
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plasma-catalytic ammonia synthesis beyond thermal equilibrium on Ru-based catalysts in non-thermal plasma†
Kevin H. R.
Rouwenhorst
 *a,
Hugo G. B.
Burbach
a,
Dave W.
Vogel‡
b,
Judit
Núñez Paulí
a,
Bert
Geerdink
a and
Leon
Lefferts
*a
*a,
Hugo G. B.
Burbach
a,
Dave W.
Vogel‡
b,
Judit
Núñez Paulí
a,
Bert
Geerdink
a and
Leon
Lefferts
*a
aCatalytic Processes & Materials, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: k.h.r.rouwenhorst@utwente.nl; l.lefferts@utwente.nl
bUniversity of Twente, The Netherlands
First published on 11th February 2021
Abstract
Recently it was proposed that plasma-catalytic NH3 synthesis with excited N2 allows for conversions beyond thermal equilibrium. We show that this is indeed possible with experimental data for Ru catalysts at temperatures above 300 °C, resulting in significant thermal activity for NH3 synthesis. The resulting NH3 concentration is determined by competition between, on the one hand, dissociative adsorption of ground-state N2 and adsorption of plasma-generated N radical species with subsequent hydrogenation to NH3, and on the other hand, thermal-catalytic decomposition of NH3. At temperatures below 300 °C, plasma-catalytic ammonia synthesis is attributed to adsorption of N radicals, generated in the plasma, with subsequent hydrogenation to NH3. These findings imply that catalysts with thermal activity are not suitable for plasma catalysis, aiming at conversion beyond equilibrium, as these also catalyze the reverse decomposition reaction.
Introduction
A circular economy without fossil-based hydrocarbons is required to decrease greenhouse gas emissions.1 With the emergence of renewable resources, such as solar panels and wind turbines, this increasingly becomes reality. However, energy storage is required, as these renewable sources are intermittent and do not match demand profiles. Various energy storage alternatives have been researched. For seasonal energy storage, chemical energy storage is the most feasible option.2 Renewable electricity can be used to produce H2via H2O by electrolysis. However, H2 is difficult to store and transport.3 Therefore, hydrogen carriers are proposed.4–8Ammonia (NH3) may be one of the hydrogen carriers of the future.2,5,9–11 NH3 can be synthesized from renewable hydrogen (H2) and nitrogen (N2), as given by eqn (1). NH3 is currently synthesized by the large-scale Haber–Bosch process, which operates at high temperatures (400–500 °C) and high pressures (100–300 bar).12 However, energy storage requires a significantly smaller scale, whereas scale-down of the Haber–Bosch process is difficult due to the severe process conditions5 and extensive heat integration. Therefore, alternative technologies are currently under development, such as electrochemical synthesis, photochemical synthesis, chemical looping, homogeneous catalysis, and bio-catalysis.13
| N2 + 3H2 ↔ 2NH3 with ΔH° = −91.8 kJ mol−1 | (1) |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N.16 In a thermal plasma, ionized and radical species dominate at a temperature of typically a few thousand K. Due to the high temperatures, thermal plasmas are not suitable for combination with a catalyst.17 In the case of a non-thermal (NT) plasma, electrons have a temperature of 10
N.16 In a thermal plasma, ionized and radical species dominate at a temperature of typically a few thousand K. Due to the high temperatures, thermal plasmas are not suitable for combination with a catalyst.17 In the case of a non-thermal (NT) plasma, electrons have a temperature of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–100000 K, whereas the molecules remain at near-ambient temperature, which is determined by translation and rotation of molecules. Most molecules are not ionized or dissociated, whereas vibrational and electronic excitation occurs.18 Thus, non-thermal plasmas can be combined with catalysts as described hereafter. NH3 synthesis in the presence of a plasma and a catalyst has been studied over the past four decades, with recently increasing focus on effective coupling between the catalyst and plasma.16–22 However, the current best reported energy efficiency is typically 25–35 gNH3 kW h−1, which is substantially lower than the required energy efficiency of 150–200 gNH3 kW h−1 to be competitive with alternative technologies for small-scale NH3 synthesis.14,23,24
000–100000 K, whereas the molecules remain at near-ambient temperature, which is determined by translation and rotation of molecules. Most molecules are not ionized or dissociated, whereas vibrational and electronic excitation occurs.18 Thus, non-thermal plasmas can be combined with catalysts as described hereafter. NH3 synthesis in the presence of a plasma and a catalyst has been studied over the past four decades, with recently increasing focus on effective coupling between the catalyst and plasma.16–22 However, the current best reported energy efficiency is typically 25–35 gNH3 kW h−1, which is substantially lower than the required energy efficiency of 150–200 gNH3 kW h−1 to be competitive with alternative technologies for small-scale NH3 synthesis.14,23,24
Recently, Mehta et al.25 postulated that catalytic NH3 synthesis can be enhanced via vibrational excitation of N2 molecules in a non-thermal plasma, without affecting the subsequent hydrogenation of N-containing surface intermediates and desorption of NH3. Plasma-activation of N2 is proposed to enhance the nitrogen dissociation rate due to the pre-activation of the N2 molecule, decreasing the apparent barrier for N2 dissociation, thereby increasing the ammonia synthesis rate.25,26 The authors also reported that plasma-catalytic NH3 synthesis can result in NH3 formation beyond thermal equilibrium,27 which the authors attributed to the plasma-activation of N2, thereby decreasing the barrier for N2 dissociation and pushing the equilibrium towards NH3 formation.
Rouwenhorst et al.28 substantiated the claim that the N2 dissociation barrier can be decreased by plasma-activation of N2 with a kinetic analysis for Ru-based catalysts in a narrow temperature range (200–330 °C) at atmospheric pressure in a dielectric barrier discharge (DBD) reactor with relatively low plasma powers between 83 and 367 J L−1. It was found that the dissociation of N2 over the catalyst is still the rate-limiting step for ammonia synthesis.28 This is supported by the similarity between the effects of electronegativity of supports and promotors on the activity of Ru-catalysts, for both thermal catalysis and plasma-catalysis. The lower electronegativity of the support and promoter leads to increased activity for NH3 synthesis due to the enhancement of N2 dissociation.28,29 The barrier for N2 dissociation was lowered from 60–115 kJ mol−1 for thermal catalysis to 20–40 kJ mol−1 for plasma-enhanced catalysis over Ru-catalysts.28 These experiments were performed at low conversion, far away from thermodynamic equilibrium and at relatively low plasma powers.
Several pathways involving species in the plasma and on the catalyst surface may contribute to plasma-catalysis, as discussed previously.28 In short, these are radical species generated in the plasma (i.e., N, H, and NHX), which may react in the plasma phase and/or on the catalyst surface to form NH3,30 while also plasma-activated molecular N2 may dissociate on the catalyst surface,25 with subsequent hydrogenation to form NH3.
The process conditions (i.e. temperature, pressure, plasma power and properties), as well as the type of catalyst probably determine the dominant pathway for NH3 formation. The goal is to get a better understanding of the dominant pathways for NH3 synthesis on Ru-based catalysts, in the temperature range between 50 °C and 500 °C and for a specific energy input (SIE) of the plasma of 11.4–19.2 kJ L−1. We will show that plasma chemistry dominates at low temperatures (<175 °C) as the empty quartz reactor, bare MgO and Ru/MgO all yield the same outlet ammonia concentration. Ru catalyzes plasma-driven NH3 synthesis exclusively at temperatures above 175 °C, allowing Nads to hydrogenate and NH3 to desorb. Between 175 °C and 300 °C, ammonia synthesis proceeds mainly via adsorption of N radicals generated in the plasma, which are subsequently hydrogenated to NH3 on the catalyst. At higher temperatures, the thermodynamic equilibrium of the reactants and the product in the ground-state is surpassed, which is attributed to a combination of catalytic ammonia synthesis with both ground-state and excited molecular N2, as well as catalytic hydrogenation of N radicals generated in the plasma, competing with thermo-catalytic ammonia decomposition.
Results
The experimental procedure can be found in the ESI.† In the upcoming section, the results of the catalytic tests for MgO, Ru/MgO and Ru–K/MgO with and without a plasma are presented. The results of catalyst characterization and plasma characterization with Lissajous plots and UV-vis spectroscopy can be found in the ESI.†Thermal catalysis
The Ru-catalysts were tested for catalytic activity in the absence of a plasma, at atmospheric pressure, a constant H2:N2 ratio of 1:1 and a total flow rate of 20 mL min−1, using typically 130 mg catalyst.
