
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Propane to olefins tandem catalysis: a selective route towards light olefins production†
Matteo
Monai
 a,
Marianna
Gambino
a,
Sippakorn
Wannakao
b and
Bert M.
Weckhuysen
*a
a,
Marianna
Gambino
a,
Sippakorn
Wannakao
b and
Bert M.
Weckhuysen
*a
aInorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl
bSCG Chemicals Co., Ltd, 1 Siam-Cement Rd, Bang sue, Bangkok 1080, Thailand
First published on 19th August 2021
Abstract
On-purpose synthetic routes for propylene production have emerged in the last couple of decades in response to the increasing demand for plastics and a shift to shale gas feedstocks for ethylene production. Propane dehydrogenation (PDH), an efficient and selective route to produce propylene, saw booming investments to fill the so-called propylene gap. In the coming years, however, a fluctuating light olefins market will call for flexibility in end-product of PDH plants. This can be achieved by combining PDH with propylene metathesis in a single step, propane to olefins (PTO), which allows production of mixtures of propylene, ethylene and butenes, which are important chemical building blocks for a.o. thermoplastics. The metathesis technology introduced by Phillips in the 1960s and mostly operated in reverse to produce propylene, is thus undergoing a renaissance of scientific and technological interest in the context of the PTO reaction. In this review, we will describe the state-of-the-art of PDH, propylene metathesis and PTO reactions, highlighting the open challenges and opportunities in the field. While the separate PDH and metathesis reactions have been extensively studied in the literature, understanding the whole PTO tandem-catalysis system will require new efforts in theoretical modelling and operando spectroscopy experiments, to gain mechanistic insights into the combined reactions and finally improve catalytic selectivity and stability for on-purpose olefins production.
 Matteo Monai | Matteo Monai was born in Cividale del Friuli, Italy, in 1989. In 2017 he received his PhD in Chemistry at the University of Trieste (Italy) with Prof. Paolo Fornasiero. From 2017, Matteo worked as a postdoctoral fellow in the group of Prof. Bert Weckhuysen at Utrecht University (the Netherlands), focusing on heterogeneous catalysts for (de)hydrogenation reactions. He was awarded the Parmaliana Prize (2017), the Eni Young Researcher of the Year Award (2019), and the Robert Karl Grasselli Award (2020). In 2021, Matteo was appointed as a tenure-track assistant professor at Utrecht University, where he works on breaking scaling relations in small molecules activation, as part of the Advanced Research Center Chemical Building Blocks Consortium (ARC CBBC). |
 Marianna Gambino | Marianna Gambino received her PhD in Materials Science from the University of Catania (Italy) in 2017 and her MSc in Chemistry from the University of Palermo (Italy) in 2013. From 2017 to 2020 she worked as a postdoctoral fellow in the group of Prof. Bert Weckhuysen at Utrecht University (the Netherlands), investigating deactivation mechanisms in heterogeneous catalysts for different industrial application, from fluid catalytic cracking to light olefins production. |
 Sippakorn Wannakao | Sippakorn Wannakao was born in Khon Kaen, Thailand, in 1986. He received his PhD in Chemistry at the Kasetsart University with Prof. Jumras Limtrakul in 2015. During his PhD, he worked as a visiting researcher at Massachusetts Institute of Technology with Prof. Alexie M. Kolpak in 2014, working on computational study of reaction mechanisms on heterogeneous catalysts and catalyst design for CO2 conversions. He worked as a postdoctoral fellow at VISTEC with Dr kanokwan kongpatpanich (2015–2017). Currently, Sippakorn is working as a researcher at SCG Chemicals, Co., Ltd, covering developments of catalysts and catalytic processes for the petrochemical industry, and bio-circular-green economy. |
 Bert M. Weckhuysen | Bert Weckhuysen was born in Aarschot, Belgium, in 1968. He obtained his PhD degree from K. U. Leuven (Belgium) in 1995 under the supervision of Prof. Robert Schoonheydt. After postdoctoral stays at Lehigh University (PA, USA) with Prof. Israel Wachs and Texas A&M University (TX, USA) with Prof. Jack Lunsford, he became in 2000 a full Professor at Utrecht University (the Netherlands). His research focuses on the development and use of in situ and operando spectroscopy for studying solid catalysts under realistic reaction conditions at different length scales. The aim is to design and apply catalytic solids for the conversion of fossil (crude oil & natural gas) and renewable (CO2, biomass & municipal waste, including plastics) feedstock into fuels, chemicals and materials. |
1. Introduction
1.1 Socio-economical context
Although seldom used as final products, light olefins are the backbone of the chemical industry. Ethylene and propylene alone are the first and second most highly produced organic chemicals worldwide (Fig. 1). They are used as feedstocks for the production of polymers (e.g. polyethylene, polypropylene, polyesters, and polyurethanes), oxygenates (e.g. ethylene glycol, acetone, and acetaldehyde), intermediates and personal care products and detergents.1,2Fig. 1 summarizes the main applications of light olefins together with their market size, trends and predicted growth. Notably, while the ethylene market is projected to grow steadily over the next years, the propylene market is set to shrink slightly, mainly due to safety regulations on manufacture and transportation.3 The 2020 COVID pandemic has impacted petrochemical and refinery markets, and put some PDH installations plans on hold, especially in Canada.4 Other α-olefins markets are instead projected to grow, pulled by the ethylene market, in particular for the production of HDPE and LDPE (high and low-density polyethylene).5 It is thus economically and socially relevant to introduce flexibility in the olefin supply chain by converting propylene to other olefins, e.g., by the metathesis reaction.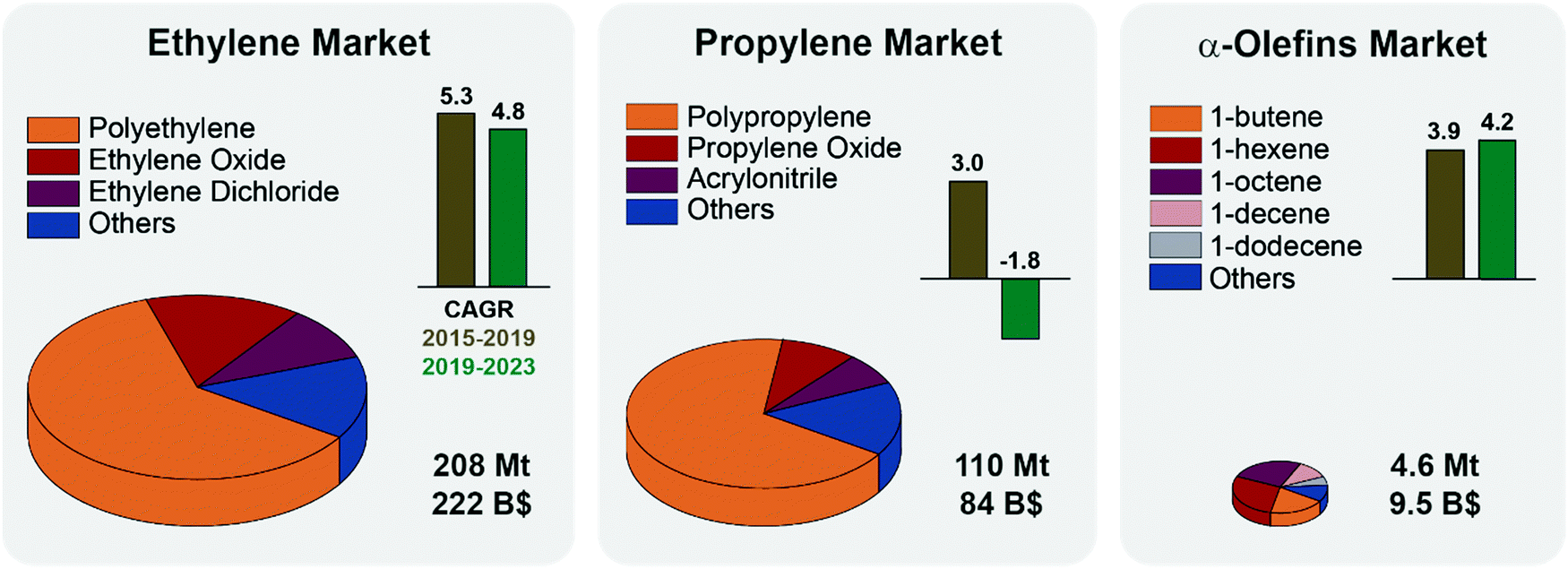 | ||
| Fig. 1 Olefin markets status and projections: the volume of each pie chart represents the size of the respective olefin market in million tonnes (Mt) in 2019. The global market value as of 2019 is also reported, in billion dollars (B$). Ethylene and propylene markets are divided by application, polymers production accounting for 61 and 68% of the total, respectively. Alpha-olefins market is an order of magnitude smaller, and here organized by olefin type (1-butene accounts for 19% of the total by weight). Compound Annual Growth Rates (CAGR) over the years 2015–2019 (brown) and the projected CAGR for 2019–2023 (green) are reported as bars. The effects of the COVID-19 outbreak are not taken into account but are set to negatively affect the markets. See ESI† for CAGR formula and summary tables. | ||
Just like molecular hydrogen, olefins are not occurring as natural resources due to their reactivity, and have to be synthesized starting from other feedstocks, almost entirely from fossil fuels. To date, olefins are mainly produced by refinery technologies such as steam cracking (SC) and fluid catalytic cracking (FCC) of naphtha, diesel or other oil byproducts.6,7 While established since the 1940s, refinery technologies have a low selectivity for light olefins, because they were developed with the purpose to produce other products, such as aromatics and gasoline (Fig. 2a and b). In the last couple of decades, on-purpose technologies to produce light olefins with higher selectivity have emerged, due to a number of technological and economical drivers, including the boom in plastic production in the ‘70–80s’ rising oil prices in the ‘90s’ and most importantly the advent of fracking technologies to produce cheap shale gas at the beginning of the 21st century.8 The shale gas share in U.S. natural gas went from 1% to 20% in the turn of 10 years, causing a drop in gas cost and a shift away from oil and coal for the production of fuels and chemicals. SC units, originally using naphtha as feedstock, began to be retrofitted to accommodate ethane or propane cracking, to increase margin thanks to lower feedstock cost while producing 80 or 40% ethylene on a weight feed basis, respectively, compared to 23% of naphtha.9,10 A similar trend is currently observed worldwide, with Asia and South America aiming to select the most optimal feed mix based on market conditions. This sets naphtha share in ethylene production to decrease from 47% in 2017 to 44% by 2027, while ethane is expected to rise from 35% to 39% in the same period.10
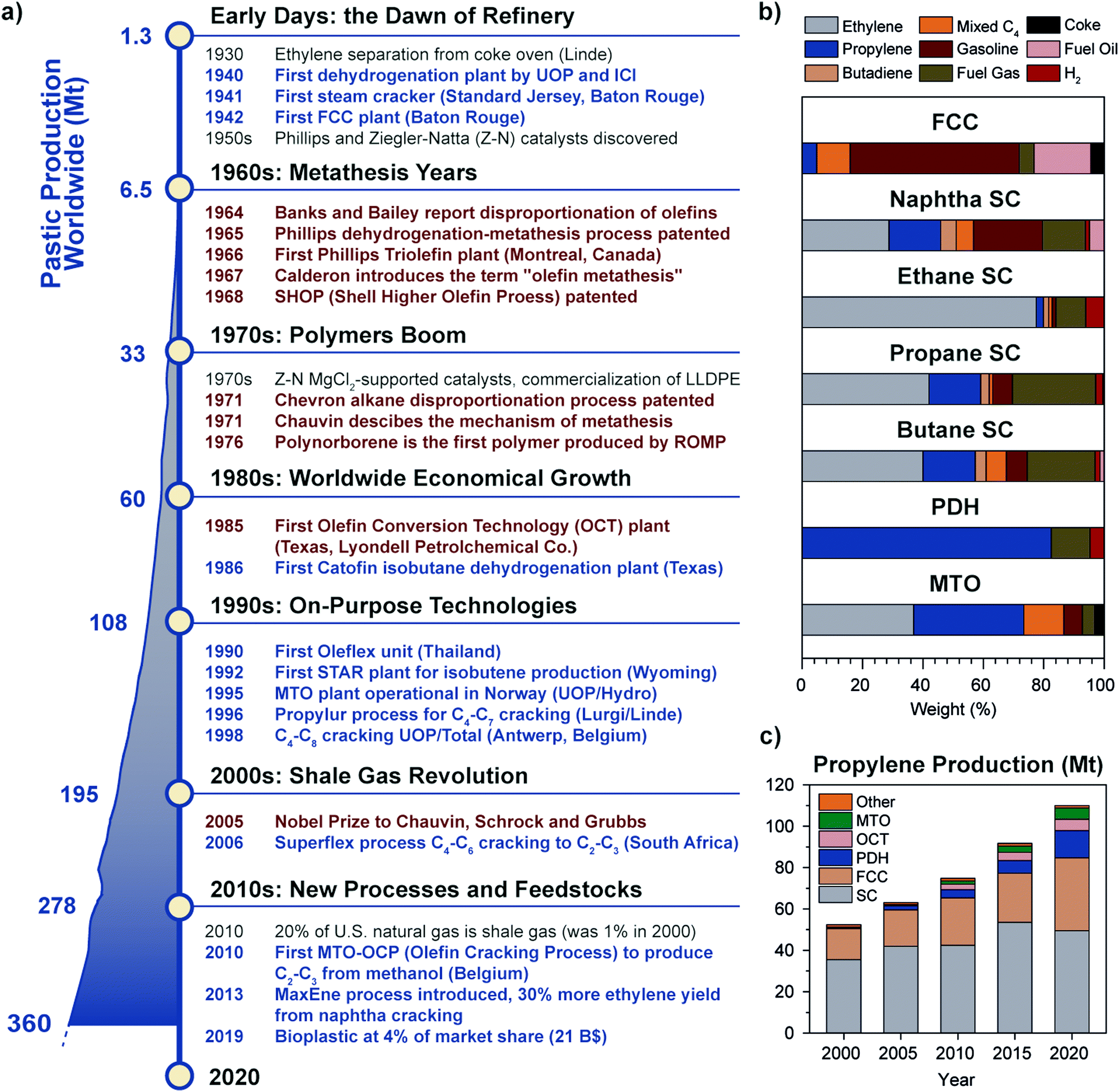 | ||
| Fig. 2 Historical perspective of olefins production and the rise of metathesis and on-purpose technologies: (a) timeline showing the main breakthroughs in olefins production (blue text) and metathesis (brown text). A detailed list of patents is provided in the ESI.† The worldwide plastic production trend in megatonnes per year from 1950 to 2018 is reported on the left (adapted from ref. 11 with permission from Elsevier); (b) product selectivity (in weight percent) of different olefin production technologies (FCC: Fluid Catalytic Cracking, SC: Steam Cracking; PDH: Propane DeHydrogenation; MTO: Methanol to Olefins), showing the advantage of on-purpose technologies to produce the desired olefin; (c) trend of propylene production in megatonnes divided by technology over the years 2000–2020 (OCT: Olefin Conversion Technology, i.e. metathesis of ethylene and butene). On-purpose technologies share increased to 23% in two decades to fill the so-called propylene gap. Adapted from ref. 7 with permission from the National Academies Press. | ||
Since propylene and butenes were historically produced as byproducts of naphtha steam cracking, and because the yield of such products from natural gas cracking is about 1 order of magnitude lower than for naphtha (Fig. 2b), the shift towards shale gas cracking negatively impacted propylene and C4 olefins production, to the point that a shortage of propylene came into being. On-purpose technologies such as propane dehydrogenation (PDH) thus became very profitable, due to the gap between propylene production and demand and the difference in price of propane and propylene (spread of 600–800 $ ton−1 on average, 2010–2019, see Fig. S1, ESI†). Two main players emerged, i.e. Chevron (Catofin technology, now Lummus) and UOP (Oleflex technology), which patented their technology in 196812 and 1970.13–15 Hundred-billion dollar investments in PDH installations during the years 2000s in China, the U.S. and the Middle East, brought propylene production by PDH to 5 million tonnes (Mt) in 2013 (5% of total), a number more than doubled in 2019 (13.5 Mt, 12% of total) (Fig. 2c).
In the last ten years, other technologies for the production of propylene, such as Methanol to Olefins or to Propylene (MTO, MTP) and Olefin Conversion Technology (OCT) underwent a similar development (5% of current production each, Fig. 2c). A map of the world showing existing and planned PDH installations is reported in Fig. 3 (see ESI† for list of installations). It should also be noted that while FCC were traditionally operated to maximize gasoline or distillate production, current catalysts formulations and improvement in process technology (high severity-FCC, HS-FCC) now allow to produce propylene with yields around 25 wt% (compared to the traditional 4–6 wt%).16
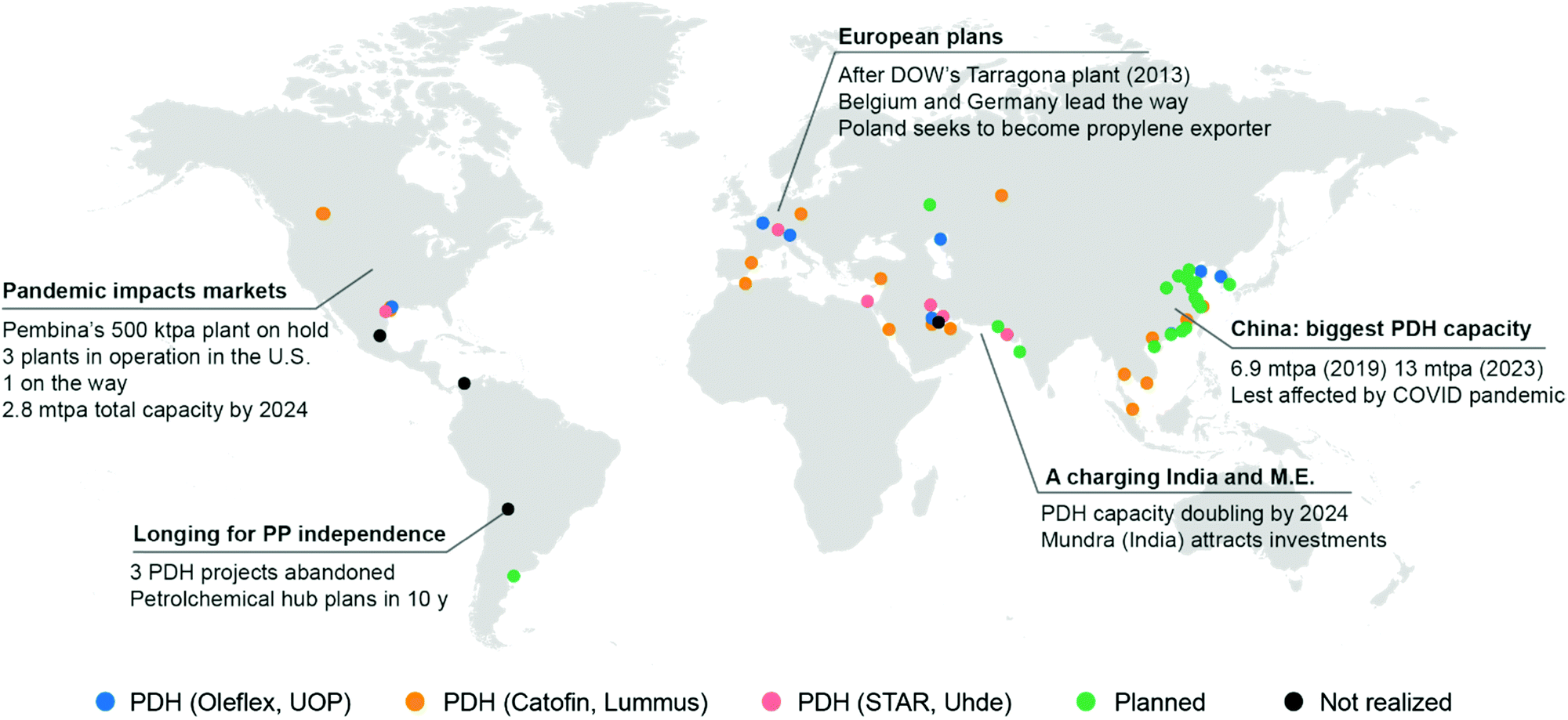 | ||
| Fig. 3 World map showing PDH (propane dehydrogenation) installations. Plants in operation or under construction are indicated by blue (UOP Oleflex), orange (Catofin, Lummus) or pink dots (STAR, Uhde). Planned installations for which the technology is not disclosed are marked with a green dot. Installations proposed but cancelled are marked in dark grey. See ESI† for a list of installations and a link to an interactive map. | ||
As can be seen in Fig. 2b, PDH is extremely selective for propylene production, which is why it is considered the technology of choice for the “propylene rush”. Nonetheless, being able to produce more than one olefin feedstock can be advantageous in a scenario of lower propylene demand (Fig. 1) and expanding market of thermoplastic, i.e., copolymers of ethylene, propylene and butene such as LDPE and HDPE. Production of butenes is also interesting per se, because of a gap between supply and demand similar to propylene's market case. Since the 1970s, the use of isobutylene for the production of the gasoline additive methyl tert-butyl ether (MTBE) caused a spike in demand, which could not be met with the existing cracking technologies. This lead to the spread of butane dehydrogenation plants in the 1980s, with a consequent 15 time increase of MTBE capacity.17 MTBE was phased out as a gasoline additive from 2000 to 2004 in the US, due to its detection in wells and ground water. Notably, no similar bans have been put in place in the rest of the world.18
Flexibility in olefins production can be achieved by combining PDH with propylene self-metathesis (Met), in the so-called Propylene to Olefins (PTO), which leads to the production of propylene, ethylene and 2-butenes (Fig. 4a).
