
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 hydrogenation to methanol and hydrocarbons over bifunctional Zn-doped ZrO2/zeolite catalysts†
Pierfrancesco
Ticali‡
 a,
Davide
Salusso‡
a,
Rafia
Ahmad
b,
Christian
Ahoba-Sam
c,
Adrian
Ramirez
b,
Genrikh
Shterk
b,
Kirill A.
Lomachenko
d,
Elisa
Borfecchia
a,
Sara
Morandi
a,
Luigi
Cavallo
b,
Jorge
Gascon
*b,
Silvia
Bordiga
*a and
Unni
Olsbye
*c
a,
Davide
Salusso‡
a,
Rafia
Ahmad
b,
Christian
Ahoba-Sam
c,
Adrian
Ramirez
b,
Genrikh
Shterk
b,
Kirill A.
Lomachenko
d,
Elisa
Borfecchia
a,
Sara
Morandi
a,
Luigi
Cavallo
b,
Jorge
Gascon
*b,
Silvia
Bordiga
*a and
Unni
Olsbye
*c
aDepartment of Chemistry, NIS Center and INSTM Reference Center, University of Turin, 10125, Turin, Italy. E-mail: silvia.bordiga@unito.it
bKing Abdullah University of Science and Technology, KAUST Catalysis Center (KCC), Thuwal 23955, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
cSMN Centre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, N-0315 Oslo, Norway. E-mail: unni.olsbye@kjemi.uio.no
dEuropean Synchrotron Radiation Facility, CS 40220, 38043 Grenoble Cedex 9, France
First published on 18th January 2021
Abstract
The tandem process of carbon dioxide hydrogenation to methanol and its conversion to hydrocarbons over mixed metal/metal oxide-zeotype catalysts is a promising path to CO2 valorization. Herein, we report three Zn-doped ZrO2 catalysts prepared by co-precipitation of Zn- and Zr-containing salts to obtain three different loadings of Zn (5, 15 and 30 wt%). In the context of bifunctional catalysts, we combined ZrZnOX with two of the most performing zeolite/zeotype catalysts for the methanol-to-hydrocarbons (MTH) reaction: H-ZSM-5 and H-SAPO-34. Catalytic testing at 250–350 °C and 20–40 bar revealed that H-ZSM-5 is more stable and more capable of converting methanol at low temperature, whereas H-SAPO-34 shows the highest C3 selectivity. The best performance was observed for the ZrZnOX sample with 30% Zn, combined with ZSM-5 at 350 °C, 30 bar and H2/CO2/N2 = 6/2/1. Under these conditions, the equilibrium methanol yield was observed after 0.4 s g−1 ml−1 over ZrZnOX alone. Mixing with ZSM-5 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio, methanol was rapidly converted to hydrocarbons, with an optimum C3 productivity of 1.5 mol kg−1 h−1 at 24000 ml h−1 g−1. An extensive surficial, textural and structural characterization of ZrZnOX alone was carried out by FT-IR spectroscopy, N2 adsorption/desorption at liquid nitrogen temperature, PXRD and XAS. Formation of a ZrZnOX tetragonal solid solution was confirmed for all the samples (PXRD, XAS). The amount of Zr4+ sites at the surface was found to decrease, while the number of oxygen vacancies increased after H2 treatment at 400 °C, coherent with an increase of Zn loading (FT-IR). DFT modelling pointed out that once a stoichiometric oxygen vacancy is induced by the presence of Zn, the formation of extra oxygen vacancies during activation is thermodynamically favored. Moreover, i) the oxygen vacancies were found to play an active role in CO2 hydrogenation, in accordance with experimental data, and ii) methanol is most likely formed via the formate pathway, and is energetically favored compared to CO formation, in agreement with the high methanol selectivity observed experimentally at low CO2 conversion. Importantly, operando-XAS, XPS, TEM and PXRD studies of the as-prepared, pretreated and tested catalysts showed that the structure and composition of the catalyst is not affected by the reaction. Indeed, a final catalytic test carried out on the regenerated ZrZnOX/H-ZSM-5 catalyst showed that the initial performances were completely restored and no Zn exchange in the zeolite was observed neither before nor after testing.
:1 weight ratio, methanol was rapidly converted to hydrocarbons, with an optimum C3 productivity of 1.5 mol kg−1 h−1 at 24000 ml h−1 g−1. An extensive surficial, textural and structural characterization of ZrZnOX alone was carried out by FT-IR spectroscopy, N2 adsorption/desorption at liquid nitrogen temperature, PXRD and XAS. Formation of a ZrZnOX tetragonal solid solution was confirmed for all the samples (PXRD, XAS). The amount of Zr4+ sites at the surface was found to decrease, while the number of oxygen vacancies increased after H2 treatment at 400 °C, coherent with an increase of Zn loading (FT-IR). DFT modelling pointed out that once a stoichiometric oxygen vacancy is induced by the presence of Zn, the formation of extra oxygen vacancies during activation is thermodynamically favored. Moreover, i) the oxygen vacancies were found to play an active role in CO2 hydrogenation, in accordance with experimental data, and ii) methanol is most likely formed via the formate pathway, and is energetically favored compared to CO formation, in agreement with the high methanol selectivity observed experimentally at low CO2 conversion. Importantly, operando-XAS, XPS, TEM and PXRD studies of the as-prepared, pretreated and tested catalysts showed that the structure and composition of the catalyst is not affected by the reaction. Indeed, a final catalytic test carried out on the regenerated ZrZnOX/H-ZSM-5 catalyst showed that the initial performances were completely restored and no Zn exchange in the zeolite was observed neither before nor after testing.
1. Introduction
Today, the use of fossil fuels, coal, oil and natural gas represents the main source of carbon dioxide, which is principally responsible for the increment of global temperature. Its concentration in the atmosphere already overtook planetary boundary estimates and is expected to keep rising, reaching 570 ppm at the end of the century.1The single carbon atom that CO2 possesses can be recovered and eventually added to other organic chemicals to obtain useful products. Technologies to recover and convert CO2 have been known since the mid-19th century, however, only from the 1970s, CO2 found its first industrial application in the synthesis of methanol from CO2-enriched syngas (CO and H2).2 To date, CO2 capture and utilization represents a promising route to control its emission while limiting fossil fuel extraction. Currently, biological, electrochemical and catalytic processes are all exploited for CO2 valorization. Concerning the catalytic processes, carbon capture and storage (CCS) technologies2 can be coupled with utilization of CO2 as a feedstock in: i) low energy processes3 such as production of urea, carbonates, carbamates and ii) high-energy processes where high-value chemicals (CH4, HCOOH, and CH3OH) are obtained.2,4
High-energy processes mostly exploit the capability of certain materials to reduce carbon dioxide to hydrocarbons and/or olefins. However, as CO2 is the most oxidized form of carbon, it is located in a thermodynamic well, which makes its chemical reduction challenging,5 such that high temperature and pressure are required, increasing the total cost of the process.
CO2 reduction can be achieved by using H2: both academic and industrial research efforts are today focused on using renewable sources of H2 to reduce the environmental impact of these processes.6,7 Hydrogenation is industrially exploited for the production of methanol, massively employed as a solvent, alternative fuel and feedstock for the chemical industry. To date, industrial-scale methanol production is still carried out from syngas over Cu/ZnO/Al2O3 catalysts developed by Imperial Chemical Industries (ICI).8 Partial substitution of the feed with CO2 causes a drastic catalyst selectivity decreases8 due to the reverse water gas shift (RWGS) reaction (eqn (1)).
Another approach is to convert methanol to hydrocarbons in the same reaction batch. For this purpose, bifunctional catalysts play a key role in carbon dioxide hydrogenation and conversion to organic compounds, but the main challenge consists of compromising the catalyst performances, such as activity, selectivity and conversion, with the energy cost of the total reaction, i.e. low pressure and temperature (1 bar, <400 °C). The main reactions involved in this process are:
| CO2 + H2 = CO + H2O (RWGS) | (1) |
| CO2 + 3H2 = CH3OH + H2O | (2) |
| CO + 2H2 = CH3OH | (3) |
| nCH3OH → CnH2n + nH2O | (4) |
Starting from the first step involved in CO2 hydrogenation to methanol, it is important to compromise the thermodynamics of the reaction,16i.e. methanol formation at low temperature and high pressure. From a global point of view, low temperature favors methanol production on the first catalyst while high temperature enhances methanol dehydration and C–C coupling in the zeolite. With this respect, high temperature moves the equilibrium of the first catalyst towards the endothermic RWGS reaction (eqn (1)).
In recent years, different research studies6,7,17–22 have been focused on using the same types of catalysts involved in the RWGS reaction but trying to promote: i) stabilization of intermediates for hydrogenation to methanol or other hydrocarbons instead of RWGS ones; ii) H2 dissociation by heterolytic splitting; and iii) inhibition of the water poisoning effect, which hampers the catalytic hydrogenation activity.21
ZrO2 has been investigated as a support material in many binary and ternary systems for CO/CO2 hydrogenation to methanol.23–29 IR and TPD studies over pristine zirconia conducted by Pokrovski et al.30 showed that CO and CO2 are mainly adsorbed as HCO−, CO32− and HCO3−, m/b-CO32−, respectively. The CO2 adsorption capacity increases with the strength of Zr4+ Lewis acid sites, O2− Lewis basic sites and higher concentration and basicity of hydroxyl groups. However former studies showed that the main CO2/ZrO2 interaction occurs through the oxide basic sites;28,31 in particular with the formation of bicarbonate b-HCO3–Zr,26 which following hydrogenation is promoted from the weak hydrophilic character of the support.32 Recent NAP-XPS and IRAS studies by Li et al.33 showed that the presence of hydroxyl groups on the ZrO2 surface is essential for the bicarbonate species formation. Doping of ZrO2 with an aliovalent cation (e.g. Zn2+) induces the formation of oxygen vacancies (VO) and, as a direct consequence, generates defects featured by coordinatively unsaturated Zr4+ sites (cus-Zr4+) which can act as strong basic and acid sites respectively.34
Carbon mono- and di-oxide activation was reported to be facilitated by the presence of neighbouring cus-Zr4+ ion sites and VO.25,35–37 Thus, VO enhance the Brønsted acidity of Zr–OH groups adjacent to cus-Zr4+ cations.38 Between the potential dopants, Zn2+ has been investigated in the formation of ZnZrOX solid solutions. CO2 adsorption and methanol selectivity are enhanced by the increase of basic sites21,28,34 while H2 activation is influenced by the synergy of Zn–Zr sites.31 Therefore, the simultaneous presence of cus-sites, VO and surface hydroxyl groups seems to be a key combination to improve CO2 adsorption, activation and hydrogenation to methanol.
Hence, both Zn and ZrO2 seem to promote several reactions related to environmental concerns. To date, a major fraction of recent works based on Zn–Zr systems deals with syngas conversion13,36,37 whereas another fraction is devoted to Zn–Zr systems involved in CO2 hydrogenation aiming for different products, such as methanol. In the case of Zn-doped ZrO2 studied by Wang et al.,31 CO2 conversion increases up to 20% at high temperatures (>320 °C) while methanol selectivity drops to less than 30%. These findings highlight that operation at high temperature thermodynamically favors side reactions, such as RWGS.16
In another recent contribution, Li et al.29 used a metal–organic approach to prepare the catalysts by means of a Schiff base, yielding a Zn-doped ZrO2 solid solution (with a 1:1 ratio). This catalyst showed a methanol selectivity of 70% with 5.7% CO2 conversion at 320 °C, V(CO2)/V(H2)/V(N2) = 24/72/4% and GHSV = 18000 mL g−1 h−1. XRD and TEM/EDS analysis confirmed the doping of Zn in the ZrO2 system, without any segregated phases. DRIFT spectroscopy was employed to investigate the produced species after CO2 hydrogenation, highlighting the formation of CHxO species on the ZnO–ZrO2 phase, confirming that methanol is one of the main products obtained by this class of catalysts.
As for the zeolite/zeotype material for a selective MTH process, in recent years many acidic catalysts have been proposed focusing on features, such as pore and channel dimensions or reaction intermediates; recalling some examples, H-ZSM-5 has been proven to favor C3 alkenes.13,39 Ongoing research efforts principally aim at: i) improving catalyst performance, e.g. by evaluating and optimizing Al dispersion in the zeolitic framework;40,41 ii) reducing coke and aromatic species formation, by understanding the influence of pore and channel dimensions in the search for optimized zeolite/zeotypes; iii) pushing temperature and pressure to a thermodynamically-favored range.
The Zn–Zr binary oxide has been combined with H-SAPO-34/H-ZSM-5/H-SSZ-13 to exploit hydrocarbon synthesis from syngas.13,36 Coupling a metal oxide for methanol production with a porous catalyst dedicated to MTH/MTO allows precise control of the elementary steps involved in the reaction (CO/CO2 chemisorption, C–C coupling and C–C cleavage).42 The weak hydrogenating nature of ZnZrO2 allows selective hydrogenation of CO/CO2 but not the eventual production of olefins/hydrocarbons.
More recent works started to investigate tandem catalysts (ZnO–ZrO2/zeolite and zeotype) for CO2 hydrogenation. Li et al.14 studied a ZnO–ZrO2 mixed metal oxide system, similar to those studied in this manuscript coupled with H-SAPO-34. A CO2 conversion of 12.6% was found at 380 °C and 3600 mL g−1 h−1, with 80% selectivity to C2=–C4=. According to their XRD and HAADF-STEM findings, the sample is a solid solution with no trace of segregated phases. By means of DRIFT spectroscopy, they studied the reaction products adsorbed on the surface of the catalyst, concluding that CHXO species are generated on the oxidic ZnZrO phase and then transferred onto SAPO zeolite for lower olefins production. Similarly, Zhou et al. studied a ZnO–ZrO2 solid solution in tandem with ZSM-5 zeolite and reported high selectivity towards aromatic products.43
Choosing what kind of zeolite/zeotype material should be used in this reaction is not straightforward. Park et al.44 compared two systems: CuZnO–ZrO2/H-ZSM-5 and CuZnO–ZrO2/H-SAPO-34. They found that the hydrocarbon distribution is strictly related to the nature of the zeolite. However, the interplay between the two catalytic functions is still not fully understood.
In this work, we investigated the catalytic properties of bifunctional catalysts obtained by physically mixing three different Zn-doped ZrO2 (ZrZnOX) with MTH-active zeolite/zeotype catalysts, H-ZSM-5 and H-SAPO-34. As such, our contribution represents a side-by-side comparison of these two materials combined with ZnZrOX.
Firstly, we thoroughly characterized the oxidic phase by infrared spectroscopy (IR), powder X-ray diffraction (PXRD), N2 adsorption/desorption and density functional theory (DFT) modelling, ultimately aiming at understanding its role in CO2 hydrogenation. Specifically, our integrated characterization approach targeted: i) the role of Zn in creating defects; ii) the detailed properties of Zn and Zr sites as revealed by IR of adsorbed CO; iii) the response of the catalyst to high-temperature treatment under model oxidative and reducing conditions. Experimental findings were corroborated by theoretical modelling, which were also used to explore different reaction pathways for CO2 hydrogenation over ZrZnOX.
Secondly, we studied both the oxidic phase alone and the combined systems by catalytic test runs under different conditions, space times and after regeneration. For all the investigated cases, we determined the CO2 conversion as well as methanol and hydrocarbon product distributions, highlighting the role of Zn in influencing the catalytic properties of the investigated systems.
Finally, we employed operando X-ray absorption spectroscopy (XAS) at Zr and Zn K-edges to directly probe the local structure and electronic properties of the ZrZnOX/ZSM-5 system before and after activation in H2, as well as to assess its stability under reaction conditions, i.e. high temperature (300 °C) and pressure (10 bar) under a CO2/H2 feed.
