
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new dynamic N2O reduction system based on Rh/ceria–zirconia: from mechanistic insight towards a practical application†
Yixiao
Wang
*ab,
Jorrit Postuma
de Boer
a and
Michiel
Makkee
 *a
*a
aCatalysis Engineering, Chemical Engineering Department, Faculty of Applied Sciences Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: m.makkee@tudelft.nl
bIdaho National Laboratory, Idaho Falls, Idaho 83415, USA. E-mail: Yixiao.wang@inl.gov
First published on 13th November 2020
Abstract
Simultaneous reduction of N2O in the presence of co-existing oxidants, especially NO, from industrial plants, is a challenging task. This study explores the applications of a hydrocarbon reduced Rh/Zr stabilized La doped ceria (Rh/CLZ) catalyst in N2O abatement from oxidant rich industrial exhaust streams e.g. NO, CO2, and O2. The reaction mechanism was studied by the temporal analysis of products. The obtained results revealed that hydrocarbon pretreatment led to the creation of ceria oxygen vacancies and the formation of carbon deposits on the Rh/CLZ catalyst surface. These ceria oxygen vacancies are the active sites for the selective reduction of N2O into N2, while the dissociated O atoms from N2O fill the ceria oxygen vacancies. The oxidation of the deposited carbon via the lattice ceria oxygen generates new ceria oxygen vacancies, thereby extending the catalytic cycle. The reduction of N2O over C3H6 reduced Rh/CLZ is a process combining oxygen vacancy healing and deposited carbon oxidation. The results obtained from fixed-bed reactor experiments demonstrated that the hydrocarbon reduced Rh/CLZ catalyst provided a unique and extraordinary N2O abatement performance in the presence of co-existing competing oxidants (reactivity order: N2O ∼ NO > O2 > CO2 ∼ H2O).
1. Introduction
N2O is a harmful gas to our environment, as it contributes to global warming and the depletion of the protective ozone layer. Human activities, e.g., agriculture, fossil fuel combustion, and industrial processes, contribute 4.7–7 million tons of N2O annually, which is about 30–40% of the total N2O emission including natural sources.1The catalytic reduction of N2O into N2 has been studied over a wide variety of catalysts, including noble-metal-supported catalysts, metal oxides, and zeolite-based catalysts.2 Several CeO2-based transition-metal catalysts (M/CeO2, M = Co, Cu, Fe, Zr, and Ni) have been applied in N2O reduction studies. Their T50 temperature for the N2O reduction varied between 300–660 °C.3–6 The impact of H2O, CO, CO2, O2, NO, and NO2 on N2O reduction is particularly important, since these substances are usually present in excess in N2O-containing gas streams. In particular, the simultaneous conversion of N2O and NO in the presence of O2 is a challenging task during N2O abatement in nitric acid plants.
A lot of research efforts have been directed towards the development of low temperature deN2O catalysts, which target N2O abatement arising from medical operating rooms, nitric acid plants, and automotive transport.7,8 In all these cases, apart from the activity at low temperatures, the tolerance to various substances present in the exhaust gases (e.g., NOx, O2, H2O, etc.) should be additionally addressed and subsequently enhanced. Few studies have addressed the simultaneous abatement of NOx and N2O. The current N2O abatement in industry is usually via a dual-bed catalytic system, in which NOx is firstly converted into N2 by either NH3-SCR or HC-SCR, while subsequently N2O is catalytically decomposed into N2 and O2.9–11 Sufficient performance has rarely been achieved in a single catalyst bed.12–14 In particular, the N2O abatement activity is strongly inhibited by the presence of NO.12
The Di-Air system, developed by Toyota Company, showed great promise in NOx abatement with regard to the current and future NOx emission standards under real driving automotive conditions (dynamic operations, high exhaust temperature, and high gas hourly space velocities (GHSV > 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L L−1 h−1)).15 The comprehensive work by Wang and Makkee has addressed the working principle and application of this Di-Air system in NO reduction.16–21 Oxygen vacancies within the ceria lattice of a reduced ceria, Pt/ceria or Rh/ceria were found to be the selective catalytic sites for the NO reduction into only N2 (100% selectivity).16 Even at low NO concentrations (ppm levels), NO could compete for oxygen vacancies with (100×) excess of O2 and CO2.21 These oxygen vacancies acted as a kind of “oxygen black hole” by catching all oxygen containing species until the holes (vacancies) were completely refilled (re-oxidized), while the captured N species would associate (recombine) into N2. In the Di-Air system, the creation of reduced (noble metal) ceria was accomplished by pulsing diesel fuel at a high frequency upstream of the catalyst bed. The amount of diesel pulsed was such that the front of the catalyst bed was in a reduced state while the back of the catalyst bed was in an oxidized state. In other words, although diesel fuel is injected, the overall catalytic bed would be in a lean (oxidized) state. During these diesel pulses carbon deposits were formed, which were oxidized in time by the lattice oxygen from the ceria catalyst and not by gaseous oxidants present in the exhaust stream such as O2 and NOx (mainly NO2).
000 L L−1 h−1)).15 The comprehensive work by Wang and Makkee has addressed the working principle and application of this Di-Air system in NO reduction.16–21 Oxygen vacancies within the ceria lattice of a reduced ceria, Pt/ceria or Rh/ceria were found to be the selective catalytic sites for the NO reduction into only N2 (100% selectivity).16 Even at low NO concentrations (ppm levels), NO could compete for oxygen vacancies with (100×) excess of O2 and CO2.21 These oxygen vacancies acted as a kind of “oxygen black hole” by catching all oxygen containing species until the holes (vacancies) were completely refilled (re-oxidized), while the captured N species would associate (recombine) into N2. In the Di-Air system, the creation of reduced (noble metal) ceria was accomplished by pulsing diesel fuel at a high frequency upstream of the catalyst bed. The amount of diesel pulsed was such that the front of the catalyst bed was in a reduced state while the back of the catalyst bed was in an oxidized state. In other words, although diesel fuel is injected, the overall catalytic bed would be in a lean (oxidized) state. During these diesel pulses carbon deposits were formed, which were oxidized in time by the lattice oxygen from the ceria catalyst and not by gaseous oxidants present in the exhaust stream such as O2 and NOx (mainly NO2).