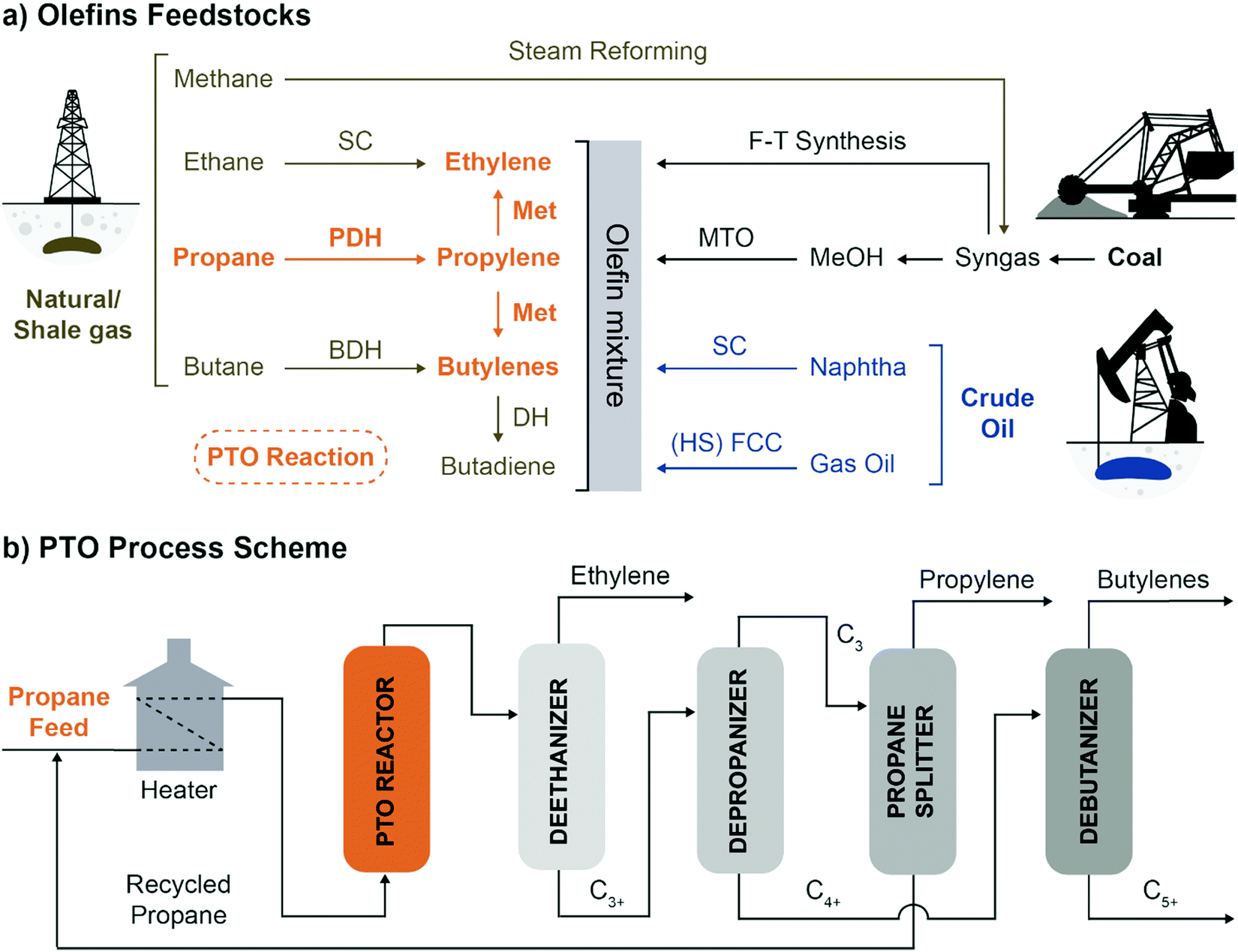 | ||
| Fig. 4 The Propane to Olefins (PTO) process in context: (a) olefins feedstocks are almost entirely fossil-fuels derivatives. PTO enables to convert propane to propylene, ethylene and butenes, introducing flexibility in Propane DeHydrogenation (PDH) plants. The main technologies to produce olefins starting from crude oil, coal or natural gas are summarized (SC: Steam Cracking, F–T: Fischer–Tropsch, Met: Metathesis, MTO: Methanol To Olefins, BDH: Butene DeHydrogenation, (HS) FCC: High Severity Fluid Catalytic Cracking); (b) schematic representation of the PTO process (adapted from patent US3445541A, ref. 19). | ||
Notably, interest in combining PDH and Met reactions dates back to the 1960s, when Phillips patented the “Combined dehydrogenation and disproportionation” technology (Fig. 2a and 4b).19 Some years later, Chevron patented a similar concept, “Disproportionation of saturated hydrocarbons”, only aimed to produce higher alkanes by re-hydrogenation of the metathesized olefins.20 So far, Phillips metathesis technology has actually been operated in reverse, in OCT plants, in which ethylene and butenes are converted to propylene,21 or in ethylene-to-propylene (ETP) plants, in which ethylene is first dimerized to butene, and then the mixture is converted to propylene.22 In principle, any PDH installation can be adapted to PTO by changing the catalyst to a mixture or dehydrogenation and metathesis catalysts, which makes a strong case for PTO to increase flexibility in the olefin supply chain. New PTO technologies are emerging to improve on the time yield and selectivity of the original Phillips process, by changing catalyst design, contact state and more of operation. Nonetheless, combining PDH and Met in such a tandem-reaction system poses intrinsic challenges, such as understanding combined deactivation mechanisms, achieve regeneration of both catalysts, and leveraging the interplay of the two catalytic components, in turn influenced by their contact state.
1.2 Scope of the review
Propane dehydrogenation, olefin metathesis and their combination in PTO have emerged as strategic technologies for the chemical industry. This review will provide the readers with a complete overview of the PDH and Met reactions and catalysts therefor, with an emphasis on their combination in PTO technologies and the relative challenges and opportunities. First, we will summarize and compare the commercial processes and patents for the three reactions. Next, we will discuss the thermodynamic boundaries of the reactions and evidence the importance of kinetic control by catalysts design. In Section 4, we will discuss catalytic aspects of the PTO reaction, focusing on PDH and Met compatibility in terms of temperature and feed composition and expected effects of contact state on catalytic performance, feasibility and catalysts stability. We further point out the need for operando characterization and theoretical calculations relevant to PTO. Finally, Section 5 provides an outlook for the development of PTO technologies, taking into account the impact and opportunities offered by recent efforts towards sustainability, such as plastic recycling and CO2 capture and valorization.To the best of our knowledge, this is the first review on emerging PTO technologies. Although PDH- and Met-oriented reviews have been published in recent years,6,23–26 the present work is uniquely positioned to critically discuss their combination in a tandem catalysis system, with a focus on mechanistic understanding of PTO heterogeneous catalysts and their relevant deactivation mechanisms. An extensive list of previous reviews on PDH and Met heterogeneous catalysts relevant to the light olefin production and interconversion field can be found in the ESI.† The present review will not address homogeneous catalysts for olefins metathesis, despite their interest for certain specialty products production due to their high activity, selectivity and compatibility with functional groups. Their cost, difficult catalyst recovery and separation and stability limit their applications in the field of simple light olefins. Advances in the field of (supported) homogeneous catalysts for industrial applications can be found in existing reviews.21,27,28
2. Commercial processes
2.1 Propane dehydrogenation
The first light alkane dehydrogenation plants were introduced already in the 1940, first by UOP and ICI, and then by Phillips, Shell and Dow a.o.29 Such early projects in light olefin dehydrogenations were shut down due to the rise of naphtha cracking and FCC technologies, which produced a variety of olefins as byproducts. Nonetheless, already in those early years, Eugene Houdry designed and commercialized (together with Petro-Tex Chemical Corporation, with the name Oxo-D) the butane dehydrogenation process operated at lower temperature which will be later renamed CATOFIN process in the 1980s. The first CATOFIN PDH plant had a 250 kt per year capacity and came in operation in 1986 in Texas,30 followed by the first Oleflex PDH plant by UOP in Thailand, which became operational in 1990 with a capacity of 315 kt per year.31 The Phillips Petroleum's STAR Krupp-Uhde (Steam Active Reforming) Process and FBD Yarsintez–Snamprogetti followed a couple of years later.6In total, nine propane dehydrogenation technologies were developed over the years, which mainly differ in three features: (i) the operation and regeneration mode, which can be cyclic or continuous; (ii) the catalyst composition, based either on Cr or Pt, and (iii) the heat management, to support the highly endothermic dehydrogenation reaction. A summary and comparison of eight of the existing technologies for PDH is reported in Table 1, their schematic representation is given in Fig. 5, and the overview of the catalytic conditions used is given in Fig. 6. KBR (Kellogg Brown & Root) recently announced a new propane dehydrogenation technology, which according to the company offers high propylene selectivity and conversions.32 However, no information could be gathered regarding the process or catalyst used and the technology is thus not discussed herein. Since the product mixture separation section downstream of the reactor(s) is quite similar for all the technologies (compressors, pressure swing adsorption, cryogenic unit, and fractionators), in this section we will focus on describing the strategies to enhance time yield and process efficiency by reactor design.
| Technology and licensor | CATOFIN CB&I-ABB Lummus | Oleflex UOP | STAR Krupp-Uhde | FBD Yarsintez–Snamprogetti | PDH Linde–BASF–Statoil (Sintef) | FLOTU/Tsinghua | FCDh™ (Dow) | SABIC | K-PROTM (KBR) |
|---|---|---|---|---|---|---|---|---|---|
| Reactor | Adiabatic fixed bed | Adiabatic moving bed | Adiabatic | Fluidized bed | Isothermal fixed bed | Bimodal fluidized bed | Fluidized bed | Fluidized bed | Fluidized bed |
| Operation | Cyclic | Continuous | Cyclic | Continuous | Cyclic | Continuous | Continuous | Continuous | Continuous |
| Catalyst | CrOx/Al2O3 + alkali | Pt–Sn/Al2O3 + alkali | Pt–Sn/Zn–Al2O3/Ca–Al2O3 | CrOx/Al2O3 + alkali | Pt/hydrotalcite Mg(Al)O; Pt–Sn/ZrO2 | Pt–Sn/SAPO-34 | Pt–Ga–K/Si–Al2O3 | Pt–Sn/K/SAPO-34 | — |
| Heat management | Pre-heaters, regeneration | Interstage heating, regenerated catalyst | Furnace and ODH | Regenerated catalyst | Furnace | Regenerated secondary catalyst | — | Integrated FBR with internal regenerator | — |
| T (°C) | 565–650 | 550–620 | DH: 550–590, ODH: ∼600 | 535–590 | ∼590 | 570–610 | ∼600 | 560–600 | ∼600 |
| P (bar) | 0.3–0.5 | 2–3 | 5–6 | 0.5–1.5 | ∼1 | 0.5–1.5 | 1 | 0.1–6 | 1.5 |
| Cycle time | 15–30 min | — | 8 h | — | 9 h | — | — | 15–30 min | — |
| Development stage | Commercial | Commercial | Commercial | Commercial | Pilot plant | Pilot plant | Retrofitting planned | — | — |
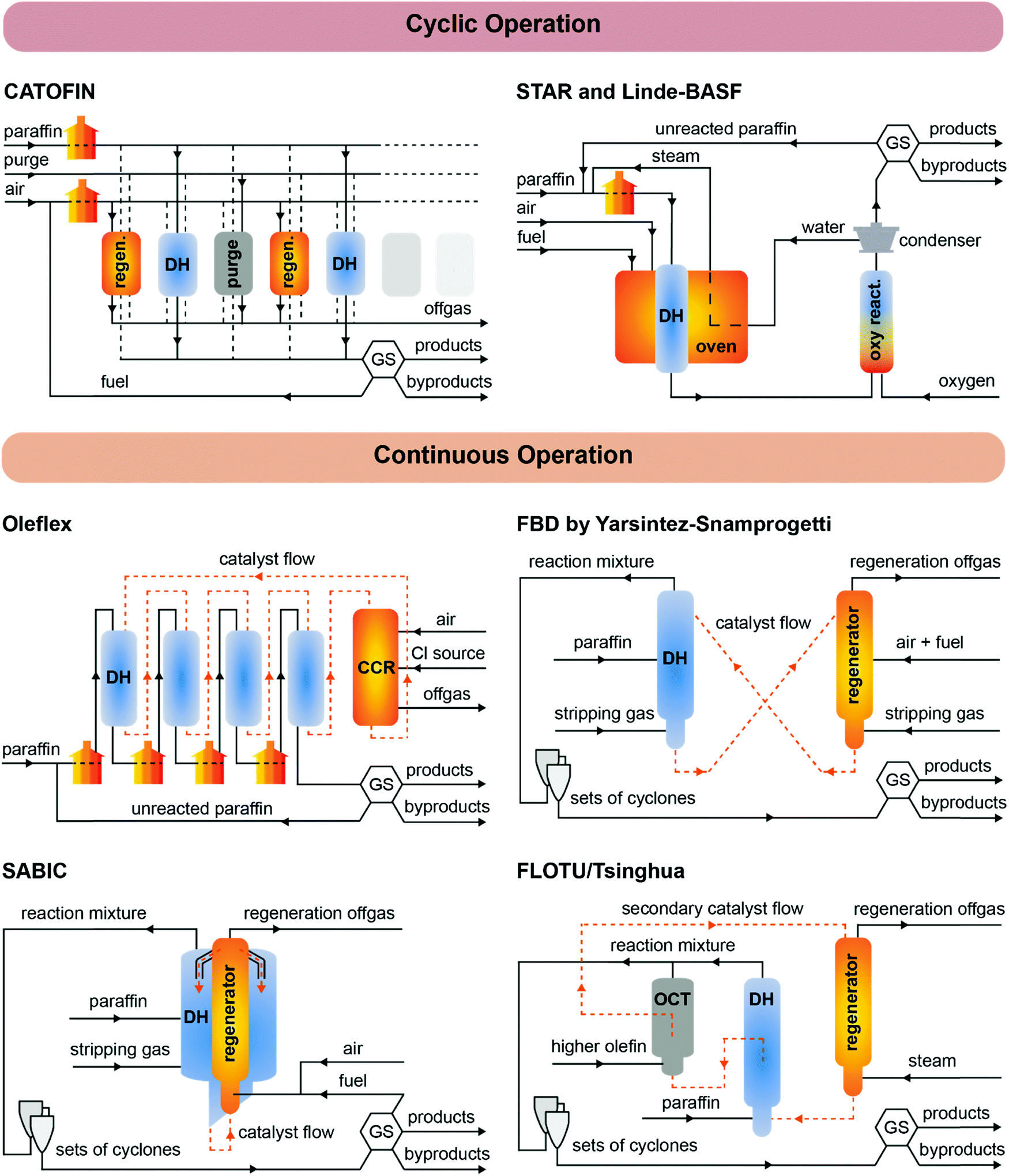 | ||
| Fig. 5 Schematic representation of commercial PDH processes. Top: CATOFIN, STAR and Linde–BASF cyclic technologies, in which the dehydrogenation (DH) reactor is subjected to alternating operation, purging and regeneration conditions. Bottom: Continuously operated technologies, in which the catalyst is continuously regenerated in a continuous catalyst regenerator (CCR). Heat sources and sinks are represented in orange and blue, respectively. GS = Gas Separation; OCT = Olefin Conversion Technology. | ||
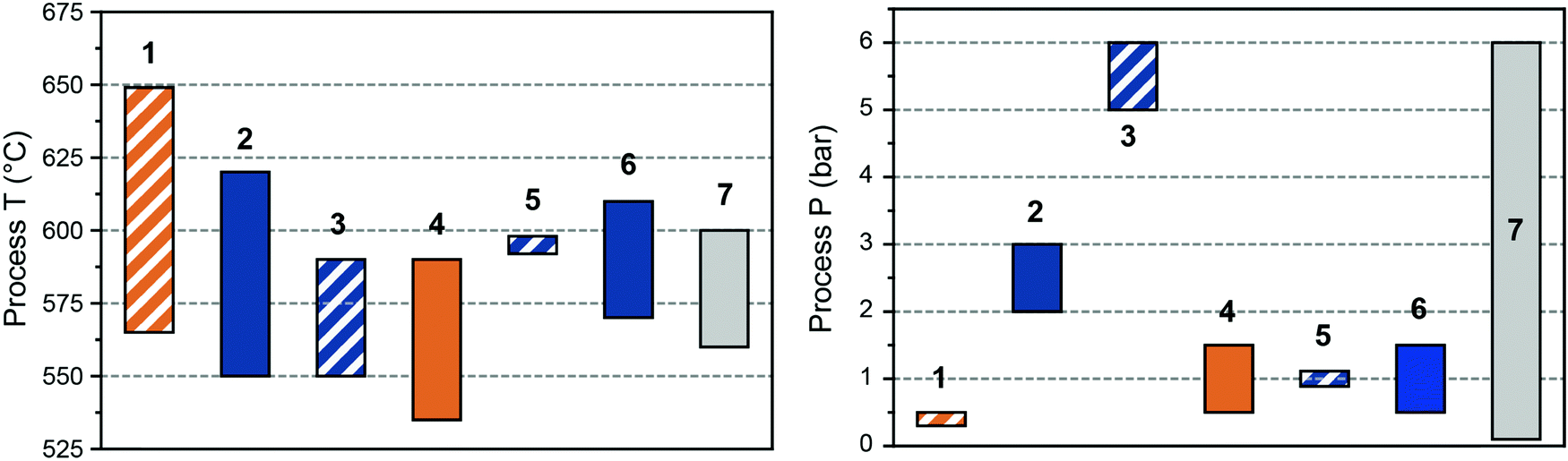 | ||
| Fig. 6 Overview of PDH processes conditions. Ranges of temperature (left) and pressure (right) used in the CATOFIN (1), Oleflex (2), Uhde STAR (3), FBD Yarsintez–Snamprogetti (4), Linde–BASF PDH (5), FLOTU/Tsinghua (6), and SABIC (7). Stripes fill: cyclic operation; solid fill: continuous operation; orange: Cr-based catalyst; blue: Pt-based catalyst; grey: Pt or Cr-based catalysts. | ||
Some general consideration can be made as to what are the key parameters to achieve high efficiencies and lower the cost of PDH: (i) high selectivity and product recovery, since propane consumption accounts for more than 80% of operation cost (i.e. 60% production cost), (ii) an efficient heat supply to utilize the catalyst during the endothermic reaction, (iii) a minimized impact of regeneration on the plant productivity, (iv) possibly operate at higher pressure (which although shifts the DH equilibrium to the left) to reduce cost of final gas compression to 15–35 bar, (v) improve energy efficiency by heat recovery.6,18,33 Integrating PDH plants with polypropylene (PP) plants can lead to numerous advantages, as outlined in ref. 33, such as combining the downstream gas separation units, using the hydrogen produced by PDH in the PP reactors and, vice versa, using the oxygen separated from the PP nitrogen production unit for e.g. regeneration operations in the PDH plant.
