
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure, reactivity, and spectroscopy of nitrogenase-related synthetic and biological clusters
Chen-Hao
Wang
and
Serena
DeBeer
 *
*
Max Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: serena.debeer@cec.mpg.de
First published on 23rd June 2021
Abstract
The reduction of dinitrogen (N2) is essential for its incorporation into nucleic acids and amino acids, which are vital to life on earth. Nitrogenases convert atmospheric dinitrogen to two ammonia molecules (NH3) under ambient conditions. The catalytic active sites of these enzymes (known as FeM-cofactor clusters, where M = Mo, V, Fe) are the sites of N2 binding and activation and have been a source of great interest for chemists for decades. In this review, recent studies on nitrogenase-related synthetic molecular complexes and biological clusters are discussed, with a focus on their reactivity and spectroscopic characterization. The molecular models that are discussed span from simple mononuclear iron complexes to multinuclear iron complexes and heterometallic iron complexes. In addition, recent work on the extracted biological cofactors is discussed. An emphasis is placed on how these studies have contributed towards our understanding of the electronic structure and mechanism of nitrogenases.
 Chen-Hao Wang | Chen-Hao Wang completed his B.Sc. in Chemistry (2012) at National Chung Hsing University, Taiwan and M.Sc. (2014) at National Taiwan University under the supervision of Prof. Ching-Wen Chiu. He earned his Ph.D. (2020) at Texas A&M University, College Station. During his doctoral studies, he carried out research on bimetallic Ru2 lattice-confined chemistry with Prof. David C. Powers. He is currently an Alexander von Humboldt Fellow at the Max Planck Institute for Chemical Energy Conversion in Mülheim an der Ruhr, Germany with Prof. Serena DeBeer. His post-doctoral research focuses on X-ray spectroscopic methods to interrogate reactivity and electronic properties of the FeMo-cofactor-related systems. |
 Serena DeBeer | Serena DeBeer is a Professor and Director at the Max Planck Institute for Chemical Energy Conversion in Mülheim an der Ruhr, Germany. She also holds an Adjunct Professor in the Department of Chemistry and Chemical Biology at Cornell University and an honorary professorship at Ruhr University in Bochum. She received her Ph.D. from Stanford University in 2002. She was a staff scientist at the Stanford Synchrotron Radiation Laboratory from 2002–2009, before moving to a faculty position at Cornell. Research in the DeBeer group focuses on the development and application of advanced X-ray spectroscopic tools for understanding mechanisms in biological, homogeneous and heterogeneous catalysis. |
1. Introduction
Iron sulfur cluster-containing proteins are found in all branches of life and carry out a wide range of essential processes, spanning from electron transfer to DNA repair to catalysis.1,2 The metallocofactors of these proteins range from simple mononuclear iron centers, dinuclear Fe2S2 clusters, tetranuclear Fe4S4 clusters, the complex P-cluster (Fe8S7) and the FeM-cofactors (MFe7S9C, where M = Mo, V or Fe) of the nitrogenase family of enzymes.3,4 Understanding how the varied structural complexity of these enzymes enables their diverse functionality has been a subject of ongoing research for decades.5–11Nitrogenase has, in particular, attracted much attention, due to both the complexity of its cofactors and the remarkable chemistry it enables. Nitrogenase enzymes are able to efficiently reduce N2 to NH3 under ambient conditions using the energy from ATP hydrolysis to drive this process.12,13 Two enzymes are utilized to affect this catalytic conversion, the iron protein, which contains an Fe4S4 cluster and serves as the reductase, and the catalytic protein, known as the MoFe, VFe or FeFe protein in the Mo-, V- and Fe-dependent forms of the enzymes, respectively. Within the catalytic MFe protein, the P-cluster (which can be viewed as two [Fe4S4] cubanes fused with a central μ6-sulfur atom) serves as an electron transfer site providing electrons to the catalytic FeM-cofactor cluster (where M = Mo, V, or Fe). Fig. 1 shows a schematic of the [Fe8S9] P-cluster and the [MoFe7S9C] FeMo-cofactor, with its unusual central μ6-carbide.9,14,15 We note that in FeV-cofactor a μ6-carbide is also present;16 however, a bridging belt sulfide has been replaced by a carbonate.17 For FeFe-cofactor, no structure is available and the exact atomic composition of this cluster awaits structural characterization. Nonetheless, for all three nitrogenase isozymes, the FeM-cofactor cluster is generally agreed to be the site of N2 reduction.4 Interestingly, the V-dependent enzyme has also been shown to perform reductive C–C bond coupling using CO and protons.18–22 While the Mo-dependent enzyme has been investigated for its CO reducing capabilities, it was found to have three orders of magnitude lower activity than the vanadium homologue.20 More recently, Fe-dependent nitrogenase has been shown to enable CO2 methanation.23 Hence, the reactivity spanned by the nitrogenase enzymes are the biological equivalents of the industrial Haber–Bosch, Fischer–Tropsch, and Sabatier processes. The ability of nitrogenases to carry out a diverse range of challenging chemical conversions has further motivated and intensified research on these fascinating enzymes.
 | ||
| Fig. 1 Chemical structures of the P-cluster and the FeM-cofactor active sites of the nitrogenase MFe proteins. | ||
Since the earliest structural studies of MoFe protein in the late 1970's,24,25 nitrogenase has served as a source of inspiration for synthetic chemists. Early studies by Holm26,27 and Coucouvanis28,29 focused on the syntheses of Fe4S4 clusters by self-assembly reaction from FeCl3/NaSH/NaSR. The addition of [MoS4]2− to the reaction mixture afforded the heterometallic [MoFe3S4]3+ single cubane clusters and [Mo2Fe6S8] double cubane clusters. These weak-field Mo–Fe–S clusters were among the first bio-inspired synthetic models of FeMo-cofactor, and interestingly predated the protein crystal structure by more than a decade. At the time, the researchers largely took inspiration from the available extended X-ray absorption fine structure (EXAFS) data on FeMo-cofactor, which provided the first hints for the coordination environment surrounding both the Mo and Fe atoms.25,30 While the crystal structure of FeMo-cofactor, revealed by Rees and coworkers in 1992,31 turned out to be more complex than either the single or double cubane models of Holm and Coucouvanis, it remains the case that these early models are among some of the closest structural models of FeMo-cofactor. The redox chemistry and electronic structure of many of these metal clusters have been studied by a range of spectroscopic methods, including Mössbauer, electron paramagnetic resonance (EPR), X-ray absorption (XAS) and X-ray emission (XES) spectroscopies.10,32–35 However, neither the single or double cubane models are able to replicate the reactivity of nitrogenase enzymes and no dinitrogen adduct has been isolated. Interestingly, however, [MoFe3S4]3+ and [VFe3S4]2+ cubanes have been shown to enable the catalytic conversion of hydrazine to ammonia, acetylene to ethylene and dimethyldiazene to methylamine, by utilizing cobaltocene and lutidinium chloride as the source of electrons and protons, respectively.29 It is noteworthy to mention that most of these reactions cannot be enabled by Fe4S4 clusters, and even in cases where this can be achieved (e.g., for acetylene to ethylene conversion), the Fe4S4 clusters are much slower than the heterometal containing [MoFe3S4]3+ and [VFe3S4]2+ cubanes, suggesting a role of the heterometal in optimizing catalytic activity. This parallels the observation that the Fe-only nitrogenases are by far the least efficient for N2 reduction.4
Nevertheless, in recent years mutation studies, high-level spectroscopy, and protein crystallography all appear to have converged on the iron atoms being the site(s) of substrate binding.36–38 This has shifted much of the attention of synthetic chemists in recent years towards understanding how molecular iron models can ultimately reduce N2. The approach has been in many ways much more reductionist than early nitrogenase-inspired synthetic chemistry. Rather than trying to build a molecule that looks like FeMo-cofactor, researchers have focused more on trying to understand the complex FeMo-cofactor cluster, by analyzing its fundamental building blocks and determining how one can systematically alter the iron electronic structure and ultimately reactivity.6,7,27,39 To this end, researchers have incorporated various light atoms to model the central carbide and have also incorporated sulfur ligation.40–44 They have examined the ability of these clusters to bind nitrogen, form metal hydrides, and reduce a range of N2 and N2-derived related substrates.45,46 Additionally, these models have also been utilized to understand potential pathways for biosynthesis of FeMo-cofactor.47
Herein, we review recent synthetic studies aimed at capturing key aspects of the FeMo-cofactor cluster, which enable its unique reactivity. We first review mononuclear iron model complexes that have been utilized as models for N2 activation. We then discuss multinuclear iron model complexes, heterometallic model complexes and finally the extracted cofactors from the nitrogenase enzymes. In all cases, we place emphasis on the key role that spectroscopy has played in characterizing these complexes and providing key signatures for studies of the enzyme.
2. Studies of mononuclear Fe sites as models for N2 activation
While FeMo-cofactor is a complex 7Fe–1Mo cluster, chemists have found great utility in utilizing simple mononuclear iron complexes in order to test hypotheses as to how FeMo-cofactor might function. In this context, the work of Peters and coworkers has been of particular interest as they have reported the first mononuclear iron complex for catalytic N2 reduction.48 The model complex utilizes a tripodal phosphine–borane ligand (3P:1B), which takes inspiration from the FeMo-cofactor 3S:1C coordination mode of each iron in FeMo-cofactor (Fig. 1). Peters and coworkers designed their model inspired by the idea that the central carbon of FeMo-cofactor might allow for greater conformational and redox flexibility during catalysis by allowing the Fe modulate the degree of interaction it has with the light atom. In their model, the Fe–B interaction acts as the adjustable “spring”, allowing a lower coordinate Fe to effectively bind the substrate.48,49 This strategy allowed N2 coordination followed by catalytic N2 reduction with the presence of the reductant. Reduction of dinitrogen to form ammonia was achieved with the presence of both the proton source and reductant under N2 atmosphere at –78 °C using a mononuclear iron complex.48 The low-valent [(BTpiPr3)Fe–N2]− adduct (compound 1, Fig. 2) could be isolated when the proton source was absent. EPR studies revealed a new rhombic S = 1/2 signal, which could be attributed to [(BTpiPr3)Fe(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH2)]+ or [(BTpiPr3)Fe(NHNH)]+ as possible intermediates during the protonation steps at low temperatures.
N–NH2)]+ or [(BTpiPr3)Fe(NHNH)]+ as possible intermediates during the protonation steps at low temperatures.
To probe the intermediate, the addition of 10 equiv. of HBArF4·2Et2O as the proton source to the low-valent [(BTpiPr3)Fe–N2]− was carried out at −136 °C.50 Analysis of CW X-band EPR at 77 K indicated the consumption of [(BTpiPr3)Fe–N2]− and the formation of a new, more rhombic, S = 1/2 signal. Fe K-edge EXAFS analysis revealed the presence of a short Fe–N bond of 1.64 Å in the frozen sample, which was confirmed by subsequent crystallography and led to the assignment of the doubly-protonated intermediate [(BTpiPr3)Fe(N–NH2)]+. To further investigate the possible formation of other iron species during the protonation step, 57Fe Mössbauer spectra were measured. 57Fe-enriched [(BTpiPr3)Fe–N2]− in diethyl ether was frozen and layered with excess HBArF4·2Et2O. The frozen sample was then annealed to ca. −110 °C and was stirred mechanically. The sample was then analyzed by Mössbauer spectroscopy, which revealed three primary iron-containing species, [(BTpiPr3)Fe–N2]− (S = 1), [(BTpiPr3)Fe(N–NH2)]+ (S = 1/2), and [(BTpiPr3)Fe]+ (S = 3/2). The presence of [(BTpiPr3)Fe–N2]− and [(BTpiPr3)Fe(N–NH2)]+ corresponded to the dinitrogen activation process, while the formation of [(L)Fe]+ suggested the oxidation of [(BTpiPr3)Fe–N2]− by HBArF4·2Et2O. To further support the assignment of a doubly protonated hydrazido (2–) complex, electron nuclear double resonance (ENDOR) spectroscopy was used to determine whether the protons were equivalent. Unlike EXAFS and Mössbauer analysis, which probes all iron-containing species in the sample, ENDOR allows the selective detection of the S = 1/2 species. The ENDOR spectra showed two distinct 1H doublets, which is consistent with an unsymmetrically protonated [–N–NH2] group. The spectroscopic data revealed that the dinitrogen activation for [(SiPiPr3)Fe–N2]− followed a distal-like mechanism since the diazene adduct, which would represent the intermediate formed in an alternating mechanism, was not observed spectroscopically.
The activity of the mononuclear iron catalyst for NH3 formation from N2 could be improved by nearly an order of magnitude by increasing acid/reductant loading. To interrogate the mechanism of N2 activation, 57Fe Mössbauer spectroscopy was utilized to characterize the iron speciation under turnover conditions.51 Mössbauer spectra of freeze-quenched reaction mixtures of [(BTpiPr3)Fe–N2]−, 150 equiv. of HBArF4, and 185 equiv. of KC8 in THF at −78 °C were collected at different time points following the initial mixing. The Mössbauer spectra revealed the presence of a diamagnetic borohyrido–hydrido Fe–N2 complex [(BTpiPr3)(μ-H)Fe(H)(N2)], which was consistent with DFT studies. [(BTpiPr3)(μ-H)Fe(H)(N2)] was the predominant iron-containing species observed by freeze-quench Mössbauer spectroscopy at early time points under turnover conditions. Also, this species was a competent pre-catalyst for dinitrogen activation under acidic and reducing conditions. Under these conditions, [(BTpiPr3)(μ-H)Fe(H)(N2)] was reduced to generate the [(BTpiPr3)Fe–N2]− with the concomitant hydrogen evolution reaction, which led to the postulate that [(BTpiPr3)(μ-H)Fe(H)(N2)] was the resting state of the turnover reaction as H2 evolution was also observed. These studies highlight the important mechanistic insights that can be obtained by performing spectroscopic studies of reaction intermediates.
In addition to boron, ligands with either carbon and silicon atom have also been investigated.52,53 The isolation of the [(BTpiPr3)Fe(N–NH2)]+ intermediate was hampered by its high reactivity and the fact that it could not be isolated in a pure form. The use of the ligand with silicon atom allowed the isolation of [(SiPiPr3)Fe(N–NH2)]+ (compound 2, Fig. 2). Single crystal X-ray diffraction analysis revealed a Fe–N distance of 1.672(2) Å, which could be assigned as a doubly-bonded Fe–N. The vibrational data of the solid [(SiPiPr3)Fe(N–NH2)]+ displayed shifts from 3207 and 3039 cm−1 to 2380 and 2241 cm−1 upon the use of DOTf as the acid. The vibrational peaks at lower energies (2380 and 2241 cm−1) were assigned to N–H and N–D stretching frequencies, respectively, with strong hydrogen bonding interaction with the triflate anion in the solid state. 57Fe-enriched [(SiPiPr3)Fe–N2]− was mixed with [H(OEt2)2][BArF24] in annealing 2-MeTHF at –135 °C and measured in situ by 57Fe Mössbauer spectroscopy. The spectra revealed [(SiPiPr3)Fe(N–NH2)]+ (S = 1/2) as the primary Fe species (∼90%) and [(SiPiPr3)Fe–N2]− (S = 1) as the minor Fe species (∼10%). The f TThe frozen THF solution of [(SiPiPr3)Fe(N–NH2)]+ was treated with stoichiometric Cp*2Co and the resulting mixture was allowed to warm to room temperature over 10 minutes. After filtration, the organic solvents were removed in vacuo, the residue was dissolved in C6D6 for NMR spectroscopy. 1H NMR spectrum revealed the signal for [(SiPiPr3)Fe(–NH2–NH2)]+. After HCl workup, N2H4 and NH3 could be detected and the ratio of the two products was found to be time-dependent with more N2H4 being converted to NH3 over time. These spectral data along with reactivity studies evidenced that the conversion from N2 to NH3 in this system followed a distal-to-alternating mechanism providing precedence for a possible hybrid mechanism in biological systems.