To the best of our knowledge, no work has been published on the application of this “oxygen black hole” concept of the Di-Air system in a deN2O application. In this study, we investigated the mechanism of the N2O reduction over a reduced Rh/CLZ catalyst with a clean surface and with carbon deposits on that surface.
The temporal analysis of products (TAP, an ultra-high vacuum pulse and response technique) was applied to study the reaction mechanism of the N2O reduction over a reduced Rh/CLZ catalyst, pre-treated with either H2 or C3H6 as a stand-in for a diesel fuel. Moreover, the reactivity of N2O versus other oxidants (O2 and NO) towards oxygen vacancies of ceria-based catalysts would be crucial for the extension of the Di-Air technology to the deN2O area, i.e., the simultaneous NOx and N2O abatement in the presence of an excess of O2. The competition between N2O and NO in an excess of O2 was further investigated under more industrially relevant conditions in a fixed-bed flow reactor.
2. Materials and methods
2.1. Materials preparation and characterization
Rh/CLZ, with a target loading of 0.5 wt%, was prepared via an incipient wetness impregnation method using a Zr stabilized La doped ceria (denoted as CLZ, a gift from Engelhard, now BASF). Rhodium(III) nitrate hydrate (Sigma Aldrich) was used and dissolved as the precursor in purified demi water. Subsequently, the samples were dried at 110 °C overnight and calcined at 550 °C for 5 h. CLZ and Rh/CLZ were characterized by ICP, XRD, TEM, XPS, Raman, and H2-TPR. Details on the characterization and instruments can be found in our previous publications.16–212.2. Catalytic testing
In the TAP experiments at 450 °C 10 mg of as-prepared CLZ and Rh/CLZ (150–212 μm) were sandwiched between inert quartz bead beds. Prior to the catalyst reduction, the catalyst was firstly oxidized by pulsing 80 vol% O2 in Ar overnight at 450 °C. The catalyst reduction was carried out by pulsing the reductant of either 80 vol% C3H6 (propene) in Ne or 66.7 vol% H2 in Ar. The re-oxidation experiment was conducted at 450 °C by pulsing either 80 vol% CO2 or 80 vol% O2 in Ar, or 80 vol% 15N2O in Kr, or co-pulsing 80 vol.% 14NO in He and 80 vol.% 15N2O in Kr.
The number of consumed oxygen species from the catalyst during the C3H6 and H2 multi-pulse experiments was calculated using eqn (1):
| nO,consumed = nH2O,out + nCO,out + 2nCO2,out | (1) |
| nC,deposited = 3nC3H6,in − 3nC3H6,out − nCO,out − nCO2,out | (2) |
Similarly, the amount of oxygen accumulation, the carbon consumption, and the nitrogen balance during the 15N2O, CO2, and O2 multi-pulse experiments were calculated using the following atomic balances:
| nO,accumulated = nN2O,in − nCO,out − 2nCO2,out − nN2O,out − 2nNO2,out − nNO,out | (3) |
| nc,accumulated = −nC,in − nCO,out − nCO2,out | (4) |
| nN,accumulated = 2nN2O,in − 2nN2O,out − 2nN2,out − nNO,out − nNO2,out | (5) |
 | (6) |
000 L L−1 h−1, at 450 °C.
3. Results and discussion
3.1. Structure, composition, and texture properties
Characterization details of the CLZ support and Rh/CLZ catalyst were reported in detail elsewhere.16–21 In brief, a 0.5 wt% Rh loading was confirmed by ICP-OES (0.0486 mmol gcat−1 Rh loading). A typical fluorite structure of CLZ was observed for both CLZ and Rh/CLZ samples by Raman as well as XRD. Rh metals or any rhodium oxides could not be observed by XRD, confirming a high Rh dispersion. Room temperature Raman results indicated that the Rh/CLZ samples had more oxygen vacancies as compared to CLZ.18 A 5 nm ceria crystal size was determined by the Scherrer equation (XRD) and was further confirmed by the analysis of the TEM micrographs. The particle size of Rh was around 2 nm as indicated in the TEM micrographs. The bulk composition of CLZ, with an atomic ratio of Ce, Zr, and La of 0.64:0.15:0.21, was determined by ICP. The BET surface area of bare (fresh and up to >2000 h time on stream) CLZ was 65 m2 g−1. The BET surface areas of Rh/CLZ (fresh and spent) were similar to that of the bare CLZ support (66 ± 2 m2 g−1).