The same design used for PDH can be applied to the catalytic dehydrogenation of butanes (pure or in mixture) to make iso, normal, or mixed butylenes. Traditionally, this process has been used for the production of gasoline additives such as methyl and ethyl tert-butyl ether (MTBE and ETBE). Such compounds were phased out in the U.S. starting from 2000 due to public health concerns, but are still in use in other parts of the world, including Europe. The butane DH process can be also used to produce isobutene for butyl rubber or other specialty applications.
CATOFIN was the first PDH technology to be developed, and it is now adopted in more than 30 projects worldwide (of which 21 since 2017), with a capacity of more than 15 Mt per year. In the CATOFIN process, adiabatic fixed-bed reactors are connected in parallel and cyclically operated in DH, purge and regeneration mode at 565–650 °C. The catalyst is based on CrOx supported on alkali-promoted alumina, but it has constantly evolved over the years and the latest catalyst generation (CATOFIN-311, undisclosed formulation) was introduced in 2019. The process is performed in short dehydrogenation–regeneration cycles (15–30 min total), and thus 5 to 8 reactors are used for continuous process operation.
The heat released by burning off carbon deposits in the course of catalyst regeneration is used to perform the endothermic dehydrogenation reactions (Fig. 5, top left). Additional heat is provided by fuel gas during the regeneration step and by burners to preheat the reaction gasses. The reaction cycle is carried on (usually for about 8 minutes) until the temperature of the reactor bed is not sufficient to obtain an adequate conversion. The cycling time is shortened over the catalyst life to maintain stable performances. Catalyst dilution with inert material is also necessary to provide an energy reservoir for the DH reaction. Nonetheless, under practical DH operations, the reactor temperature can be lowered of as much as 100 °C due to the reaction acting as a heat sink.35 In order to overcome this issue, Clariant has patented a technology in which CuO/Al2O3 (promoted by e.g., Ca ad Mn oxides) can act as a heat-generating materials (HGM) under reducing conditions, by the exothermal reduction to Cu (see Table S18 for thermodynamic values, ESI†). This offers a number of advantages, such as optimization of the catalyst bed temperature profile, lower energy and air consumption during regeneration, and longer catalyst life.
The STAR process, also cyclic but using Pt-based catalysts supported on alkali promoted Ca and Zn aluminates, was first designed and commissioned by Phillips Petroleum for the production of isobutene in Cheyenne, WY (US, 1992), and in Ensenada, Argentina (1994).36 In 1999, Uhde acquired the technology, patents and catalyst, and commissioned three PDH plants thereafter, for a total capacity of 1.3 Mt per year projected for year 2021. There are currently 3 operational facilities using the STAR technology, and 3 more PDH plants have been licensed.37 The STAR technology is composed of two stages, loaded with the same catalyst: a reformer, where the dehydrogenation reaction takes place, and an oxyreactor (optional) to shift the equilibrium towards higher olefin yields. In the first stage, reformer-like oxidative dehydrogenation reactors are placed in a furnace, which grants an increasing temperature profile along the reactor bed from 510 to 550–580 °C. Steam is co-fed with propane to convert most of the coke to CO2, allowing a much long operation time (∼7 h) before regeneration is required (1 h). No addition of sulfur to suppress coke formation, or chlorine to improve catalyst activity via Pt redispersion is needed, as in the Oleflex technology (see below). Before the second stage, the products are cooled and added with steam to adjust the steam-to-hydrocarbon ratio. In the oxyreactor, oxygen is injected right above the catalyst bed with a patented technology, to selectively combust H2 and thus shift the thermodynamic equilibrium while providing additional heat for the dehydrogenation reaction (Fig. 5, top right). The final product mixture is cooled in a heat exchanger, where heat is supplied to the feed, steam production and downstream separation operations, to improve the overall process efficiency.33
A conceptually similar technology was developed by Linde–BASF–Statoil, as announced in 1992 by Linde AG,38 but to the best of our knowledge the company has so far built two semiworks: at the BASF Ludwigshafen petrochemical complex, and at the Mongstad Statoil plant in Norway.39 The original alumina supported CrOx catalyst was developed by Engelhard de Meern, and contained promoters such as Cs (up to 10 wt%) and Zr (up to 15 wt%). A PtSn catalyst supported on hydrotalcite was then introduced, claiming record one-pass conversions of 50% with competitive selectivity (91 mol% propylene selectivity compared to 90 mol% for Oleflex).6,39 However, no information on application of such technology can be found after the year 2000.
The Oleflex process is the second-oldest PDH technology, which first came on stream in 1990 in Thailand.31 It uses a Pt–Sn-based catalyst which is transferred through a series of radial-flow moving bed reactors, to be continuously regenerated in a continuous catalyst regeneration (CCR) unit, from which it is transferred back to the first DH reactor in a cycle of 5–10 days (Fig. 5, middle left). The heat is provided by feed pre-heating, interstage heating and regenerated catalyst coming from the exothermic CCR section (about 700 °C).40 In the CCR, air and chlorine are added to burn coke and redisperse sintered Pt by formation of oxychlorinated species. Sulfur in the form of dimethyl sulfide is also added to the DH reactors to suppress coke formation and avoid embrittlement of reactor metal walls by interaction with olefins. The formed sulfur has to be naturalized (after removing coke from the reactor walls) before exposure to air during maintenance to avoid polythionic acid damage of the reactor.31 Oleflex is now one of the leaders in PDH technologies with 68 PDH projects worldwide and a capacity of 7.9 Mt per year.
The Yarsintez–Snamprogetti technology is based on a Fluidized Bed Dehydrogenation (FBD) technology, developed in the former Soviet Union in the 1960s for synthetic rubbers production. The first FBD plant was licensed to Snamprogetti by SABIC in 1994 for the dehydrogenation of butane, aimed to produce MTBE for a plant in Jubail (Saudi Arabia).41 The technology is abbreviated as FBD-3 and FBD-4, where 3 stands for propane and 4 for isobutane. The catalyst is similar to the CATOFIN chromium oxide-based catalyst, but has a lower metal loading (around 15–19 wt%).42 The catalysts particles are suspended by the gas and air flow using a distributor, and have a 10–30 min residence time in the reactor and regenerator (Fig. 5, middle right). The bed behaves like a liquid with a high level of mixing, which favors heat and mass transfer phenomena, but increases attrition. The catalysts lifetime is indeed mostly determined by catalyst loss by attrition, which in turn depends on the circulation rate and the catalyst mechanical strength.17 New catalyst can be added in the regenerator during operation to compensate such loss, without affecting yield and reactor hydrodynamics. Notably, in the FBD technology less coke is formed compared to the Oleflex case, and thus some fuel has to be added in the generator to supply the heat necessary to satisfy the overall thermal balance (Fig. 5, middle right). After the regenerator, catalysts are stripped of adsorbed oxygenates by reduction in the stripping section, and finally transferred back to the FBD reactor to close the cycle.
In the last decade, four other continuous technologies have been proposed: the SABIC integrated Fluidized Bed Reactor (FBR), Dow's Fluidized Catalytic Dehydrogenation (FCDh™) process (announced in 2016), the FLOTU/Tsinghua University technology and the K-PRO™ fluidized bed process by KBR (announced late 2018). The advantage of the SABIC FBR design is that the dehydrogenation reactor is integrated with an internal regenerator, so that continuous production is achieved by a single unit. The heat produced by burning off coke is used to run the endothermic PDH reaction isothermally, with a lower catalyst circulation and thus reduced attrition (Fig. 5, bottom left).6 Temperature loss and attrition are further reduced by using just one transfer line. The technology also allows for more flexibility in the choice of catalyst: chromium-based or Pt-based catalysts can be used by changing the regeneration frequency (8–20 min vs. 6–12 h respectively).
The FLOTU/Tsinghua University technology is also an integrated process, piloted at Tsinghua University, in Beijing (China) in late 2009, in which the dehydrogenation reactor is attached to an OCT reactor, which interconverts higher olefins to lower olefins. Only after this stage a regenerator is placed (Fig. 5, bottom right). Notably, the process uses two catalysts, with different particle size: the principal catalyst (PtSn/Al2O3–SAPO-34, i.e. silico–alumino-phosphate molecular sieve zeolite) is used for the DH reaction and stays in the DH unit, while the smaller secondary catalyst (SAPO-34) is cycled through the system, and while it is inert in the DH reaction, it serves as an OCT catalyst and as a heat transfer material (from the regeneration unit to the DH unit).43 It was proposed that a transfer of coke from the principal to the secondary catalyst helps to increase the lifetime of the principal catalyst to 6–8 h, enhancing productivity.44
The FCDh™ process by Dow is based on the company's proprietary fluidized catalytic cracking technology, and it uses a Pt–Ga–K/Si–Al2O3 catalyst. The claimed advantages of the FDCh™ process over established PDH processes are: (i) 25% lower capital and 20% less energy consumption during operation, (ii) lower emissions in terms of CO2 and NOx and (iii) higher per pass conversion and propylene selectivity.34 The company announced in 2019 that it will retrofit the FDCh™ technology into one of its mixed-feed crackers in Louisiana (USA), to produce on-purpose propylene. The KBR's K-PRO™ process uses a non-precious metal catalyst and is based on KBR's Catalytic Olefins Technology (K-COT™) process, a commercial fluidized bed technology for converting mixed olefin and paraffin streams into high value propylene.26
2.2 Olefin metathesis and PTO
Olefin metathesis, one of the latest fundamentally new organic reactions discovered, rapidly came to large-scale industrial application in the late 1960s, for the production of petrochemicals, polymers and specialty chemicals. Here, we will focus on industrial metathesis reactions over heterogeneous catalysts relevant for the PTO reaction, while broader metathesis reviews and more details on the fascinating chemistry of polymerization metathesis reactions and the Shell Higher Olefin Process (SHOP) can be found elsewhere.21,27,45The olefin metathesis reaction was discovered serendipitously by Banks and Bailey, who were looking for alternatives to HF as a catalyst to produce high-octane gasoline by olefin-isoparaffin alkylation.46 They noted that, when using a Mo-based catalyst, propene was not alkylating the co-fed paraffin, but rather split to ethylene and butene: this set the basis for the Phillips Triolefin Process, patented in 1965.47 Interestingly, the term metathesis was introduced only later, in 1967, by Calderon, and the reaction mechanism, now widely-accepted, was first described in 1971 by Chauvin (awarded the Nobel Prize in Chemistry in 2005 together with Schrock and Grubbs).48 Chevron alkane disproportionation (patent US3856876A) followed Phillips in 1971: it introduced a one-pot method to disproportionate paraffins to new hydrocarbons having higher and lower molecular weights. They proposed to combine dehydrogenation Pt/Al2O3 supported catalyst with metathesis WO3/SiO2 supported catalyst, at 200–425 °C and 7–138 bar in presence of not more than 5 wt% of olefins to avoid deactivation. This process was thus developed to produce higher weight paraffins from short chain paraffins, with limited olefins production.
The Phillips Triolefin Process instead combined the dehydrogenation of propane feed with the disproportionation of the resulting propylene in ethylene and butenes in a single bed reactor.19,49,50 The first plant was already operational in 1966 in Montreal (Canada). The technology was operated in this way until 1972 due to the lower propylene demand at the time, but it is now mostly run in reverse to produce propylene by the OCT technology offered by ABB Lummus (now CB&I).
The original patent proposed a large variety of catalysts for the dehydrogenation reaction, such as oxides or other compounds containing Pt, Fe, K, Na, Cr, Mo, W, U, Be, Mg, Cu, Ca, Th but indicated Cr2O3/Al2O3 supported catalysts as preferred ones. WO3/SiO2 or alternatively MoO3/SiO2 were listed as disproportionation catalyst. DH and Met components are physically mixed and need to be activated at high temperatures up to 850 °C. The reaction takes place between 425–650 °C, at 0–100 bar. Using Cr2O3/Al2O3 and WO3/SiO2 in 50–50 wt% at 570 °C and 14 bar leads to 40.2 mol% of total ethylene, propylene and butane selectivity, due to competitive reactions, which lead to 46.1 mol% ethane + butanes, 6.1 mol% methane and 4.8 mol% C5+ paraffins selectivity and catalysts deactivation over time, as discussed in Section 3.
The inverse, OCT process is instead run with a butene mixture and ethylene, over a fixed bed containing WO3/SiO2 metathesis catalyst and a MgO isomerization catalysts (to isomerize 1-butene to the 2-butene which is consumed in the reaction).51 Conversion above 60% and selectivity to propylene exceeding 90% can be obtained. The first plant was operational in 1985, in Channelview, Texas (U.S.), with a capacity of 135![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t propylene per year: it used part of the ethylene produced by ethane cracking to produce 2-butene on-site by dimerization over a nickel catalyst, and finally convert the two to propylene by OCT.21 In a similar way, the technology can be applied to naphtha steam crackers to adjust the final balance of ethylene and propylene. OCT currently accounts for about 10% of propylene production worldwide, with new plants being commissioned by a number of parties, especially in Asia (Fig. 2c and 3).
000 t propylene per year: it used part of the ethylene produced by ethane cracking to produce 2-butene on-site by dimerization over a nickel catalyst, and finally convert the two to propylene by OCT.21 In a similar way, the technology can be applied to naphtha steam crackers to adjust the final balance of ethylene and propylene. OCT currently accounts for about 10% of propylene production worldwide, with new plants being commissioned by a number of parties, especially in Asia (Fig. 2c and 3).
Olefin metathesis is emerging as a technology for the synthesis of 1-hexene, a high value comonomer for polyethylene production, which is conventionally obtained by ethylene trimerization. The Comonomer Production Technology (CPT), announced in 2003 by Lummus/CB&I, and demonstrated at a plant scale in 2010,52 exploits 1-butene self-metathesis to produce ethylene, 3-hexene and propylene (as a byproduct by the reaction of ethylene with butene). 3-Hexene is then isomerized to produce the final product 1-hexene. Notably, replacing ethylene with butene can reduce the feedstock costs by more than half. Moreover, butene and hexene are produced without co-production of higher alpha-olefin (LAO) cuts, as is the case in conventional wide range LAO processes, which would require a wider business management.
Recently, a new PTO technology was patented by SMH Co., Ltd (WO/2017/001445, WO/2017/001446 and their family, see ESI†), in which systems of mixture of metals such as Pt, W, K, Se, Y, and Yb on mixed oxide of Si, Al, Zr, and Mg supports were employed. Such systems can be operated under relatively milder conditions compared to the prior state-of-the-art technology. The activation temperature was reduced to 400–600 °C and a conversion temperature of 350–550 °C was preferred. A total olefin selectivity of 86–94%, including, 11–22% ethylene, 50–61% propylene, and 15–23% butenes was obtained at the operating conditions. Multi-layered catalyst (WO/2018/108443) of DH, such as Pt on SiO2–Al2O3 support, mixed metal oxide such as Mg–Ca–Al–O, and/or zeolites, and W, or Mo on oxide supports were also developed by the company. The catalyst system improves olefin selectivity due to quenching of side reactions such as, (re-)hydrogenation of ethylene, hydrogenolysis cracking of propane, and catalyst deactivation. Such effects take place when mixture of DH-Met catalysts, feed, and products are in contact at high temperature.
3. Thermodynamics of the PTO reaction
The PTO reaction is a two-step tandem reaction: first, propane is dehydrogenated to yield propylene (Scheme 1, reaction (1)), and then two propylene molecules react in a metathesis step to yield ethylene and butene (Scheme 1, reaction (2)). Scheme 1 depicts the complete reaction network including the main side reactions that are usually observed under practical PTO conditions. Propane dehydrogenation is reversible, highly endothermic and involves volume expansion, so it is favored by low pressures and high temperatures according to the Le Chatelier's principle (eqn (1) and Fig. 7a). On the other hand, the metathesis reaction is thermally neutral since it only involves scrambling of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (eqn (2), lines in Fig. 7b, other details in ESI†).8,53 Therefore, typical temperatures employed in DH are 550–750 °C, while the metathesis reaction is usually operated at 300–600 °C to achieve sensible reaction rates.