In this work, we aim to give a significant contribution to the understanding of the oxygen vacancy formation and its role in the CO2 hydrogenation pathway, elucidating the synergy between cations in ZnZrOX solid solutions. We also critically evaluated the relationship between experimental conditions (i.e. contact time) and catalyst activity towards value-added hydrocarbons at lower temperature than those usually reported in the literature.45,46
2. Experimental
2.1. Materials
Three Zn-containing ZrO2 samples were prepared by co-precipitation starting from solutions of zirconium and zinc inorganic salts following the recipe from Wang et al.31 The samples were named ZrZn-X, where X is the Zn loading determined by ICP-AES analysis (vide infra) and reported in Table 1. The typical procedure for making sample ZrZn-30, taken as an example, was by mixing 0.6 g Zn(NO3)2·6H2O and 2.15 g ZrN2O7·xH2O in 100 ml of type 2 H2O in a round bottom flask. The mixtures prepared for the three samples were then heated to 70 °C in an oil bath under reflux amidst stirring. 3.06 g (NH4)2CO3 was dissolved in 100 ml of type 2 H2O and then added to the warm precursor solutions dropwise: white precipitates immediately formed. The mixtures were further stirred at 70 °C for 2 h, cooled at ambient temperature, centrifuged and the precipitates were washed twice with type 2 H2O. The wet powders were oven-dried at 110 °C and then calcined at 500 °C for 3 h.| ZrZn-5 | ZrZn-15 | ZrZn-30 | |
|---|---|---|---|
| Zn loading (wt%) | 5 | 15 | 30 |
| SSA (m2 g−1) | 47 | 46 | 37 |
| DFT cumulative pore volume (cm3 g−1) | 0.24 | 0.21 | 0.49 |
| Space group | P42/nmc | P42/nmc | P42/nmc |
| a (= b) (Å) | 3.6049 ± 0.0008 | 3.58900 ± 0.00018 | 3.59440 ± 0.0008 |
| c (Å) | 5.0980 ± 0.0015 | 5.1020 ± 0.0005 | 5.082 ± 0.002 |
| Crystallite size (nm) | 55 ± 1 | 20 ± 1 | 12 ± 1 |
Combined systems were obtained by mechanical mixing of the ZrZn-X catalysts with a commercial H-ZSM-5 zeolite with a mass ratio of 1:1. For comparison purposes, combined systems using commercial H-SAPO-34 were also prepared by the same mechanical mixing protocol. Commercial H-ZSM-5 and H-SAPO-34 characteristics are reported in the ESI.†
2.2. Methods
Specific surface areas (SSAs) and pore size distributions (PSDs) of the ZrZn-X samples were determined by applying the Brunauer–Emmett–Teller (BET) method and the DFT method, respectively, to the adsorption/desorption isotherms of N2 at liquid nitrogen temperature obtained with a Micromeritics ASAP 2010 physisorption analyzer. PSDs were obtained applying the DFT method on cylindrical pores, using the Tarazona NLDFT approach. The adsorption/desorption isotherms were determined over a wide range of relative pressures (10−6 < p/p0 < 1). All the samples underwent an activation step to remove physisorbed species from the surface while avoiding irreversible changes of the surface or the solid structure. Each sample was studied after outgassing under vacuum at 120 °C (heating ramp of 5 °C min−1) for 5 h (residual pressure of 10−4 mbar).
Powder X-ray diffraction (PXRD) patterns of the as-prepared ZrZn-X catalysts were collected at room temperature (RT) using a glass capillary (ø = 0.3 mm) in a PW3050/60 X'Pert PRO MPD diffractometer from PANalytical working with the Bragg–Brentano geometry. Patterns from the Cu Kα1,2 X-ray source were recorded from 10 to 90° 2θ with a step size of 0.0156° and an integration time of 150 s. The Rietveld refinement method implemented in the FullProf software package47 was used to extract lattice parameters and average crystallite size from all the three samples. The PXRD patterns of ZrZn-30 alone and that physically mixed with the ZSM-5 zeolite recovered after catalytic tests (referred to as ‘tested’ in the following) were measured and refined following the same procedure mentioned above. We refer to the ESI† for the complete procedure description.
In order to characterize the Lewis acid sites, i.e. Zn2+ and Zr4+, at the surface of both pre-oxidized and pre-reduced samples, the catalysts were placed in a quartz IR cell, allowing thermal treatments and ex situ measurements of CO adsorption at liquid nitrogen temperature (LNT).
Before the IR measurements, the samples were outgassed under vacuum at 400 °C for 30 min and then oxidized or reduced. The oxidation pre-treatment was performed in dry oxygen (40 mbar) for 30 min at 400 °C. Finally, the samples were cooled to room temperature (RT) in O2.
The reduction pre-treatment was performed in the same way using H2. In this case, the hydrogen was outgassed at 400 °C and then the samples were cooled at RT under vacuum.
The unit lattice vectors and atoms of tetragonal ZrO2 were fully optimized in the beginning. We began the geometry optimization with the experimental lattice parameter values, which were optimized to a = b = 3.646 Å, and c = 5.275 Å. The most stable surface of the tetragonal ZrO2 phase was simulated by a 2 × 2 × 1 supercell model. To eliminate the artificial dipole moments within the slab model, we constructed a symmetric slab of at least 5 layers of Zr atoms. The slab was separated from its periodic image by 15 Å to avoid spurious interactions between the periodic slab models. The adsorption energy of the reactants and reaction intermediates was calculated as:
| ΔE[adsorption] = E[adsorbate + surface] − E[adsorbate] − E[clean surface] |
:1 in a mixed bed configuration was typically used. The attention was focused on the systems with H-ZSM-5 zeolite; however, for comparison purposes, combined systems with H-SAPO-34 were also tested and the results are reported in the ESI.† The gas feed composition was: 23 vol% of CO2, 69 vol% of H2 and 8 vol% of He as the internal standard. We typically aimed to have 12000 ml h−1 g−1 per channel. One of the 16 channels was always used without a catalyst as the blank. Prior to feeding the reaction mixture, all the samples were reduced in situ with a pure H2 atmosphere for 4 hours at 400 °C. The tubes were then pressurized to 30 bar using a membrane-based pressure controller. Regeneration tests were carried out in situ at atmospheric pressure and 600 °C with a 5% O2 in N2 stream for 6 hours. In some cases, the reacted gas was diluted with N2 (20 mL min−1 per reactor) in the reactor outlet and automatically supplied for online gas chromatographic (GC) analysis.
The GC is an Agilent 7890B with two sample loops. After flushing the loops for 24 min, the content is injected. One sample loop goes to the TCD channel with 2 Hayesep pre-columns and MS5A, where He, H2, CH4 and CO are separated. Gases that have longer retention times than CO2 on the Hayesep column (column 4 Hayesep Q 0.5 m G3591-80023) are back-flushed. Further separation of permanent gases is done on another Hayesep column (column 5 Hayesep Q 6 Ft G3591-80013) to separate CO2 before going to MS5A. Another sample loop goes to an Innowax pre-column (5 m, 0.20 mm OD, 0.4 μm film); in the first 0.5 min of the method, the gases coming from the pre-column are sent to the Gaspro column (Gaspro 30 M, 0.32 mm OD) followed by FID. After 0.5 min, the valve is switched and gases are sent to the Innowax column (45 m, 0.2 mm OD, 0.4 μm) followed by FID. The Gaspro column separates C1–C8, paraffins and olefins, while the Innowax column separates oxygenates and aromatics.
Conversions, CO selectivity, MeOH selectivity, hydrocarbon distribution selectivity (CO free) and C3 productivity are reported on the C1 basis and are defined as follows:
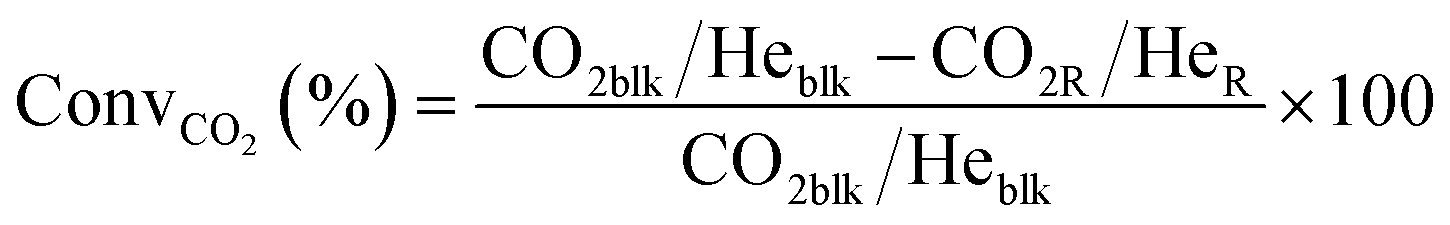
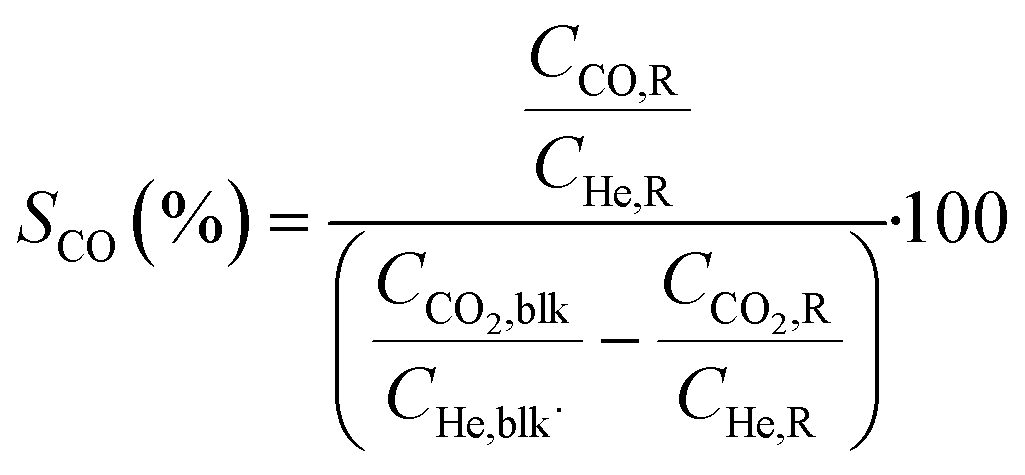
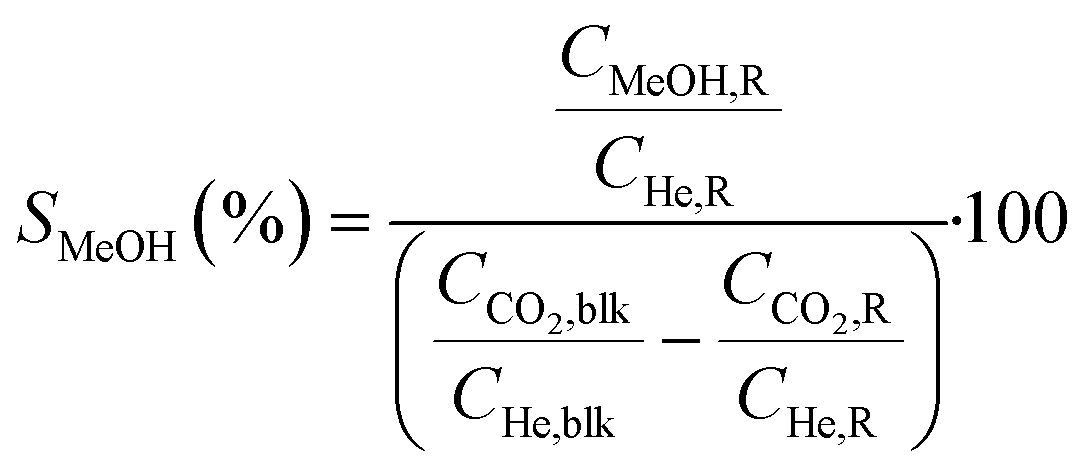
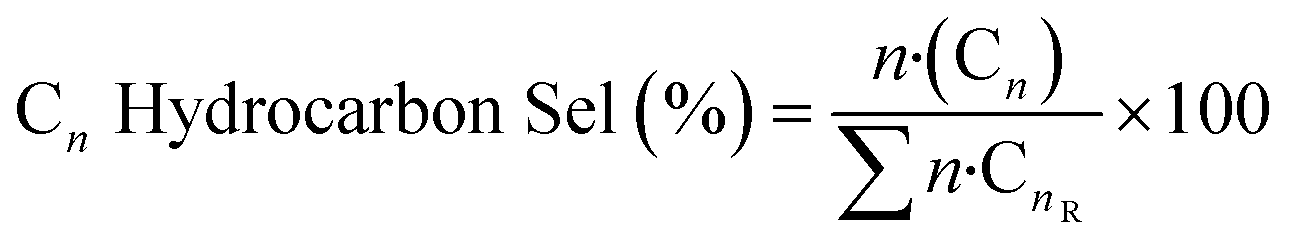
where Ciblk and CiR are the concentrations determined by GC analysis in the blank and in the reactor outlet, respectively. Carbon balance closure was better than 2.5% in all cases.
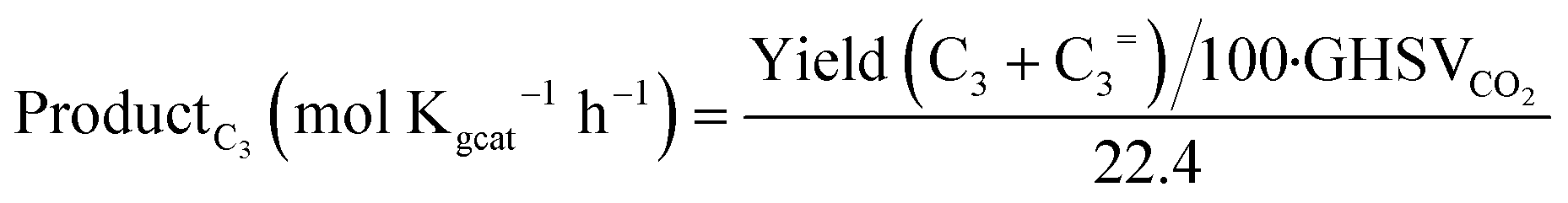 :H-ZSM-5 = 1:1 physical mixture was mortar-grounded, sieved down to 40 μm and loaded in a quartz capillary reactor (ø = 1 mm). The capillary reactor was then connected to an appropriate gas-flow setup for the CO2 hydrogenation reaction, supporting operation at high gas pressure. The temperature at the measurement position was controlled by a heat gun. The gas total flux was maintained constant (10 ml min−1) during all the measurements. The measurement protocol for the three combined systems consisted of two main parts: 1. activation: heating (RT to 400 °C, 5 °C min−1) at 1 bar in pure H2 flow; 2. reaction: feed of CO2:H2:He = 1.25:7.5:1 (mL min−1) at 300 °C (temperature showing the highest performance from the catalytic test) and 15 bar pressure.
:H-ZSM-5 = 1:1 physical mixture was mortar-grounded, sieved down to 40 μm and loaded in a quartz capillary reactor (ø = 1 mm). The capillary reactor was then connected to an appropriate gas-flow setup for the CO2 hydrogenation reaction, supporting operation at high gas pressure. The temperature at the measurement position was controlled by a heat gun. The gas total flux was maintained constant (10 ml min−1) during all the measurements. The measurement protocol for the three combined systems consisted of two main parts: 1. activation: heating (RT to 400 °C, 5 °C min−1) at 1 bar in pure H2 flow; 2. reaction: feed of CO2:H2:He = 1.25:7.5:1 (mL min−1) at 300 °C (temperature showing the highest performance from the catalytic test) and 15 bar pressure.
