
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective catalytic reduction of NO with NH3 over Cu-exchanged CHA, GME, and AFX zeolites: a density functional theory study†
Pei
Zhao
 *ab,
Bundet
Boekfa
c,
Ken-ichi
Shimizu
bd,
Masaru
Ogura
be and
Masahiro
Ehara
*ab
*ab,
Bundet
Boekfa
c,
Ken-ichi
Shimizu
bd,
Masaru
Ogura
be and
Masahiro
Ehara
*ab
aResearch Center for Computational Science, Institute for Molecular Science, Okazaki, 444-8585, Japan
bElement Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Kyoto 615-8245, Japan. E-mail: pei@ims.ac.jp; ehara@ims.ac.jp
cDepartment of Chemistry, Faculty of Liberal Arts and Science, Kasetsart University, Kamphaengsaen Campus, Nakhonpathom 73140, Thailand
dInstitute for Catalysis, Hokkaido University, Sapporo 001-0021, Japan
eInstitute of Industrial Science, The University of Tokyo, Tokyo 153-8505, Japan
First published on 18th January 2021
Abstract
Density functional theory calculations have been applied to study the selective catalytic reduction of NO by NH3 over the Cu-exchanged zeolites with cha, gme, and aft cages. The CuI, CuII, and [CuII(OH)]+ ions are considered as the active sites to study both the reduction and oxidation processes during the catalytic cycle. In the case of the reduction process, the NH2NO formation at the [CuII(OH)]+ site possesses high barriers in the three frameworks, while the lower barriers are found at the CuII site. Importantly, it is found that the barriers are largely decreased at the solvated [CuII(NH3)4]2+ site for the cha and aft frameworks, while the barrier is only slightly decreased for the gme cage. As for the oxidation, the nitrate formation has similar reaction barriers at the CuI site of the three frameworks, which are lower than the following nitrite formation. In particular, the smallest gme cage possesses the highest barrier for the nitrite formation. Calculations on the O2 activation by the NH3-solvated Cu dimer revealed that the cha and aft cages have better performance than the gme cage, and the much smaller adsorption energy of O2 in the gme cage indicates the unfavorable O2 insertion. Therefore, the selectivity caused by the cage size is identified during the reaction process, and the cha and aft cages are more favorable.
Introduction
Zeolites are inorganic microporous crystalline materials constructed of corner-sharing SiO4 and AlO4 tetrahedra, or other TO4 sites (oxygen atoms bridge the tetrahedral atoms).1 The generation of a negative charge, when Si4+ is substituted by Al3+ in the oxide framework, dictates the need for a balancing cation. This exchange capacity can lead to Cu-exchanged zeolites that are active for the selective catalytic reduction (SCR) of NO to N2 by ammonia (NH3-SCR).2,3 The small-pore Cu-exchanged CHA zeolites, in particular SSZ-13 and SAPO-34, have attracted much attention due to their high activity and hydrothermal stability.4–8 The simple structure of the Cu-CHA zeolite is also important for the fundamental SCR investigations. As for the standard SCR reaction (4NH3 + 4NO + O2 → 4N2 + 6H2O) on Cu-CHA, lots of mechanistic studies have revealed that (1) the reaction follows a redox reaction mechanism by the Cu-ion cycle between +2 and +1 oxidation states and (2) the isolated Cu ions (CuII and [CuII(OH)]+) act as the active centers.9–17The small-pore CHA zeolites have the largest openings of 8-membered rings (8r), which excels in applications dealing with smaller molecules and ions.7 Reactants and products penetrate through micropores of zeolites, which can control the selectivity accordingly. Meanwhile, the formed cages or larger intersections in the zeolite framework can influence the formed intermediates during the reaction process, which eventually results in differences in the catalytic activity. As shown in Fig. 1a, the double six-membered-ring (d6r) layers are stacked in an ABC sequence, forming the cha cage interconnected by 8r windows.18 The small-pore zeolite AFX is formed by two types of cages, gme and aft, which have the stackings of the same d6r layers in the BC and ABCB sequences, respectively (Fig. 1b).19 The cha, gme, and aft cages share the same 4r, d6r, and 8r pores, but with a different cavity size (aft > cha > gme). Recently, the Cu-exchanged AFX zeolites have been proven to be effective catalysts for NH3-SCR reactions, which are also hydrothermally stable.20–23 In the case of NH3-SCR on a Cu-CHA zeolite, it has been shown that the active Cu ions are located in the 6r and 8r of the framework,24–26 however, the Cu ions are lifted out from their original positions into the large cavities upon the adsorption of NH3, NO, or other ligands.27,28 Therefore, the SCR reaction actually takes place in the large cavities, especially for the NH3-solvated Cu ions.16,29 It has been reported that the Cu ions are detached from the zeolite structure into the pore under the exposure of NH3 below 250 °C.16,30–32 CuI can be solvated by two NH3 ligands forming a linear [CuI(NH3)2]+ complex, and CuII can form the fourfold coordinated complex of [CuII(NH3)4]2+.33,34 Meanwhile, at low Cu loadings, the observed quadratic dependence on the Cu loading indicates the reaction involves two Cu sites at low temperatures, which has been attributed to the activation of oxygen by paring of [CuI(NH3)2]+.16,29,35 So far, the related studies are mainly focused on the CHA zeolites, and the role of cavity in different small-pore zeolites remains to be studied for the NH3-SCR reaction.
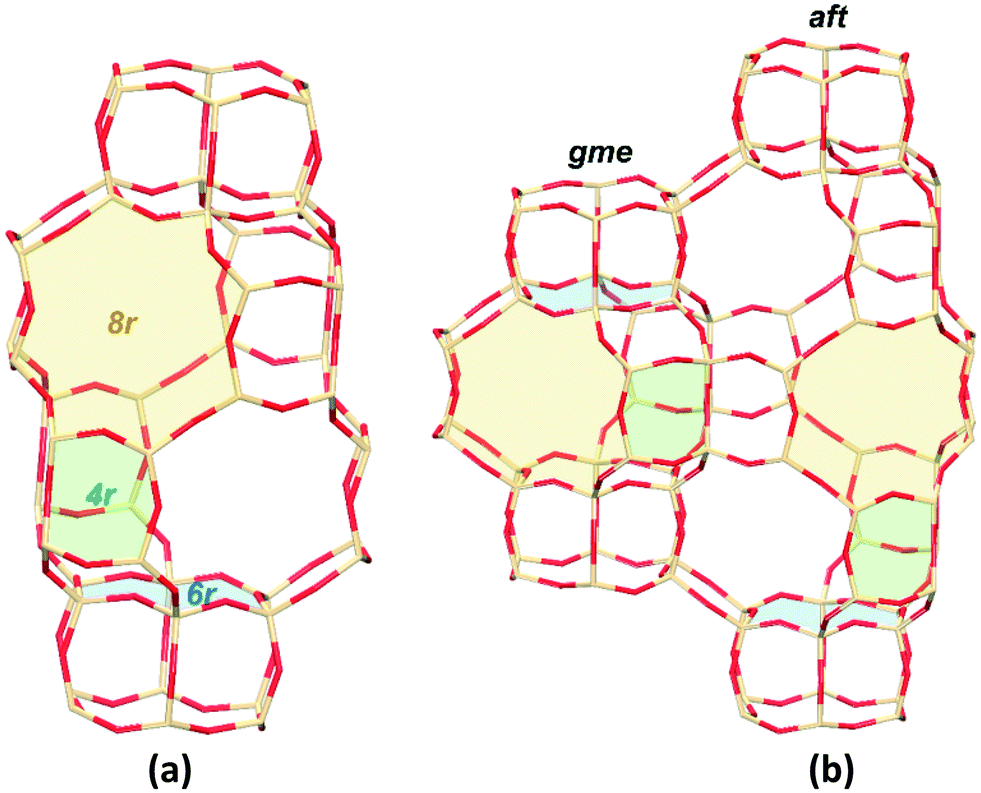 | ||
| Fig. 1 Schematic representations of CHA (a) and AFX (b) topologies. The green, blue, and yellow filled areas stand for the 4-, 6-, and 8-membered rings, respectively (color code: Si tan; O red). | ||
In this paper, we aim to unravel the impact of the cavity size on the SCR performance in the cha, gme, and aft cages by density functional theory (DFT) calculations. The Cu ions in zeolites show different ammonia SCR activities depending on the Cu environmental structure (cage and local Al number), which has led to numerous studies experimentally. Nevertheless, the activity evaluation on the Cu ions in various cages is inaccessible due to multiple environments of active Cu ions, making theoretical calculations an ideal method for the related studies. As summarized in Scheme S1,† the oxidation and reduction half-cycles of the SCR reaction are both studied, and they take place at NO + O2 and NH3 + NO atmospheres, respectively. The CuI ion or the CuII/[CuII(OH)]+ species acts as the active sites, which anchor at zeolite frameworks. The small gme cages in the large pore GME zeolites are considered to be the active Cu-ion sites for SCR reaction, which also makes our study useful for the GME zeolites. The ammonia-solvated Cu cations are also taken into account to study the NH2NO formation and the O2 activation. This work will provide a further understanding of the catalytic activity of the Cu-exchanged zeolites with cha, gme, and aft cages.