C bonds (eqn (2), lines in Fig. 7b, other details in ESI†).8,53 Therefore, typical temperatures employed in DH are 550–750 °C, while the metathesis reaction is usually operated at 300–600 °C to achieve sensible reaction rates.| C3H8 ⇌ C3H6 + H2 (ΔH0298K = 124.3 kJ mol−1) | (1) |
| 2C3H8 ⇌ C2H4 + C4H8 (ΔH0298K,trans-butene = 0.7 kJ mol−1, ΔH0298K,cis-butene = 5.0 kJ mol−1) | (2) |
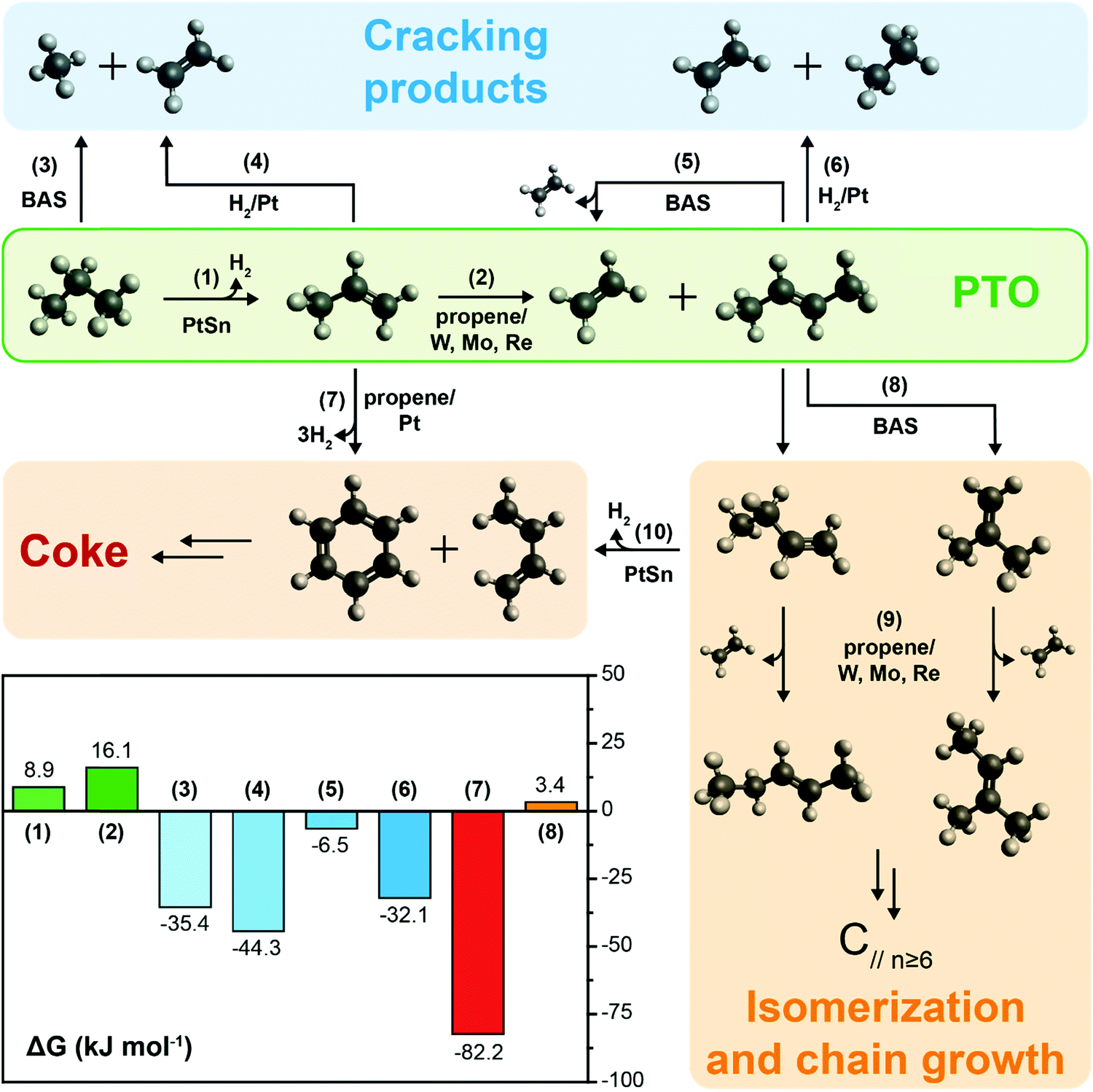 | ||
| Scheme 1 PTO reaction network and reactions free energy at 600 °C. Propane dehydrogenation (1) yields propene, which in turn undergoes self-metathesis (2) to produce ethylene and a mixture of 2-butenes (the most stable trans isomer is shown). Cracking (3, 5) and hydrogenolysis (4, 6) reactions yield a mixture of lower hydrocarbons, while propylene coupling (7) can initiate coke formation by e.g., a Diels–Adler type of addition of benzene and 1,3-dibutene. Isomerization of 2-butenes over Brønsted Acid Sites (BAS, reaction (8)) yields terminal butenes that may react further by metathesis (9) yielding linear and branched C5, which can react further inducing chain growth. The bars (bottom left) report the Gibbs free energy values at 600 °C for the main reactions involved in the network: PTO is thermodynamically unfavored against other side reactions. Kinetic control can be achieved by ensemble control on Pt by addition of Sn and inhibiting BAS. | ||
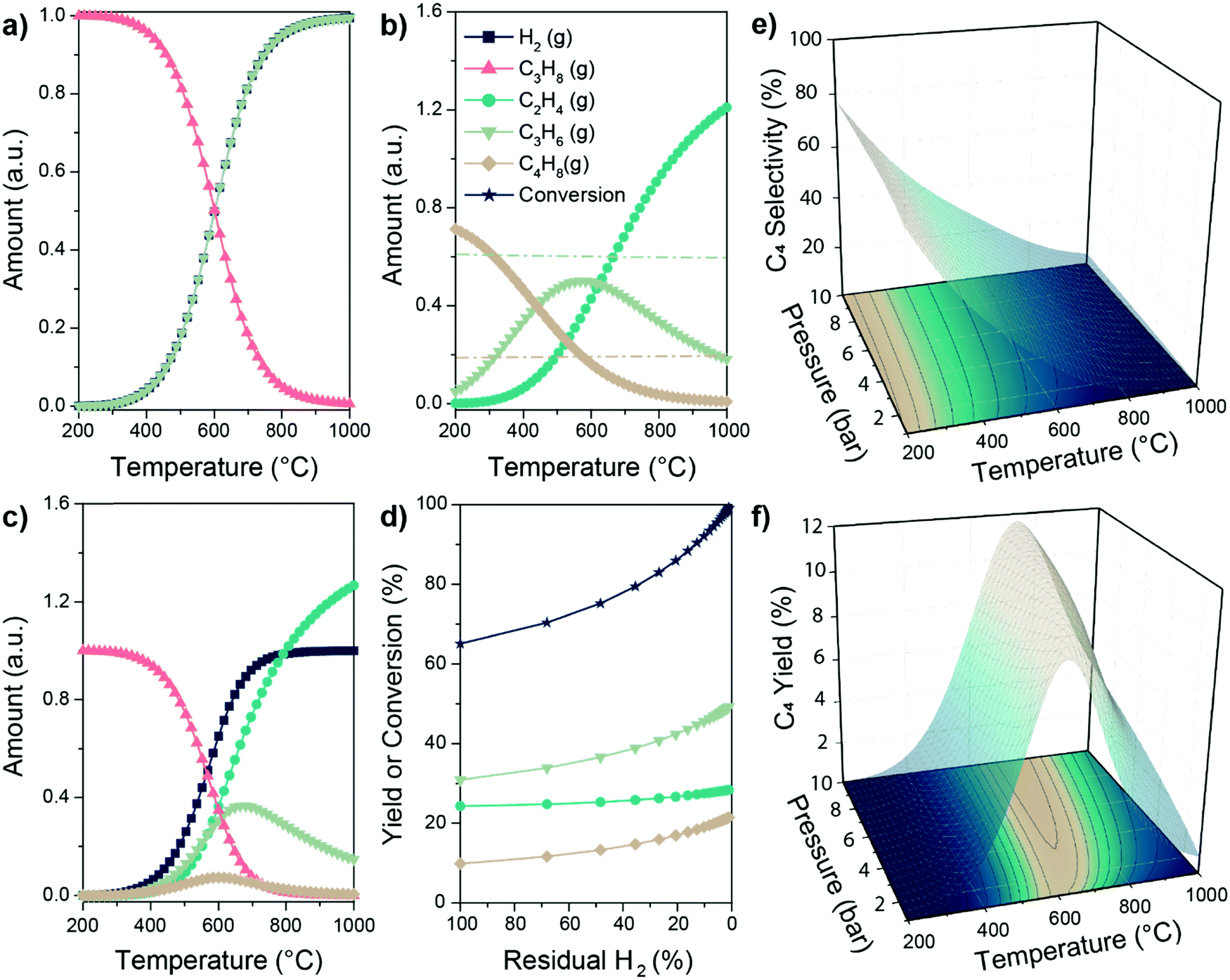 | ||
| Fig. 7 Thermodynamics boundaries of the PTO reaction. Equilibrium amounts of reagents and products involved in (a) propane dehydrogenation, (b) propylene metathesis and (c) PTO reaction according to the Gibbs free energy minimization algorithm, as a function of reaction temperature. Lines in (b) correspond to the amount of C3 (light green) and C2 or C4 (light brown) calculated according to the sole metathesis reaction (see ESI† for details); (d) increase in yield and conversion in the PTO reaction enabled by selective removal of H2 in e.g. a membrane reactor at 600 °C (40% of produced H2 removed as initial condition for each datapoint); (e and f) C4 selectivity and yield as a function of reaction temperature and pressure in the PTO reaction. Equilibrium calculations were performed through HSC Chemistry 9.1 software. For further thermodynamics calculations including all possible species, see the ESI.† | ||
So far, we discussed the thermodynamic boundaries in place for the sole species directly involved in the PTO tandem reaction. However, as summarized in Scheme 1, the following side reactions must also be considered when optimizing the reaction conditions: (3, 5) thermal or catalytic cracking, (4, 6) hydrogenolysis, (7) coke formation, (8) isomerization and (9) metathesis of isomerized olefins leading to higher olefins. Thermodynamic calculations including the main products of side reactions, such as methane, ethane, BTX (benzene, toluene, xylenes), and coke, can be found in the ESI,† (Fig. S2 and S3). Notably, byproducts formation is thermodynamically favored at relevant reaction conditions (Scheme 1), due to these general considerations: (i) C–C bonds are less thermodynamically stable than the C–H bonds, favoring cracking over dehydrogenation; (ii) olefins are more reactive than paraffines due to the presence of allylic C–H bonds and CC bonds, which can react to form to coke, in turn leading to deactivation. Therefore, a proper balance between propane conversion and stability must be found, and kinetic control over the reaction by catalyst design is pivotal in order to yield olefins with good selectivity and hinder catalyst deactivation by coking. We will focus on this aspect in the next section.
4. Catalytic aspects in the PTO reaction
In this section, the most relevant dehydrogenation and metathesis catalysts are presented and contextualized with respect to PTO technologies for light olefins production. Combining propane dehydrogenation and metathesis catalysts in PTO brings about both challenges and opportunities, related to reactor design and operation, and the chemistry and physicochemical properties of the involved materials. First, we will briefly summarize structure–activity relationships in PDH and Met catalysts. With this understanding at hand, we will then critically discuss the effect of combining PDH and Met catalysts in different contact states in the reactor (e.g., physically mixed or separated bed), in terms of (i) thermodynamic boundaries shifts and heat control, (ii) exposure to different gas mixtures and temperatures during catalysts activation, operation and regeneration, (iii) interactions between the components of the catalysts, in terms of promotion and poisoning. We emphasize what theoretical and experimental studies are needed to gain fundamental insights into catalysts design specific for PTO. We anticipate that choosing the right combination of materials and operation conditions will have a great impact on catalysts performance and lifetime in the PTO technology. Recent, detailed reviews on PDH and Met catalysts can be found elsewhere,26,56 and are out of the scope of the present review.4.1 Dehydrogenation catalysts overview
Pt-Based and chromium oxide-based supported catalysts, respectively involved in the Oleflex and CATOFIN technologies (Fig. 5 and 6), represent to date the state-of-the-art commercial catalysts for PDH commercial processes and are the main catalysts used in the existing Chevron20 and Phillips19 light alkanes dehydrogenation and metathesis technologies previously described. Both Pt-based and CrOx-based catalysts can achieve high activity and selectivity to olefins, being also adaptable to different types of reactors. The preference of one metal or the other may depend on many factors: for Pt, drawbacks may be low availability and high price, the need for a purified feedstock due to high tendency to poisoning and the required elaborate regeneration procedures; while for CrOx-based catalysts the main drawback is the formation of toxic and carcinogenic Cr6+, which raises sustainability issues about catalysts disposal.57Given their wide commercial use, the structure–performance relationships of these catalysts and their reaction mechanisms have been extensively studied in the past years, with the aim to optimize the catalyst design and enhance catalysts resistance to deactivation.8,57–60 Despite these efforts, deactivation by coking still demands continuous regeneration. In the search for alternatives, many other catalytic formulations have been explored, such as metal oxides (e.g., VOx, GaOx, FeOx), single atom catalysts (SACs) and nanocarbons, but so far progress has been incremental.23 While the reaction mechanism and the nature of active sites varies for these different catalysts, some concepts in catalyst design can be extrapolated to the general case, in terms of activity, selectivity and stability. We will here use Pt-based and CrOx catalysts as an example, and highlight the specifics of other emerging catalysts.
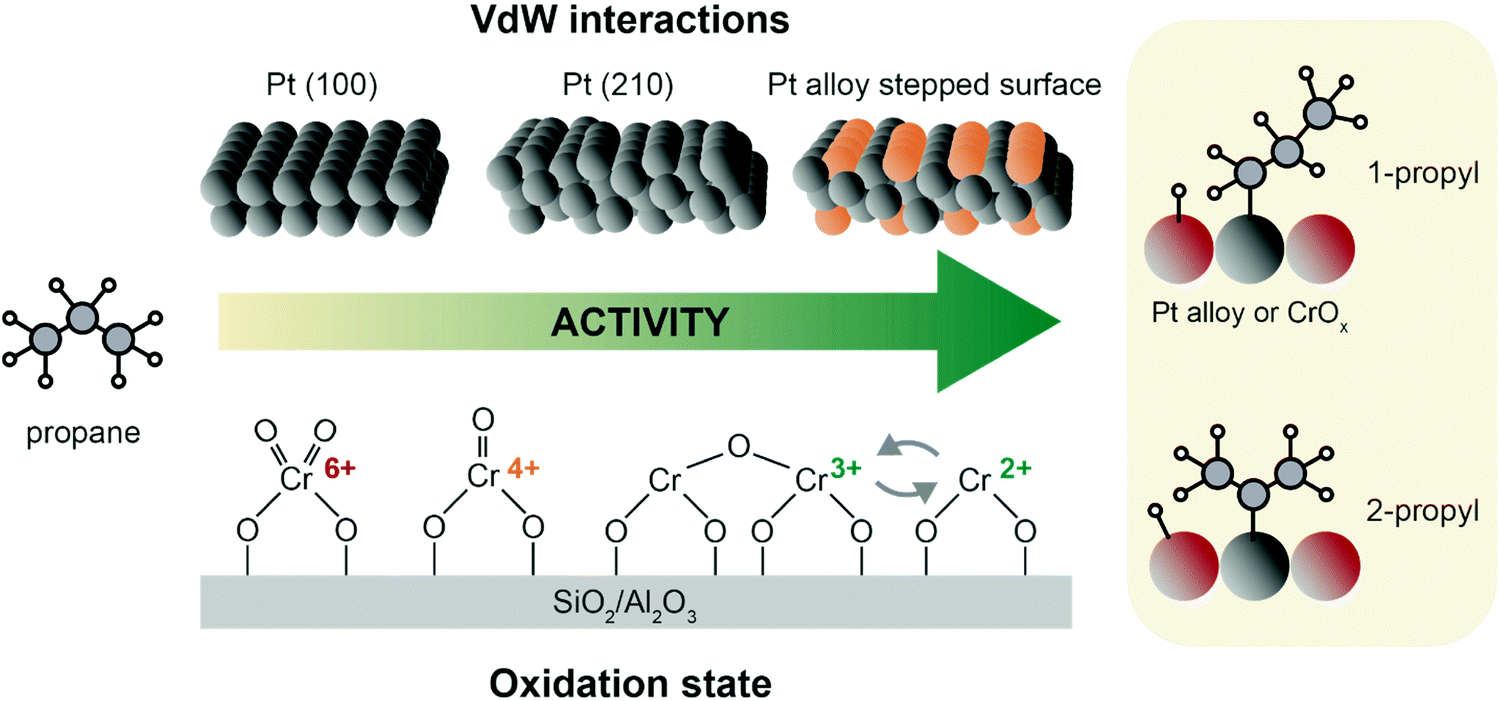 | ||
| Fig. 8 Propane adsorption and activation on archetypical PDH catalysts. On Pt, interaction with propane is governed by van der Waals interactions, and thus favored on stepped edges. Alloying with e.g. Sn favors H abstraction by a downshift of the active phase d-band.61,62 For CrOx and other oxide catalysts, activity is favored by higher CrOx dispersion and lower oxidation state, with Cr3+ and Cr2+ being the most active species.70 After H abstraction, 1-propyl and 2-propyl are formed without much preference.8 | ||
The fundamental steps in PDH over chromium oxide based catalysts are: (i) propylene adsorption, (ii) C–H cleavage and O–H bond formation, (iii) second C–H cleavage and H adsorption on Cr site, and (iv) propylene desorption and H2 formation.8,57 On CrOx and other metal oxide catalysts, propane adsorption is strongly dependent on the exposed Lewis acid sites, which in turn vary with M–O coordination, cluster sizes, and oxidation state.69 The active sites of chromium-based catalysts are defective Cr3+ and Cr2+ sites, that are usually found as Cr2+/Cr3+ mixtures depending on metal loading and type of support (Fig. 8, bottom). The precursors of these active species are Cr5+/Cr6+ that are reduced during pre-treatment or simply by in situ reduction by propane. Cr sites undergo complex changes in oxidation state during catalysts preparation: upon calcination, four-fold coordinated Cr(VI) (prevalently in mono-oxo form) precursor is formed; upon reduction a pseudo-octahedral Cr(III) site is generally obtained. However, when chromium is highly dispersed, the reduction can further proceed and highly defective Cr(II) species are also formed: this happens at low loadings and preferably on silica, which due to its electronic properties facilitates reduction more than alumina.70
Weckhuysen et al.8,60 have extensively described the factors that influence chromium oxide based catalysts in two reviews, pinpointing how chromium oxidation state, coordination and loading are key features to understand the reaction mechanism of these catalysts. The oxide loading in the catalyst can influence the catalytic performance, with the formation of large crystalline domains71 being detrimental for the reaction. Some possible active structures of CrOx are depicted in Fig. 8, where clustered and monomeric Cr3+ sites are considered to be the active site.70,72,73
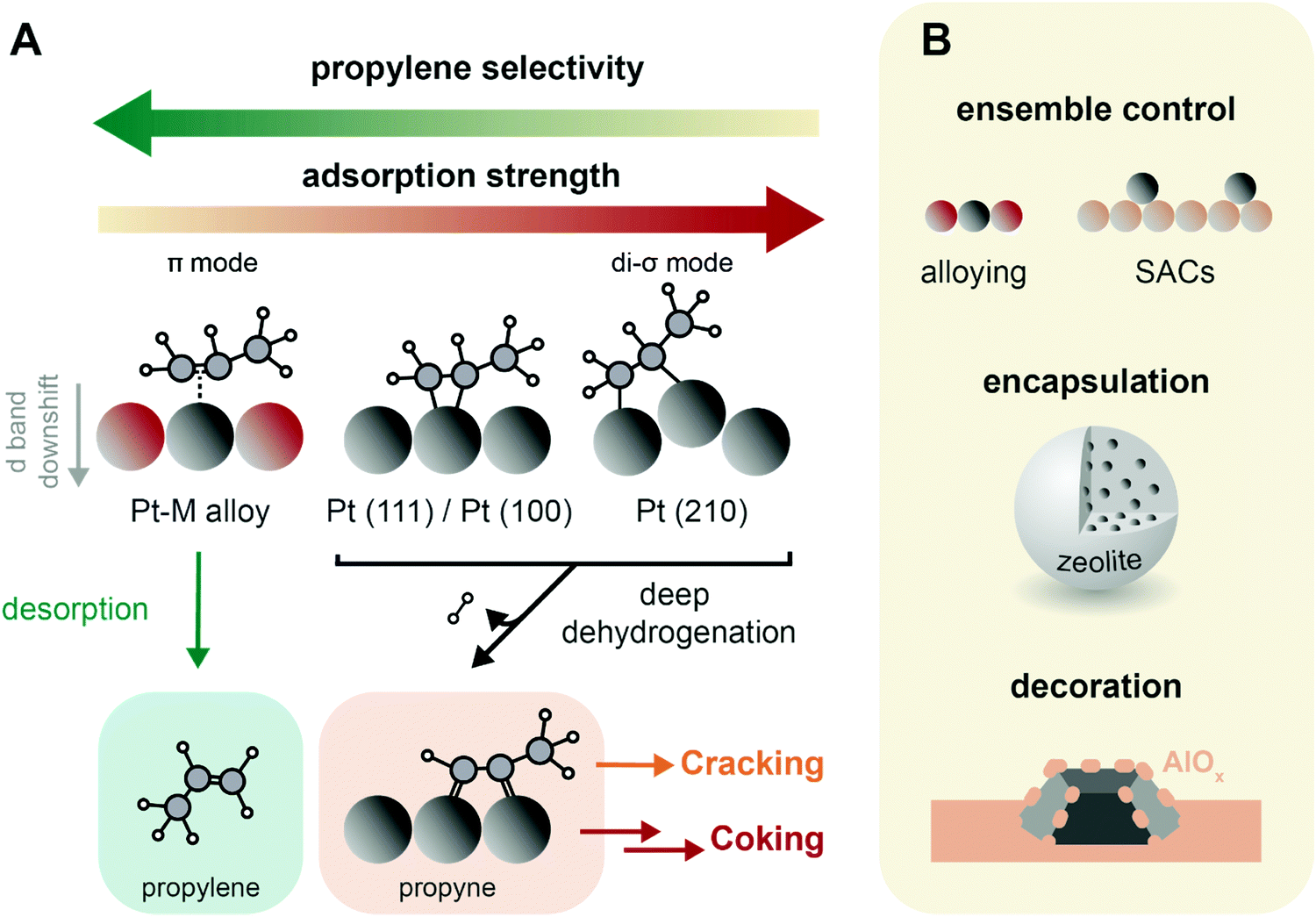 | ||
| Fig. 9 Catalyst design concepts for selectivity and stability of propane dehydrogenation (PDH) catalysts. (A) Propylene selectivity is inversely related to its adsorption strength on the catalysts active sites, where it forms di-σ and π-adsorption modes, respectively leading to a strong and weak interaction.62 The preferential pathways in this simplified scheme are indicated with arrows, and propyne is highlighted as crucial intermediate in the formation of coke and cracking products.83,85 (B) Main strategies to improve PDH catalysts stability: (i) ensemble control by e.g., alloying of Pt with Sn or producing atomically dispersed catalysts (SACs) by strong anchoring on a support such as CeO2; (ii) encapsulation in e.g. zeolites; and (iii) decoration by the support, either by strong metal-support interactions or by post-modification methods such as atomic layer deposition. | ||
When Pt nanoparticles are added with a promoter metal, the geometric and electronic structure of the active sites change, possibly enhancing catalytic performance. Alloying Pt with transition metals leads to a weaker adsorbed, π-chemisorbed or physisorbed propylene, and results in higher selectivity.62,74,80 On PtSn2(111) surfaces, propylene is adsorbed non-covalently.64 This is due to both a geometric and electronic effect. The geometric effect is due to active Pt sites separation, and/or decoration of the surface. Sn atoms are preferentially found on step edges, such as Pt(211) surfaces: this lowers the activity towards deep dehydrogenation and cracking on these edges.65,68 Electronic effects consist in a lowering of the energy of the Pt d-band center, which weakens the bond strength between the adsorbate and the metal, increasing propylene selectivity.61,81 Alloying Pt with 15% Sn increases filling of the Pt 5d-band, decreasing the adsorption strength of the molecule and therefore the occurrence of coke formation, hydrogenolysis and cracking reactions.82 The addition of promoters also results in less C* (and thus coke) formation, and in lower cracking selectivity, since the deeply dehydrogenated species propyne (CH3CCH*) was found to be the only C3 species that energetically prefers C–C bond breaking to C–H bond breaking (Fig. 9A).83 Moreover, alloying promotes the migration of polyaromatic species to the support, slows down particle growth, hinders hydrogenolysis and isomerization competitive reactions and neutralizes support acidity.8
A similar propylene desorption vs. activation concept is valid for CrOx, but structure–activity relationships are strongly dependent on CrOx clustering and interactions with the support. For example, when SBA-15 is used as support four-fold coordinated Cr3+ are more active and show higher selectivity than Cr surface sites on chromium oxide, while when alumina is used, oligomeric species show higher activity/selectivity.84 Propyne was also found to play a role in CrOx deactivation in Kinetic Monte–Carlo (kMC) simulations.85
Despite these advances, catalysts regeneration is generally required, due to the intrinsic tendency for coke formation under PDH conditions. This is done at high temperature (up to 750 °C) and oxidative environments, sometimes in the presence of Cl to help redispersing Pt and recover catalytic activity.6 Such practices may be a deal-breaker for certain metathesis catalysts in PTO operations, and regeneration strategies should be tweaked to allow for PDH activity restoration without affecting metathesis activity. We will discuss this aspect further in Section 4.3.
