
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThio-conjugation of substituted benzofurazans to peptides: molecular sieves catalyze nucleophilic attack on unsaturated fused rings†
Valentina
Verdoliva
a,
Giuseppe
Digilio
b,
Michele
Saviano
 c and
Stefania
De Luca
*a
c and
Stefania
De Luca
*a
aInstitute of Biostructures and Bioimaging, National Research Council, 80134 Naples, Italy
bDepartment of Science and Technologic Innovation, Università del Piemonte Orientale “A. Avogadro”, 15121 Alessandria, Italy
cInstitute of Crystallography, National Research Council, 70126 Bari, Italy
First published on 9th December 2020
Abstract
Bioconjugates of 2,1,3-benzoxadiazole (benzofurazan) and its derivatives have attracted considerable interest due to their biological activities and applications as fluorescent tags. A high-yield, chemoselective, and mild procedure for the S-alkylation of cysteine containing peptides by benzofurazan halogenides is reported. The key feature of this procedure is the use of activated molecular sieves (MS) to catalyze thiol activation for nucleophilic substitution under very mild conditions (room temperature and no need for added bases). To the best of our knowledge, this is the first report about thiol nucleophilic substitution performed on unsaturated and annelated systems catalyzed by activated molecular sieves. Reaction yields were remarkable even with benzofurazans having weakly activating groups or no activating groups at all. The potential of the new methodology was explored by synthesizing fluorescent, hydrophilic benzofurazan/peptide conjugates, also with peptides containing unprotected lysine residues.
Introduction
In the field of chemical biology, there is constantly increasing interest in developing bioconjugation strategies aimed at the selective modification of natural or non-natural amino acids to introduce exogenous moieties (such as drugs or tracers) into peptides.1,2 Such modifications could allow the manipulation of peptides or peptidomimetics with therapeutic potential and the introduction of probes useful for investigating biomolecular interactions. Among the 20 proteinogenic amino acids, cysteine represents one of the most viable targets for conjugation, due to its low natural abundance and to its strong nucleophilicity under slightly basic conditions (pH < 9).3–5The introduction of 2,1,3-benzoxadiazole moieties (benzofurazan) into peptide structures has shown useful application in biological and medicinal chemistry. Suitably substituted benzofurazans are commonly employed as fluorogenic reagents6–9 to label biomolecules and have also found clinical application as antibacterial and antiparasitic, as well as antiviral and antitumor agents.10–12
To date, several fluorescent 4,7-substituted benzofurazan derivatives have been investigated and employed as fluorogenic labels.13,14 Amongst them, 4-chloro-7-nitrobenzofurazan (a-Cl) is largely used as a fluorescent tag to label proteins and peptides. Indeed, its high reactivity to nucleophilic attack makes the conjugation chemistry easy to perform under slightly basic conditions.15–18 Other fluorescent/fluorogenic tags employed for biomolecule labelling rely on ionic benzofurazan derivatives. 4-Chloro-7-sulfobenzofurazan (b-Cl) and 4-fluoro-7-sulfobenzofurazan (b-F) can react specifically with a thiol group and are particularly suitable for analytical purposes. The advantage of such ionic benzofurazans is that they do not impart any hydrophobic character to the fluorescent adducts, thus enhancing the reliability of the assay.13,14 As the sulfonate group induces a weaker ring activating effect compared to the nitro group, the conjugation reaction of 7-sulfobenzofurazan halogenides (b-X) still requires basic pH (always >9) and a temperature around 60 degrees. These conditions could not be fully compliant with peptide modification. Therefore, a new synthetic protocol employing milder conditions for benzofurazan conjugation was explored.
An efficient and selective S-alkylation method to introduce modifications on a cysteine residue within peptide sequences was developed by our group. The key feature of this method is the use of activated molecular sieves (MS) as the catalyst to activate the thiol function for nucleophilic substitution under very mild conditions (room temperature and no need for any bases). It is worth remembering that cationic zeolites, such as alkaline-LTA, have been proven to be naturally endowed with mild basic properties, which is a great advantage compared to the basic strength of the commonly employed organic bases.19 By such a method, a variety of chemical functions were successfully introduced into polypeptides, including fluorophores, PEG units and lipid moieties that represent protein post-translational modifications. S-linked glycopeptides and lantipeptides were also successfully obtained.3,20–26
Here we applied our method to selectively conjugate either substituted or unsubstituted benzofurazans to cysteine containing peptides.
Results and discussion
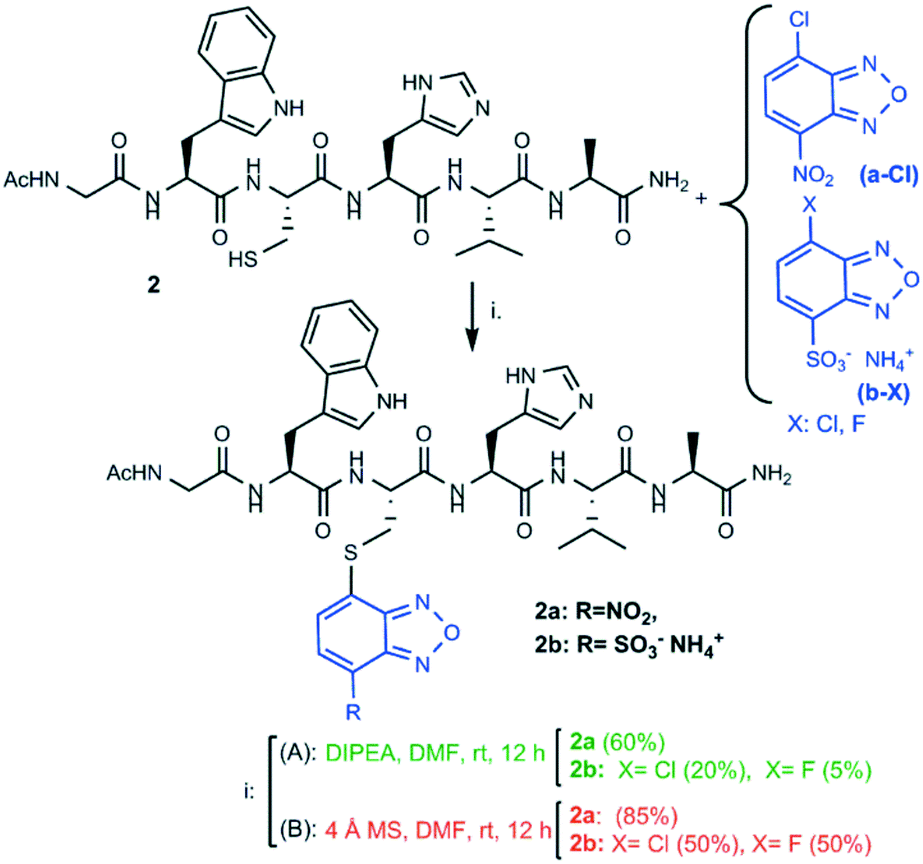
We started our investigation by reacting model peptide 1, bearing a cysteine residue at its N-terminus and no other nucleophiles, with a-Cl (4-chloro-7-nitrobenzofurazan) and b-X (4-chloro-/4-fluoro-7-sulfobenzofurazan) in the presence of activated 4 Å MS as the catalyst. MS were activated by heating at 280 °C for 4 h under vacuum, and the coupling reaction was carried out in DMF under an argon atmosphere. The benzofurazan derivative was reacted in 1.2 equiv. excess and the mixture was kept under stirring for 12 hours at room temperature. The yield of compound 1a was excellent (90%). That of compound 1b was very high as well, either by using b-Cl or b-F (85%, see Scheme 1). | ||