Computational details
Computational models
The 42T, 36T, and 54T clusters are applied for the cha, gme, and aft frameworks in our calculations (T represents the tetrahedrally coordinated Si or Al atoms), as shown in Fig. S1.† The computational models with one/two aluminum sites are both considered in our calculations to evaluate the effect of the Al content. We considered the Al atoms only have the charge-compensating effect on the Cu atom which is hosted in the same unit as the Al atoms.24 DFT calculations have generally indicated that the isolated CuI and CuII ions prefer the d6r site of the cha framework.11,12,24,36–40 Meanwhile, the two Al atoms are distributed in an AlSiSiAl configuration, which is the most favorable structure when placing both Al atoms and the CuII cation within the same d6r.38Fig. 2 shows the constructed structures with one/two aluminum sites. Due to the similar topologies, the gme and aft frameworks exhibit the same preference for the d6r site. When the OH− ligand is coordinated to the CuII ion, the preference for the d6r site diminishes, which accords with the previous reports.13,41 The [CuII(OH)]+ in the d6r is 3 kJ mol−1 lower in energy than that in the 8r site for the gme cage, while the cha and aft cages both have slightly lower energies in the 8r than the d6r. As for the structures with two Al atoms, the CuI ion is accompanied by a Brønsted proton in the framework. The computational models including the cuprous ion were used for the oxidation process, while those including the cupric ion were used for the reduction process. The zeolite cages have two Al atoms when the NH3-solvated Cu ions ([CuII(NH3)4]2+ and [CuI(NH3)2]2+) act as the active site.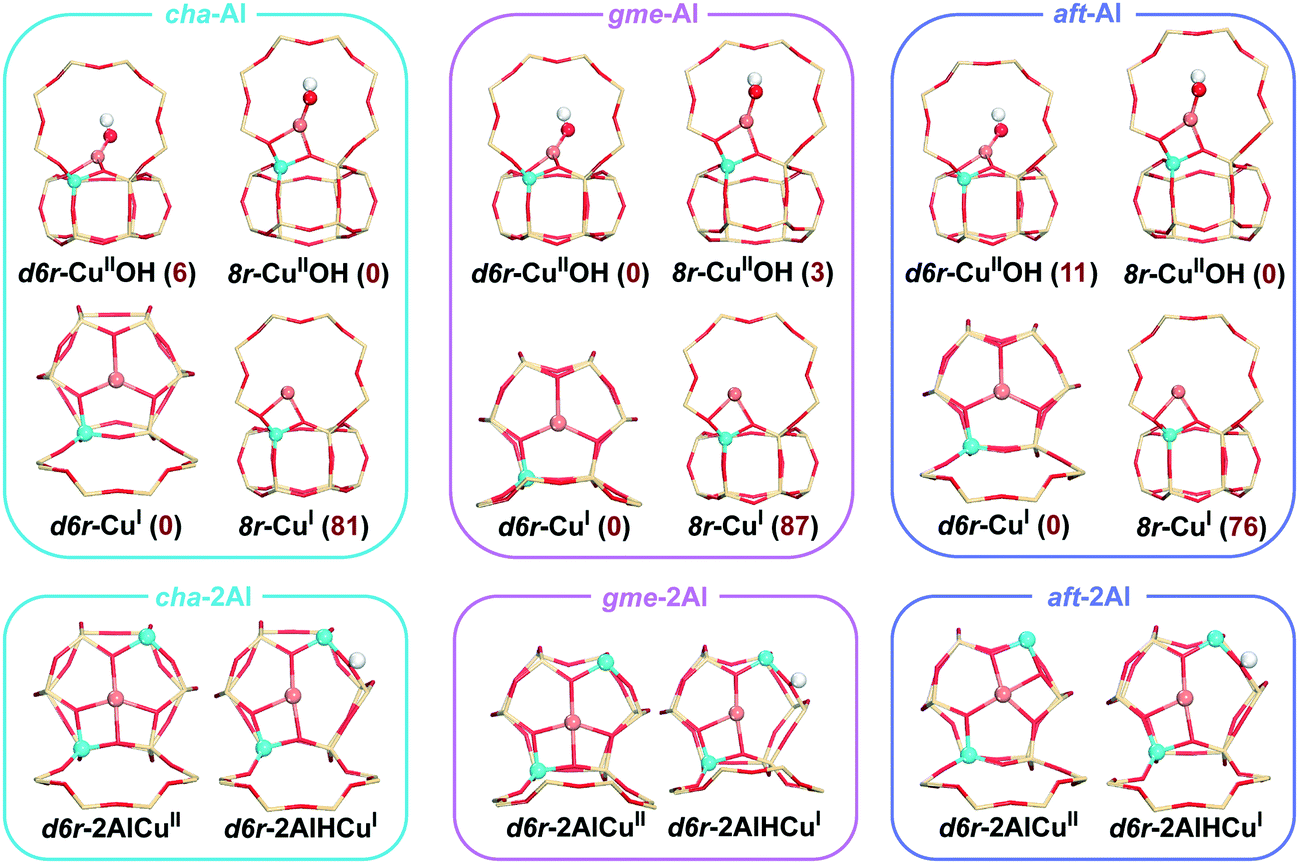 | ||
| Fig. 2 Local geometries of CuI, [CuII(OH)]+, and CuII in the d6r and 8r of cha, gme, and aft cages. Values in parenthesis are relative energies (in kJ mol−1) (color code: Al light blue; Si tan; Cu rufous; O red; H white). | ||
Computational methods
The calculations were carried out using the M06-L density functional.42 The M06-L method is reliable for the adsorption and activation energy studies on zeolites.43–46 The 6-31G (d, p) basis set was applied for the Al, Si, O, N and H atoms,47,48 and the Stuttgart effective core potential (ECP) basis set was adopted for the Cu atom.49 Only the terminal hydrogen atoms were kept fixed with X-ray structure while other atoms were allowed to relax. Based on the optimized structures, vibrational analyses were conducted to verify the stationary points to be minima or saddle points on the potential energy surface. All DFT calculations were conducted using Gaussian 09 suite of programs version E.01.50 Natural population analyses on all the structures were performed using NBO 3.1 as implemented in Gaussian 09.Results and discussion
SCR reaction cycles
As shown in Scheme S1,† the reduction of Al–CuIIOH and the oxidation of Al–CuI constitute the catalytic cycle on the 1Al site,13,21 while the reduction of 2Al–CuII and the oxidation of 2AlH–CuI form the catalytic cycle on the 2Al site.12,14 As for the reduction process, the adsorption of NO and NH3 at [CuIIOH]+ gives the NOOH–CuI–NH3 species, which generates a water molecule followed by the formation of CuI–NH2NO. The adsorption of NO and NH3 to the CuII on the 2Al site results in the formation of CuI–NH2NO by the N–H dissociation of NH3, and the H atom is transferred to the framework forming a Brønsted acid site. The CuI–NH2NO species then gives the N2 and H2O molecules. The oxidation takes place in the presence of NO and O2. The CuI ion reacts with NO and O2 to form the nitrate species, and the nitrite species is formed after the adsorption of the second NO molecule. The same nitrate and nitrite species exist on the 1Al and 2Al sites. Subsequently, the adsorption of NH3 leads to the NO2−–CuII–NH3 species on 1Al and the NO2−–CuII–NH4+ species on 2Al, which eventually can generate N2 and H2O.Reduction in Cu-cha, Cu-gme, and Cu-aft
Previous studies have proven that the presence of NO and NH3 can effectively reduce the CuII ion,6 so the NO and NH3 molecules are co-adsorbed on the Cu ion in our calculations. The reaction pathways of reduction are first studied based on CuIIOH-cha, -gme, and -aft, where the [CuII(OH)]+ unit is the active site. In this case, one ammonia molecule is adsorbed, and the [CuII(OH)]+ unit is placed at the 8r site due to the preferred stability. The reaction pathway involving NO and an NH3 molecule at the [CuII(OH)]+ site has been proposed for the cha framework, however, the detailed information such as the energy barrier is still absent.13,21,51All energies shown in these diagrams are measured with respect to the isolated zeolites and the NO/NH3 molecule. Spin multiplicities of intermediates and transition states are shown in Fig. S11–S13.† The adsorption of NO and NH3 is exothermic with adsorption energies of −225, −236, and −230 kJ mol−1 for cha, gme, and aft, as shown in Fig. 3 and S2.† Subsequently, the Cu–O bond is lengthening, the CuI–NOOH species is formed through energy barriers of 38, 32, and 47 kJ mol−1 for cha, gme, and aft, and the resulting species is similar to the proposed CuI–NO+ species.52,53 The formation of the CuI–NOOH species is slightly exothermic relative to the NO+–CuI–OH− species, which is the same as the previous report.51 The energy barriers for the H2O formation are 131, 147, and 153 kJ mol−1 for cha, gme, and aft. The following desorption of H2O is endothermic, which exhibits the same reaction energy of −48 kJ mol−1 for the three frameworks. After low energy barriers of 10, 11, and 12 kJ mol−1 for cha, gme, and aft, the N–N bond formation leads to NH2NO, which is an exothermic step. At this step, two possible intermediates of INT5 and INT5′ are located. In the case of INT5′, one N atom of the NH2NO is coordinated to the Cu atom, while the two N atoms of NH2NO are coordinated to the Cu atom in INT5. INT5′ is more stable than the INT5 for the three frameworks.
