmed and R. A. Sanchez-Delgado, Hydrogenation of quinolines, alkenes, and biodiesel by palladium nanoparticles supported on magnesium oxide, Dalton Trans., 2012, 41, 14490–14497 RSC.
- M. Fang and R. A. Sánchez-Delgado, Ruthenium nanoparticles supported on magnesium oxide: A versatile and recyclable dual-site catalyst for hydrogenation of mono- and poly-cyclic arenes, N-heteroaromatics, and S-heteroaromatics, J. Catal., 2014, 311, 357–368 CrossRef CAS.
- A. Sánchez, M. Fang, A. Ahmed and R. A. Sánchez-Delgado, Hydrogenation of arenes, N-heteroaromatic compounds, and alkenes catalyzed by rhodium nanoparticles supported on magnesium oxide, Appl. Catal., A, 2014, 477, 117–124 CrossRef.
- F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M.-M. Pohl, J. Radnik, A.-E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes, Nat. Chem., 2013, 5, 537–543 CrossRef CAS.
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Nanoscale Fe2O3-Based Catalysts for Selective Hydrogenation of Nitroarenes to Anilines, Science, 2013, 342, 1073–1076 CrossRef CAS.
- D. Formenti, C. Topf, K. Junge, F. Ragaini and M. Beller, Fe2O3/NGr@C- and Co–Co3O4/NGr@C-catalysed hydrogenation of nitroarenes under mild conditions, Catal. Sci. Technol., 2016, 6, 4473–4477 RSC.
- J. L. Fiorio, R. V. Gonçalves, E. Teixeira-Neto, M. A. Ortuño, N. López and L. M. Rossi, Accessing Frustrated Lewis Pair Chemistry through Robust Gold@N-Doped Carbon for Selective Hydrogenation of Alkynes, ACS Catal., 2018, 8, 3516–3524 CrossRef CAS.
- L. M. Martínez-Prieto, M. Puche, C. Cerezo-Navarrete and B. Chaudret, Uniform Ru nanoparticles on N-doped graphene for selective hydrogenation of fatty acids to alcohols, J. Catal., 2019, 377, 429–437 CrossRef.
- T.-N. Ye, J. Li, M. Kitano and H. Hosono, Unique nanocages of 12CaO·7Al2O3 boost heterolytic hydrogen activation and selective hydrogenation of heteroarenes over ruthenium catalyst, Green Chem., 2017, 19, 749–756 RSC.
- T. Mitsudome, M. Yamamoto, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, One-step Synthesis of Core-Gold/Shell-Ceria Nanomaterial and Its Catalysis for Highly Selective Semihydrogenation of Alkynes, J. Am. Chem. Soc., 2015, 137, 13452–13455 CrossRef CAS.
- T. Urayama, T. Mitsudome, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green, Multi-Gram One-Step Synthesis of Core–Shell Nanocomposites in Water and Their Catalytic Application to Chemoselective Hydrogenations, Chem. – Eur. J., 2016, 22, 17962–17966 CrossRef CAS.
- T. Mitsudome, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Core–Shell AgNP@CeO2 Nanocomposite Catalyst for Highly Chemoselective Reductions of Unsaturated Aldehydes, Chem. – Eur. J., 2013, 19, 5255–5258 CrossRef CAS.
- K.-i. Shimizu, Y. Miyamoto, T. Kawasaki, T. Tanji, Y. Tai and A. Satsuma, Chemoselective Hydrogenation of Nitroaromatics by Supported Gold Catalysts: Mechanistic Reasons of Size- and Support-Dependent Activity and Selectivity, J. Phys. Chem. C, 2009, 113, 17803–17810 CrossRef CAS.
- L. Luza, C. P. Rambor, A. Gual, J. Alves Fernandes, D. Eberhardt and J. Dupont, Revealing Hydrogenation Reaction Pathways on Naked Gold Nanoparticles, ACS Catal., 2017, 7, 2791–2799 CrossRef CAS.
- W. Wan, X. Nie, M. J. Janik, C. Song and X. Guo, Adsorption, Dissociation, and Spillover of Hydrogen over Au/TiO2 Catalysts: The Effects of Cluster Size and Metal-Support Interaction from DFT, J. Phys. Chem. C, 2018, 122, 17895–17916 CrossRef CAS.