The discussed structure–activity relationships concepts can be applied to a wide range of PDH catalysts, but their implementation in catalyst design requires a specific knowledge on the type and distribution of active sites. The reducibility of the oxide is known to play a role, as upon H adsorption on Cr3+ and O a charge transfer occurs from O–H to Cr, which is reduced to Cr2+.89 For VOx-based catalysts, similarly to CrOx, oligomeric and monomeric vanadium clusters with a tetrahedral structure are believed to be the most active.90 For ZrO2-based catalysts, active sites can be created by introduction of vacancies, where unsaturated Zr atoms are able to homolytically split C–H bonds with a low activation energy.91 Over Ga/ZSM-5 catalysts, active species were identified as [GaH]2+ ions in proximity with a framework Brønsted-acid site.92 For nanocarbons, oxygen functional groups are believed to be key for activity.93 The diversity of proposed active sites gives an idea of the complex quest of developing better PDH catalysts by rational design, which shall be based on insights from in situ and operando characterization and theoretical calculations.94
4.2 Metathesis catalysts overview
WOx, MoOx and ReOx are the most widely studied active phases of heterogeneous metathesis catalysts.95 ReOx-Based catalysts are generally the most active ones, achieving conversion at room temperature (e.g. Re2O7/Al2O3), and being able to convert functionalized olefins if activated with organometallic promoters.96 Nonetheless, ReOx catalysts were never used in the petrochemical industry, due to their fast deactivation by poisoning upon exposure to oxidating agents and other common impurities in industrial streams. MoOx catalysts are somewhat intermediate between rhenium and tungsten, can operate at 50–250 °C and are mainly used in the Shell Higher Olefin Process (SHOP).97 However, they also tend to deactivate due to poisoning and high mobility of Mo oxides.98 Despite being the less active of the three (conventional operating temperatures are 300–500 °C), WOx-based supported catalysts (typically WO3/SiO2) are the most used in the combined dehydrogenation and metathesis PTO industrial technologies, due to their resistance to deactivation and to small oxygenates concentration in the feedstock, fast regeneration and high resistance to embrittlement over time.56,95,99–101C bonds in olefins into alkylidene (carbene) intermediates, which react with a second olefin molecule to yield the metathesis products and a new carbene species. The most widely accepted mechanism for metathesis was proposed by Herisson and Chauvin (Fig. 10B), and it involves a [2+2] cycloaddition forming a metallacyclobutane intermediate, and a cycloreversion. The reaction was shown to proceed with different RDS, depending on the available metal sites, adsorbate molecule (ethene or trans-2-butene) and temperature.102,103 In any case, the metal alkylidenes active sites are formed in situ, via partial, 2-electron reduction of the metal (M6+) oxide by the olefin to low-valent M4+ (Fig. 10A). Pre-treatments under inert or reducing conditions (e.g., He, H2 or alkanes) have been shown to favor the formation of the active sites by removing strongly bound oxygen.104–106 This has direct implications on the way the catalyst should be activated and regenerated during PTO, as discussed in Section 4.3.
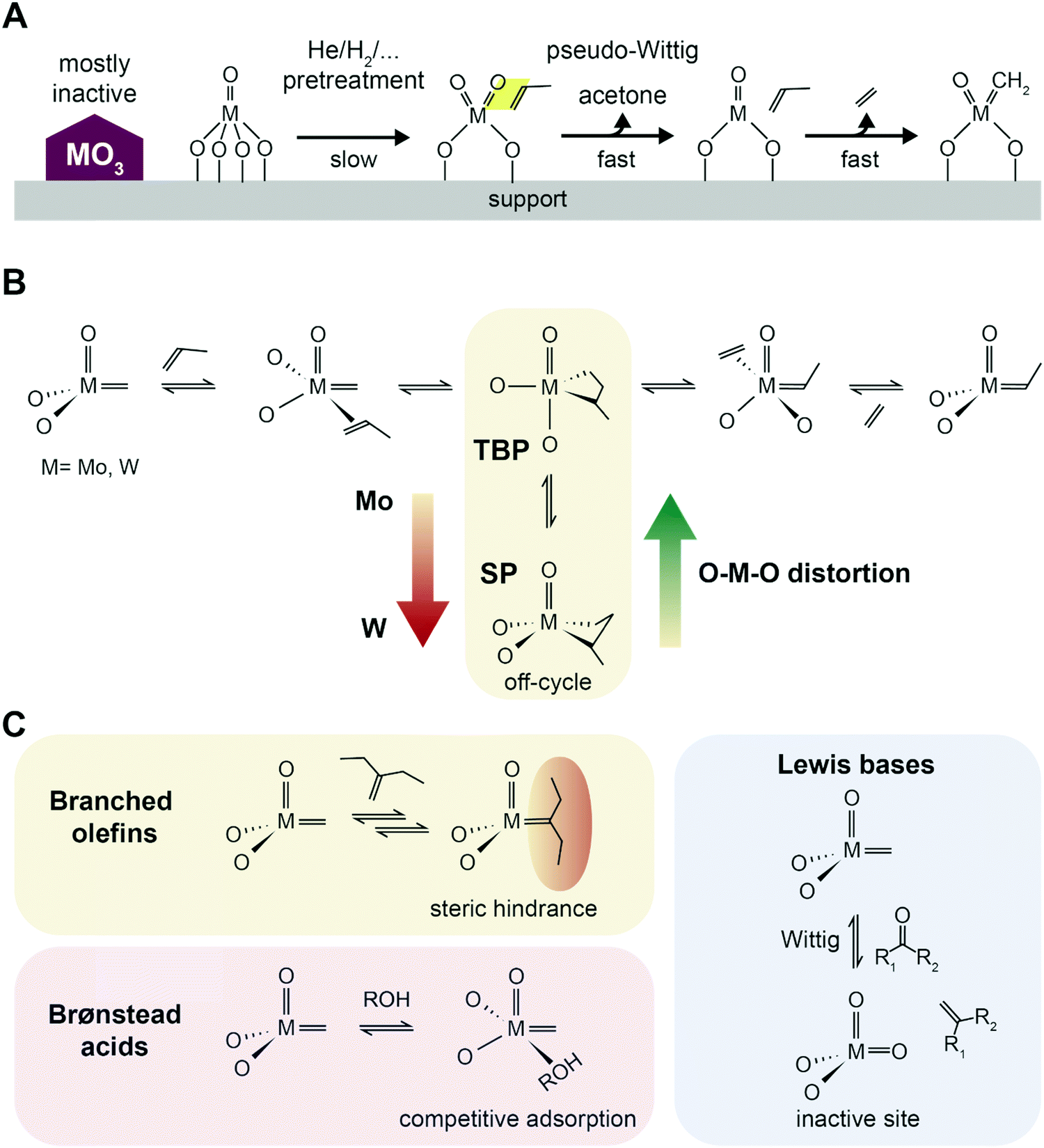 | ||
| Fig. 10 Initiation, propagation and poisoning of catalytic metathesis. (A) Proposed scheme of active site formation by reaction with propylene in a pseudo-Wittig oxygen removal, and formation of alkylidene species. High temperature pretreatments under reducing or inert conditions speed up activation. Adapted with permission from ref. 105. Copyright 2016 American Chemical Society. (B) Proposed Chauvin's reaction scheme of propylene metathesis with the TBP–SP isomerization (TBP = trigonal-bipyramidal geometry, SP = square pyramidal geometry) based on insights from Surface Organometallic Chemistry (SOMC). Adapted with permission from ref. 97 – published by The Royal Society of Chemistry. (C) Effect of poisoning agents commonly present in industrial streams: branched olefins, causing deactivation by steric hindrance on the active site; Brønsted acids competing adsorption; and Lewis bases inducing deactivation by oxidation of the active site via a Wittig reaction.100 | ||
As for chromium-oxide based catalysts for alkanes dehydrogenation, supported metal oxide catalysts for olefin metathesis, such as WOx/SiO2, are very complex systems, due to the changes in geometry and oxidation state of the metal center under different reaction environments.95,105 Several mechanisms of active site formation have been proposed, which will not be discussed herein.97,107 The in situ activation, however, results in active sites accounting for only a few percentage (5–10%) of the total metal sites, which is due to many factors and makes the investigation of active site formation challenging. First of all, the formation of alkylidenes is slightly thermodynamically unfavored, and second, the initiation is believed to occur with participation by proton transfer from the acid OH groups of the support.108 It is thus widely accepted that to achieve better activity, the metathesis catalyst should be well dispersed and in the form of defective clusters, to avoid formation of largely inactive crystalline trioxide phases99 In the case of W, WOx surface sites can be simultaneously found together with crystalline WO3 nanoparticles depending on surface coverage, with the optimal loading for high tungsten dispersion being below 8 wt% on a 332 m2 g−1 surface area SiO2.95
The stability of the metallacyclobutane (MCB) intermediate is another general descriptors of metathesis activity identified based on insights from Surface Organometallic Chemistry (SOMC), which allow for precise design of supported catalysts.28,109 Tungsten catalysts tend to form stable and detectable MCB, while on Mo-based catalysts MCB are rarely reported. Based on computational and SOMC solid state nuclear magnetic resonance (SS-NMR) results, this was attributed to the fact that MCB, formed in the trigonal-bipyramidal geometry, can more easily isomerize to the more stable, off-cycle square pyramidal geometry on W than on Mo (Fig. 10B). Notably, such intermediates were not yet identified on ill-defined heterogeneous catalysts, but recent development in SS-NMR will likely allow to observe the specific signals of the MCB intermediates in the near future and help in rational synthesis of better supported metathesis catalysts.110
dimerization to C4 and oligomerization to C4–C6 alkanes.95 Non-stoichiometric metal oxide phases have been shown to have higher selectivity towards primary metathesis products,99 but higher temperatures and acidic supports such as silica favor isomerization and reduce selectivity.100 Dehydroaromatization to benzene and hydrogenolysis to CH4 and ethylene were also reported at higher temperature. Therefore, controlling and optimizing reaction conditions is here just as important as catalyst design.
WOx/SiO2 catalysts were shown to be stable against poisoning from branched olefines, which usually result in deactivation by steric hindrance around the metal center, mostly due to the higher operating temperature employed.100 Oxygenates containing a Lewis base group such as a carbonyl group (e.g., acetic acid, aldehydes, ethyl acetate) can coordinate to the alkylidene active site and oxidize it back in a Wittig-type reaction, forming a CC double bond in the leaving molecule. The resulting W6+ oxide is a non-active site which has to be re-activated by the olefin (as described above in the Activity section). Brønsted acids such as water or ROH (e.g., butanol) will instead result in reversible deactivation by competitive adsorption.100 The effect of contaminants of common PDH streams should thus also be explored in the context of the PTO reaction.
Finally, particle growth leads to deactivation by loss of surface sites and transformation in crystalline MO3 phases. This process is promoted by high temperature, and is strongly affected by gas composition and interaction with the support. This is extremely relevant for PTO operations and catalysts regeneration procedures after coking.107 Mo and Re are particularly prone to deactivation by coalescence because of the high volatility of their oxides (vaporizing as Re2O7 dimers111 or MoO3 trimers112) and of the oxide–hydroxide MoO2(OH)2, already around 500 °C. Tungsten oxides on the other hand are not as volatile, and can resist higher operating temperatures. Accordingly, Mo was observed to be very mobile on SiO2, but with contrasting results showing coalescence or redispersion depending on the amount of water in the feed, with dehydration resulting in spreading of Mo oxide clusters, as revealed by in situ EXAFS and Raman spectroscopy.98
As in the case of PDH catalysts, emerging strategies for thermal stabilization involve incorporation of the active phase in zeolites, which resulted in higher intrinsic activity for MoOx@silicate when compared to MoOx/SiO2.113 Similar observations were made in the case of WOx@silicate.114 Notably, the introduction of heteroatoms in the silicate (such as Nb) lead to even higher metathesis activity, which showed correlation with the calculated OWO angle.115 This is in accordance to SOMC insights showing that distortion of metal complexes using different ligands resulted in unfavored MCB isomerization, and thus less off-cycle square planar MCBs.97 These results further suggest that mixed oxides of WOx may also result in better Met activity, which may be especially relevant in the case of intimate contact state between PDH and Met catalysts in PTO technologies.