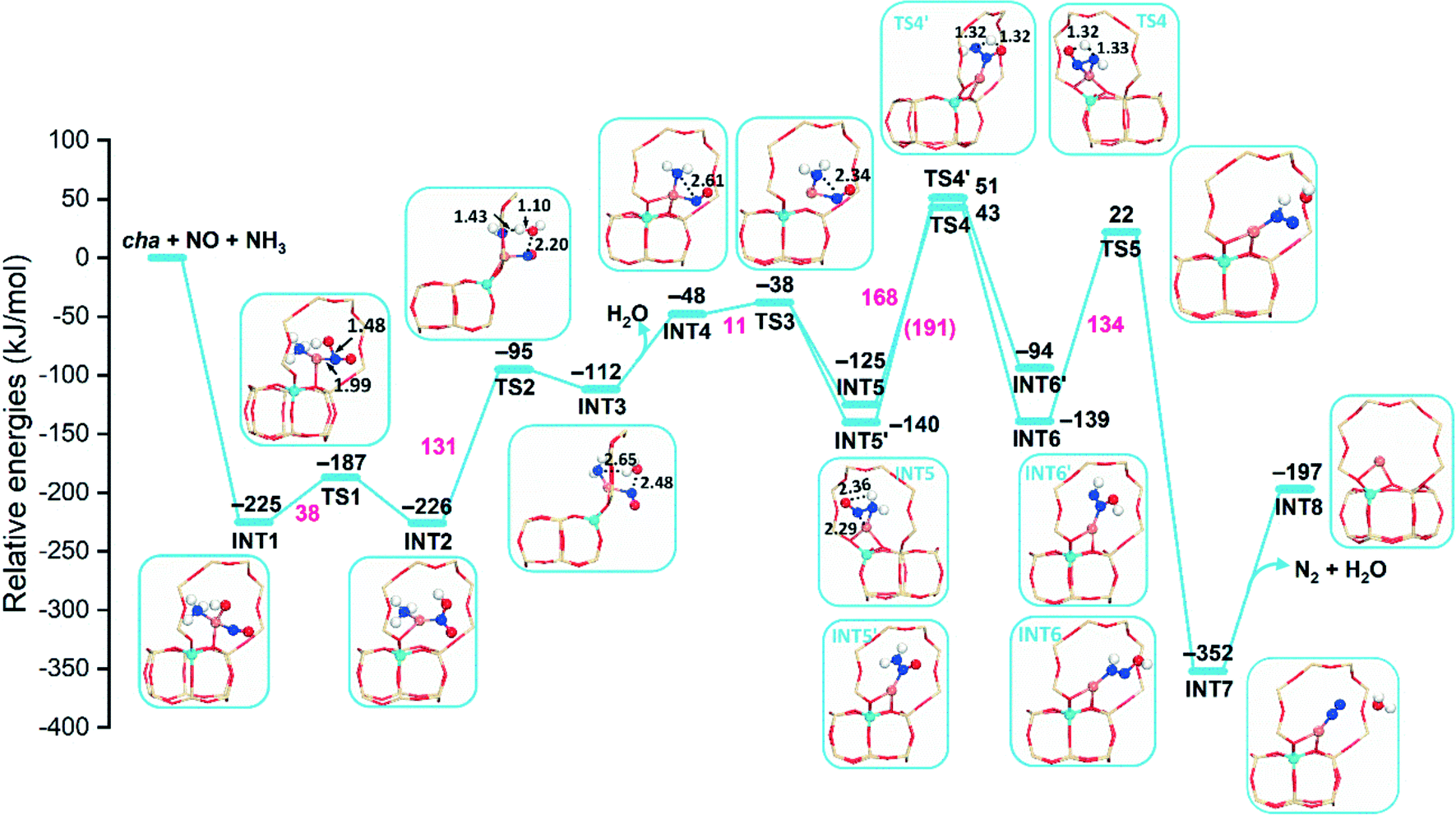 | ||
| Fig. 3 Reaction energy diagrams and optimized structures of intermediates and transition states for reduction by NO and NH3 on CuIIOH–Al-cha. The values of energetics and energy barriers are shown in black and magenta, respectively (color code: Al light blue; Si tan; Cu rufous; N mazarine; O red; H white). | ||
In the case of cha and aft, the following energy barriers for the H transfer from INT5 (168 and 164 kJ mol−1 for cha and aft) are lower than those from INT5′ (191 and 196 kJ mol−1 for cha and aft). As for the gme cage, two close energy barriers are identified, that is, 152 and 156 kJ mol−1 from INT5′ and INT5, which are lower than those on the cha and aft cages. Then, the HONNH species is generated, which are endothermic and exothermic for INT6′ and INT6, respectively. A high energy barrier is also found for the following formation of N2 and H2O, which are 155, 134, and 138 kJ mol−1 for the gme, cha, and aft cages. The bare CuI ion is formed after the desorption of N2 and H2O. The CuI ion is placed at the 8r site in the energy profiles, which can migrate to the d6r site due to the higher stability (Fig. 2). Note that the observed high energy barriers are not surprising for the intramolecular proton transfer due to the formation of an unstable four-membered ring, as indicated in Fig. 3 and S2† (TS4 and TS4′). The so-called hydrogen push–pull mechanism has been proposed for the NH2NO decomposition, which occurs at the Brønsted acid site with lower energy barriers.15,35,54 We studied the NH2NO decomposition at Brønsted acid site in the gme cage, which has lower reaction barriers (more details will follow in a subsequent section). Overall, the formation of NH2NO needs to overcome high barriers for the three cages when the [CuII(OH)]+ unit serves as the active site.
When the CuII ion is considered as the active site, the CuII-cha, -gme, and -aft models with two Si atoms separating the Al pair are applied. As presented in Scheme S1,† the formation of CuI–NH2NO is accompanied by the generation of a Brønsted acid proton on the framework O, which can easily adsorb the second NH3. Therefore, two NH3 molecules are applied in this case. As shown in Fig. 4 and S3,† with the assistance of the second NH3 molecule, the respective barriers for the CuI–NH2NO formation are 70, 73, 63 kJ mol−1 for cha, gme, and aft, which are much lower compared to those on the [CuII(OH)]+ active site. The following intramolecular H transfer still exhibits high energy barriers, that is, 172, 162, and 164 kJ mol−1 for cha, gme, and aft, respectively. However, when it comes to the following formation of N2 and H2O, the energy barriers are decreased to 89, 77, 81 kJ mol−1 for cha, gme, and aft, respectively, which should be ascribed to the stabilization of OH− by the NH3 molecule. Based on the results, the CuII ion at the d6r site is more favorable as the active site compared to the [CuII(OH)]+ site, and the involvement of the second NH3 molecule also facilitates the formation of NH2NO.
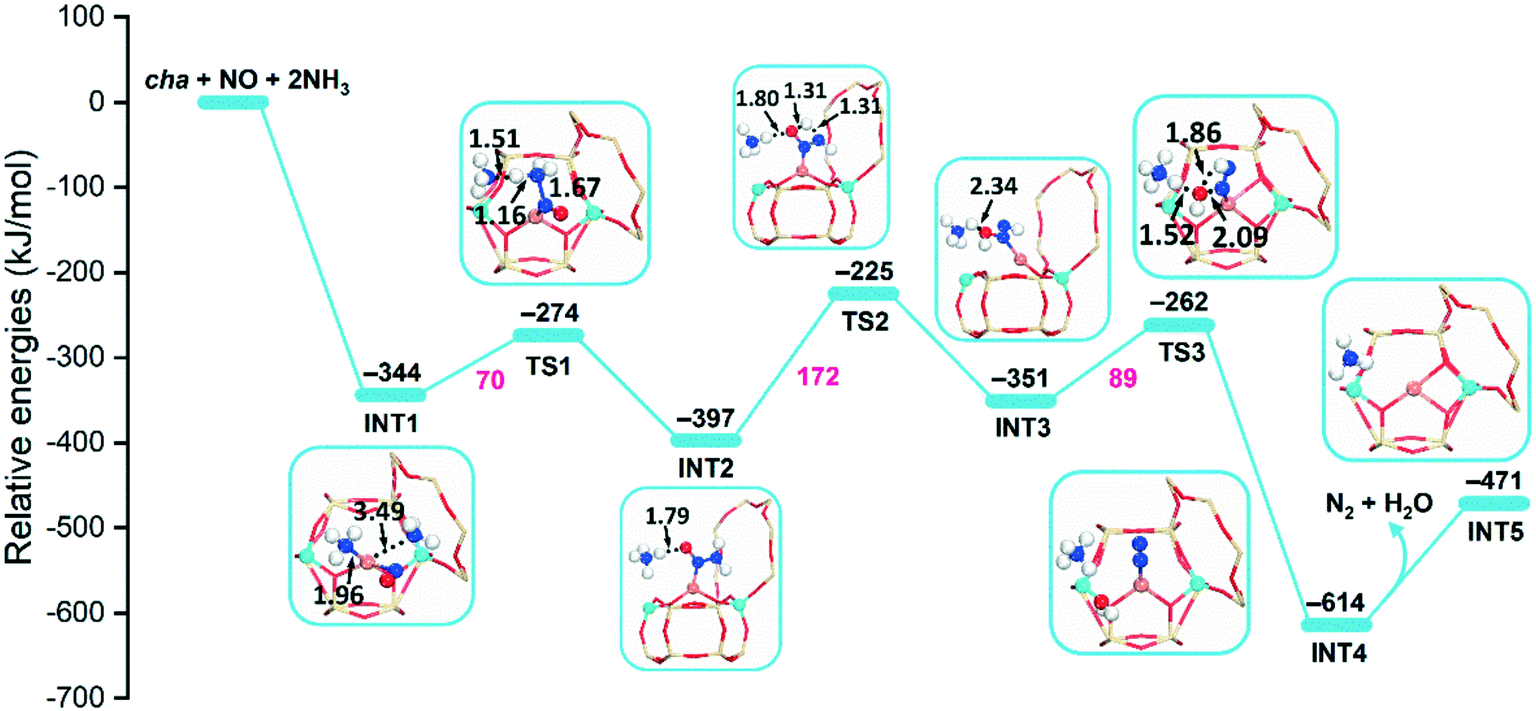 | ||
| Fig. 4 Reaction energy diagrams and optimized structures of intermediates and transition states for reduction by NO and NH3 on CuII–2Al-cha. | ||
To further study the influence of NH3 on the formation of the NH2NO species, four NH3 molecules are adsorbed to the CuII active site forming the square planner [CuII(NH3)4]2+ amino complex, which is a dominant species at low temperatures.13 The formation of [CuII(NH3)4]2+ are strongly exothermic, which exhibit similar adsorption energies of −494, −493, −489 kJ mol−1 for cha, gme, and aft, respectively. Fig. 5 shows energy profiles for the formation of NH2NO. The adsorption of NO is exothermic, which have adsorption energies of −26, −23, and −32 kJ mol−1 for cha, gme, and aft, respectively. Upon the adsorption of NO, the resulting structures have the triplet ground state for the three frameworks, which are slightly more stable than the singlet state by 8, 2, and 8 kJ mol−1 for cha, gme, and aft, respectively. The following formation of NH2NO is accompanied by the formation of the linear [CuI(NH3)2]+ complex observed in experiments.13 Furthermore, the observed energy barriers are 40 and 38 kJ mol−1 for cha and aft, which are much lower than those at the bare CuII site. However, the energy barrier of 66 kJ mol−1 in gme is only slightly lower than that at the bare CuII site, which should be attributed to the small inner space in gme. As summarized in Table S1,† the gme cage has the largest deformation energies than the other two cages. The deformation energy was calculated as the energy difference between the zeolite cage from INT/TS and the free zeolite cage. Therefore, because of the limited cage size, the NH2NO formation at the [CuII(NH3)4]2+ site is less preferred in the gme cage than the other two cages.