Incident X-ray energy at both Zr and Zn K-edges was scanned by two quick-XAS monochromators, each mounted on a cam-driven tilt table that oscillates periodically around a fixed Bragg angle. A Si(111) monochromator was used to measure the Zn K-edge (9659 eV) while Si(220) was employed for the Zr K-edge (17998 eV). Time-resolved data throughout the applied protocol were initially obtained as the average of 50 scans for an exposure time of 12.5 s and a total time/scan of 25 seconds. The reported XAS spectra representative of the as-prepared and activated catalysts, as well as of the catalysts under reaction conditions, are obtained upon further averaging of the time-resolved spectra obtained in the last 10 min of acquisition for each protocol step, after checking for the complete stabilization of the spectral features. Incident (I0) and transmitted (I1) beams were measured by two sets of ionization chambers. An energy step of 2 eV was used for the two edges. The energy sampling was intensified using a step of 0.2 eV in the main edge region for Zn (range: 9530–9780 eV) and of 0.4 eV for Zr (range: 17970–18120 eV). A third set of ionization chambers (I2) was employed to measure simultaneously the transmitted intensity after Zr and Zn metal foils, for energy calibration purposes. Pure hexagonal ZnO and tetragonal ZrO2 powders, used as reference compounds, were measured at the same beamline, in the form of self-supporting pellets with optimized mass for transmission XAS at Zn and Zr K-edge, respectively. For the sake of comparison, a reference monoclinic ZrO2 was also measured in the form of an optimized self-supporting pellet at the BM23 beamline of the European Synchrotron Radiation Facility (ESRF).54
The Athena software from the Demeter package55 was used to align in energy and normalize the XAS spectra to unity edge jump, as well as to extract the χ(k) EXAFS function and calculate its Fourier transform.
3. Results and discussion
3.1. Chemical, textural and structural characterization of the ZrZn-X catalysts
As for zirconium and zinc contents in the ZrZn-X samples determined by ICP-AES analysis, the approximate compositions were calculated as weight percentage of Zn and reported in Table 1 along with specific surface areas (SSA) and pore volumes. As already mentioned in the Experimental section, the samples were named ZrZn-X, where X is the Zn loading.The SSAs of ZrZn-5 and ZrZn-15 are approximatively the same, whereas that of ZrZn-30 is about 20% lower. The adsorption/desorption isotherms56,57 and the pore size distributions (PSDs) of the as-prepared ZrZn-X catalysts are displayed in the ESI,† Fig. S1 and S2,† respectively.
All the samples exhibit the hysteresis loop characteristic of mesoporous materials with similar PSDs with a maximum at about 3 nm. However, all these samples show a broad PSD covering a wide range of pore widths from 3 to 12 nm. Moreover, the pore volumes are comparable for ZrZn-5 and ZrZn-15, whereas it is twice as high for ZrZn-30.
The PXRD patterns of the as-prepared ZrZn-X samples are reported in Fig. 1. Intensities were not rescaled but only offset-shifted. All the three samples show a diffraction pattern typical of crystalline zirconia, but the diffractograms of cubic and tetragonal ZrO2 are not distinguishable between each other. However, XAS measurements discussed in the following (see section 3.5) evidenced features characteristic of tetragonal zirconia. Moreover, reflections of monoclinic ZrO2 are absent, also at 2θ < 20° as shown in Fig. S3.†
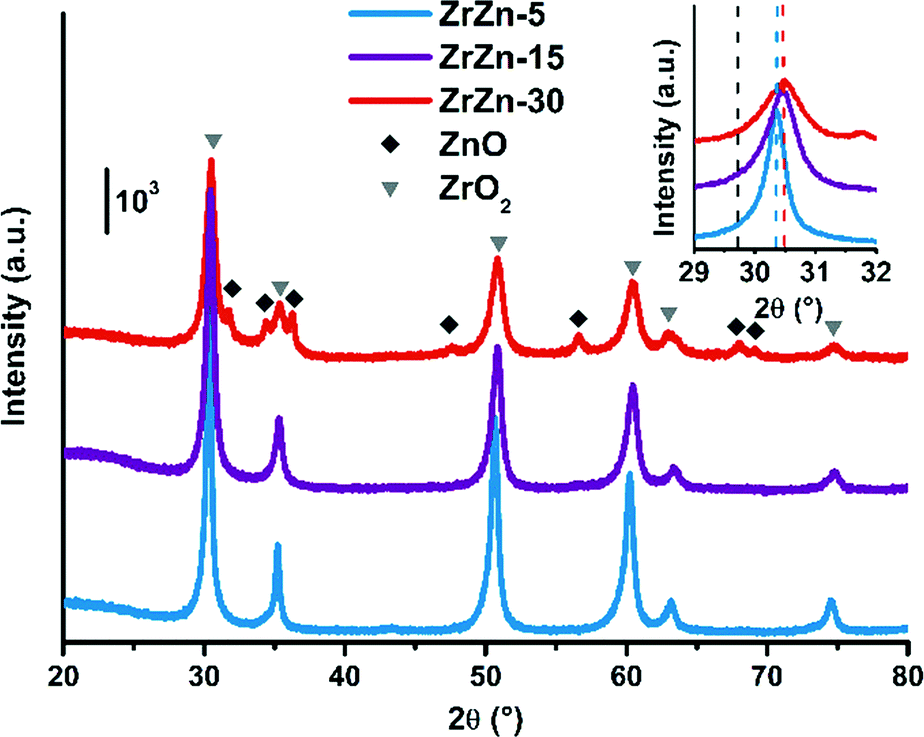 | ||
| Fig. 1 PXRD patterns of the as-prepared ZrZn-X samples. Triangles and diamonds indicate respectively peak positions of cubic/tetragonal ZrO2 and hexagonal ZnO. Inset: Magnification of the ZrO2 (101) reflection with the peak position for a pure ZrO2 (ref. 60) (dashed black line) compared to that observed for the ZrZn-X samples (dashed coloured lines). | ||
The inset in Fig. 1 underlines a shift to higher 2θ values of the (101) reflection with respect to a pure ZrO2 reference. This shift was previously observed by Wang et al.31 and explained considering a shrinking of the ZrO2 unit cell when the largest Zr4+ (0.82 Å)58 is substituted by Zn2+ (0.74 Å).
The peak shift trend is consistent with the Zn concentration found from ICP analysis (i.e., the higher the Zn loading, the more pronounced the shift results). Moreover, comparison of the diffractograms highlights a peak broadening effect, slightly enhanced as the Zn loading increases. The three samples were measured with the same instrumental parameters and the background position was the same for the three diffractograms (Fig. S3†). Hence, we can safely verify that the amorphous fraction is the same. The crystallite size obtained from Rietveld refinement decreases as the loading of Zn increases.
However, the SSA value does not reflect this trend (Table 1). The SEM images of the catalysts (Fig. S17†) show that they consist of particles with small aggregated crystallites. Therefore, the area exposed is correlated to the dimensions of these aggregates rather than to the crystallite size.
Extra reflections are present only in sample ZrZn-30 (diamond symbols in Fig. 1). They are indexed considering an additional ZnO (ref. 59) wurtzite phase. The ZrO2:ZnO phase ratio was extracted by Rietveld refinement. Using hexagonal ZnO (P63mc) and tetragonal ZrO2 (P42/nmc60) as input parameters, we found that ZrZn-30 is composed of 85 wt% of ZrO2 and 15 wt% of ZnO (Table S1†). Consequently, part of Zn is not incorporated in the host lattice but is segregated as a second phase, justifying also why the (101) reflection for ZrZn-30 is very close to the one for ZrZn-15. Nonetheless, as evidenced by EXAFS results presented in section 3.5, a slightly higher amount of Zn is expected to enter the ZrO2 lattice in ZrZn-30 with respect to ZrZn-15. With our co-precipitation technique, we could therefore achieve a maximum Zn doping of ZrO2 of about 15 wt%. Finally, the decreased area and the increased pore volume observed for ZrZn-30 could be also correlated to the presence of segregated ZnO.
3.2. Spectroscopic characterization of the ZrZn-X catalysts
Fig. S4† compares the spectra recorded in oxygen and in hydrogen at 400 °C for the three catalysts. For all the samples under both conditions, absorption bands in the regions 4000–3000 cm−1, 2500–2000 cm−1 and 1700–1000 cm−1 are present. These bands are related to surface hydroxyls, CO2 encapsulated in closed pores and carbonate/nitrate species, respectively. Encapsulated CO2 and carbonates/nitrates stem directly from the precursors used for the synthesis. More detailed discussion about these species is reported in the ESI.†
Focusing on the effect of the interaction with H2, in Fig. S4† an increase of the sample absorbance in a large spectroscopic region passing from oxygen to hydrogen is evident, in particular for ZrZn-15 and ZrZn-30. This is due to the increase of a very broad absorption band, whose shape is discernable by subtracting the spectrum recorded in oxygen from the spectrum recorded in hydrogen. The result of this subtraction for the different samples is reported in Fig. 2. The very broad bands evidenced by the grey dotted lines are related to the photo-ionization of mono-ionized oxygen vacancies.61–63 On these electronic absorption bands, negative vibrational peaks that complicate the shape of the spectra are superimposed.
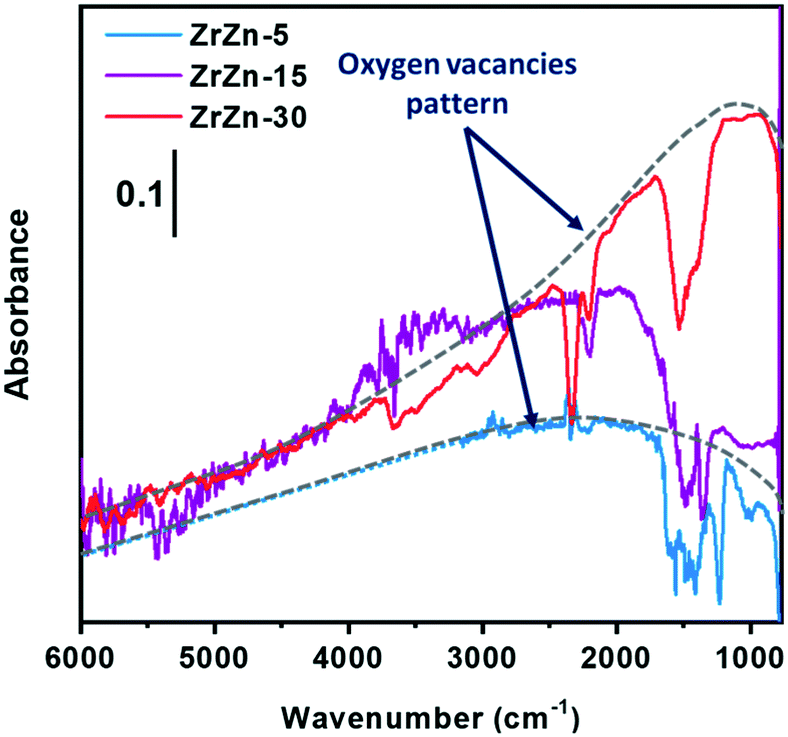 | ||
| Fig. 2 FT-IR difference spectra of ZrZn-X catalysts at 400 °C in H2 (subtrahend spectrum is that recorded in oxygen at 400 °C). The ZrZn-5 spectrum is cut at 3155 cm−1 because, beyond that frequency, data are affected by the low signal-to-noise ratio; the cut part has been substituted with a dotted blue line, which aims to reproduce the trend. | ||
Before discussing the origin of the negative peaks, we focus on the broad electronic absorption. It is well known that ZnO is a semiconducting material due to the presence of lattice defects, i.e. oxygen vacancies (VO).61,62 The two electronic levels at 0.05 and 0.18 eV below the conduction band (C.B.) are associated with the VO of ZnO. Neutral vacancies show two trapped electrons that occupy the above-mentioned levels. The first ionization energy is so low that the major part of VO are mono-ionized (VO+), being the excited electrons in the C.B. The second ionization of VO can be promoted by IR radiation (photo-ionization of mono-ionized oxygen vacancies): in the spectrum of pure ZnO, a broad absorption band appears centered at the energy corresponding to 0.18 eV, i.e. at about 1450 cm−1, after reduction treatments. Interaction with hydrogen can create VO+ following two routes: i) the filling with an electron of the pre-existing bi-ionized VO (VO2+) by consuming adsorbed oxygen species, such as O2−, O−, and O22−; ii) the creation of new VO+ extracting lattice oxygen ions from the surface. This last pathway occurs only at high temperature, with the temperature threshold depending on the specific material. The IR technique is not able to distinguish the two routes to VO+ formation.
Concerning our case, the VO+ absorption bands reported in Fig. 2 for ZrZn-30 and ZrZn-5 show a maximum centered at about 1100 and 2200 cm−1, respectively, which can be associated with mono-ionized oxygen vacancies at 0.14 and 0.27 eV under the C.B.61–63 This result evidences the influence of the different Zn loadings on the associated energy level of the mono-ionized oxygen vacancies with respect to the C.B. In particular, ZrZn-30 shows a VO+ ionization energy (0.14 eV) very close to that of pure ZnO (0.18 eV), being the sample with the highest Zn content in the ZrO2 lattice, as evidenced by the EXAFS results presented in section 3.5.
We cannot exclude that the presence of a segregated ZnO extra-phase, as shown by PXRD (Fig. 1), could also slightly influence the position of the oxygen vacancy band. As for ZrZn-5, the VO+ ionization energy (0.27 eV) is higher than that of pure ZnO: the effect of the ZrO2 lattice and the low amount of Zn induce the formation of VO+ with electronic levels deeper in the band gap.
The identification of the correct position of the maximum absorption related to VO+ for ZrZn-15 is complicated by the superimposed, above mentioned “negative vibrational peaks”. These peaks are well visible for all the samples, but only for ZrZn-15 their presence hamper the identification of the actual shape of the VO+ absorption. The negative peaks are related to the vibrational modes of encapsulated CO2 and carbonate/nitrate species and they arose from the subtraction operation, since these vibrational bands show lower intensity in hydrogen than in oxygen. It is important to underline that their intensities return to the original ones when the samples were exposed to oxygen after interaction with hydrogen. So, carbonates/nitrates and, even more reasonably, encapsulated CO2 are not partially removed from the samples by the interaction with H2, but their intensity loss has another origin. In particular, it is possible to consider a coupling process occurring between the electronic absorption of VO+ and the surface species vibrations. Genzel and Martin,64 using a continuum model made up of a phonon term and a free electron term, provided an explanation for a similar phenomenon when plasmon absorptions occurred in small particles of conducting and semiconducting materials. When the concentration of the free carriers is high enough to cause the plasmon frequency to overcome the phonon frequency, a plasmon–phonon coupling process occurs leading to the decrease/disappearance of any band of a purely vibrational nature. In our case, the coupling process occurs between vibrational modes of surface species and IR absorption of electrons trapped in mono-ionized oxygen vacancies, as already observed and reported in the literature for semiconducting oxides, such as SnO2, ZnO, WO3, and MoO3.65,66
As for the intensity of the electronic band, ZrZn-30 and ZrZn-15 show a significant absorption related to VO+, whereas ZrZn-5 does not, due to the different amount of VO+ generated. As demonstrated by quantitative analysis, the Zn loading decreases in the order: ZrZn-30 > ZrZn-15 > ZrZn-5, so that the infrared absorption shown in Fig. 2 is in line with the chemical composition and with the Zn content in the ZrO2 lattice shown by PXRD and EXAFS results. These results are corroborated by modelling calculations (see section 3.3). Moreover, the highest amount of VO+ observed for ZrZn-30 can be correlated to the best catalytic performances of this catalyst among all the samples (see section 3.4). Finally, these IR results highlight the importance of pre-reducing the samples before the catalytic run in order to create a high concentration of reactive oxygen vacancies.