4.3 Catalysts challenges and opportunities in the PTO technology
When combining propane dehydrogenation and propylene metathesis in one industrial process one must consider the effect of the operating conditions of the reaction (i.e., temperature, exposure to feedstock, other gases, products), activation and regeneration procedures, and contact state and relative amount of the two materials in the reactor. The contact state is the first factor to consider as it influences all the other aspects. In Fig. 11, the possible contact states between PDH and Met are summarized from most separated to closest: (a) separate reactors, where the PDH and Met are run in tandem; (b) separate PDH-Met bed, in which the first part of a reactor is filled with PDH catalyst bodies (usually in extrudates form); (c) physically mixed bed, in which the PDH-only and Met-only catalyst bodies are mixed together; (d) mixed in extrudate, where PDH and Met powdered catalysts are mixed together in the same catalyst body, possibly with core–shell structures; and (e) mixed on support, where PDH and Met active phases are mixed on the same powdered support (e.g., by co-impregnation of precursors).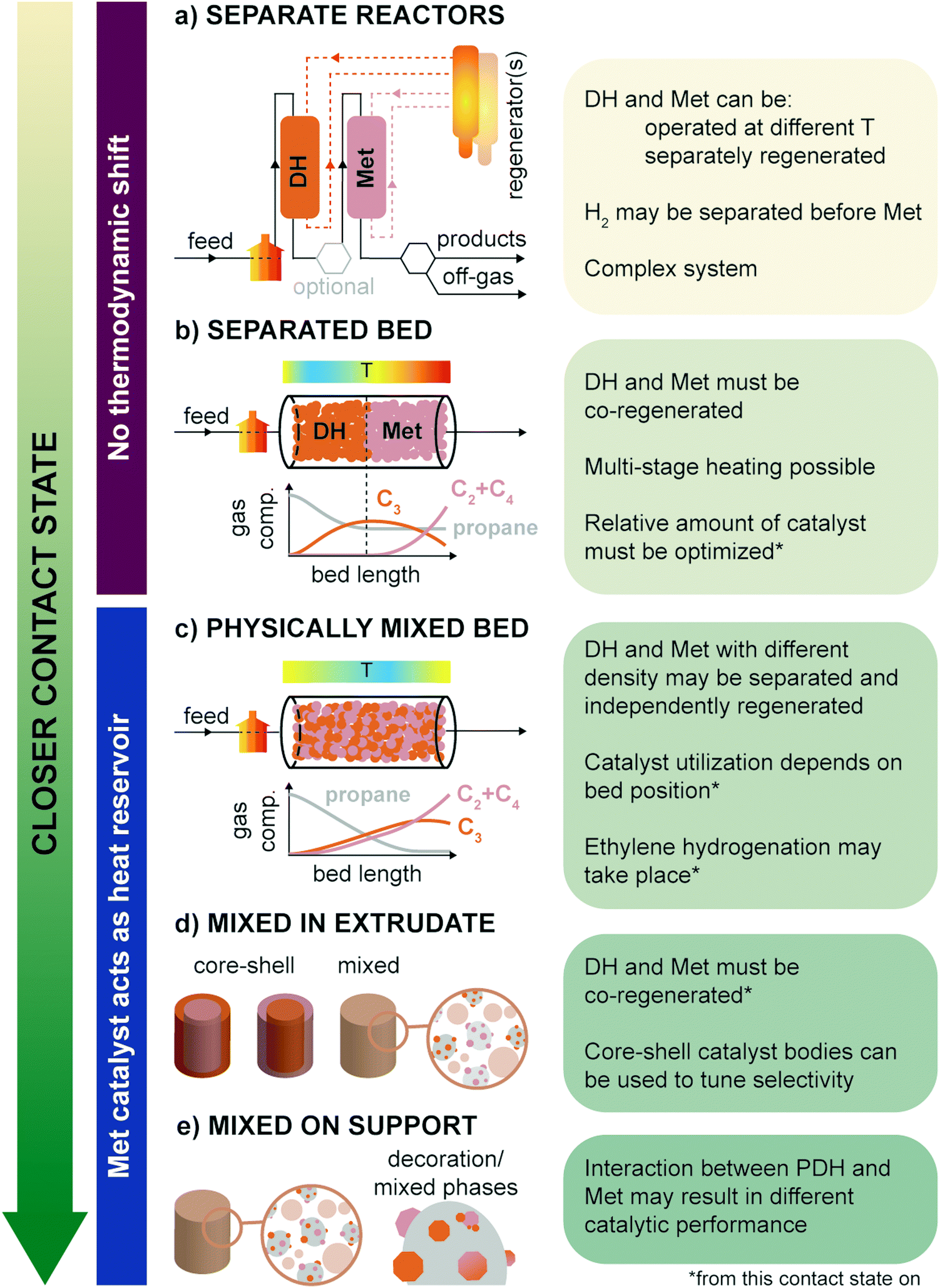 | ||
| Fig. 11 The possible catalysts contact state in the propane-to-olefins reaction are listed from most separated configuration to the most intimate one. Pros and cons of each contact state are highlighted. | ||
The separate reactors mode (Fig. 11a) allows for the most flexibility but is also the most complex. Here, PDH and Met may be (i) run at different temperatures, (ii) separately or jointly regenerated, depending on the catalysts regeneration system, and (iii) exposed to controlled streams, by e.g., additional separation of H2 before the Met bed. Being able to independently operate and regenerate the PDH and Met catalysts would allow to use more active Mo-based catalysts, which would otherwise be deactivated by particle growth at the high PDH temperature and in the oxidizing environment found in PDH regenerators. On the other hand, the effect of H2 was shown to be beneficial for Met catalysts operation, so its separation from the gas feed is not an added value.106 For example, H2 was found to lower coke deposition and increase the number of reduced catalytic active sites in ReOx catalysts.56
A remarkable drawback of the separate reactors mode is that the PDH reaction runs independently of the Met reaction, and thus there is no shift in the PDH thermodynamic equilibrium, as discussed in Section 3. A similar case can be made for the separated bed mode (Fig. 11b): since propylene is only converted in the second half of the reactor, the PDH equilibrium is unaltered, and it governs the amount of propane which can be converted. Moreover, a sufficient amount of catalyst or an adequate gas flow must be chosen to reach equilibrium and avoid high propane slips to the Met zone. The relative amount of PDH and Met catalyst is a general concern for all operating modes, and requires optimization for a particular installation and catalysts intrinsic activity.
To keep the PDH and Met bed separate, practical regeneration should resemble the CATOFIN process, with multiple reactors in cyclic operation (reaction conditions/purge/regeneration stages). This means that the two catalysts must be regenerated together, limiting (at least in principle) the Met catalysts choice to W-based catalysts. Multi-stage heating and change in regeneration procedures may however overcome this problem. Differential heating would also avoid the formation of strong thermal gradients along the reactor. Notably, in existing PDH technologies heat is provided by the pre-heated gas flow and by the catalyst itself (heated during the regeneration treatment). Since PDH is an extremely endothermic reaction and Met is almost thermally neutral, heat will be consumed much more in the first part of the reactor, leaving the Met zone hotter, and favoring isomerization and coking.
A more intimate contact of PDH and Met catalysts, such as in physically mixed bed (Fig. 11c), overcomes many of these problems and is thus to be preferred in PTO technologies. In this case, the PDH thermodynamic equilibrium is shifted by the in situ conversion of produced propylene over the Met catalyst. This should also result in a more homogeneous thermal profile along the reactor bed, with the Met catalyst working as a heat reservoir, in place of (or in addition to) commonly used inert materials. Moreover, provided that PDH and Met catalyst bodies are produced with different density, they may be separately cycled through different regenerators using a concept similar to FLOTU/Tsinghua bi-catalyst technology (Fig. 5). On the downside, at this and closer contact states, the PDH catalyst is exposed not only to the presence of propylene, but also of other olefins, such as ethylene and butene. As a result, ethylene selectivity can be lower than expected due to thermodynamically favored re-hydrogenation of ethylene to ethane, by reaction with H2 produced during dehydrogenation. On the other hand, the Met catalyst portion at the reactor mouth is exposed almost solely to propane and is thus not efficiently used. Despite these relatively minor drawbacks, we expect this and the next two operation modes to be the most relevant for future PTO technologies.
PDH and Met catalysts powders may also be combined in a single catalyst body, either randomly mixed or in the form of core–shell spheres or extrudates (Fig. 11d, mixed in extrudate). In this configuration, the two catalysts are in close contact, and may not be separated at any operational stage. Core–shell extrudates have been shown to control catalyst activity and selectivity in e.g. branched olefin production in Fischer Tropsch, due to control of migration of the reactants to the tandem active sites, and are thus interesting to implement in PTO technologies.116 Recently, Sripinun et al. have shown that indeed catalyst bodies composed of a dehydrogenation catalysts shell and a metathesis catalysts (Met@PHD) core outperform in the PTO reaction physically mixed single-catalyst extrudates, mixed in extrudate, and inverted (dehydrogenation catalyst core and metathesis shell – PDH@Met) alternatives, yielding propylene with 43 mol% yield, improving by 25–35% from the base case.117 Reaction simulations based on reaction kinetics and mathematical models in gPROMS were developed and used to orient catalyst bodies preparation, finally hindering side reactions such as hydrogenolysis and ethylene hydrogenation, and allowing to reduce the amount of (Pt-based and thus expensive) dehydrogenation catalyst needed by 59%.
Another interesting option is the use of catalytic formulations where PDH and Met catalysts are supported on the same support (Fig. 11e, mixed on support): in this case it is important to understand how the two components can influence each other and how much they are compatible, by studying for instance the possible formation of Pt–W alloys, the formation of Cr–W mixed oxides upon annealing or the electron transfer between different components. The metathesis activity of WOx was indeed shown to be increased by the addition of promoters such as Nb2O5 (patent no. CN1618515A), while were deactivated by impurities such as Fe and alkali in the range of 100 ppm. Notably, Fe and Nb are both good catalysts/promoters in PDH.26 Mixed phases and alloying can be monitored by in situ and operando X-ray absorption spectroscopy, drawing inspiration from work on Pt–In: changes in Pt–In alloys were monitored under O2–H2 cycles, investigating the redox behavior and the sintering mechanism of the alloy under typical regeneration–reactivation conditions used in PDH.118 The results showed that during oxidation, the Pt–In alloy simultaneously undergoes decomposition and Pt oxidation, the latter being the rate determining step, and that oxidation requires higher thermal activation compared to reduction. On the other side, the reduction step has a lower activation energy and involves the reduction of platinum and indium oxides with subsequent formation of the highly stable Pt13In9 alloy. The authors also observed that after 60 catalytic cycles sintering of Pt nanoparticle takes place, as also observed for PtSn alloys. Synchrotron X-ray characterization techniques can especially provide a unique tool to collect precious information about bimetallic catalyst structure, alloying mechanism and stability under different reaction atmospheres, that are of utmost importance for tweaking catalyst design.
The following trends were established: (i) at low loadings and high surface areas, an increasing isoelectric point of the support leads to higher monochromate/dichromate ratios; (ii) polychromates tend to form at higher loadings and lower surface areas; (iii) anchoring on hydroxyl groups during calcination can lead to different structures depending on all the discussed parameters; (iv) supported Cr ions are mobile, and the oxide support can be envisioned as a ligand controlling the redox properties of the supported Cr ions.122,123
The support has an influence also on the oxidation state of the reduced Cr form: at 4 wt% loading, Cr2+ is mainly obtained as reduced form on silica, while a Cr2+/Cr3+ mixture is obtained on mixed alumina/silica supports and Cr3+ species are obtained on alumina. The authors ascribed the support effect on reduction to the fact that the presence of silica might favor reducibility compared to alumina, leading to a stronger interaction of the active site with the support. Similarly, the isoelectric point (IEP) of the support affects the amount of surface dichromate, which increases as the IEP decreases.124 Indeed, modifications of CrOx/SiO2 catalysts have been recently proposed either where chromium oxide is modified with zirconia or ceria, or where the support is different from silica, in order to enhance catalytic performance by tuning Cr redox properties.125,126
Therefore, compatibility between dehydrogenation and metathesis catalysts (i.e., formation of secondary phases) and anchoring over a suitable support, is another field that to the best of our knowledge has been unexplored so far and is important to study, especially when considering different catalyst contact states in the reactor. The parameters involved prevent us from establishing an overall “compatibility table”, as this will depend on all the previously discussed factors. It is thus necessary to address in future studies how such factors influence catalyst deactivation, products selectivity and competitive reactions. This would be of great interest to design catalysts that are stable towards deactivation.
• ReOx/alumina was regenerated by treatments with H2O2, or bases (e.g., NaOH, KOH, or NH4OH);
• WOx and MoOx catalysts were regenerated by water at 50 °C or by steam at 170 °C (e.g., patent no. WO2009013964)
• H2, syngas or even the reaction products of C2–5 alkane dehydrogenation was shown to be as or more effective than coke combustion in O2-containing gasses (e.g., patent no WO2002000341A2);
• 550–700 °C treatments in olefin-containing atmosphere (e.g. propylene) were reported to increase the activity of silica-supported WOx and MOx catalysts of 2–3 orders of magnitude.127
• MoOx catalysts were activated by CH4 atmosphere at 600 °C or above for 30 min.128
From the above list, it is clear that the PDH product mixture should be actually beneficial for Met catalysts (e.g., H2 and propylene, CH4 produced by cracking,) and that much more should be studied in terms of activation and regeneration of Met catalysts. For instance, pre-reduced WOx catalysts were recently shown to be active for PDH themselves.129 The reduction temperature during pretreatment was also shown to influence the Cr2+/Cr3+ ratio in Cr-based PDH catalysts, depending on the support.130 New operando studies have started to unravel the complex interdependence of type of support, Cr loading and pre-treatment conditions on the catalysts structure and activity. Further work should be devoted in understanding redox cycling during subsequent catalysts reactivation and regeneration, to guide the development of better catalysts and fine-tuned modes of plant operation. We foresee that the development of better PTO technologies will be a driver for such future investigation.
5. Conclusions and perspectives
Thanks to favorable market conditions and the development of on-purpose PDH technologies, the propane value chain has seen a booming growth, to become a main branch of the (poly)olefins industry. With many more PDH installation planned in the coming 5 years, especially in China, interest is growing also in alternative methods for propane valorization, targeting specific olefinic products. PTO is a promising technology to expand the capabilities of the PDH reaction to higher alpha olefins, but the method is still in its infancy and increasing the olefins yield will be crucial for commercialization. Operating the PDH and Met reactions in tandem adds a layer of complexity, but can improve the overall olefins yield by shifting the equilibrium via in situ propylene conversion. While the standalone Met reaction does not present major obstacles, the PDH step introduces some intrinsic limitations, such as low equilibrium conversion and competing coking and cracking reactions.Despite fast advances in PDH technologies in the last 5 years, practical alternatives to the classic Pt–Sn/Al2O3 (Oleflex) and CrOx/Al2O3 (Catofin) catalysts are still lacking. Pt and Cr-based catalysts have been optimized by using different supports, introducing promoters, and changing their preparation methods, but their stability under PDH conditions is still relatively low, so that constant regeneration is required. Metal oxides such as VOx, GaOx, FeOx single atom catalysts and nanocarbons have been proposed and studied as PDH catalysts, but so far they did not outperform traditional Pt- or Cr-based materials.23 The rich chemistry of V (many oxidation states), Ga (hydrides formation) and nanocarbons (heteroatoms and functionalities) represents a new frontier in exploration of PDH catalyst design, and will require efforts in theoretical calculations, simulations and operando characterization to develop appropriate structure–activity relationships.131 Recent studies showed that Fe, Co and Sn are also promising alternative for PDH, with Sn showing surprisingly stable performance under PDH.132
While side reactions and deactivation for PDH are well known in literature, for metathesis catalysts detailed literature exists about homogeneous catalysts,48,133,134 but not so much for heterogeneous supported catalysts in terms of structure–properties correlations. In situ and operando spectroscopic studies are relatively new,56 but were proven crucial for rational catalysts design to approach the activity of organometallic catalysts.95,101,105 A series of in situ and operando spectroscopic studies related to WOx, ReOx and MoOx supported on silica or alumina were carried out, with the aim to shed light on the structure–performance relationships of the catalysts at work.95,101,102,105,107,135,136 Moreover, precise design of these supported catalysts can be obtained using advanced strategies for surface sites engineering, such as Surface Organometallic Chemistry (SOMC).28,109 Advances in SS-NMR and operando vibrational and electronic spectroscopy, on the other hand, can provide deep insights on reaction intermediates and help unravel activation, reaction and deactivation mechanisms.97
For the metathesis reaction, WOx/SiO2 is the optimal catalyst due to its resistance to trace quantities of oxygenates in the feedstock, the long catalyst lifetime compared to MoOx and ReOx supported metathesis catalysts, the easy regeneration to remove coke deposits and the absence of catalyst embrittlement upon periodic regeneration.56,95 However, about 80 wt% of W worldwide is mined in China, which makes tungsten a strategic metal, and its sustainability critical.137 Fine-tuning W oxidation state and carbide formation using promoters and support effects will be key to make the catalysts more durable under PTO conditions and to reduce the amount of W needed. Increasing W recycling rates (already at 37%) and exploring alternatives, such as Mo and Re or combinations thereof, should also be pursued to increase resilience of Met and PTO technologies.
Catalyst design is an important part in the development of more sustainable and efficient chemical processes, but it has to be complemented with smart reactor design and operations. In PDH, thermodynamic constraints are heavily limiting the possibly achievable yield of propylene. According to the Le Chatelier principle, removing products from the reactor will shift the equilibrium of any reaction to the right. Therefore, using H2 membrane reactors in PDH, one could dramatically increase the yield of propylene. The main issue with this approach is that membranes have limited thermal stability, but recent advances have extended the temperature window for their operation, and the European MACBETH consortium is currently working to apply H2 membrane technology to PDH.138 According to some of the partners, sustainability, investment and operating costs can also be reduced using membrane reactors. Emerging membrane materials (such as metal or covalent organic frameworks and polymers with intrinsic microporosity) are also promising for olefin/paraffin separation, crucial for DH and other petrochemical processes, as alternative to the state-of-the-art cryogenic distillation, which is an extremely energy-intensive process.139
Oxidative dehydrogenation (OPDH) of propane 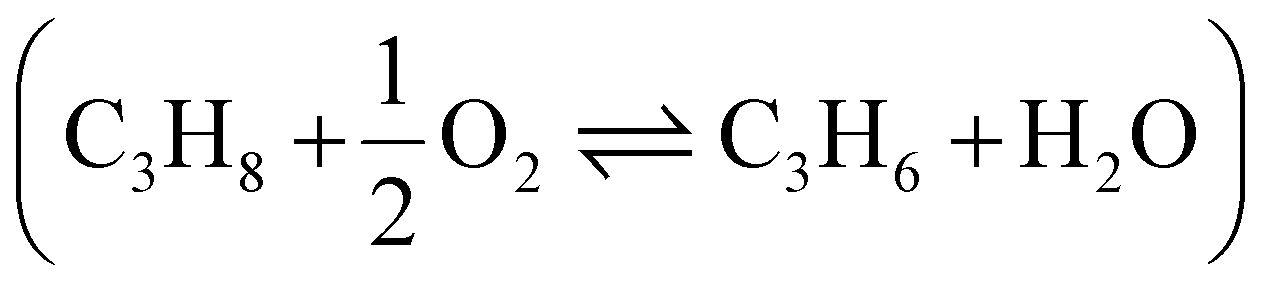 is another emerging technology with the potential to overcome the equilibrium limitations of nonoxidative alkane dehydrogenation.140 Oxidizing hydrogen atoms to water makes the reaction exothermic (ΔH0298 = −117 kJ mol−1) and thus not equilibrium limited, and it allows to produce olefins at lower temperatures and higher pressures, resulting in lower coke formation. Nonetheless, paraffin overoxidation to CO and CO2 is a major drawback.72,73,140 Using CO2 as an oxidizer has recently been proposed improve carbon utilization, while avoiding over-oxidation.55 OPDH is however competing with dry reforming of propane (3CO2 + C3H8 ⇌ 6CO + 4H2), so that selective catalysts are needed. Boron-based catalysts have been proposed for the OPDH reaction using oxygen as an oxidizer (e.g., B-substituted zeolites141,142 and hexagonal BN or BN nanotubes143). Future studies should focus on the application of such catalysts in combination with CO2 as an oxidant and in deriving structure–activity relationship principles to guide catalyst design. Finally, combining CO2 valorization to olefin production is interesting to evaluate in the context of CO2-neutral plastic production, in line with worldwide goals to increase plastic sustainability.144,145
is another emerging technology with the potential to overcome the equilibrium limitations of nonoxidative alkane dehydrogenation.140 Oxidizing hydrogen atoms to water makes the reaction exothermic (ΔH0298 = −117 kJ mol−1) and thus not equilibrium limited, and it allows to produce olefins at lower temperatures and higher pressures, resulting in lower coke formation. Nonetheless, paraffin overoxidation to CO and CO2 is a major drawback.72,73,140 Using CO2 as an oxidizer has recently been proposed improve carbon utilization, while avoiding over-oxidation.55 OPDH is however competing with dry reforming of propane (3CO2 + C3H8 ⇌ 6CO + 4H2), so that selective catalysts are needed. Boron-based catalysts have been proposed for the OPDH reaction using oxygen as an oxidizer (e.g., B-substituted zeolites141,142 and hexagonal BN or BN nanotubes143). Future studies should focus on the application of such catalysts in combination with CO2 as an oxidant and in deriving structure–activity relationship principles to guide catalyst design. Finally, combining CO2 valorization to olefin production is interesting to evaluate in the context of CO2-neutral plastic production, in line with worldwide goals to increase plastic sustainability.144,145
Combining emerging PDH technologies with Met in the PTO reaction holds promise to achieve better performances. However, it is unknown how Met catalysts will react to such different compositions in the feed. Unraveling the role of competitive reactions and catalyst deactivation mechanisms occurring during the PTO process will be fundamental to overcome the existing challenges. To this end, cooperation between academia and industry is the key point to make these technological advances possible: a synergistic approach is required to gain an insight into the fundamental science of these heterogeneous catalysts, establish structure–property correlations and apply this knowledge to large scale processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. M. W. thanks SCG Chemicals for financial support for this project. This work was supported by the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), a NWO Gravitation program funded by the Ministry of Education, Culture and Science of the government of the Netherlands, as well as by the Advanced Research Consortium Chemical Building Block Consortium (ARC CBBC).References
- Ethylene (ET), 2020 World Market Outlook and Forecast up to 2029, https://mcgroup.co.uk/researches/ethylene, accessed April 13, 2020.