Among these ligands, SiPiPr3, BTpiPr3 and CPiPr3 (see compounds 1, 2, and 3 in Fig. 2), the most flexible axial bonding, BTpiPr(Fe), showed the highest dinitrogen activation reactivity under a common set of reaction conditions. SiPiPr3(Fe) only yielded stoichiometric ammonia and CPiPr3(Fe) fell in between in terms of both flexibility (i.e., the Fe–B bond was more flexible than the Fe–C bond when a nitrene intermediate was formed) and reactivity. With similar ligand sets, Co,54 Ru,55 and Os55 analogues were found to exhibit activity for dinitrogen fixation. In addition to the reactivity studies, these model complexes have provided insight into a wide range of intermediates relevant to nitrogen reduction. Through EPR, Mössbauer, XAS, single-crystal X-ray diffraction, and DFT studies, the expansive studies of Peters and coworkers have been key in establishing potential spectroscopic fingerprints for key intermediates corresponding to both distal and alternating pathways for N2 reduction.
In addition to N2-to-NH3 conversion, reduction of the cyanide to methane and ammonia was studied.56 Paramagnetic [(SiPiPr3)Fe–CN] underwent one-electron reduction by Na(Hg) to afford paramagnetic [(SiPiPr3)Fe–CN]−. EPR analysis showed the solution spin states of S = 1 and 1/2 respectively. [(SiPiPr3)Fe–CN] was readily protonated by [H(OEt2)2][BArF24] to afford the hydrogen isocyanide complex [(SiPiPr3)Fe–CNH]+. The protonated complex had a spin state of S = 1, which complicated the characterization of the acid-derived proton by 1H NMR spectroscopy. Single-crystal X-ray diffraction allowed the analysis of the Fourier difference map in which the proton could be located on the terminal nitrogen of the isocyanide. To investigate the intermediate during the cyanide reduction, [(SiPiPr3)Fe–CN]− was treated with HOTf to afford [(SiPiPr3)Fe–C(NH2)]+, which was isolated and characterized with X-ray diffraction. The intermediate [(SiPiPr3)Fe–C(NH2)]+ suggested a mechanism related to the distal mechanism of N2 reduction. These studies also showed that these mononuclear iron complexes were capable of binding a range of substrates, potentially analogous to the Fe sites of FeMo-cofactor. It is noteworthy to mention that among these intermediates (Fe–NNR, FeN–NR2, Fe–CNR, and Fe![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–NR2, where R = H, Me), the bond dissociation energies for the N–H bonds range from 37 to 65 kcal mol−1.57 The weakening of the N–H bonds can be attributed to the varying degrees of NN or CN bond weakening that occurs concomitant with H-atom addition.
C–NR2, where R = H, Me), the bond dissociation energies for the N–H bonds range from 37 to 65 kcal mol−1.57 The weakening of the N–H bonds can be attributed to the varying degrees of NN or CN bond weakening that occurs concomitant with H-atom addition.
In an effort to more closely capture the iron ligation sphere in FeMo-cofactor, Holland and coworkers synthesized an Fe(II) tris(thiolate) adduct ([L(Fe)–N2]2−, compound 4, Fig. 2) which modelled the Fe–S/C coordination in FeMo-cofactor.58,59 The group focused on the hypothesis that one of the Fe–S bonds dissociates upon reduction/protonation to provide an open coordination at the iron site for substrate binding.37,60 [L(Fe)–N2]2− could be synthesized by reduction of [L(Fe)–SAr*]− (compound 5, Fig. 2) in an N2 atmosphere. Similarly, reduction of [L(Fe)]− in an N2 atmosphere also afforded [L(Fe)–SAr*]−. It is noteworthy to mention that the reduction of [L(Fe)–SAr*]− involves Fe–S dissociation, which is consistent with proposed substrate binding mechanism in FeMo-cofactor iron sites. Single-crystal X-ray diffraction of [L(Fe)–N2]2− revealed Fe–S bond distances of 2.32 and 2.35 Å and a Fe–C bond distance of 2.04 Å, which were in line with averaged Fe–S and Fe–C bond distances (2.26 Å and 2.01 Å respectively) of the resting state of FeMo-cofactor. Superconducting quantum interference device (SQUID) measurements revealed that [L(Fe)–N2]2− had a high-spin (S = 1) electronic configuration in solid state, which was consistent with DFT calculations. Addition of a weak acid to [L(Fe)–N2]2− afforded NH3 and N2H4 in low yields. [L(Fe)–SAr*]− could be reduced by one electron in N2 or Ar without Fe–S dissociation at –70 °C to afford [L(Fe)–SAr*]2−. EPR measurements showed that [L(Fe)–SAr*]2− had two signals with slightly different rhombicities, but both with an S = 3/2 spin state. The reason for the two components in the EPR spectrum was unclear, but it was suggested that two conformers of [L(Fe)–SAr*]2− were present in the frozen sample which could not be resolved at 80 K at zero field. It is noteworthy to mention that EPR spectroscopy is much more sensitive to small perturbations in zero-field splitting than Mössbauer spectroscopy.61–64 Upon warming the sample to room temperature over 15 minutes, a decrease in S = 3/2 signal and an increase in S = 1/2 were observed (the S = 1/2 signal was present at the beginning of the experiment, suggesting small amount of thermal decomposition when [L(Fe)–SAr*]2− was transferred to the EPR tube), which was indicative of the formation of a new species, [L(Fe)]−. Fe K-edge EXAFS spectra of [L(Fe)–SAr*]−, [L(Fe)–SAr*]2−, and [L(Fe)]− were collected and the fits to the experimental data were analyzed. Fe–S bond distances in [L(Fe)–SAr*]2− (2.32 Å) were longer than those in [L(Fe)]− (2.25 and 2.08 Å), suggesting [L(Fe)–SAr*]2− had a high-spin electronic configuration and [L(Fe)]− was low-spin. Reduction of [L(Fe)–SAr*]− by 2.4 equiv. of KC8 at −65 °C under argon atmosphere resulted in Fe–S dissociation to afford [L(Fe)]2−. Single-crystal X-ray diffraction revealed the iron atom in [L(Fe)]2− adopted a η6 coordination to the central arene ring. 57Fe Mössbauer analysis suggested [L(Fe)]2− had an S = 1 spin state as a high-spin Fe(0) complex. Dinitrogen binding was observed by Mössbauer spectroscopy when [L(Fe)]2− (generated in situ from [L(Fe)–SAr*]2− under argon) was exposed to N2 atmosphere to afford [L(Fe)(N2)]2−. Further reduction could be achieved by the treatment of [L(Fe)(N2)]2− with 20 equiv. of a variety of proton sources, affording 1% or less of hydrazine and ammonia with alternating mechanism. It is noteworthy to mention that the catalytic reaction was not achieved due to the protonation of the thiolate ligand followed by the degradation of the complex via ligand dissociation. Holland's model chemistry resembles a single Fe–S–C site of FeMo-cofactor, and the spectroscopic methods provide insightful information in terms of the electronic configuration of the intermediates and the dinitrogen activation mechanism.
2.1 Studies of multinuclear Fe sites as models for N2 activation
In the previous section, we focused on mononuclear iron complexes which mimic certain aspects of FeMo-cofactor's geometric and electronic properties and in some cases, also its reactivity. However, modeling the complexity of FeMo-cofactor with high fidelity clearly requires the synthesis of larger, more complex multinculear clusters, which allow the roles of multiple iron sites, as well as that of the central carbide atom, to be more rigorously evaluated.In contrast to mononuclear iron complexes, Holland and coworkers isolated a dinitrogen complex K2[(L)Fe(N2)Fe(L)] (compound 6, Fig. 3) by reducing (L)FeCl65 (compound 7, Fig. 3) or [(L)FeCl]266 (compound 8, Fig. 3), where L was a diketiminate ligand, in which the R group was varied, as shown in Fig. 3. Single-crystal X-ray diffraction analysis revealed that the N–N bond of dinitrogen was more activated in K2[(L)Fe(N2)Fe(L)] compared to that in [(L)Fe(N2)Fe(L)] (R = t-Bu, N–N bond distances were 1.233(6) and 1.182(5) Å respectively).
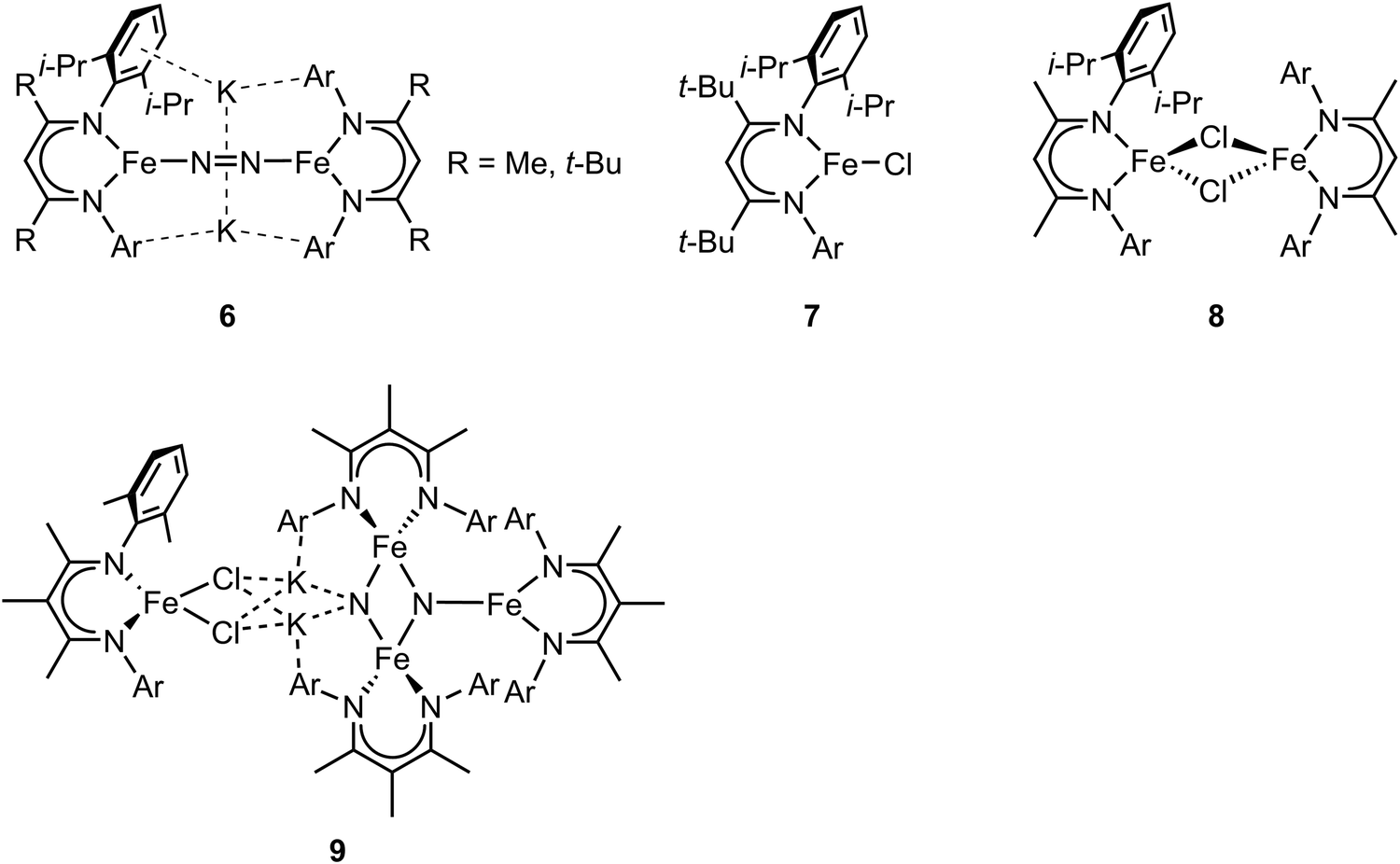 | ||
| Fig. 3 Chemical structures of compound 6–9. Adapted from ref. 65–67. | ||
While the potassium activated dimer clearly showed a longer N–N bond, it could not be completely cleaved in this complex. In order to achieve N–N bond cleavage, the low-coordinate dimeric iron complex was supported by a less bulky ligand.67 Under N2 atmosphere, reduction of [L′FeCl]2 by KC8 afforded an iron tetramer (compound 9, Fig. 3), where the dinitrogen was activated to form a bis-nitride. Single-crystal X-ray diffraction revealed that one of the nitrides was attached to two potassium cations and two iron atoms as the other nitride coordinated to three iron atoms. The zero-field Mössbauer spectrum revealed that the bis-nitrido complex had three different iron environments. The decreased steric hindrance around the iron atoms allowed the three-electron reduction by three iron atoms to break the triply-bonded dinitrogen, evidenced by the Fe3N2 core. The analysis of the isomer shifts indicated the presence of two equivalent high-spin Fe(III) nitrides, a high-spin three coordinate Fe(II) site, and a high-spin four coordinate Fe(II) site. Stoichiometric ammonia formation was observed when the Fe4N2 complex was worked up in acidic conditions. In contrast to the monometallic complexes, in which the formation of the metal nitride requires protonation via a distal mechanism, the end-on bimetallic dinitrogen activation allows the formation of two metal nitrides without protonation, resembling the proposed mechanism in the Haber–Bosch process.68