- H. Chen, D. A. Cullen and J. Z. Larese, Highly Efficient Selective Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol over Gold Supported on Zinc Oxide Materials, J. Phys. Chem. C, 2015, 119, 28885–28894 CrossRef CAS.
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, Rapid synthesis of unsaturated alcohols under mild conditions by highly selective hydrogenation, Chem. Commun., 2013, 49, 7034–7036 RSC.
- M. Tamura, D. Yonezawa, T. Oshino, Y. Nakagawa and K. Tomishige, In Situ Formed Fe Cation Modified Ir/MgO Catalyst for Selective Hydrogenation of Unsaturated Carbonyl Compounds, ACS Catal., 2017, 7, 5103–5111 CrossRef CAS.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Characterization of Re–Pd/SiO2 Catalysts for Hydrogenation of Stearic Acid, ACS Catal., 2015, 5, 7034–7047 CrossRef CAS.
- Y. Zhu, L. Tian, Z. Jiang, Y. Pei, S. Xie, M. Qiao and K. Fan, Heteroepitaxial growth of gold on flowerlike magnetite: An efficacious and magnetically recyclable catalyst for chemoselective hydrogenation of crotonaldehyde to crotyl alcohol, J. Catal., 2011, 281, 106–118 CrossRef CAS.
- A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Selective Deoxygenation of Epoxides to Alkenes with Molecular Hydrogen Using a Hydrotalcite-Supported Gold Catalyst: A Concerted Effect between Gold Nanoparticles and Basic Sites on a Support, Angew. Chem., Int. Ed., 2011, 50, 2986–2989 CrossRef CAS.
- A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Unique catalysis of gold nanoparticles in the chemoselective hydrogenolysis with H2: cooperative effect between small gold nanoparticles and a basic support, Chem. Commun., 2012, 48, 6723–6725 RSC.
- Y. Shen, K. Yin, C. An and Z. Xiao, Design of a difunctional Zn-Ti LDHs supported PdAu catalyst for selective hydrogenation of phenylacetylene, Appl. Surf. Sci., 2018, 456, 1–6 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Photochemical route for synthesizing atomically dispersed palladium catalysts, Science, 2016, 352, 797–800 CrossRef CAS.
- S. Kunz, Supported, Ligand-Functionalized Nanoparticles: An Attempt to Rationalize the Application and Potential of Ligands in Heterogeneous Catalysis, Top. Catal., 2016, 59, 1671–1685 CrossRef CAS.
- L. M. Martínez-Prieto and B. Chaudret, Organometallic Ruthenium Nanoparticles: Synthesis, Surface Chemistry, and Insights into Ligand Coordination, Acc. Chem. Res., 2018, 51, 376–384 CrossRef.
- L. M. Martínez-Prieto and P. W. N. M. van Leeuwen, Ligand Effects in Ruthenium Nanoparticle Catalysis, in Recent Advances in Nanoparticle Catalysis, ed. P. W. N. M. van Leeuwen and C. Claver, Springer International Publishing, Cham, 2020, pp. 407–448 Search PubMed.
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang, Z. Zhao, Z. Zhou, G. Fu and N. Zheng, Interfacial electronic effects control the reaction selectivity of platinum catalysts, Nat. Mater., 2016, 15, 564–569 CrossRef CAS.
- I. Cano, M. J. L. Tschan, L. M. Martinez-Prieto, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, Enantioselective hydrogenation of ketones by iridium nanoparticles ligated with chiral secondary phosphine oxides, Catal. Sci. Technol., 2016, 6, 3758–3766 RSC.
- C. A. Schoenbaum, D. K. Schwartz and J. W. Medlin, Controlling the Surface Environment of Heterogeneous Catalysts Using Self-Assembled Monolayers, Acc. Chem. Res., 2014, 47, 1438–1445 CrossRef CAS.
- K. V. S. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Asymmetric nanocatalysis: N-heterocyclic carbenes as chiral modifiers of Fe3O4/Pd nanoparticles, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef CAS.
- A. R. Puigdollers, S. Tosoni and G. Pacchioni, Turning a Nonreducible into a Reducible Oxide via Nanostructuring: Opposite Behavior of Bulk ZrO2 and ZrO2 Nanoparticles Toward H2 Adsorption, J. Phys. Chem. C, 2016, 120, 15329–15337 CrossRef.