- Propylene: 2020 World Market Outlook and Forecast up to 2029, https://mcgroup.co.uk/researches/propylene, accessed April 13, 2020.
- 2020 Global Ethylene Market and Propylene Industry Research Analysis by TBRC, https://www.prnewswire.com/news-releases/2020-global-ethylene-market-and-propylene-industry-research-analysis-by-tbrc-301014325.html, accessed April 13, 2020.
- L. Morey, Plug pulled on CKPC's $4.5B PDH project, Sherwood Park News, 2020, https://www.sherwoodparknews.com/news/local-news/plug-pulled-on-ckpcs-4-5b-pdh-project, accessed November 26, 2020.
- Alpha Olefin Market – Industry Analysis and Forecast (2019–2026) – By Product, By Application And By Region, https://www.maximizemarketresearch.com/market-report/alpha-olefin-market/13674/, accessed April 13, 2020.
- Z. Nawaz, Light alkane dehydrogenation to light olefin technologies: a comprehensive review, Rev. Chem. Eng., 2015, 31, 413–436, DOI:10.1515/revce-2015-0012.
- The Changing Landscape of Hydrocarbon Feedstocks for Chemical Production, National Academies Press, ed. J. Alper, Washington, DC, 2016 DOI:10.17226/23555.
- J. J. H. B. Sattler, J. Ruiz-martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides, Chem. Rev., 2014, 114, 10613–10653, DOI:10.1021/cr5002436.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Shale Gas Revolution: An Opportunity for the Production of Biobased Chemicals?, Angew. Chem., Int. Ed., 2013, 52, 11980–11987, DOI:10.1002/anie.201305058.
- Analysis: Naphtha's challenge in the age of petrochemical feedstock boom, https://www.hellenicshippingnews.com/analysis-naphthas-challenge-in-the-age-of-petrochemical-feedstock-boom/, accessed April 13, 2020.
- J. Zalasiewicz, C. N. Waters, J. A. Ivar do Sul, P. L. Corcoran, A. D. Barnosky, A. Cearreta, M. Edgeworth, A. Gałuszka, C. Jeandel, R. Leinfelder, J. R. McNeill, W. Steffen, C. Summerhayes, M. Wagreich, M. Williams, A. P. Wolfe and Y. Yonan, The geological cycle of plastics and their use as a stratigraphic indicator of the Anthropocene, Anthropocene, 2016, 13, 4–17, DOI:10.1016/j.ancene.2016.01.002.
- E. Clippinger and B. F. Mulaskey, Group viii noble metal,tin and solid inorganic refractory metal oxide catalyst composites and their use in hydrocarbon dehydrogenations, US pat., US3531543A, 1970 Search PubMed.
- R. E. Rausch, Dehydrogenation with a catalytic composite containing platinum,rhenium and tin, US pat., US3584060, 1971 Search PubMed.
- J. C. Hayes, Multicomponent dehydrogenation catalyst, US pat., US3878131, 1975 Search PubMed.
- T. Imai and H. Abrevaya, Dehydrogenation catalyst compositions and method of their preparation, US pat., US4595673, 1986 Search PubMed.
- R. S. Parthasarathi and S. S. Alabduljabbar, HS-FCC High-severity fluidized catalytic cracking: a newcomer to the FCC family, Appl. Petrochem. Res., 2014, 4, 441–444, DOI:10.1007/s13203-014-0087-5.
- D. Sanfilippo, Dehydrogenation of paraffins: Key technology for petrochemicals and fuels, CATTECH, 2000, 4, 56–73, DOI:10.1023/a:1011947328263.
- D. Sanfilippo and I. Miracca, Dehydrogenation of paraffins: Synergies between catalyst design and reactor engineering, Catal. Today, 2006, 111, 133–139, DOI:10.1016/j.cattod.2005.10.012.
- L. F. Heckelsberg and R. L. Banks, Dehydrogenation and disproportionation of hydrocarbons, US pat., US3445541A, 1969, https://patents.google.com/patent/US3445541A/en Search PubMed.
- R. Burnett, Disproportionation of saturated hydrocarbons employing a catalyst that comprises platinum and tungsten, US pat., US3856876A, 1974, https://patents.google.com/patent/US3856876A/en Search PubMed.
- J. Mol, Industrial applications of olefin metathesis, J. Mol. Catal. A: Chem., 2004, 213, 39–45, DOI:10.1016/j.molcata.2003.10.049.
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, New Trends in Olefin Production, Engineering, 2017, 3, 171–178, DOI:10.1016/J.ENG.2017.02.006.
- Z.-P. Hu, D. Yang, Z. Wang and Z.-Y. Yuan, State-of-the-art catalysts for direct dehydrogenation of propane to propylene, Chinese, J. Catal., 2019, 40, 1233–1254, DOI:10.1016/S1872-2067(19)63360-7.
- J. M. Basset, C. Copéret, D. Soulivong, M. Taoufik and J. Thivolle-Cazat, From olefin to alkane metathesis: A historical point of view, Angew. Chem., Int. Ed., 2006, 45, 6082–6085, DOI:10.1002/anie.200602155.
- N. Gholampour, M. Yusubov and F. Verpoort, Investigation of the preparation and catalytic activity of supported Mo, W, and Re oxides as heterogeneous catalysts in olefin metathesis, Catal. Rev.: Sci. Eng., 2016, 58, 113–156, DOI:10.1080/01614940.2015.1100871.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Propane dehydrogenation: catalyst development, new chemistry, and emerging technologies, Chem. Soc. Rev., 2021, 50, 3315–3354, 10.1039/D0CS00814A.
- C. Deraedt, M. D’Halluin and D. Astruc, Metathesis reactions: Recent trends and challenges, Eur. J. Inorg. Chem., 2013, 4881–4908, DOI:10.1002/ejic.201300682.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Bridging the Gap between Industrial and Well-Defined Supported Catalysts, Angew. Chem., Int. Ed., 2018, 57, 6398–6440, DOI:10.1002/anie.201702387.
- M. P. Atkins and G. R. Evans, Review of current processes and innovations, Erdoel, Erdgas, Kohle, 1995, 111, 271–274 CAS.
- D. E. Resasco and G. L. Haller, Catalysis: Catalytic Dehydrogenation of Lower Alkanes, Royal Society of Chemistry, 2007, ch. 9, vol. 11 Search PubMed.
- P. Chaiyavech, Commercialization of the World's First Oleflex Unit, J. R. Inst. Thail., 2002, 27, 1–7 Search PubMed.
- B. Hapes, KBR Announces a New Propane Dehydrogenation Technology, Wire Release, 2018, https://www.kbr.com/en/insights-events/press-release/kbr-announces-new-propane-dehydrogenation-technology, accessed November 26, 2020.
- M. Heinritz-Adrian, S. Wenzel and F. Youssef, Advanced propane dehydrogenation, Pet. Technol. Q., 2008, 13, 1–8 Search PubMed.
- T. Degnan, Fluidized bed propane dehydrogenation technologies challenge established fixed and moving bed processes, Focus Catal. Res., 2019, 4, 1–2, DOI:10.1016/j.focat.2019.03.001.
- V. Fridman and M. A. Urbancic, Dehydrogenation process with heat generating material, US pat., US2015/0259265 A1, 2015 Search PubMed.
- References: Organic chemicals, Dehydrogenation – The STAR process, https://www.thyssenkrupp-industrial-solutions.com/en/references/chemical-industry/organic-chemicals, accessed May 13, 2020.
- Plasteurope.com, Agreement with ThyssenKrupp Industrial Solutions on development of PDH technology, 2020, https://www.plasteurope.com/news/UPDATE_-_BASF_t245569/, accessed November 26, 2020.
- Linde set to license C3/C4 dehydrogenation process, 1992, https://www.icis.com/explore/resources/news/1992/03/09/15012/linde-set-to-license-c3-c4-dehydrogenation-process/.
- N. Basta, ACHEMA 2000: Linde AG Updates Its Propane-to-Propylene Process Development, 2000, https://www.hydrocarbononline.com/doc/achema-2000-linde-ag-updates-its-propane-to-p-0001.
- UOP Oleflex process for light olefin production, https://pet-oil.blogspot.com/2012/10/uop-oleflex-process-for-light-olefin.html, accessed May 14, 2020.
- Snamprogetti wins Sabic MTBE contract, 1994, https://www.icis.com/explore/resources/news/1994/01/08/27434/snamprogetti-wins-sabic-mtbe-contract/.
- I. Miracca and L. Piovesan, Light paraffins dehydrogenation in a fluidized bed reactor, Catal. Today, 1999, 52, 259–269, DOI:10.1016/S0920-5861(99)00080-2.
- Z. Nawaz, Y. Chu, W. Yang, X. Tang, Y. Wang and F. Wei, Study of propane dehydrogenation to propylene in an integrated fluidized bed reactor using Pt-Sn/Al-SAPO-34 novel catalyst, Ind. Eng. Chem. Res., 2010, 49, 4614–4619, DOI:10.1021/ie902043w.
- Z. Nawaz and F. Wei, Integrated bi-modal fluidized bed reactor for butane dehydrogenation to corresponding butylenes, Chem. Eng. J., 2014, 238, 249–253, DOI:10.1016/j.cej.2013.07.103.
- W. Keim, Oligomerization of ethylene to α-olefins: Discovery and development of the shell higher olefin process (SHOP), Angew. Chem., Int. Ed., 2013, 52, 12492–12496, DOI:10.1002/anie.201305308.
- R. L. Banks and G. C. Bailey, Olefin Disproportionation. A New Catalytic Process, Ind. Eng. Chem. Prod. Res. Dev., 1964, 3, 170–173, DOI:10.1021/i360011a002.
- L. F. Heckelsberg, Conversion of Olefins, US pat., US3365513, 1965, https://dl.acm.org/doi/10.1145/178951.178972 Search PubMed.
- Y. Chauvin, Olefin Metathesis: The Early Days (Nobel Lecture), Angew. Chem., Int. Ed., 2006, 45, 3740–3747, DOI:10.1002/anie.200601234.
- M. Gierada and J. Handzlik, Active sites formation and their transformations during ethylene polymerization by the Phillips CrOx/SiO2 catalyst, J. Catal., 2017, 352, 314–328, DOI:10.1016/j.jcat.2017.05.025.
- M. Gierada and J. Handzlik, Computational insights into reduction of the Phillips CrOx/SiO2 catalyst by ethylene and CO, J. Catal., 2018, 359, 261–271, DOI:10.1016/j.jcat.2018.01.014.
- G. Parkinson, Reviving up for alkylation, Chem. Eng., 2001, 108, 27–33 CAS.
- Lummus/CB&I Hexene-1 from C4 Process (Comonomer Production Technology), 2010, https://ihsmarkit.com/products/chemical-technology-pep-reviews-lummus-cb-i-hexene-from-c4-2010.html.
- F. Kapteijn, E. Homburg and J. C. Mol, Thermodynamics of the metathesis of propene into ethene and 2-butene, J. Chem. Thermodyn., 1983, 15, 147–152, DOI:10.1016/0021-9614(83)90153-2.
- M. Taghizadeh and F. Aghili, Recent advances in membrane reactors for hydrogen production by steam reforming of ethanol as a renewable resource, Rev. Chem. Eng., 2019, 35, 377–392, DOI:10.1515/revce-2017-0083.
- E. Gomez, S. Kattel, B. Yan, S. Yao, P. Liu and J. G. Chen, Combining CO2 reduction with propane oxidative dehydrogenation over bimetallic catalysts, Nat. Commun., 2018, 9, 1398, DOI:10.1038/s41467-018-03793-w.
- S. Lwin and I. E. Wachs, Olefin metathesis by supported metal oxide catalysts, ACS Catal., 2014, 4, 2505–2520, DOI:10.1021/cs500528h.
- G. Wang, X. Zhu and C. Li, Recent Progress in Commercial and Novel Catalysts for Catalytic Dehydrogenation of Light Alkanes, Chem. Rec., 2020, 20, 604–616, DOI:10.1002/tcr.201900090.
- J. C. Bricker, Advanced Catalytic Dehydrogenation Technologies for Production of Olefins, Top. Catal., 2012, 55, 1309–1314, DOI:10.1007/s11244-012-9912-1.
- B. V. Vora, Development of dehydrogenation catalysts and processes, Top. Catal., 2012, 55, 1297–1308, DOI:10.1007/s11244-012-9917-9.
- B. M. Weckhuysen and R. A. Schoonheydt, Alkane dehydrogenation over supported chromium oxide catalysts, Catal. Today, 1999, 51, 223–232, DOI:10.1016/S0920-5861(99)00047-4.
- M. L. Yang, Y. A. Zhu, X. G. Zhou, Z. J. Sui and D. Chen, First-principles calculations of propane dehydrogenation over PtSn catalysts, ACS Catal., 2012, 2, 1247–1258, DOI:10.1021/cs300031d.
- M. L. Yang, J. Zhu, Y. A. Zhu, Z. J. Sui, Y. Da Yu, X. G. Zhou and D. Chen, Tuning selectivity and stability in propane dehydrogenation by shaping Pt particles: A combined experimental and DFT study, J. Mol. Catal. A: Chem., 2014, 395, 329–336, DOI:10.1016/j.molcata.2014.08.008.
- A. J. Bukvic, A. L. Burnage, G. J. Tizzard, A. J. Martínez-Martínez, A. I. McKay, N. H. Rees, B. E. Tegner, T. Krämer, H. Fish, M. R. Warren, S. J. Coles, S. A. Macgregor and A. S. Weller, A Series of Crystallographically Characterized Linear and Branched σ-Alkane Complexes of Rhodium: From Propane to 3-Methylpentane, J. Am. Chem. Soc., 2021, 143, 5106–5120, DOI:10.1021/jacs.1c00738.
- Y. Wang, P. Hu, J. Yang, Y. A. Zhu and D. Chen, C–H bond activation in light alkanes: A theoretical perspective, Chem. Soc. Rev., 2021, 50, 4299–4358, 10.1039/d0cs01262a.
- I. Takigawa, K. I. Shimizu, K. Tsuda and S. Takakusagi, Machine-learning prediction of the d-band center for metals and bimetals, RSC Adv., 2016, 6, 52587–52595, 10.1039/c6ra04345c.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Modification of the surface electronic and chemical properties of Pt(111) by subsurface 3d transition metals, J. Chem. Phys., 2004, 120, 10240–10246, DOI:10.1063/1.1737365.
- L. Nykänen and K. Honkala, Density functional theory study on propane and propene adsorption on Pt(111) and PtSn alloy surfaces, J. Phys. Chem. C, 2011, 115, 9578–9586, DOI:10.1021/jp1121799.
- M. S. Pereira and M. A. C. Nascimento, Theoretical study of the dehydrogenation reaction of ethane catalyzed by zeolites containing non-framework gallium species: The 3-step mechanism × the 1-step concerted mechanism, Chem. Phys. Lett., 2005, 406, 446–451, DOI:10.1016/j.cplett.2005.02.108.
- V. Z. Fridman and R. Xing, Investigating the CrOx/Al2O3 dehydrogenation catalyst model: II. Relative activity of the chromium species on the catalyst surface, Appl. Catal., A, 2017, 530, 154–165, DOI:10.1016/j.apcata.2016.11.024.
- B. M. Weckhuysen, R. A. Schoonheydt, J. M. Jehng, I. E. Wachs, S. J. Cho, R. Ryoo, S. Kijlstra and E. Poels, Combined DRS-RS-EXAFS-XANES-TPR study of supported chromium catalysts, J. Chem. Soc., Faraday Trans., 1995, 91, 3245–3253, 10.1039/FT9959103245.
- A. Hakuli, A. Kytökivi and A. O. I. Krause, Dehydrogenation of i-butane on CrOx/Al2O3 catalysts prepared by ALE and impregnation techniques, Appl. Catal., A, 2000, 190, 219–232, DOI:10.1016/S0926-860X(99)00310-5.
- M. Botavina, C. Barzan, A. Piovano, L. Braglia, G. Agostini, G. Martra and E. Groppo, Insights into Cr/SiO2 catalysts during dehydrogenation of propane: An operando XAS investigation, Catal. Sci. Technol., 2017, 7, 1690–1700, 10.1039/c7cy00142h.
- M. A. Botavina, Y. A. Agafonov, N. A. Gaidai, E. Groppo, V. Cortés Corberán, A. L. Lapidus and G. Martra, Towards efficient catalysts for the oxidative dehydrogenation of propane in the presence of CO2: Cr/SiO2 systems prepared by direct hydrothermal synthesis, Catal. Sci. Technol., 2016, 6, 840–850, 10.1039/c5cy00998g.
- M. L. Yang, Y. A. Zhu, C. Fan, Z. J. Sui, D. Chen and X. G. Zhou, DFT study of propane dehydrogenation on Pt catalyst: Effects of step sites, Phys. Chem. Chem. Phys., 2011, 13, 3257–3267, 10.1039/c0cp00341g.
- Z. J. Zhao, C. C. Chiu and J. Gong, Molecular understandings on the activation of light hydrocarbons over heterogeneous catalysts, Chem. Sci., 2015, 6, 4403–4425, 10.1039/c5sc01227a.
- M. L. Yang, Y. A. Zhu, C. Fan, Z. J. Sui, D. Chen and X. G. Zhou, Density functional study of the chemisorption of C1, C2 and C3 intermediates in propane dissociation on Pt(111), J. Mol. Catal. A: Chem., 2010, 321, 42–49, DOI:10.1016/j.molcata.2010.01.017.
- R. M. Watwe, R. D. Cortright, J. K. Nørskov and J. A. Dumesic, Theoretical Studies of Stability and Reactivity of C2 Hydrocarbon Species on Pt Clusters, Pt(111), and Pt(211), J. Phys. Chem. B, 2000, 104, 2299–2310, DOI:10.1021/jp993202u.
- R. D. Cortright, R. M. Watwe and J. A. Dumesic, Ethane hydrogenolysis over platinum selection and estimation of kinetic parameters, J. Mol. Catal. A: Chem., 2000, 163, 91–103, DOI:10.1016/S1381-1169(00)00402-7.
- G. Peng, D. Gerceker, M. Kumbhalkar, J. A. Dumesic and M. Mavrikakis, Ethane dehydrogenation on pristine and AlOx decorated Pt stepped surfaces, Catal. Sci. Technol., 2018, 8, 2159–2174, 10.1039/c8cy00398j.
- A. Valcárcel, J. M. Ricart, A. Clotet, F. Illas, A. Markovits and C. Minot, Theoretical study of dehydrogenation and isomerisation reactions of propylene on Pt(111), J. Catal., 2006, 241, 115–122, DOI:10.1016/j.jcat.2006.04.022.
- J. A. Singh, J. Nelson, R. C. Vicente, B. C. Scott and S. L. van Bokhoven, Electronic structure of alumina-supported monometallic Pt and bimetallic PtSn catalysts under hydrogen and carbon monoxide environment, Phys. Chem. Chem. Phys., 2010, 12, 5668–5677 RSC.