| Scheme 1 Incorporation of halogen-sulfo/nitro-benzofurazans in peptide 1. | ||
These results prompted us to continue with a deeper investigation to assess whether the nucleophilicity of the cysteine was affected by its relative position within the peptide sequence. Moreover, two potentially sensitive nucleophilic amino acids (Trp and His) were inserted into the sequence in close proximity to the cysteine residue. Model peptide 2 was conjugated with a-Cl and b-X (X = Cl, F). To better assess the catalytic efficiency of molecular sieves with respect to the standard chemistry for thiol activation, the thio-conjugation was performed either in the presence of DIPEA (strategy A) or in the presence of activated MS (strategy B), and the yields of the two synthetic strategies were compared. As shown in Scheme 2, the standard conditions (strategy A: 3 equiv. DIPEA, rt and 12 h) provided compound 2a in quite high yield, as expected, while they provided compound 2b in low or very low yield (20% and 5% by using b-Cl or b-F, respectively). The yield was assessed by the ratio between the HPLC peak area of the desired final product and the total area relative to peptides (including the unreacted peptide, the product, and by-products).
 | ||
| Scheme 2 Incorporation of halogen-sulfo/nitro-benzofurazans in peptide 2. | ||
The same conjugation reactions (Scheme 2) performed according to strategy B gave compound 2a with an enhanced yield (85%). Enhanced yields were also achieved for compound 2b (50%), by using both b-Cl or b-F as the coupling reagent (Scheme 2). This represents a huge improvement compared to the yields obtained by following strategy A. Compared to the yield (85%) obtained for compound 1b, we observed a lower yield (50%) for compound 2b, even though the same benzofurazan halogenides b-X (X = Cl, F) were used as reagents. Likely, the more hindered position of the cysteine residue in peptide 2 limits the final functionalization yield.
A detailed structural characterization of the reaction products was carried out by NMR to confirm that benzofurazan conjugation occurred at the cysteine sulfur atom and not at the potentially competing nucleophilic nitrogen atoms of tryptophan or histidine side chains. First, full proton resonance assignment was achieved by means of the sequence-specific method based on the iterative analysis of 2D-TOCSY and 2D-ROESY NMR spectra. Then, the 2D-ROESY NMR spectra were analyzed further for short distances involving the benzofurazan ring protons and neighboring amino acid side chain protons (representative 2D-NMR spectra for compound 2b are shown in Fig. 1; see also the ESI†). The formation of the S-conjugation product was unambiguously confirmed by the detection of intense ROE peaks (i.e. short interproton distances) between the cysteine Hα/Hβ protons and the benzofurazan H5 ring proton. No ROE signals between benzofurazan ring protons and side chain protons of tryptophan and histidine were detected.
 | ||
| Fig. 1 Overlay between the 2D-TOCSY (black peaks) and the 2D-ROESY (red peaks) NMR spectra of compound 2b (600 MHz, DMSO-d6, 298 K) showing the expansion over the fingerprint region. 2D-TOCSY strips with amino acid assignment are shown. ROE cross-peaks connecting the benzofurazan ring H5 proton and cysteine Hα and Hβ protons are indicated. 2D NMR spectra with full proton resonance assignments for all compounds considered are reported in the ESI.† | ||
Next, the accessibility to the thiol nucleophilic attack of benzofurazans bearing a halogen substituent at either position 4 or 5 was checked. The S-conjugation of peptide 1 and peptide 2 was performed with the substrates shown in Schemes 3 and 4. These benzofurazan substrates lack any electron-withdrawing group activating the substrate. For comparison, the same reactions were repeated in the presence of DIPEA as the base (strategy A: 3 equiv. DIPEA, rt and 12 h).
 | ||
| Scheme 3 Incorporation of 4-halogen-benzofurazan in peptide sequences. | ||
 | ||
| Scheme 4 Incorporation of 5-halogen-benzofurazan in peptide sequences. | ||
The yields for all the conjugates obtained by following the DIPEA protocol (strategy A) were poor or negligible. Meanwhile, the MS activation protocol (strategy B) provided the final products with yields ranging from 10% (2c, X = Cl) to 65% (1c, X = F) (Scheme 3). The NMR analysis confirmed that S-conjugation occurred at the expected position on the benzofurazan ring (i.e. at position 4 for 1c/2c and at position 5 for 1d/2d). This was confirmed by the detection of the expected pattern of ROE contacts between cysteine Hα/Hβ protons and the benzofurazan H5 proton (in compounds 1c and 2c), or the benzofurazan H4 proton (in compounds 1d and 2d). In addition, 2D 1H,13C HMBC NMR spectra showed the expected heteronuclear 3JHC correlation between Cys Hβ and the quaternary C4 carbon in compound 2c, or that between Cys Hβ and the quaternary C5 carbon for compound 2d (see the ESI†).
Concerning the 4-halogenobenzofurazans (c-X), the fluoro group was more reactive than the chloro group toward the sulfhydryl attack, for both peptides (Scheme 3). This trend is in agreement with the hypothesized mechanism of the nucleophilic substitution of halogen-benzofurazans, which proceeds through a two-step mechanism of SNAr type, even though they are not aromatic substrates. It is worth noting that the same trend was not so clearly observed for the 7-sulfo-benzofurazan rings, likely because the ring activating effect of the sulfonic group flattens the effect of the leaving halogen, especially when the products are obtained in high yields (Scheme 1).