The Ru/MgO catalyst showed little thermal activity for NH3 synthesis under the conditions used. Formation of ammonia never reached the detection limit of the gas analyzer, i.e. ∼0.07 mol% equivalent to a catalyst activity of 1440 μmol NH3 h−1 gcat−1. This is in line with results of Aika et al.,31 reporting an NH3 synthesis rate as low as 60 μmol NH3 h−1 gcat−1 over Ru/MgO for H2:N2 = 3:1 at 315 °C. Indeed, that is far below the detection limit of the gas analyzer used in this work. Fig. 1 presents the result for co-feeding 0.5 or 1.0 mol% NH3. Again, no ammonia formation could be detected. However, the Ru/MgO catalyst is active for NH3 decomposition at 400 °C and above, at which temperatures decomposition is thermodynamically possible. Thermodynamic equilibrium is approached at 500 °C.
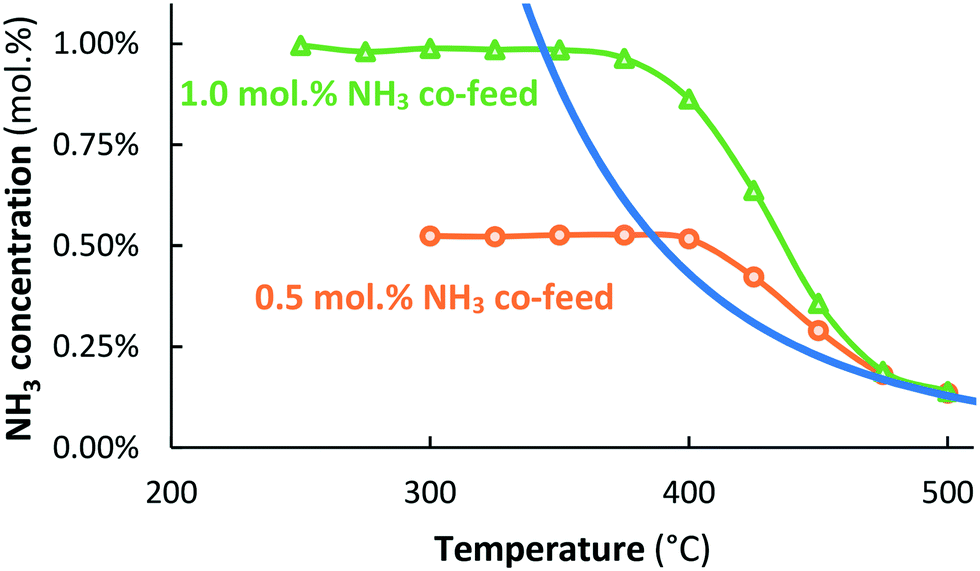 | ||
Fig. 1 Activity for thermal-catalytic NH3 decomposition with 0.5 mol% NH3 co-feed ( orange circles) and 1.0 mol% NH3 co-feed ( orange circles) and 1.0 mol% NH3 co-feed ( green triangles) over Ru/MgO. Total flow rate 20 mL min−1, H2:N2 = 1:1, catalyst loading 130 mg (250–300 μm). green triangles) over Ru/MgO. Total flow rate 20 mL min−1, H2:N2 = 1:1, catalyst loading 130 mg (250–300 μm). | ||
The potassium-promoted Ru/MgO catalyst (Ru–K/MgO) is more active than the unpromoted Ru/MgO catalyst, for both NH3 synthesis and NH3 decomposition (see Fig. 2), in agreement with the literature.31,32 NH3 is formed over the Ru–K/MgO catalyst at about 310 °C and above. The NH3 synthesis rate over Ru–K/MgO is about 1750 μmol NH3 h−1 gcat−1 at 315 °C, in reasonable agreement with literature values for the NH3 synthesis rate (560–1060 μmol NH3 h−1 gcat−1) over Ru–K/MgO at the same temperature and a H2:N2 ratio of 3:1.31 The higher NH3 synthesis rate reported here can be attributed to the lower H2:N2 ratio of 1:1, preventing too high hydrogen coverage, which suppresses adsorption of nitrogen.33
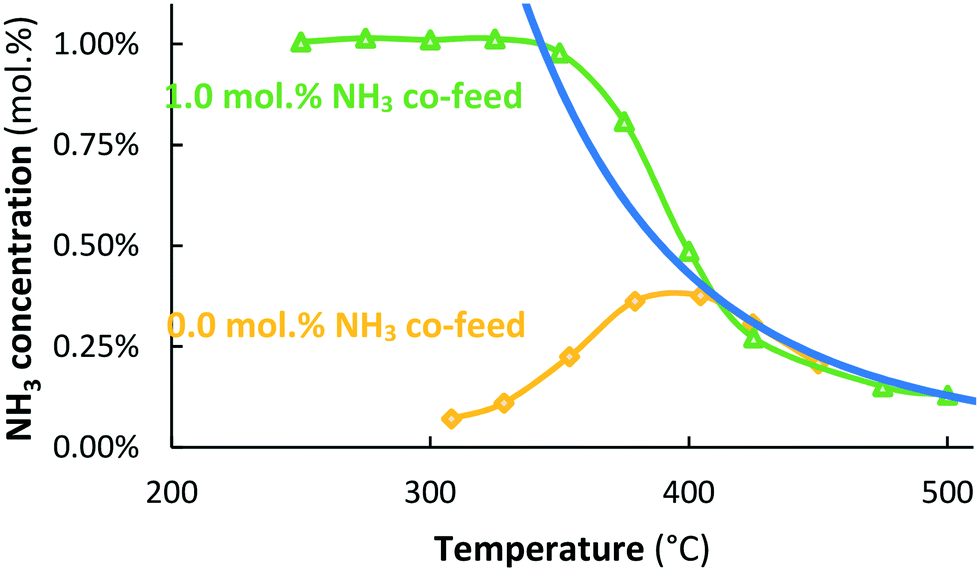 | ||
Fig. 2 Activity for thermal-catalytic NH3 synthesis with 0.0 mol% NH3 co-feed ( yellow diamonds) and NH3 decomposition with 1.0 mol% NH3 co-feed ( yellow diamonds) and NH3 decomposition with 1.0 mol% NH3 co-feed ( green triangles) over Ru–K/MgO. Total flow rate 20 mL min−1, H2:N2 = 1:1, catalyst loading 130 mg (250–300 μm). green triangles) over Ru–K/MgO. Total flow rate 20 mL min−1, H2:N2 = 1:1, catalyst loading 130 mg (250–300 μm). | ||
An Arrhenius plot based on the data between 320 and 355 °C results in an apparent activation barrier for NH3 synthesis of 92 kJ mol−1 (see Fig. S5†), in line with the literature for NH3 synthesis over Ru-catalysts.31,32 This barrier is attributed to the nitrogen dissociation step, the rate-limiting step for NH3 synthesis over Ru-catalysts.29
Fig. 2 also shows that Ru–K/MgO becomes active for NH3 decomposition at 350 °C when co-feeding 1.0 mol% NH3, at significantly lower temperatures than needed for NH3 decomposition over Ru/MgO (Fig. 1). The recombination of N atoms to form N2 is the rate limiting step for NH3 decomposition over Ru-catalysts.34,35 Summarizing, the potassium-promoted catalyst is significantly more active for both NH3 synthesis and NH3 decomposition, as expected.32,36
Plasma-catalysis
In case the plasma is illuminated, all reactor packings (MgO, Ru/MgO and Ru–K/MgO) show conversion to NH3, as shown in Fig. 3. An empty reactor without a packed bed, but with the spacer and quartz wool, shows an outlet NH3 concentration of 0.14–0.17 mol% independent of temperature, indicating that NH3 is formed via chemical reactions in the plasma via radicals.20 The presence of a packed bed of MgO particles does not influence the conversion as compared to the empty reactor, indicating that the MgO surface does not play a significant role in the conversion of plasma-activated species to NH3. This agrees well with observations in the literature.37 Also, the presence of MgO seems not to influence the plasma significantly.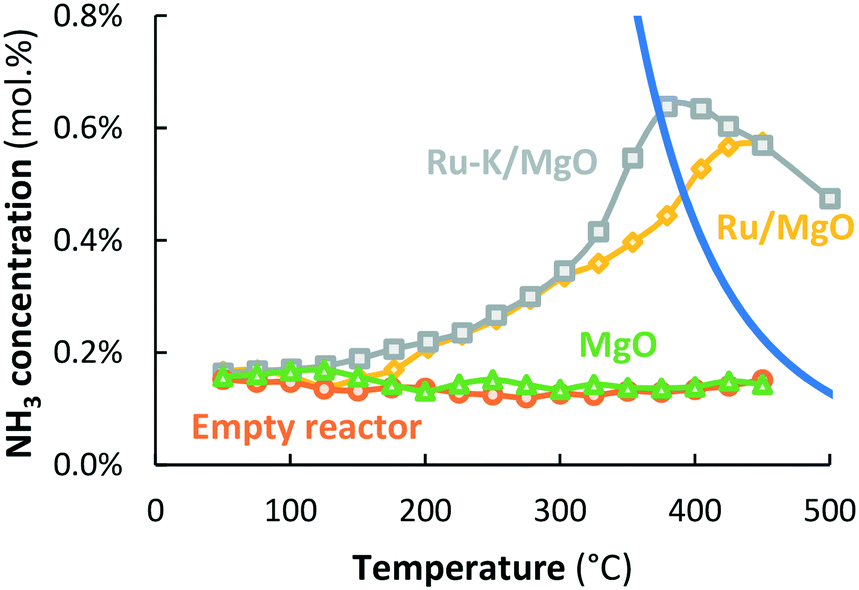 | ||
Fig. 3 Activity for plasma-catalytic NH3 synthesis (and decomposition) for an empty reactor (only the spacer & quartz wool,  orange circles), MgO ( orange circles), MgO ( green triangles), Ru/MgO ( green triangles), Ru/MgO ( yellow diamonds), and Ru–K/MgO ( yellow diamonds), and Ru–K/MgO ( grey squares). Total flow rate 20 mL min−1, H2:N2 = 1:1 (no NH3 co-feed), catalyst loading 130 mg (250–300 μm), plasma power 3.8 W (SIE = 11.4 kJ L−1). grey squares). Total flow rate 20 mL min−1, H2:N2 = 1:1 (no NH3 co-feed), catalyst loading 130 mg (250–300 μm), plasma power 3.8 W (SIE = 11.4 kJ L−1). | ||
Below 175 °C, the conversion obtained with Ru/MgO is similar to the conversion with bare MgO, implying that plasma chemistry is the dominant NH3 formation mechanism at low temperature, rather than any catalytic contribution over the Ru surface. This is in line with the fact that ammonia desorption from Ru/Al2O3, Ru/SiO2, and Ru/AC requires at least 180 °C,38 although weaker adsorption of ammonia on Ru/MgO is suggested by Xie et al.39 and Zhang et al.40 based on TPD experiments, as well as by Szmigiel et al.41 based on temperature programmed reaction experiments with adsorbed Nads with H2. In any case, hydrogenation of N or NHX surface species and/or desorption of ammonia limit the reaction at temperatures below 175 °C, based on the temperature programmed reaction experiments performed by Szmigiel et al.41
At temperatures above 175 °C, the conversion to NH3 over Ru/MgO increases with increasing temperature. Consequently, the presence of Ru increases the rate of formation of ammonia compared to bare MgO, demonstrating a catalytic effect of Ru. Furthermore, NH3 formation surpasses the thermodynamic equilibrium at temperatures above 400 °C.