3.2. Transient N2O reduction
Fig. 1A shows the reactant and product evolution during a 15N2O pulse experiment over H2 reduced Rh/CLZ at 450 °C. A 15N2O conversion of 100% was observed, while 15N2 was observed as the only N containing product from pulse numbers of 0 to 3400 (Fig. 1A, reduced state of Rh/CLZ). 15NO was not observed during the whole experiment. There was no indication of any 15N species accumulation on the catalyst (Fig. 1B), which suggested that N2O was instantaneously reduced into N2 with 100% selectivity. Oxygen atoms were observed to accumulate incrementally within the catalyst and 99% of the oxygen vacancies were refilled during the first 3400 15N2O pulses. The results shown in Fig. 1 suggested that the N2O reduction over the reduced Rh/CLZ catalyst was an oxygen vacancy refilling process, which was also evidenced by in situ Raman and XPS results from the study by Bueno-López et al.22 Gradually a 15N2O breakthrough was observed after pulse number 3400 (Fig. 1A), corresponding to a 15N2O conversion of roughly 95%. From pulse number 3400 onwards, the Rh/CLZ catalyst became completely oxidized and O2 evolution was observed. From this point on the N2O reduction proceeded via adsorbed O species recombination forming gas phase O2 thereby regenerating two active sites, e.g., reduced Rh metal sites and ceria oxygen vacancy sites. A slightly lower 15N2O conversion was observed when the catalyst was in a fully oxidized state (Fig. 1A), as compared to the reduced state. This was likely caused by a slower ‘O’ association into O2 over the oxidized Rh/CLZ surface. The O2(g) formation process consisted of a surface ‘O’ association step and an O2(g) desorption step. In order to elucidate the slow O2(g) formation step, O2 was pulsed over an oxidized Rh/CLZ surface at the same temperature as the N2O pulse experiment. As shown in Fig. S1,† a clear O2 response was observed during the O2 pulses, while no clear O2 desorption curve was observed during the N2O pulses over an oxidized Rh/CLZ surface. Therefore, a slow ‘O’ association step was likely the cause of the slow O2 desorption over the oxidized Rh/CLZ surface during N2O reduction. A similar dynamic trend was observed with the H2 pre-reduced CLZ bare support under the same reaction conditions (Fig. S2, ESI†) although the time until 100% 15N2O conversion was shorter due to the significantly lower reduction degree for the H2 reduction of CLZ. In this case, a full conversion of 15N2O into 15N2 was observed when the CLZ was in a reduced state. After that, the N2 production decreased with a lower N2O reduction activity (only 12% conversion of N2O). The results obtained for the reduced CLZ (Fig. S2, ESI†) indicated that the N2O reduction was an oxygen vacancy refilling process as well. From Fig. 1 and S2† it follows that the total amount of N2O converted over the reduced catalysts was equal to the total amount of oxygen vacancies created during the H2 reduction. Therefore, the role of Rh is to increase the CLZ support reduction degree by the H2 reduction process. The presence of Rh did not noticeably alter the N2O reduction rate, since 100% N2O conversion was observed over reduced Rh/CLZ and CLZ. However, over oxidized CLZ the presence of Rh led to a significant improvement in the N2O reduction activity, as the conversion of N2O over oxidized Rh/CLZ was approximately 8 times that over oxidized CLZ. From in situ XPS results obtained for a Rh/CeO2 system by Parres-Esclapez et al.,22 it was known that the reduced rhodium sites could be re-oxidized afterwards by either N2O or ceria lattice oxygen. These vacant oxygen positions in ceria were subsequently oxidized by N2O. The active sites for the N2O chemisorption and reduction were not only located on rhodium, but were also present on the ceria. Additionally, Rh was a powerful promoter in enhancing the surface oxygen diffusion and lowering the oxygen activation barrier,23,24 and therefore, Rh could promote a faster surface oxygen association and desorption of gas-phase O2 on the oxidized catalyst surfaces during the N2O reduction. Rh could be a distinctive mechanistic feature for the promotion of the N2O reduction process.25
 | ||
| Fig. 1 A) Product and reactant evolution and B) O and N balance versus pulse number during the 15N2O multi-pulse experiment over H2 reduced Rh/CLZ at 450 °C. | ||
Transient N2 formation during the 15N2O pulses was compared between the H2 reduced Rh/CLZ and CLZ samples as shown in Fig. 2. In these experiments exclusively 15N2 was observed as a reaction product. No observable N2 flux difference was observed, which suggested that N2O most likely reacted on the same reaction sites. These active sites were most likely the surface oxygen vacancies on the reduced CLZ support. The reduction of N2O led to the oxidation of Ce3+ to Ce4+, while N2 was released. If two active sites should exist, i.e., oxygen vacancies on the reduced CLZ support and Rh, then two distinguishable responses would have been expected26–28 rather than a single peak response that was observed in the current experiment. The hypothesis that only oxygen vacancy active sites were used on the reduced CLZ even in the presence of Rh explained the observed 100% 15N2O conversion over both reduced CLZ and Rh/CLZ. The results presented in Fig. 1, S2† and 2 all indicated that the oxygen vacancies on CLZ were the only active sites for the N2O reduction. During the N2O reduction, the O species refilled the CLZ lattice oxygen vacancies and N2 desorbed to the gas phase. The role of Rh was the promotion of the deep CLZ reduction at lower temperatures, however this deep reduction had an insignificant impact on the N2O reduction, when the catalyst was in a reduced state, i.e., the presence of ceria oxygen vacancies. The presence of Rh started to promote the N2O reduction only when the catalyst was in an oxidized state, i.e., the absence of ceria oxygen vacancies.
 | ||
| Fig. 2 Comparison of the height normalized intensity of 15N2 between the H2 reduced Rh/CLZ sample and reduced CLZ sample during the 15N2O pulses. | ||
The investigation of the impact of deposited carbon on N2O reduction is presented in Fig. 3. Fig. 3A shows the reactant and product evolution achieved over C3H6 reduced Rh/CLZ at 450 °C versus the incremental pulse number. Full 15N2O conversion was observed until pulse number 4500, while 15N2 evolved as the dominant product. From pulse number 45000 onward, a progressive, but small decline in the 15N2O conversion to 95% was observed. This decline was accompanied by the formation of O2, while hardly any CO and/or CO2 was formed at this stage. No 15NO was observed during the whole experiment.