In addition to the catalytic insights provided by the Holland group's complexes described above, these complexes also served as invaluable references for the development of valence-to-core X-ray emission spectroscopy (VtC XES) as a probe of dintrogen bond actiavtion.69 XES is element specific which utilizes high-energy X-rays to generate a 1s core hole on a metal atom and the photons emitted are measured when the core vacancy is filled by electron in higher lying orbitals.70,71 Since the intensity of XES spectra follows the dipole selection rule (the amount of metal np character present in the donor orbital can be expressed by the oscillator strength), transitions from metal 2p orbitals (Kα emission) and 3p orbitals (Kβ emission) are both possible and give intense features. Moreover, any filled ligand-based valence orbitals that possess appreciable metal p character will also display emission features (Kβ′′ and Kβ2,5 “valence-to-core” (VtC) emission), albeit with relatively low intensity. A series of iron–N2 complexes (compounds 6, 9, [L′FeCl]2, and [(L)Fe(N2)Fe(L)]) were examined by VtC XES to interrogate the degree of dinitrogen activation (Fig. 4). It was found that the energy of the VtC XES feature at ∼7100 eV was proportional to the to the N–N stretching frequency and inversely proportional to the N–N bond length. These experiments showed that VtC XES is able to probe N–N bond length changes as small as 0.02 Å. Due to the ability of XES be measured in a dispersive mode, it also opens up a new spectroscopic means to detect small molecule activation during transition metal catalysis.72
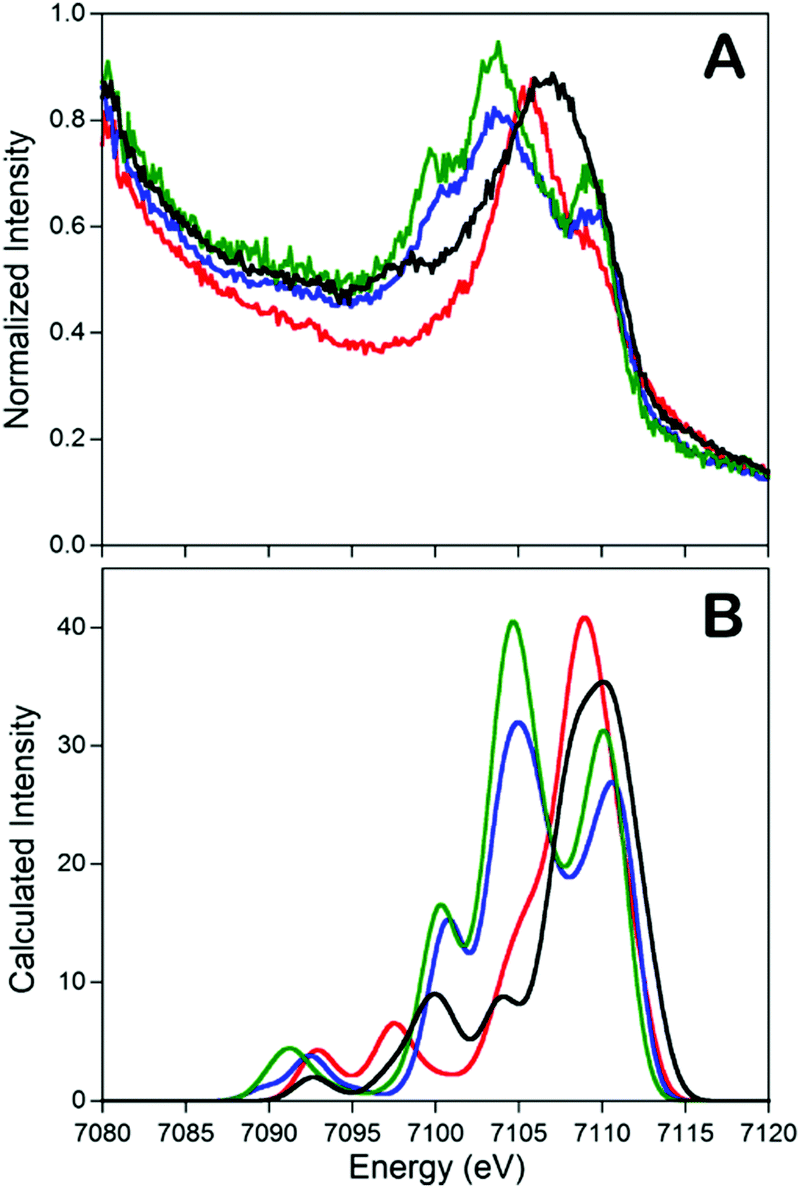 | ||
Fig. 4 Experimental (A) and calculated (B) valence-to-core XES spectra of compounds 6 ( ), 9 (—), [L’FeCl]2 ( ), 9 (—), [L’FeCl]2 ( ), and [(L)Fe(N2)Fe(L)] ( ), and [(L)Fe(N2)Fe(L)] ( ). The feature at ∼7100 eV is a key feature to determine the degree of dinitrogen activation. Reprinted with permission from ref. 69. Copyright (2013) American Chemical Society. ). The feature at ∼7100 eV is a key feature to determine the degree of dinitrogen activation. Reprinted with permission from ref. 69. Copyright (2013) American Chemical Society. | ||
In contrast to end-on Fe–N2 or linear Fe–N2–Fe, examples of non-linear Fe–N2–Fe complexes are rare and less investigated.73,74 Tomson and coworkers utilized a macrocycle as the ligand to construct a constrained environment around the iron atoms and interrogated its impacts on the structural and electronic configuration. The dinucleating macrocycle was used to force the geometry of the bimetallic iron complex which was inaccessible by typical ligands.75 Chemical reduction of [(L)(PR3)Fe(μ-Cl)Fe(PR3)(L)]+ with 2 equiv. of KC8 under N2 atmosphere afforded the dinitrogen complex [(L)(PR3)Fe(μ-N2)Fe(PR3)(L)] (compound 10, Fig. 3). Single-crystal X-ray analysis revealed N–N distance of 1.135(3) (R = Me) and 1.139(4) (R = Ph) Å, indicating a slight degree of N–N bond activation. A Fe–CtN2–Fe angle of ∼151° (CtN2 = the middle point of the N–N bond) was observed due to the constrained environment. DFT calculations revealed that the nonlinear Fe2N2 unit had an additional interaction between Fe d and N2 π* compared to linear Fe2N2 unit, but the mixing was too low to activate dinitrogen significantly. This study opens an avenue for potentially regulating the geometry of the M–N2–M motif by using a bulky ligand to constrain the distance between the iron sites. Further, the importance of the substrate geometry with respect to the active site metals is highlighted in order to achieve optimal dinitrogen activation.
It has been proposed that the sulfides and/or the carbide of the FeMo-cofactor cluster play essential roles in enabling conformational flexibility during catalysis.37,52,58,60,76,77 This could occur through actual Fe–ligand bond breaking processes or simply through a weakening of the Fe–ligand interactions upon substrate binding. The large size of the FeMo-cofactor cluster, may help stabilize these processes, by having the transformation localized at a single iron site. Inspired by this idea, Holland and coworkers isolated a [4Fe–3S]2− cluster (compound 11, Fig. 5) in which three of the irons are four-coordinate and one of the iron atoms is a planar three-coordinate site, ligated by only sulfides78,79 with an average Fe–S bond length of 2.17(1) Å. The EPR measurements of the [4Fe–3S]2− cluster revealed an S = 1/2 ground state from antiferromagnetic coupling between the four paramagnetic iron centers. Zero-field Mössbauer measurements at 80 K of powder samples of [4Fe–3S]2− cluster displayed three quadrupole doublets in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio (Fig. 6). Density functional theory calculations were used to locate the Fe(I) atom. The simulated Mössbauer parameters agreed with the experimental data within 0.10 mm s−1 only when the central iron was assigned as Fe(I). Oxidation of the [4Fe–3S]2− cluster by AgPF6 afforded an all-ferrous [4Fe–3S]− cluster and the calculated ligand-field splitting diagram revealed that the splitting of the [4Fe–3S]− cluster between (dx2−y2, dxy) and (dxz, dyz) of the central iron was relatively large, rationalizing the low-spin configuration. The electronic configuration of [4Fe–3S]− cluster was further probed by Mössbauer measurements, where the spectrum showed a superposition of two doublets in a 3:1 ratio, corresponding to peripheral and central iron site respectively (Fig. 6). A characteristic lower isomer shift of 0.37 mm s−1 attributed to the central Fe(II) of the [4Fe–3S]− cluster fell outside of the reported Fe(II) sites in Fe–S clusters (0.6–0.8 mm s−1),80–83 highlighting its low-spin electronic configuration. EPR measurements showed that the all-ferrous [4Fe–3S]− cluster exhibited a featureless perpendicular mode spectrum. The central iron site of the [4Fe–3S]− cluster exhibited biomimetic reactivity for the N–N reduction of hydrazine and the isolated product was an amide-bound iron–sulfur [4Fe–3S]−–NH2 cluster. Infrared spectrum of [4Fe–3S]−–NH2 cluster exhibited amide N–H stretch. 1H NMR measurements further revealed the signal for the N–H proton, which disappeared when the deuterated analogue was used. Zero-field Mössbauer measurements at 80 K of powder samples of the [4Fe–3S]−–NH2 cluster (containing three Fe(II) and one Fe(III)) displayed two overlapping quadrupole doublets in a 3:1 ratio with isomer shifts of 0.76 and 0.74 mm s−1, respectively (Fig. 7). The observed isomer shifts were inconsistent with the previously reported four-coordinate Fe(III)–S sites (∼0.3 mm s−1). Thus, the hole was delocalized over the peripheral iron sites (an average of 2.33), as the central iron was assigned as high-spin Fe(II). To investigate and compare the oxidation states of the [4Fe–3S]2−, [4Fe–3S]−, and [4Fe–3S]−–NH2 clusters, the first derivative of the Fe K-edge XAS spectra were interrogated and the [4Fe–3S]−–NH2 cluster was found to have the highest average oxidation state, while the [4Fe–3S]2− cluster had the lowest oxidation state in the series (Fig. 8). The shoulder at ∼7115 eV was assigned to 1s to 4pz transition at the central trigonal-planar iron atom, shifting to a higher energy by 0.41 eV upon oxidation of Fe(I)-containing [4Fe–3S]2− cluster to all-ferrous [4Fe–3S]− cluster. These XAS data were consistent with the oxidation of the central iron atoms assigned by the Mössbauer measurements. The presented spectroscopic signatures are important for the identification of unsaturated iron sites, which may be of relevance to intermediate states in FeMo-cofactor. The biomimetic hydrazine reduction also highlights the reactivity of the unsaturated iron site, providing precedence for a related mechanism in the enzyme.
:1:1 ratio (Fig. 6). Density functional theory calculations were used to locate the Fe(I) atom. The simulated Mössbauer parameters agreed with the experimental data within 0.10 mm s−1 only when the central iron was assigned as Fe(I). Oxidation of the [4Fe–3S]2− cluster by AgPF6 afforded an all-ferrous [4Fe–3S]− cluster and the calculated ligand-field splitting diagram revealed that the splitting of the [4Fe–3S]− cluster between (dx2−y2, dxy) and (dxz, dyz) of the central iron was relatively large, rationalizing the low-spin configuration. The electronic configuration of [4Fe–3S]− cluster was further probed by Mössbauer measurements, where the spectrum showed a superposition of two doublets in a 3:1 ratio, corresponding to peripheral and central iron site respectively (Fig. 6). A characteristic lower isomer shift of 0.37 mm s−1 attributed to the central Fe(II) of the [4Fe–3S]− cluster fell outside of the reported Fe(II) sites in Fe–S clusters (0.6–0.8 mm s−1),80–83 highlighting its low-spin electronic configuration. EPR measurements showed that the all-ferrous [4Fe–3S]− cluster exhibited a featureless perpendicular mode spectrum. The central iron site of the [4Fe–3S]− cluster exhibited biomimetic reactivity for the N–N reduction of hydrazine and the isolated product was an amide-bound iron–sulfur [4Fe–3S]−–NH2 cluster. Infrared spectrum of [4Fe–3S]−–NH2 cluster exhibited amide N–H stretch. 1H NMR measurements further revealed the signal for the N–H proton, which disappeared when the deuterated analogue was used. Zero-field Mössbauer measurements at 80 K of powder samples of the [4Fe–3S]−–NH2 cluster (containing three Fe(II) and one Fe(III)) displayed two overlapping quadrupole doublets in a 3:1 ratio with isomer shifts of 0.76 and 0.74 mm s−1, respectively (Fig. 7). The observed isomer shifts were inconsistent with the previously reported four-coordinate Fe(III)–S sites (∼0.3 mm s−1). Thus, the hole was delocalized over the peripheral iron sites (an average of 2.33), as the central iron was assigned as high-spin Fe(II). To investigate and compare the oxidation states of the [4Fe–3S]2−, [4Fe–3S]−, and [4Fe–3S]−–NH2 clusters, the first derivative of the Fe K-edge XAS spectra were interrogated and the [4Fe–3S]−–NH2 cluster was found to have the highest average oxidation state, while the [4Fe–3S]2− cluster had the lowest oxidation state in the series (Fig. 8). The shoulder at ∼7115 eV was assigned to 1s to 4pz transition at the central trigonal-planar iron atom, shifting to a higher energy by 0.41 eV upon oxidation of Fe(I)-containing [4Fe–3S]2− cluster to all-ferrous [4Fe–3S]− cluster. These XAS data were consistent with the oxidation of the central iron atoms assigned by the Mössbauer measurements. The presented spectroscopic signatures are important for the identification of unsaturated iron sites, which may be of relevance to intermediate states in FeMo-cofactor. The biomimetic hydrazine reduction also highlights the reactivity of the unsaturated iron site, providing precedence for a related mechanism in the enzyme.