Ru–K/MgO has a similar activity profile to Ru/MgO. However, the onset temperature for the catalytic conversion is lower (125 °C), which can be attributed to repulsion between adsorbed NHX species and potassium, and subsequently enhancement of NH3 desorption caused by the potassium promoter at such low temperatures. As discussed above, distinction between effects via, on the one hand, the rate of hydrogenation of NHX species and on the other hand, the rate of desorption of ammonia cannot be made and is not important for the discussion here. There is ample proof that alkali promotion (K, Cs) on Fe and Ru catalysts promotes hydrogenation and/or ammonia desorption from Fe (ref. 42) and Ru.41,43,44
The conversions on Ru–K/MgO and Ru/MgO are similar up to 300 °C. In the temperature window above 300 °C, however, the conversion on Ru–K/MgO is higher than that on Ru/MgO. This is in line with the observation that thermal-catalytic NH3 synthesis on Ru–K/MgO is significant at 325 °C and above, as shown in Fig. 2. Thus, dissociative adsorption of molecular N2 contributes to plasma catalysis in this temperature window, because K also promotes N2 dissociation, in line with the literature29,45,46 as well as our previous results.28 The highest energy yield obtained for Ru–K/MgO at 390 °C is 1.23 gNH3 kW h−1, which is far below the target of 150–200 gNH3 kW h−1.
Plasma-catalysis beyond thermal equilibrium
The NH3 concentration on Ru/MgO goes through a maximum at about 420 °C, after which the conversion decreases (see Fig. 3). Apparently, the Ru/MgO catalyst is active for thermal NH3 decomposition above 390 °C, in line with Fig. 1. A similar result is obtained with Ru–K/MgO at somewhat lower temperature, i.e. above 370 °C, whereas the ammonia concentrations obtained with Ru/MgO and Ru–K/MgO at 450 °C and above are the same. The conversion decreases further at higher temperatures. This is in line with theoretical calculations performed by Mehta et al.27 for catalysts with an intermediate N binding energy, for which conversions beyond the thermal equilibrium are predicted upon plasma-activation of N2. N2 dissociation is the rate-determining step for NH3 synthesis over such catalysts, and plasma-activation enhances the rate of N2 dissociation towards NH3 formation.Fig. 4 shows the effect of co-feeding of 0.5 and 1.0 mol% NH3 to Ru/MgO and Ru–K/MgO. The results without addition of ammonia (see Fig. 3) are repeated in Fig. 4 for easy comparison. The plasma-catalytic conversion on Ru/MgO and Ru–K/MgO is different in the temperature window where thermodynamic equilibrium is not yet achieved, as Ru–K/MgO is more active for NH3 synthesis. Furthermore, the results in Fig. 4 confirm that Ru–K/MgO is more active for ammonia decomposition than Ru/MgO, as decomposition is observed when increasing the temperature just beyond thermodynamic equilibrium. In the case of Ru/MgO, significantly higher temperatures are required before observing significant ammonia decomposition. Above 450 °C, both Ru/MgO and Ru–K/MgO approach the same conversion above thermal equilibrium, independent of the co-feed NH3 concentration. This will be discussed below.
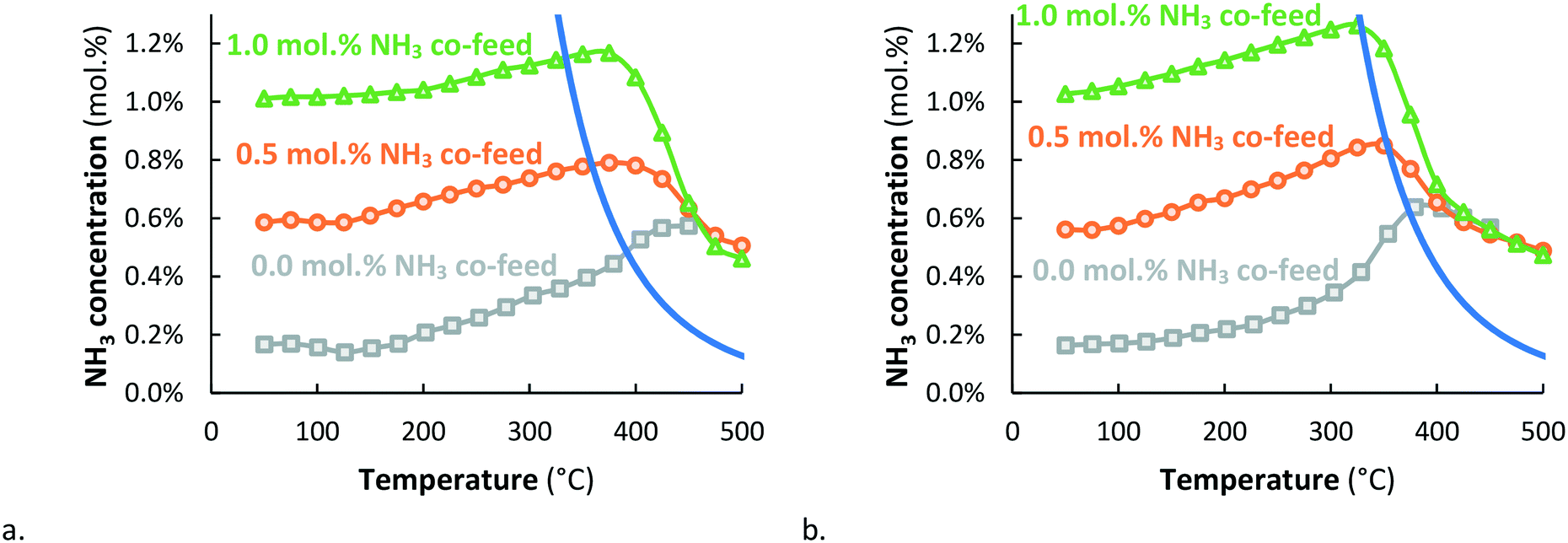 | ||
Fig. 4 Activity for plasma-catalytic NH3 synthesis (and NH3 decomposition) for Ru/MgO (a.) & Ru–K/MgO (b.) for various NH3 co-feed concentrations (0.0 mol% ( grey squares), 0.5 mol% ( grey squares), 0.5 mol% ( orange circles) and 1.0 mol% ( orange circles) and 1.0 mol% ( green triangles)). Total flow rate 20 mL min−1, H2:N2 = 1:1, catalyst loading 130 mg (250–300 μm), plasma power 3.8 W (SIE = 11.4 kJ L−1). green triangles)). Total flow rate 20 mL min−1, H2:N2 = 1:1, catalyst loading 130 mg (250–300 μm), plasma power 3.8 W (SIE = 11.4 kJ L−1). | ||
Effect of plasma power
Fig. 5 shows that the ammonia concentration over Ru–K/MgO at temperatures above 450 °C depends on the plasma power, which was varied between 3.8 W and 6.4 W. Additional experiments at 4.8 W and 6.4 W confirm that the NH3 concentration obtained at temperatures above 400 °C does not depend on co-feeding of ammonia (see Fig. S6 and S7†), very similar to the result presented in Fig. 4 at 3.8 W power.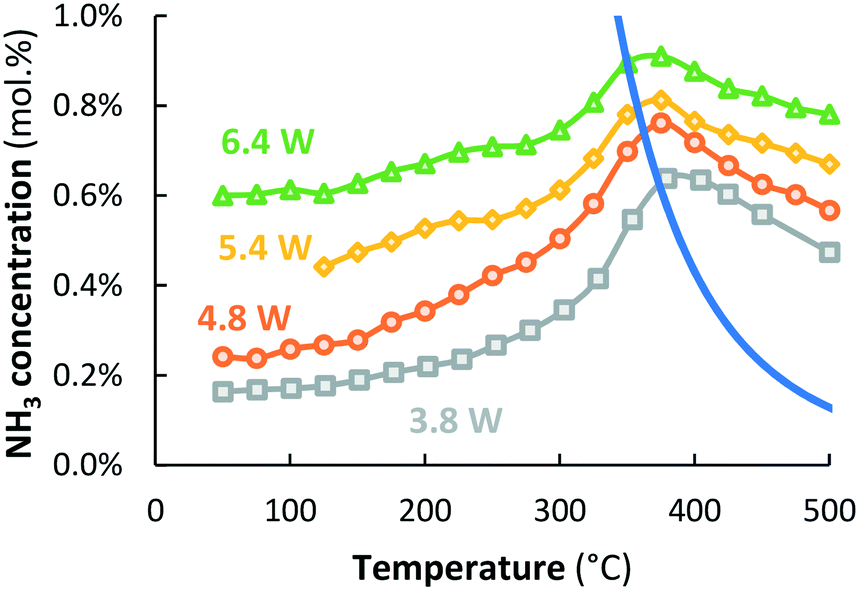 | ||
Fig. 5 Activity for plasma-catalytic NH3 synthesis (and NH3 decomposition) for Ru/MgO for various plasma powers (3.8 W (SIE = 11.4 kJ L−1,  grey squares), 4.8 W (SIE = 14.4 kJ L−1, grey squares), 4.8 W (SIE = 14.4 kJ L−1,  orange circles), 5.4 W (SIE = 16.3 kJ L−1, orange circles), 5.4 W (SIE = 16.3 kJ L−1,  yellow diamonds) and 6.4 W (SIE = 19.2 kJ L−1, yellow diamonds) and 6.4 W (SIE = 19.2 kJ L−1,  green triangles)). Total flow rate 20 mL min−1, H2:N2 = 1:1 (no NH3 co-feed), catalyst loading 130 mg (250–300 μm). green triangles)). Total flow rate 20 mL min−1, H2:N2 = 1:1 (no NH3 co-feed), catalyst loading 130 mg (250–300 μm). | ||
Discussion