 | ||
| Fig. 3 A) Reactant and product evolution and B) 15N, O, and C balance versus pulse number during the 15N2O multi-pulse experiment over C3H6 reduced Rh/CLZ at 450 °C; (a)–(c) represent the catalyst stage of the reduced catalyst with carbon deposits, oxidized catalyst with carbon deposits, and oxidized catalyst, respectively. | ||
A small amount of CO evolution was observed during the first 15000 15N2O pulses, during this time frame 80% of the ceria oxygen vacancies were refilled, while only 10% of the deposited carbon was consumed (Fig. 3B). This indicated that the carbonaceous residues, left on the surface after the C3H6 pre-reduction, did not directly participate in the reduction of 15N2O into 15N2. The formation of 15N2 indicated that 15N2–O was dissociated on reduced CLZ sites, the O atom of 15N2O refilled the ceria oxygen vacancies and at the same time the remaining adsorbed 15N2 species desorbed as 15N2. A significant role of the direct reaction between 15N2O and deposited carbon could be ruled out since the formation of a 15N2 molecule would yield one CO molecule, according to eqn (7).
| N2O + C → N2 + CO | (7) |
000 onward in the form of CO2. Oxygen accumulation dropped to zero starting from pulse number 20000, at that point almost 100% of the oxygen vacancies were refilled. The direct interaction of N2O with deposited carbon, leading to the formation of CO2 and N2, could be ruled out, since an identical N2 response with a single characteristic peak was observed for both the C3H6 reduced and the H2 reduced Rh/CLZ samples (Fig. S3, ESI†). The deposited carbon consumption decreased after pulse number 450000. The 15N species accumulation on the catalyst surface was insignificant. The ratio between N2 and CO2 was around 2 in the time interval between pulse numbers 10000 and 450000, which clearly demonstrated that for the formation of one CO2 molecule two 15N2O molecules had to be reduced forming two 15N2 molecules. Such a phenomenon suggested that the oxidation of one deposited carbon atom to CO2 created two oxygen vacancies, which allowed for the reduction of two 15N2O molecules into two 15N2 molecules. The formation of CO2 started when the catalyst was largely oxidized, which suggested that CO2 formed via a CO intermediate, which was subsequently oxidized into CO2 by a ceria lattice oxygen. In previous studies,18,19 we have demonstrated that CO could reduce oxidized ceria up to almost one hypothetical ceria layer under identical reaction conditions.
Over H2 reduced Rh/CLZ 15N2O reduction proceeded for approximately 3400 pulses (Fig. 1), while over C3H6 reduced Rh/CLZ this proceeded for approximately 45000 pulses (Fig. 3); this remarkable difference indicated that the deposited carbon acted as a reductant buffer. N2O was reduced over ceria oxygen vacancies, which led to the re-oxidation of these oxygen vacancies while N2 was released at the same time. When most ceria oxygen vacancies were filled, ceria lattice oxygen became capable of oxidizing the carbon deposits into CO and CO2, thereby regenerating the ceria oxygen vacancies. The total amount of deposited carbon determined the additional ceria oxygen vacancies the Rh/CLZ system could provide, besides the ceria oxygen vacancies present after the reduction. The benefit of using hydrocarbons as reductants arose from the extended time interval in which 100% N2O conversion was observed. In a previous publication we have demonstrated by means of an 18O2 pulse experiment over C3H8 reduced Rh/CLZ at 450 °C that only lattice oxygen was responsible for the oxidation of deposited carbon, since only C16O and C16O2 oxidation products containing exclusively 16O from the CLZ lattice were observed.18
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 15N bond cleavage was absent, since 14N15N products were not observed. The transient kinetic data of 15N2 and 14N2 showed that 15N2 formation was faster than 14N2 formation, as 15N2 was observed prior to 14N2 (Fig. 4B). The formation of 14N2 required the cleavage of the 14NO bond and the subsequent association of two 14N species, which was expected to proceed more slowly than 15N2 formation via the direct cleavage of the 15NO bond of 15N2O.
15N bond cleavage was absent, since 14N15N products were not observed. The transient kinetic data of 15N2 and 14N2 showed that 15N2 formation was faster than 14N2 formation, as 15N2 was observed prior to 14N2 (Fig. 4B). The formation of 14N2 required the cleavage of the 14NO bond and the subsequent association of two 14N species, which was expected to proceed more slowly than 15N2 formation via the direct cleavage of the 15NO bond of 15N2O.
 | ||
| Fig. 4 A) Reactant and product evolution during 15N2O and 14NO co-pulses over H2 reduced Rh/CLZ at 450 °C and B) the response of 15N2 and 14N2 averaged by the first 5000 pulses. | ||
As shown in Fig. 5A, all pulsed 15NO converted to 15N2 until the catalyst became oxidized. 15NO is a powerful oxidant and is capable of filling all ceria oxygen vacancies (those on reduced CLZ and those created by the oxidation of carbon deposits by the CLZ lattice oxygen). The results presented in Fig. 4A suggested that the 15N2O reduction activity was not affected by the presence of 14NO when the catalyst was in a reduced state. However, the 15N2O reduction activity was dramatically inhibited by NO when the catalyst switched to an oxidized state.