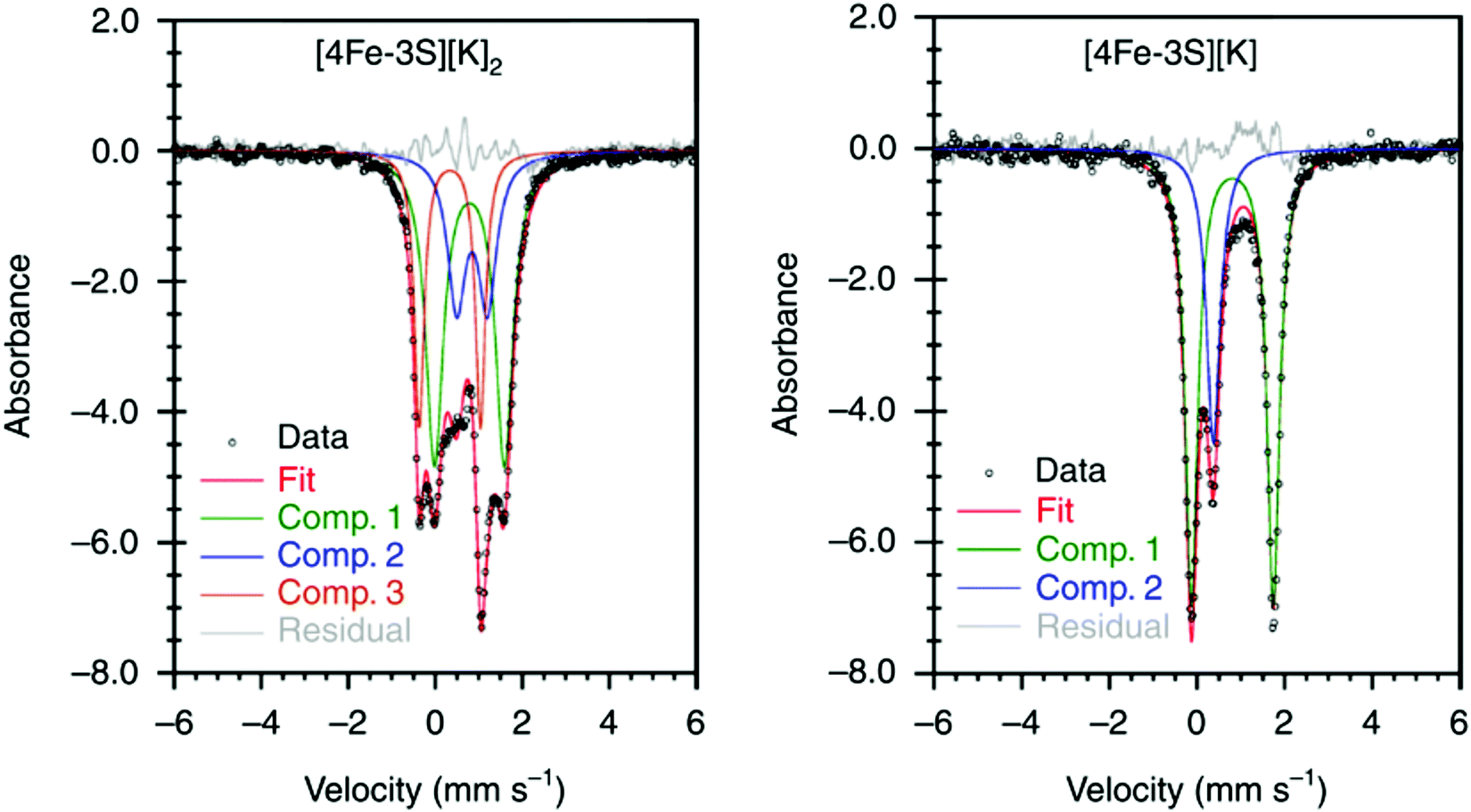 | ||
Fig. 6 Mössbauer spectra of [4Fe–3S][K]2 (left) and [4Fe–3S][K] (right) at 80 K. The spectrum of [4Fe–3S][K]2 shows three components in a 2:1:1 ratio for three high-spin Fe(II) sites and a central Fe(I) site. The minor component (Comp. 1,  ) of the spectrum of [4Fe–3S][K] shows the unusual low-spin Fe(II) site at the center of the cluster. Reprinted and adapted with permission from ref. 78. Copyright (2019) Springer Nature. ) of the spectrum of [4Fe–3S][K] shows the unusual low-spin Fe(II) site at the center of the cluster. Reprinted and adapted with permission from ref. 78. Copyright (2019) Springer Nature. | ||
 | ||
| Fig. 7 Mössbauer spectrum of [4Fe–3S][K]–NH2 at 80 K. Reprinted and adapted with permission from ref. 78. Copyright (2019) Springer Nature. | ||
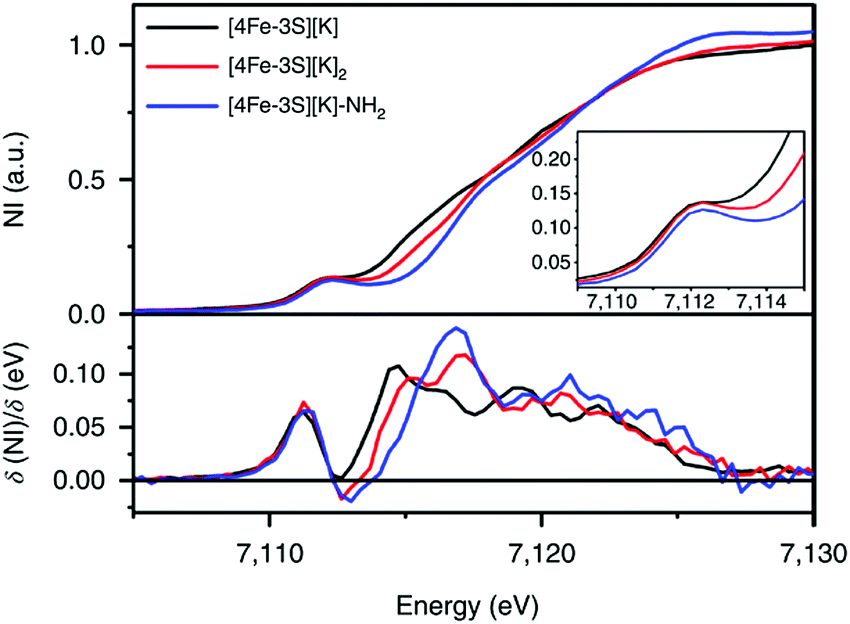 | ||
Fig. 8 Fe K-edge XAS spectra of [4Fe-3S][K] (—), [4Fe-3S][K]2 ( ) and [4Fe–3S][K]–NH2 ( ) and [4Fe–3S][K]–NH2 ( ) (top) and first derivatives (bottom). The inset shows the region between 7109 and 7115 eV. NI, normalized intensity. Reprinted and adapted with permission from ref. 78. Copyright (2019) Springer Nature. ) (top) and first derivatives (bottom). The inset shows the region between 7109 and 7115 eV. NI, normalized intensity. Reprinted and adapted with permission from ref. 78. Copyright (2019) Springer Nature. | ||
The oxidation state of the iron sites are important to substrate binding, as described in the previous text. The exact distribution of oxidation states in FeMo-cofactor has been a longstanding question in nitrogenase research. In recent years, combined Mo/Fe XAS,84,85 Mössbauer,86 X-ray magnetic circular dichroism (XMCD),87,88 and DFT studies,84,89 converged on a 4Fe(II):3Fe(III) oxidation state distribution in FeMo-cofactor in the E0 state; however, it is important to note that these data do not provide insight into how the electrons are spatially distributed. To this end, spatially resolved anomalous dispersion refinement (SpReAD) of MoFe protein in the resting form, has shown that the Fe1/3/7 positions are more reduced than the other four irons.35,90 Crystallographic studies have also shown that CO binds at the Fe2/6 site of FeMo-cofactor, replacing a belt sulfide. This finding is somewhat counterintuitive as the CO binds at what has been shown to be is the more oxidized face of FeMo-cofactor, at least in the resting form. However, it is important to note that no SpReAD data are available on CO-bound FeMo-cofactor, and it is possible that redox reorganization could occur within the cluster. This idea is supported by recent synthetic model studies of Agapie and coworkers in which a series of tetranuclear iron complexes with various oxidation states were synthesized as models to study redox chemistry and concomitant structural change upon CO binding.91 [LFe3O(PhIm)3Fe]n+ (n = 3, 2, 1, 0), corresponding to FeIIFeIII3 through FeII4 (compound 12, Fig. 9), were synthesized. Bond lengths were determined by single-crystal X-ray diffraction and the analysis focused on the Fe4(μ4-O) motif, which could be viewed as an apical iron atom and three basal iron atoms. Bond length analysis showed that upon oxidation from [LFe3O(PhIm)3Fe]2+ to [LFe3O(PhIm)3Fe]3+, a contraction of one of the basal Fe–O (2.092(2) to 1.983(4) Å) was observed, consistent with the oxidation of the basal tri-iron core. In contrast, reduction of [LFe3O(PhIm)3Fe]2+ to [LFe3O(PhIm)3Fe]+ resulted in the elongation of the apical Fe–O (1.813(2) to 1.883(4) Å), consistent with the reduction of the apical Fe. 57Fe Mössbauer spectrum at 80 K of [LFe3O(PhIm)3Fe]2+ revealed four quadrupole doublets, corresponding to four inequivalent iron atoms. The best fit to the Mössbauer spectrum revealed a basal FeII2FeIII core with an apical high-spin FeII. Compared to the spectrum of [LFe3O(PhIm)3Fe]2+, the signal of the basal core FeII of the [LFe3O(PhIm)3Fe]3+ near 3 mm s−1 decreased in intensity, which was consistent with the oxidation of the triiron core. The parameters showed that the apical iron of [LFe3O(PhIm)3Fe]3+ was a high-spin FeIII. Upon the reduction of [LFe3O(PhIm)3Fe]2+ to form [LFe3O(PhIm)3Fe]+, there was no change in the relative intensity of the Lorentzian feature near 3 mm s−1 but an increase in the isomer shift (from 0.19 to 0.89 mm s−1) of the quadrupole doublet, which was assigned to the reduction of the apical iron. These Mössbauer data was consistent with the X-ray crystallographic analysis. To provide further support for the high-spin configuration of the four-coordinate apical iron of [LFe3O(PhIm)3Fe]2+ and [LFe3O(PhIm)3Fe]3+, variable temperature (VT) magnetic susceptibility and variable temperature-variable field (VTVH) magnetization data were obtained to interrogate the nature of the spin ground state and the exchange coupling with a focus on [LFe3O(PhIm)3Fe]2+, which had the shortest apical Fe–O in the series. The fits revealed that the apical iron was FeIII which afforded an S = 4 ground state (the apical FeIII had a strong antiferromagnetic interaction with basal iron atoms, resulting in ferromagnetic alignment of the spins on basal iron sites at low temperatures, S = (−5/2) (apical FeIII) + 5/2 (basal FeIII) + 2 × 2 (two basal FeII) = 4). The reactivity of [LFe3O(PhIm)3Fe]2+ with CO in dichloromethane was invesigated by infrared spectroscopy at 195 K. Infrared spectra revealed a signal at 1944 cm−1 (mono-adduct, [LFe3O(PhIm)3Fe(CO)]2+) and two weaker signals at 2014 and 1980 cm−1 (bis-adduct, [LFe3O(PhIm)3Fe(CO)2]2+). These three signals disappeared upon warming to room temperature, indicating the loss of CO. The zero-field Mössbauer spectrum at 80 K of the frozen CO-saturated solution of [LFe3O(PhIm)3Fe]2+ in 2,6-lutidine revealed a significant loss of basal FeII signal intensity. The fit showed that the basal core changed from [FeII2FeIII] to [FeIIFeIII2] upon binding of CO. The simulated parameters suggested that the ([LFe3O(PhIm)3Fe(CO)n]2+) exhibited an S = 1 bipyramidal FeII–CO complex following internal electron transfer. The redox reorganization in this model chemistry captures features which may key to substrate binding by FeMo-cofactor in various reduced states, as also recently discussed by Henthorn et al. in spectroscopic studies of CO-bound selenated FeMo-cofactor.92
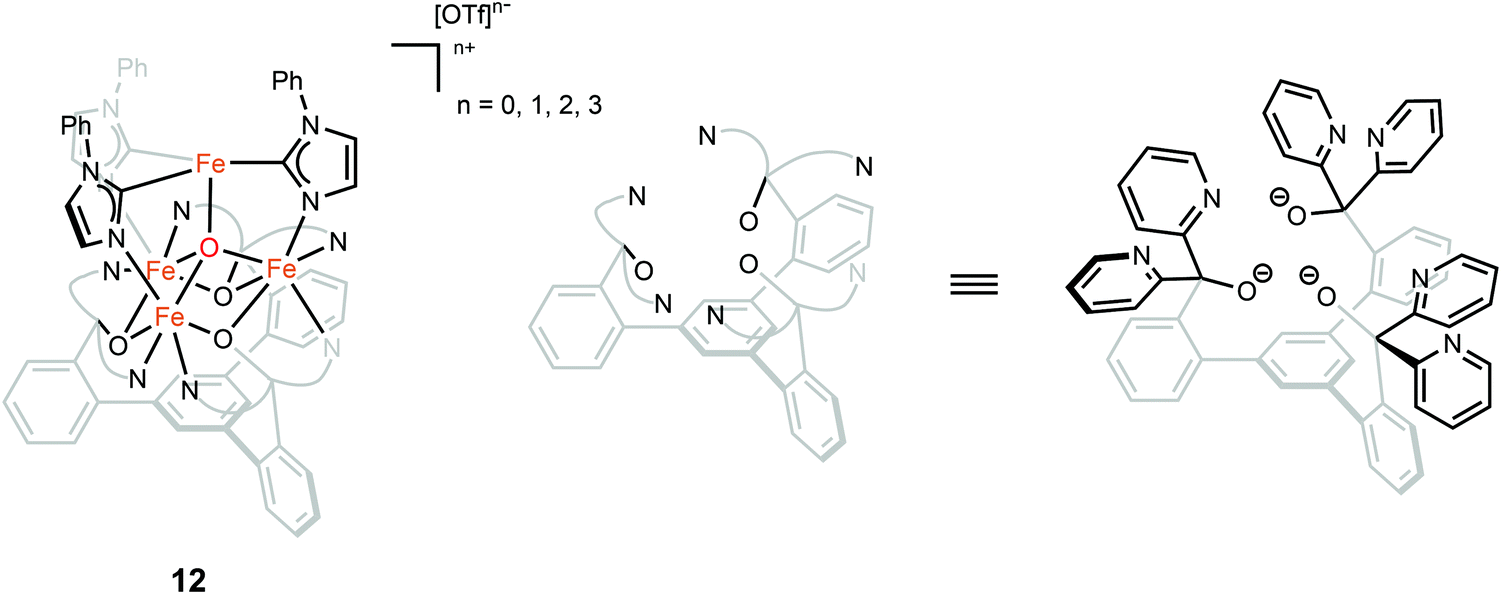 | ||
| Fig. 9 Chemical structure of compound 12. Adapted from ref. 91. | ||
To better understand the intermediates involved in FeMo-cofactor chemistry, the synthesis of models relevant to more reduced E states, which may bind hydrides and/or N2, is essential. Inspired by this, Peters group have interrogated the model chemistry of various bimetallic iron complexes, which show interactions with these important substrates. The low-spin dimeric iron complex, [(L)FeII(μ-H)2FeII(N2)(L)] (compound 13, Fig. 10), was reduced by one electron under N2 atmosphere to afford the mixed-valent [(L)FeI(N2)(μ-H)2FeII(N2)(L)]− as a rare hydrido-bridged bis-dinitrogen complex.93 Cyclic voltammetry (CV) experiments revealed that the complex, [(L)Fe(μ-H)2Fe(N2)(L)]−, a one-electron reduction complex of compound 13, had a 106-enhancement on N2 binding affinity compared to compound 13. Despite its N2 affinity, catalytic ammonia formation from dinitrogen was not achieved.
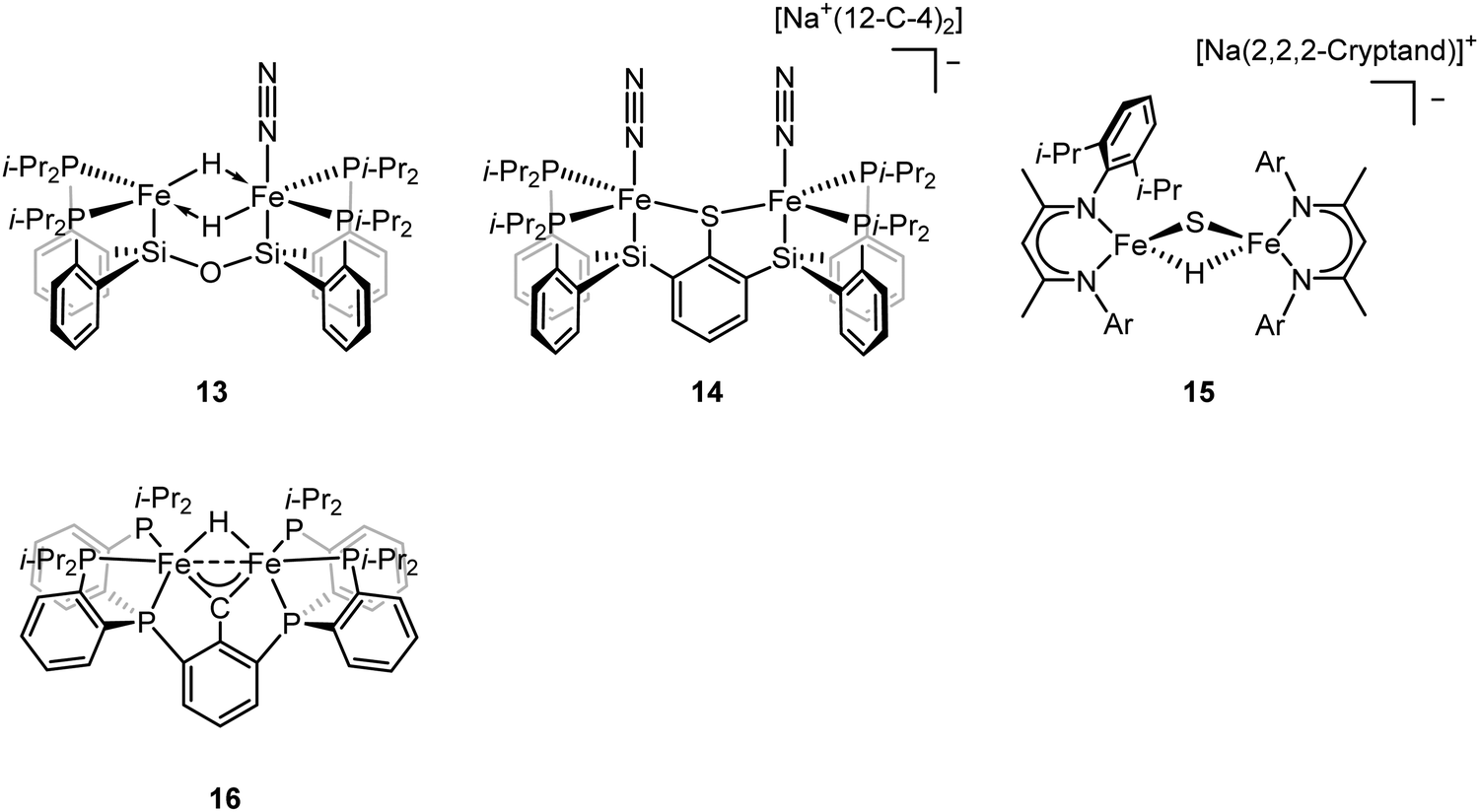
The same group further extended the model chemistry to the Fe–S–Fe scaffold by synthesizing a [(Cl)FeII(μ-SAr)FeII(Cl)]− supported by a binucleating ligand.94 After halide abstraction by one equiv. of NaBPh4 and reduction by excess KC8 under N2 atmosphere, a bis-dinitrogen [(L)(N2)FeI(μ-SAr)FeI(N2)(L)]− complex was isolated (compound 14, Fig. 10). Upon oxidation, [(L)(N2)FeII(μ-SAr)FeI(N2)(L)] and [(L)(N2)FeII(μ-SAr)FeII(N2)(L)]+ were isolated. Infrared spectra revealed the stretching frequency of N–N shifted from 2017 and 1979 cm−1 in [(L)(N2)FeI(μ-SAr)FeI(N2)(L)]−, to 2070 and 1983 cm−1 in [(L)(N2)FeII(μ-SAr)FeI(N2)(L)], and to 2129 cm−1 in [(L)(N2)FeII(μ-SAr)FeII(N2)(L)]+, suggesting the lowest-valent diiron complex had the best ability to activate dinitrogen. Based on the dinitrogen activation trend observed by infrared spectroscopy, [(L)(N2)FeI(μ-SAr)FeI(N2)(L)]− was used as a pre-catalyst for dinitrogen-to-ammonia reaction. Reactivity studies revealed that ammonia formation was observed when excess of reductant and proton source were added to [(L)(N2)FeI(μ-SAr)FeI(N2)(L)]− at −78 °C under N2 atmosphere.