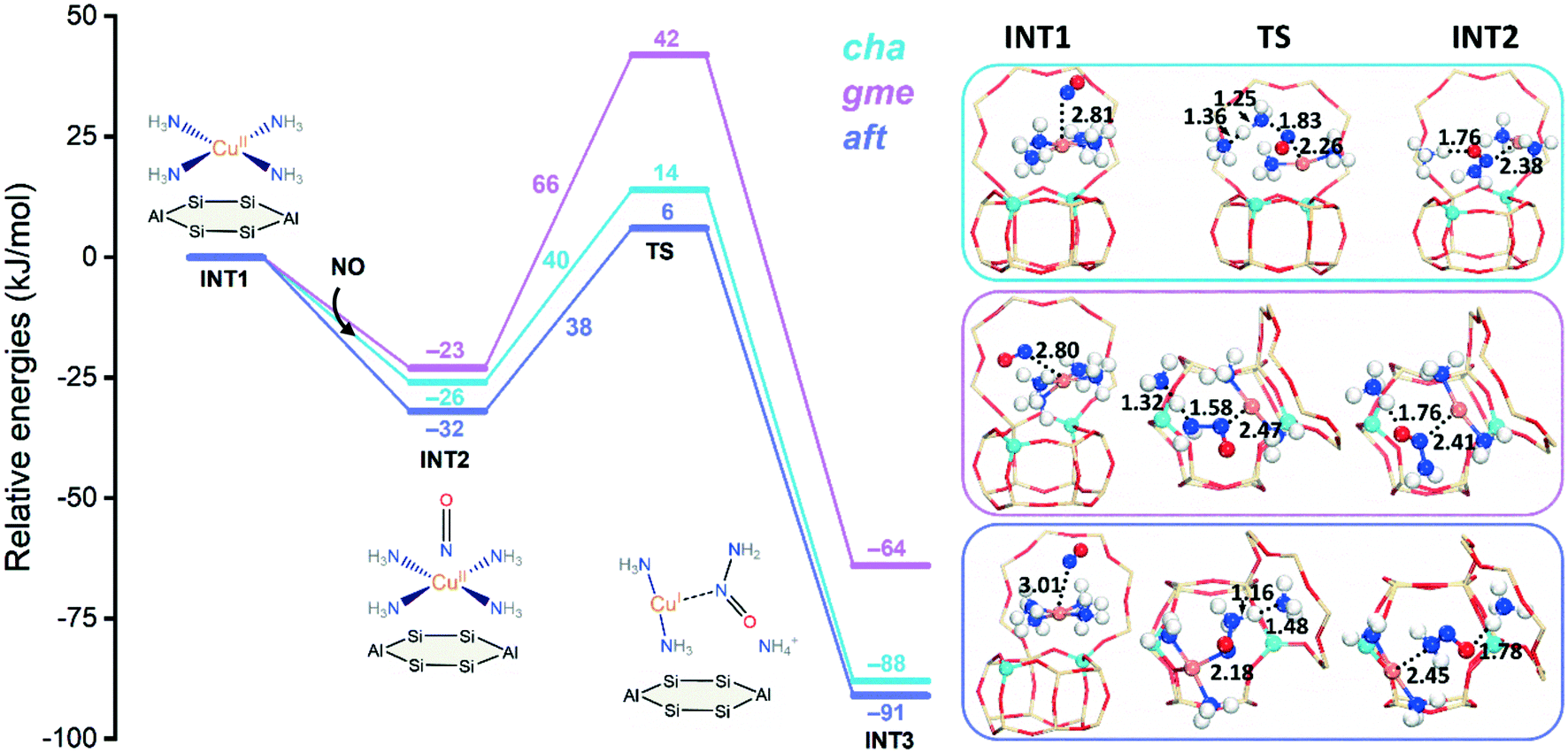 | ||
| Fig. 5 Reaction energy diagrams and optimized structures of intermediates and transition states for reduction at the [CuII(NH3)4]2+ site of CuII–2Al-cha, -gme, and aft. | ||
Fig. 6 compares reaction energy barriers for the NH2NO formation at the [CuII(OH)]+, CuII, and [CuII(NH3)4]2+ sites in the three cages. The reaction between NO and NH3 at [CuII(OH)]+ has very high energy barriers in all cages, in which the gme and aft cages have slightly higher barriers. The lower barriers are found at the CuII site for the reaction between NO and 2NH3 molecules, and the similar barriers indicate small differences caused by the cage size. At the [CuII(NH3)4]2+ site, upon adsorption of NO, the smallest gme cage has the highest energy barrier due to the largest deformation energies compared to the other two cages. Therefore, compared to [CuII(OH)]+, the bare CuII site is more favorable for reduction, and the selectivity caused by cage size is not identified so much at the CuII site. For the NH3-solvated [CuII(NH3)4]2+ site at low temperatures, the smallest gme cage is not preferred.
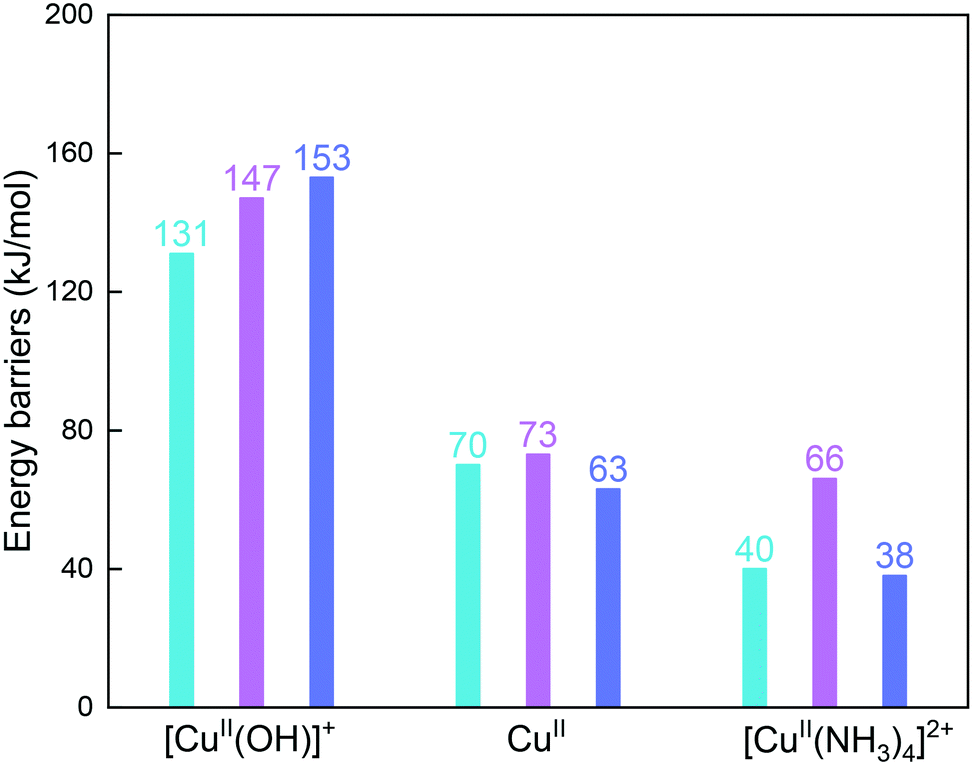 | ||
| Fig. 6 Reaction energy barriers for the formation of NH2NO at the [CuII(OH)]+, CuII, and [CuII(NH3)4]2+ sites for cha (blue), gme (pink), and aft (purple) cages (the energy barrier for the H2O formation at [CuII(OH)]+ is shown, which is the highest energy barrier during the NH2NO formation). | ||
As mentioned above, the intramolecular NH2NO decomposition has high energy barriers due to the formation of an unstable four-membered ring in TS. The proton exchange between NH2NO and a Brønsted acid site can significantly reduce the reaction barriers, that is, the hydrogen push–pull mechanism, which has been explored in NH3-SCR over V2O5 (ref. 54) and zeolites.15,35 We examined the NH2NO decomposition at the Brønsted acid site in the gme cage. As shown in Fig. 7, the adsorption of NH2NO at Brønsted acid site is exothermic by −92 kJ mol−1. In subsequent steps, the NH2NO species adjusts its configurations by donating and receiving H, which eventually generates N2 and H2O. The observed reaction barriers are much lower than those obtained from the intramolecular NH2NO decomposition. Therefore, the NH2NO species will migrate to the Brønsted acid site to proceed the subsequent decomposition.