 | ||
| Fig. 5 Gas evolution during 15NO (A), CO2 (B), and O2 (C) pulses over C3H6 pre-reduced Rh/CLZ at 450 °C. | ||
CO2 fully converted into CO when the catalyst was in a significantly reduced state (Fig. 5B). However, when the catalyst was almost completely (re-)oxidized, the CO2 reactivity suddenly dropped down while most of the deposited carbon was still on the Rh/CLZ surface (Fig. 5B). CO2 hardly consumed any deposited carbon. A quasi-equilibrium between CO, CO2, Ce3+, and Ce4+ appeared to limit the achievable oxidation degree of reduced ceria.19 Therefore, it can be concluded that CO2 was a mild oxidant as compared to NO and N2O, as it could hardly oxidize the deposited carbon. In the field of the dry CO2 reforming of methane (DRM) reaction, the oxygen vacancies of a ceria support provided the catalytic sites for the CO2 reduction to CO. The oxygen transport from the ceria lattice to the metal (Rh) largely reduced the carbon deposition during the DRM reaction.29,30Fig. 5C shows the results obtained in an O2 pulse experiment over C3H6 pre-reduced Rh/CLZ. O2 was fully converted while CO and CO2 formed which originated from the oxidation of deposited carbon (Fig. 5C). O2 broke through when the catalyst became oxidized. Therefore, O2 was a strong oxidant, which can compete with NO and N2O for oxygen vacancies. However, NO was a more reactive and competitive reactant towards the oxygen vacancies as compared to O2 as evidenced in previously published experiments in which 500–2000 ppm NO and 5% O2 were co-fed over C3H6 reduced Rh/CLZ at 450 °C.21 The current study presented in Fig. 4A suggested that N2O was comparably reactive towards ceria oxygen vacancies as NO did, and therefore, N2O was a more reactive and competitive reactant towards the oxygen vacancies as compared to O2.
In our previous publication, H2 pulses over an oxidized ceria (CLZ) led to the formation of H2O, yielding less than one monolayer of reduced ceria. This indicated the presence of a quasi-equilibrium established between H2, H2O, Ce3+, and Ce4+, which limited the deeper reduction of ceria by H2 or complete re-oxidation of reduced ceria by H2O.19 Therefore, H2O was a weaker oxidant towards oxygen vacancies. As a consequence, the presence of H2O would not affect the NO and N2O reduction over a reduced ceria. Ceria-based catalysts are among others the best candidates for the water gas shift reaction.31,32 Oxygen vacancies on the ceria surface played an essential role in the water dissociation, yielding H2 while the oxygen atoms filled the ceria oxygen vacancies during the WGS reaction. The reaction of CO with ceria lattice oxygen led to the formation of CO2 thereby recreating a ceria oxygen vacancy. The WGS reaction was an equilibrium-limited reaction. The water dissociation would produce H2 and oxidize the reduced ceria while the formed CO2 from CO would create the ceria oxygen vacancy. Therefore, the O reactivity of CO2 and H2O was expected to be relatively small and CO2 and H2O would not inhibit NO and N2O reduction into N2 to a large extent.
3.3. Catalytic fixed-bed reactor evaluation with regard to a potential industrial application
To confirm the results obtained in the TAP experiments, similar experiments were performed over Rh/CLZ in a flow reactor at atmospheric pressure and industrial exhaust concentrations. Similar to the TAP experiment, the flow of 1.25% C3H6/He in a fixed-bed reactor at 450 °C for 2 h led to the formation of H2O, CO2, CO, H2 and carbon on the catalyst surface. The quantification of the oxygen and carbon balance was performed, according to eqn (1) and (2), respectively, and showed a reduction of around 3 CLZ layers and deposition of 8.2 × 1017 carbon atoms per mgcat. N2O reduction over the C3H6 pre-reduced Rh/CLZ was investigated under a gas mixture of 2000 ppm N2O/He, (2000 ppm N2O + 5% O2)/He, and (2000 N2O ppm + 2000 ppm NO)/He. Both MS and FTIR were used to detect the gas evolution. m/z = 28 can be attributed to either CO or N2, and m/z = 44 to either CO2 or N2O. Gas species which contributed to the vibration peaks in the FT-IR spectrum can be seen in Table 1.| Wavenumber/cm−1 | Gas species |
|---|---|
| 2350 | CO2 |
| 2235 and 2208 | N2O |
| 2174 and 2116 | CO |
| 1908 and 1850 | NO |
| 1601 and 1628 | NO2 |
Fig. 6 shows the results of the exposure of a C3H6 reduced Rh/CLZ catalyst to 2000 ppm N2O at 450 °C with a GHSV of 67000 L L−1 h−1. In Fig. 6A, m/z = 28 was observed, which could be attributed to the formation of N2 and CO. The formation of CO was confirmed by FT-IR (Fig. 6B). The CO yield increased up to a maximum of 2500 ppm, after which it declined to zero (Fig. 6B and C). After CO had vanished (t = 1000 s), m/z = 28 was still observed in the MS (Fig. 6A). Therefore, in addition to CO, N2 also contributed to m/z = 28. m/z = 44 was observed between 400 s and 1500 s, which could be attributed to the formation of CO2 and the slip of N2O. The formation of the latter could be excluded, during this time interval, FT-IR results indicated the absence of peaks at 2235 and 2208 cm−1 and the presence of a peak at 2350 cm−1, which confirmed the formation of CO2 and excluded the presence (slip) of N2O in the reactor effluent. The formation of CO and CO2 indicated the oxidation of deposited carbon by the reduction of N2O. No NO or NO2 formation was observed during the whole experiment. N2O was completely converted into N2 as evidenced by FTIR where no N2O and/or NO2 peaks were observed within the detection limit of 1 ppm. The observation of N2 in the MS indicated an extremely selective reduction of N2O into N2. O2 arising from N2O started to break through roughly from 1400 s onward, while the CO2 yield started to decrease. The breakthrough of O2 implied that the catalyst was largely oxidized and coincided with the disappearance of CO and CO2 from the FTIR spectrum, indicating that all deposited carbon was oxidized. These observations indicated that the N2O reduction over C3H6 pre-reduced Rh/CLZ consisted of the refilling of the oxygen vacancies and the oxidation of the carbon deposits. Overall, the results presented in Fig. 6 clearly demonstrate that the HC pre-reduced Rh/CLZ catalyst exhibited excellent N2O reduction performance, which was in line with the conclusion from the TAP study (Fig. 3).
 | ||
| Fig. 6 Gas evolution during the exposure of C3H6 reduced Rh/CLZ to 2000 ppm N2O in He at 450 °C. A) MS responses, B) FT-IR spectral responses, and C) quantification of (B). | ||
In order to explore the performance of Rh/CLZ in real industrial applications, a good catalytic activity for only N2O is not sufficient. The N2O reduction activity has to be studied in the presence of potential inhibitors in the exhaust stream under atmospheric pressure. NO and O2 are the most challenging inhibitors as they both can compete with N2O for the oxygen vacancies. Fig. 7 and 8 summarize the results obtained in the presence of O2 and NO.