Meanwhile, the reactivity order involving the leaving group for the 5-halogenobenzofurazans was in the order Br > Cl, i.e. the opposite of that typically observed for nucleophilic substitution on activated benzofurazan rings. In fact, halogen-benzofurazans have to be treated as halogen-activated unsaturated derivatives where the electron-withdrawing furazan group exerts an activating effect. In other words, dealing with the reaction mechanism, there is evidence that in some cases the order of halogen substituted benzofurazans followed that expected for the SNAr mechanism (F > Cl ∼ Br), in other cases it did not. The reaction mechanism is still unclear, and it deserves further investigations. An additional consideration concerns the higher yield of the conjugates obtained from benzofurazans substituted at position 5 (d-X) (Scheme 4). This could be explained by the better accessibility of position 5 to the nucleophile (d-X) compared to that of position 4 (c-X), which is peri within the annelated system (see Table 1).27
| Entrya | Peptide | Yield (%) DIPEA | Yield (%) MS |
|---|---|---|---|
| a Twelve hours reaction time at room temperature; ny: negligible yield. | |||
| 1a | AcC(a)GVANH2 | — | Cl: 90 |
| 1b | AcC(b)GVANH2 | — | X = Cl: 85 |
| X = F: 85 | |||
| 1c | AcC(c)GVANH2 | ny | X = F: 65 |
| X = Cl: 50 | |||
| 1d | AcC(d)GVANH2 | ny | X = Br: 85 |
| X = Cl: 30 | |||
| 2a | AcGWC(a)HVANH2 | Cl: 60 | Cl: 85 |
| 2b | AcGWC(b)HVANH2 | X = Cl: 20 | X = Cl: 50 |
| X = F: 5 | X = F: 50 | ||
| 2c | AcGWC(c)HVANH2 | X = F: 5 | X = F: 50 |
| X = Cl: ny | X = Cl: 10 | ||
| 2d | AcGWC(d)HVANH2 | X = Br: ny | X = Br: 40 |
| X = Cl: ny | X = Cl: 20 | ||
| 3d | AcC(d)GMVANH2 | — | Br: 90 |
| 4d | AcC(d)GWVANH2 | — | Br: 50 |
| 5d | AcC(d)GTVANH2 | — | Br: 75 |
| 6d | AcC(d)GVHANH2 | — | Br: 60 |
| 7d | AcC(d)GKVANH2 | — | X = Br: 80 |
| 7b | AcC(b)GKVANH2 | — | X = Cl: 85 |
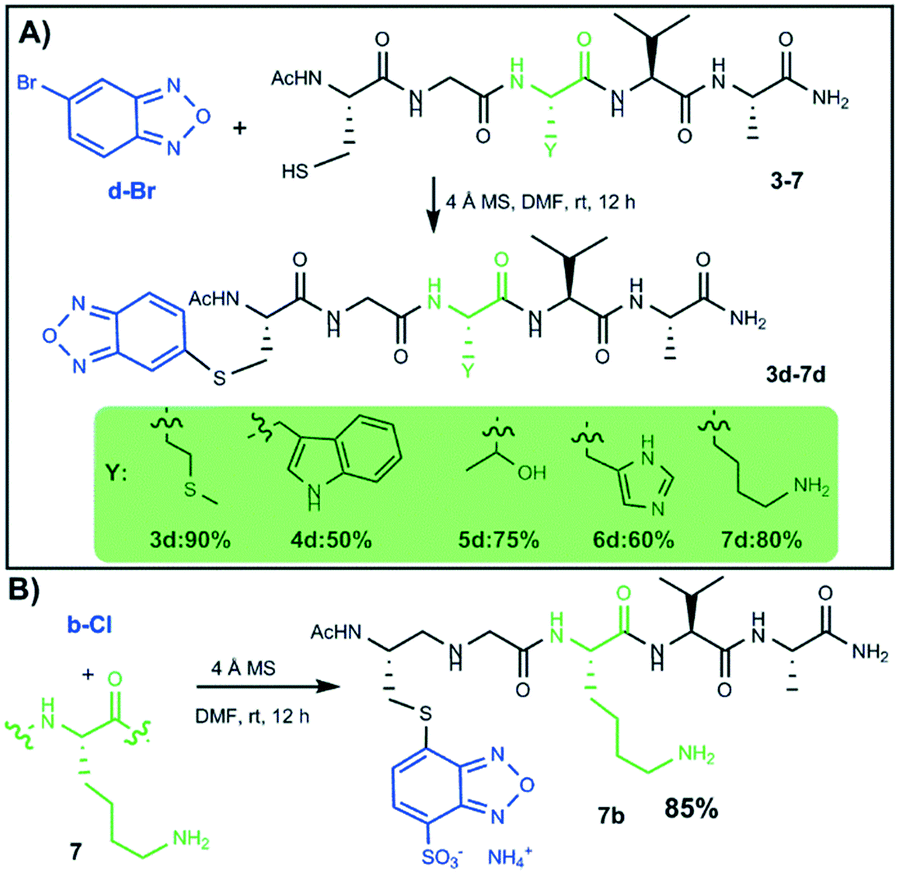
As the conjugates obtained from 5-bromobenzofurazan (d-Br) were collected in high yields (Scheme 4), a further study aimed at assessing the chemoselectivity for cysteine thiol was carried out on peptide sequences containing either Trp, Met, His, Thr, or Lys as potential nucleophile competitors (Scheme 5, panel A).
 | ||
| Scheme 5 Chemoselectivity of benzofurazans for cysteine in peptides containing potentially competing nucleophilic groups. | ||
All sequences proved to react selectively with 5-bromobenzofurazan (d-Br) through the cysteine thiol, as confirmed by NMR analysis (see the ESI†), and all peptides provided high conjugation yields. Remarkably, a very high chemoselectivity was also found when the competitor was the amino group of lysine that, amongst the side chain functional groups that are typically found in natural peptides, is by far the most potent competitor of the cysteine sulfhydryl group for nucleophilic attack. As a matter of fact, the synthesis of compound 7d (Scheme 5, panel A) gave the mono-S-alkylated peptide in high yield (80%), and no di-alkylation product (i.e. both on cysteine and lysine) was found at all by means of mass spectrometry.