- J. Hagen, Industrial Catalysis: A Practical Approach, Wiley-VCH, Weinheim, 2nd edn, 2015 Search PubMed.
- Z. Lian, S. Ali, T. Liu, C. Si, B. Li and D. S. Su, Revealing the Janus Character of the Coke Precursor in the Propane Direct Dehydrogenation on Pt Catalysts from a kMC Simulation, ACS Catal., 2018, 8, 4694–4704, DOI:10.1021/acscatal.8b00107.
- M. Santhosh Kumar, N. Hammer, M. Rønning, A. Holmen, D. Chen, J. C. Walmsley and G. Øye, The nature of active chromium species in Cr-catalysts for dehydrogenation of propane: New insights by a comprehensive spectroscopic study, J. Catal., 2009, 261, 116–128, DOI:10.1016/j.jcat.2008.11.014.
- M. Huš, D. Kopač and B. Likozar, Kinetics of non-oxidative propane dehydrogenation on Cr2O3 and the nature of catalyst deactivation from first-principles simulations, J. Catal., 2020, 386, 126–138, DOI:10.1016/j.jcat.2020.03.037.
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, A combined in situ time-resolved UV-Vis, Raman and high-energy resolution X-ray absorption spectroscopy study on the deactivation behavior of Pt and PtSn propane dehydrogenation catalysts under industrial reaction conditions, J. Catal., 2010, 276, 268–279, DOI:10.1016/j.jcat.2010.09.018.
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Subnanometer Bimetallic Platinum–Zinc Clusters in Zeolites for Propane Dehydrogenation, Angew. Chem., 2020, 132, 19618–19627, DOI:10.1002/ange.202003349.
- J. Zhu, R. Osuga, R. Ishikawa, N. Shibata, Y. Ikuhara, J. N. Kondo, M. Ogura, J. Yu, T. Wakihara, Z. Liu and T. Okubo, Ultrafast Encapsulation of Metal Nanoclusters into MFI Zeolite in the Course of Its Crystallization: Catalytic Application for Propane Dehydrogenation, Angew. Chem., Int. Ed., 2020, 59, 19669–19674, DOI:10.1002/anie.202007044.
- Q. Y. Chang, Q. Yin, F. Ma, Y. A. Zhu, Z. J. Sui, X. G. Zhou, D. Chen and W. K. Yuan, Tuning adsorption and catalytic properties of α-Cr2O3 and ZnO in propane dehydrogenation by creating oxygen vacancy and doping single Pt atom: A comparative first-principles study, Ind. Eng. Chem. Res., 2019, 58, 10199–10209, DOI:10.1021/acs.iecr.9b01143.
- H. H. Kristoffersen and H. Metiu, Interaction between Monomeric Vanadium Oxide Clusters Supported on Titania and Its Influence on Their Reactivity, J. Phys. Chem. C, 2016, 120, 13610–13621, DOI:10.1021/acs.jpcc.6b04216.
- T. Otroshchenko, S. Sokolov, M. Stoyanova, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, ZrO2-Based Alternatives to Conventional Propane Dehydrogenation Catalysts: Active Sites, Design, and Performance, Angew. Chem., Int. Ed., 2015, 54, 15880–15883, DOI:10.1002/anie.201508731.
- E. A. Pidko, V. B. Kazansky, E. J. M. Hensen and R. A. van Santen, A comprehensive density functional theory study of ethane dehydrogenation over reduced extra-framework gallium species in ZSM-5 zeolite, J. Catal., 2006, 240, 73–84, DOI:10.1016/j.jcat.2006.03.011.
- Z. P. Hu, C. Chen, J. T. Ren and Z. Y. Yuan, Direct dehydrogenation of propane to propylene on surface-oxidized multiwall carbon nanotubes, Appl. Catal., A, 2018, 559, 85–93, DOI:10.1016/j.apcata.2018.04.017.
- J. J. H. B. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Platinum-promoted Ga/Al2O3 as highly active, selective, and stable catalyst for the dehydrogenation of propane, Angew. Chem., Int. Ed., 2014, 53, 9251–9256, DOI:10.1002/anie.201404460.
- S. Lwin, Y. Li, A. I. Frenkel and I. E. Wachs, Nature of WOx Sites on SiO2 and Their Molecular Structure–Reactivity/Selectivity Relationships for Propylene Metathesis, ACS Catal., 2016, 6, 3061–3071, DOI:10.1021/acscatal.6b00389.
- C. Copéret, Design and understanding of heterogeneous alkene metathesis catalysts, Dalton Trans., 2007, 5498, 10.1039/b713314f.
- C. Copéret, Z. J. Berkson, K. W. Chan, J. de Jesus Silva, C. P. Gordon, M. Pucino and P. A. Zhizhko, Olefin metathesis: what have we learned about homogeneous and heterogeneous catalysts from surface organometallic chemistry?, Chem. Sci., 2021, 12, 3092–3115, 10.1039/d0sc06880b.
- M. de Boer, A. J. van Dillen, D. C. Koningsberger, J. W. Geus, M. A. Vuurman and I. E. Wachs, Remarkable spreading behavior of molybdena on silica catalysts. An in situ EXAFS-Raman study, Catal. Lett., 1991, 11, 227–239, DOI:10.1007/BF00764089.
- A. Spamer, T. I. Dube, D. J. Moodley, C. Van Schalkwyk and J. M. Botha, Application of a WO3/SiO2 catalyst in an industrial environment: Part II, Appl. Catal., A, 2003, 255, 133–142, DOI:10.1016/S0926-860X(03)00535-0.
- C. Van Schalkwyk, A. Spamer, D. J. Moodley, T. Dube, J. Reynhardt, J. M. Botha and H. C. M. Vosloo, Factors that could influence the activity of a WO3/SiO2 catalyst: Part III, Appl. Catal., A, 2003, 255, 143–152, DOI:10.1016/S0926-860X(03)00536-2.
- K. Amakawa, J. Kröhnert, S. Wrabetz, B. Frank, F. Hemmann, C. Jäger, R. Schlögl and A. Trunschke, Active Sites in Olefin Metathesis over Supported Molybdena Catalysts, ChemCatChem, 2015, 7, 4059–4065, DOI:10.1002/cctc.201500725.
- S. Lwin and I. E. Wachs, Reaction Mechanism and Kinetics of Olefin Metathesis by Supported ReOx/Al2O3 Catalysts, ACS Catal., 2016, 6, 272–278, DOI:10.1021/acscatal.5b02233.
- Z. Cheng and C. S. Lo, Propagation of Olefin Metathesis to Propene on WO3 Catalysts: A Mechanistic and Kinetic Study, ACS Catal., 2015, 5, 59–72, DOI:10.1021/cs500531b.
- S. Zhang, D. F. Consoli, S. K. Shaikh and Y. Román-Leshkov, Effects of WO3 nanoparticle size on ethylene-butene metathesis activity, Appl. Catal., A, 2019, 580, 53–58, DOI:10.1016/j.apcata.2019.04.019.
- J. G. Howell, Y. P. Li and A. T. Bell, Propene Metathesis over Supported Tungsten Oxide Catalysts: A Study of Active Site Formation, ACS Catal., 2016, 6, 7728–7738, DOI:10.1021/acscatal.6b01842.
- K. Gayapan, S. Sripinun, J. Panpranot, P. Praserthdam and S. Assabumrungrat, Effects of calcination and pretreatment temperatures on the catalytic activity and stability of H2-treated WO3/SiO2 catalysts in metathesis of ethylene and 2-butene, RSC Adv., 2018, 8, 28555–28568, 10.1039/c8ra04949a.
- S. Lwin, Y. Li, A. I. Frenkel and I. E. Wachs, Activation of Surface ReOx Sites on Al2O3 Catalysts for Olefin Metathesis, ACS Catal., 2015, 5, 6807–6814, DOI:10.1021/acscatal.5b01944.
- K. Amakawa, S. Wrabetz, J. Kröhnert, G. Tzolova-Müller, R. Schlögl and A. Trunschke, In situ generation of active sites in olefin metathesis, J. Am. Chem. Soc., 2012, 134, 11462–11473, DOI:10.1021/ja3011989.
- S. R. Docherty, L. Rochlitz, P.-A. Payard and C. Copéret, Heterogeneous alkane dehydrogenation catalysts investigated via a surface organometallic chemistry approach, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC.
- C. Copéret, W. C. Liao, C. P. Gordon and T. C. Ong, Active Sites in Supported Single-Site Catalysts: An NMR Perspective, J. Am. Chem. Soc., 2017, 139, 10588–10596, DOI:10.1021/jacs.6b12981.
- B. Mitra, X. Gao, I. E. Wachs, A. M. Hirt and G. Deo, Characterization of supported rhenium oxide catalysts: Effect of loading, support and additives, Phys. Chem. Chem. Phys., 2001, 3, 1144–1152, 10.1039/b007381o.
- P. E. Blackburn, M. Hoch and H. L. Johnston, The Vaporization of Molybdenum and Tungsten Oxides, J. Phys. Chem., 1958, 62, 769–773, DOI:10.1021/j150565a001.
- A. Uchagawkar, A. Ramanathan, Y. Hu and B. Subramaniam, Highly dispersed molybdenum containing mesoporous silicate (Mo-TUD-1) for olefin metathesis, Catal. Today, 2020, 343, 215–225, DOI:10.1016/j.cattod.2019.03.073.
- J. F. Wu, A. Ramanathan, A. Biancardi, A. M. Jystad, M. Caricato, Y. Hu and B. Subramaniam, Correlation of Active Site Precursors and Olefin Metathesis Activity in W-Incorporated Silicates, ACS Catal., 2018, 8, 10437–10445, DOI:10.1021/acscatal.8b03263.
- J. F. Wu, A. Ramanathan, R. Kersting, A. Jystad, H. Zhu, Y. Hu, C. P. Marshall, M. Caricato and B. Subramaniam, Enhanced Olefin Metathesis Performance of Tungsten and Niobium Incorporated Bimetallic Silicates: Evidence of Synergistic Effects, ChemCatChem, 2020, 12, 2004–2013, DOI:10.1002/cctc.201902131.
- J. Bao, J. He, Y. Zhang, Y. Yoneyama and N. Tsubaki, A core/shell catalyst produces a spatially confined effect and shape selectivity in a consecutive reaction, Angew. Chem., Int. Ed., 2008, 47, 353–356, DOI:10.1002/anie.200703335.
- S. Sripinun, K. Lorattanaprasert, K. Gayapan, K. Suriye, S. Wannakao, P. Praserthdam and S. Assabumrungrat, Design of hybrid pellet catalysts of WO3/Si-Al and PtIn/hydrotalcite via dehydrogenation and metathesis reactions for production of olefins from propane, Chem. Eng. Sci., 2021, 229, 116025, DOI:10.1016/j.ces.2020.116025.
- M. Filez, H. Poelman, E. A. Redekop, V. V. Galvita, K. Alexopoulos, M. Meledina, R. K. Ramachandran, J. Dendooven, C. Detavernier, G. Van Tendeloo, O. V. Safonova, M. Nachtegaal, B. M. Weckhuysen and G. B. Marin, Kinetics of Lifetime Changes in Bimetallic Nanocatalysts Revealed by Quick X-ray Absorption Spectroscopy, Angew. Chem., Int. Ed., 2018, 57, 12430–12434, DOI:10.1002/anie.201806447.
- W. Tolek, K. Suriye, P. Praserthdam and J. Panpranot, Effect of preparation method on the Pt-In modified Mg(Al)O catalysts over dehydrogenation of propane, Catal. Today, 2020, 358, 100–108 CrossRef CAS.
- W. Tolek, K. Suriye, P. Praserthdam and J. Panpranot, Enhanced Stability and Propene Yield in Propane Dehydrogenation on PtIn/Mg(Al)O Catalysts with Various In Loadings, Top. Catal., 2018, 61, 1624–1632, DOI:10.1007/s11244-018-1008-0.
- F. D. Hardcastle and I. E. Wachs, Raman spectroscopy of chromium oxide supported on Al2O3, TiO2 and SiO2: a comparative study, J. Mol. Catal., 1988, 46, 173–186, DOI:10.1016/0304-5102(88)85092-2.
- S. De Rossi, G. Ferraris, S. Fremiotti, E. Garrone, G. Ghiotti, M. C. Campa and V. Indovina, Propane Dehydrogenation on Chromia/Silica and Chromia/Alumina Catalysts, J. Catal., 1994, 148, 36–46, DOI:10.1006/jcat.1994.1183.
- B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Surface Chemistry and Spectroscopy of Chromium in Inorganic Oxides, Chem. Rev., 1996, 96, 3327–3350, DOI:10.1021/cr940044o.
- B. M. Weckhuysen, A. A. Verberckmoes, A. L. Buttiens and R. A. Schoonheydt, Diffuse reflectance spectroscopy study of the thermal genesis and molecular structure of chromium-supported catalysts, J. Phys. Chem., 1994, 98, 579–584, DOI:10.1021/j100053a037.
- S. Han, Y. Zhao, T. Otroshchenko, Y. Zhang, D. Zhao, H. Lund, T. H. Vuong, J. Rabeah, U. Bentrup, V. A. Kondratenko, U. Rodemerck, D. Linke, M. Gao, H. Jiao, G. Jiang and E. V. Kondratenko, Unraveling the Origins of the Synergy Effect between ZrO2 and CrOx in Supported CrZrOx for Propene Formation in Nonoxidative Propane Dehydrogenation, ACS Catal., 2020, 10, 1575–1590, DOI:10.1021/acscatal.9b05063.
- K. H. Kang, T. H. Kim, W. C. Choi, Y. K. Park, U. G. Hong, D. S. Park, C. J. Kim and I. K. Song, Dehydrogenation of propane to propylene over CrOy-CeO2-K2O/γ-Al2O3 catalysts: Effect of cerium content, Catal. Commun., 2015, 72, 68–72, DOI:10.1016/j.catcom.2015.09.009.
- K. Ding, A. Gulec, A. M. Johnson, T. L. Drake, W. Wu, Y. Lin, E. Weitz, L. D. Marks and P. C. Stair, Highly Efficient Activation, Regeneration, and Active Site Identification of Oxide-Based Olefin Metathesis Catalysts, ACS Catal., 2016, 6, 5740–5746, DOI:10.1021/acscatal.6b00098.
- P. Michorczyk, A. Wegrzyniak, A. Wegrzynowicz and J. Handzlik, Simple and Efficient Way of Molybdenum Oxide-Based Catalyst Activation for Olefins Metathesis by Methane Pretreatment, ACS Catal., 2019, 9, 11461–11467, DOI:10.1021/acscatal.9b03714.
- Y. Yun, J. R. Araujo, G. Melaet, J. Baek, B. S. Archanjo, M. Oh, A. P. Alivisatos and G. A. Somorjai, Activation of Tungsten Oxide for Propane Dehydrogenation and Its High Catalytic Activity and Selectivity, Catal. Lett., 2017, 147, 622–632, DOI:10.1007/s10562-016-1915-2.
- B. M. Weckhuysen, L. M. De Ridder and R. A. Schoonheydt, A quantitative diffuse reflectance spectroscopy study of supported chromium catalysts, J. Phys. Chem., 1993, 97, 4756–4763, DOI:10.1021/j100120a030.
- M. Filez, E. A. Redekop, J. Dendooven, R. K. Ramachandran, E. Solano, U. Olsbye, B. M. Weckhuysen, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Formation and Functioning of Bimetallic Nanocatalysts: The Power of X-ray Probes, Angew. Chem., Int. Ed., 2019, 58, 13220–13230, DOI:10.1002/anie.201902859.
- G. Wang, H. Zhang, Q. Zhu, X. Zhu, X. Li, H. Wang, C. Li and H. Shan, Sn-containing hexagonal mesoporous silica (HMS) for catalytic dehydrogenation of propane: An efficient strategy to enhance stability, J. Catal., 2017, 351, 90–94, DOI:10.1016/j.jcat.2017.04.018.
- R. H. Grubbs, Olefin-Metathesis Catalysts for the Preparation of Molecules and Materials (Nobel Lecture), Angew. Chem., Int. Ed., 2006, 45, 3760–3765, DOI:10.1002/anie.200600680.
- R. R. Schrock, Multiple metal-carbon bonds for catalytic metathesis reactions (Nobel lecture), Angew. Chem., Int. Ed., 2006, 45, 3748–3759, DOI:10.1002/anie.200600085.
- K. Amakawa, L. Sun, C. Guo, M. Hävecker, P. Kube, I. E. Wachs, S. Lwin, A. I. Frenkel, A. Patlolla, K. Hermann, R. Schlögl and A. Trunschke, How strain affects the reactivity of surface metal oxide catalysts, Angew. Chem., Int. Ed., 2013, 52, 13553–13557, DOI:10.1002/anie.201306620.
- A. Chakrabarti, M. E. Ford, D. Gregory, R. Hu, C. J. Keturakis, S. Lwin, Y. Tang, Z. Yang, M. Zhu, M. A. Bañares and I. E. Wachs, A decade+ of operando spectroscopy studies, Catal. Today, 2017, 283, 27–53, DOI:10.1016/j.cattod.2016.12.012.
- A. H. Tkaczyk, A. Bartl, A. Amato, V. Lapkovskis and M. Petranikova, Sustainability evaluation of essential critical raw materials: Cobalt, niobium, tungsten and rare earth elements, J. Phys. D: Appl. Phys., 2018, 51, 203001 CrossRef.
- M. Binotti, G. Di Marcoberardino and G. Manzolini, Bionico – biogas membrane reformer for deceNtralIzed hydrogen production, Impact, 2020, 46–48 CrossRef.
- J. Hou, P. Liu, M. Jiang, L. Yu, L. Li and Z. Tang, Olefin/paraffin separation through membranes: From mechanisms to critical materials, J. Mater. Chem. A, 2019, 7, 23489–23511, 10.1039/c9ta06329c.
- L. M. Madeira and M. F. Portela, Catalytic oxidative dehydrogenation of n -butane, Catal. Rev., 2002, 44, 247–286, DOI:10.1081/CR-120001461.
- H. Zhou, X. Yi, Y. Hui, L. Wang, W. Chen, Y. Qin, M. Wang, J. Ma, X. Chu, Y. Wang, X. Hong, Z. Chen, X. Meng, H. Wang, Q. Zhu, L. Song, A. Zheng and F.-S. Xiao, Isolated boron in zeolite for oxidative dehydrogenation of propane, Science, 2021, 372, 76–80, DOI:10.1126/science.abe7935.
- N. R. Altvater, R. W. Dorn, M. C. Cendejas, W. P. McDermott, B. Thomas, A. J. Rossini and I. Hermans, B-MWW Zeolite: The Case Against Single-Site Catalysis, Angew. Chem., Int. Ed., 2020, 59, 6546–6550, DOI:10.1002/anie.201914696.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Selective oxidative dehydrogenation of propane to propene using boron nitride catalysts, Science, 2016, 354, 1570–1573, DOI:10.1126/science.aaf7885.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Beyond Mechanical Recycling: Giving New Life to Plastic Waste, Angew. Chem., Int. Ed., 2020, 59, 15402 CrossRef CAS PubMed.
- Z. Zhao, K. Chong, J. Jiang, K. Wilson, X. Zhang and F. Wang, Low-carbon roadmap of chemical production: A case study of ethylene in China, Renewable Sustainable Energy Rev., 2018, 97, 580–591, DOI:10.1016/j.rser.2018.08.008.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cs00357g |
| This journal is © The Royal Society of Chemistry 2021 |