The discussion aims to identify the dominant mechanistic pathways for plasma-driven NH3 synthesis, in the presence and absence of an active catalyst and under various process conditions.Activity trends for plasma-catalysis
Various authors reported that the presence of a transition metal catalyst enhances ammonia synthesis in a non-thermal plasma.23,39,47–53 On the other hand, the reactivity of the support is not always considered in plasma-catalytic systems. However, some authors have reported on the difference in conversion for a supported metal catalyst and the bare support. Peng et al.50,51 reported on plasma-driven conversion of an empty reactor, a bare support, and a supported metal catalyst (with a promoter) at near ambient temperature. The plasma-driven conversion decreased in the order Ru–Cs/MgO > Ru/MgO ≈ MgO > empty reactor.50 Similarly, Wang et al.48 reported on plasma-conversion over various metal catalysts supported on Al2O3 in a DBD reactor, as well as an empty reactor under near-ambient conditions. The plasma-driven conversion decreased in the order Ni/Al2O3 ≈ Cu/Al2O3 > Fe/Al2O3 > bare Al2O3 > empty reactor.48 The plasma-driven conversion in a plasma-reactor packed with Al2O3 support increases with increasing temperature with an activity decreasing in the order Co/Al2O3 ≈ Ni/Al2O3 ≈ Ru/Al2O3 > Al2O3, as reported by Barboun et al.49In the current work, a Ru metal loading of 2 wt% is used to minimize the potential effects of the metal nanoparticles on the plasma characteristics. Patil et al.53 showed that high metal loadings of 10 wt% on oxide supports may result in changes in the discharge characteristics. On the other hand, Herrera et al.54 concluded that the impact of metal nanoparticles on the discharge characteristics is not significant for 5 wt% metal loadings on Al2O3. In the current work, there is no significant effect of the Ru metal loading, as supported by the similarity in the Lissajous figures for MgO and Ru–K/MgO packing (see Fig. S3†).
As shown in Fig. 3, the conversion is constant with temperature for the empty reactor (i.e., quartz wool only), as well as for MgO at a SIE of 11.4 kJ L−1. Thus, the MgO support has no significant influence on the plasma-chemical reactions to NH3, resulting in typically 0.15 mol% NH3, as shown in Fig. 3 and 6 for easy comparison. The fact that NH3 forms in the plasma phase or on the reactor wall implies that N, H, and NHX radials are present in the plasma, as previously reported by various authors.55,56
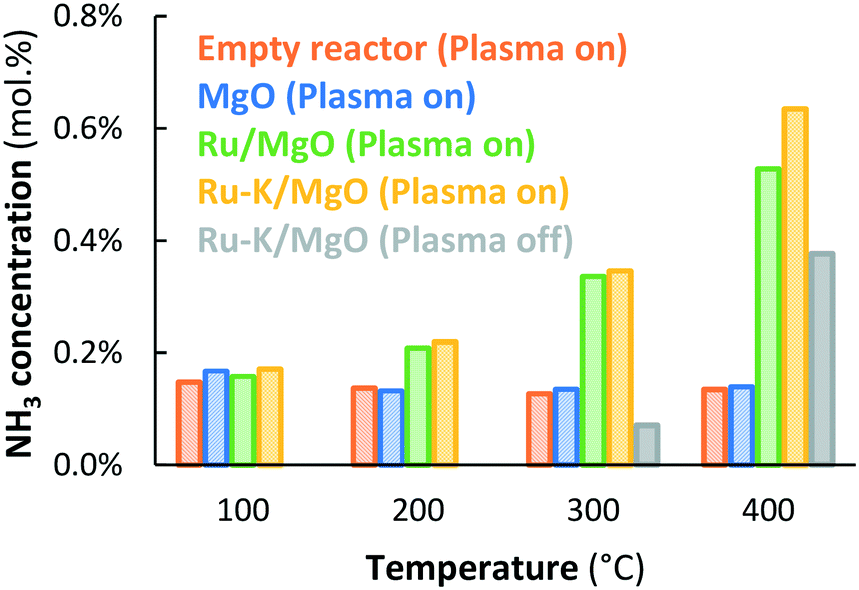 | ||
Fig. 6 NH3 outlet concentration for plasma-driven NH3 synthesis and thermal-catalytic NH3 synthesis as a function of temperature. From left to right: The empty quartz reactor ( orange striped – plasma on), MgO packing ( orange striped – plasma on), MgO packing ( blue striped – plasma on), Ru/MgO catalyst ( blue striped – plasma on), Ru/MgO catalyst ( green spotted – plasma on), and Ru–K/MgO catalyst ( green spotted – plasma on), and Ru–K/MgO catalyst ( yellow checkerboard – plasma on; yellow checkerboard – plasma on;  grey single color – plasma off). Total flow rate 20 mL min−1, H2:N2 = 1:1, no NH3 co-feed, catalyst loading 130 mg (250–300 μm), plasma power 3.8 W (SIE = 11.4 kJ L−1). grey single color – plasma off). Total flow rate 20 mL min−1, H2:N2 = 1:1, no NH3 co-feed, catalyst loading 130 mg (250–300 μm), plasma power 3.8 W (SIE = 11.4 kJ L−1). | ||
NH3 synthesis is catalyzed on the Ru metal in the presence of a plasma when operating above the apparent onset hydrogenation of Nads and subsequent NH3 desorption from the Ru/MgO and Ru–K/MgO catalysts, 175 °C and 125 °C, respectively (see Fig. 3). The activity of Ru/MgO and Ru–K/MgO in the presence of a plasma is similar in the temperature window between 200 °C and 300 °C (see Fig. 3). Furthermore, the catalysts are not thermally active for NH3 synthesis in the temperature window below 300 °C in the absence of a plasma, due to kinetic limitations for N2 dissociation.28,29 The fact that potassium does not influence ammonia formation between 200 °C and 300 °C (see Fig. 3) implies that ammonia synthesis in this temperature regime cannot proceed via dissociation of ground-state N2 or plasma-activated N2, as potassium would enhance the dissociation rate of N2.29,46 Thus, the reaction proceeds dominantly via N radicals rather than molecular N2. This is further supported by density functional theory (DFT) calculations performed by Engelmann et al.30
The N radicals may react on the catalyst surface along two pathways. Firstly, the N radicals may adsorb on the Ru surface, followed by hydrogenation on the surface and NH3 desorption. Secondly, an Eley–Rideal type of reaction, e.g. N + Hads → NHads, without adsorbing the N radical first, may contribute, as proposed by Engelmann et al.30,55 with DFT calculations and Yamijala et al.57 with ab initio calculations.