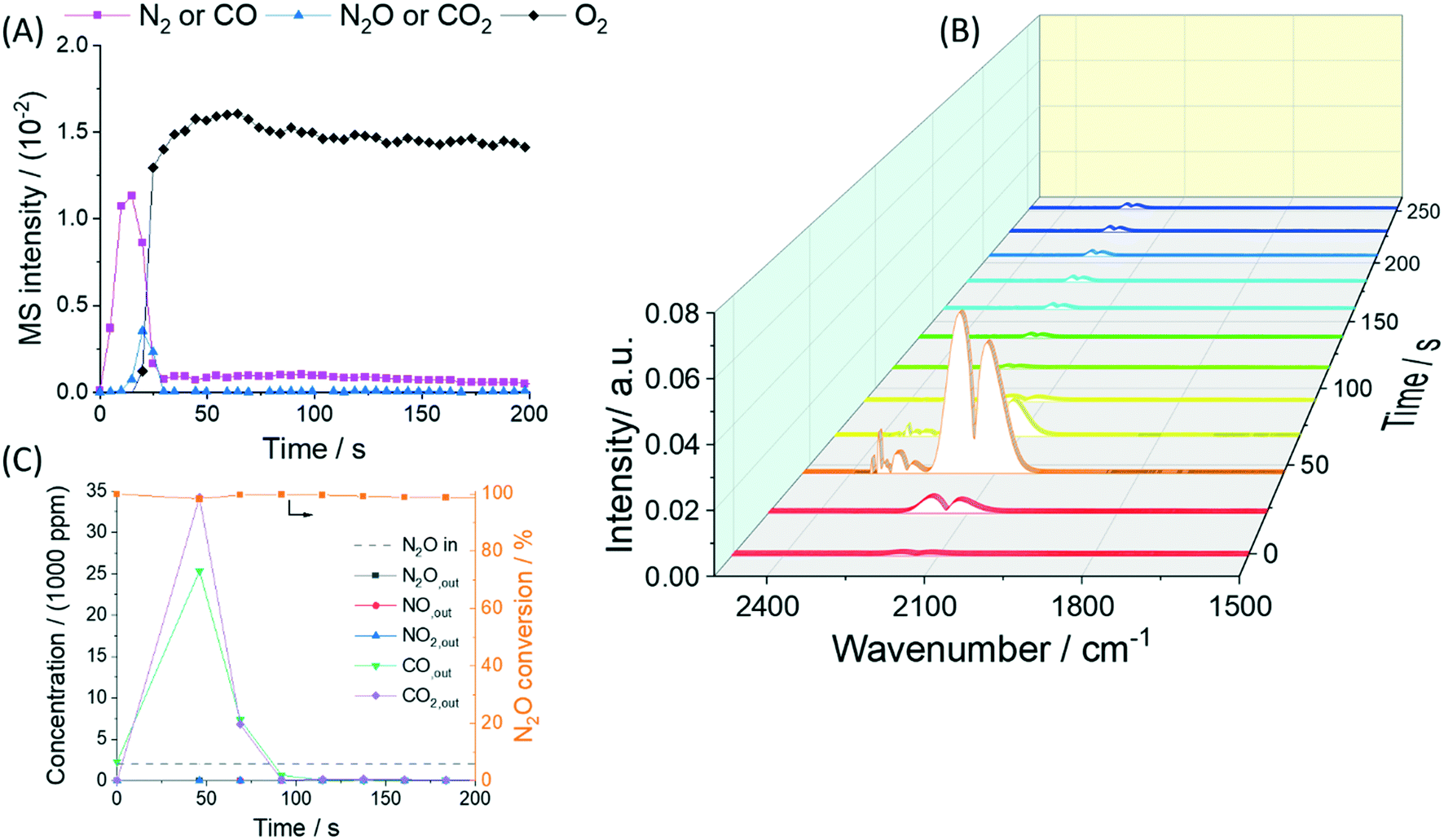 | ||
| Fig. 7 Gas evolution during the exposure of C3H6 reduced Rh/CLZ to 2000 ppm N2O + 5 vol% O2 in He at 450 °C. A) MS responses, B) FT-IR spectral responses, and C) quantification of (B). | ||
 | ||
| Fig. 8 Gas evolution during the exposure of C3H6 reduced Rh/CLZ to 2000 ppm N2O + 2000 ppm NO in He at 450 °C. A) MS responses, B) FT-IR spectral responses, and C) quantification of (B). | ||
The influence of O2 addition to the N2O (2000 ppm) gas feed on N2O reduction is shown in Fig. 7. O2 (m/z = 32) started to break through after approximately 20 s, while N2O was not observed (detection limit of 1 ppm) until 160 s. From that point on around 25 ppm N2O was detected by FT-IR. The N2O breakthrough time was 8× later than that of O2 (50000 ppm), which indicated that a small concentration of N2O (2000 ppm) was able to compete with an excess of O2. NO and NO2 were not detected anytime in the reactor effluent. This clearly suggested that N2O could be selectively reduced into N2 in the presence of O2. The observation of 25 ppm of N2O after O2 breakthrough (Fig. 7C), i.e., 98.8% N2O conversion, suggested that the presence of O2 inhibited the catalytic reduction of N2O to a very small extent when the catalyst became oxidized. These results indicated that the reduction of N2O into N2 over reduced Rh/CLZ was not affected by the addition of O2. N2O was much more competitive towards the oxygen vacancies as compared to O2.