These results prompted us to assess whether such a thiol-specificity was maintained also by 4-chloro-7-sulfobenzofurazan b-Cl (also known as Sbf), whose chemoselectivity in basic aqueous media under heating has already been established.13 As depicted in Scheme 5 (panel B), the reaction of b-Cl with peptide 7 (containing both cysteine and lysine) gave the mono-S-alkylated product 7b with an 85% yield.
The fluorescence emission spectrum of the purified product was characterized by an emission peak at 520 nm (excitation at 380 nm, see Fig. 2), that is typical and fully consistent with a Sbf-thiol derivative. Again, no di-alkylated or N-alkylated products were found, either by HPLC-MS or fluorescence spectroscopy (fluorescence emission spectra of Sbf-amine derivatives should give a typical emission peak at 590 nm when excited at 411 nm, which was not detected).
 | ||
| Fig. 2 Fluorescence emission spectrum (λex = 380 nm) of sulfobenzofurazan derivative 7b in water (the inset shows the spectrum of b-Cl). | ||
Experimental section
Materials and methods
Fmoc protected amino acids, Rink amide MBHA resin, N-hydroxybenzotriazole (HOBT) and benzotriazol-1-yl-oxy-tris-pyrrolidino-phosphonium (PyBOP) were purchased from Calbiochem-Novabiochem (Laufelfingen, Switzerland); piperidine and diisopropylethylamine (DIPEA) were purchased from Fluka (Milwaukee, WI); all solvents were purchased from Aldrich (St Louis, MI) or Fluka (Milwaukee, WI) and were used without further purification, unless otherwise stated. Molecular sieves, type 4 Å (beads, diameter 1.6 mm) were purchased from Aldrich and activated by heating at 280 °C for 4 h under vacuum. The benzofurazans [(4-chloro-benzofurazan, 4-chloro-7-sulfobenzofurazan ammonium salt, 4-chloro-7-nitrobenzofurazan), (4-fluoro-benzofurazan, 5-chloro-benzofurazan, 5-bromo-benzofurazan, 4-fluoro-7-sulfobenzofurazan ammonium salt)] were purchased from Sigma Aldrich and from Santa Cruz Biotechnology (Heidelberg).For all the RP-HPLC procedures the system solvent used was: H2O 0.1% TFA (A) and CH3CN 0.1% TFA (B) and detection was performed at 210 nm and 280 nm. Analytical RP-HPLC runs were carried out on a HP Agilent Series 1100 apparatus using a Phenomenex (Torrance, California) Kinetex column (5 μm C18 100 A – 60 × 4.60 m) with a flow rate of 1.0 mL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) min−1and a linear gradient starting from 5% to 70% B in 10 min; preparative RP-HPLC was carried out on a HP Agilent Series 1200 apparatus using a Phenomenex (Torrance, California) Gemini column (5 μm NX-C18 110 Å – 150 × 21.2 mm, AXIA™) with a flow rate of 15 mLmin−1 and a linear gradient starting from 5% to 70% B in 20 min.
min−1and a linear gradient starting from 5% to 70% B in 10 min; preparative RP-HPLC was carried out on a HP Agilent Series 1200 apparatus using a Phenomenex (Torrance, California) Gemini column (5 μm NX-C18 110 Å – 150 × 21.2 mm, AXIA™) with a flow rate of 15 mLmin−1 and a linear gradient starting from 5% to 70% B in 20 min.
LC-ESI-TOF-MS analyses were performed with an Agilent 1290 Infinity LC system coupled to an Agilent 6230 TOF LC/MS system (Agilent Technologies, CernuscoSulNaviglio, Italy). The system solvent used was: H2O 0.05% TFA (A) and CH3CN 0.05% TFA (B); Phenomenex (Torrance, California) Jupiter column (3 μm C18 300 Å – 150 × 2.0 mm); linear gradient starting from 5% to 70% B in 20 min and detection at 210 nm and 280 nm.
NMR spectra were acquired with a Bruker Avance spectrometer operating at 14 T (corresponding to a proton Larmor frequency of 600 MHz), equipped with an inverse Z-gradient 5 mm BBI probe. The temperature was set to 298.0 K, and controlled within ±0.1 K by means of a BTO2000 VTU system. Samples were dissolved in 600 μL of DMSO-d6 (99.9 atom%). The residual solvent resonance at 2.54 ppm was used as a secondary reference for chemical shift calibration. Resonance assignment was based on the analysis of homonuclear 2D-TOCSY and 2D-ROESY NMR spectra. The 2D-TOCSY spectra were acquired with the Bruker mlevphpr pulse program (homonuclear Hartman–Hahn transfer by means of the MLEV17 sequence28) in the phase-sensitive mode according to the States-TPPI scheme. Typical acquisition parameters included: 2 s relaxation delay, 32–64 scans, 16 dummy scans, 25 Hz bandwidth for the water suppression presaturation pulse (if required), 2048 × 256–400 data points, 13 ppm spectral width (in F2 and F1), and 100 ms mixing time. Data were treated with squared cosine window functions (both along F2 and F1) prior to complex FT. The 2D-ROESY spectra were acquired with the Bruker roesyphpr.2 pulse program in the phase-sensitive mode by the States TPPI scheme.29 Typical acquisition and processing parameters were similar to those for the 2D-TOCSY spectra, but with a mixing time of 300 ms at 2.5 kHz spin-lock field strength, and 64 scans. 2D 1H,13C 2D HSQC NMR spectra were acquired by means of the Bruker hsqcetgp pulse program in the phase sensitive mode using Echo/Antiecho-TPPI gradient selection and with decoupling during acquisition. Acquisition parameters included 1024 × 128 data points, 14 ppm (1H) and 165 ppm (13C) spectral window, and 16–64 scans. 2D 1H,13C 2D HMBC NMR spectra to detect long range 1H/13C couplings were acquired by means of the Bruker hmbcgplpndqf pulse program in the phase-insensitive mode with a low-pass J-filter to suppress one-bond correlations and no decoupling during acquisition. Acquisition parameters included 2048 × 128 data points, 14 ppm (1H) and 220 ppm (13C) spectral window, 32–128 scans, and 60 ms delay for the evolution of long range couplings. The spectra were processed using the Bruker Topspin 4 software package. Sequence specific assignment was carried out using the Computer Aided Resonance Assignment software package (CARA: R.L.J Keller “The Computer Aided Resonance Assignment”, 2004 CANTINA Verlag, Goldau, Switzerland).