Ammonia synthesis during plasma-catalysis on Ru–K/MgO is significantly faster than on Ru/MgO in the temperature window between 300 °C and 400 °C (see Fig. 3 and 6), in which Ru–K/MgO is also thermally active (see Fig. 2). Molecular N2 can dissociate thermally, suggesting that dissociation of plasma-activated molecular N2 is even more facile.28 The potassium promoter enhances the N2 dissociation rate,29,46 explaining the higher activity for Ru–K/MgO as compared to Ru/MgO. Thus, the resulting activity is a mix of a molecular mechanism via N2 dissociation of both ground-state N2 and probably plasma-activated N2, as well as a reaction pathway via N radicals generated in the plasma phase, as discussed above.
The contribution of ground-state N2 and plasma-activated N2 to NH3 synthesis depends not only on the catalyst activity for N2 dissociation, but also on the plasma power.28 In our previous work, we showed that dissociation of plasma-activated, molecular N2 and subsequent hydrogenation is dominant over Ru-catalysts for low plasma powers in the range of 0.1–0.4 kJ L−1 at 200–300 °C.28 In contrast, the plasma power in our current work is much higher, typically 11–19 kJ L−1, implying substantially higher concentrations of N radicals.
N-Recombination to N2, i.e. the rate limiting step for NH3 decomposition over Ru-catalysts,34 is fast over Ru–K/MgO at temperatures above 350 °C in the absence of a plasma (see Fig. 2). Therefore, the thermo-catalytic ammonia concentration is controlled by thermodynamic equilibrium at 350 °C. In the presence of a plasma, higher ammonia concentrations are attained than would be expected based on thermodynamic equilibrium (see Fig. 3–5), which will be discussed hereafter.
Beyond thermal equilibrium
Plasma-driven conversions surpassing thermodynamic equilibrium are frequently reported for CO2 splitting,58 dry reforming of methane (DRM)59 and non-oxidative coupling of methane (NOCM),60 mostly at temperatures where thermal reactions do not contribute at all. The results in Fig. 3–5 show surpassing thermodynamic equilibrium at temperatures at which thermo-catalytic ammonia synthesis as well as thermo-catalytic ammonia decomposition proceeds significantly, as schematically presented in Fig. 7. The outlet ammonia concentration is the result of the competition between three reactions, i.e. on the one hand NH3 synthesis via molecular N2, either in the ground-state (rf,th) or in any excited state (rf,pl) and on the other hand NH3 decomposition of the ground-state NH3 (rb,th) exclusively (see eqn (2)). Note that no distinction can be made between excited nitrogen via vibrational excitation, electronic excitation or dissociation to N radicals for r,pl.| RNH3prod = rf,th + rf,pl − rb,th | (2) |
:N2 ratio is not significantly influenced by the low concentration of added ammonia. The observation that the ammonia concentration is influenced by the level of pre-activation of N2 is in agreement with the trends predicted by the model of Mehta et al.27 (see the ESI†). In any case, the plasma-driven reaction (rf,pl) is apparently faster than thermal ammonia decomposition (rb,th), resulting in plasma-catalytic ammonia synthesis beyond equilibrium. This observation also rules out that thermal effects induced by the plasma dominate, because a temperature increase would decrease the ammonia concentration, according to the thermodynamic equilibrium.
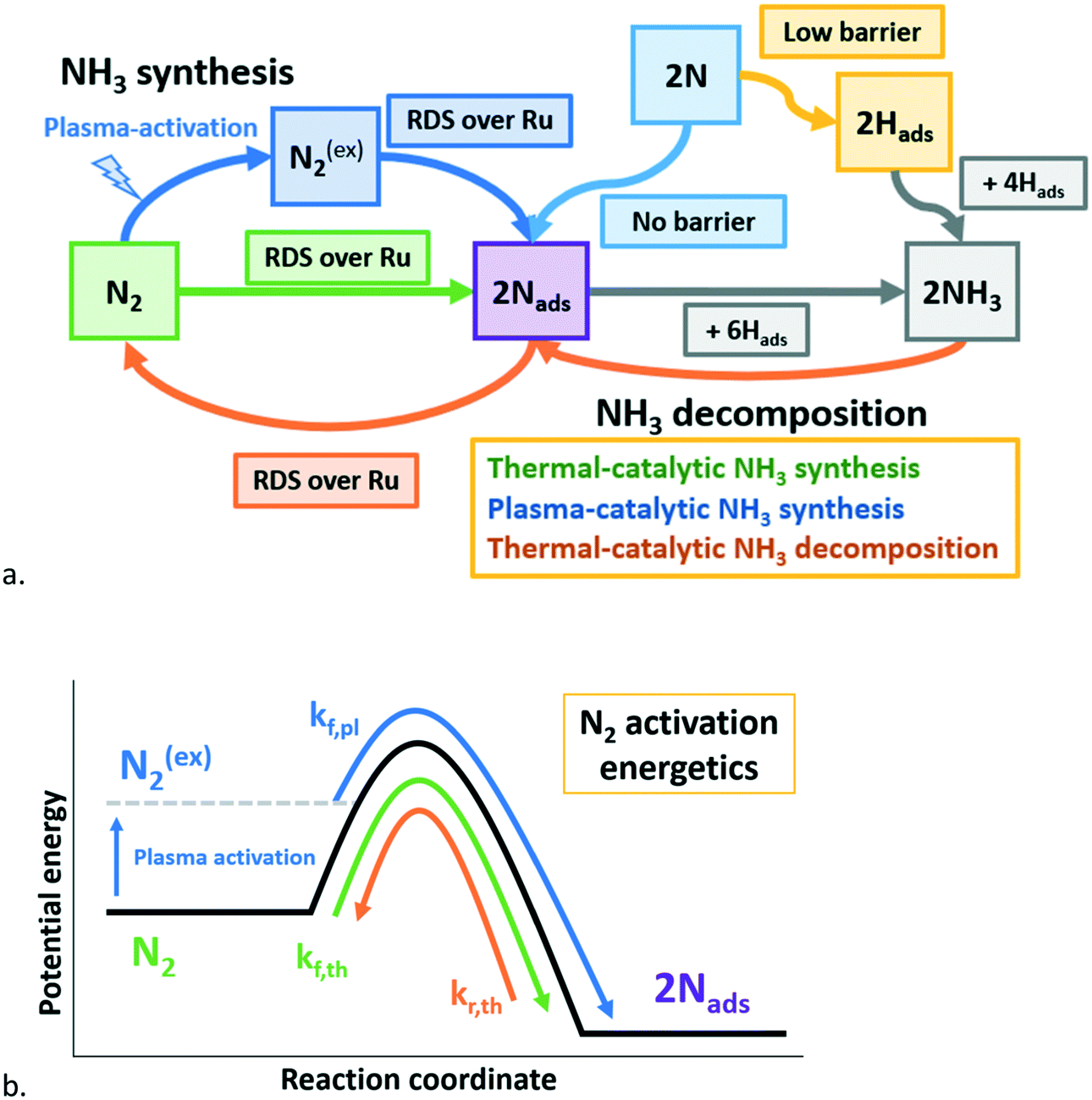 | ||
| Fig. 7 a. Schematic representation of plasma-catalytic ammonia synthesis above the onset temperature for thermal-catalysis, including reactions with ground-state N2 (thermal catalysis), reactions with plasma-excited N2 (plasma-enhanced catalysis), and reactions with N radicals (adsorption of N on empty sites and Eley–Rideal reaction with Hads).30 b. Schematic free energy for thermal-catalytic NH3 synthesis (green), plasma-catalytic NH3 synthesis (blue), and thermal-catalytic (& plasma-catalytic) NH3 decomposition (orange). Based on ref. 27. See also eqn (2). RDS: rate determining step. | ||
It is reasonable to assume that N2 and/or H2 is much more activated by the plasma than ammonia, due to the low concentration of NH3 in all experiments (<1.0 mol% compared to typically 49 mol% H2 and N2). The rate-limiting step for thermal NH3 decomposition on Ru is either N2 recombination or NHads dissociation to Nads and Hads on the surface.34 Thus, it is unlikely that plasma-activation of NH3 affects catalytic NH3 decomposition. Furthermore, the contribution of plasma induced ammonia decomposition is not significant at the plasma power applied (3.8 W), as shown in Fig. S9 in the ESI.† Therefore, decomposition of activated ammonia is not included in eqn (2) and Fig. 7b. At higher plasma power, however, ammonia may decompose in micro-discharges.55,61,62
Fig. 8 shows good correlations between the NH3 concentration measured at 450 °C and plasma power. Remarkably, the ammonia concentration also correlates linearly with the concentration of excited N2 molecules in the plasma, as measured with UV vis spectroscopy (Fig. S4†). The level of excitation of individual N2 molecules increases with power, thereby decreasing the activation barrier for dissociation (Fig. 7a). Upon further increasing the plasma power, excitation of N2 molecules eventually leads to dissociation to N radicals with further increasing plasma power.