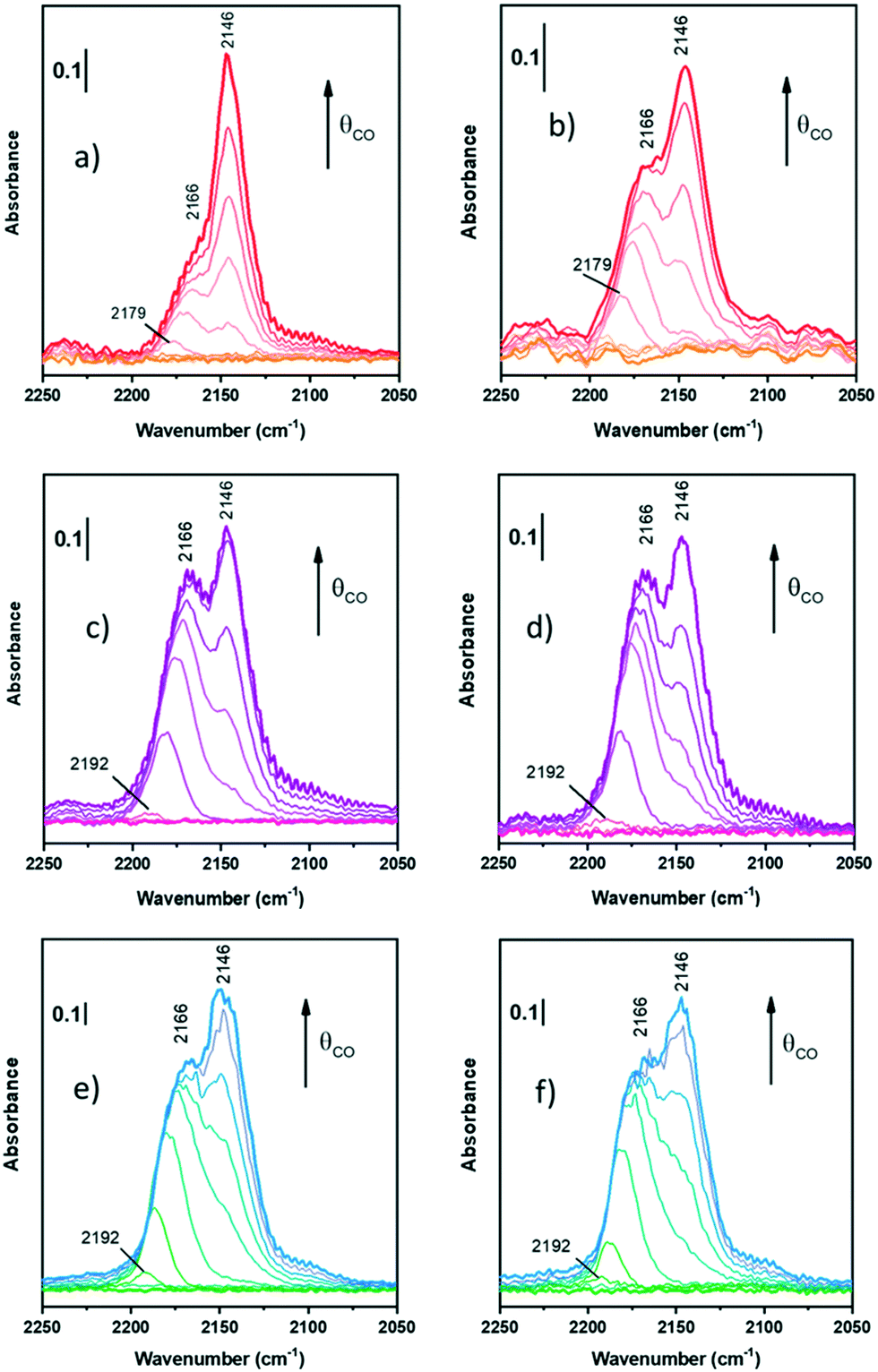 | ||
| Fig. 3 FT-IR spectra of CO adsorption at LNT on oxidized ZrZn-30 (a), ZrZn-15 (c), and ZrZn-5 (e) and reduced ZrZn-30 (b), ZrZn-15 (d), and ZrZn-5 (f) at increasing doses up to 20 mbar. | ||
Starting from the oxidized samples (Fig. 3a, c and e), two main peaks are highlighted between 2200 and 2100 cm−1. The first peak at 2166 cm−1 can be related to coordinatively unsaturated Zr4+ carbonyls67 (cus-Zr4+–CO), i.e. Zr atoms located on edges or steps. Zr4+ carbonyls on regular facets should show absorption bands between 2148 and 2142 cm−1.67 However, when CO is adsorbed on metal cations with a dominant σ-donation, the higher the unsaturation of the adsorption site, the higher the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O). In particular, Zr4+ is a 4d0 cation, thus it lacks π-backdonation and σ-bonding is the only contribution to the bond with CO.
O). In particular, Zr4+ is a 4d0 cation, thus it lacks π-backdonation and σ-bonding is the only contribution to the bond with CO.
The peak related to cus-Zr4+–CO also features a red-shift from 2179–2192 cm−1 (according to the sample) to 2166 cm−1. Different explanations can be proposed for the frequency shift vs. θCO, but the most observed are usually four: i) “through space” dipole–dipole interaction between parallel vibrating molecules;68 ii) “through solid” via the vibrational coupling mechanism across binding electrons;69 iii) the “chemical effect”, another “through solid” phenomenon due to adsorbed molecules;69,70 and iv) the “electrostatic” or “solvent” effect caused by adsorbed molecules perturbing each other.71 The first and the second effect are dynamic, whereas the third and the fourth ones are static. Typically, among dynamic effects, the second one is negligible when adsorption is characterized by small adsorption enthalpy, or in general when ν(CO) is very close to that of free CO (2143 cm−1). Among the static effects, the fourth one is often small or negligible72 and it usually appears at higher pressures or for densely packed CO, where it assumes a solvent-like behavior, hence not in this case, since we observed this effect at low pressures.
Among the remaining effects, in our case we can exclude the dipole–dipole coupling since it is the dominant factor for CO adsorbed on sites on extended regular facets (dipolar coupling occurs between “equal” oscillators, i.e. CO molecules, and defects interrupt dipole–dipole coupling) and it causes in all cases a blue-shift on increasing coverage. Hence, the observed red-shift is due to the “chemical effect”, as a result of the reduction of CO σ-donation on increasing coverage. As a matter of fact, for metal cations with dominant σ-donation, the higher the θCO, the higher the electron density on the binding sites. As a consequence, on increasing coverage the σ-donation contribution of all adsorbed CO molecules becomes smaller and smaller and thereby a decrease of ν(CO) is observed.73
The second peak at 2146 cm−1 is assigned to CO adsorbed on Zr4+ with a lower coordinative unsaturation.67,74 This band does not show any shift on increasing CO coverage: this is reasonably due to the compensation between the dipole–dipole coupling effect (blue shift) and the chemical one (red-shift). As a consequence, the peak remains stationary and this observation confirms its assignment to CO on Zr4+ sites of regular facets.
Differently from Zr4+ sites, Zn2+ sites are not visible. Zinc is in a lower amount and, reasonably, its carbonyl band can be totally hidden by Zr4+–CO bands. Indeed, according to some authors,74,75 Zn2+–CO is characterized by peaks between 2190 and 2160 cm−1, where the absorption frequency changes according to the chemical environment. For the sake of clarity, it is possible that all peaks at 2192 (Fig. 3c and e) and 2179 cm−1 (Fig. 3a) observed as first peaks during CO adsorption can be associated with Zn2+–CO, but there is neither evidence nor references to prove it in systems like the ZrZn-X samples studied in this work.
On the reduced samples (Fig. 3b, d and f), all peaks can be assigned as reported for the oxidized ones.
Nevertheless, comparing the spectra of all the samples, normalized for the specific surface area and pellet thickness, many features are evident (Fig. 4). First of all, for the oxidized samples, the markedly lower intensity of the band at 2166 cm−1, related to coordinatively unsaturated Zr4+, is well evident for ZrZn-30 with respect to the other samples. The lower amount of defect sites for ZrZn-30 can be related to the BET results: the lower surface area of this sample with respect to ZrZn-5 and ZrZn-15 is reasonably reflected in a minor amount of cus-Zr4+ sites. Moreover, by comparing the total integrated intensity of the bands in the region 2200–2100 cm−1, it decreases in the order: ZrZn-5 > ZrZn-15 > ZrZn-30. This is in agreement with the chemical analysis and the EXAFS results (vide infra) on the Zn content in the ZrO2 lattice: on increasing the Zn loading, the amount of surface Zr4+ sites decreases.
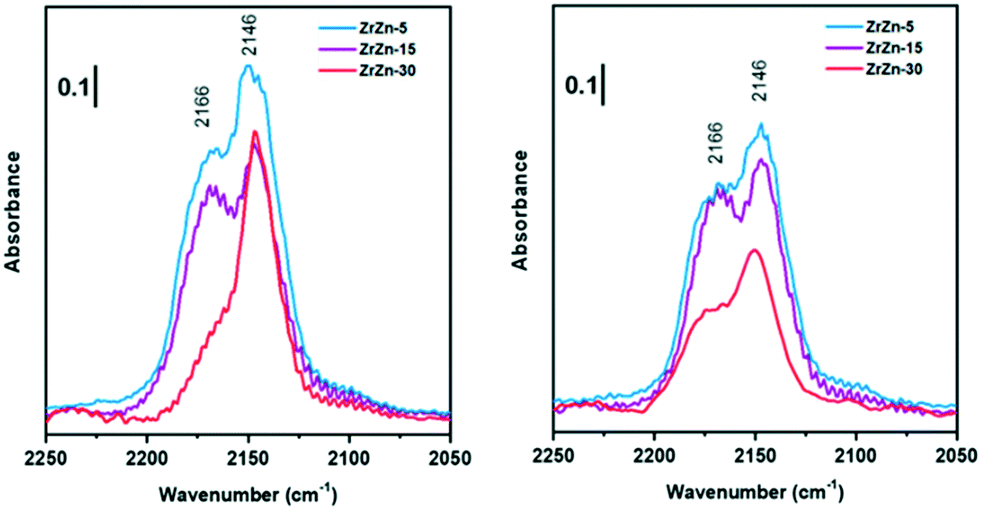 | ||
| Fig. 4 Comparison between FT-IR spectra collected at the highest CO coverage for oxidized (left) and reduced (right) ZrZn-X catalysts. | ||
After reduction, there is not a significant variation in spectra except for ν(CO) at 2166 cm−1 on ZrZn-30. Comparing CO adsorption on oxidized and reduced ZrZn-30, the intensity of the band at 2166 cm−1 appears significantly increased after reduction. This phenomenon can be ascribed to oxygen vacancy formation: after reduction at 400 °C in H2 the presence of oxygen vacancies is responsible for an increased surface disorder, which causes a growth of cus-Zr4+ concentration (2166 cm−1) and thereby a correlated reduction in the amount of Zr4+ on facets (2146 cm−1).
3.3. Molecular modelling of the ZrZn-X catalysts
We performed DFT calculations to characterize the catalyst structure and the involved reaction mechanism for CO2 hydrogenation on a ZnZrOX solid solution. For the sake of simplicity, we model the system as a five layered ZrO2 slab presenting variable ZnO–ZrO2 composition on the surface. Beginning with a tetragonal unit cell of ZrO2, we constructed low index facets (100), (101), and (111). The surface energies of the slabs were computed as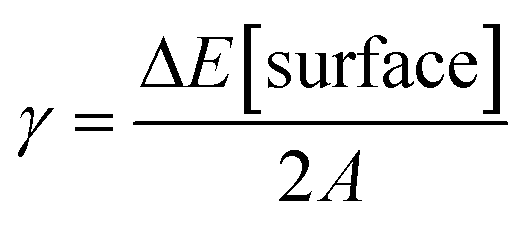 , with ΔE[surface] defined in eqn (5):
, with ΔE[surface] defined in eqn (5):| ΔE[surface] = E[slab] − NZrO2 × E[bulk] | (5) |
According to calculations, the (101) surface has the lowest surface energy, 0.1 J m−2, followed by the (100) and (111) surfaces, 0.7 and 2.8 J m−2 (see Fig. S5†). We thus focused on the (101) surface to investigate the formation energy of O vacancies in the presence of H2, as thermal O vacancies are highly unlikely on ZrO2, on ZnZrOX solid solutions using eqn (6):
| E[vac] = E[pristine] − E[system with O vacancy] + EH2O − EH2 | (6) |
Using eqn (6), we first calculated the energy required to form an O vacancy on a pristine ZrO2 (101) surface, 3.28 eV. In line with earlier reports,76,77 this indicates that no O vacancy can be expected at thermodynamic equilibrium on the pristine (101) facets of ZrO2 under the reactivity conditions used in this work. To include the effect of Zn doping, we replaced one ZrO2 unit on the surface with one ZnO unit and a “stoichiometric” O vacancy, which is a vacancy introduced to balance the charge difference created by replacing one Zr4+ with one Zn2+ in the lattice. To quantify the formation energy of stoichiometric O vacancies, we computed the substitution energy of ZrO2 units by ZnO units, E[sub], using eqn (7):
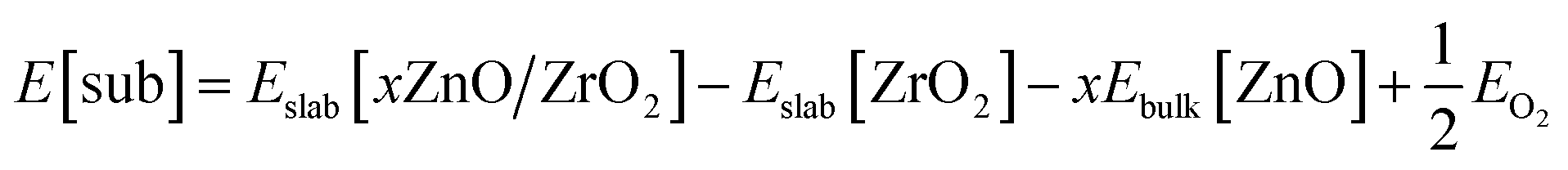 | (7) |
Fig. 5 summarizes the formation energies of stoichiometric and additional O vacancies for x ranging from 1 to 3. Considering that the ZrO2 supercell we used has 8 Zr atoms, the doping we considered corresponds to 12.5, 25.0 and 37.5% Zn atoms on the surface. Calculations indicate that substitution of a single ZrO2 unit by ZnO, with creation of a vacancy, is thermodynamically favored by −0.46 eV, a value that reduces slightly to −0.29 eV per ZnO unit and −0.87 eV when 3 ZrO2 units are replaced with 3 ZnO. This indicates that ZrO2 can tolerate high amounts of Zn substitution at the surface. As for the formation of O vacancies, in addition to the stoichiometric ones, our calculations indicate that the system with just one Zn doped on the surface is not prone to further O vacancy formation, with an E[vac] = 1.53 eV, although this value is remarkably lower than that calculated on a pristine ZrO2 surface, 3.28 eV. However, the chances of formation of additional O vacancies increase with increasing number of Zn atoms on the surface, with an E[vac] = 0.9 eV only, when 3 out of the 8 ZrO2 units on the surface are replaced with ZnO. Overall, this is in qualitative agreement with the experimental evidence that increasing amounts of O vacancies are experimentally observed at increasing Zn content.
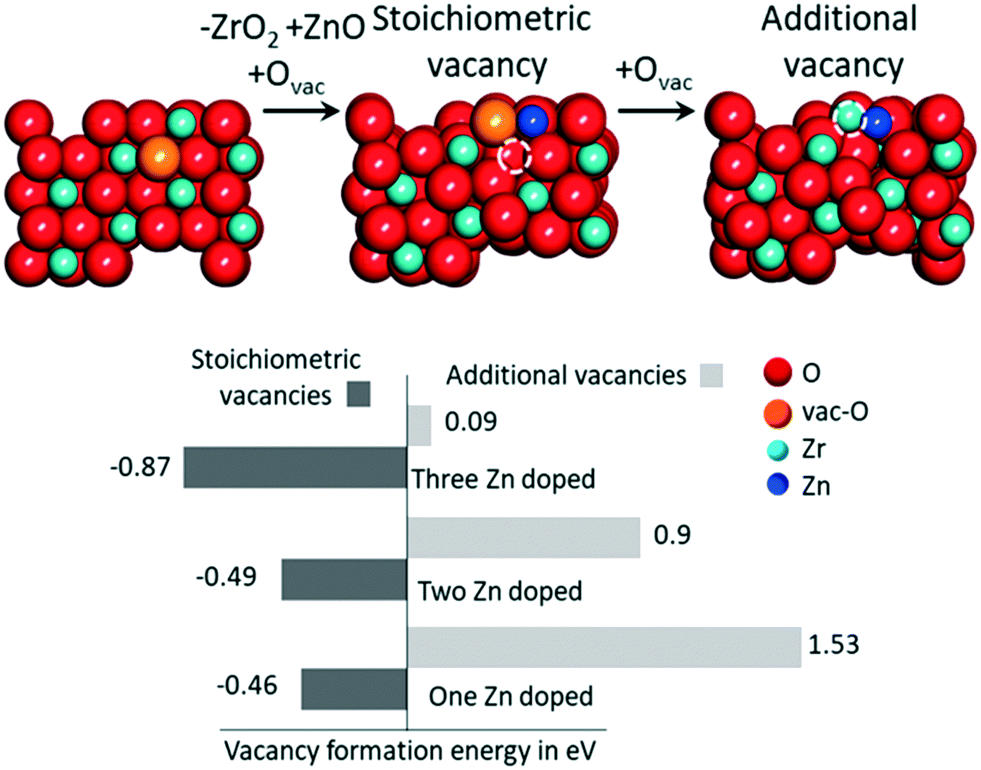 | ||
| Fig. 5 The O vacancy formation energy trend with increasing number of Zn atoms on the surface of ZrO2 (101). The schematic illustrates the way “stoichiometric” and additional vacancies are modeled. The red, blue and dark blue colors represent oxygen, zirconium and zinc atoms, respectively. The orange atom and dashed circle show the O atom to be removed and the O vacancy formed, respectively. | ||
To investigate the catalytic behavior, we used the model composed of one ZnO unit replacing a surface ZrO2 unit, with generation of a stoichiometric O vacancy. We first calculated CO2 adsorption on the O vacancy near the Zn atom, which resulted in an adsorption energy of −0.50 eV. Dissociation of the adsorbed CO2 molecule with release of a CO molecule is thermodynamically unfavored by 0.71 eV, indicating that these surface O vacancies cannot be CO2 traps generating CO (Fig. S6†).
We were not able to locate any other energetically favored CO2 adsorption geometry. Adsorption of molecular hydrogen occurs at the Zn atom, with an adsorption energy of −0.20 eV. However, dissociation of molecular hydrogen into 2H* is favored, with an energy gain of 0.39 eV. The dissociated hydrogen is present as Hδ+ and Hδ− species on the O and Zn sites, respectively. Simultaneous adsorption of CO2 and 2H* is favored by −0.73 eV, which is slightly less than the sum of the adsorption energies of isolated CO2 and 2H*, −1.09 eV. The completely optimized geometries of the Zn-doped ZrO2 (101) with CO2, H2, 2H* and CO2 + 2H* are shown in Fig. S7.†
Possible thermodynamic profiles for the conversion of adsorbed CO2 and dissociated H2 on the ZnO/ZrO2 surface are reported in Fig. 6. Considering that the formate and CO pathways have been proposed to be involved in methanol formation,17 we evaluated the free energies of the most important intermediates involved in the two pathways. The starting point is CO2 adsorbed on the O vacancy near the Zn site, and dissociated H2 adsorbed on the Zn site and on a nearby O atom.
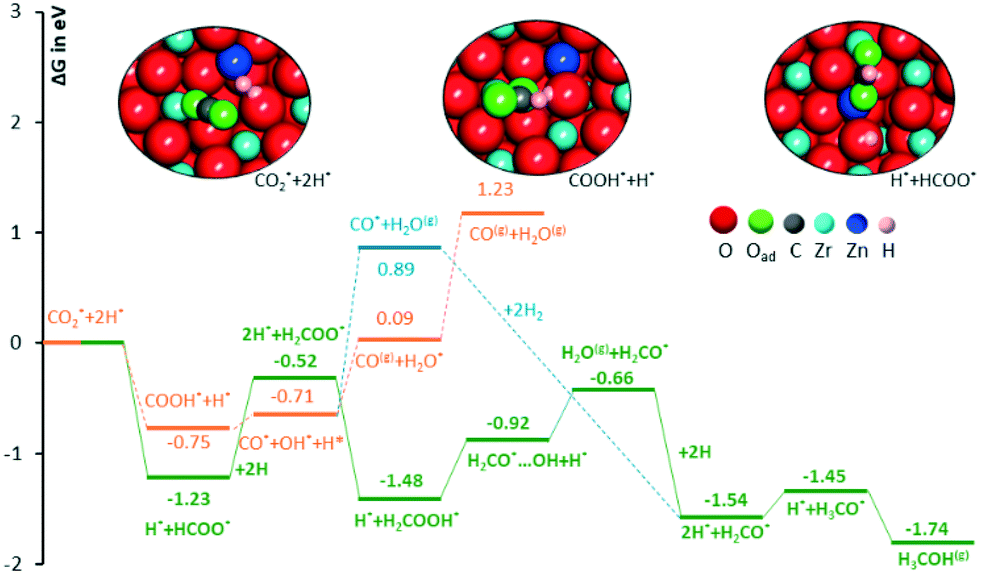 | ||
| Fig. 6 Free energy diagram for comparing the intermediates involved in CO2 hydrogenation to methanol via the formate and CO pathway. The three insets represent the completely optimized structures of reactants and intermediates with HCOO* and COOH*, respectively. The red, grey, pink, blue, green and dark blue colors represent oxygen, carbon, hydrogen, zirconium, O atom of the adsorbate and zinc atoms, respectively. | ||
The first possibility we examined is the transfer of the H* on Zn to the C atom of *CO2 along the formate pathway, leading to formation of HCOO* through a highly exergonic step, by −1.23 eV. Subsequent hydrogenation of HCOO* leads first to an adsorbed formaldehyde molecule with liberation of a water molecule, H2CO* + H2O(gas), and finally to adsorbed methoxide, CH3O*. All intermediates along the formate pathway are at free energies below the starting 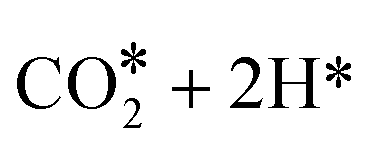 species, and the overall energy span between the highest and the lowest energy intermediates is smaller than 1.22 eV, indicating a viable reaction pathway under the reaction conditions used in this work. The second possibility we examined is reactivity along the carbon monoxide pathway, which starts with conversion of
species, and the overall energy span between the highest and the lowest energy intermediates is smaller than 1.22 eV, indicating a viable reaction pathway under the reaction conditions used in this work. The second possibility we examined is reactivity along the carbon monoxide pathway, which starts with conversion of 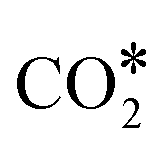 to CO*, followed by its hydrogenation to form methanol.31 As shown in Fig. 7, the first intermediate along this pathway, COOH*, at −0.75 eV, is less stable than the first intermediate along the formate pathway, HCOO*, resting at −1.23 eV.
to CO*, followed by its hydrogenation to form methanol.31 As shown in Fig. 7, the first intermediate along this pathway, COOH*, at −0.75 eV, is less stable than the first intermediate along the formate pathway, HCOO*, resting at −1.23 eV.
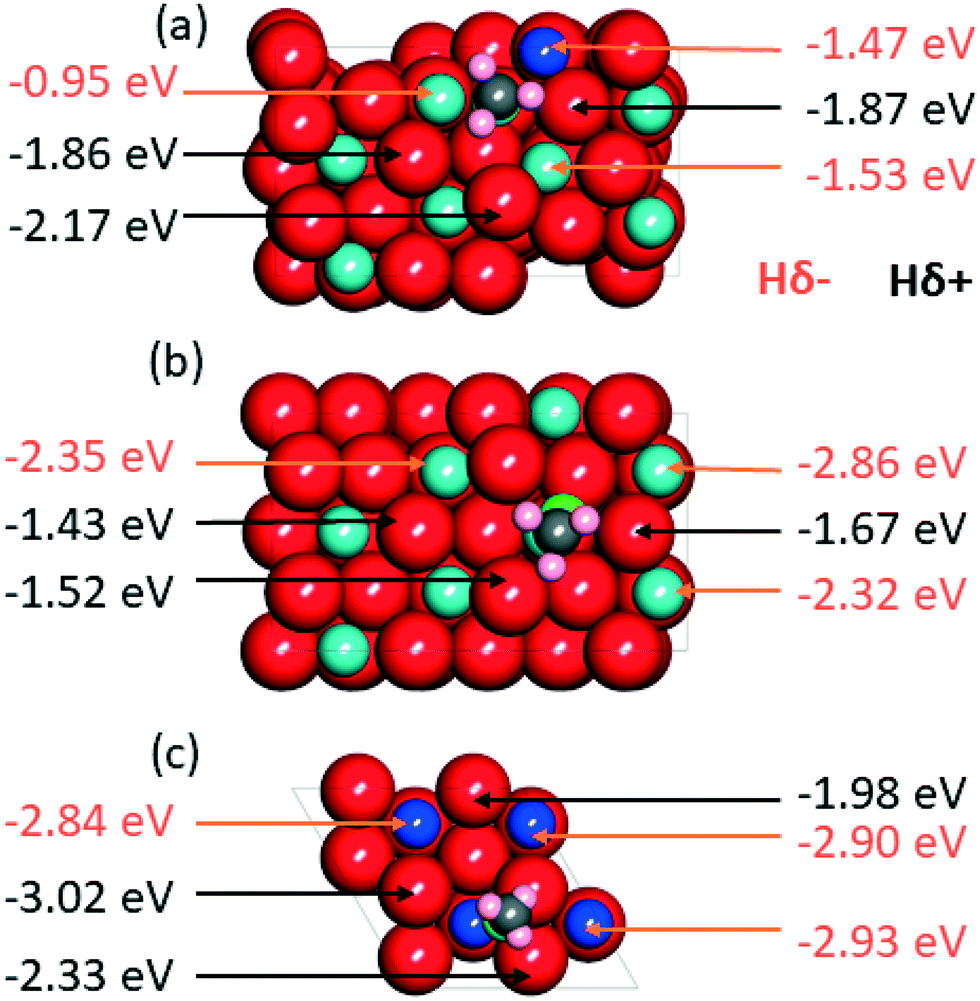 | ||
| Fig. 7 A top view of the space filling atom model, with adsorbed CH3O*, of (a) Zn doped ZrO2 (101), (b) ZrO2 (101), and (c) ZnO (111) surface comparing the adsorption energies of Hδ+ and Hδ− species. The red, grey, pink, blue, green and dark blue colors represent oxygen, carbon, hydrogen, zirconium, O atom of the adsorbate and zinc atoms, respectively. | ||
Evolution of this intermediate to CO* + H2O* first, followed by CO dissociation to CO(gas) + H2O*, is an endergonic sequence, with CO(gas) + H2O* above the starting 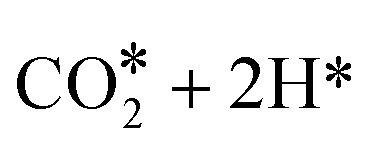 species. Similarly, H2O dissociation leaving CO*, from which hydrogenation to CH3O* can occur, is even more expensive, with CO* + H2O(gas) at 0.89 eV above the starting
species. Similarly, H2O dissociation leaving CO*, from which hydrogenation to CH3O* can occur, is even more expensive, with CO* + H2O(gas) at 0.89 eV above the starting 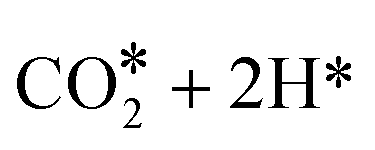 species.
species.
Furthermore, all intermediates involved in dissociation of CO2 to CO are less stable than the intermediates formed by subsequent hydrogenation of formate species. This is consistent with the experimental CH3OH selectivity (vide infra) and it suggests that the formate reaction pathway is operative.
Having clarified the pathway leading to CH3O*, we investigate methanol versus methane selectivity, which has been shown to depend on a competition between the transfer of a Hδ+ to the O atom of CH3O*, liberating methanol, and the transfer of a Hδ− species to the C atom of CH3O*, dissociating the C–O bond and liberating methane.78,79 To shed light on this point, we explored the relative stabilities of Hδ+ and Hδ− species on Zn doped ZrO2, and pristine ZrO2 and ZnO (Fig. 7). According to calculations, in the presence of CH3O* on Zn doped ZrO2, Hδ+ species have stronger binding energies compared to Hδ− species, which can explain the catalyst selectivity towards methanol production.78 On the other hand, on pristine ZrO2 (101) and ZnO (111), Hδ− species have stronger binding energies than Hδ+ species, which should imply that Zn doped ZrO2 has better selectivity towards methanol formation than both its pristine counterparts.
3.4. Catalytic tests on the ZrZn-X catalysts and the combined ZrZn-X/zeolite systems
We first studied the stand-alone ZrZn-X catalysts with different Zn-loadings (ZrZn-5, ZrZn-15 and ZrZn-30) in the CO2 conversion to methanol (MeOH), the initial step in the CO2 ‘cascade’ conversion over the bifunctional catalysts. In particular, we screened the effect of reaction pressure (20, 30 and 40 bar), temperature (250 °C, 300 °C and 350 °C) and CO addition (10% in the feed), as this gas is likely to be recycled with the unreacted CO2 and H2 in a perspective process.80,81 The results are summarized in Fig. 8.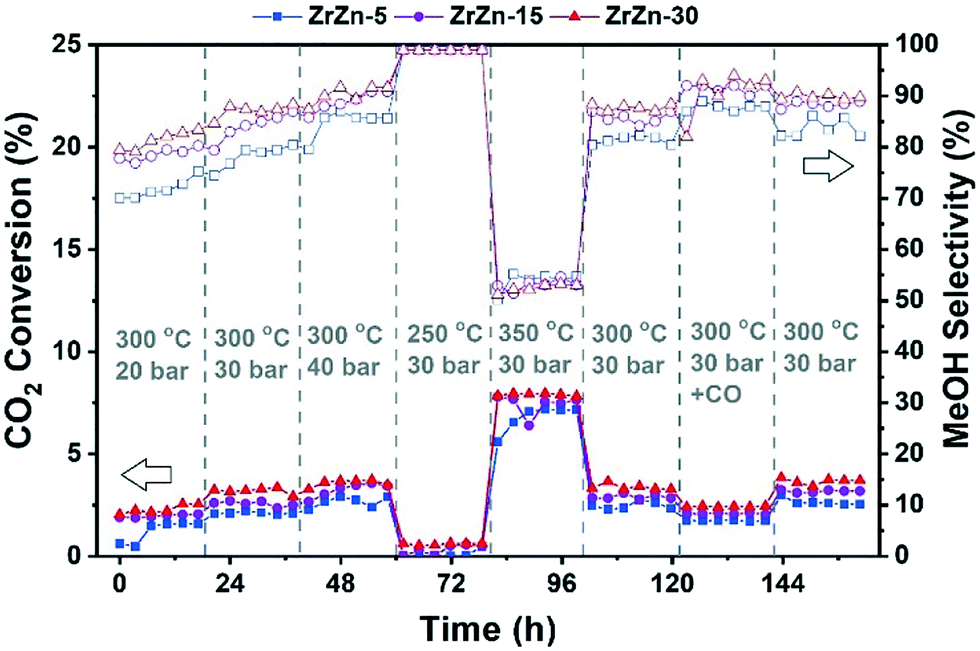 | ||
| Fig. 8 Catalytic performance of all ZrZn-X stand-alone samples for the CO2 conversion to MeOH. CO2:H2 1:3, 12000 mL h−1 g−1. | ||
We can observe that increasing the pressure results in higher conversion and methanol selectivity for the three catalysts, in good agreement with the process thermodynamics.78,82 The main byproduct in all cases is CO with small traces of CH4 (selectivity <1%) being as well detected. Similarly, decreasing the temperature increases the methanol selectivity to almost 100% with the CO2 conversion being drastically reduced. On the other hand, increasing the temperature to 350 °C significantly increases the conversion with the MeOH selectivity being reduced to ca. 50%. CO addition slightly increases MeOH selectivity, again in line with previous observations.82 Interestingly, despite the multiple conditions tested, no deactivation was observed for any of the samples after more than 150 hours under reaction conditions. From the reported results, ZrZn-30 appears as the optimal catalyst composition, displaying the highest activity and selectivity regardless of the reaction conditions. We attribute this superior performance of the ZrZn-30 sample to the already discussed higher amount of oxygen vacancies in the sample.17,83
Afterwards, we studied the combination of ZrZn-X catalysts with the two most common zeolites for the CO2 cascade conversion:84,85 ZSM-5 and H-SAPO-34. Similar to the above tests, we evaluated the effect of reaction pressure, temperature and CO addition. The results are summarized in Fig. 9 for ZSM-5 and in Fig. S8† for H-SAPO-34. We can observe that the CO2 conversion for these bifunctional catalysts goes in line with the one observed for the stand-alone ZrZn-X samples, increasing with both pressure and temperature. Similar conversion values and CO selectivity are also obtained regardless of the reaction conditions or zeolite component (see Fig. S9† for a detailed comparison). However, when looking at the hydrocarbon distribution, we can observe that the zeolite component plays a critical role. In particular, the H-SAPO-34 based catalyst displayed a higher C3 selectivity among hydrocarbons (up to 60%) but it is rapidly deactivated, especially at 350 °C where it lost almost all activity in less than 20 hours and unreacted methanol became the main reaction product (see Fig. S8 and S9†). Moreover, it seems that an operation temperature of 250 °C is too low for MeOH conversion to occur in H-SAPO-34.45,86 This catalytic behavior is consistent with the fast deactivation and higher selectivity observed for H-SAPO-34 in the MTH reaction.87,88 On the other hand, the ZSM-5 based catalyst displayed a more stable performance, with a C3 selectivity of ca. 40%. However, we need to point out that a slight deactivation is also observed at 350 °C for the ZSM-5 based catalysts. Finally, CO addition seems to slightly enhance the C3 selectivity, in line with the recent results by Tan et al.89 who observed an increase in the hydrocarbon selectivity by CO co-feeding. Altogether, we can consider the ZrZn-30/ZSM-5 combined system, tested at 350 °C and 30 bar, as the most promising candidate/reaction conditions, displaying the highest conversion and stability with a C3 selectivity close to 35% among hydrocarbons.
 | ||
| Fig. 9 Catalytic performance of the combined ZrZn-X/ZSM-5 systems for CO2 conversion to hydrocarbons. CO2:H2 1:3, 12000 mL h−1 g−1. Please note that the secondary Y axis refers to the C3 selectivity among hydrocarbons (CO free). | ||
Further catalyst studies were performed under these optimal reaction conditions (350 °C, 30 bar), using two other ZSM-5 samples with Si/Al = 25 and 360 (ESI,† section 5.3). When testing the ZrZn-30 catalyst alone, methanol and CO were the only carbon-containing products (Fig. S10†). Product selectivity favored methanol at the shortest contact times, suggesting that the rate of CO2 conversion to methanol (eqn (2)) is faster than the reverse water gas shift reaction (eqn (1)), in agreement with the results of the computational study (section 3.3). The methanol yield reached equilibrium after 0.4 s g−1 ml−1 contact time. The CO2 conversion and hence, CO selectivity increased with a further increase in contact time. Due to water formed in the reverse water gas shift reaction, the methanol equilibrium yield decreased with increasing contact time.
When mixing ZrZn-30 with the two H-ZSM-5 catalysts in a 1:1 ratio, a range of hydrocarbon products, as well as dimethyl ether (DME) were observed, in addition to CO and methanol (Fig. S11†). The methanol yield was low, substantially below equilibrium, and decreased with increasing acid site density in H-ZSM-5. This result suggests that CO2 hydrogenation to methanol is the rate-limiting step of hydrocarbon formation in the bifunctional ZrZn-30:H-ZSM-5 = 1:1 mixed catalysts.
Considering next CO2 conversion versus contact time, it did not change significantly with the addition of H-ZSM-5, as already observed in Fig. 8 and 9. However, the CO selectivity decreased with the addition of H-ZSM-5, and decreased further with an increase in the acid site density of H-ZSM-5 (Fig. S12†). This result may suggest that CO, like the hydrocarbons, is a (competing) secondary product from methanol, or that CO, like methanol, is converted to hydrocarbons over H-ZSM-5. The recent literature suggests that both hypotheses are plausible.90,91
Surprisingly, when considering next the hydrocarbon distribution over mixed catalysts, the aromatics selectivity is typically below 10% and only at 6000 ml h−1 g−1 is a significant fraction observed for the main ZSM-5 catalyst tested here (Fig. 10).
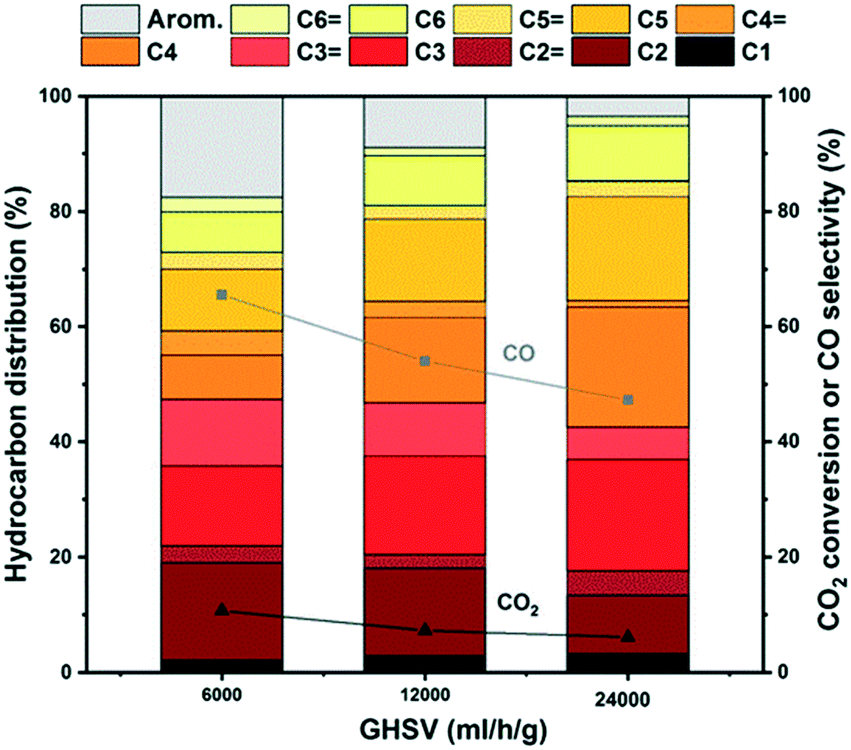 | ||
| Fig. 10 Hydrocarbon distribution of the ZrZn-30/ZSM-5 combined system for CO2 conversion to hydrocarbons at different space times. CO2:H2 1:3, 350 °C, 30 bar. | ||
We attribute these results to the high space time employed in this work that suppresses the aromatization cycle, in line with the results by Cui et al. who observed an increase of aromatics selectivity from ca. 20% to 75% by reducing the space time by one order of magnitude.92 These results are supported by testing other ZSM-5 catalysts mixed with ZrZn-30 (section S5.3). A higher acid site density in H-ZSM-5 led to more saturated aliphatic products, and less aromatic products, compared to a lower acid site density (Fig. S13 and S14†). These results suggest an intricate, joint behavior of the two catalyst functions that warrants further investigations in future contributions.
Additionally, if we look in detail at the CO free hydrocarbon distribution (Table S4† and Fig. 11), we can observe that, apart from the above-mentioned aromatics influence, the space time also affects the olefin/paraffin ratio. At high space times, paraffins are the predominant fraction, while at lower space times the olefins start to increase. These trends can be counter-intuitive and the opposite trend should be expected since olefins are the primary products of the HC pool reaction and the thermodynamic equilibrium of alkane dehydrogenation reactions lies far to the alkane side for C2 and C3. However, if we look in detail at the reaction kinetics over ZSM-5,93,94 we can observe that at very high space times (like the ones in our study) the slope of olefin increase is higher than the ones for paraffins plus C5+ hydrocarbons, therefore in line with our experimental observations. Last but not least, the productivities displayed here are among the highest reported for state-of-the-art catalysts84 despite the low conversion, probably owing to the high space times employed in our study and the associated absence of aromatics.
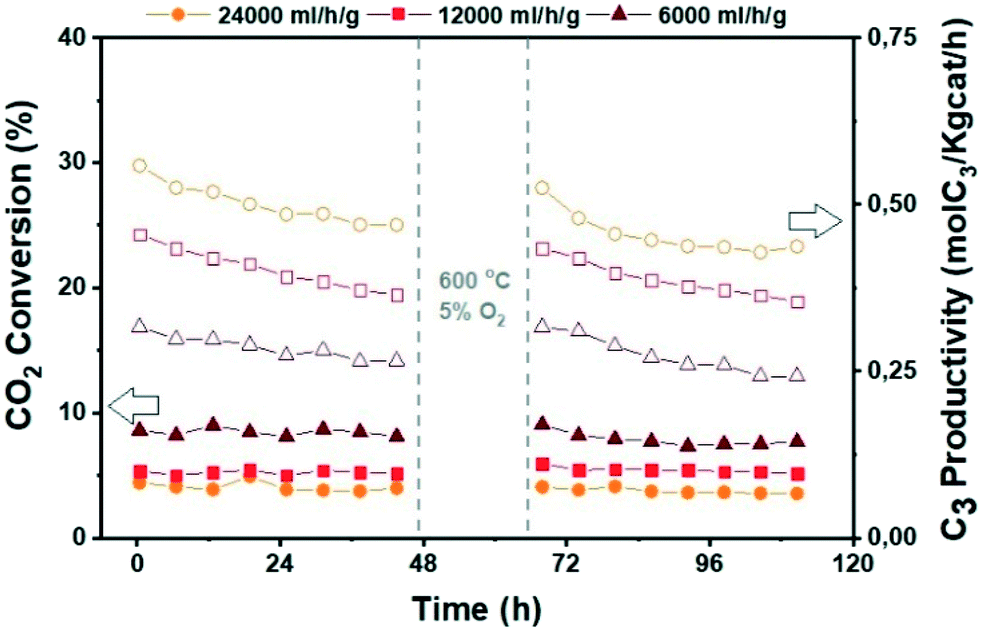 | ||
| Fig. 11 Catalytic performance of the ZrZn-30/ZSM-5 combined system before and after regeneration for CO2 conversion to hydrocarbons at different space times. CO2:H2 1:3, 350 °C, 30 bar. | ||
Finally, since deactivation can play a role especially in view of industrial implementation of the investigated bifunctional catalysts, we studied the effect of in situ regeneration at 600 °C with a 5% O2 in N2 stream for the ZrZn-30/ZSM-5 combined system at different space times. The results are summarized in Fig. 11. We can observe that the in situ regeneration worked for all the samples and the initial activity was regained after the regeneration cycle at 600 °C. Moreover, increasing the space time drastically increases the C3 productivity despite the CO2 conversion decrease, achieving a maximum of 1.5 mol kg−1 h−1 at 24000 ml h−1 g−1.
3.5. XAS measurements on the combined ZrZn-X/ZSM-5 systems
Focusing on ZrZn-X/ZSM-5 combined systems, we finally applied in situ and operando XAS to monitor the average electronic properties and local structure of Zr and Zn metal centres, in the presence of the zeolite functionality and under realistic activation and process conditions. This becomes especially relevant, in view of recent findings highlighting inter-phase ion exchange phenomena in combined systems obtained by physically mixing acid zeolites and Zn-containing hydrogenation catalysts.10 To obtain fully comparable information at Zr and Zn K-edges, we measured the two absorption edges quasi-simultaneously during the same experiment, exploiting the unique capability of the ROCK beamline53 of the SOLEIL synchrotron (see section 2.2.5 for details).Considering the Zn K-edge XAS spectra in Fig. 12a, we observe how the as-prepared samples exclusively contain Zn2+ species. Indeed, the edge energy position is substantially equivalent for the three ZrZn-X/ZSM-5 combined systems and overlapped with that of the ZnO model compound. Notably, ZrZn-30/ZSM-5 shows an overall XANES line-shape and specific post-edge features characteristic of ZnO (e.g. peaks at 9714 and 9738 eV), which are instead not detected in the other samples. This is explained considering the presence of the ZnO extra phase unveiled by PXRD in the ZrZn-30 catalyst. The inset of Fig. 12a shows the FT-EXAFS spectra of the two combined systems featuring the higher Zn-loadings, whereas for the lowest-loading ZrZn-5/ZSM-5 system, the low S/N ratio in the EXAFS region unfortunately prevented a reliable data interpretation. Both the samples show a very similar first coordination shell peak stemming from O nearest neighbours (NNs). The first-shell peaks are comparable with that of the reference ZnO in terms of the R-space position, while they display slightly lower intensity, consistent with distortions in the local coordination environment of substitutional Zn ions in the ZrO2 lattice. The two samples show more pronounced differences in the second-shell region of the EXAFS spectra. In particular, for ZrZn-30/ZSM-5 we recognize a well-defined peak matching the position of the second-shell feature in ZnO, arising from Zn next nearest neighbour (NNN) atoms. The lower peak intensity with respect to what is observed for the model compound can be connected with the simultaneous presence of substitutional Zn ions in the ZrO2 lattice, as well as with possible defectiveness of the segregated ZnO particles. Conversely, only a broad and much weaker peak is observed for ZrZn-15/ZSM-5 in the second-shell region, pointing to rather high structural disorder in the NNN distribution for substitutional Zn ions in ZrO2.
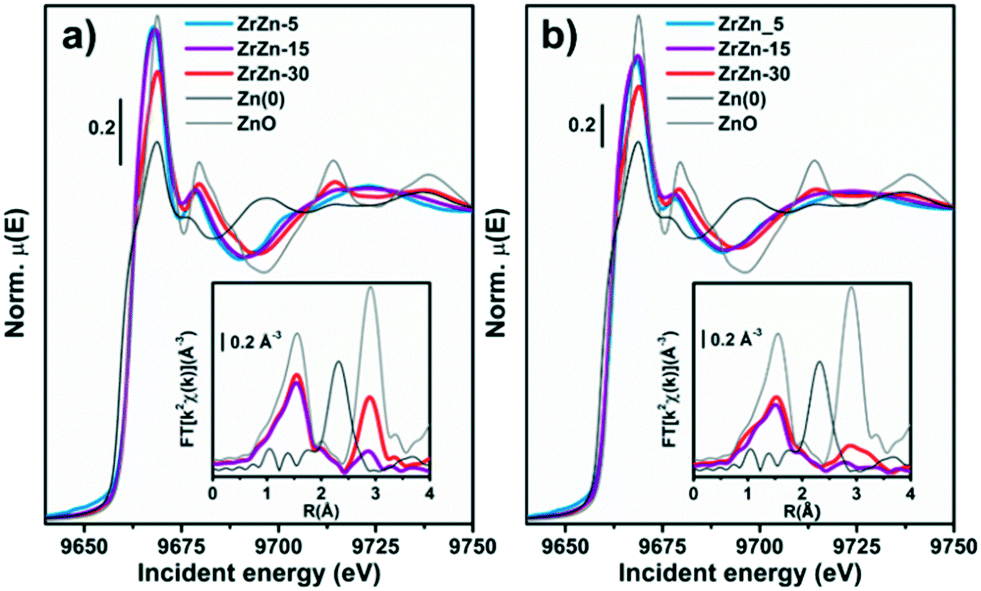 | ||
| Fig. 12 K-edge XANES (main panel) and phase-uncorrected FT-EXAFS (bottom inset) spectra of the three samples and of Zn metal and ZnO model compounds, collected at (a) RT under He flux and (b) after the activation process (400 °C, H2). The EXAFS spectra reported in the bottom insets have been obtained transforming the corresponding k2χ(k) EXAFS function in the 2.5–13.0 Å−1 range. | ||
During activation (Fig. 12b) the Zn K-edge XANES features are substantially unchanged, underpinning two important facts: i) the average oxidation state of Zn does not change (no edge-shift is observed nor any evidence for the formation of Zn0 phases) and ii) Zn2+ does not diffuse in the zeolite (typical spectral features of Zn-exchanged zeolites95 are not observed). Considering the corresponding FT-EXAFS spectra (Fig. 12b, inset), the first-shell peak undergoes a slight intensity decrease, consistent with the increased thermal contribution to Debye–Waller factors at 400 °C. The second shell peaks, connected with NNN contributions, appear more strongly affected. The two samples maintain the same intensity trend as in their as-prepared state, with ZrZn-30/ZSM-5 showing the highest intensity; however, in both cases an important dampening/broadening effect is observed. Also in this case, increased Debye–Waller factors at 400 °C contribute to EXAFS signal dampening. However, it is clear that activation also triggered an increase in the local disorder around Zn2+ sites – both those hosted in the ZrO2 lattice and those segregated as ZnO extra-phases, in agreement with IR results demonstrating oxygen vacancy formation during thermal treatment in H2 up to 400 °C.
Under quasi-simultaneous acquisition conditions, Zr K-edge XAS (Fig. 13a) allowed us to obtain structural insights on the ZrO2 matrix complementary to the ones accessed by PXRD analysis (see section 3.1). In particular, we were able to discriminate tetragonal from cubic and monoclinic structures, as the XANES features for the two configurations are strongly influenced by the ZrO8 polyhedron distortion. Li et al.96 reported three important features in the spectra ascribable to tetragonal ZrO2: i) the pre-edge peak associated with the 1s → 4d transition, ii) white-line peak splitting, absent in the monoclinic ZrO2 and iii) broad post-edge resonance around 35 eV after the edge. All these spectroscopic fingerprints, further corroborated by the Zr K-edge XAS spectra of reference monoclinic and tetragonal ZrO2 reported in Fig. 13, are observable in the XANES of the three investigated ZrZn-X/ZSM-5 combined systems, pointing to the presence of a tetragonal ZrO2 phase. In particular the 1s → 4d pre-edge feature, which is very evident in the XANES first derivative (Fig. S15†), is a fingerprint of t-ZrO2 where Zr is eight-fold coordinated. In the perfect ZrO8 pyrochlore-like structure, this s–d transition would not be detectable, as it is dipole forbidden.
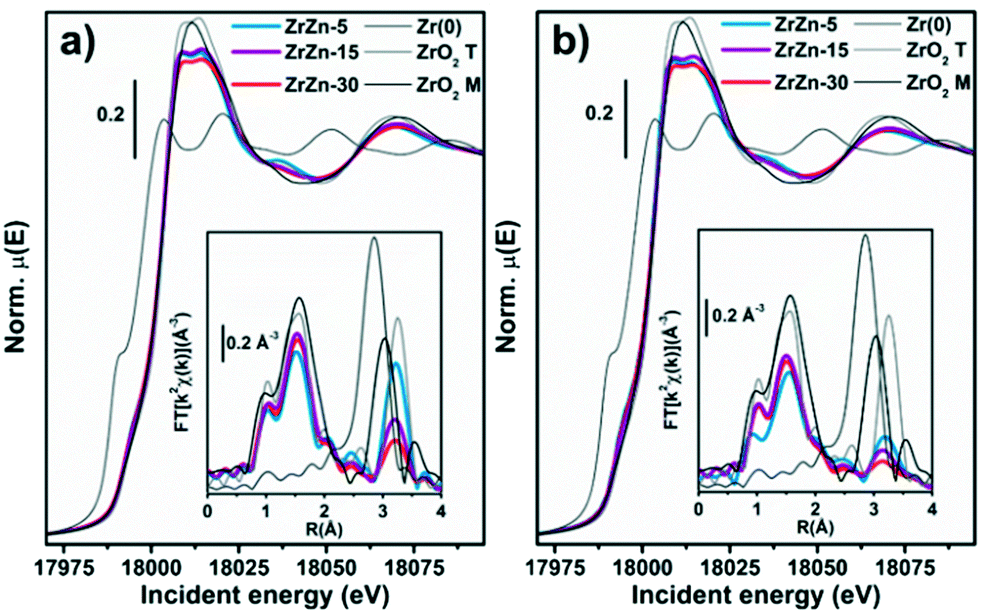 | ||
| Fig. 13 K-edge XANES (main panel) and phase-uncorrected FT-EXAFS (bottom inset) spectra of the three samples and of Zr metal and ZrO2 model compounds, collected at (a) RT under He flux and (b) after the activation process (400 °C, H2). The EXAFS spectra reported in the bottom insets have been obtained transforming the corresponding k2χ(k) EXAFS function in the 2.5–13.0 Å−1 range. | ||
However, it is well documented97,98 that in t-ZrO2 four oxygens are closer while four are farther from the Zr cation. This leads to visualization of the Zr atom as coordinated with two different tetrahedra of oxygen atoms.96,99 Since the centrosymmetricity is broken, the dipole forbidden 1s → 4d transition gains in intensity being observable in the Zr K pre-edge.96,100 Moreover, two features in the FT-EXAFS, highlighted by dashed lines in Fig. S15,† can be ascribed to t-ZrO2: i) the Zr–Zr second shell position and ii) the relatively intense high-R peak in the 6–7 Å range in the phase-uncorrected spectra. As shown by Li et al.96,100 with careful EXAFS fit of various ZrO2 polymorphs, in c-ZrO2 the average Zr–Zr distance is almost 0.1 Å shorter than that in t-ZrO2. Therefore, the Zr–Zr second feature of c-ZrO2 should be located at a lower R value than that of the reference t-ZrO2. Besides, the intense feature around 6.7 Å (Fig. S15† inset) is related to collinear multiple scattering between Zr atoms which is present in t-ZrO2 but absent in c-ZrO2.96,101
Previous works explained the stabilization of tetragonal ZrO2 considering the substitution of the Zr atom with either Zn (ref. 31) or Hf.102 For clarity, we outline that a trace of Hf L3-edge (9561 eV) was observed during the Zn K-edge XAS measurements, in line with chemical analysis results: the small amount of Hf present in the investigated samples plausibly also contributes to promoting the tetragonal ZrO2 structure. Phase-uncorrected FT-EXAFS, reported in the inset of Fig. 13a, show a first-shell peak stemming from O NN, consistent for all the three samples with the one observed for the ZrO2 model compound.
Conversely, with the increase of Zn content (ZrZn-30 > ZrZn-15 > ZrZn-5) the intensity of the second-shell peak is progressively attenuated, while its position is substantially unaltered, always closely resembling the one observed for the tetragonal ZrO2 model compound. In agreement with PXRD results, this intensity trend stems from Zn entering the ZrO2 lattice, causing destructive interference among scattering paths involving Zr and Zn NNNs. We note a pronounced intensity decrease while moving from ZrZn-5 to ZrZn-15, while a further increase of Zn-loading in ZrZn-30 only results in a slight additional attenuation of the second-shell peak. This observation further supports that in ZrZn-15 we are close to the upper threshold for the incorporation of Zn in the ZrO2 lattice.
However, in ZrZn-30, a slightly higher amount of Zn still enters the ZrO2 matrix, as proven by the additional weakening of the second-shell peak. Afterwards, excess Zn segregates as hexagonal ZnO, silent in Zr K-edge XAS but detectable in Zn K-edge XAS and PXRD. Activation (Fig. 13b) does not cause substantial modifications in the ZrO2 structure, nor detectable reduction phenomena involving Zr atoms. Tetragonal features of ZrO2 are still evident, while the structural disorder in the NNN distribution around Zr centres further increases, translating into an abatement of the second-shell peaks, as observed under the same conditions in Zn K-edge FT-EXAFS.
Under reaction conditions (Fig. S16†), XAS data collected at both absorption edges showed that the catalyst structural stability is preserved. In particular: i) the ZrO2 matrix maintains the tetragonal structure; ii) Zn does not exchange in the zeolite; iii) both Zr4+ and Zn2+ do not undergo detectable reduction phenomena. The absence of structural changes after the reaction was also observed from the PXRD measurement of the tested catalysts, reported in the following section. Structural modifications possibly involving surface metal sites upon interaction with the CO2/H2 feed are not detectable, as the measured XAS signal includes mainly bulk information.
3.6. Structural characterization of the fresh/tested ZrZn-30/ZSM-5 combined system
In order to investigate possible structural modification induced by the physical mixing and the reaction conditions, we measured the PXRD pattern of the ZrZn-30/ZSM-5 physical mixture before and after 120 hours of catalytic test. Each reflection was ascribed to the corresponding crystalline phase by measuring the PXRD for the single components: i) ZrZn-30 catalyst alone after a reaction cycle and ii) commercial ZSM-5. The diffraction pattern of the ZrZn-30/ZSM-5 combined system in Fig. 14, measured before and after the catalytic test, does not present any differences related to potential structural changes, i.e. crystallite defects such as dimensions, stress or strain related to peak broadening. We can clearly distinguish the reflections from each crystal phase, i.e. orthorhombic zeolite, tetragonal zirconia and hexagonal zinc oxide. Rietveld refinement was conducted only on the diffraction pattern of the ZrZn-30 catalyst measured after the reaction (Fig. 14 inset). The same refinement strategy used for the fresh catalyst was applied. Taking into account the error of the technique, the results in Fig. 14 and Table S2† evidence structural features very similar to those observed for the fresh sample, while the zeolite crystallinity is preserved as the peaks show similar FWHM (Fig. 14 inset). The increase of the intensity between ZrZn-30 alone and its physical mixture with the zeolite is related to the decrease of the total absorption coefficient as in the second case, half of the capillary contains the zeolite.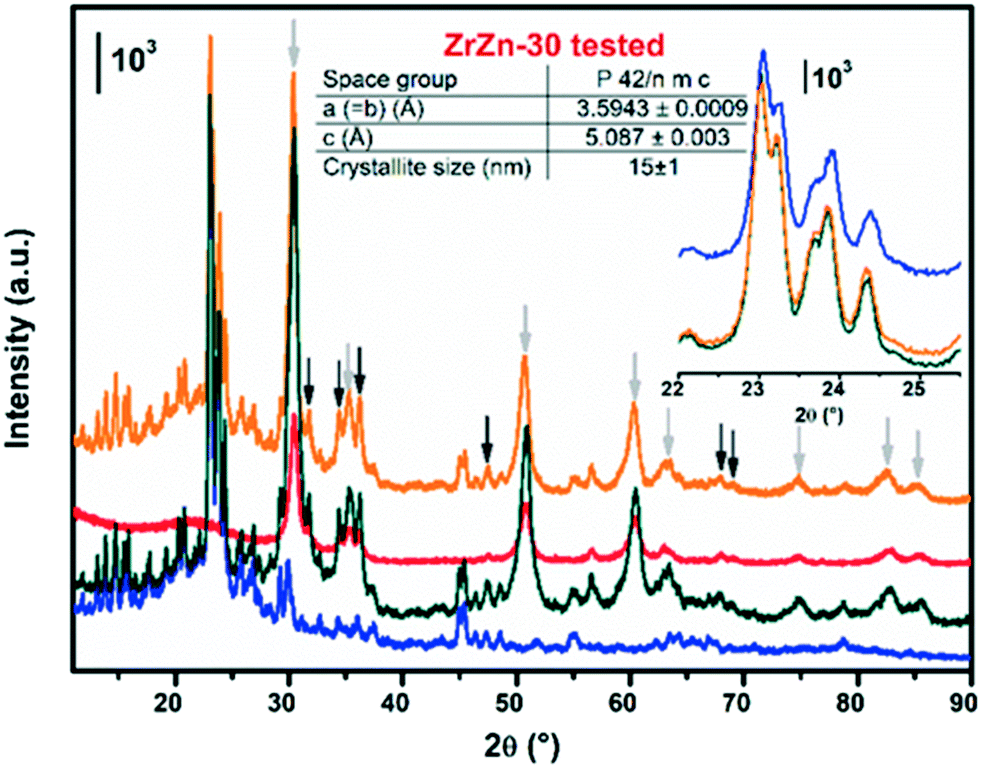 | ||
| Fig. 14 Stacked representation of the PXRD measured for i) commercial ZSM-5 zeolite alone, in blue; ii) fresh physical mixture ZrZn-30/ZSM-5, in dark-green, iii) ZrZn-30 catalyst after 120 hours of catalytic test, in red; iv) physical mixture of ZrZn-30/ZSM-5 after 120 hours of catalytic test, in orange. For the sake of clarity in the latter pattern, the reflections corresponding to tetragonal ZrO2 and hexagonal ZnO are indicated by light and dark grey arrows, respectively. Structure results obtained from Rietveld refinement of the tested ZrZn-30 catalyst alone are reported in the table. A detail of the zeolite Bragg peaks is reported in the inset. | ||
We further investigated the possible surface composition changes before and after the reaction by performing XPS analysis on both fresh and tested samples (Fig. S18 and S19†). We can observe that there are no appreciable shifts in the binding energies of both Zn and Zr after 48 hours of catalytic test. Furthermore, the binding energy of Zr at the Zr3p5/2 level is 182.37 eV, lower than the 182.7 eV assigned to the pure ZrO2.103 This shift has already been reported in the presence of oxygen vacancies due to the substitution of Zn4+ by Zn2+,104 in line with our FT-IR observations. We also need to remark that, similar to the PXRD in Fig. 14, the intensity difference between ZrZn-30 alone and its physical mixture with the zeolite is related to the decrease of loading.
We next investigated the possible morphological changes in our system via high-angle annular dark-field transmission microscopy (HAADF) in conjunction with energy-dispersive X-ray spectroscopy (EDXS). We can observe that in the fresh ZrZn-30 sample both Zn and Zr elements are closely incorporated and that an excess of Zn segregates as ZnO, in line again with the above XANES and PXRD characterization. (Fig. S20†). Similarly, imaging of the tested sample (Fig. 15) shows no structural changes after the reaction with an intimate mixture of both ZrZn-30 and ZSM-5 components in the final bifunctional system. However, we need to point out that a small migration of Zn was observed in some of the zeolite particles (∼0.2 wt%, Fig. S21†).
 | ||
| Fig. 15 HAADF STEM-EDXS images of the tested ZrZn-30/ZSM-5 catalyst after 48 hours of catalytic test. | ||
Conclusions
In this work we have synergized catalytic tests under different conditions, multi-technique characterization and computational modelling to advance the understanding of bifunctional Zn-doped-ZrO2/zeolite catalysts for CO2 hydrogenation to methanol and conversion to value-added hydrocarbons.Three Zn-containing ZrO2 samples were prepared by co-precipitation (ZrZn-X, X = Zn wt%, i.e. 5, 15, 30). The formation of the expected ZrZnOX solid solution was confirmed by both XAS characteristic features and powder diffractograms. In particular, the crystalline structure of tetragonal zirconia was recognized with diffraction peak shifts consistent with the Zn loading in the structure. A ZnO extra phase is present for the ZrZn-30 sample, evidencing that the co-precipitation technique employed allows achievement of a maximum Zn doping of ZrO2 of about 15 wt%, even though EXAFS results show that a slightly higher amount of Zn enters the ZrO2 lattice in ZrZn-30 with respect to ZrZn-15. CO adsorption (at LNT) followed by FT-IR spectroscopy shows that the amount of Zr4+ sites at the surface decreases coherently with the increase of Zn loading found from chemical composition and X-ray results.
Interaction with hydrogen at increasing temperature causes the formation of mono-ionized oxygen vacancies, as evidenced by FT-IR spectroscopy. In agreement with FT-IR results, DFT calculations show that chances of oxygen vacancy formation with the assistance of hydrogen increase with increasing Zn content. In particular, DFT modelling points out that once a stoichiometric oxygen vacancy is induced by the presence of Zn, the formation of extra oxygen vacancies during activation is thermodynamically favored. Moreover, DFT modelling points out that i) the oxygen vacancies play an active role in CO2 hydrogenation, ii) the presence of neighboring Zn and Zr sites enhances methanol selectivity thanks to the proximity of CH3O* and Hδ+, strongly bonded than Hδ−, and iii) methanol is most likely formed via the formate pathway.
Coherently, the catalytic performances of the stand-alone ZrZn-X samples showed the same trend shown by the oxygen vacancy amount, i.e. CO2 conversion and CH3OH selectivity increases with the Zn loading. While we cannot exclude that segregated ZnO in the most active catalyst (ZrZn-30) also plays a role, the reported characterization, modelling and testing results consistently suggest that the main contribution to the catalyst activity comes from the ZrZnOx solid solution. Combined systems were obtained by mechanical mixing of the ZrZn-X catalysts with a commercial zeolite (H-ZSM-5 and H-SAPO-34) with a mass ratio of 1:1. Comparing ZrZn-30/ZSM-5 and ZrZn-30/H-SAPO-34 combined systems, we pointed out that the ZSM-5-containing system is more promising than the one with H-SAPO-34. Indeed, the highest conversion and stability with circa 30% C3 selectivity among hydrocarbons were found testing ZrZn-30/ZSM-5 at 350 °C and 30 bar. At T < 350 °C, alkylbenzene dealkylation is unlikely to occur.45,46 However, in this work we highlight an unusual aliphatics selectivity (and stability) at lower temperature and shorter contact time, more favored with H-ZSM-5 than H-SAPO-34 as the zeotype has lower activity/stability at low temperature.
Finally, the structure of the catalysts is not affected by the reaction conditions as shown by the operando XAS and the structural/textural characterization of the tested samples; indeed a final catalytic test carried out on the regenerated catalyst shows that the initial performances are completely restored.
In conclusion, in the light of our findings, we can affirm that, for a bifunctional catalyst, both chemical (oxidic phase composition, Brønsted acid site density, and pore dimensions) and kinetic factors (temperature, pressure and space time velocity) must be considered in order to drive the reaction towards the desired products and, therefore, to achieve a good reaction yield.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No. 837733. This work was supported by a public grant overseen by the French National Research Agency (ANR) as part of the “Investissements d'Avenir” program (reference: ANR-10-EQPX-45). The authors are grateful to C. La Fontaine, V. Briois, A. L. Bugaev and A. Lazzarini for the help with the XAS experiment at ROCK, Soleil.References
- J. Rockström, W. Steffen, K. Noone, Å. Persson, F. S. Chapin, E. F. Lambin, T. M. Lenton, M. Scheffer, C. Folke, H. J. Schellnhuber, B. Nykvist, C. A. de Wit, T. Hughes, S. van der Leeuw, H. Rodhe, S. Sörlin, P. K. Snyder, R. Costanza, U. Svedin, M. Falkenmark, L. Karlberg, R. W. Corell, V. J. Fabry, J. Hansen, B. Walker, D. Liverman, K. Richardson, P. Crutzen and J. A. Foley, Nature, 2009, 461, 472–475 CrossRef.
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, J. CO2 Util., 2014, 5, 66–81 CrossRef CAS.
- Z. Jiang, T. Xiao, V. L. Kuznetsov and P. P. Edwards, Philos. Trans. R. Soc., A, 2010, 368, 3343–3364 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- J. B. Hansen and P. E. Højlund Nielsen, Methanol Synthesis, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley, Germany, 2008 Search PubMed.
- J. R. Gallagher, D. J. Childers, H. Zhao, R. E. Winans, R. J. Meyer and J. T. Miller, Phys. Chem. Chem. Phys., 2015, 17, 28144–28153 RSC.
- C. Ahoba-Sam, E. Borfecchia, A. Lazzarini, A. Bugaev, A. A. Isah, M. Taoufik, S. Bordiga and U. Olsbye, Catal. Sci. Technol., 2020, 10, 4373–4385 RSC.
- M. Gentzen, D. E. Doronkin, T. L. Sheppard, A. Zimina, H. Li, J. Jelic, F. Studt, J. D. Grunwaldt, J. Sauer and S. Behrens, Angew. Chem., Int. Ed., 2019, 58, 15655–15659 CrossRef CAS.
- M. W. Tew, H. Emerich and J. A. Van Bokhoven, J. Phys. Chem. C, 2011, 115, 8457–8465 CrossRef CAS.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- G. Bonura, M. Migliori, L. Frusteri, C. Cannilla, E. Catizzone, G. Giordano and F. Frusteri, J. CO2 Util., 2018, 24, 398–406 CrossRef CAS.
- W. J. Thomas and S. Portalski, Ind. Eng. Chem., 1958, 50, 967–970 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- A. S. Malik, S. F. Zaman, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, Appl. Catal., A, 2018, 560, 42–53 CrossRef CAS.
- A. S. Malik, S. F. Zaman, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, Catal. Today, 2020, 357, 573–582 CrossRef CAS.
- H. Bahruji, M. Bowker, W. Jones, J. Hayward, J. Ruiz Esquius, D. J. Morgan and G. J. Hutchings, Faraday Discuss., 2017, 197, 309–324 RSC.
- K. Li and J. G. Chen, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 357, 1296–1299 CrossRef.
- Y. Amenomiya, Appl. Catal., A, 1987, 30, 57–68 CrossRef CAS.
- X. Dong, F. Li, N. Zhao, F. Xiao, J. Wang and Y. Tan, Appl. Catal., B, 2016, 191, 8–17 CrossRef CAS.
- H. Gu, J. Ding, Q. Zhong, Y. Zeng and F. Song, Int. J. Hydrogen Energy, 2019, 44, 11808–11816 CrossRef CAS.
- I. A. Fisher and A. T. Bell, J. Catal., 1999, 184, 144–156 CrossRef.
- G. Wang, D. Mao, X. Guo and J. Yu, Int. J. Hydrogen Energy, 2019, 44, 4197–4207 CrossRef CAS.
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Appl. Catal., A, 2008, 350, 16–23 CrossRef CAS.
- W. Li, K. Wang, J. Huang, X. Liu, D. Fu, J. Huang, Q. Li and G. Zhan, ACS Appl. Mater. Interfaces, 2019, 11, 33263–33272 CrossRef CAS.
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17, 4297–4303 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, 1–11 Search PubMed.
- A. Wokaun, Phys. Chem. Chem. Phys., 1999, 1, 5071–5080 RSC.
- H. Li, C. Rameshan, A. V. Bukhtiyarov, I. P. Prosvirin, V. I. Bukhtiyarov and G. Rupprechter, Surf. Sci., 2019, 679, 139–146 CrossRef CAS.
- L. H. Chagas, P. C. Zonetti, C. R. V. Matheus, C. R. K. Rabello, O. C. Alves and L. G. Appel, ChemCatChem, 2019, 11, 5625–5632 CrossRef CAS.
- O. E. Everett, P. C. Zonetti, O. C. Alves, R. R. de Avillez and L. G. Appel, Int. J. Hydrogen Energy, 2020, 45, 6352–6359 CrossRef CAS.
- X. Liu, W. Zhou, Y. Yang, K. Cheng, J. Kang, L. Zhang, G. Zhang, X. Min, Q. Zhang and Y. Wang, Chem. Sci., 2018, 9, 4708–4718 RSC.
- A. V. Kirilin, J. F. Dewilde, V. Santos, A. Chojecki, K. Scieranka and A. Malek, Ind. Eng. Chem. Res., 2017, 56, 13392–13401 CrossRef CAS.
- M. D. Rhodes, K. A. Pokrovski and A. T. Bell, J. Catal., 2005, 233, 210–220 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS.
- S. Wang, P. Wang, Z. Qin, Y. Chen, M. Dong, J. Li, K. Zhang, P. Liu, J. Wang and W. Fan, ACS Catal., 2018, 8, 5485–5505 CrossRef CAS.
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang, S. Wang, G. Wang, M. Dong, W. Fan and J. Wang, ACS Catal., 2016, 6, 7311–7325 CrossRef CAS.
- K. Cheng, J. Kang, Q. Zhang and Y. Wang, Sci. China Chem., 2017, 60, 1382–1385 CrossRef CAS.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2020, 10, 302–310 CrossRef CAS.
- Y. K. Park, K. C. Park and S. K. Ihm, Catal. Today, 1998, 44, 165–173 CrossRef CAS.
- H. Schulz, Catal. Today, 2010, 154, 183–194 CrossRef CAS.
- F. Bleken, M. Bjørgen, L. Palumbo, S. Bordiga, S. Svelle, K. P. Lillerud and U. Olsbye, Top. Catal., 2009, 52, 218–228 CrossRef CAS.
- J. Rodríguez-Carvajal, Newsl. Comm. Powder Diffr. IUCr, 2001, vol. 26, pp. 12–19 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 1–13 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- C. La Fontaine, S. Belin, L. Barthe, O. Roudenko and V. Briois, Synchrotron Radiat. News, 2020, 33, 20–25 CrossRef.
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet, S. Gatla, R. Hino, I. Kantor, T. Mairs, M. Munoz, S. Pasternak, F. Perrin and S. Pascarelli, J. Synchrotron Radiat., 2015, 22, 1548–1554 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- K. S. W. Sing and R. T. Williams, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- B. Y. R. D. Shannon, M. H. N. H. Baur, O. H. Gibbs, M. Eu and V. Cu, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. C. Abrahams and J. L. Bernstein, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1233–1236 CrossRef CAS.
- G. Teufer, Acta Crystallogr., 1962, 15, 1187 CrossRef CAS.
- Ü. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doǧan, V. Avrutin, S. J. Cho and H. Mork
![[o with combining cedilla]](https://www.rsc.org/images/entities/char_006f_0327.gif) , J. Appl. Phys., 2005, 98, 1–103 CrossRef.
, J. Appl. Phys., 2005, 98, 1–103 CrossRef. - W. Göpel and U. Lampe, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 6447–6462 CrossRef.
- S. Morandi, A. Fioravanti, G. Cerrato, S. Lettieri, M. Sacerdoti and M. C. Carotta, Sens. Actuators, B, 2017, 249, 581–589 CrossRef CAS.
- L. Genzel and T. P. Martin, Surf. Sci., 1973, 34, 33–49 CrossRef CAS.
- G. Ghiotti, A. Chiorino and F. Prinetto, Sens. Actuators, B, 1995, 25, 564–567 CrossRef CAS.
- S. Morandi, M. C. Paganini, E. Giamello, M. Bini, D. Capsoni, V. Massarotti and G. Ghiotti, J. Solid State Chem., 2009, 182, 3342–3352 CrossRef CAS.
- C. Morterra, E. Giamello, L. Orio and M. Volante, J. Phys. Chem., 1990, 94, 3111–3116 CrossRef CAS.
- B. N. J. Persson and R. Ryberg, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 6954–6970 CrossRef CAS.
- M. Moskovits and J. E. Hülse, Surf. Sci., 1978, 78, 397–418 CrossRef CAS.
- G. D. Mahan and A. A. Lucas, J. Chem. Phys., 1978, 68, 1344–1348 CrossRef CAS.
- G. L. Griffin and J. T. Yates, J. Chem. Phys., 1982, 77, 3744–3750 CrossRef CAS.
- D. Scarano and A. Zecchina, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 3611–3624 RSC.
- C. Morterra, V. Bolis, B. Fubini, L. Orio and T. B. Williams, Surf. Sci., 1991, 251–252, 540–545 CrossRef CAS.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 CAS.
- D. Scarano, S. Bertarione, G. Spoto, A. Zecchina and C. Otero Areán, Thin Solid Films, 2001, 400, 50–55 CrossRef CAS.
- L. R. del Silva-Calpa, P. C. Zonetti, C. P. Rodrigues, O. C. Alves, L. G. Appel and R. R. de Avillez, J. Mol. Catal. A: Chem., 2016, 425, 166–173 CrossRef.
- A. A. Safonov, A. A. Bagatur'yants and A. A. Korkin, Microelectron. Eng., 2003, 69, 629–632 CrossRef CAS.
- A. Bavykina, I. Yarulina, A. J. Al Abdulghani, L. Gevers, M. N. Hedhili, X. H. Miao, A. R. Galilea, A. Pustovarenko, A. Dikhtiarenko, A. Cadiau, A. Aguilar-Tapia, J. L. Hazemann, S. M. Kozlov, S. Oud-Chikh, L. Cavallo and J. Gascon, ACS Catal., 2019, 9, 6910–6918 CrossRef CAS.
- J. L. Snider, V. Streibel, M. A. Hubert, T. S. Choksi, E. Valle, D. C. Upham, J. Schumann, M. S. Duyar, A. Gallo, F. Abild-Pedersen and T. F. Jaramillo, ACS Catal., 2019, 9, 3399–3412 CrossRef CAS.
- E. S. Van-Dal and C. Bouallou, J. Cleaner Prod., 2013, 57, 38–45 CrossRef CAS.
- D. Milani, R. Khalilpour, G. Zahedi and A. Abbas, J. CO2 Util., 2015, 10, 12–22 CrossRef CAS.
- Y. Slotboom, M. J. Bos, J. Pieper, V. Vrieswijk, B. Likozar, S. R. A. Kersten and D. W. F. Brilman, Chem. Eng. J., 2020, 389, 124181 CrossRef CAS.
- N. Rui, Z. Wang, K. Sun, J. Ye, Q. Ge and C. J. Liu, Appl. Catal., B, 2017, 218, 488–497 CrossRef CAS.
- A. Dokania, A. Ramirez, A. Bavykina and J. Gascon, ACS Energy Lett., 2019, 4, 167–176 CrossRef CAS.
- Z. Q. Ma and M. D. Porosoff, ACS Catal., 2019, 9, 2639–2656 CrossRef CAS.
- H. Schulz, Catal. Lett., 2018, 148, 1263–1280 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjorgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS.
- L. Tan, P. P. Zhang, Y. Cui, Y. Suzuki, H. J. Li, L. S. Guo, G. H. Yang and N. Tsubaki, Fuel Process. Technol., 2019, 196, 106174 CrossRef CAS.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- Q. Song, Y. Men, J. Wang, S. Liu, S. Chai, W. An, K. Wang, Y. Li and Y. Tang, Int. J. Hydrogen Energy, 2020, 45, 9592–9602 CrossRef.
- X. Cui, P. Gao, S. G. Li, C. G. Yang, Z. Y. Liu, H. Wang, L. S. Zhong and Y. H. Sun, ACS Catal., 2019, 9, 3866–3876 CrossRef CAS.
- J. S. Martinez-Espin, M. Mortén, T. V. W. Janssens, S. Svelle, P. Beato and U. Olsbye, Catal. Sci. Technol., 2017, 7, 2700–2716 RSC.
- T. Cordero-Lanzac, A. Ateka, P. Pérez-Uriarte, P. Castaño, A. T. Aguayo and J. Bilbao, Ind. Eng. Chem. Res., 2018, 57, 13689–13702 CrossRef CAS.
- I. Pinilla-herrero, E. Borfecchia, J. Holzinger, U. V. Mentzel, F. Joensen, K. A. Lomachenko, S. Bordiga, C. Lamberti, G. Berlier, U. Olsbye, S. Svelle, J. Skibsted and P. Beato, J. Catal., 2018, 362, 146–163 CrossRef CAS.
- P. Li, I. W. Chen and J. E. Penner-Hahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10063–10073 CrossRef CAS.
- P. Li, I. W. Chen and J. E. Penner-Hahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10082–10089 CrossRef CAS.
- C. J. Howard, R. J. Hill and B. E. Reichert, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 44, 116–120 CrossRef.
- P. Li, I.-W. Chen and J. E. Penner-Hahn, J. Am. Ceram. Soc., 1994, 77, 1281–1288 CrossRef CAS.
- P. Li, I. W. Chen and J. E. Penner-Hahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10074–10081 CrossRef CAS.
- B. W. Veal, A. G. McKale, A. P. Paulikas, S. J. Rothman and L. J. Nowicki, Physica B+C, 1988, 150, 234–240 CrossRef CAS.
- D. Y. Cho, H. S. Jung and C. S. Hwang, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 1–7 CrossRef.
- Y. Liu, C. Xia, Q. Wang, L. Zhang, A. Huang, M. Ke and Z. Song, Catal. Sci. Technol., 2018, 8, 4916–4924 RSC.
- C. Wang, G. Garbarino, L. F. Allard, F. Wilson, G. Busca and M. Flytzani-Stephanopoulos, ACS Catal., 2016, 6, 210–218 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01550d |
| ‡ Authors equally contributed. |
| This journal is © The Royal Society of Chemistry 2021 |