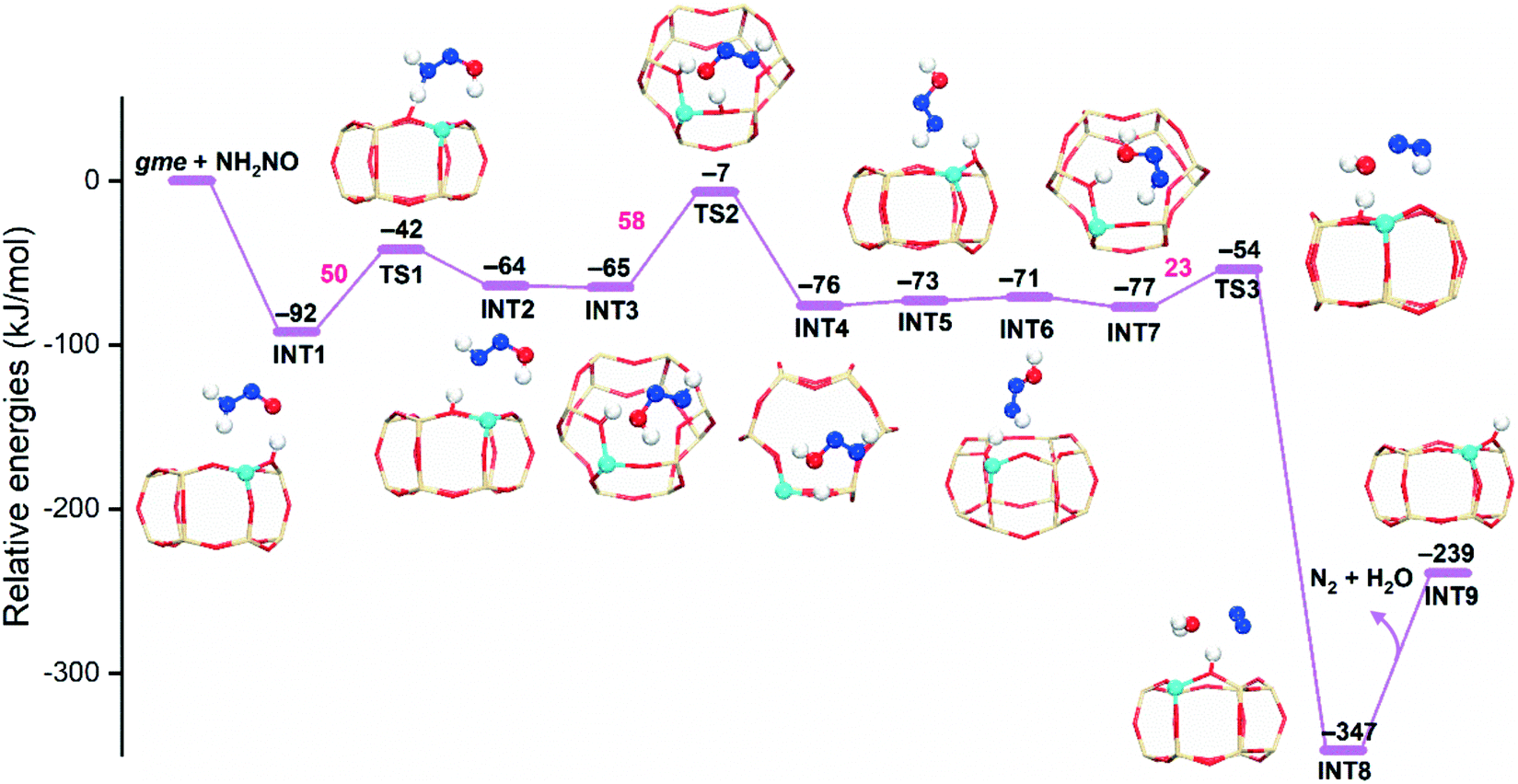 | ||
| Fig. 7 Reaction energy diagrams and local structures of intermediates and transition states for NH2NO decomposition assisted by Brønsted acid sites (AlH-gme). | ||
Oxidation in Cu-cha, Cu-gme, and Cu-aft
Fig. 8 presents the energy profiles for the NO oxidation in the CuI-cha, -gme, and -aft cages with one Al atom in the d6r site. The adsorption of NO and O2 to the CuI ion is exothermic with adsorption energies of −154, −160, and −161 kJ mol−1 for cha, gme, and aft. Upon the adsorption, the O2 molecule is activated through the weakening of the O–O bond, which is accompanied by the lengthening of the Cu–N bond. After moderate energy barriers of 55, 51, and 52 kJ mol−1 for cha, gme, and aft, one O atom of O2 is transferred to the NO molecule. Subsequently, the nitrate (NO3−) is formed by transferring the other O atom of O2 to the NO2 unit with the energy barriers of 17, 19, and 20 kJ mol−1 for cha, gme, and aft, which is strongly exothermic. The species has a chelating bidentate nitrate configuration on a monomeric Cu ion, which has been confirmed by the Fourier transformed EXAFS spectra.13 Overall, the similar energy barriers are observed for the formation of the nitrate in the three frameworks.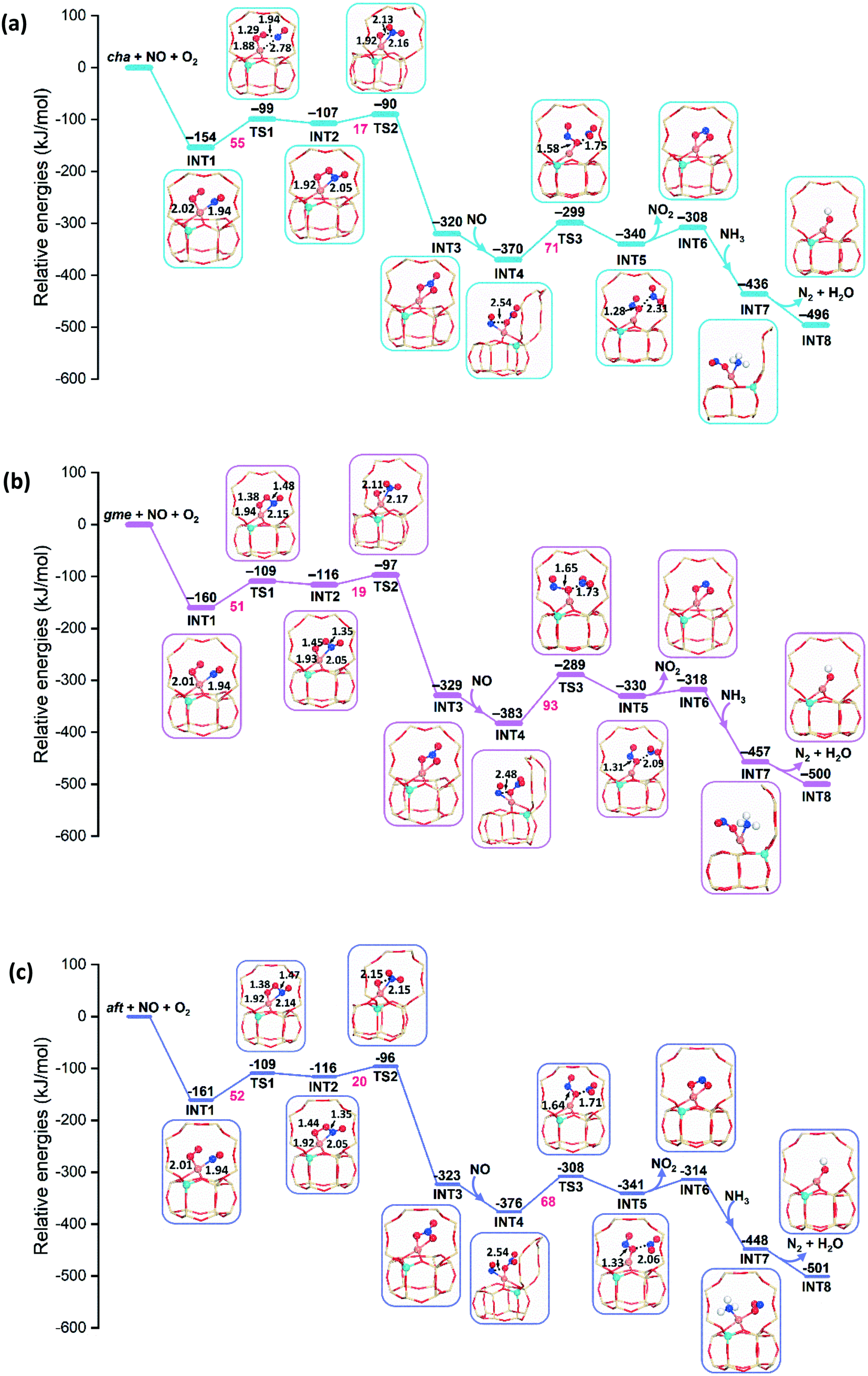 | ||
| Fig. 8 Reaction energy diagrams and local geometries of intermediates and transition states for the oxidation by NO and O2 on CuI–Al-cha (a), -gme (b), and -aft (c). | ||
In the next step, the second NO molecule is adsorbed to the Cu ion with similar energetics of −370, −383, and −376 kJ mol−1 for cha, gme, and aft. Subsequently, one O atom of the nitrate is transferred to the NO molecule through energy barriers of 71, 93, and 68 kJ mol−1 for cha, gme, and aft, where the smallest gme cage has the highest barrier than the other two frameworks (also see Fig. 9). Analyses on the deformation energy of the zeolite cage revealed that the largest deformation energy of TS3 in the gme cage should contribute to the highest energy barrier (Table S2†). The resulting intermediates (INT5) are endothermic (−340, −330, and −341 kJ mol−1 for cha, gme, and aft) with respect to the former step, which is the same situation as the previous study of Cu-CHA.13 Therefore, a higher energy barrier is found for the formation of nitrite than that for the nitrate formation, which is different from the previous study.13 Interestingly, the calculated energy barrier of 71 kJ mol−1 is very close to the apparent activation energy of 69 kJ mol−1 for the oxidation on Cu-CHA.13 Overall, as summarized in Fig. 9a, the highest energy barrier is observed for TS3 during the oxidation in all three types of frameworks, especially, the smallest gme cage possesses the highest energy barrier. Then, the aadsorption of NH3 to the Cu ion are strongly exothermic, and the adsorption energies are −436, −457, and −448 kJ mol−1 for cha, gme, and aft, respectively. The reaction pathway for the formation of H2O and N2 is still unclear, therefore, only the energetics of INT8 relative to INT7, H2O and N2 are shown in energy profiles.