ENDOR and EPR spectroscopy of FeMo-cofactor, freeze-quenched during catalytic turnover with N2, has revealed an N2-bound intermediate with S = 1/2 signal, which was assigned as a substrate-bound intermediate. To understand the nature of this intermediate, it was trapped at low temperature and the frozen sample was allowed to relax to the resting E0 state. Upon cryoannealing, the concentration of [H2] and [N2] was varied and the rate of decay of the trapped intermediate was monitored. ENDOR studies revealed [H2] and [N2] dependence on the EPR signal ascribed to the intermediate. The decay was faster as [H2] was increased, and the decay was slower as [N2] was increased, suggesting the intermediate to be E4(2N2H) (E4(4H) + N2 ⇌ E4(2N2H) + H2).95 The spectroscopic data and the kinetic profile suggested that the E4 state might comprise [Fe(μ-H)(μ-S)Fe] motif. As the formation of the bridging metal hydrides in FeMo-cofactor has been suggested to be an essential part of the mechanism, where reductive elimination of H2 is believed to occur upon N2 binding at the E4 state, Holland and coworkers synthesized a diiron sulfide hydride complex [(L)FeII(μ-H)(μ-S)FeII(L)]− (compound 15, Fig. 10) to interrogate the properties of the metal hydride.96 The reactivity studies revealed that [(L)FeII(μ-H)(μ-S)FeII(L)]− reduced the multiple C–O bond of CO2 to generate [(L)FeII(μ-CHOO)(μ-S)FeII(L)]−, which was further supported by Mössbauer spectroscopy, showing a single quadrupole doublet for the equivalent high-spin Fe(II). Single-crystal X-ray diffraction revealed that the reaction of [(L)FeII(μ-H)(μ-S)FeII(L)]− and m-tolylacetylene resulted in a terminally bound alkynyl complex [(L)FeII(CC–C6H4CH3)(μ-S)FeII(L)]−, suggesting that the hydride was a better base than a nucleophile under the reaction condition. It also suggested that the FeMo-cofactor-bound hydride may inhibit the nucleophilic attack on a terminal alkyne and instead favor the elimination of H2 which has been proposed for the hypothetical N2 binding.8,361H NMR revealed that the reaction of [(L)FeII(μ-H)(μ-S)FeII(L)]− with 2 equiv. of KC8 under N2 atmosphere led to the formation of [(L)FeI(μ-S)FeI(L)]2− and [(L)Fe0(μ-N2)Fe0(L)]2−, which had been characterized previously.66,97 The formation of the formally diiron(0)–N2 complex implied another reduction event occurred during the reaction as the KC8 was only sufficient to reduce [(L)FeII(μ-H)(μ-S)FeII(L)]− to the diiron(I) complex. Gas chromatography of the headspace of the reaction revealed that H2 formed during the reaction, suggesting the reductive elimination was possible, at least under this reaction condition. However, other unidentified species formed during this process so it was not possible to conclude that the reductive elimination was the second reduction event. Nevertheless, it could be inferred that [(L)FeII(μ-H)(μ-S)FeII(L)]− bound N2 and formed H2 thereafter, consistent with reductive elimination of H2 upon N2 binding which is proposed as an essential component in the mechanism of all nitrogenases.
To understand the impact of electronic structure of carbide on substrate binding and activation, synthetic analogues are of interest. Both Rauchfuss and Rose have recently isolated iron carbonyl clusters with central carbides and sulfides as the ligands.98–101 However, these clusters all contain low-valent, low-spin iron and thus are not of direct electronic structural relevance to FeMo-cofactor. While thus far it has not been possible to fuse two iron sulfur cubanes with a carbide (as realized in the biosynthesis of FeMo-cofactor47,102), synthetic chemists have attempted to approach this question in a more stepwise fashion utilizing high-spin iron sites which are electronically analogous to those in FeMo-cofactor and incorporating light atoms into dinuclear and larger clusters. Agapie and coworkers synthesized [Fe(μ-H)(μ-CAr)Fe] as a mimic of the Fe–C–Fe site (compound 16, Fig. 10).1031H NMR spectrum revealed Curie behavior between 198–298 K, indicating an open-shell configuration. Variable-temperature magnetic susceptibility measurements showed that the χT value was nearly constant between 5–298 K, suggesting a thermally isolated S = 1 ground state. It is noteworthy to mention that the iron carbyne complex is low-spin. Reaction between [Fe(μ-H)(μ-CAr)Fe] and equimolar of H2 and N2 led to the slow formation of a diamagnetic species as the single product, which could be formulated as [Fe(H)2(H2)Fe(H)2(H2)] based on 1H NMR, 31P NMR, and single-crystal X-ray diffraction analyses. Exposure of [Fe(H)2(H2)(μ-CAr)Fe(H)2(H2)] to N2 afforded the bis-dinitrogen complex [Fe(H)2(N2)Fe(H)2(N2)]. To understand the mechanism, the independent synthesis to characterize the intermediate was carried out. 1H NMR and single-crystal X-ray diffraction analyses suggested the protonation of the carbyne to form the diiron carbene complex. Further protonation afforded μ-N2 intermediate [Fe(H)(μ-N2)Fe(H)] before the formation of [Fe(H)2(N2)Fe(H)2(N2)]. The reactivity studies revealed that [Fe(μ-H)(μ-CAr)Fe] did not bind to N2, implying the robustness of the Fe(μ-X)2Fe (X = C, H, or S) core. The protonation of the carbyne prior to N2 binding highlighted the carbide flexibility in FeMo-cofactor in terms of N2-binding step. Detailed analysis on the electronic configuration of [Fe(μ-H)(μ-CAr)Fe] was investigated by the same group.104 The oxidation and reduction of [Fe(μ-H)(μ-CAr)Fe] afforded [Fe(μ-H)(μ-CAr)Fe]+ and [Fe(μ-H)(μ-CAr)Fe]− respectively, which could be viewed as En and En+2 states of the nitrogenase. [Fe(μ-H)(μ-CAr)Fe]+ was a stable and isolable complex. EPR analysis revealed that the iron sites were antiferromagnetically coupled, leading to a doublet ground state for the dimer. [Fe(μ-H)(μ-CAr)Fe]− was a reactive species and thus in situ characterization was necessary. Q-band ESE-EPR measurements of [Fe(μ-H)(μ-CAr)Fe]+ and [Fe(μ-H)(μ-CAr)Fe]− suggested a spin state S = 1/2 for each. The EPR signal for [Fe(μ-H)(μ-CAr)Fe]− maintained at −78 °C for 30–60 min. Storing for more than an hour or upon warming above −78 °C led to the loss of its EPR signal. Hyperfine sublevel correlation (HYSCORE) measurements of the 13C-labeled [Fe(μ-H)(μ-CAr)Fe]+ and [Fe(μ-H)(μ-CAr)Fe]− revealed a strong coupling interaction of the bridging carbon atom with the diiron motif. The comparison of the orientation of the g tensor suggested a decrease in Fe–C covalency upon reduction. Analysis on Fe K-edge X-ray absorption together with time dependent DFT calculations revealed that the rising edge energy of the Fe K-edge X-ray absorption shifted from 7118.0 eV for [Fe(μ-H)(μ-CAr)Fe]− to 7118.4 eV for [Fe(μ-H)(μ-CAr)Fe] and to 7119.2 eV for [Fe(μ-H)(μ-CAr)Fe]+, consistent with the more oxidized iron site in each complex. The intensity of the pre-edge at ∼7111 eV increased upon oxidation, indicating a higher probability for Fe 1s → Fe-based 3d transitions as more holes were presented in a more oxidized complex. In contrast, the intensity of the pre-edge at ∼7113–7115 eV decreased upon oxidation, suggesting less Fe 1s → 3d/carbyne transitions. The intensity loss of this feature indicated a higher Fe–C covalency. The isomer shifts measured by 57Fe Mössbauer spectroscopy were used to interrogate the Fe–C covalency. The Mössbauer measurements for these three diiron complexes were nearly identical (0.25 mm s−1 for [Fe(μ-H)(μ-CAr)Fe]−, 0.25 mm s−1 for [Fe(μ-H)(μ-CAr)Fe], and 0.23 mm s−1 for [Fe(μ-H)(μ-CAr)Fe]+). Although the relative degree of the covalency were indiscernible by Mössbauer spectroscopy, they all exhibited a high Fe–C covalency. The spectroscopic data highlight the impact of carbon atom on the electronic configuration in the model chemistry.
The exact role of the carbide in FeMo-cofactor and how it modulates the electronic structure of this cluster relative to other iron sulfur clusters remain open questions.14,15,105 This has motivated efforts to synthetically and spectroscopically investigate the effect of the light atom inclusion in Fe–S clusters. Using a combination of XAS spectroscopy and DFT calculations a series of iron sulfur clusters were investigated.106 These included [Fe2S2Cl4]2−,107 [Fe2S(NtBu)Cl4]2−, [Fe4S4Cl4]2−,108 [Fe4S3(NtBu)Cl4]2−,42,109 [Fe4S2(NtBu)2Cl4]2−,42,109 [Fe4S(NtBu)3Cl4]−,42,109 and [Fe4(NtBu)4Cl4].110 Using ligand K-edge XAS (at both the S and Cl K-edges), the pre-edge features were examined to understand the modulation in metal ligand covalency as a function of light atom introduction into the Fe–S clusters. While strong modulations in the electronic structure were observed for the dimers, the tetramers showed only subtle perturbations upon inclusion of a single ligand (−NtBu). The studies were then extended to hypothetical clusters, and it was suggested that strong electron donors, such as a carbide, were required to modulate the electronic configurations of tetramers or larger clusters. On this basis, it was hypothesized that the carbide might play not only a structural role in FeMo-cofactor, but also modulated the electronic structure to enable optimal reactivity.
The biosynthesis of the FeM-cofactors of nitrogenase involves modification of iron–sulfur (Fe–S) precursors which are enabled by the activity of radical S-adenosylmethionine (SAM) enzymes. The central carbide originates from the transfer of a methyl group from SAM to a precursor [Fe4S4] cluster.32,33,47,111 This suggests that [Fe4S4]–alkyl clusters are important intermediates in the formation of the FeM-cofactors, as well as in other radical reactions mediated by SAM enzymes.112–114 However, the role of the alkyl has not been studied extensively in model chemistry. To understand how an electron-donating and strong-field ligand (i.e., alkyl) perturbs the electronic configuration of the [Fe4S4] clusters, Suess and coworkers isolated the [Fe4S4]2+–Et cluster (compound 17, Fig. 11) by adding Et2Zn to the [Fe4S4]2+–Cl cluster.115–117 Single-crystal X-ray diffraction revealed the Fe–C bond length in Fe–Et was 2.05 Å, which was comparable to that in a tris(thioether)borate-ligated Fe2+–Me complex (2.03 Å).11857Fe Mössbauer spectra of [Fe4S4]2+–Cl and [Fe4S4]2+–Et clusters at 90 K and the calculations revealed that the positive charge was more localized on the apical iron site in the [Fe4S4]2+–Et cluster due to the electron-donating ethyl group on the apical site. Reversible formation of alkyl radicals was reported by Suess and coworkers by using the electron-donating ligands to promote homolysis of [Fe4S4]2+–alkyl cluster (compound 18, Fig. 11).119 The mechanistic studies revealed that the homolysis rate was dependent on the substituent of the ligand; homolysis rate was increased when a more electron-donating ligand was used (i.e., reaction with DMAP had a higher rate than that with pyridine), forming the R–R coupled hydrocarbons as the final products. This facile process even generated primary octyl radicals at room temperature. To further understand the possible intermediates related to the alkylated clusters, the synthesis of [Fe4S4]3+–Et cluster was attempted. However, one-electron oxidation did not afford target compound. Instead, a tris(imidazolinimine)borate ligand was used and the [Fe4S4]3+–Me cluster (compound 19, Fig. 11) was synthesized by one-electron oxidation of the [Fe4S4]2+–Me cluster.120 EPR spectrum of [Fe4S4]3+–Me cluster revealed an S = 1/2 ground state with giso > 2. 57Fe Mössbauer spectra and calculations revealed that the apical iron site in the [Fe4S4]3+–Me cluster exhibited charge localization due to the methyl group, consistent with the observed data for the [Fe4S4]2+–Et cluster, which reflected the covalent Fe–C had impacts on the electronic configuration of the [Fe4S4] clusters.