- (a) O. Diwald, P. Hofmann and E. Knözinger, H2 chemisorption and consecutive UV stimulated surface reactions on nanostructured MgO, Phys. Chem. Chem. Phys., 1999, 1, 713–721 RSC; (b) M. Sterrer, T. Berger, S. Stankic, O. Diwald and E. Knözinger, Spectroscopic Properties of Trapped Electrons on the Surface of MgO Nanoparticles, ChemPhysChem, 2004, 5, 1695–1703 CrossRef CAS.
- S. Zhang, Z.-Q. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C.-R. Chang and Y. Qu, Solid frustrated-Lewis-pair catalysts constructed by regulations on surface defects of porous nanorods of CeO2, Nat. Commun., 2017, 8, 15266 CrossRef CAS.
- M. S. Frei, M. Capdevila-Cortada, R. García-Muelas, C. Mondelli, N. López, J. A. Stewart, D. Curulla Ferré and J. Pérez-Ramírez, Mechanism and microkinetics of methanol synthesis via CO2 hydrogenation on indium oxide, J. Catal., 2018, 361, 313–321 CrossRef CAS.
- K. K. Ghuman, L. B. Hoch, T. E. Wood, C. Mims, C. V. Singh and G. A. Ozin, Surface Analogues of Molecular Frustrated Lewis Pairs in Heterogeneous CO2 Hydrogenation Catalysis, ACS Catal., 2016, 6, 5764–5770 CrossRef CAS , and references therein.
- K. K. Ghuman, T. E. Wood, L. B. Hoch, C. A. Mims, G. A. Ozin and C. V. Singh, Illuminating CO2 reduction on frustrated Lewis pair surfaces: investigating the role of surface hydroxides and oxygen vacancies on nanocrystalline In2O3−x(OH)y, Phys. Chem. Chem. Phys., 2015, 17, 14623–14635 RSC.
- M. Ghoussoub, S. Yadav, K. K. Ghuman, G. A. Ozin and C. V. Singh, Metadynamics-Biased ab Initio Molecular Dynamics Study of Heterogeneous CO2 Reduction via Surface Frustrated Lewis Pairs, ACS Catal., 2016, 6, 7109–7117 CrossRef CAS.
- K. K. Ghuman, L. B. Hoch, P. Szymanski, J. Y. Y. Loh, N. P. Kherani, M. A. El-Sayed, G. A. Ozin and C. V. Singh, Photoexcited Surface Frustrated Lewis Pairs for Heterogeneous Photocatalytic CO2 Reduction, J. Am. Chem. Soc., 2016, 138, 1206–1214 CrossRef CAS.
- L. He, T. E. Wood, B. Wu, Y. Dong, L. B. Hoch, L. Reyes, D. Wang, C. Kübel, C. Qian, J. Jia, K. Liao, P. G. O'Brien, A. Sandhel, J. Y. Y. Loh, P. Szymanski, N. P. Kherani, T. C. Sum, C. A. Mims and G. A. Ozin, Spatial Separation of Charge Carriers in In2O3–x(OH)y Nanocrystal Superstructures for Enhanced Gas-Phase Photocatalytic Activity, ACS Nano, 2016, 10, 5578–5586 CrossRef CAS.
- L. Wan, Q. Zhou, X. Wang, T. E. Wood, L. Wang, P. N. Duchesne, J. Guo, X. Yan, M. Xia, Y. F. Li, A. A. Jelle, U. Ulmer, J. Jia, T. Li, W. Sun and G. A. Ozin, Cu2O nanocubes with mixed oxidation-state facets for (photo)catalytic hydrogenation of carbon dioxide, Nat. Catal., 2019, 2, 889–898 CrossRef CAS.
- R. García-Muelas, F. Dattila, T. Shinagawa, A. J. Martín, J. Pérez-Ramírez and N. López, J. Phys. Chem. Lett., 2018, 9, 7153–7159 CrossRef.
- Z. K. Sweeney, J. L. Polse, R. G. Bergman and R. A. Andersen, Organometallics, 1999, 18, 5502–5510 CrossRef CAS.
Footnote |
| † Dedicated to the memory of Prof. Paul C. J. Kamer, who passed away on November 19, 2020. |
| This journal is © The Royal Society of Chemistry 2021 |