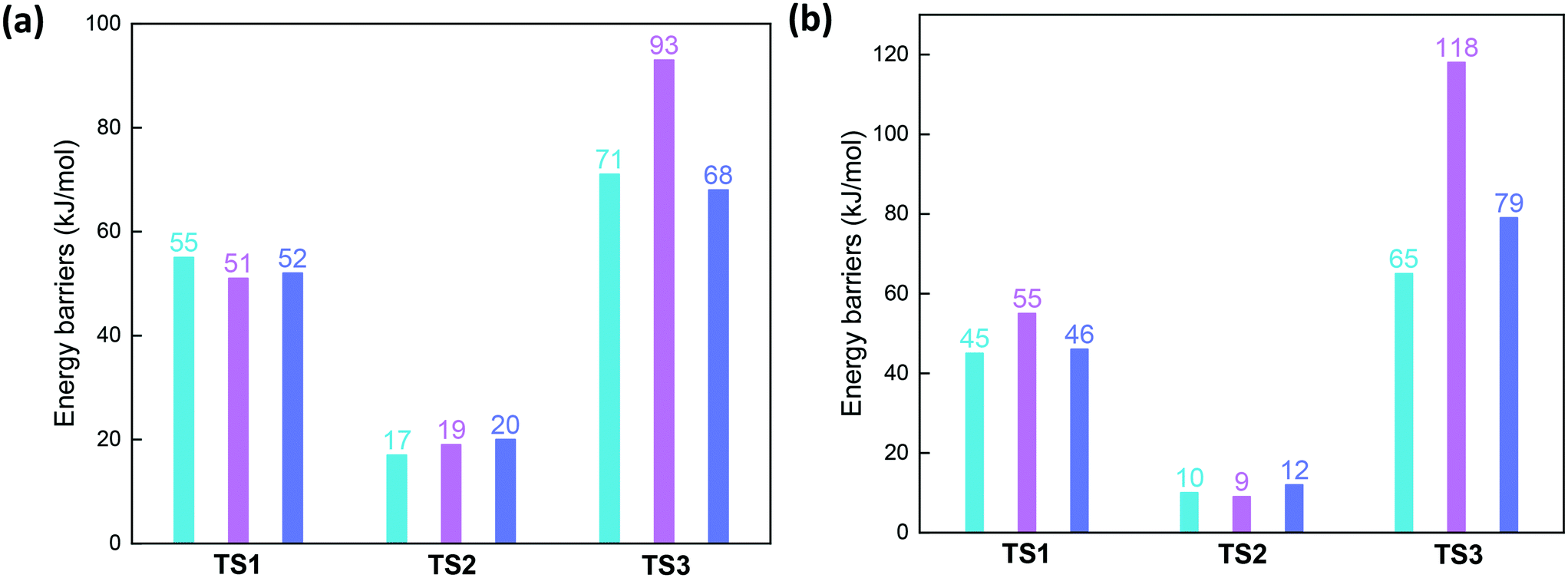 | ||
| Fig. 9 Reaction barriers of three transition states during oxidation by NO and O2 on CuI–Al-cha, -gme, and -aft (a) as well as CuI–2AlH-cha, -gme, and -aft (b). | ||
The models with two Al atoms accompanied by one Brønsted acid site are also considered for the oxidation process, which can simulate the oxidation of catalyst with acidic properties. As shown in Fig. S4,† the adsorption energies of NO and O2 are −142, −154, and −139 kJ mol−1 for cha, gme, and aft, which are smaller compared to those on the 1Al site. Meanwhile, it is found that energy barriers for the first O transfer in the cha and aft cages are slightly decreased to 45 and 46 kJ mol−1, while that in the gme cage is slightly increased to 55 kJ mol−1 due to the more stable complex adsorbing NO and O2. The next energy barriers for the formation of nitrate are decreased to 10, 9, and 12 kJ mol−1 for cha, gme, and aft, which should be partially ascribed to the formation of the hydrogen bonds (Fig. S5†). Note that the energetics of INT1, TS1, INT2, and TS2 on the 2Al site are less stable than those on the 1Al site for aft, while the other two frameworks have comparable values on 1Al and 2Al sites. The deformation energy of INT2 on aft is 163 kJ mol−1, which is even larger than those on cha (147 kJ mol−1) and gme (150 kJ mol−1) (Table S3†). The formation of the nitrate species possesses similar reaction energies to those on 1Al site, that is, −321, −332, and −319 kJ mol−1 for cha, gme, and aft.
After the second NO molecule is adsorbed, the O transfer from the nitrate to the NO has the energy barriers of 65, 118, and 79 kJ mol−1 for cha, gme, and aft (Fig. 9b and S4†), in which gme and aft exhibit higher barriers than those on the 1Al site. As shown in Fig. 10, the INT4a and INT4b are considered to represent the different positions of the adsorbed molecules relative to the Brønsted proton. The INT4a structures are more stable due to the hydrogen bond interaction. The INT4b configurations are shown in the energy profiles due to the resulting lower barriers. It is also found that the energy difference of INT4a and INT4b increases with the increasing cage size, that is, 7, 12, and 23 kJ mol−1 for gme, cha, and aft, respectively. The energetics of the located TS3 structures are −280, −312, and −284 kJ mol−1 for gme, cha, and aft, thus, the highest energy barrier of 118 kJ mol−1 is observed on the gme cage (Fig. S4†). Fig. S6† also shows the distances between the adsorbed molecules and the Brønsted proton in INT5 and INT6. When an NH3 molecule is introduced, the reaction energies are −503, −505, and −490 kJ mol−1 for cha, gme, and aft, which are more exothermic due to the formation of NH4+ at the Brønsted acid site. Then, the NH4NO2 species can be formed, which can be easily decomposed into N2 and H2O under SCR operation conditions.55–57 Therefore, the results on the 2Al site still reveal that the oxidation process on the gme cage needs the largest activation energy compared to the cha and aft cages (Fig. 9b). The results on the bare CuII ions will be useful for the high temperature oxidation.
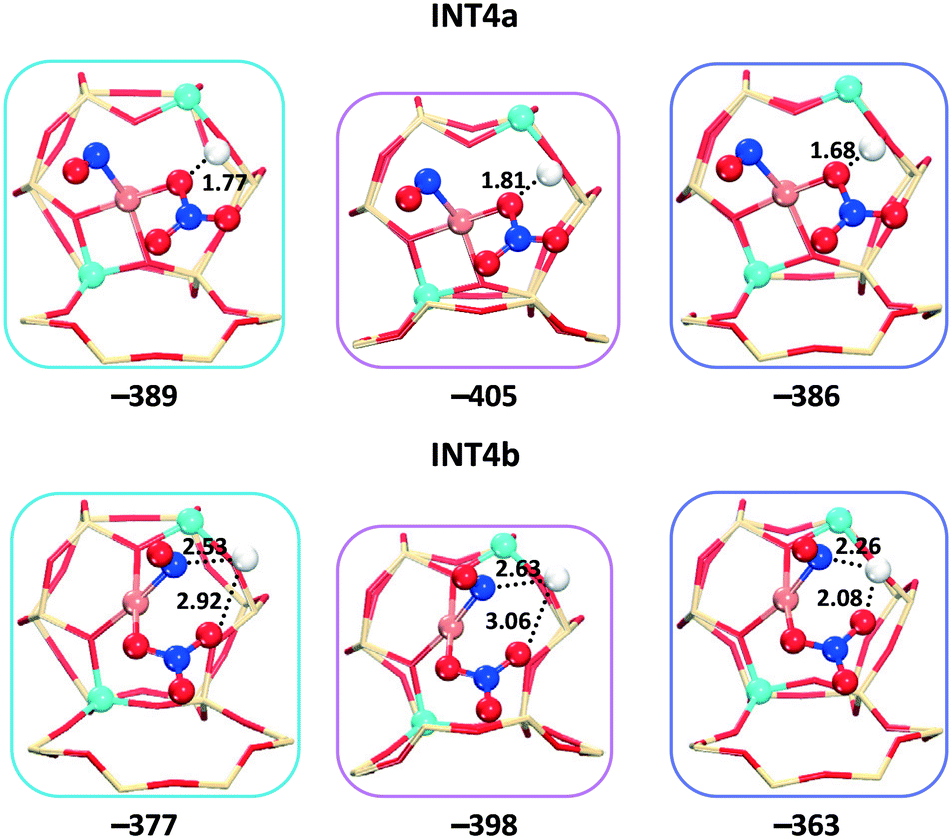 | ||
| Fig. 10 Local geometries of the two intermediates (INT4a and INT4b) on cha (blue), gme (pink), and aft (purple). The distances between the adsorbed molecules and the Brønsted proton are shown in angstrom. The values beneath each structure are reaction energies with respect to the isolated reactants. | ||
We also evaluated the O2 activation by paring of [CuI(NH3)2]+ in the cha, aft, and gme cages, which is an important step for the low temperature oxidation with low Cu loadings. Due to the large inner space in the cha and aft cages, the two [CuI(NH3)2]+ species inside these cages may have two different alignments with respect to the d6r: parallel to the d6r (para-site) and perpendicular to the d6r (per-site). Fig. 11 and S8† show the two different alignments for INT1, INT2, and INT4 in cha and aft. The gme cage can only have the [CuI(NH3)2]+ pair at the para-site due to the restricted space, as shown in Fig. S7.† Note that previous studies only considered the per-site to place the two [CuI(NH3)2]+ species.35,58 It is found that the INT1 structures with the per-site are over 50 kJ mol−1 higher in energy compared to those with the para-site. The following INT2 and INT4 structures also have the same preference for the para-site. In order to explain the stabilization of the inner species at the para-site, we calculated the interaction energies between the inner species and the outer cages of INT1 and INT4. As shown in Table S4,† the separated fragments (the outer cage and the inner species) from the per-site structures are more stable than those from the para-site ones, however, stronger interaction energies between the two fragments were identified for the para-site case. Therefore, the higher stability of the para-site structures originates from the stronger interactions between the inner species and the outer cages.