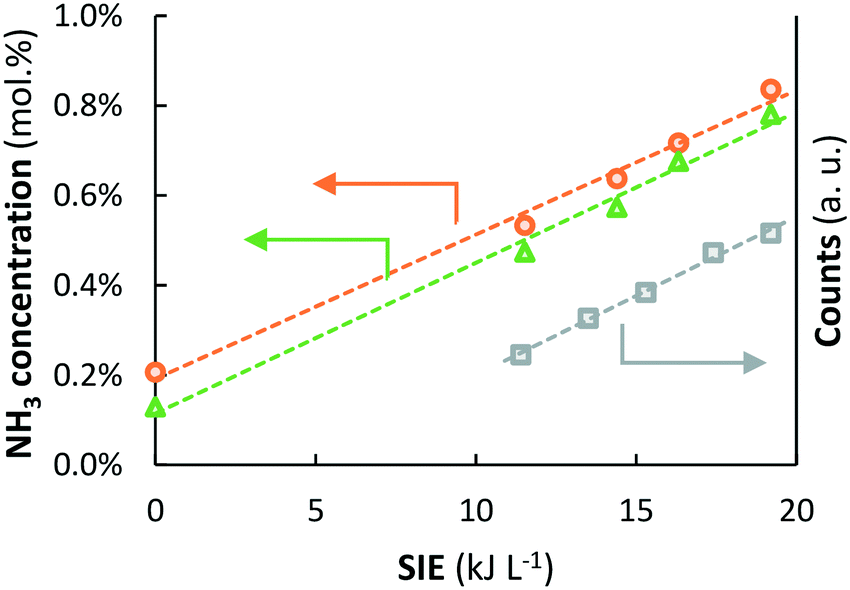 | ||
Fig. 8 Left axis: NH3 outlet concentration as a function of the SIE at 450 °C ( orange circles) and 500 °C ( orange circles) and 500 °C ( green triangles) over Ru–K/MgO (based on data from Fig. 5). Total flow rate 20 mL min−1, H2:N2 = 1:1 (no NH3 co-feed), catalyst loading 130 mg (250–300 μm), plasma power 3.8–6.4 W (SIE = 11.4–19.2 kJ L−1). Right axis: Intensity of the peak at 337 nm in the UV vis spectrum (transition from N2(C3Πu(v = 0)) to N2(B3Πg(v = 0)), green triangles) over Ru–K/MgO (based on data from Fig. 5). Total flow rate 20 mL min−1, H2:N2 = 1:1 (no NH3 co-feed), catalyst loading 130 mg (250–300 μm), plasma power 3.8–6.4 W (SIE = 11.4–19.2 kJ L−1). Right axis: Intensity of the peak at 337 nm in the UV vis spectrum (transition from N2(C3Πu(v = 0)) to N2(B3Πg(v = 0)),  grey squares) as a function of the SIE. The density of NH radicals is also measured at 336.7 nm. However, the density of NH is orders of magnitude lower than that of plasma-activated N2.55 See Section S2.2† for the interpretation of UV-vis measurements and Fig. S4† for the UV-vis spectra. grey squares) as a function of the SIE. The density of NH radicals is also measured at 336.7 nm. However, the density of NH is orders of magnitude lower than that of plasma-activated N2.55 See Section S2.2† for the interpretation of UV-vis measurements and Fig. S4† for the UV-vis spectra. | ||
Catalysts suited for thermal operation are not necessarily the optimal choice for plasma catalysis, in agreement with a theoretical argument formulated in the latest roadmap for plasma catalysis.63 This notion should have a major impact in the field, as very frequently thermal catalysts are used in plasma catalysis research. It is now experientially demonstrated that the activity for the reverse reaction is undesired and different catalysts should be considered when approaching or surpassing thermodynamic equilibrium based on ground-state molecules.
Conclusion
Plasma-catalytic NH3 synthesis has been assessed over a wide temperature window (50–500 °C). A distinction was made between plasma-chemical and plasma-catalytic effects by performing measurements with an empty quartz reactor, MgO support and MgO supported Ru-catalysts. At low temperatures (<175 °C), plasma chemistry dominates, resulting in the same ammonia outlet concentration for the empty quartz reactor, the MgO support, and the MgO supported Ru-catalysts. Plasma-driven NH3 synthesis is catalyzed by Ru at temperatures above 175 °C. The potassium promoter has no influence on the plasma-catalytic activity at temperatures with insignificant thermal activity, i.e. typically between 175 °C and 300 °C, indicating that the pathway via adsorption of N radicals is dominant.At temperatures with significant thermal activity for ammonia synthesis, i.e. above 300 °C for Ru–K/MgO, the plasma enhances the catalytic NH3 synthesis rate. The plasma-catalytic NH3 synthesis rate is then a combination of the catalytic hydrogenation of N radicals on the Ru surface and the catalytic NH3 formation via N2 dissociation of both ground-state molecular N2 and plasma-activated molecular N2.
At elevated temperatures, typically above 400 °C, plasma-catalysis results in ammonia concentrations beyond thermodynamic equilibrium for ground-state N2. Therefore, plasma-activated molecular N2 and N radicals enhance the formation of ammonia, increasing the rate of formation of ammonia more than the activity of the catalyst to decompose ammonia. With increasing plasma power, the density of plasma-activated molecular N2 and N radicals increases, thereby increasing the conversion beyond equilibrium.
Author contributions
K. H. R. R., H. G. B. B. and J. N. P. performed NH3 synthesis experiments. K. H. R. R. and B. G. performed plasma characterization experiments. K. H. R. R. and D. W. V. performed modelling work. K. H. R. R. and L. L. co-wrote the manuscript. All the authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is co-financed by TKI-Energie from Toeslag voor Topconsortia voor Kennis en Innovatie (TKI) from the Ministry of Economic Affairs and Climate Policy, the Netherlands. The authors acknowledge K. Altena-Schildkamp for N2 chemisorption and CO chemisorption measurements. The authors acknowledge T. M. L. Velthuizen for XRF analysis.References
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer and Y. M. Chiang, et al., Net-Zero Emissions Energy Systems, Science, 2018, 360(6396), eaas9793, DOI:10.1126/science.aas9793.
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Ammonia for Power, Prog. Energy Combust. Sci., 2018, 69, 63–102, DOI:10.1016/j.pecs.2018.07.001.
- J. Andersson and S. Grönkvist, Large-Scale Storage of Hydrogen, Int. J. Hydrogen Energy, 2019, 44(23), 11901–11919, DOI:10.1016/j.ijhydene.2019.03.063.
- M. Götz, J. Lefebvre, F. Mörs, A. McDaniel Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Power-to-Gas: A Technological and Economic Review, Renewable Energy, 2016, 85, 1371–1390, DOI:10.1016/j.renene.2015.07.066.
- K. H. R. Rouwenhorst, A. G. J. Van Der Ham, G. Mul and S. R. A. Kersten, Islanded Ammonia Power Systems: Technology Review & Conceptual Process Design, Renewable Sustainable Energy Rev., 2019, 114, 109339, DOI:10.1016/j.rser.2019.109339.
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, Ammonia as a Renewable Energy Transportation Media, ACS Sustainable Chem. Eng., 2017, 5(11), 10231–10239, DOI:10.1021/acssuschemeng.7b02219.
- A. Goeppert, G. A. Olah and G. K. Surya Prakash, Toward a Sustainable Carbon Cycle: The Methanol Economy, in Green Chemistry: An Inclusive Approach, Elsevier Inc., 2017, pp. 919–962, DOI:10.1016/B978-0-12-809270-5.00031-5.
- J. W. Makepeace, T. He, C. Weidenthaler, T. R. Jensen, F. Chang, T. Vegge, P. Ngene, Y. Kojima, P. E. de Jongh and P. Chen, et al., Reversible Ammonia-Based and Liquid Organic Hydrogen Carriers for High-Density Hydrogen Storage: Recent Progress, Int. J. Hydrogen Energy, 2019, 44(15), 7746–7767, DOI:10.1016/j.ijhydene.2019.01.144.
- J. Guo and P. Chen, Catalyst: NH3 as an Energy Carrier, Chem, 2017, 3(5), 709–712, DOI:10.1016/j.chempr.2017.10.004.
- W. H. Avery, A Role for Ammonia in the Hydrogen Economy, Int. J. Hydrogen Energy, 1988, 13(12), 761–773, DOI:10.1016/0360-3199(88)90037-7.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Ammonia as a Possible Element in an Energy Infrastructure: Catalysts for Ammonia Decomposition, Energy Environ. Sci., 2012, 5(4), 6278–6289, 10.1039/C2EE02865D.
- A. Hellman, K. Honkala, S. Dahl, C. H. Christensen and J. K. Nørskov, Ammonia Synthesis: State of the Bellwether Reaction, in Comprehensive Inorganic Chemistry (II), Elsevier Ltd, 2013, DOI:10.1016/B978-0-08-097774-4.00725-7.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Ammonia, 4. Green Ammonia Production, Ullmann's Encyclopedia of Industrial Chemistry, 2020, DOI:10.1002/14356007.w02_w02.
- H.-H. Kim, Y. Teramoto, A. Ogata, H. Takagi and T. Nanba, Plasma Catalysis for Environmental Treatment and Energy Applications, Plasma Chem. Plasma Process., 2016, 36(1), 45–72, DOI:10.1007/s11090-015-9652-7.
- K. H. R. Rouwenhorst, Y. Engelmann, K. Van't Veer, R. S. Postma, A. Bogaerts and L. Lefferts, Plasma-Driven Catalysis: Green Ammonia Synthesis with Intermittent Electricity, Green Chem., 2020, 22(19), 6258–6287, 10.1039/D0GC02058C.
- P. Mehta, P. Barboun, D. B. Go, J. C. Hicks and W. F. Schneider, Catalysis Enabled by Plasma Activation of Strong Chemical Bonds: A Review, ACS Energy Lett., 2019, 4(5), 1115–1133, DOI:10.1021/acsenergylett.9b00263.
- A. Bogaerts and E. C. Neyts, Plasma Technology: An Emerging Technology for Energy Storage, ACS Energy Lett., 2018, 3(4), 1013–1027, DOI:10.1021/acsenergylett.8b00184.
- E. C. Neyts, K. Ostrikov, M. K. Sunkara and A. Bogaerts, Plasma Catalysis: Synergistic Effects at the Nanoscale, Chem. Rev., 2015, 115(24), 13408–13446, DOI:10.1021/acs.chemrev.5b00362.
- P. Peng, P. Chen, C. Schiappacasse, N. Zhou, E. Anderson, D. Chen, J. Liu, Y. Cheng, R. Hatzenbeller and M. Addy, et al., A Review on the Non-Thermal Plasma-Assisted Ammonia Synthesis Technologies, J. Cleaner Prod., 2018, 177, 597–609, DOI:10.1016/j.jclepro.2017.12.229.
- J. Hong, S. Prawer and A. B. Murphy, Plasma Catalysis as an Alternative Route for Ammonia Production: Status, Mechanisms, and Prospects for Progress, ACS Sustainable Chem. Eng., 2018, 6(1), 15–31, DOI:10.1021/acssuschemeng.7b02381.
- P. Peng, C. Schiappacasse, N. Zhou, M. Addy, Y. Cheng, Y. Zhang, K. Ding, Y. Wang, P. Chen and R. Ruan, Sustainable Non-thermal Plasma-assisted Nitrogen Fixation - Synergistic Catalysis, ChemSusChem, 2019, 1–12, DOI:10.1002/cssc.201901211.
- S. Li, J. A. M. Jimenez, V. Hessel and F. Gallucci, Recent Progress of Plasma-Assisted Nitrogen Fixation Research: A Review, Processes, 2018, 6(12), 248, DOI:10.3390/pr6120248.
- H.-H. Kim, Y. Teramoto, A. Ogata, H. Takagi and T. Nanba, Atmospheric-Pressure Nonthermal Plasma Synthesis of Ammonia over Ruthenium Catalysts, Plasma Processes Polym., 2017, 14(6), 1–9, DOI:10.1002/ppap.201600157.
- K. H. R. Rouwenhorst and L. Lefferts, Feasibility Study of Plasma-Catalytic Ammonia Synthesis for Energy Storage Applications, Catalysts, 2020, 10(9), 999, DOI:10.3390/catal10090999.
- P. Mehta, P. Barboun, F. A. Herrera, J. Kim, P. Rumbach, D. B. Go, J. C. Hicks and W. F. Schneider, Overcoming Ammonia Synthesis Scaling Relations with Plasma-Enabled Catalysis, Nat. Catal., 2018, 1(4), 269–275, DOI:10.1038/s41929-018-0045-1.
- K. H. R. Rouwenhorst and L. Lefferts, On the Mechanism for the Plasma-Activated N2 Dissociation on Ru Surfaces, J. Phys. D: Appl. Phys., 2021 Search PubMed , submitted.
- P. Mehta, P. Barboun, Y. Engelmann, D. B. Go, A. Bogaerts, W. F. Schneider and J. C. Hicks, Plasma-Catalytic Ammonia Synthesis Beyond the Equilibrium Limit, ACS Catal., 2020, 10(12), 6726–6734, DOI:10.1021/acscatal.0c00684.
- K. H. R. Rouwenhorst, H.-H. Kim and L. Lefferts, Vibrationally Excited Activation of N2 in Plasma-Enhanced Catalytic Ammonia Synthesis: A Kinetic Analysis, ACS Sustainable Chem. Eng., 2019, 7(20), 17515–17522, DOI:10.1021/acssuschemeng.9b04997.
- K.-I. Aika, Role of Alkali Promoter in Ammonia Synthesis over Ruthenium Catalysts—Effect on Reaction Mechanism, Catal. Today, 2017, 286, 14–20, DOI:10.1016/j.cattod.2016.08.012.
- Y. Engelmann, K. van't Veer, Y. Gorbanev, E. C. Neyts, W. F. Schneider and A. Bogaerts, Plasma Catalysis for Ammonia Synthesis: Contributions of Eley-Rideal Reactions, ACS Catal., 2021 Search PubMed , submitted.
- K. Aika, T. Takano and S. Murata, Preparation and Characterization of Chlorine-Free Ruthenium Catalysts and the Promoter Effect in Ammonia Synthesis. 3. A Magnesia-Supported Ruthenium Catalyst, J. Catal., 1992, 136(1), 126–140, DOI:10.1016/0021-9517(92)90112-U.
- M. Muhler, F. Rosowski, O. Hinrichsen, A. Hornung and G. Ertl, Ruthenium as Catalyst for Ammonia Synthesis, Stud. Surf. Sci. Catal., 1996, 101, 317–326, DOI:10.1016/S0167-2991(96)80242-4.
- H. Liu, Ammonia Synthesis Catalyst 100 Years: Practice, Enlightenment and Challenge, Chin. J. Catal., 2014, 35(10), 1619–1640, DOI:10.1016/S1872-2067(14)60118-2.
- F. Hayashi, Y. Toda, Y. Kanie, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, Ammonia Decomposition by Ruthenium Nanoparticles Loaded on Inorganic Electride C12A7:E−, Chem. Sci., 2013, 8, 3124–3130, 10.1039/c3sc50794g.
- J. C. Ganley, F. S. Thomas, E. G. Seebauer and R. I. Masel, A Priori Catalytic Activity Correlations: The Difficult Case of Hydrogen Production from Ammonia, Catal. Lett., 2004, 96(3–4), 117–122, DOI:10.1023/B:CATL.0000030108.50691.d4.
- K.-I. Aika, K. Shimazaki, Y. Hattori, A. Ohya, S. Ohshima, K. Shirota and A. Ozaki, Support and Promoter Effect of Ruthenium Catalyst. I. Characterization of Alkali-Promoted Ruthenium/Alumina Catalysts for Ammonia Synthesis, J. Catal., 1985, 92(2), 296–304, DOI:10.1016/0021-9517(85)90264-7.
- B. S. Patil, A. S. R. van Kaathoven, F. Peeters, N. Cherkasov, Q. Wang, J. Lang and V. Hessel, Deciphering the Synergy between Plasma and Catalyst Support for Ammonia Synthesis in a Packed DBD Reactor, J. Phys. D: Appl. Phys., 2020, 53(14), 144003, DOI:10.1088/1361-6463/ab6a36.
- R. Pandya, R. Mane and C. V. Rode, Cascade Dehydrative Amination of Glycerol to Oxazoline, Catal. Sci. Technol., 2018, 8(11), 2954–2965, 10.1039/c8cy00185e.
- Q. Xie, S. Zhuge, X. Song and M. Lu, Non-Thermal Atmospheric Plasma Synthesis of Ammonia in a DBD Reactor Packed with Various Catalysts, J. Phys. D: Appl. Phys., 2019, 53(6), 064002, DOI:10.1088/1361-6463/ab57e5.
- J. Zhang, L. Wang, C. Li, S. Jin and C. Liang, Selective Hydrogenolysis of Dibenzofuran over Highly Efficient Pt/MgO Catalysts to o-Phenylphenol, Org. Process Res. Dev., 2018, 22(1), 67–76, DOI:10.1021/acs.oprd.7b00339.
- D. Szmigiel, H. Bielawa, M. Kurtz, O. Hinrichsen, M. Muhler, W. Raróg, S. Jodzis, Z. Kowalczyk, L. Znak and J. Zieliński, The Kinetics of Ammonia Synthesis over Ruthenium-Based Catalysts: The Role of Barium and Cesium, J. Catal., 2002, 205(1), 205–212, DOI:10.1006/jcat.2001.3431.
- D. R. Strongin and G. A. Somorjai, The Effects of Potassium on Ammonia Synthesis over Iron Single-Crystal Surfaces, J. Catal., 1988, 109(1), 51–60, DOI:10.1016/0021-9517(88)90184-4.
- S. Dahl, J. Sehested, C. J. H. Jacobsen, E. Törnqvist and I. Chorkendorff, Surface Science Based Microkinetic Analysis of Ammonia Synthesis over Ruthenium Catalysts, J. Catal., 2000, 192(2), 391–399, DOI:10.1006/jcat.2000.2857.
- O. Hinrichsen, F. Rosowski, M. Muhler and G. Ertl, The Microkinetics of Ammonia Synthesis Catalyzed by Cesium-Promoted Supported Ruthenium, Chem. Eng. Sci., 1996, 51(10), 1683–1690, DOI:10.1016/0009-2509(96)00027-9.
- G. Ertl, Mechanisms of Heterogeneous Catalysis, in Reactions at Solid Surfaces, John Wiley & Sons, Inc., Hoboken (NJ), 2009, pp. 123–139 Search PubMed.
- J. J. Mortensen, B. Hammer and J. K. Nørskov, Alkali Promotion of N2 Dissociation over Ru(0001), Phys. Rev. Lett., 1998, 80(19), 4333–4336, DOI:10.1103/PhysRevLett.80.4333.
- S. Li, T. van Raak and F. Gallucci, Investigating the Operation Parameters for Ammonia Synthesis in Dielectric Barrier Discharge Reactors, J. Phys. D: Appl. Phys., 2020, 53(1), 014008, DOI:10.1088/1361-6463/ab4b37.
- Y. Wang, M. Craven, X. Yu, J. Ding, P. Bryant, J. Huang and X. Tu, Plasma-Enhanced Catalytic Synthesis of Ammonia over a Ni/Al2O3 Catalyst at Near-Room Temperature: Insights into the Importance of the Catalyst Surface on the Reaction Mechanism, ACS Catal., 2019, 9, 10780–10793, DOI:10.1021/acscatal.9b02538.
- P. M. Barboun, P. Mehta, F. Herrera, D. B. Go, W. F. Schneider and J. C. Hicks, Distinguishing Plasma Contributions to Catalyst Performance in Plasma-Assisted Ammonia Synthesis, ACS Sustainable Chem. Eng., 2019, 7(9), 8621–8630, DOI:10.1021/acssuschemeng.9b00406.
- P. Peng, Y. Li, Y. Cheng, S. Deng, P. Chen and R. Ruan, Atmospheric Pressure Ammonia Synthesis Using Non-Thermal Plasma Assisted Catalysis, Plasma Chem. Plasma Process., 2016, 36(5), 1201–1210, DOI:10.1007/s11090-016-9713-6.
- P. Peng, Y. Cheng, R. Hatzenbeller, M. Addy, N. Zhou, C. Schiappacasse, D. Chen, Y. Zhang, E. Anderson and Y. Liu, et al., Ru-Based Multifunctional Mesoporous Catalyst for Low-Pressure and Non-Thermal Plasma Synthesis of Ammonia, Int. J. Hydrogen Energy, 2017, 42(30), 19056–19066, DOI:10.1016/j.ijhydene.2017.06.118.
- T. Mizushima, K. Matsumoto, H. Ohkita and N. Kakuta, Catalytic Effects of Metal-Loaded Membrane-like Alumina Tubes on Ammonia Synthesis in Atmospheric Pressure Plasma by Dielectric Barrier Discharge, Plasma Chem. Plasma Process., 2007, 27(1), 1–11, DOI:10.1007/s11090-006-9034-2.
- B. S. Patil, N. Cherkasov, N. V. Srinath, J. Lang, A. O. Ibhadon, Q. Wang and V. Hessel, The Role of Heterogeneous Catalysts in the Plasma-Catalytic Ammonia Synthesis, Catal. Today, 2020, 362, 2–10, DOI:10.1016/j.cattod.2020.06.074.
- F. Herrera, G. H. Brown, P. Barboun, N. Turan, P. Mehta, W. Schneider, J. Hicks and D. B. Go, The Impact of Transition Metal Catalysts on Macroscopic Dielectric Barrier Discharge (DBD) Characteristics in an Ammonia Synthesis Plasma Catalysis Reactor, J. Phys. D: Appl. Phys., 2019, 52(22), 224002, DOI:10.1088/1361-6463/ab0c58.
- K. Van't Veer, Y. Engelmann, F. Reniers and A. Bogaerts, Plasma-Catalytic Ammonia Synthesis in a DBD Plasma: Role of the Micro-Discharges and Their Afterglows, J. Phys. Chem. C, 2020, 124(42), 22871–22883, DOI:10.1021/acs.jpcc.0c05110.
- J. Hong, S. Pancheshnyi, E. Tam, J. J. Lowke, S. Prawer and A. B. Murphy, Kinetic Modelling of NH3 Production in N2-H2 Non-Equilibrium Atmospheric-Pressure Plasma Catalysis, J. Phys. D: Appl. Phys., 2017, 50(15), 154005, DOI:10.1088/1361-6463/aa6229.
- S. S. R. K. C. Yamijala, G. Nava, Z. A. Ali, D. Beretta, B. M. Wong and L. Mangolini, Harnessing Plasma Environments for Ammonia Catalysis: Mechanistic Insights from Experiments and Large-Scale Ab Initio Molecular Dynamics, J. Phys. Chem. Lett., 2020, 11(24), 10469–10475, DOI:10.1021/acs.jpclett.0c03021.
- Y. Uytdenhouwen, K. M. Bal, E. C. Neyts, V. Meynen, P. Cool and A. Bogaerts, How Process Parameters and Packing Materials Tune Chemical Equilibrium and Kinetics in Plasma-Based CO2 Conversion, Chem. Eng. J., 2019, 372, 1253–1264, DOI:10.1016/j.cej.2019.05.008.
- J. Kim, M. S. Abbott, D. B. Go and J. C. Hicks, Enhancing C-H Bond Activation of Methane via Temperature-Controlled, Catalyst-Plasma Interactions, ACS Energy Lett., 2016, 1(1), 94–99, DOI:10.1021/acsenergylett.6b00051.
- K. C. Szeto, S. Norsic, L. Hardou, E. Le Roux, S. Chakka, J. Thivolle-cazat, A. Baudouin, C. Papaioannou, J.-M. Basset and M. Taoufik, Non-Oxidative Coupling of Methane Catalysed by Supported Tungsten Hydride onto Alumina and Silica–Alumina in Classical and H2 Permeable Membrane Fixed-Bed Reactors, Chem. Commun., 2010,(22), 3985–3987, 10.1039/c0cc00007h.
- P. Navascués, J. M. Obrero-Pérez, J. Cotrino, A. R. González-Elipe and A. Gómez-Ramírez, Unraveling Discharge and Surface Mechanisms in Plasma-Assisted Ammonia Reactions, ACS Sustainable Chem. Eng., 2020, 8(39), 14855–14866, DOI:10.1021/acssuschemeng.0c04461.
- P. Navascués, J. M. Obrero-Pérez, J. Cotrino, A. R. González-Elipe and A. Gómez-Ramírez, Isotope Labelling for Reaction Mechanism Analysis in DBD Plasma Processes, Catalysts, 2019, 9(1), 1–12, DOI:10.3390/catal9010045.
- A. Bogaerts, X. Tu, J. C. Whitehead, G. Centi, L. Lefferts, O. Guaitella, F. Azolina-Jury, H.-H. Kim, A. B. Murphy and W. F. Schneider, et al., The 2020 Plasma Catalysis Roadmap, J. Phys. D: Appl. Phys., 2020, 53, 1–51, DOI:10.1088/1361-6463/ab9048.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy02189j |
| ‡ Current address: Breemarsweg 248, 7553 HW, Hengelo, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2021 |