Fig. 8 evaluates the effect of adding NO to the N2O gas feed. N2O and NO roughly broke through at the same time while CO formation decreased, which indicated that N2O and NO compete equally for the active sites. The presence of NO did not affect the reduction of N2O into N2, while the deposited carbon was oxidized. Only 100 ppm of NO2 was observed when NO appeared in the reactor effluent as noticed in the FT-IR spectrum (Fig. 8C). This NO2 likely formed due to the reaction of NO with surface oxygen species in the N2O reduction through steps (8)–(10):
| N2O + * → N2 + *_O | (8) |
| NO + * → NO_* | (9) |
| NO + *_O → NO2_* | (10) |
Fig. 9 summarizes the observed N2O conversion for the different gas feeds over O2 pre-oxidized and C3H6 pre-reduced Rh/CLZ. For N2O, the catalyst displayed 100% N2O conversion over both O2 pre-oxidized and C3H6 pre-reduced samples. For N2O + O2 (excess), the N2O conversion dropped from 100% to 98.8% when the catalyst switched from a reduced to an oxidized state. For N2O + NO, the conversion of N2O dropped from 100% to 37% when the catalyst switched from a reduced into an oxidized state. The inhibition of the N2O reduction by NO was a common issue in the N2O abatement, since the majority of explored catalysts had a very low tolerance towards NO. In summary, the above experiment clearly demonstrated that a C3H6 pre-reduced Rh/CLZ catalyst exhibited a unique and extraordinary N2O reduction performance, when the Rh/CLZ was in a reduced state. Again, carbon deposits extended the time frame during which the Rh/CLZ catalyst remained reduced.
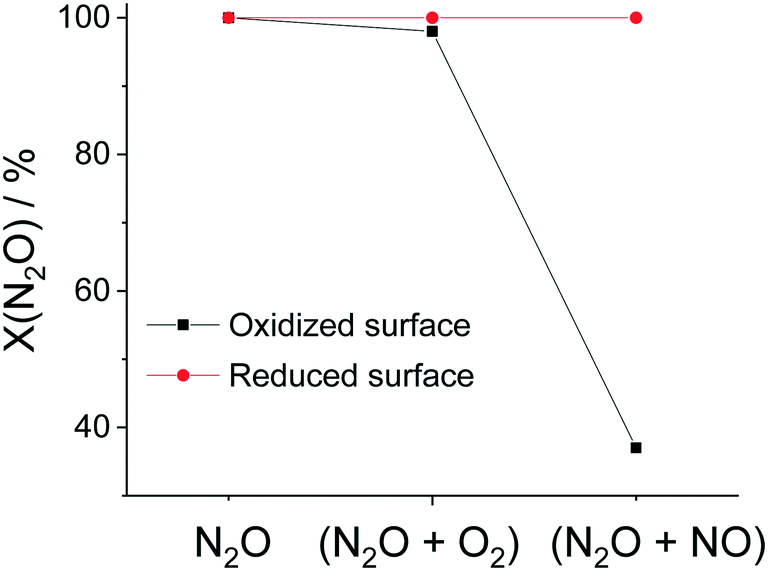 | ||
| Fig. 9 N2O conversion over O2 pre-oxidized and C3H6 pre-reduced Rh/CLZ in 2000 ppm N2O in He, 2000 ppm N2O + 5% O2 in He, and 2000 ppm N2O + 2000 ppm NO in He. Conditions: atmospheric pressure, 450 °C, and GHSV = 67000 L L−1 h−1. | ||
Besides our previous publication,17 the experiment of (5% CO2 + 2000 ppm NO)/He over C3H6 pre-reduced Rh/CLZ in a fixed bed flow reactor indicated that NO by far was a more powerful reductant in the competition for the oxygen vacancies as compared to CO2. Around 90% of the deposited carbon was consumed by NO via the lattice oxygen of the ceria. NO was selectively reduced into N2 regardless of the CO2 presence.17 The presence of CO2 did not affect the NO reactivity and selectivity over the reduced CLZ and Rh/CLZ catalysts. The presence of CO2 would, therefore, not affect both the N2O and NO reduction into N2 over reduced Rh/CLZ.
4. Conclusion
This work shows that a C3H6 pre-reduced Rh/CLZ catalyst exhibits a unique and extraordinary performance in the reduction of N2O in the presence of other oxidants, e.g., most importantly O2 and NO. The reductive pretreatment with C3H6 created oxygen vacancies and carbon deposits on the Rh/CLZ surface. These oxygen vacancies were the catalytic sites for an extremely selective reduction of N2O into N2, in which the oxygen vacancies were replenished. The deposited carbon acted as a buffer reductant and was responsible for the generation of new oxygen vacancies. This new N2O reduction system could be cycled by short pulses of hydrocarbons upstream of the catalyst bed, which allowed regeneration of the oxygen vacancies and deposited carbon. Our work clearly indicated that the Di-Air DeNOx system could be applied in simultaneous NOx and N2O reduction under oxygen rich conditions, using a single Rh/CLZ catalyst bed, under industrial relevant conditions.Conflicts of interest
There are no conflicts to be declared.References
- E. A. Davidson and D. Kanter, Environ. Res. Lett., 2014, 9, 105012 CrossRef
.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS
- P. Esteves, Y. Wu, C. Dujardin, M. Dongare and P. Granger, Catal. Today, 2011, 176, 453–457 CrossRef CAS
- A. Adamski, W. Zając, F. Zasada and Z. Sojka, Catal. Today, 2012, 191, 129–133 CrossRef CAS
- H. Zhou, P. Hu, Z. Huang, F. Qin, W. Shen and H. Xu, Ind. Eng. Chem. Res., 2013, 52, 4504–4509 CrossRef CAS
- F. Perez-Alonso, I. Melián-Cabrera, M. L. Granados, F. Kapteijn and J. G. Fierro, J. Catal., 2006, 239, 340–346 CrossRef CAS
- S. Gudyka, G. Grzybek, J. Gryboś, P. Indyka, B. Leszczyński, A. Kotarba and Z. Sojka, Appl. Catal., B, 2017, 201, 339–347 CrossRef CAS
- G. Delahay, M. Mauvezin, B. Coq and S. Kieger, J. Catal., 2001, 202, 156–162 CrossRef CAS
- J. Pérez-Ramırez, J. García-Cortés, F. Kapteijn, M. Illán-Gómez, A. Ribera, C. S. M. de Lecea and J. A. Moulijn, Appl. Catal., B, 2000, 25, 191–203 CrossRef
- M. K. Kim, P. S. Kim, B. K. Cho, I.-S. Nam and S. H. Oh, Catal. Today, 2012, 184, 95–106 CrossRef CAS
- M. Inger, J. Rajewski, M. Ruszak and M. Wilk, Chem. Pap., 2019, 1–8 Search PubMed
- M. Kögel, R. Mönnig, W. Schwieger, A. Tissler and T. Turek, J. Catal., 1999, 182, 470–478 CrossRef
- F. Schuricht and W. Reschetilowski, Microporous Mesoporous Mater., 2012, 164, 135–144 CrossRef CAS
- M. C. Campa, D. Pietrogiacomi, C. Scarfiello, L. R. Carbone and M. Occhiuzzi, Appl. Catal., B, 2019, 240, 367–372 CrossRef CAS
- Y. Bisaiji, K. Yoshida, M. Inoue, K. Umemoto and T. Fukuma, SAE Int. J. Fuels Lubr., 2012, 5, 380–388 CrossRef CAS
- Y. Wang, J. P. de Boer, F. Kapteijn and M. Makkee, ChemCatChem, 2016, 8, 102–105 CrossRef CAS
- Y. Wang and M. Makkee, Appl. Catal., B, 2018, 221, 196–205 CrossRef CAS
- Y. Wang, F. Kapteijn and M. Makkee, Appl. Catal., B, 2018, 231, 200–212 CrossRef CAS
- Y. Wang and M. Makkee, Appl. Catal., B, 2018, 223, 125–133 CrossRef CAS
- Y. Wang and M. Makkee, Appl. Catal., B, 2019, 259, 117895 CrossRef CAS
- Y. Wang, R. Oord, D. Van Den Berg, B. M. Weckhuysen and M. Makkee, ChemCatChem, 2017, 9, 2935–2938 CrossRef CAS
- S. Parres-Esclapez, I. Such-Basañez, M. Illán-Gómez, C. S.-M. de Lecea and A. Bueno-López, J. Catal., 2010, 276, 390–401 CrossRef CAS
-
H. Abderrahim and D. Duprez, in Studies in Surface Science and Catalysis, ed. A. Crucq and A. Frennet, Elsevier, 1987, vol. 30, pp. 359–368 Search PubMed
- C. Descorme and D. Duprez, Appl. Catal., A, 2000, 202, 231–241 CrossRef CAS
- E. V. Kondratenko, V. A. Kondratenko, M. Santiago and J. Pérez-Ramírez, J. Catal., 2008, 256, 248–258 CrossRef CAS
- Y. Wang, M. R. Kunz, S. Siebers, H. Rollins, J. Gleaves, G. Yablonsky and R. Fushimi, Catalysts, 2019, 9, 104 CrossRef
- Y. Wang, R. Kunz, P. D. D. Constales, G. Yablonsky and R. R. Fushimi, J. Phys. Chem. A, 2019, 123, 8717–8725 CrossRef CAS
- Y. Wang, M. R. Kunz, Z. Fang, G. Yablonsky and R. R. Fushimi, Ind. Eng. Chem. Res., 2019, 58, 10238–10248 CrossRef CAS
- I. V. Yentekakis, G. Goula, M. Hatzisymeon, I. Betsi-Argyropoulou, G. Botzolaki, K. Kousi, D. I. Kondarides, M. J. Taylor, C. M. A. Parlett, A. Osatiashtiani, G. Kyriakou, J. P. Holgado and R. M. Lambert, Appl. Catal., B, 2019, 243, 490–501 CrossRef CAS
- G. Goula, G. Botzolaki, A. Osatiashtiani, C. Parlett, G. Kyriakou, R. M. Lambert and I. V. Yentekakis, Catalysts, 2019, 9, 541 CrossRef CAS
- D. Pal, R. Chand, S. Upadhyay and P. Mishra, Renewable Sustainable Energy Rev., 2018, 93, 549–565 CrossRef CAS
- J. Vecchietti, A. Bonivardi, W. Xu, D. Stacchiola, J. J. Delgado, M. Calatayud and S. E. Collins, ACS Catal., 2014, 4, 2088–2096 CrossRef CAS
- M. Zabilskiy, P. Djinović, E. Tchernychova and A. Pintar, Appl. Catal., B, 2016, 197, 146–158 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy02035d |
| This journal is © The Royal Society of Chemistry 2021 |