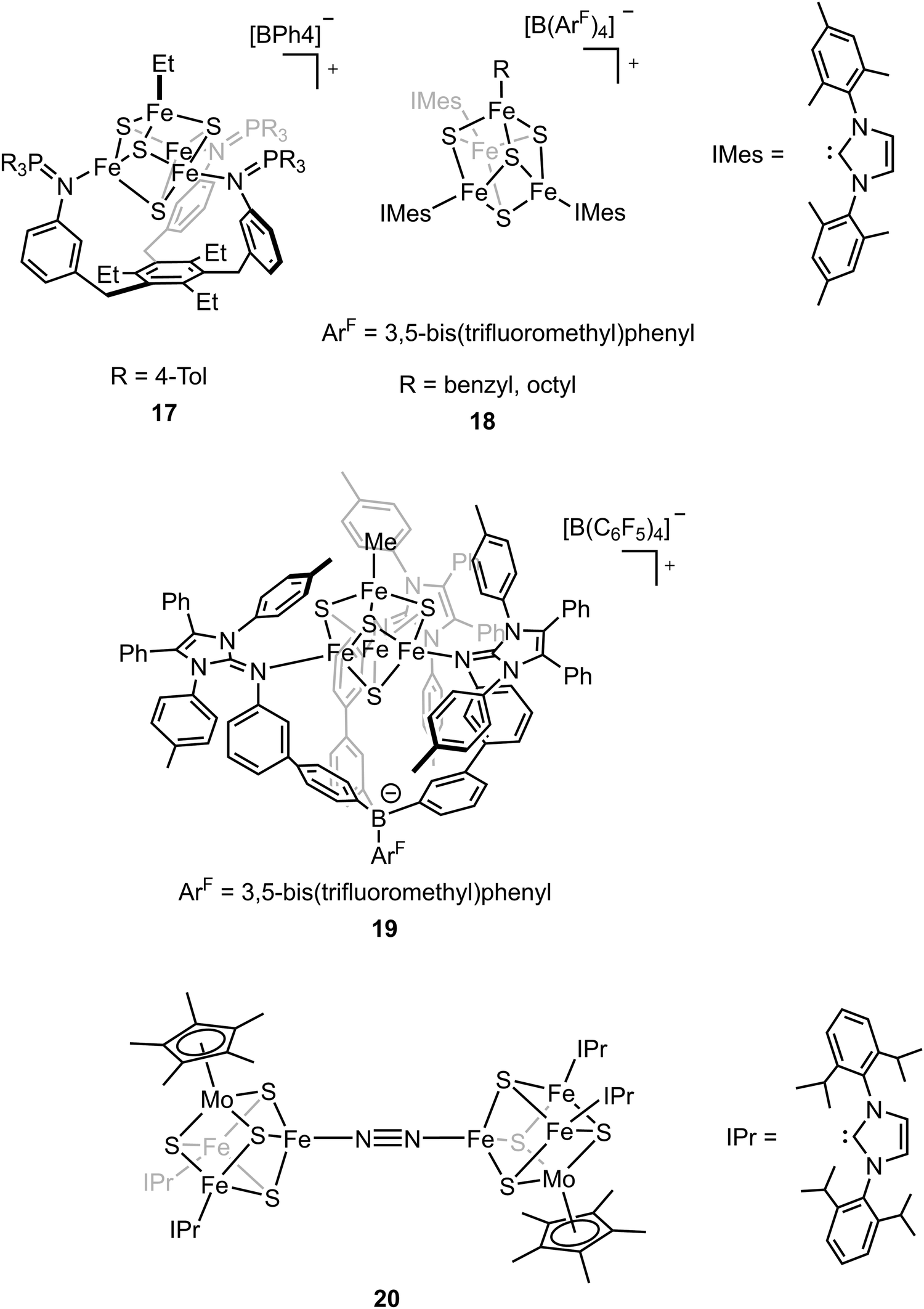
The belt sulfide (S2B) has been proposed as a potential leaving group to generate a low-coordinate iron site for N2 binding. Efforts have been made in cluster synthetic chemistry (e.g., the reduction of Fe–Cl to generate a low-valent Fe site) but the unwanted side reactions (e.g., dimerization of two [MoFe3S4] clusters) hamper the N2 binding as the iron site is not protected by the protein. The same group recently utilized the sterically demanding N-heterocyclic carbenes (NHCs) as the protective ligands to outcompete the side reaction and the reduced clusters bound N2 to form the bridging complex (compound 20, Fig. 11).121 Single-crystal X-ray diffraction studies revealed that the N–N bond distance (1.145(3) Å) was elongated in the bridging complex. Mössbauer spectra as well as the bond length analysis revealed the increasing cluster covalency upon N2 binding. Interestingly, the average Fe–S distances for all the iron sites decreased, suggesting the contribution of all the iron sites for N2 binding. The findings illustrate that the covalent interactions are important for cluster N2 binding chemistry.
2.2 Studies of heterometallic sites as models for N2 activation.
In the previous section, we focused on the recent iron model chemistry aimed at understanding FeMo-cofactor. However, as FeMo-cofactor incorporates Mo into the cluster, the potential role of the heterometal in modulating the electronic structure and reactivity remain a question of ongoing interest. As discussed in the introduction, there has been a longstanding interest in the synthesis of heterometallic clusters to model FeMo-cofactor in order to capture its unique electronic and structural properties, beginning with the early work of Holm and Coucouvanis. As some of the synthetic models capture some aspects of FeMo-cofactor, none of them fully resemble FeMo-cofactor in terms of chemical structure and reactivity on dinitrogen reduction. In the following section, recent progress on this field will be discussed.To systematically synthesize various heterometallic clusters, ligand metathesis was utilized to incorporate Mo and W into clusters by Holm and coworkers.44 This approach afforded versatile heterometallic complete and incomplete cubanes which captured many of the key geometric and electronic features of the active sites in the FeMo-cofactor (Fig. 12). Although reactivity of these clusters was lacking, they have provided key structural and spectroscopic reference points for understanding FeMo-cofactor. Of particular interest is the fact that both FeMo-cofactor and the MoFe3 cubanes produced by Holm and coworkers have been shown to contain a Mo(III) atom in an unusual non-Hund configuration. This was initially suggested by DFT studies84 and most recently received experimental support from Mo L-edge XMCD studies.88
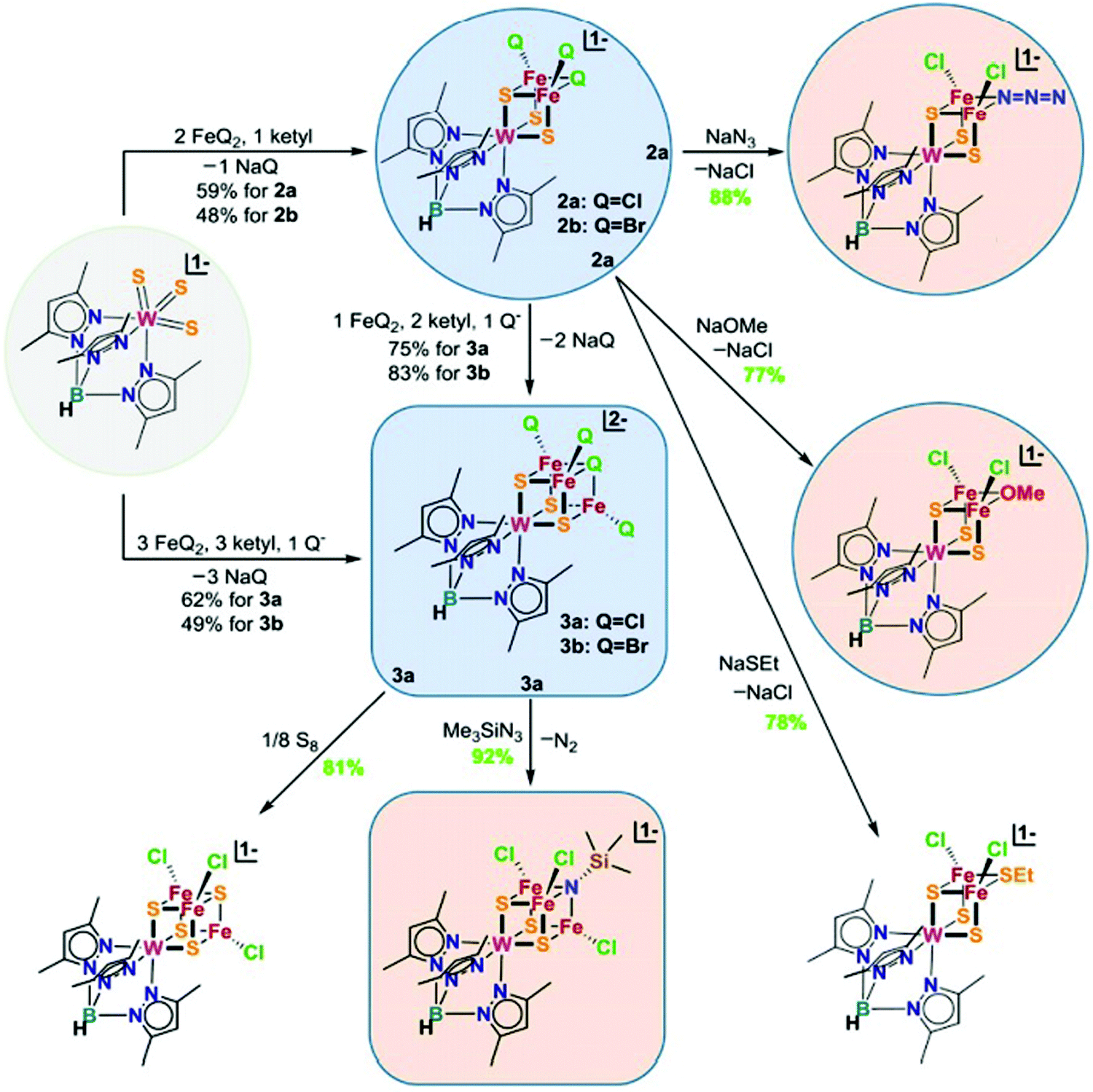 | ||
| Fig. 12 Chemical structures of heterometallic cubanes synthesized by ligand metathesis. Q = halides. Reprinted and adapted with permission from ref. 44. Copyright (2018) National Academy of Sciences. | ||
To synthesize the heterometallic cluster [Mo3S4M], a synthetic route has been proposed which involves the reaction between the incomplete cluster [Mo3S4] (e.g., [Cp*Mo3S4]+) and [MLn].122–124 However this strategy only applies to Groups 8–10 metals and Group 6 carbonyls. Ohki and coworkers proposed a new protocol which utilized a reduced incomplete cluster [Mo3S4] (e.g., [Cp*Mo3S4]−) and early transition metals (e.g., TiCl3(THF)3) to afford heterometallic [Cp*3Mo3S4Ti]125 and this strategy allowed the synthesis of versatile heterometallic single-cubane clusters [Cp*3Mo3S4M] (M = Ti, Mn, Fe, Co, Ni) (Fig. 13).126 Although the afforded heterometallic clusters do not capture the properties of FeMo-cofactor, it serves as a useful tool to broaden the scope of the synthetic heterometallic clusters.
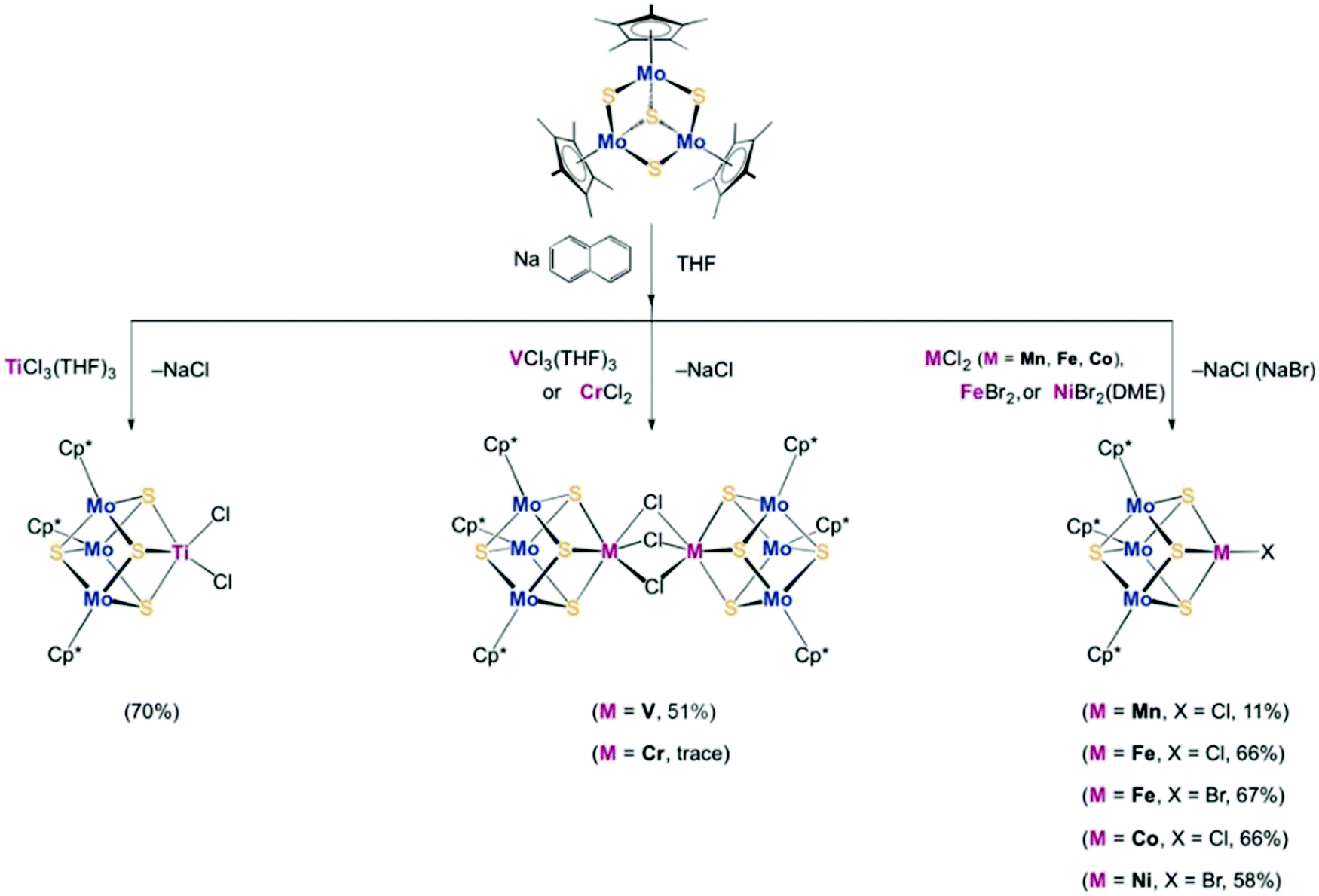 | ||
| Fig. 13 [Cp*3Mo3S4M] (M = Ti, Mn, Fe, Co, Ni) single-cubane as a starting material to synthesize heterometallic clusters. Reprinted and adapted with permission from ref. 126. Copyright (2018) Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
While many synthetic model complexes related to FeMo-cofactor have been synthesized and characterized, none of them catalyze dinitrogen activation to form ammonia under ambient conditions. In contrast to reducing N2 under the strong reducing conditions employed in much of the synthetic chemistry described above, photochemical ammonia production has been realized by using ultraviolet light in semiconductor films (e.g., titania,127–129 diamond,130,131 and fullerenes132). Recently, light-driven reduction of dinitrogen to form ammonia in aqueous solution under ambient temperatures and pressures was reported by Kanatzidis and coworkers using the solid-state chalcogels composed of [Mo2Fe6S8(SPh)3]3+ and [Sn2S6]4− clusters (Fig. 14).133 The immobilized FeMoS active sites were proposed to feature a high spatial density proximal to other active sites, allowing efficient multielectron transformations during chemical events. In addition, Mo-free chalcogels were investigated which contained only Fe4S4 clusters.134 Interestingly, the Mo-free variants showed even higher N2 reduction efficiency, suggesting that Mo is not necessary for optimal catalysis, at least in the tested photochemical conversion.