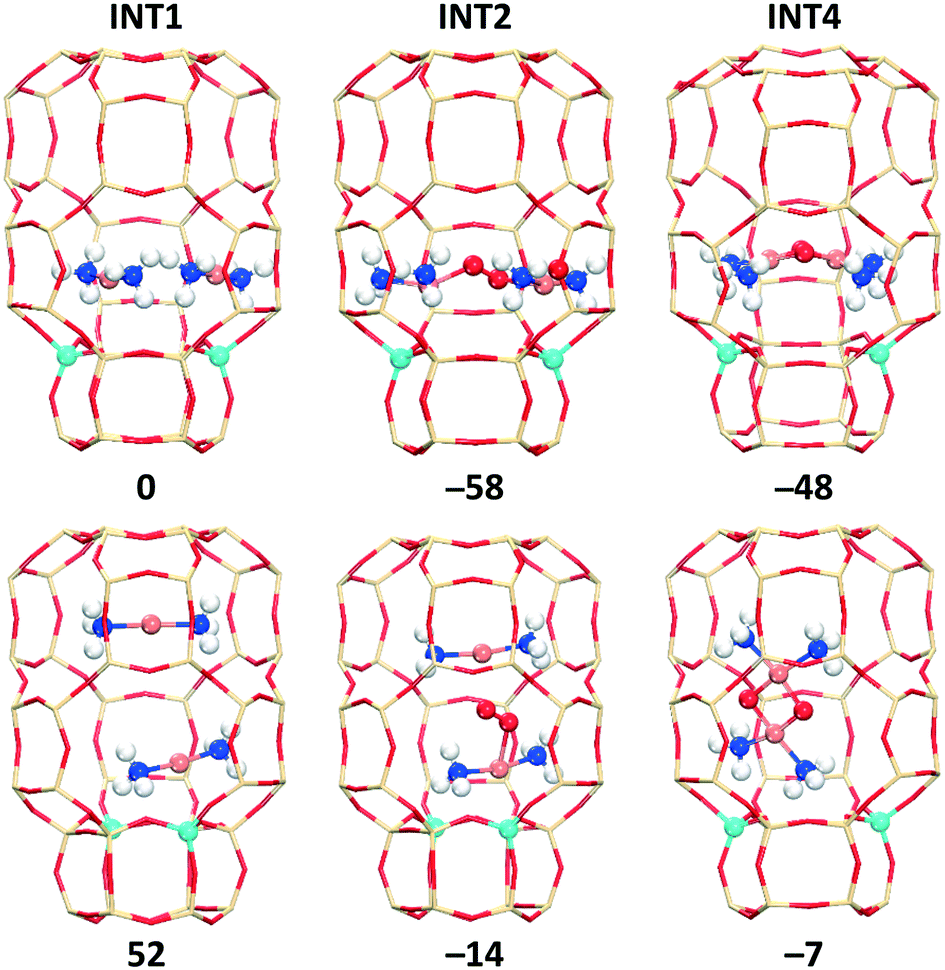 | ||
| Fig. 11 Optimized structures and relative energies (kJ mol−1) of INT1, INT2, and INT4 for the cha cage (top: para-site; bottom: per-site). The energies of INT2 and INT4 are obtained with respect to the para-site INT1 and O2. | ||
Fig. 12 shows energy profiles of the O2 activation for cha, aft, and gme, in which the para-site structures are shown. The O2 insertion is the most favorable in the largest aft cage (−67 kJ mol−1), followed by the cha cage (−58 kJ mol−1), while the corresponding energy is only −17 kJ mol−1 in the smallest gme cage. The following INT3 also follows the same sequence in reaction energy, aft > cha > gme. Fig. S10† shows the potential energy surface between INT2 and INT3 for the cha cage because the corresponding TS search fails. Interestingly, a new intermediate with a lower energy than INT2 is identified along the potential energy surface, in which the O–O distance is in the range of 1.36–1.38 Å. Meanwhile, it is found that the significant energy barrier between INT2 and INT3 does not exist and the spin-crossing occurs via minimum energy seam of crossing (MESX) in the vicinity of INT3 as shown in Fig. S10.† As for the transition state between INT3 and INT4, reaction barriers are almost the same for cha (47 kJ mol−1) and aft (48 kJ mol−1), while a lower barrier is found in gme (35 kJ mol−1). Note that the TS in gme is the most unstable one, and the lower barrier in gme should be partially ascribed to the lower stability of INT3. The resulting INT4 is the most exothermic in gme (−71 kJ mol−1), followed by aft (−56 kJ mol−1) and cha (−48 kJ mol−1). The most exothermic INT4 in gme should be due to the strongest interaction than cha and aft (Table S4†). Even though the gme cage has the largest reaction energy for the INT4 formation, the smallest adsorption energy (INT2) in gme suggests the encapsulation of O2 into the small cage is more difficult compared to the other two cages, which can inhibit the process to proceed. In general, it can be concluded that the aft cage has the similar activity for the O2 activation to the cha cage.
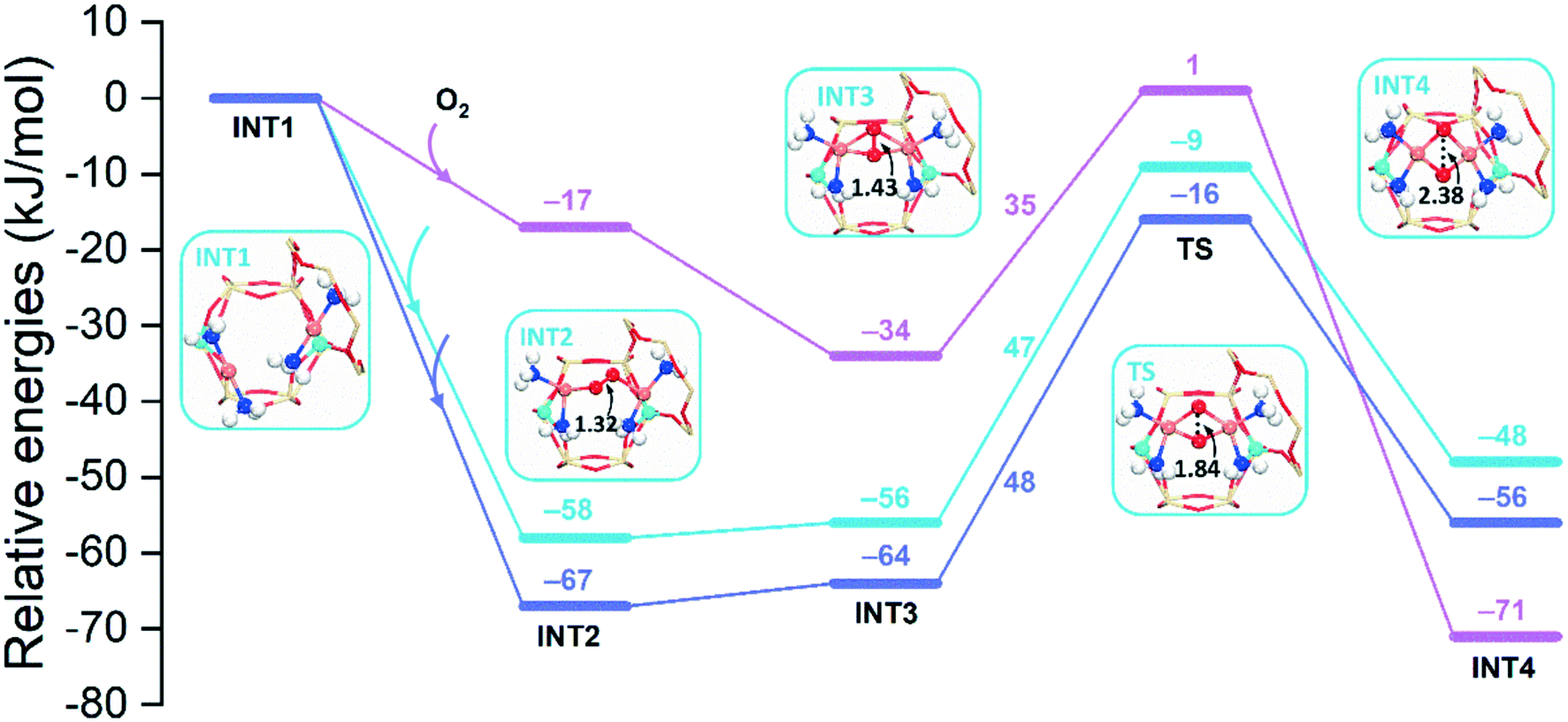 | ||
| Fig. 12 Reaction energy diagrams for the O2 activation by the 2[NH3–CuI–NH3] dimer in 2Al-cha (blue), -gme (pink), and -aft (purple). Local geometries of intermediates and transition states for cha are shown. | ||
As for all intermediates and transition states, natural electron configurations and natural population analysis (NPA) partial charges of Cu are summarized in ESI† (Tables S5–S10 and Fig. S11–S13). The results revealed that the electrons from the 3d orbital have more significant changes than those from the 4s orbital when the oxidation state of Cu changes during the reaction.