Fluorescence spectra were recorded at room temperature on a Varian Cary Eclipse spectrofluorimeter, equipped with a 1.0 cm quartz cell, with an excitation wavelength of 380 nm and an emission range of 400–600 nm. Equal excitation and emission bandwidths were used throughout experiments, with a recording speed of 120 nm min−1 and automatic selection of the time constant.
Peptide synthesis
Peptide synthesis was carried out manually by a solid-phase method using the standard Fmoc-protecting group strategy. Appropriate Fmoc-amino acid derivatives [Fmoc-Ala-OH, Fmoc-Val-OH, Fmoc-Gly-OH, Fmoc-Cys(Trt)-OH, Fmoc-Met-OH, Fmoc-His(Trt)-OH, Fmoc-Trp(Boc)-OH, Fmoc-thr(tBu)-OH, Fmoc-Lys(Boc)OH] were employed and a Rink amide MBHA resin (0.7 mmol g−1 substitution; 50 μmol scale) was used as a solid support, as it releases peptides amidated at the C-terminus upon acid treatment. All Fmoc-amino acids were activated by an in situ PyBop/HOBt//DIPEA activation procedure. Amino acid coupling steps were monitored by the Kaiser test after 60 min coupling cycles. Fmoc-deprotection was performed with 20% piperidine in DMF for 5 + 10 min. The peptide N-terminus was acetylated by treatment with a mixture of acetic anhydride (4.7%) and pyridine (4%) in DMF for 10 min. The cleavage from the solid support and the simultaneous deprotection of all side chains were performed by suspending the fully protected compound-resins in TFA/H2O/TIS (97:2:1) for 3 h. The peptides were isolated by precipitation in cold diethyl ether and centrifuged to form a pellet.
General procedure for post-synthetic peptide S-alkylation
Each solution of acetylated peptide in DMF (5 mg mL−1) was kept under an argon atmosphere for 5 min. Then, the solution was transferred, using a syringe, in a 10 mL round-bottom flask covered with a rubber top and containing 4 Å molecular sieves (3–3.5 g), previously activated at 280 C° for 4 h under vacuum (10−4 mbar). After a few minutes, the benzofurazans (1.2 equiv.) were added to the reaction mixture that was stirred at room temperature for 12 h. The mixture was centrifuged to eliminate the sieves and the supernatant was concentrated under vacuum. RP-HPLC and mass spectrometry analyses were performed on the obtained crude and each recovered final product fully characterized by NMR spectroscopy.Yield (1a) after RP-HPLC purification: 50% (3.4 mg); preparative HPLC tR = 17.844 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C21H29N8O8S+ 553.1824, found 553.1920 ([M + H]+). Yellow solid.
Compound 1a1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.61 (d, 1H), (8.56 t, 1H), 8.54 (d, 1H), 8.02 (d, 1H), 7.86 (d, 1H), 7.67 (d, 1H), 7.23 (s, 1H), 6.99 (s, 1H), 4.76 (dt, 1H), 4.23–4.21 (m, o, 2H), 3.87 (dd, 1H), 3.79–3.76 (m, o, 2H), 3.53 (dd, 1H), 2.03 (m, 1H), 1.91 (s, 3H), 1.25 (d, 3H), 0.90 (d, 3H), 0.86 (d, 3H).
Yield (1b) after RP-HPLC purification: 22% (1.3 mg); preparative HPLC tR = 12.728 min (close to the tR of the substrate employed b-Cl); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C21H30N7O9S2+ 588.1541, found 588.1545. Yellow solid.
Yield (1b) after RP-HPLC purification: 22% (1.8 mg); preparative HPLC tR = 13.043 min (co-eluted with the substrate employed b-F); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C21H30N7O9S2+ 588.1541, found 588.1514. Yellow solid.
Compound 1b1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.53 (t, 1H), 8.42 (d, 1H), 8.00 (d, 1H), 7.78 (d, 1H), 7.67 (d, 1H), 7.47 (d, 1H), 7.21 (s, 1H), 6.98 (s, 1H), 4.63 (dt, 1H), 4.23–4.21 (m, o, 2H), 3.83 (dd, 1H), 3.74 (dd, 1H), 3.61 (dd, 1H), 3.38 (m, overlapping with the water residual signal), 2.02 (m, 1H), 1.88 (s, 3H), 1.24 (d, 3H), 0.89 (d, 3H), 0.85 (d, 3H).
Yield (2c) after RP-HPLC purification: 32% (2.0 mg); preparative HPLC tR = 17.068 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C21H30N7O6S+ 508.1973, found 508.1949. White solid.
Yield (1c) after RP-HPLC purification: 28% (2.1 mg); preparative HPLC tR = 17.123 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C21H30N7O6S+ 508.1973, found 508.1949. White solid.
Compound 1c1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.49 (t, 1H), 8.43 (d, 1H), 8.01 (d, 1H), 7.87 (d, 1H), 7.80 (d, 1H), 7.58–7.55 (AB system, 2H), 7.22 (s, 1H), 6.99 (s, 1H), 4.63 (dt, 1H), 4.23–4.20 (m, o, 2H), 3.83 (dd, 1H), 3.74 (dd, 1H), 3.61 (dd, 1H), 3.36 (dd, overlapping with the water residual signal), 2.03 (m, 1H), 1.88 (s, 3H), 1.24 (d, 3H), 0.89 (d, 3H), 0.85 (d, 3H).
13C-NMR δ, ppm, (from HSQC/HMBC, 150 MHz, DMSO-d6, 298 K): 174.8, 171.0, 170.7, 170.5, 169.3, 149.7, 149.3, 133.7, 128.3, 126.7, 112.8, 58.4, 52.3, 48.8, 43.0, 33.8, 31.2, 23.3, 19.8, 18.8, 18.6.
Yield (1d) after RP-HPLC purification: 48% (3.0 mg); preparative HPLC tR = 17.181 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C21H30N7O6S+ 508.1973, found 508.1955. White solid.
Yield (1d) after RP-HPLC purification: 18% (1.1 mg); preparative HPLC tR = 17.190 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C21H30N7O6S+ 508.1973, found 508.2059. White solid.
Compound 1d1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.58 (t, 1H), 8.46 (d, 1H) 8.02 (d, 1H), 8.00 (d, 1H), 7.89 (d, 1H), 7.81 (d, 1H), 7.48 (d, H1), 7.22 (s, 1H), 6.99 (s, 1H), 4.68 (dt, 1H), 4.24–4.22 (m,o, 2H), 3.86 (dd, 1H), 3.77 (dd, 1H), 3.52 (dd, 1H), 3.33 (dd, overlapping with the water residual signal), 2.03 (m, 1H), 1.91 (s, 3H), 1.25 (d, 3H), 0.90 (d, 3H), 0.86 (d, 3H).
13C-NMR δ, ppm, (from HSQC/HMBC, 150 MHz, DMSO-d6, 298 K): 174.8, 171.1, 171.0, 170.8, 170.5, 169.6, 149.9, 148.6, 144.1, 136.9, 134.2, 127.7, 124.3, 121.4, 119.1, 118.7, 116.3, 111.8, 110.3, 107.9, 58.4, 54.0, 51.8, 48.7, 42.7, 33.7, 31.1, 28.3, 23.2, 19.6, 18.9, 18.8.
Yield (2a) after RP-HPLC purification: 30% (1.4 mg); preparative HPLC tR = 18.069 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H46N13O10S+ 876.3206, found 876.3182. Yellow solid.
Yield (2a) after RP-HPLC purification: 47% (3.0 mg); preparative HPLC tR = 18.302 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H46N13O10S+ 876.3206, found 876.3658. Yellow solid.
Compound 2a1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 10.85 (d, H1), 8.97 (br), 8.64 (m, o, 1H), 8.62 (m, o, 1H), 8.47 (d, 1H), 8.17–8.10 (m, o, 3H), 7.90 (d, 1H), 7.60 (d, 1H), 7.58 (d, 1H), 7.37 (br), 7.33 (d, 1H), 7.27 (br, 1H), 7.16 (d, 1H), 7.06 (t, 1H), 7.01 (s, 1H), 6.97(t, 1H), 4.71–4.70 (m,o, 2H), 4.57 (m, 1H), 4.24 (m, 1H), 4.18 (m, 1H), 3.76 (dd, o, 1H), 3.70 (dd, o, 1H), 3.60 (dd, o, 1H), 3.58 (dd, o, 1H), 3.17 (dd, 1H), 3.10 (br, 1H), 3.03 (br, 1H), 2.97 (dd, 1H), 2.05 (m, 1H), 1.84 (s, 3H), 1.25 (d, 3H), 0.89 (d, 3H), 0.86 (d, 3H).
Yield (2b) after RP-HPLC purification: 12% (0.7 mg); preparative HPLC tR = 15.329 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O11S2+ 911.2923, found 911.2884. Yellow solid.
Yield (2b) after RP-HPLC purification: 26% (1.5 mg); preparative HPLC tR = 15.388 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O11S2+ 911.2923, found 911.3159. Yellow solid.
Yield (2b) after RP-HPLC purification: 0.5%; preparative HPLC tR = 21.112 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O11S2+ 911.2923, found 11.2831. Yellow solid.
Yield (2b) after RP-HPLC purification: 31% (1.8 mg); preparative HPLC tR = 15.462 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O11S2+ 911.2923, found 911.2884. Yellow solid.
Compound 2b1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 10.85 (d, 1H), 8.98 (s, br, 1H), 8.53 (d, 1H), 8.48 (d, 1H), 8.15–8.10 (m, o, 3H), 7.83 (d, 1H), 7.70 (d, 1H), 7.60 (d, 1H), 7.43 (d, 1H), 7.40 (s, 1H), 7.34 (d, 1H), 7.27 (s, 1H), 7.18 (d, 1H), 7.07 (t, 1H), 7.00 (s, o, 1H), 6.99 (t, o, 1H), 4.69 (q, 1H), 4.59 (dt, 1H), 4.54 (q, 1H), 4.23 (m, 1H), 4.19 (dd, 1H), 3.77 (dd, 1H), 3.63 (dd, o, 1H), 3.59 (dd, o, 1H), 3.41 (dd, overlapping with the water residual signal, 1H), 3.17 (dd, 1H), 3.12 (dd, 1H), 3.02 (dd, o, 1H), 2.97 (dd, o, 1H), 2.04 (m, 1H), 1.85 (s, 3H), 1.24 (d, 3H), 0.89 (d, 3H), 0.86 (d, 3H).
Yield (2c) after RP-HPLC purification: 0.5%. Preparative HPLC tR = 17.725 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O8S+ 831.3355, found 831.3327. White solid.
Yield (2c) after RP-HPLC purification: 30% (1.7 mg); preparative HPLC tR= 17.670 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O8S+ 831.3355, found 831.3279. White solid.
Yield (2c) after RP-HPLC purification: 0%.
Yield (2c) after RP-HPLC purification: 7% (∼0.2 mg); preparative HPLC tR = 17.726 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O8S+ 831.3355, found 831.3321 ([M + H]+). White solid.
Compound 2c1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 10.87 (d, 1H), 8.96 (br, 1H), 8.55 (d, 1H), 8.44 (d, 1H), 8.17 (d, br, 1H), 8.13 (t, 1H), 8.10 (d, 1H), 7.88 (d, 1H), 7.85 (d, 1H), 7.59 (d, 1H), 7.56 (d, 1H), 7.51 (d, 1H), 7.37 (br, 1H), 7.35 (d, 1H), 7.28 (s, 1H), 7.17 (d, 1H), 7.07 (t, 1H), 7.01 (s, 1H), 6.98 (t, 1H), 4.70 (q, 1H), 4.58–4.56 (m, o, 2H), 4.24 (m, 1H), 4.19 (dd, 1H), 3.78 (dd, 1H), 3.61 (m, o, 2H), 3.42 (dd, overlapping with the water residual signal), 3.16 (dd, o, 1H), 3.12 (dd, 1H), 3.01 (dd, 1H), 2.96 (dd, 1H), 2.04 (m, 1H), 1.85 (s, 3H), 1.24 (d, 3H), 0.89 (d, 3H), 0.86 (d, 3H).
Yield (2d-Br) after RP-HPLC purification: 0%.
Yield (2d) after RP-HPLC purification: 24% (1.1 mg); preparative HPLC tR = 17.838 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O8S+ 831.3355, found 831.3337 ([M + H]+). White solid.
Yield (2d-Cl) after RP-HPLC purification: 0%.
Yield (2d) after RP-HPLC purification: 5% (∼0.3 mg); preparative HPLC tR = 17.944 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C38H47N12O8S+ 831.3355, found 831.3344 ([M + H]+). White solid.
Compound 2d1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 10.87 (s, 1H), 8.97 (br, 1H), 8.57 (br, 1H), 8.52 (d, 1H), 8.17–8.11 (m, o, 3H), 8.01 (d, 1H), 7.87 (s, 1H), 7.83 (d, 1H), 7.61 (d, 1H), 7.46 (d, 1H), 7.36 (d, 1H), 7.27 (br, 1H), 7.17 (s, 1H), 7.08 (t, 1H), 7.02–6.99 (m, o, 2H), 4.70 (br, 1H), 4.63–4.62 (m, o, 2H), 4.26 (m, 1H), 4.19 (br, 1H), 3.78 (dd, 1H), 3.61 (dd, 1H), 3.49 (dd, overlapping with the water residual signal), 3.18 (dd, 1H), 3.13 (br, o), 3.01 (br, o), 2.97 (dd, o, 1H), 2.05 (m, 1H), 1.85 (s, 3H), 1.25 (d, 3H), 0.90 (d, 3H), 0.87 (d, 3H).
Yield (3d) after RP-HPLC purification: 49% (2.4 mg); preparative HPLC tR = 18.577 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C26H39N8O7S2+ 639.2378, found 639.2380.
Compound 3d1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.61 (t, 1H), 8.49 (d, 1H), 8.00 (d, 1H), 7.97 (d, 1H), 7.91–7.88 (m, o, 3H), 7.48 (dd, 1H), 7.29 (s, 1H), 7.01 (s, 1H), 4.65 (dt, 1H), 4.46 (dt, 1H), 4.23 (m, 1H), 4.17 (dd, 1H), 3.78 (ABX system, 2H), 3.53 (dd, 1H), 3.33 (dd, overlapping with the residual water signal), 2.47 (m, overlapping with solvent), 2.07 (s, 3H), 2.03 (m, 2H), 1.95 (m, 1H), 1.91 (s, 3H), 1.83 (m, 1H), 1.23 (d, 3H), 0.88 (d, 3H), 0.86 (d, 3H).
Yield (4d) after RP-HPLC purification: 29% (1.8 mg); preparative HPLC tR = 19.847 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C32H40N9O7S+ 694.2766, found 694.2780. White solid.
Compound 4d1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 10.81 (d, 1H), 8.53 (t, 1H), 8.43 (d, 1H), 8.04 (d, 1H), 8.01 (d, 1H), 7.97 (d, 1H), 7.91 (d, 1H), 7.87 (s, 1H), 7.62 (d, 1H), 7.45 (dd, 1H), 7.33 (d, 1H), 7.29 (s, 1H), 7.15 (d, 1H), 7.07 (t, 1H), 7.02 (s, 1H), 6.99 (t, 1H), 4.67–4.66 (m.o, 2H), 4.26 (m, 1H), 4.19 (dd, 1H), 3.82 (dd, 1H), 3.64 (dd, 1H), 3.48 (dd, 1H), 3.29 (dd, 1H), 3.19 (dd, 1H), 2.98 (dd, 1H), 2.05 (m, 1H), 1.89 (s, 3H), 1.25 (d, 3H), 0.89 (d, 3H), 0.87 (d, 3H).
13C-NMR δ, ppm, (from HSQC/HMBC, 150 MHz, DMSO-d6, 298 K): 174.8, 171.1, 171.0, 170.8, 170.5, 169.6, 149.9, 148.6, 144.1, 136.9, 134.2, 127.7, 124.3, 121.4, 119.1, 118.7, 116.3, 111.8, 110.3, 107.9, 58.4, 54.0, 51.8, 48.7, 42.7, 33.7, 31.1, 28.3, 23.2, 19.6, 18.9, 18.8.
Yield (5d) after RP-HPLC purification: 35% (2.0 mg); preparative HPLC tR = 16.782 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C25H37N8O8S+ 609.2450, found m/z 609.2418 ([M+H]+). White solid.
Compound 5d1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.63 (t, 1H), 8.50 (d, 1H), 8.00 (d, 1H), 7.94 (d, 1H), 7.90 (s, 1H), 7.78 (d, o, 1H), 7.77 (d, o, 1H), 7.48 (dd, 1H), 7.22 (s, 1H), 7.00 (s, 1H), 5.01 (d, 1H), 4.68 (dt, 1H), 4.37 (dd, 1H), 4.21 (m, o, 2H), 4.04 (m, 1H), 3.88 (dd, 1H), 3.82 (dd, 1H), 3.52 (dd, 1H), 3.33 (m, overlapping with the water residual signal), 2.06 (m, 1H), 1.91 (s, 3H), 1.23 (d, 3H), 1.07 (d, 3H), 0.90 (d, 3H), 0.87 (d, 3H).
Yield (6d) after RP-HPLC purification: 30% (1.8 mg); preparative HPLC tR = 29.905 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C27H37N10O7S+ 645.2562, found 645.2568. White solid.
Compound 6d1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.94 (br, exchange), 8.60 (t, 1H), 8.49 (d, 1H), 8.17 (br, 1H), 8.15 (d, 1H), 8.00 (d, 1H), 7.92 (d, 1H), 7.87 (s, 1H), 7.48 (dd, 1H), 7.33 (br, exchange), 7.27 (s, 1H), 7.02 (s, 1H), 4.70 (br, o, 1H), 4.65 (dt, 1H), 4.24 (m, 1H), 4.17 (dd, 1H), 3.80 (dd, 1H), 3.75 (dd, 1H), 3.53 (dd, 1H), 3.33 (m, overlapping with the water residual signal), 3.09 (br, 1H), 2.98 (br, 1H), 2.05 (m, 1H), 1.91 (s, 3H), 1.25 (d, 3H), 0.90 (d, 3H), 0.87 (d, 3H).
Yield (7d) after RP-HPLC purification: 45% (3.1 mg); preparative HPLC tR = 15.055 min; HRMS (ESI-TOF) m/z: [M + H]+ calculated for C27H42N9O7S+ 636.2943, found 636.2976. White solid.
Compound 7d1H-NMR δ, ppm (600 MHz, DMSO-d6, 298 K). 8.58 (t, 1H), 8.49 (d, 1H), 8.01 (d, 1H), 7.95 (d, 1H), 7.89–7.87 (m, o, 3H), 7.64 (br, exchange), 7.48 (dd, 1H), 7.32 (s, 1H), 7.02 (s, 1H), 4.66 (dt, 1H), 4.38 (dt, 1H), 4.22 (m, 1H), 4.17 (dd, 1H), 3.78 (ABX system, 2H), 3.50 (dd, 1H), 3.32 (overlapping with the water residual signal), 2.79 (m, 2H), 2.04 (m, 1H), 1.91 (s, 3H), 1.71–1.55 (m, o, 5H), 1.32 (m, 1H), 1.23 (d, 3H), 0.88 (d, 3H), 0.86 (d, 3H).
Yield (7b) after RP-HPLC purification: 42% (4.0. mg); preparative HPLC tR = 12.248 min (co-eluted with the substrate employed b-Cl); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C27H42N9O10S2+ 716.2503, found 716.2564. Yellow solid.
Conclusion
In conclusion, a clean, mild and efficient procedure to selectively introduce benzofurazan moieties into peptide sequences via S-conjugation was developed. It relies on the moderate catalytic activity of activated molecular sieves to promote the reaction.We showed that our protocol can be successfully applied to a number of different benzofurazan halogenides, even those lacking activating groups. Hydrophilic fluorescent peptide-based probes were prepared, proving the potential and the reliability of the developed synthetic method.
As a final remark, the excellent chemoselectivity for cysteine over lysine provided by the mild catalytic conditions paves the way for dual-labelled peptides, as S-alkylated peptides can be subjected to sequential N-alkylation to add another functional group. The developed protocol can be potentially used to synthesize multi-functionalized peptides on a scale allowing applications beyond analytical purposes. This matter certainly deserves further studies, considering the ease of handling of the reaction mixture and a simple final work up that characterizes the modification of peptide molecules under the reported reaction conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like thank Leopoldo Zona, Luca De Luca and Maurizio Amendola for the technical assistance.Notes and references
- (a) C. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC; (b) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed; (c) Q.-Y. Hu, F. Berti and R. Adamo, Chem. Soc. Rev., 2016, 45, 1691–1719 RSC.
- (a) J. N. DeGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed; (b) N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed; (c) C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740–4753 CrossRef CAS PubMed.
- E. Calce and S. De Luca, Chem. – Eur. J., 2017, 23, 224–233 CrossRef CAS PubMed.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- K. Renault, J. W. Fredy, P. Renard and C. Sabot, Bioconjugate Chem., 2018, 29, 2497–2513 CrossRef CAS PubMed.
- K. Imai, T. Toyo'oka and Y. Watanabe, Anal. Biochem., 1983, 128, 471–473 CrossRef CAS PubMed.
- S. Uchiyama, T. Santa and K. Imai, Anal. Chem., 2001, 73, 2165–2170 CrossRef CAS PubMed.
- S. Uchiyama, T. Santa, S. Suzuki, H. Yokosu and K. Imai, Anal. Chem., 1999, 71, 5367–5371 CrossRef CAS PubMed.
- S. Uchiyama, T. Santa, T. Fukushima, H. Homma and K. Imai, J. Chem. Soc., Perkin Trans. 2, 1998, 10, 2165–2174 RSC.
- A. P. Piccionello and A. Guarcello, Curr. Bioact. Compd., 2010, 6, 266–283 CrossRef CAS.
- D. Rotili, A. De Luca, D. Tarantino, S. Pezzola, M. Forgione, B. M. Della Rocca and A. M. Caccuri, Eur. J. Med. Chem., 2015, 89, 156–171 CrossRef CAS PubMed.
- I. Almi, S. Belaidi, N. Melkemi and D. Bouzidi, J. Bionanosci., 2018, 12, 1–9 CrossRef.
- J. L. Andrews, P. Ghosh, B. Ternai and M. W. Whitehouse, Arch. Biochem. Biophys., 1982, 214, 386–396 CrossRef CAS PubMed.
- Y. Numasawa, K. Okabe, S. Uchiyama, T. Santa and K. Imai, Dyes Pigm., 2005, 67, 189–195 CrossRef CAS.
- L.-Y. Niu, H.-R. Zheng, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Analyst, 2014, 139, 1389–1395 RSC.
- Y. Yang and X. Guan, Anal. Chem., 2015, 87, 649–655 CrossRef CAS PubMed.
- Y. Li, Y. Yang and X. Guan, Anal. Chem., 2012, 84, 6877–6883 CrossRef CAS PubMed.
- A. Koshiyama and K. Imai, Analyst, 2010, 135, 2119–2124 RSC.
- V. Verdoliva, M. Saviano and S. De Luca, Catalysts, 2019, 9, 248 CrossRef , 1–22.
- L. Monfregola and S. De Luca, Amino Acids, 2011, 41, 981–990 CrossRef CAS PubMed.
- L. Monfregola, M. Leone, E. Calce and S. De Luca, Org. Lett., 2012, 14, 1664–1667 CrossRef CAS PubMed.
- E. Calce, M. Leone, L. Monfregola and S. De Luca, Org. Lett., 2013, 15, 5354–5357 CrossRef CAS PubMed.
- E. Calce and S. De Luca, Amino Acids, 2016, 48, 2267–2271 CrossRef CAS PubMed.
- E. Calce, M. Leone, L. Monfregola and S. De Luca, Amino Acids, 2014, 46, 1899–1905 CrossRef CAS PubMed.
- E. Calce, M. Leone, F. A. Mercurio, L. Monfregola and S. De Luca, Org. Lett., 2015, 17, 5646–5649 CrossRef CAS PubMed.
- E. Calce, G. Digilio, V. Menchise, M. Saviano and S. De Luca, Chem. – Eur. J., 2018, 24, 6231–6238 CrossRef CAS PubMed.
- D. Dal Monte, E. Sandri, L. Di Nunno, S. Florio and P. E. Todesco, J. Chem. Soc. B, 1971, 0, 2209–2213 RSC.
- A. Bax and D. G. Davis, J. Magn. Reson., 1985, 65, 355–360 CAS.
- T.-L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1992, 114, 3157–3159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, HPLC profiles, NMR, MS and fluorescence spectra, and NMR chemical shift tables of benzofurazan/peptide conjugates. See DOI: 10.1039/d0cy02004d |
| This journal is © The Royal Society of Chemistry 2021 |