 | ||
| Fig. 14 Representation of Mo2Fe6S8–Sn2S6 chalcogel. Blue: Mo; Red: S; yellow: S; black: Sn. Reprinted with permission from ref. 133. Copyright (2015) American Chemical Society. | ||
3. Reactivity of extracted FeMo-cofactor
In the previous sections, we have discussed various synthetic models to capture the electronic properties, structural properties, and reactivity of the FeMo-cofactor cluster of nitrogenase. Despite all the attempts, a cluster that fully models FeMo-cofactor has yet to be realized. To further interrogate the chemical properties of nitrogenase, isolated FeMo-cofactor, FeV-cofactor and FeFe-cofactor have been extracted by strongly coordinating organic solvents (e.g., N-methylformamide (NMF)) from MoFe protein.11,135,136 It is noteworthy to mention that extracted cofactors have yet to be structurally characterized by single-crystal X-ray diffraction and our understanding of the cofactors geometric and electronic structures have relied heavily on spectroscopy. Among the three extracted cofactors, extracted FeFe-cofactor has been characterized by EXAFS and 57Fe Mössbauer spectroscopy.137 Its EPR-silent nature in the resting state limits further characterization of its ground state electronic properties. Extracted FeV-cofactor, in contrast, has been characterized more recently by both EPR and EXAFS, however, a more detailed characterization has yet to be realized.138 Extracted FeMo-cofactor is by far the most studied and we would like to briefly review its characterization in this section. Extracted FeMo-cofactor has been characterized by EXAFS139 and EPR studies on extracted FeMo-cofactor reveal a broadened S = 3/2 X-band spectrum, which has been attributed to the increased D-strain in the extracted cofactor relative to that contained within wild-type MoFe protein.138,140 It has been proposed that slight geometric variations change the distribution of D-strain, and the change will be more pronounced in the solvated form.25,141 The underlying structural rigidity in MoFe protein may play a significant role in terms of reactivity. Here it is of interest to note that small-angle X-ray scattering (SAXS) results have suggested extracted FeMo-cofactor is not monomeric in DMF, but rather forms oligomers in DMF and the addition of coordinating thiols does not change the aggregation behavior in DMF.142 However, as FeMo-cofactor can be reinserted into apo MoFe protein and recover its reactivity, it appears that the structural integrity should be reasonably retained or at least recovered upon incorporation into MoFe protein.12,135Hu, Ribbe and coworkers studied the activity of extracted FeMo-cofactor and FeV-cofactor on CO and CN− reduction with Eu(II) as the reductant.143 The reactivity studies revealed that both of the extracted cofactors reduced CO and CN− to hydrocarbons with FeMo-cofactor exhibiting slightly better activity. It is noteworthy that there were discrepancies in the CO reduction reactivity between the solvent-extracted/Eu(II)-driven and protein-bound/ATP-driven reactions. The total amount of hydrocarbons formed by the extracted FeMo-cofactor and FeV-cofactor were 67.9% and 0.05% of the totals produced by the protein-bound FeMo-cofactor and FeV-cofactor respectively.20,143 The drastic decrease in extracted FeV-cofactor's CO reduction activity rendered extracted FeMo-cofactor, which was only 0.1% as active as FeV-cofactor within proteins, slightly more active than extracted FeV-cofactor, highlighting the significance of the protein environment during chemical reactions in nature. The same group revealed that NMF-extracted FeMo-cofactor, FeV-cofactor, and FeFe-cofactor were capable of reducing CO, CN−, and CO2 to form hydrocarbons with SmI2 as the reductant.144 Unfortunately, while hydrocarbon formation by reduction of carbon monoxide or cyanide has been reported, extracted FeMo-cofactor, FeV-cofactor and FeFe-cofactor exhibit no N2 reduction reactivity. It should also be noted that all the above-mentioned CN−, CO, and CO2 reductions are sub-stoichiometric and efficient catalytic reduction of these substrates has yet to be achieved.
To understand the impact of the protein environment on the reactivity, synthetic clusters have been inserted into proteins in order to understand the contribution of the protein environment. In this context, it is of interest to note this strategy has been very successful for hydrogenases but thus far limited for nitrogenases.145–152 Hu, Ribbe and coworkers inserted a synthetic iron cluster, [Fe6S9(SEt)2]4−, into the catalytic component of the nitrogenase (designated as NifDKapo) with this biohybrid designated as NifDKFe.149 The reactivity studies revealed that C2H4 and C2H2 reduction were achieved when NifDKFe was used in the presence of Eu(II). [Fe6S9(SEt)2]4− and NifDKapo exhibited no reactivity, highlighting the importance of both the metal cofactor and the protein environment in terms of reactivity. An extracted cofactor is possibly reinserted into another protein as a “hybrid protein” but detailed experiments and characterization have yet to be realized.111,153 We note, however, that there are many examples in which synthetic complexes have been inserted into proteins (i.e., artificial metalloenzymes) or synthetic scaffolds (e.g., zeolites), and the environment around the complexes has been shown to play an important role in tuning the chemo-, regio-, site-, and enantioselectivity during catalysis.154–159
High-resolution single-crystal X-ray diffraction has revealed that carbon monoxide, a molecule that inhibits FeMo-cofactor, replaces the belt sulfur atom at 2B position (S2B) in FeMo-cofactor.37 The lability of the belt sulfide in FeMo-cofactor inspired the idea to substitute S2B with Se due to its spectroscopic properties by the addition of KSeCN under proton-reducing turnover conditions.60 Selective substitution of the bridging sulfurs by selenium in FeMo-cofactor served as a tool in which the Fe–Se environment could be interrogated by utilizing high-energy resolution fluorescence detected X-ray absorption spectroscopy (HERFD XAS). Our group, together with the Rees group, utilized Se HERFD XAS to reveal that 2B and 3A/5A bridging positions of Se-substituted FeMo-cofactor were electronically distinct. The Fe2/Fe6 edge consistent with an antiferromagnetically coupled diferric pair, while the Fe3/Fe4/Fe5/Fe7 face of the cofactor exhibited more localized ferrous character. These differences were attributed to asymmetry in the electrostatic and hydrogen bonding interactions with the belt sulfides, suggesting a more localized electronic configuration. Measurements of the extracted selenated cofactor would be an ideal way to test this hypothesis, however, thus far has not been possible. Finally, it is also of interest to note that Se HERFD XAS studies of CO-bound selenated FeMo-cofactor, show that the Fe3/Fe4/Fe5/Fe7 face of the CO-bound cofactor appears more oxidized from the perspective of the coordinated Se ligands than the resting state. This is counterintuitive, since the cluster has to be reduced to bind CO. This observation provides support for redox organization occurring within FeMo-cofactor in order to facilitate substrate binding. Interestingly, the potential importance of redox reorganization in FeMo-cofactor was first hypothesized based on model chemistry.91 Thus, the subsequent discovery of its relevance to FeMo-cofactor further highlights the synergy that exists between the synthetic and enzymatic systems.
4. Summary and outlook
In this review, we have discussed various synthetic models that have been used to understand the mechanism, potential intermediates, reactivity, and electronic properties of nitrogenases. Studies of these molecular models have provided key spectroscopic fingerprints which have been, and will continue to be, essential in correlated studies with the enzyme. These molecular model studies have also shown that a single iron site is capable of catalytic N2 reduction,48,51–53 albeit only at low temperature and under harsh reducing conditions. Interestingly, dinuclear and multinuclear iron model complexes have been shown to activate, and in one case even cleave, the strong triple bond of N2.67,75,93 However, catalytic N2 reduction has only been observed in multinuclear clusters through photochemical initiation and in contrast to the enzyme, Mo has been found to decrease, rather than increase, activity.133,134 Hence, it is clear that a significant gap remains between synthetic modelling and the actual FeM-cofactor clusters.Interestingly, while the FeM-cofactor clusters can be extracted into organic solvent, research in this area remains limited due to the unstable nature of these clusters outside the protein environment.20,143 It is noteworthy to mention that the extracted FeMo-cofactor can be reinserted to its protein and the reactivity is restored. This indicates that the protein environment plays a prominent role in optimizing the active site for catalysis. In the field of nitrogenase research, it appears there remains ample room to further explore protein engineering and artificial metalloenzyme approaches, which may allow for the present gaps between synthetic modelling and the biological systems to be bridged. While great advances have been made in our understanding of nitrogenase, with substantial contributions from synthetic chemistry, it is clear there remains much to explore in molecular, biological, and biohybrid systems.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge the Max Planck Society for funding. C.-H. W. acknowledges funding from the Alexander von Humboldt Foundation. This work is also supported by the DFG project DE 1877/1-2 (S. D.) within the SPP 1927 “Iron–Sulfur for Life”. Open Access funding provided by the Max Planck Society.References
- H. Beinert, J. Biol. Inorg. Chem., 2000, 5, 2–15 CrossRef CAS PubMed.
- R. Lill and U. Mühlenhoff, Annu. Rev. Biochem., 2008, 77, 669–700 CrossRef CAS PubMed.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- R. R. Eady, Chem. Rev., 1996, 96, 3013–3030 CrossRef CAS PubMed.
- R. H. Holm, Chem. Soc. Rev., 1981, 10, 455–490 RSC.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- S. C. Lee and R. H. Holm, Chem. Rev., 2004, 104, 1135–1158 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- C. Van Stappen, L. Decamps, G. E. Cutsail, R. Bjornsson, J. T. Henthorn, J. A. Birrell and S. DeBeer, Chem. Rev., 2020, 120, 5005–5081 CrossRef CAS PubMed.
- H. L. Rutledge and F. A. Tezcan, Chem. Rev., 2020, 120, 5158–5193 CrossRef CAS PubMed.
- J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS.
- B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385–402 CrossRef CAS PubMed.
- K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974–977 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed.
- J. A. Rees, R. Bjornsson, J. Schlesier, D. Sippel, O. Einsle and S. DeBeer, Angew. Chem., Int. Ed., 2015, 54, 13249–13252 CrossRef CAS PubMed.
- D. Sippel and O. Einsle, Nat. Chem. Biol., 2017, 13, 956–960 CrossRef CAS PubMed.
- C. C. Lee, Y. Hu and M. W. Ribbe, Science, 2010, 329, 642 CrossRef CAS PubMed.
- Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, J. Biol. Chem., 2011, 286, 19417–19421 CrossRef CAS.
- Y. Hu, C. C. Lee and M. W. Ribbe, Science, 2011, 333, 753–755 CrossRef CAS PubMed.
- L. C. Seefeldt, Z.-Y. Yang, S. Duval and D. R. Dean, Biochim. Biophys. Acta. Bioenerg., 2013, 1827, 1102–1111 CrossRef CAS PubMed.
- C. C. Lee, A. W. Fay, T.-C. Weng, C. M. Krest, B. Hedman, K. O. Hodgson, Y. Hu and M. W. Ribbe, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13845–13849 CrossRef CAS PubMed.
- Y. Zheng, D. F. Harris, Z. Yu, Y. Fu, S. Poudel, R. N. Ledbetter, K. R. Fixen, Z.-Y. Yang, E. S. Boyd, M. E. Lidstrom, L. C. Seefeldt and C. S. Harwood, Nat. Microbiol., 2018, 3, 281–286 CrossRef CAS PubMed.
- S. P. Cramer, K. O. Hodgson, W. O. Gillum and L. E. Mortenson, J. Am. Chem. Soc., 1978, 100, 3398–3407 CrossRef CAS.
- S. P. Cramer, W. O. Gillum, K. O. Hodgson, L. E. Mortenson, E. I. Stiefel, J. R. Chisnell, W. J. Brill and V. K. Shah, J. Am. Chem. Soc., 1978, 100, 3814–3819 CrossRef CAS.
- R. H. Holm, Acc. Chem. Res., 1977, 10, 427–434 CrossRef CAS.
- S. C. Lee, W. Lo and R. H. Holm, Chem. Rev., 2014, 114, 3579–3600 CrossRef CAS.
- D. Coucouvanis, Acc. Chem. Res., 1981, 14, 201–209 CrossRef CAS.
- S. M. Malinak and D. Coucouvanis, Prog. Inorg. Chem., 2001, 599–662, DOI:10.1002/9780470166512.ch6.
- A. M. Flank, M. Weininger, L. E. Mortenson and S. P. Cramer, J. Am. Chem. Soc., 1986, 108, 1049–1055 CrossRef CAS.
- M. Georgiadis, H. Komiya, P. Chakrabarti, D. Woo, J. Kornuc and D. Rees, Science, 1992, 257, 1653–1659 CrossRef CAS PubMed.
- R. Cammack, in Adv. Inorg. Chem., ed. R. Cammack, Academic Press, 1992, vol. 38, pp. 281–322 Search PubMed.
- H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653–659 CrossRef CAS.
- N. S. Lees, R. L. McNaughton, W. V. Gregory, P. L. Holland and B. M. Hoffman, J. Am. Chem. Soc., 2008, 130, 546–555 CrossRef CAS.
- T. Spatzal, J. Schlesier, E.-M. Burger, D. Sippel, L. Zhang, S. L. A. Andrade, D. C. Rees and O. Einsle, Nat. Commun., 2016, 7, 10902 CrossRef CAS.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2013, 46, 587–595 CrossRef CAS PubMed.
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed.
- T. M. Buscagan and D. C. Rees, Joule, 2019, 3, 2662–2678 CrossRef CAS PubMed.
- I. Čorić and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 7200–7211 CrossRef PubMed.
- Y. Ohki, Y. Sunada, M. Honda, M. Katada and K. Tatsumi, J. Am. Chem. Soc., 2003, 125, 4052–4053 CrossRef CAS PubMed.
- Y. Ohki, Y. Ikagawa and K. Tatsumi, J. Am. Chem. Soc., 2007, 129, 10457–10465 CrossRef CAS PubMed.
- X.-D. Chen, J. S. Duncan, A. K. Verma and S. C. Lee, J. Am. Chem. Soc., 2010, 132, 15884–15886 CrossRef CAS PubMed.
- S. Ohta, Y. Ohki, T. Hashimoto, R. E. Cramer and K. Tatsumi, Inorg. Chem., 2012, 51, 11217–11219 CrossRef CAS PubMed.
- G. Xu, Z. Wang, R. Ling, J. Zhou, X.-D. Chen and R. H. Holm, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5089–5092 CrossRef CAS PubMed.
- L. C. Seefeldt, Z.-Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed.
- K. Tanifuji and Y. Ohki, Chem. Rev., 2020, 120, 5194–5251 CrossRef CAS PubMed.
- S. Burén, E. Jiménez-Vicente, C. Echavarri-Erasun and L. M. Rubio, Chem. Rev., 2020, 120, 4921–4968 CrossRef PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- J. S. Anderson, M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 534–537 CrossRef CAS PubMed.
- J. S. Anderson, G. E. Cutsail, J. Rittle, B. A. Connor, W. A. Gunderson, L. Zhang, B. M. Hoffman and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 7803–7809 CrossRef CAS PubMed.
- T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341–5350 CrossRef CAS PubMed.
- S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105–1115 CrossRef CAS PubMed.
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 4243–4248 CrossRef CAS PubMed.
- T. J. Del Castillo, N. B. Thompson, D. L. M. Suess, G. Ung and J. C. Peters, Inorg. Chem., 2015, 54, 9256–9262 CrossRef CAS PubMed.
- J. Fajardo and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 16105–16108 CrossRef CAS PubMed.
- J. Rittle and J. C. Peters, Angew. Chem., Int. Ed., 2016, 55, 12262–12265 CrossRef CAS PubMed.
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 3161–3170 CrossRef CAS PubMed.
- I. Čorić, B. Q. Mercado, E. Bill, D. J. Vinyard and P. L. Holland, Nature, 2015, 526, 96–99 CrossRef PubMed.
- A. L. Speelman, I. Čorić, C. Van Stappen, S. DeBeer, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2019, 141, 13148–13157 CrossRef CAS PubMed.