Conclusions
By means of DFT calculations, the SCR activity in the Cu-exchanged CHA, GME, and AFX zeolites are evaluated through the Cu-cha, -gme, and -aft cages, and the oxidation and reduction processes are both studied. We considered CuI, CuII, and [CuII(OH)]+ as the active sites which are placed in the frameworks with the 1Al or 2Al site. As for the reduction process, the [CuII(OH)]+ (1Al) and the CuII (2Al) are constructed to proceed the reaction. The results have revealed that the NH2NO formation at the [CuII(OH)]+ site has high energy barriers in the three considered frameworks, while the lower barriers are found at the CuII site for them. Additionally, the barriers are largely decreased at the solvated [CuII(NH3)4]2+ site for the cha and aft frameworks, while the barrier is only slightly decreased for gme. The formed NH2NO species then migrates to the Brønsted acid site for the formation of N2 and H2O. The CuI (1Al) and the CuI plus a Brønsted acid site (2Al) are constructed to study the oxidation process. The results on both models have revealed that the nitrate formation has similar energy barriers in the three frameworks, which are lower than the following nitrite formation. Particularly, the smallest gme cage possesses the highest energy barrier for the nitrite formation, which should be ascribed to the largest deformation caused by the limited space. It is also found that the Brønsted acid proton can slightly affect the stability of the intermediates and transition states, which has little influence on the general trend. As for the O2 activation by paring of [CuI(NH3)2]+, cha and aft have similar performance, whereas gme is not favorable due to the limited space for the insertion of O2. Overall, the reactivity differences in the three cages have revealed the cage size is important to proceed this reaction, which may provide a way to tune the reaction pathway by the cage size selectivity. Additionally, it can be concluded that the AFX zeolite has the comparable SCR catalytic activity to the CHA one, even though further studies are demanded.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Research Association of Automotive Internal Combustion Engines (AICE) project, Japan. M. E. acknowledges Grants from Japan Society for the Promotion of Science (Grant numbers of JP16H06511, JP20H02718).References
- R. M. Barrer, Zeolites, 1981, 1, 130–140 CrossRef CAS.
- M. Iwamoto and H. Hamada, Catal. Today, 1991, 10, 57–71 CrossRef CAS.
- G. Centi and S. Perathoner, Appl. Catal., A, 1995, 132, 179–259 CrossRef CAS.
- B. Chen, R. Xu, R. Zhang and N. Liu, Environ. Sci. Technol., 2014, 48, 13909–13916 CrossRef CAS.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. F. Peden, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS.
- Y. Xin, Q. Li and Z. Zhang, ChemCatChem, 2018, 10, 29–41 CrossRef CAS.
- F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, J. Catal., 2013, 300, 20–29 CrossRef CAS.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- S. A. Bates, A. A. Verma, C. Paolucci, A. A. Parekh, T. Anggara, A. Yezerets, W. F. Schneider, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2014, 312, 87–97 CrossRef CAS.
- C. Paolucci, A. A. Verma, S. A. Bates, V. F. Kispersky, J. T. Miller, R. Gounder, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11828–11833 CrossRef CAS.
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS.
- Y. Mao, Z. Wang, H.-F. Wang and P. Hu, ACS Catal., 2016, 6, 7882–7891 CrossRef CAS.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS.
- M. Moreno-González, R. Millán, P. Concepción, T. Blasco and M. Boronat, ACS Catal., 2019, 9, 2725–2738 CrossRef.
- L. S. Dent and J. V. Smith, Nature, 1958, 181, 1794–1796 CrossRef CAS.
- R. F. Lobo, S. I. Zones and R. C. Medrud, Chem. Mater., 1996, 8, 2409–2411 CrossRef CAS.
- S. V. Priya, T. Ohnishi, Y. Shimada, Y. Kubota, T. Masuda, Y. Nakasaka, M. Matsukata, K. Itabashi, T. Okubo, T. Sano, N. Tsunoji, T. Yokoi and M. Ogura, Bull. Chem. Soc. Jpn., 2017, 91, 355–361 CrossRef.
- A. H. Clark, R. J. G. Nuguid, P. Steiger, A. Marberger, A. W. Petrov, D. Ferri, M. Nachtegaal and O. Kröcher, ChemCatChem, 2020, 12, 1429–1435 CrossRef CAS.
- N. Martín, C. Paris, P. N. R. Vennestrøm, J. R. Thøgersen, M. Moliner and A. Corma, Appl. Catal., B, 2017, 217, 125–136 CrossRef.
- H. Kubota, C. Liu, T. Toyao, Z. Maeno, M. Ogura, N. Nakazawa, S. Inagaki, Y. Kubota and K.-i. Shimizu, ACS Catal., 2020, 10, 2334–2344 CrossRef CAS.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563 RSC.
- A. Godiksen, F. N. Stappen, P. N. R. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS.
- U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, ACS Catal., 2013, 3, 413–427 CrossRef CAS.
- J. Szanyi, J. H. Kwak, H. Zhu and C. H. F. Peden, Phys. Chem. Chem. Phys., 2013, 15, 2368–2380 RSC.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Science, 2017, 357, 898–903 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS.
- A. Marberger, A. W. Petrov, P. Steiger, M. Elsener, O. Kröcher, M. Nachtegaal and D. Ferri, Nat. Catal., 2018, 1, 221–227 CrossRef CAS.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS.
- E. Borfecchia, C. Negri, K. A. Lomachenko, C. Lamberti, T. V. W. Janssens and G. Berlier, React. Chem. Eng., 2019, 4, 1067–1080 RSC.
- C. Negri, E. Borfecchia, M. Cutini, K. A. Lomachenko, T. V. W. Janssens, G. Berlier and S. Bordiga, ChemCatChem, 2019, 11, 3828–3838 CrossRef CAS.
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, ACS Catal., 2020, 10, 5646–5656 CrossRef CAS.
- F. Göltl, P. Sautet and I. Hermans, Catal. Today, 2016, 267, 41–46 CrossRef.
- J. S. McEwen, T. Anggara, W. F. Schneider, V. F. Kispersky, J. T. Miller, W. N. Delgass and F. H. Ribeiro, Catal. Today, 2012, 184, 129–144 CrossRef CAS.
- F. Göltl, R. E. Bulo, J. Hafner and P. Sautet, J. Phys. Chem. Lett., 2013, 4, 2244–2249 CrossRef.
- F. Göltl and J. Hafner, J. Chem. Phys., 2012, 136, 064501 CrossRef.
- U. Deka, A. Juhin, E. A. Eilertsen, H. Emerich, M. A. Green, S. T. Korhonen, B. M. Weckhuysen and A. M. Beale, J. Phys. Chem. C, 2012, 116, 4809–4818 CrossRef CAS.
- R. Zhang, J.-S. McEwen, M. Kollár, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, ACS Catal., 2014, 4, 4093–4105 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef.
- C. Kumsapaya, K. Bobuatong, P. Khongpracha, Y. Tantirungrotechai and J. Limtrakul, J. Phys. Chem. C, 2009, 113, 16128–16137 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. C, 2008, 112, 6860–6868 CrossRef CAS.
- P. Zhao, B. Boekfa, T. Nishitoba, N. Tsunoji, T. Sano, T. Yokoi, M. Ogura and M. Ehara, Microporous Mesoporous Mater., 2020, 294, 109908 CrossRef CAS.
- T. J. Goncalves, P. N. Plessow and F. Studt, ChemCatChem, 2019, 11, 4368–4376 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Peterssonet al., Gaussian 09, Revision E.01, Gaussian, Inc, Wallingford Ct, 2013 Search PubMed.
- J. Rudolph and C. R. Jacob, ACS Omega, 2019, 4, 7987–7993 CrossRef CAS.
- C. Prestipino, G. Berlier, F. X. Llabrés i Xamena, G. Spoto, S. Bordiga, A. Zecchina, G. Turnes Palomino, T. Yamamoto and C. Lamberti, Chem. Phys. Lett., 2002, 363, 389–396 CrossRef CAS.
- F. X. Llabrés i Xamena, P. Fisicaro, G. Berlier, A. Zecchina, G. T. Palomino, C. Prestipino, S. Bordiga, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2003, 107, 7036–7044 CrossRef.
- M. Anstrom, N.-Y. Topsøe and J. A. Dumesic, J. Catal., 2003, 213, 115–125 CrossRef CAS.
- F. Liu, Y. Yu and H. He, Chem. Commun., 2014, 50, 8445–8463 RSC.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Appl. Catal., B, 2014, 156–157, 428–437 CrossRef CAS.
- C. Liu, H. Kubota, T. Toyao, Z. Maeno and K.-i. Shimizu, Catal. Sci. Technol., 2020, 10, 3586–3593 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The computational models of cha (42T), gme (36T) and aft (54T); the reaction mechanism for the SCR reaction in the Cu-zeolite; reaction energy diagrams and optimized structures of intermediates and transition states for reduction by NO and NH3 on CuIIOH–Al-cha and CuIIOH–Al-aft; reaction energy diagrams and optimized structures of intermediates and transition states for reduction by NO and NH3 on CuII–2Al-gme and -aft; deformation energies of intermediates and transition states with respect to the pristine structures of CuII–2Al-cha, -gme, and -aft; reaction energy diagrams and local structures of intermediates and transition states for oxidation by CuI–2AlH-cha, -gme, and -aft; local geometries of the INT1, TS1, INT2, TS2 and INT3 of cha, gme, and aft at the 2Al site; local geometries of the TS3, INT5, and INT6 of cha, gme, and aft at the Al site; deformation energies of intermediates and transition states for the oxidation with respect to the pristine structures of CuI–Al-cha, -gme, and -aft; deformation energies of intermediates and transition states for the oxidation with respect to the pristine structures of CuI–2AlH-cha, -gme, and -aft; optimized structures and relative energies of INT1, INT2, and INT4 for gme and aft; interaction energies between the [zeolite]2− cage and the inner 2[CuI(NH3)2]+/2[OCuI(NH3)2]+ ion in INT1 and INT4 as well as relative energies of the outer cage and the inner ion for cha and aft; local geometries of intermediates and transition states for the O2 activation by the 2[NH3-CuI-NH3] dimer in 2Al-gme, and -aft; potential energy surface between INT2 and INT3 for the O2 activation by the 2[NH3–CuI–NH3] dimer in 2Al-cha; natural electron configuration of Cu in Cu-cha, Cu-gme, and Cu-aft; NPA charge of Cu in Cu-cha, Cu-gme, and Cu-aft. See DOI: 10.1039/d0cy02342f |
| This journal is © The Royal Society of Chemistry 2021 |