- T. Spatzal, K. A. Perez, J. B. Howard and D. C. Rees, eLife, 2015, 4, e11620 CrossRef PubMed.
- S. A. Stoian, J. M. Smith, P. L. Holland, E. Münck and E. L. Bominaar, Inorg. Chem., 2008, 47, 8687–8695 CrossRef CAS PubMed.
- J. Krzystek, A. Ozarowski, S. A. Zvyagin and J. Telser, Inorg. Chem., 2012, 51, 4954–4964 CrossRef CAS PubMed.
- M. Yousif, D. Wannipurage, C. D. Huizenga, E. Washnock-Schmid, N. J. Peraino, A. Ozarowski, S. A. Stoian, R. L. Lord and S. Groysman, Inorg. Chem., 2018, 57, 9425–9438 CrossRef CAS PubMed.
- L. Bucinsky, M. Breza, M. Malček, D. C. Powers, S. J. Hwang, J. Krzystek, D. G. Nocera and J. Telser, Inorg. Chem., 2019, 58, 4907–4920 CrossRef PubMed.
- J. M. Smith, R. J. Lachicotte, K. A. Pittard, T. R. Cundari, G. Lukat-Rodgers, K. R. Rodgers and P. L. Holland, J. Am. Chem. Soc., 2001, 123, 9222–9223 CrossRef CAS PubMed.
- J. M. Smith, A. R. Sadique, T. R. Cundari, K. R. Rodgers, G. Lukat-Rodgers, R. J. Lachicotte, C. J. Flaschenriem, J. Vela and P. L. Holland, J. Am. Chem. Soc., 2006, 128, 756–769 CrossRef CAS PubMed.
- M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- C. J. Pollock, K. Grubel, P. L. Holland and S. DeBeer, J. Am. Chem. Soc., 2013, 135, 11803–11808 CrossRef CAS PubMed.
- P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS.
- N. Lee, T. Petrenko, U. Bergmann, F. Neese and S. DeBeer, J. Am. Chem. Soc., 2010, 132, 9715–9727 CrossRef CAS PubMed.
- G. E. Cutsail III, N. L. Gagnon, A. D. Spaeth, W. B. Tolman and S. DeBeer, Angew. Chem., Int. Ed., 2019, 58, 9114–9119 CrossRef PubMed.
- T. M. Figg, P. L. Holland and T. R. Cundari, Inorg. Chem., 2012, 51, 7546–7550 CrossRef CAS PubMed.
- K. Grubel, W. W. Brennessel, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 16807–16816 CrossRef CAS PubMed.
- T. Liu, M. R. Gau and N. C. Tomson, J. Am. Chem. Soc., 2020, 142, 8142–8146 CrossRef CAS PubMed.
- D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, Science, 2018, 359, 1484–1489 CrossRef CAS PubMed.
- A. L. Nagelski, M. S. Fataftah, M. M. Bollmeyer, S. F. McWilliams, S. N. MacMillan, B. Q. Mercado, K. M. Lancaster and P. L. Holland, Chem. Sci., 2020, 11, 12710–12720 RSC.
- D. E. DeRosha, V. G. Chilkuri, C. Van Stappen, E. Bill, B. Q. Mercado, S. DeBeer, F. Neese and P. L. Holland, Nat. Chem., 2019, 11, 1019–1025 CrossRef CAS PubMed.
- D. E. DeRosha, N. A. Arnet, B. Q. Mercado and P. L. Holland, Inorg. Chem., 2019, 58, 8829–8834 CrossRef CAS PubMed.
- H. C. Angove, S. J. Yoo, B. K. Burgess and E. Münck, J. Am. Chem. Soc., 1997, 119, 8730–8731 CrossRef CAS.
- E. J. Leggate, E. Bill, T. Essigke, G. M. Ullmann and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10913–10918 CrossRef CAS PubMed.
- L. Deng and R. H. Holm, J. Am. Chem. Soc., 2008, 130, 9878–9886 CrossRef CAS PubMed.
- A. Albers, S. Demeshko, K. Pröpper, S. Dechert, E. Bill and F. Meyer, J. Am. Chem. Soc., 2013, 135, 1704–1707 CrossRef CAS PubMed.
- R. Bjornsson, F. A. Lima, T. Spatzal, T. Weyhermüller, P. Glatzel, E. Bill, O. Einsle, F. Neese and S. DeBeer, Chem. Sci., 2014, 5, 3096–3103 RSC.
- J. Kowalska and S. DeBeer, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1406–1415 CrossRef CAS PubMed.
- R. Bjornsson, F. Neese and S. DeBeer, Inorg. Chem., 2017, 56, 1470–1477 CrossRef CAS.
- J. K. Kowalska, B. Nayyar, J. A. Rees, C. E. Schiewer, S. C. Lee, J. A. Kovacs, F. Meyer, T. Weyhermüller, E. Otero and S. DeBeer, Inorg. Chem., 2017, 56, 8147–8158 CrossRef CAS.
- J. K. Kowalska, J. T. Henthorn, C. Van Stappen, C. Trncik, O. Einsle, D. Keavney and S. DeBeer, Angew. Chem., Int. Ed., 2019, 58, 9373–9377 CrossRef CAS PubMed.
- T. V. Harris and R. K. Szilagyi, Inorg. Chem., 2011, 50, 4811–4824 CrossRef CAS.
- B. B. Wenke, T. Spatzal and D. C. Rees, Angew. Chem., Int. Ed., 2019, 58, 3894–3897 CrossRef CAS PubMed.
- C. H. Arnett, M. J. Chalkley and T. Agapie, J. Am. Chem. Soc., 2018, 140, 5569–5578 CrossRef CAS PubMed.
- J. T. Henthorn, R. J. Arias, S. Koroidov, T. Kroll, D. Sokaras, U. Bergmann, D. C. Rees and S. DeBeer, J. Am. Chem. Soc., 2019, 141, 13676–13688 CrossRef CAS PubMed.
- J. Rittle, C. C. L. McCrory and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13853–13862 CrossRef CAS.
- S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 7310–7313 CrossRef CAS.
- D. Lukoyanov, Z.-Y. Yang, N. Khadka, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2015, 137, 3610–3615 CrossRef CAS PubMed.
- N. A. Arnet, T. R. Dugan, F. S. Menges, B. Q. Mercado, W. W. Brennessel, E. Bill, M. A. Johnson and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13220–13223 CrossRef CAS PubMed.
- M. M. Rodriguez, B. D. Stubbert, C. C. Scarborough, W. W. Brennessel, E. Bill and P. L. Holland, Angew. Chem., Int. Ed., 2012, 51, 8247–8250 CrossRef CAS PubMed.
- L. Liu, T. B. Rauchfuss and T. J. Woods, Inorg. Chem., 2019, 58, 8271–8274 CrossRef CAS PubMed.
- L. Liu, T. J. Woods and T. B. Rauchfuss, Eur. J. Inorg. Chem., 2020, 3460–3465 CrossRef CAS PubMed.
- C. Joseph, J. P. Shupp, C. R. Cobb and M. J. Rose, Catalyst, 2020, 10, 1317 CrossRef CAS.
- C. Joseph, C. R. Cobb and M. J. Rose, Angew. Chem., Int. Ed., 2021, 60, 3433–3437 CrossRef CAS PubMed.
- L. M. Rubio and P. W. Ludden, Annu. Rev. Microbiol., 2008, 62, 93–111 CrossRef CAS PubMed.
- C. H. Arnett and T. Agapie, J. Am. Chem. Soc., 2020, 142, 10059–10068 CrossRef CAS.
- C. H. Arnett, I. Bogacz, R. Chatterjee, J. Yano, P. H. Oyala and T. Agapie, J. Am. Chem. Soc., 2020, 142, 18795–18813 CrossRef CAS PubMed.
- K. M. Lancaster, Y. Hu, U. Bergmann, M. W. Ribbe and S. DeBeer, J. Am. Chem. Soc., 2013, 135, 610–612 CrossRef CAS PubMed.
- C. J. Pollock, L. L. Tan, W. Zhang, K. M. Lancaster, S. C. Lee and S. DeBeer, Inorg. Chem., 2014, 53, 2591–2597 CrossRef CAS PubMed.
- Y. Do, E. D. Simhon and R. H. Holm, Inorg. Chem., 1983, 22, 3809–3812 CrossRef CAS.
- G. B. Wong, M. A. Bobrik and R. H. Holm, Inorg. Chem., 1978, 17, 578–584 CrossRef CAS.
- X.-D. Chen, W. Zhang, J. S. Duncan and S. C. Lee, Inorg. Chem., 2012, 51, 12891–12904 CrossRef CAS PubMed.
- A. K. Verma, T. N. Nazif, C. Achim and S. C. Lee, J. Am. Chem. Soc., 2000, 122, 11013–11014 CrossRef CAS.
- A. J. Jasniewski, C. C. Lee, M. W. Ribbe and Y. Hu, Chem. Rev., 2020, 120, 5107–5157 CrossRef CAS PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS PubMed.
- B. J. Landgraf, E. L. McCarthy and S. J. Booker, Annu. Rev. Biochem., 2016, 85, 485–514 CrossRef CAS PubMed.
- G. L. Holliday, E. Akiva, E. C. Meng, S. D. Brown, S. Calhoun, U. Pieper, A. Sali, S. J. Booker and P. C. Babbitt, in Methods Enzymol., ed. V. Bandarian, Academic Press, 2018, vol. 606, pp. 1–71 Search PubMed.
- A. McSkimming and D. L. M. Suess, Inorg. Chem., 2018, 57, 14904–14912 CrossRef CAS PubMed.
- A. C. Brown and D. L. M. Suess, Inorg. Chem., 2019, 58, 5273–5280 CrossRef CAS PubMed.
- M. Ye, N. B. Thompson, A. C. Brown and D. L. M. Suess, J. Am. Chem. Soc., 2019, 141, 13330–13335 CrossRef CAS PubMed.
- C. V. Popescu, M. T. Mock, S. A. Stoian, W. G. Dougherty, G. P. A. Yap and C. G. Riordan, Inorg. Chem., 2009, 48, 8317–8324 CrossRef CAS PubMed.
- A. C. Brown and D. L. M. Suess, J. Am. Chem. Soc., 2020, 142, 14240–14248 CrossRef CAS PubMed.
- A. McSkimming, A. Sridharan, N. B. Thompson, P. Müller and D. L. M. Suess, J. Am. Chem. Soc., 2020, 142, 14314–14323 CrossRef CAS PubMed.
- A. McSkimming and D. L. M. Suess, Nat. Chem., 2021 DOI:10.1038/s41557-021-00701-6.
- B. Rink, M. Brorson and I. J. Scowen, Organometallics, 1999, 18, 2309–2313 CrossRef CAS.
- K. Herbst, M. Monari and M. Brorson, Inorg. Chem., 2001, 40, 2979–2985 CrossRef CAS PubMed.
- I. Takei, K. Suzuki, Y. Enta, K. Dohki, T. Suzuki, Y. Mizobe and M. Hidai, Organometallics, 2003, 22, 1790–1792 CrossRef CAS.
- Y. Ohki, K. Uchida, M. Tada, R. E. Cramer, T. Ogura and T. Ohta, Nat. Commun., 2018, 9, 3200 CrossRef PubMed.
- Y. Ohki, K. Uchida, R. Hara, M. Kachi, M. Fujisawa, M. Tada, Y. Sakai and W. M. C. Sameera, Chem. – Eur. J., 2018, 24, 17138–17147 CrossRef CAS PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- K. Hoshino, Chem. – Eur. J., 2001, 7, 2727–2731 CrossRef CAS.
- M. Liras, M. Barawi and V. A. de la Peña O’Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed.
- J. R. Christianson, D. Zhu, R. J. Hamers and J. R. Schmidt, J. Phys. Chem. B, 2014, 118, 195–203 CrossRef CAS PubMed.
- Y. Nishibayashi, M. Saito, S. Uemura, S.-I. Takekuma, H. Takekuma and Z.-I. Yoshida, Nature, 2004, 428, 279–280 CrossRef CAS PubMed.
- A. Banerjee, B. D. Yuhas, E. A. Margulies, Y. Zhang, Y. Shim, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 2030–2034 CrossRef CAS PubMed.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- V. K. Shah and W. J. Brill, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 3249–3253 CrossRef CAS PubMed.
- A. W. Fay, C.-C. Lee, J. A. Wiig, Y. Hu and M. W. Ribbe, in Nitrogen Fixation: Methods and Protocols, ed. M. W. Ribbe, Humana Press, Totowa, NJ, 2011 DOI:10.1007/978-1-61779-194-9_16, pp. 239–248.
- E. Krahn, B. Weiss, M. Kröckel, J. Groppe, G. Henkel, S. Cramer, A. Trautwein, K. Schneider and A. Müller, J. Biol. Inorg. Chem., 2002, 7, 37–45 CrossRef CAS PubMed.
- A. W. Fay, M. A. Blank, C. C. Lee, Y. Hu, K. O. Hodgson, B. Hedman and M. W. Ribbe, J. Am. Chem. Soc., 2010, 132, 12612–12618 CrossRef CAS PubMed.
- M. R. Antonio, B. K. Teo, W. H. Orme-Johnson, M. J. Nelson, S. E. Groh, P. A. Lindahl, S. M. Kauzlarich and B. A. Averill, J. Am. Chem. Soc., 1982, 104, 4703–4705 CrossRef CAS.
- G. N. George, R. C. Prince and R. E. Bare, Inorg. Chem., 1996, 35, 434–438 CrossRef CAS PubMed.
- A. R. Bizzarri, M. Bacci and S. Cannistraro, Biophys. Chem., 1993, 46, 117–129 CrossRef CAS PubMed.
- D. Eliezer, P. Frank, N. Gillis, W. E. Newton, S. Doniach and K. O. Hodgson, J. Biol. Chem., 1993, 268, 20953–20957 CrossRef CAS.
- C. C. Lee, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2012, 51, 1947–1949 CrossRef CAS PubMed.
- C. C. Lee, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2015, 54, 1219–1222 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- K. Tanifuji, C. C. Lee, Y. Ohki, K. Tatsumi, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2015, 54, 14022–14025 CrossRef CAS PubMed.
- S. Shima, D. Chen, T. Xu, M. D. Wodrich, T. Fujishiro, K. M. Schultz, J. Kahnt, K. Ataka and X. Hu, Nat. Chem., 2015, 7, 995–1002 CrossRef CAS PubMed.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- K. Tanifuji, C. C. Lee, N. S. Sickerman, K. Tatsumi, Y. Ohki, Y. Hu and M. W. Ribbe, Nat. Chem., 2018, 10, 568–572 CrossRef CAS PubMed.
- C. C. Lee, K. Tanifuji, M. Newcomb, J. Liedtke, Y. Hu and M. W. Ribbe, ChemBioChem, 2018, 19, 649–653 CrossRef CAS PubMed.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306–307 CrossRef CAS.
- T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- M. T. Reetz, Acc. Chem. Res., 2019, 52, 336–344 CrossRef CAS PubMed.
- K. R. Miller, S. Biswas, A. Jasniewski, A. H. Follmer, A. Biswas, T. Albert, S. Sabuncu, E. L. Bominaar, M. P. Hendrich, P. Moënne-Loccoz and A. S. Borovik, J. Am. Chem. Soc., 2021, 143, 2384–2393 CrossRef CAS PubMed.
- R. Weindl, R. Khare, L. Kovarik, A. Jentys, K. Reuter, H. Shi and J. A. Lercher, Angew. Chem., Int. Ed., 2021, 60, 9301–9305 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |